 Adrien Moreau1 Dagmar I. Keller2
Adrien Moreau1 Dagmar I. Keller2 Hai Huang1 Véronique Fressart3 Christian Schmied4
Hai Huang1 Véronique Fressart3 Christian Schmied4 Quadiri Timour5
Quadiri Timour5 Mohamed Chahine1,6*
Mohamed Chahine1,6*- 1 Centre de Recherche, Institut Universitaire en Santé Mentale de Québec, Quebec City, QC, Canada
- 2 Department of Internal Medicine and Cardiology, University Hospital Zurich, Zurich, Switzerland
- 3 UF Cardiogénétique et Myogénétique, Groupe Hospitalier Pitié-Salpêtrière, Paris, France
- 4 Department of Cardiology, University Hospital Zurich, Zurich, Switzerland
- 5 INSERM ERI22, Université Claude Bernard de Lyon, Lyon, France
- 6 Department of Medicine, Université Laval, Quebec City, QC, Canada
The human cardiac sodium channel Nav1.5 encoded by the SCN5A gene plays a critical role in cardiac excitability and the propagation of action potentials. Nav1.5 dysfunctions due to mutations cause cardiac diseases such as the LQT3 form of long QT syndrome, conduction disorders, and Brugada syndrome (BrS). They have also recently been associated with dilated cardiomyopathy. BrS is characterized by coved ST-segment elevation on surface ECGs and lethal ventricular arrhythmias in an apparently structurally normal heart. Nav1.5 mutations that cause BrS result in a loss of channel function. Our aim was to functionally characterize two novel Nav1.5 mutations (A124D and V1378M) in BrS patients. Wild-type (WT) and mutant Nav1.5 channels were expressed in tsA201 cells in the presence of the β1-auxiliary subunit. The patch-clamp technique and immunocytochemistry approaches were used to study the mutant channels and their cellular localization. The two mutant channels displayed a dramatic reduction in current density but had normal gating properties. The reduction in current density was caused by the retention of channel proteins in the endoplasmic reticulum (ER). Mutant channel retention could be partially reversed by incubating transfected cells at 25°C and by treating them with mexiletine (for V1378M but not A124D), or with curcumin or thapsigargin, two drugs that target ER resident proteins. It is likely that the clinical phenotypes observed in these two BrS patients were related to a surface expression defect caused by ER retention.
Introduction
The cardiac Nav1.5 channel encoded by the SCN5A gene plays a critical role in cardiac excitability and conduction. It is responsible for the rapid upstroke of the action potential (AP) caused by the rapid entry of Na+ ions into cardiac myocytes. Dysfunctions of this channel cause diseases such as type 3 long QT syndrome (LQT), Brugada syndrome (BrS), and conduction disorders. More recently, SCN5A mutations have been associated with dilated cardiomyopathy (Amin et al., 2010). BrS is characterized by a coved-type ST-segment elevation in V1 through V3 derivations on surface ECGs and a right bundle branch block pattern in a morphologically normal heart. This pathology leads to high mortality due to malignant ventricular arrhythmias (Bhar-Amato et al., 2010). Coved-type ST-segment elevations are characteristic of BrS type 1 ECGs, whereas saddle-back ST-segment elevations correspond to BrS type 2 and 3 ECGs (Wilde et al., 2002). Type 1 BrS ECGs can be unmasked by potent Na+ channel blockers and other factors such as fever (Keller et al., 2006).
BrS is an inherited arrhythmic syndrome related to mutations in the SCN5A gene in 10–30% of cases (Antzelevitch, 2006; Zimmer and Surber, 2008). Others genes such as SNC1B and GDP1L as well as genes that code for calcium channel subunits are also linked to this syndrome (Antzelevitch, 2006; London et al., 2007; Watanabe et al., 2008; Zimmer and Surber, 2008).
A number of SCN5A mutations that lead to BrS cause trafficking defects (Baroudi et al., 2001; Valdivia et al., 2004). Various treatments can correct these defects in vivo, including incubating transfected cells at low temperature (25°C) or treating the cells with mexiletine (class I antiarrhythmic), or curcumin and thapsigargin, two drugs that act on the SERCA pump in the endoplasmic reticulum (ER) membrane (Baroudi et al., 2002; Valdivia et al., 2004; Keller et al., 2005). All these treatments target various aspect of the retention mechanism of channels such as heat shock proteins (HSP) and calnexin, a reticular chaperone (Zhou et al., 1999; Egan et al., 2004).
We studied the biophysical properties and cellular localization of two mutations in BrS patients. Two highly conserved residues, alanine 124 and valine 1378, were substituted by an aspartic acid and a methionine, respectively. We investigated how these two mutations, which are located in different regions on the channel, caused a loss of function. We observed a dramatic reduction in Na+ currents following the transient expression of mutant Nav1.5 channels in tsA201 cells. A reduction in the incubation temperature and a number of chemical treatments partially restored the Na+ currents, suggesting that the mutations caused ER retention.
Materials and Methods
Clinical Evaluations and Molecular Genetics
Written informed consent was obtained from the two participating index patients in accordance with the study protocol, which was approved by the ethics committee of the University Hospital of Basel, Switzerland. The investigation conformed to the principles set out in the Declaration of Helsinki. Index patients underwent detailed clinical assessments, including 12-lead ECGs and specific examinations related to the clinical presentation of BrS.
Genomic DNA was extracted from peripheral lymphocytes isolated from the two patients. The coding exons of SCN5A were amplified by PCR using primers designed with intronic flanking sequences according to the gene sequence described by Wang et al. (1996). Denaturating high performance liquid chromatography (dHPLC) was performed on DNA amplification products using at least one temperature condition. Abnormal dHPLC profiles were analyzed by cycle sequencing both strands of the exons using a big dye termination mix and an automated laser fluorescent DNA sequencer (ABI Prism 377, Applied Biosystems, Foster City, CA, USA). No mutations on SCN5A gene were found in the chromosomes of 200 normal control subjects.
Cell Cultures
The tsA201 cell line is a modified human embryonic kidney HEK-293 cell line stably transfected by the simian virus 40 large T antigen that can promote the replication of viral promoter-containing constructs (Huang et al., 2011). The cells were transfected with WT or mutant human Nav1.5 cDNA (2–5 μg) and the human β1-subunit (2–5 μg) using the calcium-phosphate method as previously described (Deschenes et al., 2000) and were grown at 37°C in a 5% CO2 humidified atmosphere in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml of penicillin G, and 10 mg/ml of streptomycin (GIBCO-BRL Life Technologies, Burlington, ON, Canada). The human Na+ channel β1-subunit and CD8 were inserted in the pIRES bicistronic vector in the form of pCD8-IRES-β1. For the patch-clamp experiments, the cells were incubated for 2 min in an extracellular solution containing anti-CD8-coated beads 2 days after transfection (Dynabeads CD8, Dynal Biotech, Oslo, Norway).
Patch-Clamp Electrophysiology
Macroscopic Na+ currents from transfected cells were recorded using the whole-cell configuration of the patch-clamp technique as previously described (Huang et al., 2011).
Solutions and Reagents
The patch pipettes were filled with a solution containing 35 mM NaCl, 105 mM CsF, 10 mM EGTA, and 10 mM Cs-HEPES. The pH was adjusted to 7.4 using 1 N CsOH. The bath solution contained 150 mM NaCl, 2 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM Na–HEPES. The pH was adjusted to pH 7.4 using 1 N NaOH. The liquid junction potential between the patch pipette and the bath solution was corrected by −7 mV. Mexiletine, curcumin, and thapsigargin (Sigma) were used at 400 μM (overnight incubation), 25 μM (4-h incubation), and 1 μM (2-h incubation), respectively. Curcumin and thapsigargin were diluted in DMSO. Consequently, when indicated cells were incubated in 0.25% of DMSO.
Immunocytochemistry
Transfected tsA201 cells were fixed in a PBS/4% paraformaldehyde/4% sucrose solution for 20 min and were then permeabilized using 0.1% Triton in PBS/1% BSA. They were then incubated with rabbit anti-FLAG Ab-1 primary antibody (1:1000) to detect FLAG-tagged Nav1.5 channels (Fisher Scientific). The tag epitope was inserted in the S5 to S6 extracellular loop of domain I in the WT and mutant channels. A mouse anti-calnexin antibody (1:200, StressGen Biotechnologies Corp., Victoria, BC, Canada) was used to label the ER. The permeabilized cells were then incubated with F(ab′)2-conjugated goat anti-rabbit (1:250) and F(ab′)2-conjugated goat anti-mouse (1:250) secondary antibodies (Invitrogen). The cells were also incubated with DAPI (5 μg/ml) to label the nucleus. The cells were examined using a confocal Zeiss LSM 510 microscope equipped with a 63× oil immersion objective with a 1.4 numerical aperture and the appropriate lasers.
Statistical Analysis
Data are expressed as means ± SEMs. When indicated, a t-test or one-way ANOVA was performed using software in SigmaPlot (Jandel Scientific Software). Differences were considered statistically significant at p < 0.05 (*/#), p < 0.01 (**/##), and p < 0.001 (***/###).
Results
Clinical Phenotypes of Brugada Syndrome
Patient 1
A 32-year-old male patient was admitted to the emergency care unit due to syncope during walking with spontaneous recovery. At the time, the patient suffered from a febrile infection with gastric symptoms. The 12-lead ECG with a BrS type 1 pattern at admission with a fever (39°C) is shown in the upper panel of Figure 1A. The administration of an antipyretic changed the BrS type 1 ECG to a BrS type 3 ECG pattern (Figure 1A, lower panel). The patient’s history revealed that he had had long-standing, multiple, neurally mediated, vasovagal syncopes in the past. To differentiate the syncope from one of neurally mediated origin, we performed a head up tilt table test (Figure 1C), which was mixed positive after 3 min at a tilt angle of 60° (a syncope with a drop in blood pressure to 75/49 mmHg and a 13-s asystole that disappeared spontaneously when the patient was returned to the supine position (Moya et al., 2009). No ventricular arrhythmias were observed during tilt table test or recovery phase. No implantable cardioverter-defibrillator (ICD) was implanted. Follow-up to date has been uneventful. There was no family history of sudden cardiac death (SCD), but the patient’s mother and brother had also suffered from vasovagal syncopes. Their ECGs were normal, but had never been recorded while they had a fever or after a flecainide challenge. They refused genotyping.
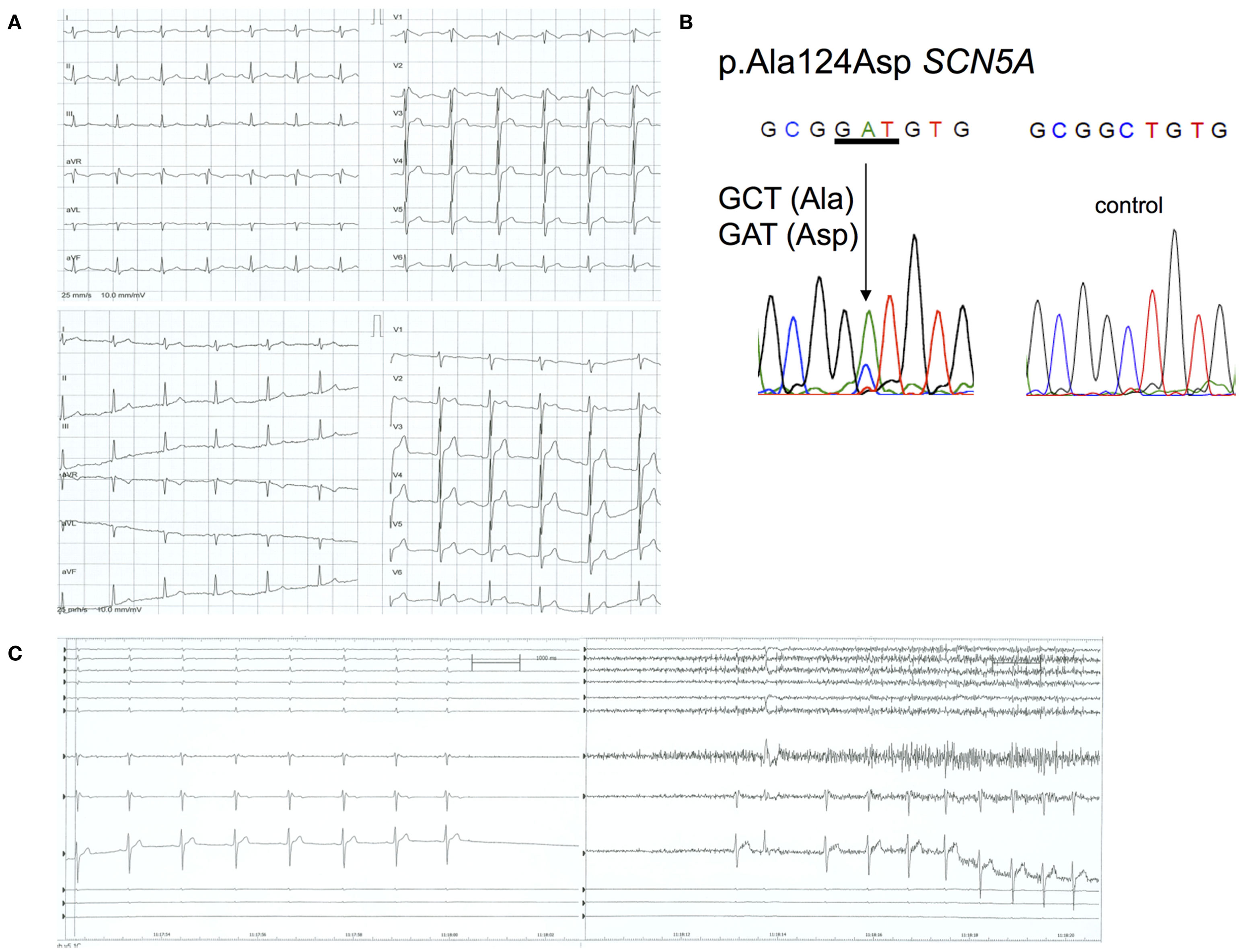
Figure 1. ECG, tilt table, and genotype: Patient 1. (A) Upper panel: 12-lead ECG while febrile (39°C) showing a BrS type 1 ECG with coved ST-segment elevation in the right precordial leads; lower panel: 12-lead ECG while afebrile showing a BrS type 3 ECG with saddle-back ST-segment elevation in the right precordial leads. (B) c.371 C > A resulted in a heterozygous p.A124D mutation in SCN5A. (C) Asystole during tilt table testing, with spontaneous recovery of the sinus rhythm in the supine position.
Patient 2
A 60-year-old male patient was admitted to the hospital due to a first episode of violent syncope with a basilar skull fracture and subarachnoid hemorrhage. The transthoracic echocardiography was normal, and left heart catheterization revealed normal coronary arteries. A 12-lead ECG showed a typical BrS type 1 pattern with coved ST-segment elevation (Figure 2A). The patient had no fever during the syncope or ECG recording. The family history revealed that a paternal uncle had unexpectedly died at the age of 64. An ICD was implanted in the patient. Follow-up to date has been uneventful. Further family screening was not performed. The patient has no children.
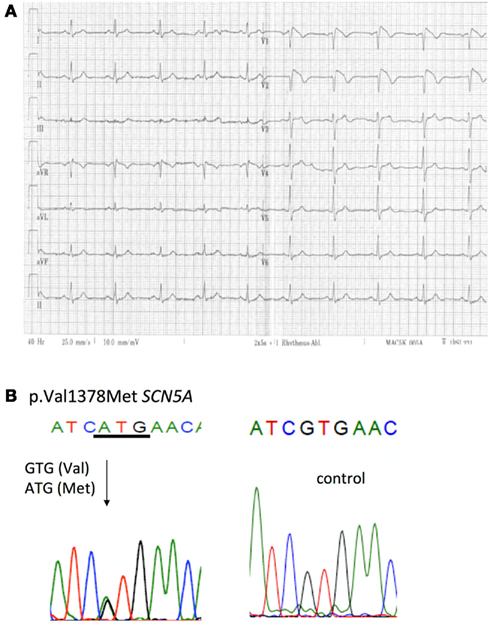
Figure 2. ECG and genotype: Patient 2. (A) 12-lead ECG showing a BrS type 1 ECG with coved ST-segment elevation in the right precordial leads. (B) c.4132 G > A resulted in a heterozygous p.V1378M mutation in SCN5A.
Identification of the P.A124D and P.V1378M Mutations in SCN5A
We identified two novel missense mutation (p.A124D and p.V1378M) in the SCN5A genes of the two index patients (Figures 1B and 2B). Patient 1 had a heterozygous C to A base change at position 371 in exon 3 of SCN5A that resulted in the substitution of an alanine (GCT) for an asparagine (GAT) in residue 124 (Figure 1B). The mutation was located in the C-terminal region of the Nav1.5 channel (Figure 3). Patient 2 had a heterozygous G to A base change at position 4132 in exon 23 that resulted in the substitution of a valine (GTG) for a methionine (ATG) in residue 1378 (Figure 2B). The mutation was located in the third domain between the fifth and sixth transmembrane segment of the Nav1.5 channel (Figure 3). Both mutations affected residues that are highly conserved in Na+ channels in many species (data not shown).
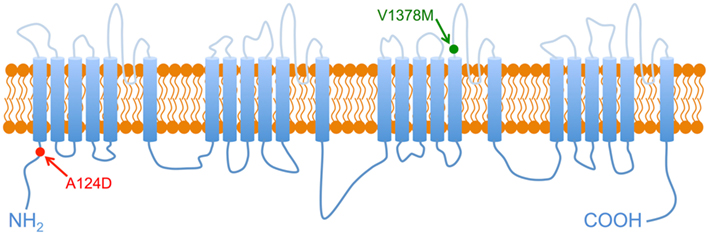
Figure 3. Nav1.5 2-D structure. Schematic representation of Nav1.5 structure, arrows point to the localization of the two mutations A124D and V1378M.
Biophysical Properties of the A124D and V1378M Mutants
To understand the clinical phenotypes of the two patients, the biophysical properties of the WT and mutant channels were studied. Macroscopic Na+ currents were recorded from transfected tsA201 cells. A dramatic reduction in current amplitudes was observed with both mutants. Examples of whole-cell current traces (A) and I–V relationship curves (B) are shown in Figure 4. The non-significant variation of the reversal potentials of WT (29.9 ± 1.2 mV, n = 10), A124D (35.6 ± 2.4 mV, n = 14), and V1378M (30.4 ± 1.8 mV, n = 15) indicated that the mutations had no impact on channel selectivity, as was also evidenced by the I–V curves (Figure 4B). The current reduction was quantified by measuring the peak current density, which was 563.4 ± 217.0 pA/pF (n = 10) for the WT channel, and 132.2 ± 17.0 pA/pF (n = 14) and 183.6 ± 15.6 pA/pF (n = 15) for the A124D and V1378M channels, respectively (Figure 5A).
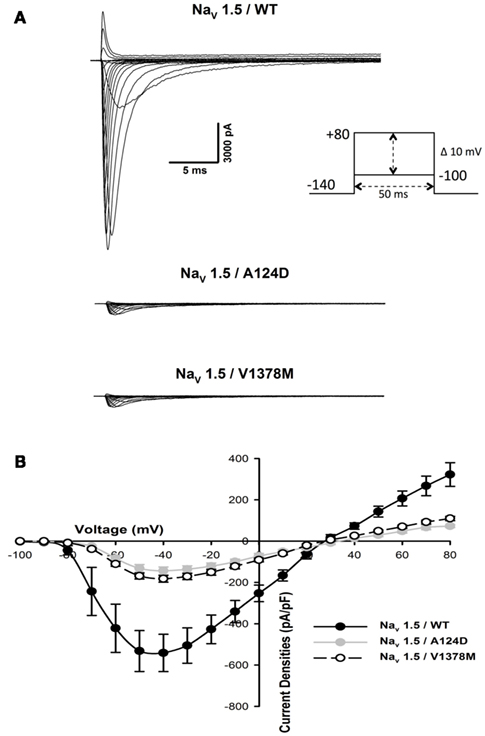
Figure 4. Expression of WT, A124D, and V1378M Nav1.5 channels in tsA201 cells. (A) Representative whole-cell current traces of WT (top), A124D (middle), and V1378M (bottom) channels. Currents were elicited using a voltage-clamp protocol where depolarizing pulses were applied for 50 ms from −100 to +80 mV in 10 mV increments. (B) Current–voltage (I–V) relationships of WT (•, n = 10), A124D ( , n = 14), and V1378M (◯, n = 15) channels. All currents were normalized to cellular membrane capacitance to give the current density (pA/pF).
, n = 14), and V1378M (◯, n = 15) channels. All currents were normalized to cellular membrane capacitance to give the current density (pA/pF).
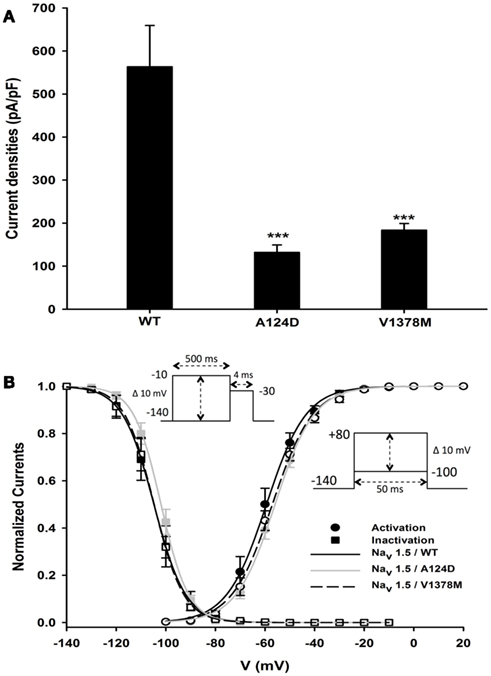
Figure 5. Current densities and gating properties of WT, A124D, and V1378M Nav1.5 channels. (A) Histogram summarizing the current densities of the WT, A124D, and V1378M Nav1.5 channels. Current densities were significantly lower for A124D (132.2 ± 36.8 pA/pF, n = 14) and V1378M (183.6 ± 33.5 pA/pF, n = 15) than for WT (563.4 ± 217.0 pA/pF, n = 10; ***, p < 0.001). (B) Voltage dependence of steady-state activation (•) or inactivation (●) for the WT (-), A124D ( ), and V1378M (- - -) Nav1.5 channels. Activation curves were generated using a standard Boltzmann distribution [G(V)/Gmax = 1/(1 + exp(−(V − V1/2)/k))] to give the V1/2 and k values listed in Table 1. Inactivation currents were obtained by applying conditioning pre-pulses to membrane potentials ranging from a holding potential of −140 to −10 mV for 500 ms in 10 mV increments and were then measured using a 4-ms pulse to −30 mV at each step (see protocol in inset). The recorded inactivation values were fitted to a standard Boltzmann equation [I(V)/Imax = 1/(1 + exp((V − V1/2)/k)) + C] to give the values listed in Table 1.
), and V1378M (- - -) Nav1.5 channels. Activation curves were generated using a standard Boltzmann distribution [G(V)/Gmax = 1/(1 + exp(−(V − V1/2)/k))] to give the V1/2 and k values listed in Table 1. Inactivation currents were obtained by applying conditioning pre-pulses to membrane potentials ranging from a holding potential of −140 to −10 mV for 500 ms in 10 mV increments and were then measured using a 4-ms pulse to −30 mV at each step (see protocol in inset). The recorded inactivation values were fitted to a standard Boltzmann equation [I(V)/Imax = 1/(1 + exp((V − V1/2)/k)) + C] to give the values listed in Table 1.
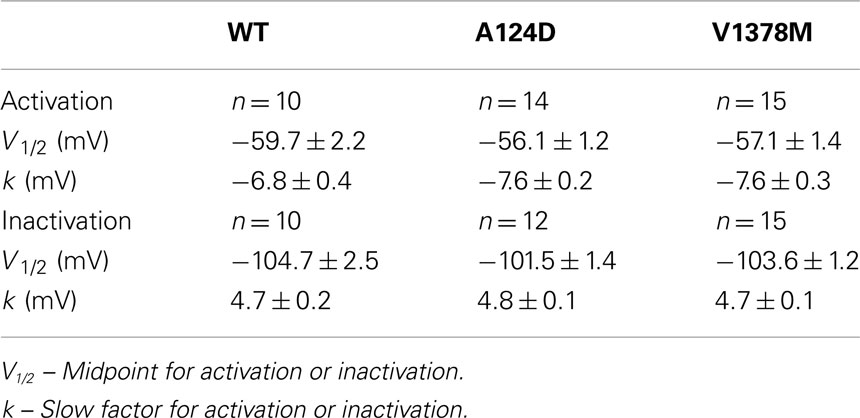
Table 1. Biophysical properties of the WT, A124D, and V1378M Nav1.5 channels.
The WT and mutant channels had similar gating properties (Figure 5B; Table 1). There were no statistically significant differences in activation and inactivation parameters (Table 1).
A124D and V1378M Current Densities Were Rescued by Low Temperature and Drug Treatments
tsA201 cells expressing A124D or V1378M mutant channels were incubated at 25°C for 24 h or with curcumin, thapsigargin, or mexiletine in an attempt to restore Na+ currents. The results are shown in Figure 6, Tables 2 and 3. All the treatments are known to restore trafficking defects induced by mutations, albeit in different ways. Curcumin and thapsigargin were diluted in dimethylsulfoxide (DMSO). No differences were observed between DMSO-treated cells and control cells. Nevertheless, mutant cells incubated with DMSO alone were used as controls. The peak current densities of the DMSO-treated A124D and control cells were 102.4 ± 16.8 pA/pF (n = 11) and 132.2 ± 17.0 pA/pF (n = 14), respectively. The peak current densities of the DMSO-treated and untreated V1378M cells were 163.1 ± 20.8 pA/pF (n = 8) and 183.6 ± 15.6 pA/pF (n = 15), respectively. Curcumin and thapsigargin partially restored the currents. The peak current density of the A124D mutant was 200.0 ± 24.6 pA/pF (n = 16) with curcumin and 214.3 ± 19.9 pA/pF (n = 7) with thapsigargin. The peak current density of the V1378M mutant was 337.6 ± 31.2 pA/pF (n = 8) with curcumin and 262.4 ± 19.9 pA/pF (n = 13) with thapsigargin.
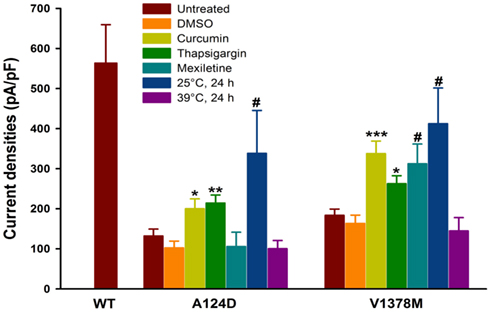
Figure 6. Maximum current densities of WT, A124D, and V1378M Nav1.5 channels in tsA201 cells following various treatments. Histogram summarizing the current densities of the WT, A124D, and V1378M mutant channels treated or not with 0.25% DMSO, 25 μM curcumin (4 h), 1 μM thapsigargin (2 h), 400 μM mexiletine (24 h), at 25°C for 24 h or at 39°C for 24 h. The current densities of WT, 563.4 ± 217.0 pA/pF (n = 10), for A124D, 132.2 ± 17.0 pA/pF (n = 14) without DMSO, 102.4 ± 16.8 pA/pF (n = 11) with DMSO, 200.0 ± 24.6 pA/pF (n = 16) with curcumin, 214.3 ± 19.9 pA/pF (n = 7) with thapsigargin, 105.8 ± 35.5 pA/pF (n = 5) with mexiletine, and 338.6 ± 107.0 pA/pF (n = 6) when the cells were incubated at 25°C and 100.2 ± 20.6 pA/pF (n = 7) when the cells were incubated at 39°C. Significant differences were observed between DMSO-treated and curcumin- or thapsigargin-treated A124D cells (*p < 0.05; **p < 0.01) but not between DMSO-treated and control cells. A significant difference was observed between untreated cells grown at 37°C and cells grown at 25°C (#p < 0.05). There was no difference between untreated and mexiletine-treated or 39°C-incubated cells. Untreated, DMSO-, curcumin-, thapsigargin-, mexiletine-, 25°C-, and 39°C-treated V1378M cells had current densities of 183.6 ± 15.6 pA/pF (n = 15), 163.1 ± 20.8 pA/pF (n = 8), 337.6 ± 31.2 pA/pF (n = 8), 262.4 ± 19.9 pA/pF (n = 13), 312.4 ± 49.1 pA/pF (n = 5), 412.5 ± 88.9 pA/pF (n = 6), and 144.6 ± 33.4 pA/pF (n = 7) respectively. Significant differences were observed between the DMSO-treated and curcumin- or thapsigargin-treated V1378M mutant (*p < 0.05; ***p < 0.001) but not between the untreated and DMSO-treated or 39°C-incubated cells. Significant differences were observed between the untreated cells and mexiletine- or 25°C-treated V1378M mutant (#p < 0.05).

Table 2. Biophysical properties of the A124D mutant Nav1.5 channel following various treatments.

Table 3. Biophysical properties of the V1378M Nav1.5 mutant channel following various treatments.
The peak current density of the V1378M mutant was restored partially by mexiletine (312.4 ± 49.1 pA/pF, n = 5) while that of the A124D mutant was not (105.8 ± 35.5 pA/pF, n = 5). After a 24-h incubation at 25°C, the current densities of the A124D and V1378M mutants were 338.6 ± 107.0 pA/pF (n = 6) and 412.5 ± 88.9 pA/pF (n = 6), respectively. The low temperature treatment was the most effective in restoring peak current densities.
To mimic the fever observed in our patient, cells expressing mutant channels were incubated for 24-h at 39°C. The peak current density was not significantly modified after incubation neither for A124D (100.2 ± 20.6 pA/pF, n = 7) nor for V1378M (144.6 ± 33.4 pA/pF, n = 7) mutants.
Immunolabeling of WT and Mutant Nav1.5 channels
Immunocytochemistry was used to clearly understand the impact of the mutations on the localization of Nav1.5 channel proteins. The channels were tagged with a Flag epitope to improve the specificity of the labeling. Patch-clamp experiments were performed to ensure that the epitope had no effect on the biophysical properties of the WT and mutant Nav1.5 channels (data not shown). The WT channels localized on the membrane (green arrow) and in the ER (red arrows) whereas the mutant channels (A124D and V1378M) co-localized on the ER (red arrows; Figure 7A).
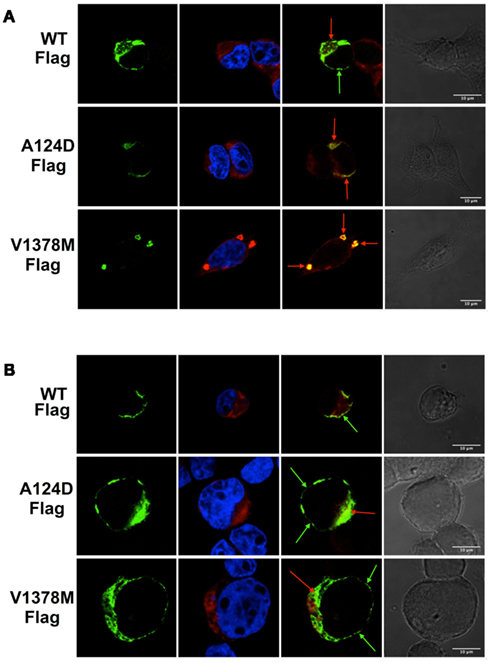
Figure 7. Immunostaining of tsA201 cells expressing the Flag-tagged WT and mutant Nav1.5 cardiac channels. Images were taken with a confocal Zeiss microscope equipped with 63× oil immersion objective and appropriate lasers. Column 1 shows Flag-staining (green), which indicates Nav1.5 channel staining. Column 2 shows nuclear (blue) and ER (red) staining. Column 3 shows a merged image of Flag-tagged Nav1.5 staining (green) and ER staining (red). Column 4 shows a light transmitted image. The scale bar on the last column is the same for all the line images. For all the groups (A,B), Nav1.5/WT-Flag is represented on the upper panel, Nav1.5/A124D-Flag on the middle panel, and Nav1.5/V1378M-Flag on the lower panel. Red arrows point to ER-localized Flag-Nav1.5 channels, and green arrows point to membrane-localized Flag-Nav1.5 channels. (A) Immunocytochemistry performed under normal conditions. (B) Immunocytochemistry performed after a 24-h incubation at 25°C.
After incubating the cells at 25°C for 24 h, the immunolabeling was repeated in order to study the impact of the low temperature treatment. The WT channels remained localized on the cell membrane (green arrows, upper panel) while the two mutant channels localized on the cell surface (green arrows) and co-localized on the ER (red arrows; Figure 7B). The two mutations resulted in the Nav1.5 mutant channels being retained in the ER while the incubation at 25°C almost completely restored the trafficking defect.
Discussion
Our results clearly showed that while the two SCN5A mutations (A124D and V1378M) caused dramatically reduced current densities, the kinetics and gating properties of the mutant channels were unaffected. This suggests that these mutant channels could be made fully functional by restoring their cell surface expression. In order to identify the cause of the reduction in current density, cells expressing mutant or WT channels were incubated under several conditions. The treatments that we investigated (incubation at low temperature and with mexiletine, thapsigargin, or curcumin) are known to restore protein trafficking defects, albeit by different molecular mechanisms (Baroudi et al., 2002; Egan et al., 2002; Valdivia et al., 2004). The reduction in the current densities of the mutant channels was caused by their retention in the ER but the functionality of the channels was restored when they reached the membrane (Figures 6 and 7).
Alanine at position 124 and valine at position 1378 are highly conserved in voltage gated Na+ channels. Given this, we hypothesized that mutating these two amino acids might affect the protein structure, leading to misfolding, and reticular retention.
Dramatic reductions in Na+ current densities induce significant ionic imbalances in cardiomyocytes. As a result, other ionic currents such as potassium currents (It0) can become pathologically prominent and lead to BrS symptoms (Antzelevitch, 2006).
Interestingly, some WT channels localized in the ER (Figure 7A). This is in agreement with the results of a study by Zimmer et al. (2002), who reported that a large proportion of Nav1.5 Na+ channels localized inside the cell (HEK-293 or cardiomyocytes), notably to enable adaptive responses.
While mexiletine is known to correct Nav1.5 trafficking defects, the A124D and V1378M mutants did not react in the same way to a 24-h incubation with 400 μM mexiletine. This drug had no effect on the current density of A124D channels while the current density of V1378M channels was partially restored. This antiarrhythmic drug binds to Nav1.5 channels on a local anesthetic binding site in the sixth transmembrane segment of the fourth homologous domain (Weiser et al., 1999). We hypothesized that when mexiletine binds to V1378M channels in the ER, it acts as a molecular chaperone and allows the export of the retained channels. On the other hand, the A124D mutation in the N-terminus is probably too far from local anesthetic binding site for mexiletine to be effective, which could explain our results. This suggests that the ability of mexiletine to rescue trafficking defects may depend on the location of the mutation. Our findings further indicated that mexiletine may be a spatially limited chaperone and, as such, cannot restore channel functionality if the mutation is too far from its binding site.
Whereas the V1378M mutation was associated with a classic BrS clinical phenotype, the A124D mutation was associated with syncope and a BrS type 1 ECG when the patient was febrile and that resolved into a BrS type 3 ECG when the patient was afebrile. In addition, the patient had a past history of neurally mediated vasovagal syncope. It has recently been proposed that some BrS patients have an impaired balance of the autonomic nervous system and that their syncopal episodes have origins other than ventricular arrhythmias (Yokokawa et al., 2010). Whether the drugs we used in in vitro conditions have an influence on the autonomic nervous system in vivo remains unclear. The index patient exhibited symptoms when febrile; we therefore tested the effect of higher temperature mimicking fever. The higher temperatures experienced during fever can activate HSPs (Tulapurkar et al., 2009). However, incubating transfected cells for 24-h at 39°C did not significantly impact the peak current density (Figure 6). We suggest that fever might trigger the clinical manifestation of the disease by yet to uncover mechanism.
Interestingly, our findings point to potential new therapeutic approaches. Broad spectrum treatments that target the entire ER are not the best way to treat trafficking defects since they may alter more than one cellular function. Specific therapies using mexiletine-like drugs that directly target misfolded Na+ channel proteins may be a better approach. However, a patient-specific approach will be required in order to choose the appropriate drug for a specific mutation.
Limitations of the Study
It is important to note some limitations of this study. Although drugs are useful research tools to characterize and understand the pathogenic processes, they cannot be used for clinical applications because of their potential side effects. First, curcumin and thapsigargin are two drugs that target the SERCA pump in the ER membrane. These drugs inhibit lectin chaperones such as calnexin inducing a partial shutdown of the ER quality control. Second, high concentrations of mexiletine are needed for efficient protein trafficking rescue. Under physiological conditions such a concentration of Na+ channel blocker may mask the benefits of the rescue. The ideal pharmacological strategy would be to specifically rescue the trafficking defect of mutated channels without affecting normal Na+ channel gating and permeability.
Conclusion
The present study showed that the BrS phenotypes observed in patients carrying the A124D and V1378M mutations can explained by a dramatic reduction in Na+ currents. These mutations might affect the structure of the channel, causing misfolded proteins and leading to the retention of these channels in the ER. Immunocytochemical analyses and drug treatments provided evidence for ER retention. The ability of mexiletine to restore Na+ currents by correcting the trafficking defect might depend on the location of the mutations. Lastly, correcting trafficking defects may be a valuable therapeutic approach for treating BrS patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the Heart and Stroke Foundation of Quebec (HSFQ) and the Canadian Institutes of Health Research (CIHR, MT-13181). We thank Dr. Heinz Schlaepfer, MD, for providing some of the clinical data.
References
Amin, A. S., Asghari-Roodsari, A., and Tan, H. L. (2010). Cardiac sodium channelopathies. Pflugers Arch. 460, 223–237.
Baroudi, G., Acharfi, S., Larouche, C., and Chahine, M. (2002). Expression and intracellular localization of an SCN5A double mutant R1232W/T1620M implicated in Brugada syndrome. Circ. Res. 90, E11–E16.
Baroudi, G., Pouliot, V., Denjoy, I., Guicheney, P., Shrier, A., and Chahine, M. (2001). Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ. Res. 88, E78–E83.
Bhar-Amato, J., Nunn, L., and Lambiase, P. (2010). A review of the mechanisms of ventricular arrhythmia in Brugada syndrome. Indian Pacing Electrophysiol. J. 10, 410–425.
Deschenes, I., Baroudi, G., Berthet, M., Barde, I., Chalvidan, T., Denjoy, I., Guicheney, P., and Chahine, M. (2000). Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc. Res. 46, 55–65.
Egan, M. E., Glockner-Pagel, J., Ambrose, C., Cahill, P. A., Pappoe, L., Balamuth, N., Cho, E., Canny, S., Wagner, C. A., Geibel, J., and Caplan, M. J. (2002). Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat. Med. 8, 485–492.
Egan, M. E., Pearson, M., Weiner, S. A., Rajendran, V., Rubin, D., Glockner-Pagel, J., Canny, S., Du, K., Lukacs, G. L., and Caplan, M. J. (2004). Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 304, 600–602.
Huang, H., Priori, S. G., Napolitano, C., O’Leary, M. E., and Chahine, M. (2011). Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of Nav1.5 channels. Am. J. Physiol. Heart Circ. Physiol. 300, H288–H299.
Keller, D. I., Huang, H., Zhao, J., Frank, R., Suarez, V., Delacretaz, E., Brink, M., Osswald, S., Schwick, N., and Chahine, M. (2006). A novel SCN5A mutation, F1344S, identified in a patient with Brugada syndrome and fever-induced ventricular fibrillation. Cardiovasc. Res. 70, 521–529.
Keller, D. I., Rougier, J. S., Kucera, J. P., Benammar, N., Fressart, V., Guicheney, P., Madle, A., Fromer, M., Schlapfer, J., and Abriel, H. (2005). Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc. Res. 67, 510–519.
London, B., Michalec, M., Mehdi, H., Zhu, X., Kerchner, L., Sanyal, S., Viswanathan, P. C., Pfahnl, A. E., Shang, L. L., Madhusudanan, M., Baty, C. J., Lagana, S., Aleong, R., Gutmann, R., Ackerman, M. J., McNamara, D. M., Weiss, R., and Dudley, S. C. Jr. (2007). Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116, 2260–2268.
Moya, A., Sutton, R., Ammirati, F., Blanc, J. J., Brignole, M., Dahm, J. B., Deharo, J. C., Gajek, J., Gjesdal, K., Krahn, A., Massin, M., Pepi, M., Pezawas, T., Ruiz Granell, R., Sarasin, F., Ungar, A., Van Dijk, J. G., Walma, E. P., and Wieling, W. (2009). Guidelines for the diagnosis and management of syncope (version 2009). Eur. Heart J. 30, 2631–2671.
Tulapurkar, M. E., Asiegbu, B. E., Singh, I. S., and Hasday, J. D. (2009). Hyperthermia in the febrile range induces HSP72 expression proportional to exposure temperature but not to HSF-1 DNA-binding activity in human lung epithelial A549 cells. Cell Stress Chaperones 14, 499–508.
Valdivia, C. R., Tester, D. J., Rok, B. A., Porter, C. B., Munger, T. M., Jahangir, A., Makielski, J. C., and Ackerman, M. J. (2004). A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 62, 53–62.
Wang, Q., Li, Z., Shen, J., and Keating, M. T. (1996). Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics 34, 9–16.
Watanabe, H., Koopmann, T. T., Le Scouarnec, S., Yang, T., Ingram, C. R., Schott, J. J., Demolombe, S., Probst, V., Anselme, F., Escande, D., Wiesfeld, A. C., Pfeufer, A., Kaab, S., Wichmann, H. E., Hasdemir, C., Aizawa, Y., Wilde, A. A., Roden, D. M., and Bezzina, C. R. (2008). Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268.
Weiser, T., Qu, Y., Catterall, W. A., and Scheuer, T. (1999). Differential interaction of R-mexiletine with the local anesthetic receptor site on brain and heart sodium channel alpha-subunits. Mol. Pharmacol. 56, 1238–1244.
Wilde, A. A., Antzelevitch, C., Borggrefe, M., Brugada, J., Brugada, R., Brugada, P., Corrado, D., Hauer, R. N., Kass, R. S., Nademanee, K., Priori, S. G., and Towbin, J. A. (2002). Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 106, 2514–2519.
Yokokawa, M., Okamura, H., Noda, T., Satomi, K., Suyama, K., Kurita, T., Aihara, N., Kamakura, S., and Shimizu, W. (2010). Neurally mediated syncope as a cause of syncope in patients with Brugada electrocardiogram. J. Cardiovasc. Electrophysiol. 21, 186–192.
Zhou, Z., Gong, Q., and January, C. T. (1999). Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Biol. Chem. 274, 31123–31126.
Zimmer, T., Biskup, C., Dugarmaa, S., Vogel, F., Steinbis, M., Bohle, T., Wu, Y. S., Dumaine, R., and Benndorf, K. (2002). Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the a subunit in HEK293 cells and dog cardiac myocytes. J. Membr. Biol. 186, 1–12.
Keywords: sodium channels, Brugada syndrome, ventricular fibrillation, cardiac arrhythmias, Nav1.5, mexiletine
Citation: Moreau A, Keller DI, Huang H, Fressart V, Schmied C, Timour Q and Chahine M (2012) Mexiletine differentially restores the trafficking defects caused by two Brugada syndrome mutations. Front. Pharmacol. 3:62. doi: 10.3389/fphar.2012.00062
Received: 21 February 2012; Paper pending published: 22 March 2012;
Accepted: 27 March 2012; Published online: 20 April 2012.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Stephane Hatem, Université Pierre et Marie Curie, FrancePatrick Bois, Institut de Physiologie et Biologie Cellulaires, France
Copyright: © 2012 Moreau, Keller, Huang, Fressart, Schmied, Timour and Chahine. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mohamed Chahine, Centre de Recherche, Institut Universitaire en Santé Mentale de Québec, 2601 Chemin de la Canardière, Quebec City, QC, Canada G1J 2G3. e-mail:bW9oYW1lZC5jaGFoaW5lQHBoYy51bGF2YWwuY2E=