


- Institute of Neuroscience of the National Research Council and Department of Biomedical Sciences, University of Padova, Padova, Italy
Epilepsies comprise a family of multifactorial neurological disorders that affect at least 50 million people worldwide. Despite a long history of neurobiological and clinical studies the mechanisms that lead the brain network to a hyperexcitable state and to the intense, massive neuronal discharges reflecting a seizure episode are only partially defined. Most epilepsies of genetic origin are related to mutations in ionic channels that cause neuronal hyperexcitability. However, idiopathic epilepsies of unclear origin represent the majority of these brain disorders. A large body of evidence suggests that in the epileptic brain neurons are not the only players. Indeed, the glial cell astrocyte is known to be morphologically and functionally altered in different types of epilepsy. Although it is unclear whether these astrocyte dysfunctions can have a causative role in epileptogenesis, the hypothesis that astrocytes contribute to epileptiform activities recently received a considerable experimental support. Notably, currently used antiepileptic drugs, that act mainly on neuronal ion channels, are ineffective in a large group of patients. Clarifying astrocyte functions in the epileptic brain tissue could unveil astrocytes as novel therapeutic targets. In this review we present first a short overview on the role of astrocytes in the epileptic brain starting from the “historical” observations on their fundamental modulation of brain homeostasis, such as the control of water content, ionic equilibrium, and neurotransmitters concentrations. We then focus our review on most recent studies that hint at a distinct contribution of these cells in the generation of focal epileptiform activities.
Introduction
Epilepsies are caused by genetic or acquired factors, such as brain injury, post-infection lesions, and tumors and affect at least 50 million people worldwide (Ngugi et al., 2010; Thurman et al., 2011). An epileptic seizure is the clinical manifestation of an abnormal and excessive discharge that initially involves neurons from a restricted brain site (the epileptogenic focus) and then propagates to nearby neuronal populations eventually spreading to the entire brain. Most currently used antiepileptic drugs (AEDs) target either voltage-sensitive ion channels or GABAergic signaling reducing neuronal excitability or increasing inhibition in the brain network, respectively (Rogawski and Loscher, 2004). These drugs are, however, ineffective in a large portion of epileptic patients and their anticonvulsant action is often accompanied by relevant side effects. Novel therapeutic targets are, therefore, highly demanded.
A different relatively unexploited strategy would be to target the glia cell astrocytes. These cells are key players in the control of the brain tissue homeostasis by regulating water content and K+ extracellular levels and by providing neurons with nutrients and trophic factors. Astrocytes play a prominent role in neurovascular coupling mechanism (Zonta et al., 2003; Takano et al., 2006; for recent reviews, see Carmignoto and Gomez-Gonzalo, 2010; Petzold and Murthy, 2011) and are also largely responsible for maintaining physiological levels of glutamate (Rothstein et al., 1994, 1996; Huang et al., 2004) and GABA in the extracellular space (Conti et al., 2004; Madsen et al., 2010). In addition, astrocytes participate with microglial cells in the defensive response against endo and exogenous insults by releasing various cytokines and pro-inflammatory mediators that ultimately have a proconvulsant action (Turrin and Rivest, 2004; Vezzani and Granata, 2005; Vezzani et al., 2008; Fabene et al., 2010).
While the astrocytic contribution to these processes is well recognized since many decades, more recent works have greatly extended the role of astrocytes from the control of brain tissue homeostasis to the modulation of synaptic transmission and network activities. At the start of the nineties a number of pioneering studies showed that glutamate applications in cell cultures and brain slice preparations evoked Ca2+ elevations and propagating Ca2+ waves in astrocytes (Dani et al., 1992). Subsequent observations revealed that Ca2+ elevations in astrocytes triggered glutamate release that evoked AMPA and NMDA receptor-mediated Ca2+ increases in nearby neurons (Nedergaard, 1994; Parpura et al., 1994; Pasti et al., 1997). The indication of an astrocyte-to-neuron signaling represented a turning point in astrocyte research and paved the way to studies in brain slice preparations and in vivo which essentially confirmed the existence of a complex bidirectional signaling between neurons and astrocytes (Zonta and Carmignoto, 2002). We now know that astrocytes express a large variety of receptors that can be activated by various neurotransmitters, such as glutamate (Porter and McCarthy, 1996; Pasti et al., 1997), GABA (Kang et al., 1998), noradrenaline (Duffy and MacVicar, 1995; Kulik et al., 1999), acetylcholine (Shelton and McCarthy, 2000; Araque et al., 2002), ATP, and also by cannabinoids released by neuronal dendrites (Navarrete and Araque, 2008, 2010). These molecules evoke Ca2+ elevations in astrocytes that, in turn, release other transmitters including glutamate, D-serine, and ATP. By means of these transmitters, termed gliotransmitters, astrocytes affect neuronal excitability, and modulate synaptic transmission. Beside these gliotransmitters astrocytes can release cytokines such as IL-1β and TNFα, as well as other molecules such as TGFβ, GDNF, BDNF, nitric oxide, and neurosteroids that can also affect neuronal functions. The ability of astrocytes to listen and talk to synapses by exerting both excitatory and inhibitory actions on neurons (Pasti et al., 1997; Araque et al., 1999; Brockhaus and Deitmer, 2002; Zhang et al., 2003; Pascual et al., 2005; Panatier et al., 2006, 2011; Serrano et al., 2006; Jourdain et al., 2007; Perea and Araque, 2007; Di Castro et al., 2011; Shigetomi et al., 2011; Min and Nevian, 2012; Navarrete et al., 2012) is revolutionizing our original view that information processing in the brain is exclusively based on billions of neurons interacting dynamically in the neuronal network. On the other hand, clues for a distinct contribution of astrocytes in the highest functions of the mammalian brain during phylogeny are the increasing complexity and size of protoplasmic astrocytes with respect to the unchanged features of cortical neurons, and the relative expansion in the number of astrocytes with respect to that of neurons (Oberheim et al., 2006, 2009). All in all, the astrocyte is now proposed to represent an intrinsic element of the neuronal circuit that composes a tripartite synapse with the pre- and the post-synaptic neuronal membrane (Araque et al., 1999; Carmignoto, 2000; Haydon and Carmignoto, 2006; Halassa et al., 2009; Perea et al., 2009).
The discovery that astrocytes modulate neuronal communication and are involved in the control of brain network activities suggest that astrocytes have the potential to be involved in the development of a number of brain disorders, from acute brain injury or infection to chronic diseases including Alzheimer’s diseases, Parkinson disease, amyotrophic lateral sclerosis, and epilepsy (Maragakis and Rothstein, 2006; Seifert et al., 2006; Blackburn et al., 2009). The aforementioned observations on Ca2+-dependent release of glutamate in astrocytes providing direct excitation to neighboring neurons is of particular interest in the context of epileptogenesis. Accordingly, alterations in this astrocyte-derived excitatory pathway in concert with impaired astrocytic glutamate and K+ uptake may result in a hyperexcitable neuron-astrocyte network which favors neuronal synchronization and ultimately predisposes neurons to seizures. In this review we present an overview of the “historical” observations that suggested a role of astrocytes in the epileptic brain and we also critically discuss the most recent results that hint at a distinct contribution of astrocytes in the generation of focal epileptiform activities.
Astrocytes in the Epileptic Brain
It has been known for a long time that early in the development of many brain disorders, astrocytes exhibit significant morphological and functional changes (Hamby and Sofroniew, 2010). This condition, termed reactive gliosis, comprises increased number, altered morphology with hypertrophic soma and processes, spatial overlapping and different functional changes of astrocytes. Gliosis is frequent in the human epileptic brain and seizures have been shown to frequently initiate within or very near gliotic brain tissues (McKhann et al., 2000). Gliosis is frequently observed in temporal lobe epilepsies (TLE), a severe form of partial epilepsy with focal onset in temporal structures such as hippocampus, entorhinal cortex, and amygdala. Surgical ablation of the epileptogenic region may be the only treatment available in the most severe forms of TLE that are resistant to all the currently available AEDs. Studies from these tissues, as well as from post-mortem tissues or from animal models, have shown relevant alterations of neuronal and astrocytic morphology. It is important to point out that it is still unclear whether an altered glial morphology can contribute to the generation of the disorder or be a consequence of an established damage of the brain tissue. This especially holds for human tissues from patients that may be suffering from the pathology since many years, rather than for animal models in which investigations can be more easily performed at different steps of the disease.
The most frequent and striking alteration in TLE is hippocampal sclerosis which is characterized by a massive gliosis, neuronal loss, synaptic, and microvascular reorganization. Gliosis, that can be revealed by increased expression of glial fibrillary acidic protein (GFAP, an intermediate filament protein), has been described also in other forms of epilepsies like post-traumatic and infection-induced epilepsy (Seifert et al., 2006). In animal models, including the pilocarpine- and the kainate-induced epilepsy, gliosis is also early observed during the period that precedes recurrent seizure onset, suggesting a causative role of astrocytes in epileptogenesis (Represa et al., 1995; Belluardo et al., 1996; Bouilleret et al., 1999; Borges et al., 2003; Rizzi et al., 2003).
While we have only a limited knowledge of the functional changes occurring in reactive astrocytes, we can reasonably advance the hypothesis that an astrocyte dysfunction contributes or even plays a central role in epileptogenesis. Notably, although astrocyte abnormalities have been, in general, linked to proconvulsant actions, astrocyte signals can also exert an anticonvulsants action in a context-dependent manner. In the following sections we will examine to what extent an impairment of a distinct astrocytic function may contribute to modulate brain network excitability ultimately favoring, or opposing, the generation and spread of seizure-like discharges. In view of the recognized role played by gliotransmission in various forms of synaptic plasticity (Serrano et al., 2006; Florian et al., 2011; Panatier et al., 2011; Min and Nevian, 2012; Navarrete et al., 2012), it is worth underlying that the cognitive deficits observed in a number of neurological disorders, including epilepsy, may be due, at least in part, to an impairment of the reciprocal signaling between neurons and astrocytes.
Potassium Homeostasis
A correct K+ homeostasis is a crucial factor in the control of neuronal excitability. Because even small elevations in the extracellular K+ concentrations ([K+]o) can significantly increase neuronal network activity, it was hypothesized that [K+]o is a causal factor for epileptiform activity. Indeed, perfusion of brain slices with high K+ is effectively used to induce epileptic-like discharges in neurons. Moreover, a bulk of experimental studies performed in different in vitro and in vivo models of epilepsy showed that seizure activity itself can lead to a significant extracellular K+ increase up to 10–12 mM (Pedley et al., 1976; Heinemann et al., 1977, 1986; Lopantsev et al., 2009). A number of early studies also revealed a high and rapid permeability of glial cells to K+ ions, even in response to small neuronal activities (Kuffler et al., 1966; Orkand et al., 1966). In these initial studies the so-called “spatial potassium buffering hypothesis” was advanced. According to this hypothesis, astrocytes either singularly or connected through gap-junctions, were proposed to redistribute local [K+]o increases either to neighboring regions or directly into the blood vessels (Orkand et al., 1966; Kofuji and Newman, 2004). Accordingly, astrocytes are thought to play a predominant role in the clearance of the K+ ions which accumulate in the extracellular space during the intense neuronal firing that characterizes epileptic discharges. Inward rectifying K+ channels (Kir) are the main responsible for the high astrocytic K+ permeability. Of the 16 distinct Kir channel subunits which have been identified, type 4.1 (Kir4.1) and, to a lesser extent, type 2.3 (Kir2.3) are the most intensively studied (Neusch et al., 2006; Olsen and Sontheimer, 2008). Notably, astrocytes from different brain regions abundantly express Kir4.1 channels (Takumi et al., 1995; Ishii et al., 1997; Poopalasundaram et al., 2000; Higashi et al., 2001; Li et al., 2001). Unlike other K+ channels that allow large K+ efflux from depolarized cells, such as those that underline membrane repolarization during action potential firing in neurons, Kir channels have the peculiarity to mediate small K+ efflux from depolarized cells and large influx at hyperpolarized potentials. Most importantly, due to a high open probability at resting potential (Ransom and Sontheimer, 1995) and a channel conductance that is proportional to [K+]o (Sakmann and Trube, 1984; Newman, 1993), Kir4.1 channels can be effectively activated by large [K+]o increases and mediate a large K+ influx.
Several studies showed altered Kir4.1 expression and activity in epilepsy, both in humans and experimental animal models, as well as in other disorders including brain injury. A reduction of astrocytic Kir4.1 channels expression was observed in several studies on tissues from TLE patients (Bordey and Sontheimer, 1998; Hinterkeuser et al., 2000; Kivi et al., 2000; Schroder et al., 2000). Kir currents recorded in astrocytes from human sclerotic hippocampi were also reduced (Heinemann et al., 2000; Hinterkeuser et al., 2000). It is, however, unclear whether these changes are a reaction of the tissue to the epileptiform activities or whether they are directly involved in the genesis of the disease. The use of transgenic animals helped to address this issue and results obtained confirmed the importance of Kir channels during epileptogenesis. In particular, Kir4.1 knock-out mice exhibited an impaired K+ homeostasis in the retina (Kofuji et al., 2000) and the brain stem (Neusch et al., 2006), and showed stress-induced seizures (Djukic et al., 2007). In a recent in vivo study in Kir4.1 conditional knock-out mice, the central role of Kir4.1 channels in the [K+]o regulation in the hippocampal region was also reported (Chever et al., 2010). Another compelling, although indirect, evidence for the Kir involvement in the generation of epileptiform activities is the finding that mutations or polymorphisms of KCNJ10, the gene encoding Kir4.1 subunits, have been associated with increased seizure susceptibility and epileptic phenotypes in mice (Ferraro et al., 2004; Inyushin et al., 2010). Humans KCNJ10 mutations are also associated to seizure susceptibility, idiopathic generalized epilepsy (Buono et al., 2004; Lenzen et al., 2005) or to complex syndromes that encompass epilepsy (Bockenhauer et al., 2009; Scholl et al., 2009; Reichold et al., 2010). Notably, a reduced Kir4.1-mediated K+ buffering, due to conditional knock-out of Kir4.1 channels or polymorphism of KCNJ10, has been linked to a reduced glutamate clearance (Djukic et al., 2007; Kucheryavykh et al., 2007; Inyushin et al., 2010) that further contributes to increase network excitability. Altogether, these observations support the view that either a reduced Kir channel expression or a functional impairment of these channels due to neuronal injury or gene polymorphisms can increase network activity leading to a higher susceptibility to proconvulsant stimuli or to generation of a full epileptic activity. As crucial regulators of [K+] and network excitability, astrocytic Kir channels represent potential targets for a novel therapeutic approach for epilepsies.
Water Homeostasis
Water homeostasis is another aspect of great importance in brain pathophysiology in which astrocytes play a pivotal role. Different conditions such as brain ischemia, injury, or hepatic encephalopathy can result in cerebral edema that is mainly due to water accumulation in astrocytes. Astrocyte swelling leads to a reduction of the extracellular space that enhances network excitability and favors epileptiform discharges (Dudek et al., 1990; Andrew, 1991; Roper et al., 1992; Chebabo et al., 1995; Pan and Stringer, 1996), possibly due to an increase of ephaptic interactions between neurons. Astrocyte swelling can also result in an increased release of gliotransmitters, such as glutamate, via volume-sensitive organic anion channels (VSOACs), that further enhances network excitability. Water influx in astrocytes is meditated by the water channel aquaporin 4 (AQP4) that is, indeed, highly expressed in these cells (reviewed by Badaut et al., 2002, 2007) with a prevalent localization in perivascular endfeet (Nielsen et al., 1997; Higashi et al., 2001; Nagelhus et al., 2004). The peculiar anchoring of AQP4 on perivascular endfeet seems to rely on α-syntrophin, a dystrophin complex protein (reviewed by Amiry-Moghaddam et al., 2004) that is important to link intracellular water content to blood circulation. Interestingly, AQP4 channels colocalize with Kir4.1 (Nagelhus et al., 2004) and AQP4 deficient or knock-out mice show a slowed K+ influx. These latter mice also show prolonged seizures (Amiry-Moghaddam et al., 2003; Binder and Steinhauser, 2006). These and other studies support the hypothesis that AQP4 and Kir4.1 channels cooperate in the regulation of water and K+ homeostasis in brain tissue. Like Kir4.1 channels, AQP4 are also altered in the sclerotic human hippocampi with an overall increase of expression (Lee et al., 2004), but a redistribution from the perivascular site to the perisynaptic one (Lee et al., 2004). Similar results are also observed in epileptic tissue with focal cortical dysplasia (FCD), a cortical malformation that frequently generates epileptic foci (Medici et al., 2011). In another recent study, a decreased expression of AQP4 has been observed in the kainic acid model of epileptogenesis in wild-type mice together with an increase of seizures occurrence in AQP4−/− mice (Lee et al., 2012). An altered distribution of AQP4 may thus reduce the transfer of water from astrocytes into blood vessels. This would lead to astrocyte swelling, reduced clearance of both extracellular K+ and glutamate as well as an increase in glutamate release, also through volume-sensitive anion channels, triggering a deleterious loop that can further facilitate cell swelling. A recent work has shown a critical role of AQP4 also for astrocytic Ca2+ signaling (Thrane et al., 2011). In particular, the authors showed that hypo-osmotic stress triggers Ca2+ increases in astrocytes and this response depends on AQP4 and, partially, on P2Y receptors. Consistent with an important role of AQ-4 in astrocytic Ca2+ signaling and astrocyte-neuron interactions, AQ-4 knock-out mice exhibited a cognitive defect and a reduction of a distinct form of hippocampal LTP (Skucas et al., 2011). Altogether, the currently available data suggest a strong involvement of astrocytic water channels in epileptogenesis (for review see Binder et al., 2012).
Glutamate and GABA
According to a classical paradigm epileptiform activities may arise from a brain network that becomes hyperexcitable by an imbalance in glutamatergic and inhibitory GABAergic transmission (Prince and Wilder, 1967; Bradford, 1995). This simple view has been recently revisited taking into account that GABAergic transmission may favor epileptic activity by synchronizing large neuronal populations and even depolarize, at least under certain conditions, the neuronal membrane thereby enhancing neuronal excitability (Cossart et al., 2005; Fritschy, 2008; Avoli and de Curtis, 2011). Although the ultimate action of GABAergic transmission may be context-dependent, seizure threshold is strongly affected by the dynamics of both glutamate and GABA signals. The tonic extracellular level of these neurotransmitters can also contribute to control network excitability. As regards glutamate, the large amount of this neurotransmitter, released into the extracellular space by the activity of both glutamatergic synapses and astrocytes, is internalized by an efficient reuptake system that includes five different excitatory amino acid transporters, EAAT1-5. EAAT1 and EAAT2 are selectively expressed in astrocytes (GLAST and GLT-1 in rodents, respectively) and they represent the main regulators of extracellular glutamate levels in the brain (Rothstein et al., 1994; Tanaka et al., 1997). An additional factor that contributes to set the extracellular glutamate concentration in the brain is the glutamate metabolism. According to the glutamate-glutamine cycle, glutamate is mainly synthesized in astrocytes starting from the glucose internalized by astrocyte endfeet in contact with cerebral blood arterioles. Glucose is then converted to glutamate (via α-ketoglutarate) and successively to glutamine by the specific astrocytic enzyme glutamine synthetase (GS). Glutamine released from astrocytes is captured by glutamatergic neurons through specific transporters and finally deaminated to glutamate by phosphate activated glutaminase. In astrocytes the extracellular glutamate is internalized and converted again into glutamine that is then transferred to neurons to start a new cycle.
High extracellular glutamate levels have been reported in human epileptic tissues (During and Spencer, 1993; Glass and Dragunow, 1995; Cavus et al., 2005). These observations lead to the hypothesis that an increased glutamatergic transmission may be an early event that contributes to the generation of distinct types of epilepsy. Controversial results have been, however, reported in studies investigating the EAAT levels from tissues of TLE patients (reviewed by Eid et al., 2008b; Seifert et al., 2010). While some studies revealed a down regulation of astrocytic glutamate transporters (Mathern et al., 1999; Proper et al., 2002; Sarac et al., 2009), others failed to observe similar alterations (Tessler et al., 1999; Eid et al., 2004). The potential of astrocytic EEAT transporters to participate in seizure generation was, however, demonstrated in a transgenic mouse line in which a deletion of EATT2 in astrocytes resulted in epileptiform activities (Tanaka et al., 1997). In addition, in an animal model of focal epilepsy induced by a blood brain barrier (BBB) disruption that mimics post-traumatic epilepsy, a reduced glutamate and K+ clearance capacity was reported (David et al., 2009; see also below).
A more general consensus regards the loss of GS in the hippocampus from TLE patients that showed elevated extracellular glutamate levels (Eid et al., 2004; van der Hel et al., 2005). Reduced GS levels and activity are thought to increase extracellular glutamate levels because the intracellular conversion of glutamate to glutamine favors the uptake of glutamate from extracellular space (Otis and Jahr, 1998). It was recently shown that inhibition of GS by methionine sulfoximine induced recurrent seizures in animal models (Eid et al., 2008a; Wang et al., 2009). Notably, astrocytic glutamine is not only the main source of glutamate in pyramidal neurons, but it also produces GABA in GABAergic interneurons. Among the different interneurons, the fast-spiking subtype demands a very efficient GABA recycling system due to the very high frequency of action potential firing in these cells. Indeed, the inhibition of astrocytic GS in experimental animal models leads to reduced inhibitory synaptic currents and GABA release without similar changes in the glutamatergic pathway (Liang et al., 2006; Kam and Nicoll, 2007; Ortinski et al., 2010). Consistent with this view, reduced levels and activity of GS in the epileptic brain were observed to favor seizure generation by increasing extracellular glutamate and decreasing GABA release at presynaptic terminals. Interestingly, in a recent study in different genetic models of absence seizures – a form of thalamic epilepsy with excessive GABA levels – a reduced GABA uptake in astrocytes was found to be a causal factor for this epileptic phenotype, pointing out at an astrocytic dysfunction as a main cause also for this type of epilepsy (Cope et al., 2009). Although some controversial aspects need to be addressed, results from these studies support the hypothesis that altered glutamate/GABA metabolism and uptake in astrocytes can play an important role in the epileptic brain.
While the interactions between GABAergic interneurons and astrocytes have been poorly addressed in the context of epilepsy, it is noteworthy that astrocytes are also a source of neurosteroids, a class of potent endogenous modulators of GABAA receptors with sedative, anxiolytic, and anticonvulsive properties (Lambert et al., 2003; Reddy, 2010). Neurosteroids such as allopregnanolone (3α-hydroxy-5α-pregnane-20-one) and allotetrahydrodeoxycorticosterone (THDOC; 3α,21-dihydroxy-5α-pregnan-20-one), can either originate in peripheral organs or be synthesized de novo in the brain by both neurons and astrocytes (Mensah-Nyagan et al., 1999; Agis-Balboa et al., 2006). Interestingly, the cytochrome P450 cholesterol side-chain cleavage enzyme (P450scc) that catalyzes the initial step of neurosteroid biosynthesis in the brain was found to be increased in hippocampal astrocytes during the latent period after pilocarpine-induced status epilepticus (SE) in rats (Biagini et al., 2006, 2009). In these studies longer SE was correlated to higher P450scc expression in astrocytes and longer latent period, while blockade of neurosteroid synthesis accelerated seizure onset, suggesting that activated astrocytes may temporarily prevent spontaneous seizure onset by producing neurosteroids. These data raise the hypothesis of an anticonvulsant action of astrocytes and press for additional studies aimed to clarify their role in the production of neurosteroids and of other modulators of neuronal excitability.
Gliotransmission: Glutamate Release
The excessive brain network excitability and the hypersynchronous discharges that are observed in epileptic disorders are believed to represent abnormalities intrinsic to neurons. In the generation of epileptiform activity non-neuronal mechanisms may, however, also contribute (Konnerth et al., 1986; Dudek et al., 1998; Jefferys, 2003). Astrocytes can represent, indeed, an extrasynaptic source of glutamate that triggers synchronized activities in groups of neurons from different brain regions (Angulo et al., 2004; Fellin et al., 2004, 2007). Paired patch-clamp recording experiments from two pyramidal neurons revealed that the release of glutamate from astrocytes – in response to Ca2+ elevations evoked by different agents or direct mechanical stimulation – induced in both neurons N-methyl-D-aspartic acid (NMDA) receptor-mediated slow inward currents (SICs) that could occur with a high level of synchrony (Angulo et al., 2004; Fellin et al., 2004). Ca2+ imaging experiments also revealed that astrocyte Ca2+ elevations could be followed by episodes of simultaneous Ca2+ elevations in small groups of adjacent neurons, a response that we termed a “domain response” (Fellin et al., 2004). It resulted that stimuli commonly used to activate a Ca2+ signal in astrocytes, such as agonists of type I metabotropic glutamate receptors (mGluRs), purinergic, and prostaglandin E2 receptors, triggered in the presence of TTX glutamate release from astrocytes and synchronous activity in neurons (Fellin et al., 2004, 2006b). Given that the Ca2+ dependent release of glutamate from astrocytes could promote local synchronous activities in hippocampal neurons, it was reasonable to propose that an excessive activation of Ca2+ elevations in astrocytes may result in a massive glutamate release and represent a relevant non-neuronal mechanism of hypersynchronous discharges in neurons. In support of this view, studies performed both on brain slices and in vivo showed that during epileptiform activity, the frequency of Ca2+ oscillations in astrocytes is significantly increased (Tian et al., 2005; Fellin et al., 2006a), while it is reduced by anticonvulsant drugs (Tian et al., 2005). Moreover, mGluR expression in hippocampal astrocytes from animal models of temporal lobe epilepsy was also found to be increased (Aronica et al., 2000; Ulas et al., 2000). Furthermore, glutamate released from rodent astrocytes during SE contributes to neuronal death (Ding et al., 2007). This process could be averted by inhibiting glia-neuron signaling and by applying mGluR5 and NR2B NMDA receptor antagonists which suppress astrocytic Ca2+ increase and block extrasynaptic NMDA receptors activated by glia-derived glutamate (Ding et al., 2007). These observations support the hypothesis that an increase excitability in the neuron–astrocyte network favors neuronal synchronization and ultimately predisposes neurons to seizures.
In five different experimental models of chemically induced epilepsy, 70–90% of paroxysmal depolarizing shifts (PDSs), i.e., interictal spikes, were found to be insensitive to tetrodotoxin applications (Kang et al., 2005; Tian et al., 2005). Given that astrocytic glutamate plays a prominent role in the generation of such a large fraction of PDSs, SICs coincide, according to these authors, with PDSs and seizure activity may have an astrocytic basis (Tian et al., 2005). This conclusion, however, fueled a controversial debate on the role of astrocytes in epileptogenesis since it contradicts a large number of previous studies showing that TTX efficiently blocks PDS (D’Ambrosio, 2006; Seifert et al., 2006; Wetherington et al., 2008). The conclusion by Tian et al., was also disputed by subsequent studies which further confirmed the sensitivity of PDS to TTX (Fellin and Haydon, 2005; Gomez-Gonzalo et al., 2010). On the other hand, it is rather unlikely that astrocyte-mediated SICs in pyramidal neurons represent PDSs. SICs have been demonstrated to be mediated exclusively by NMDA receptors (Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005), while PDSs were reported to be, at least in large part, insensitive to D-AP5. As reviewed by Wetherington et al. (2008) a huge amount of data strongly support the conclusion that SICs and PDSs, in spite of a similar time course, are distinct events and derive from a different cellular source.
The results reported above do not rule out the possibility, however, that astrocytic glutamate is an important factor in epileptiform activities (Fellin et al., 2006a). A recent study from our group helped to clarify the effective weight of the astrocyte contribution to the generation of epileptic discharges (Gomez-Gonzalo et al., 2010). We found that astrocytes could contribute to seizure-like ictal discharge, but not to interictal discharge arising spontaneously in our picrotoxin/low Mg2+ cortical slice model. Indeed, astrocyte Ca2+ elevations were observed to regularly accompany the generation of the ictal discharge, but these events were observed only rarely in a few astrocytes during the interictal discharges (Figure 1). To further define the role of astrocytes in the generation of epileptiform activities, we also used additional models of epileptic seizures, including a new model of stimulus-evoked seizures (Losi et al., 2010). In the new model, an episode of hyperactivity that is induced in a small group of neurons by local NMDA applications in the presence of the proconvulsant 4-amino pyridine (4-AP), generates an ictal, seizure-like discharge that from this epileptogenic site propagates to adjacent neuronal populations. The unique advantage of the new model is that we know in advance when and where a focal seizure will occur, giving us the opportunity to investigate the earlier cellular events that develop at the epileptogenic site during the preictal, transition phase, and predispose neurons to seizures. We found that neuronal hyperactivities at these restricted brain sites are accompanied by an early Ca2+ elevation in a large number of astrocytes (see Figure 1) that contributes to drive neurons toward the threshold of seizure discharge generation (Gomez-Gonzalo et al., 2010). Indeed, the onset of Ca2+ elevations in astrocytes from the epileptogenic site occurred a few seconds before the initial development of focal, seizure-like discharges. Most importantly, we were able to show that the early astrocyte activation was not a mere consequence of the episode of high neuronal activity triggered by NMDA and it rather represents a crucial step in the generation of the ictal discharge. After the Ca2+ chelator BAPTA was introduced into the astrocyte syncytium by patching individual astrocytes with a BAPTA-containing pipette, the Ca2+ elevations in astrocytes were strongly reduced and the NMDA stimulation that was normally successful failed to evoke an ictal discharge. According to results obtained from different control experiments, the effect of BAPTA on ictal discharge generation was specifically linked to the inhibition of astrocyte Ca2+ signals, and, possibly, to the inhibition of gliotransmitter release, such as glutamate and D-serine, that can favor synchronous neuronal activities.
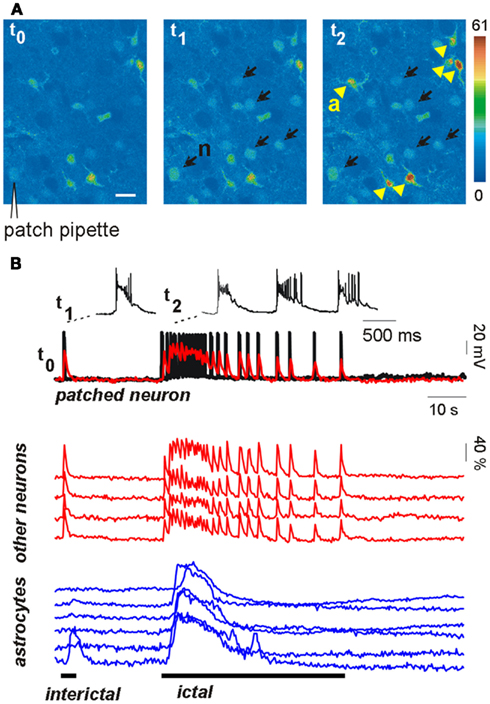
Figure 1. Astrocytes are activated during focal seizure propagation in cortical slices. (A) Image sequence of the fluorescence signal change in layer V–VI neurons (n, black arrows) and astrocytes (a, yellow arrowheads) from the entorhinal cortex of a young rat after slice loading with the Ca2+ dye Oregon-Green BAPTA1-AM during a propagating ictal discharge. (B) Simultaneous patch-clamp recording from one neuron [black trace; the patch-pipette is indicated in (A)] and ΔF/F0 traces of the Ca2+ signal in the same neuron (superimposed red trace) and other neighboring neurons (red traces) and astrocytes (blue traces). The Ca2+ signal from all neurons precisely matched the electrical activity of the patched neuron during both the interictal event (single event at trace onset) and the subsequent ictal discharge. Astrocytes were strongly activated during the ictal discharge while they were poorly activated by the interictal discharge (adapted from Gomez-Gonzalo et al., 2010).
These results suggest that when an episode of hyperactivity in a group of neurons consistently engages nearby astrocytes, a feedback signal from these latter cells enlarges the population of neurons recruited into a coherent activity. If this signal operates on a brain network prone to seizures, neurons are driven toward the ictal discharge threshold. It is worthwhile to emphasize here that when the astrocyte contribution was reduced, for example following BAPTA intracellular applications, the ictal discharge was inhibited, but it could be recovered by applying a higher NMDA stimulation (Gomez-Gonzalo et al., 2010). By activating directly a larger number of neurons, this higher stimulus evokes a level of correlated activity that is sufficient for seizure-like discharge generation, bypassing the astrocyte contribution in the recruitment process. Thus, astrocytes are not an absolute requirement, but they can provide an important and even, under certain circumstances, crucial contribution to ID generation.
Gliotransmission: ATP Release
Another gliotransmitter that plays a relevant role in the control of epileptic activity is ATP. The effect of ATP can be even more complex than that of glutamate and exert in the neuronal network both pro and anticonvulsant actions. ATP is released from astrocytes either through a Ca2+-dependent (Coco et al., 2003; Pascual et al., 2005) or a Ca2+-independent mechanism, the latter involving connexin and pannexin hemichannels (for a review, see Burnstock et al., 2011). Astrocytic ATP and its main metabolite adenosine (Ado) exert a plethora of actions on astrocytes, neurons, microglia, oligodendrocytes, and blood vessels through different purinergic receptors. ATP is responsible for the slow Ca2+ waves that propagate across the astrocytic syncytium by acting on P2Y receptors (Cotrina et al., 1998; Koizumi, 2010).
While ATP can directly excite neurons through P2 receptors with pro-epileptic effects (for a review, see Kumaria et al., 2008), it might also have an anticonvulsant action due to activation of P2Y receptors on inhibitory interneurons (see below) or for the effects of its metabolite Ado. This strongly inhibits excitatory neurotransmitter release through activation of presynaptic A1 receptors and is considered, indeed, a potent endogenous anticonvulsant (for reviews see Li et al., 2007; Boison, 2008, 2010). Ado derived from astrocytic ATP is also at the basis of some distinct forms of synaptic plasticity such as the heterosynaptic depression in the hippocampus (Zhang et al., 2003, p. 6691; Pascual et al., 2005, p. 7468; Serrano et al., 2006, p. 8330). In light of these findings astrocytic ATP may initially favor (via P2 receptors) and then inhibit epileptic activity after its conversion to Ado (via A1 receptors). Noteworthy is also that astrocytes are major regulators of extracellular Ado levels since these cells are not only its major source but they also predominantly express adenosine kinase (ADK), an enzyme that efficiently degrades Ado (Gouder et al., 2004; Boison, 2005). In the epileptic brain, including the hippocampus from patients with temporal lobe epilepsy, ADK is increased resulting in decreased extracellular Ado concentrations that favor network excitability (Gouder et al., 2004; Aronica et al., 2011). Notably, cell grafts releasing Ado and genetic reduction of ADK were shown to prevent seizures and reduce epileptogenesis in animal models of epilepsy (Huber et al., 2001; Li et al., 2007, 2008). These observations suggest that ADK can be considered not only a diagnostic marker of epilepsies, but also a potential target for the development of a new therapy that could prevent epileptic seizures (Boison, 2008; Theofilas et al., 2011).
Interestingly, a recent study showed that a local Ca2+ decrease in the extracellular space initiates a Ca2+ wave in astrocytes specifically mediated by astrocytic ATP release through connexin 43 hemichannels (Torres et al., 2012). This astrocytic ATP enhances inhibitory transmission by acting on the P2Y1 receptors expressed in a subset of hippocampal interneurons (Torres et al., 2012). Given that during epileptic activity the extracellular Ca2+ is markedly reduced, the consequent release of astrocytic ATP could potentiate inhibitory transmission thereby working as an anticonvulsant feedback mechanism that opposes seizure propagation.
All in all, it can be predicted that ATP gliotransmission may soon be recognized as target for developing new antiepileptic therapies.
Inflammation
A large variety of brain insults including stroke, acute infections, trauma, febrile seizures, and cancer are associated with a high risk of developing epilepsy and all these conditions are known to trigger a rapid inflammatory reaction in the brain tissue. The activation of inflammatory pathways appears, indeed, to play an important role in the etiopathogenesis of several forms of epilepsy (Turrin and Rivest, 2004; Vezzani and Granata, 2005; Vezzani et al., 2008; Fabene et al., 2010). Among the inflammatory agents that are highly expressed in the epileptogenic tissue are the pro-inflammatory cytokines IL-1β and TNFα. Main sources of these cytokines are microglia and astrocytes. The mechanism at the basis of the IL-1β proconvulsant action is not completely understood. Recent works suggest that IL-1β acts on neuronal IL-1β receptor 1 (IL-1R1), the IL-1R accessory protein, myeloid differentiation primary response protein complex (MyD88) and Src family kinases to ultimately enhance NMDA-mediated Ca2+ influx by increasing the phosphorylation of the NR2B subunit (Viviani et al., 2003; Balosso et al., 2008). Other mechanisms are, however, possible. In a recent study in brain slices IL-1β was reported to induce hyperexcitability independently of NMDA receptors (Rossi et al., 2012). The authors report an increased occurrence of AMPA receptor-mediated spontaneous excitatory synaptic currents due to the action of IL-1β on transient receptor potential vanilloid 1 (TRPV1) channels.
It is worth underlying that IL-1β and TNFα were also reported to act on astrocytes, significantly reducing glutamate uptake and increasing glutamate release in these cells (Ye and Sontheimer, 1996; Hu et al., 2000; Bezzi et al., 2001; Santello et al., 2011) raising the possibility that the proconvulsant action of these cytokines can be linked, at least in part, to a direct effect on astrocytes. In a series of recent work it was also shown that in neocortex the disruption of the BBB, an event frequently associated with neuroinflammation, leads to a rapid astrocyte activation with increased GFAP expression. This is due to albumin diffusion which is selectively captured by astrocytes through a mechanism involving TGF-β receptors. Indeed, an increased BBB permeability is proposed to contribute to the generation of an epileptogenic focus (Seiffert et al., 2004; Ivens et al., 2007; Tomkins et al., 2007; Friedman et al., 2009). Importantly, the rapid gliosis that follows a disruption of the BBB was associated with a reduced expression of Kir channels, gap-junctional proteins, and glutamate transporters in astrocytes that lead to an increased network excitability by reducing K+ and glutamate uptake (David et al., 2009).
The high-mobility group box-1 (HMGB1) is an additional inflammatory agent (Müller et al., 2004; Pedrazzi et al., 2007; Sims et al., 2010) recently revealed to be involved in seizure generation (Maroso et al., 2010). HMGB1 is produced by both neurons and glia and its action appears to be mediated by the toll-like receptor 4 (TLR4), a receptor that recognizes several pathogenic microbial molecules (Maroso et al., 2010).
Although many molecular aspects remain to be elucidated, these studies provide significant support to the hypothesis that astrocytes are directly involved in the inflammatory reaction that favor the development of epilepsy.
Conclusions and Perspectives
Astrocytes are key players in the regulation of brain tissue homeostasis and neuronal excitability. A large body of evidence showed the impairment of several astrocytic functions in the epileptic brain, such as the control of water and ionic equilibrium, neurotransmitter concentrations, cerebral blood flow, and the ability to release modulators and cytokines. Although a dysregulation of a number of astrocyte-specific functions has been identified and linked to epilepsies, the precise role of gliotransmission in initiation and propagation of seizures is unclear and many key questions remain unanswered. Among these questions it is relevant to clarify if and under which conditions the different gliotransmitters exert a pro or an anticonvulsant action (see Figure 2). Very likely, the signaling of astrocytes to principal neurons and distinct interneurons through glutamate release may be context-dependent and needs selective tools to be elucidated. ATP-mediated gliotransmission is another promising field for future experiments given its role in mediating Ca2+ waves in the astrocytic syncytium and its wide effects on neuronal functions. Accordingly, a clarification of ATP release mechanisms could potentially allow the development of selective tools to modulate gliotransmission and network excitability. As to epileptogenesis, an important issue is whether astrocytes favor the generation of epileptic events or may, instead, oppose it by releasing, for example, molecules like neurosteroids and trophic factors. To address these questions a great help is provided by new experimental approaches that use molecular genetic tools to selectively affect specific signaling pathways in astrocytes as well as by the application of Ca2+ imaging techniques in in vivo animal models of epilepsy.
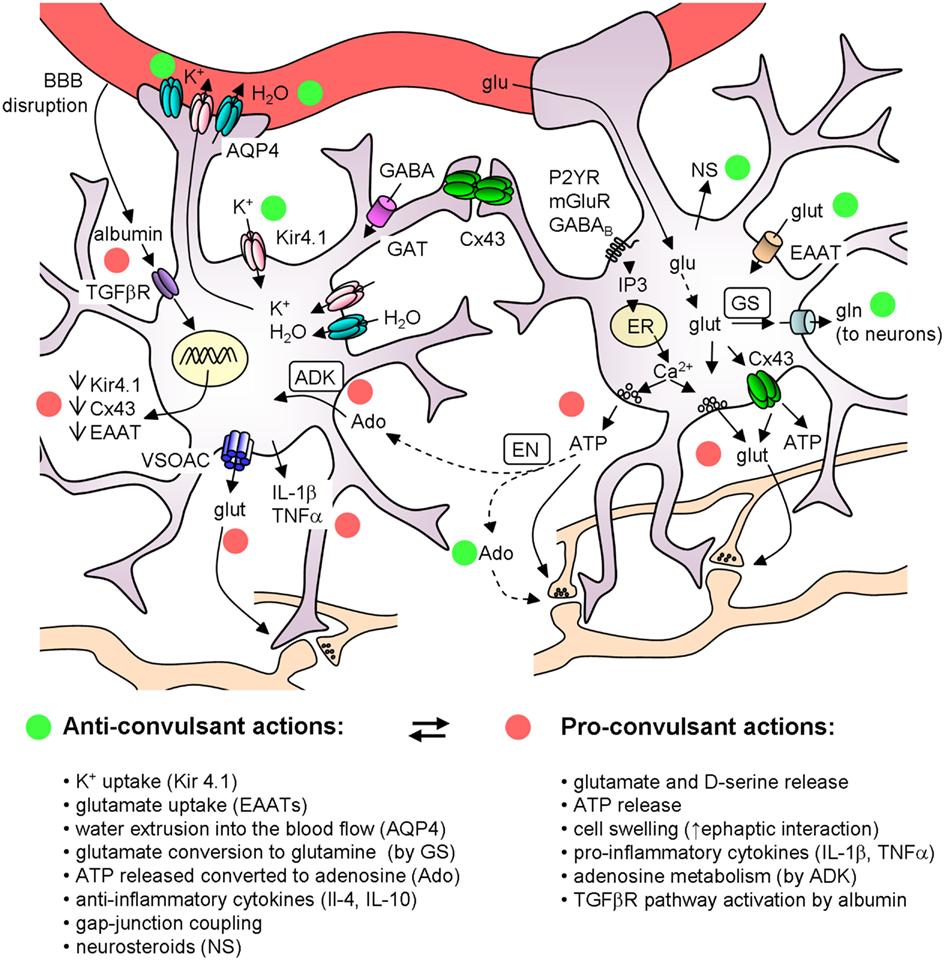
Figure 2. Schematic of the main astrocyte actions that can affect epileptiform activities. The colored circles mark the anticonvulsant (green) and the proconvulsant (red) actions of astrocytes. Note that (i) ATP can have per se a proconvulsant action, but after its conversion to Ado it is anticonvulsant; (ii) astrocytic cytokines can have either pro- or anti-inflammatory actions. ADK, adenosine kinase; Ado, adenosine; AQP4, aquaporin 4; BBB, blood brain barrier; Cx43, connexin 43; EAAT, excitatory amino acid transporters; EN, ectonucleotidase; ER, endoplasmatic reticulum; GAT, GABA transporters; glu, glucose; glut, glutamate; gln, glutamine; GS, glutamine syntethase; NS, neurosteroids; VSOACs, volume-sensitive organic anion channels.
Clarification of the astrocyte role in brain disorders represents one of the most difficult, but also one of the most fascinating challenges in neurobiological research in the years to come.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The original work described in this review was supported by European Union HEALTH-F2-2007-202167, Telethon Italy (GGP10138B; GGP07274) and Cariparo Foundation.
References
Agis-Balboa, R. C., Pinna, G., Zhubi, A., Maloku, E., Veldic, M., Costa, E., and Guidotti, A. (2006). Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 103, 14602–14607.
Amiry-Moghaddam, M., Frydenlund, D. S., and Ottersen, O. P. (2004). Anchoring of aquaporin-4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience 129, 999–1010.
Amiry-Moghaddam, M., Williamson, A., Palomba, M., Eid, T., De Lanerolle, N. C., Nagelhus, E. A., Adams, M. E., Froehner, S. C., Agre, P., and Ottersen, O. P. (2003). Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of α-syntrophin-null mice. Proc. Natl. Acad. Sci. U.S.A. 100, 13615–13620.
Andrew, R. D. (1991). Seizure and acute osmotic change: clinical and neurophysiological aspects. J. Neurol. Sci. 101, 7–18.
Angulo, M. C., Kozlov, A. S., Charpak, S., and Audinat, E. (2004). Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J. Neurosci. 24, 6920–6927.
Araque, A., Martín, E. D., Perea, G., Arellano, J. I., and Buño, W. (2002). Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J. Neurosci. 22, 2443–2450.
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215.
Aronica, E., Van Vliet, E. A., Mayboroda, O. A., Troost, D., Da Silva, F. H., and Gorter, J. A. (2000). Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur. J. Neurosci. 12, 2333–2344.
Aronica, E., Zurolo, E., Iyer, A., De Groot, M., Anink, J., Carbonell, C., Van Vliet, E. A., Baayen, J. C., Boison, D., and Gorter, J. A. (2011). Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 52, 1645–1655.
Avoli, M., and de Curtis, M. (2011). GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog. Neurobiol. 95, 104–132.
Badaut, J., Brunet, J. F., and Regli, L. (2007). Aquaporins in the brain: from aqueduct to “multi-duct”. Metab. Brain Dis. 22, 251–263.
Badaut, J., Lasbennes, F., Magistretti, P. J., and Regli, L. (2002). Aquaporins in brain: distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 22, 367–378.
Balosso, S., Maroso, M., Sanchez-Alavez, M., Ravizza, T., Frasca, A., Bartfai, T., and Vezzani, A. (2008). A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1β. Brain 131, 3256–3265.
Belluardo, N., Mudo, G., Jiang, X. H., and Condorelli, D. F. (1996). Induction of astroglial gene expression by experimental seizures in the rat: spatio-temporal patterns of the early stages. Glia 16, 174–186.
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq, E., Vescovi, A., Bagetta, G., Kollias, G., Meldolesi, J., and Volterra, A. (2001). CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710.
Biagini, G., Baldelli, E., Longo, D., Pradelli, L., Zini, I., Rogawski, M. A., and Avoli, M. (2006). Endogenous neurosteroids modulate epileptogenesis in a model of temporal lobe epilepsy. Exp. Neurol. 201, 519–524.
Biagini, G., Longo, D., Baldelli, E., Zoli, M., Rogawski, M. A., Bertazzoni, G., and Avoli, M. (2009). Neurosteroids and epileptogenesis in the pilocarpine model: evidence for a relationship between P450scc induction and length of the latent period. Epilepsia 50(Suppl. 1), 53–58.
Binder, D. K., Nagelhus, E. A., and Ottersen, O. P. (2012). Aquaporin-4 and epilepsy. Glia 60, 1203–1214.
Binder, D. K., and Steinhauser, C. (2006). Functional changes in astroglial cells in epilepsy. Glia 54, 358–368.
Blackburn, D., Sargsyan, S., Monk, P. N., and Shaw, P. J. (2009). Astrocyte function and role in motor neuron disease: a future therapeutic target? Glia 57, 1251–1264.
Bockenhauer, D., Feather, S., Stanescu, H. C., Bandulik, S., Zdebik, A. A., Reichold, M., Tobin, J., Lieberer, E., Sterner, C., Landoure, G., Arora, R., Sirimanna, T., Thompson, D., Cross, J. H., Van’t Hoff, W., Al Masri, O., Tullus, K., Yeung, S., Anikster, Y., Klootwijk, E., Hubank, M., Dillon, M. J., Heitzmann, D., Arcos-Burgos, M., Knepper, M. A., Dobbie, A., Gahl, W. A., Warth, R., Sheridan, E., and Kleta, R. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 360, 1960–1970.
Boison, D. (2005). Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist 11, 25–36.
Boison, D. (2008). The adenosine kinase hypothesis of epileptogenesis. Prog. Neurobiol. 84, 249–262.
Boison, D. (2010). Adenosine dysfunction and adenosine kinase in epileptogenesis. Open Neurosci. J. 4, 93–101.
Bordey, A., and Sontheimer, H. (1998). Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 32, 286–303.
Borges, K., Gearing, M., McDermott, D. L., Smith, A. B., Almonte, A. G., Wainer, B. H., and Dingledine, R. (2003). Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp. Neurol. 182, 21–34.
Bouilleret, V., Ridoux, V., Depaulis, A., Marescaux, C., Nehlig, A., and Le Gal La Salle, G. (1999). Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience 89, 717–729.
Brockhaus, J., and Deitmer, J. W. (2002). Long-lasting modulation of synaptic input to Purkinje neurons by Bergmann glia stimulation in rat brain slices. J. Physiol. 545, 581–593.
Buono, R. J., Lohoff, F. W., Sander, T., Sperling, M. R., O’Connor, M. J., Dlugos, D. J., Ryan, S. G., Golden, G. T., Zhao, H., Scattergood, T. M., Berrettini, W. H., and Ferraro, T. N. (2004). Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res. 58, 175–183.
Burnstock, G., Krugel, U., Abbracchio, M. P., and Illes, P. (2011). Purinergic signalling: from normal behaviour to pathological brain function. Prog. Neurobiol. 95, 229–274.
Carmignoto, G. (2000). Reciprocal communication systems between astrocytes and neurones. Prog. Neurobiol. 62, 561–581.
Carmignoto, G., and Gomez-Gonzalo, M. (2010). The contribution of astrocyte signalling to neurovascular coupling. Brain Res. Rev. 63, 138–148.
Cavus, I., Kasoff, W. S., Cassaday, M. P., Jacob, R., Gueorguieva, R., Sherwin, R. S., Krystal, J. H., Spencer, D. D., and Abi-Saab, W. M. (2005). Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol. 57, 226–235.
Chebabo, S. R., Hester, M. A., Aitken, P. G., and Somjen, G. G. (1995). Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res. 695, 203–216.
Chever, O., Djukic, B., McCarthy, K. D., and Amzica, F. (2010). Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777.
Coco, S., Calegari, F., Pravettoni, E., Pozzi, D., Taverna, E., Rosa, P., Matteoli, M., and Verderio, C. (2003). Storage and release of ATP from astrocytes in culture. J. Biol. Chem. 278, 1354–1362.
Conti, F., Pinelli, A., and Melone, M. (2004). GABA transporters in the mammalian cerebral cortex: localization, development and pathological implications. Brain Res. Rev. 45, 196–212.
Cope, D. W., Di Giovanni, G., Fyson, S. J., Orban, G., Errington, A. C., Lorincz, M. L., Gould, T. M., Carter, D. A., and Crunelli, V. (2009). Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 15, 1392–1398.
Cossart, R., Bernard, C., and Ben-Ari, Y. (2005). Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 28, 108–115.
Cotrina, M. L., Lin, J. H.-C., Alves-Rodrigues, A., Liu, S., Li, J., Azmi-Ghadimi, H., Kang, J., Naus, C. C. G., and Nedergaard, M. (1998). Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. U.S.A. 95, 15735–15740.
D’Ambrosio, R. (2006). Does glutamate released by astrocytes cause focal epilepsy? Epilepsy Curr. 6, 173–176.
Dani, J. W., Chernjavsky, A., and Smith, S. J. (1992). Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 8, 429–440.
David, Y., Cacheaux, L. P., Ivens, S., Lapilover, E., Heinemann, U., Kaufer, D., and Friedman, A. (2009). Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J. Neurosci. 29, 10588–10599.
Di Castro, M. A., Chuquet, J., Liaudet, N., Bhaukaurally, K., Santello, M., Bouvier, D., Tiret, P., and Volterra, A. (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 14, 1276–1284.
Ding, S., Fellin, T., Zhu, Y., Lee, S. Y., Auberson, Y. P., Meaney, D. F., Coulter, D. A., Carmignoto, G., and Haydon, P. G. (2007). Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 27, 10674–10684.
Djukic, B., Casper, K. B., Philpot, B. D., Chin, L. S., and McCarthy, K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365.
Dudek, F. E., Obenaus, A., and Tasker, J. G. (1990). Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci. Lett. 120, 267–270.
Dudek, F. E., Yasumura, T., and Rash, J. E. (1998). ‘Non-synaptic’ mechanisms in seizures and epileptogenesis. Cell Biol. Int. 22, 793–805.
Duffy, S., and MacVicar, B. A. (1995). Adrenergic calcium signaling in astrocyte networks within the hippocampal slice. J. Neurosci. 15, 5535–5550.
During, M. J., and Spencer, D. D. (1993). Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341, 1607–1610.
Eid, T., Ghosh, A., Wang, Y., Beckstrom, H., Zaveri, H. P., Lee, T. S., Lai, J. C., Malthankar-Phatak, G. H., and De Lanerolle, N. C. (2008a). Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131, 2061–2070.
Eid, T., Williamson, A., Lee, T. S., Petroff, O. A., and De Lanerolle, N. C. (2008b). Glutamate and astrocytes – key players in human mesial temporal lobe epilepsy? Epilepsia 49(Suppl. 2), 42–52.
Eid, T., Thomas, M. J., Spencer, D. D., Runden-Pran, E., Lai, J. C., Malthankar, G. V., Kim, J. H., Danbolt, N. C., Ottersen, O. P., and De Lanerolle, N. C. (2004). Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37.
Fabene, P. F., Bramanti, P., and Constantin, G. (2010). The emerging role for chemokines in epilepsy. J. Neuroimmunol. 224, 22–27.
Fellin, T., D’Ascenzo, M., and Haydon, P. G. (2007). Astrocytes control neuronal excitability in the nucleus accumbens. ScientificWorldJournal 7, 89–97.
Fellin, T., Gomez-Gonzalo, M., Gobbo, S., Carmignoto, G., and Haydon, P. G. (2006a). Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J. Neurosci. 26, 9312–9322.
Fellin, T., Pozzan, T., and Carmignoto, G. (2006b). Purinergic receptors mediate two distinct glutamate release pathways in hippocampal astrocytes. J. Biol. Chem. 281, 4274–4284.
Fellin, T., and Haydon, P. G. (2005). Do astrocytes contribute to excitation underlying seizures? Trends Mol. Med. 11, 530–533.
Fellin, T., Pascual, O., Gobbo, S., Pozzan, T., Haydon, P. G., and Carmignoto, G. (2004). Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43, 729–743.
Ferraro, T. N., Golden, G. T., Smith, G. G., Martin, J. F., Lohoff, F. W., Gieringer, T. A., Zamboni, D., Schwebel, C. L., Press, D. M., Kratzer, S. O., Zhao, H., Berrettini, W. H., and Buono, R. J. (2004). Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm. Genome 15, 239–251.
Florian, C., Vecsey, C. G., Halassa, M. M., Haydon, P. G., and Abel, T. (2011). Astrocyte-derived adenosine and A1 receptor activity contribute to sleep loss-induced deficits in hippocampal synaptic plasticity and memory in mice. J. Neurosci. 31, 6956–6962.
Friedman, A., Kaufer, D., and Heinemann, U. (2009). Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 85, 142–149.
Fritschy, J. M. (2008). Epilepsy, E/I balance and GABAA receptor plasticity. Front. Mol. Neurosci. 1:5. doi:10.3389/neuro.02.005.2008
Glass, M., and Dragunow, M. (1995). Neurochemical and morphological changes associated with human epilepsy. Brain Res. Brain Res. Rev. 21, 29–41.
Gomez-Gonzalo, M., Losi, G., Chiavegato, A., Zonta, M., Cammarota, M., Brondi, M., Vetri, F., Uva, L., Pozzan, T., De Curtis, M., Ratto, G. M., and Carmignoto, G. (2010). An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. 8, e1000352. doi:10.1371/journal.pbio.1000352
Gouder, N., Scheurer, L., Fritschy, J. M., and Boison, D. (2004). Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J. Neurosci. 24, 692–701.
Halassa, M. M., Fellin, T., and Haydon, P. G. (2009). Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 57, 343–346.
Hamby, M. E., and Sofroniew, M. V. (2010). Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7, 494–506.
Haydon, P. G., and Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009–1031.
Heinemann, U., Gabriel, S., Jauch, R., Schulze, K., Kivi, A., Eilers, A., Kovacs, R., and Lehmann, T. N. (2000). Alterations of glial cell function in temporal lobe epilepsy. Epilepsia 41(Suppl. 6), S185–S189.
Heinemann, U., Konnerth, A., Pumain, R., and Wadman, W. J. (1986). Extracellular calcium and potassium concentration changes in chronic epileptic brain tissue. Adv. Neurol. 44, 641–661.
Heinemann, U., Lux, H. D., and Gutnick, M. J. (1977). Extracellular free calcium and potassium during paroxsmal activity in the cerebral cortex of the cat. Exp. Brain Res. 27, 237–243.
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T., and Kurachi, Y. (2001). An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931.
Hinterkeuser, S., Schroder, W., Hager, G., Seifert, G., Blumcke, I., Elger, C. E., Schramm, J., and Steinhauser, C. (2000). Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur. J. Neurosci. 12, 2087–2096.
Hu, S., Sheng, W. S., Ehrlich, L. C., Peterson, P. K., and Chao, C. C. (2000). Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7, 153–159.
Huang, Y. H., Sinha, S. R., Tanaka, K., Rothstein, J. D., and Bergles, D. E. (2004). Astrocyte glutamate transporters regulate metabotropic glutamate receptor-mediated excitation of hippocampal interneurons. J. Neurosci. 24, 4551–4559.
Huber, A., Padrun, V., Deglon, N., Aebischer, P., Mohler, H., and Boison, D. (2001). Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc. Natl. Acad. Sci. U.S.A. 98, 7611–7616.
Inyushin, M., Kucheryavykh, L. Y., Kucheryavykh, Y. V., Nichols, C. G., Buono, R. J., Ferraro, T. N., Skatchkov, S. N., and Eaton, M. J. (2010). Potassium channel activity and glutamate uptake are impaired in astrocytes of seizure-susceptible DBA/2 mice. Epilepsia 51, 1707–1713.
Ishii, M., Horio, Y., Tada, Y., Hibino, H., Inanobe, A., Ito, M., Yamada, M., Gotow, T., Uchiyama, Y., and Kurachi, Y. (1997). Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J. Neurosci. 17, 7725–7735.
Ivens, S., Kaufer, D., Flores, L. P., Bechmann, I., Zumsteg, D., Tomkins, O., Seiffert, E., Heinemann, U., and Friedman, A. H. (2007). TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547.
Jefferys, J. G. R. (2003). Models and mechanisms of experimental epilepsies. Epilepsia 44(Suppl. 12), 44–50.
Jourdain, P., Bergersen, L. H., Bhaukaurally, K., Bezzi, P., Santello, M., Domercq, M., Matute, C., Tonello, F., Gundersen, V., and Volterra, A. (2007). Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 10, 331–339.
Kam, K., and Nicoll, R. (2007). Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J. Neurosci. 27, 9192–9200.
Kang, J., Jiang, L., Goldman, S. A., and Nedergaard, M. (1998). Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692.
Kang, N., Xu, J., Xu, Q., Nedergaard, M., and Kang, J. (2005). Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 94, 4121–4130.
Kivi, A., Lehmann, T. N., Kovacs, R., Eilers, A., Jauch, R., Meencke, H. J., Von Deimling, A., Heinemann, U., and Gabriel, S. (2000). Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur. J. Neurosci. 12, 2039–2048.
Kofuji, P., Ceelen, P., Zahs, K. R., Surbeck, L. W., Lester, H. A., and Newman, E. A. (2000). Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J. Neurosci. 20, 5733–5740.
Kofuji, P., and Newman, E. A. (2004). Potassium buffering in the central nervous system. Neuroscience 129, 1045–1056.
Koizumi, S. (2010). Synchronization of Ca2+ oscillations: involvement of ATP release in astrocytes. FEBS J. 277, 286–292.
Konnerth, A., Heinemann, U., and Yaari, Y. (1986). Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. I. Development of seizurelike activity in low extracellular calcium. J. Neurophysiol. 56, 409–423.
Kucheryavykh, Y. V., Kucheryavykh, L. Y., Nichols, C. G., Maldonado, H. M., Baksi, K., Reichenbach, A., Skatchkov, S. N., and Eaton, M. J. (2007). Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55, 274–281.
Kuffler, S. W., Nicholls, J. G., and Orkand, R. K. (1966). Physiological properties of glial cells in the central nervous system of amphibia. J. Neurophysiol. 29, 768–787.
Kulik, A., Haentzsch, A., Luckermann, M., Reichelt, W., and Ballanyi, K. (1999). Neuron-glia signaling via α1 adrenoceptor-mediated Ca2+ release in Bergmann glial cells in situ. J. Neurosci. 19, 8401–8408.
Kumaria, A., Tolias, C. M., and Burnstock, G. (2008). ATP signalling in epilepsy. Purinergic Signal. 4, 339–346.
Lambert, J. J., Belelli, D., Peden, D. R., Vardy, A. W., and Peters, J. A. (2003). Neurosteroid modulation of GABAA receptors. Prog. Neurobiol. 71, 67–80.
Lee, D. J., Hsu, M. S., Seldin, M. M., Arellano, J. L., and Binder, D. K. (2012). Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp. Neurol. 235, 246–255.
Lee, T. S., Eid, T., Mane, S., Kim, J. H., Spencer, D. D., Ottersen, O. P., and De Lanerolle, N. C. (2004). Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol. 108, 493–502.
Lenzen, K. P., Heils, A., Lorenz, S., Hempelmann, A., Hofels, S., Lohoff, F. W., Schmitz, B., and Sander, T. (2005). Supportive evidence for an allelic association of the human KCNJ10 potassium channel gene with idiopathic generalized epilepsy. Epilepsy Res. 63, 113–118.
Li, L., Head, V., and Timpe, L. C. (2001). Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia 33, 57–71.
Li, T., Lan, J. Q., and Boison, D. (2008). Uncoupling of astrogliosis from epileptogenesis in adenosine kinase (ADK) transgenic mice. Neuron Glia Biol. 4, 91–99.
Li, T., Quan Lan, J., Fredholm, B. B., Simon, R. P., and Boison, D. (2007). Adenosine dysfunction in astrogliosis: cause for seizure generation? Neuron Glia Biol. 3, 353–366.
Liang, S. L., Carlson, G. C., and Coulter, D. A. (2006). Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J. Neurosci. 26, 8537–8548.
Lopantsev, V., Both, M., and Draguhn, A. (2009). Rapid plasticity at inhibitory and excitatory synapses in the hippocampus induced by ictal epileptiform discharges. Eur. J. Neurosci. 29, 1153–1164.
Losi, G., Cammarota, M., Chiavegato, A., Gomez-Gonzalo, M., and Carmignoto, G. (2010). A new experimental model of focal seizures in the entorhinal cortex. Epilepsia 51, 1493–1502.
Madsen, K. K., White, H. S., and Schousboe, A. (2010). Neuronal and non-neuronal GABA transporters as targets for antiepileptic drugs. Pharmacol. Ther. 125, 394–401.
Maragakis, N. J., and Rothstein, J. D. (2006). Mechanisms of disease: astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2, 679–689.
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., Rossetti, C., Molteni, M., Casalgrandi, M., Manfredi, A. A., Bianchi, M. E., and Vezzani, A. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419.
Mathern, G. W., Mendoza, D., Lozada, A., Pretorius, J. K., Dehnes, Y., Danbolt, N. C., Nelson, N., Leite, J. P., Chimelli, L., Born, D. E., Sakamoto, A. C., Assirati, J. A., Fried, I., Peacock, W. J., Ojemann, G. A., and Adelson, P. D. (1999). Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 52, 453–472.
McKhann, G. M. II, Schoenfeld-McNeill, J., Born, D. E., Haglund, M. M., and Ojemann, G. A. (2000). Intraoperative hippocampal electrocorticography to predict the extent of hippocampal resection in temporal lobe epilepsy surgery. J. Neurosurg. 93, 44–52.
Medici, V., Frassoni, C., Tassi, L., Spreafico, R., and Garbelli, R. (2011). Aquaporin 4 expression in control and epileptic human cerebral cortex. Brain Res. 1367, 330–339.
Mensah-Nyagan, A. G., Do-Rego, J. L., Beaujean, D., Luu-the, V., Pelletier, G., and Vaudry, H. (1999). Neurosteroids: expression of steroidogenic enzymes and regulation of steroid biosynthesis in the central nervous system. Pharmacol. Rev. 51, 63–81.
Min, R., and Nevian, T. (2012). Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat. Neurosci. 15, 746–753.
Müller, S., Ronfani, L., and Bianchi, M. E. (2004). Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J. Intern. Med. 255, 332–343.
Nagelhus, E. A., Mathiisen, T. M., and Ottersen, O. P. (2004). Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 129, 905–913.
Navarrete, M., and Araque, A. (2008). Endocannabinoids mediate neuron-astrocyte communication. Neuron 57, 883–893.
Navarrete, M., and Araque, A. (2010). Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68, 113–126.
Navarrete, M., Perea, G., De Sevilla, D. F., Gomez-Gonzalo, M., Nunez, A., Martin, E. D., and Araque, A. (2012). Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10, e1001259. doi:10.1371/journal.pbio.1001259
Nedergaard, M. (1994). Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science 263, 1768–1771.
Neusch, C., Papadopoulos, N., Muller, M., Maletzki, I., Winter, S. M., Hirrlinger, J., Handschuh, M., Bahr, M., Richter, D. W., Kirchhoff, F., and Hulsmann, S. (2006). Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J. Neurophysiol. 95, 1843–1852.
Newman, E. A. (1993). Inward-rectifying potassium channels in retinal glial (Müller) cells. J. Neurosci. 13, 3333–3345.
Ngugi, A. K., Bottomley, C., Kleinschmidt, I., Sander, J. W., and Newton, C. R. (2010). Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia 51, 883–890.
Nielsen, S., Nagelhus, E. A., Amiry-Moghaddam, M., Bourque, C., Agre, P., and Ottersen, O. P. (1997). Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 17, 171–180.
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H., Wang, F., Xu, Q., Wyatt, J. D., Pilcher, W., Ojemann, J. G., Ransom, B. R., Goldman, S. A., and Nedergaard, M. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287.
Oberheim, N. A., Wang, X., Goldman, S., and Nedergaard, M. (2006). Astrocytic complexity distinguishes the human brain. Trends Neurosci. 29, 547–553.
Olsen, M. L., and Sontheimer, H. (2008). Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J. Neurochem. 107, 589–601.
Orkand, R. K., Nicholls, J. G., and Kuffler, S. W. (1966). Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 29, 788–806.
Ortinski, P. I., Dong, J., Mungenast, A., Yue, C., Takano, H., Watson, D. J., Haydon, P. G., and Coulter, D. A. (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591.
Otis, T. S., and Jahr, C. E. (1998). Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J. Neurosci. 18, 7099–7110.
Pan, E., and Stringer, J. L. (1996). Influence of osmolality on seizure amplitude and propagation in the rat dentate gyrus. Neurosci. Lett. 207, 9–12.
Panatier, A., Theodosis, D. T., Mothet, J.-P., Toquet, B., Pollegioni, L., Poulain, D. A., and Oliet, S. H. R. (2006). Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784.
Panatier, A., Vallee, J., Haber, M., Murai, K. K., Lacaille, J. C., and Robitaille, R. (2011). Astrocytes are endogenous regulators of Basal transmission at central synapses. Cell 146, 785–798.
Parpura, V., Basarsky, T. A., Liu, F., Jeftinija, K., Jeftinija, S., and Haydon, P. G. (1994). Glutamate-mediated astrocyte-neuron signalling. Nature 369, 744–747.
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J. Y., Takano, H., Moss, S. J., McCarthy, K., and Haydon, P. G. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116.
Pasti, L., Volterra, A., Pozzan, T., and Carmignoto, G. (1997). Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 17, 7817–7830.
Pedley, T. A., Fisher, R. S., Futamachi, K. J., and Prince, D. A. (1976). Regulation of extracellular potassium concentration in epileptogenesis. Fed. Proc. 35, 1254–1259.
Pedrazzi, M., Patrone, M., Passalacqua, M., Ranzato, E., Colamassaro, D., Sparatore, B., Pontremoli, S., and Melloni, E. (2007). Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J. Immunol. 179, 8525–8532.
Perea, G., and Araque, A. (2005). Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J. Neurosci. 25, 2192–2203.
Perea, G., and Araque, A. (2007). Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317, 1083–1086.
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431.
Petzold, G. C., and Murthy, V. N. (2011). Role of astrocytes in neurovascular coupling. Neuron 71, 782–797.
Poopalasundaram, S., Knott, C., Shamotienko, O. G., Foran, P. G., Dolly, J. O., Ghiani, C. A., Gallo, V., and Wilkin, G. P. (2000). Glial heterogeneity in expression of the inwardly rectifying K+ channel, Kir4.1, in adult rat CNS. Glia 30, 362–372.
Porter, J. T., and McCarthy, K. D. (1996). Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci. 16, 5073–5081.
Prince, D. A., and Wilder, B. J. (1967). Control mechanisms in cortical epileptogenic foci. “Surround” inhibition. Arch. Neurol. 16, 194–202.
Proper, E. A., Hoogland, G., Kappen, S. M., Jansen, G. H., Rensen, M. G., Schrama, L. H., Van Veelen, C. W., Van Rijen, P. C., Van Nieuwenhuizen, O., Gispen, W. H., and De Graan, P. N. (2002). Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125, 32–43.
Ransom, C. B., and Sontheimer, H. (1995). Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J. Neurophysiol. 73, 333–346.
Reddy, D. S. (2010). Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. 186, 113–137.
Reichold, M., Zdebik, A. A., Lieberer, E., Rapedius, M., Schmidt, K., Bandulik, S., Sterner, C., Tegtmeier, I., Penton, D., Baukrowitz, T., Hulton, S. A., Witzgall, R., Ben-Zeev, B., Howie, A. J., Kleta, R., Bockenhauer, D., and Warth, R. (2010). KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. U.S.A. 107, 14490–14495.
Represa, A., Niquet, J., Pollard, H., and Ben-Ari, Y. (1995). Cell death, gliosis, and synaptic remodeling in the hippocampus of epileptic rats. J. Neurobiol. 26, 413–425.
Rizzi, M., Perego, C., Aliprandi, M., Richichi, C., Ravizza, T., Colella, D., Veliskova, J., Moshe, S. L., De Simoni, M. G., and Vezzani, A. (2003). Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiol. Dis. 14, 494–503.
Rogawski, M. A., and Loscher, W. (2004). The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 5, 553–564.
Roper, S. N., Obenaus, A., and Dudek, F. E. (1992). Osmolality and nonsynaptic epileptiform bursts in rat CA1 and dentate gyrus. Ann. Neurol. 31, 81–85.
Rossi, S., Furlan, R., De Chiara, V., Motta, C., Studer, V., Mori, F., Musella, A., Bergami, A., Muzio, L., Bernardi, G., Battistini, L., Martino, G., and Centonze, D. (2012). Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann. Neurol. 71, 76–83.
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., Kanai, Y., Hediger, M. A., Wang, Y., Schielke, J. P., and Welty, D. F. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686.
Rothstein, J. D., Martin, L., Levey, A. I., Dykes-Hoberg, M., Jin, L., Wu, D., Nash, N., and Kuncl, R. W. (1994). Localization of neuronal and glial glutamate transporters. Neuron 13, 713–725.
Sakmann, B., and Trube, G. (1984). Voltage-dependent inactivation of inward-rectifying single-channel currents in the guinea-pig heart cell membrane. J. Physiol. 347, 659–683.
Santello, M., Bezzi, P., and Volterra, A. (2011). TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69, 988–1001.
Sarac, S., Afzal, S., Broholm, H., Madsen, F. F., Ploug, T., and Laursen, H. (2009). Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 117, 291–301.
Scholl, U. I., Choi, M., Liu, T., Ramaekers, V. T., Hausler, M. G., Grimmer, J., Tobe, S. W., Farhi, A., Nelson-Williams, C., and Lifton, R. P. (2009). Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847.
Schroder, W., Hinterkeuser, S., Seifert, G., Schramm, J., Jabs, R., Wilkin, G. P., and Steinhauser, C. (2000). Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia 41(Suppl. 6), S181–S184.
Seifert, G., Carmignoto, G., and Steinhauser, C. (2010). Astrocyte dysfunction in epilepsy. Brain Res. Rev. 63, 212–221.
Seifert, G., Schilling, K., and Steinhauser, C. (2006). Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat. Rev. Neurosci. 7, 194–206.
Seiffert, E., Dreier, J. P., Ivens, S., Bechmann, I., Tomkins, O., Heinemann, U., and Friedman, A. (2004). Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J. Neurosci. 24, 7829–7836.
Serrano, A., Haddjeri, N., Lacaille, J. C., and Robitaille, R. (2006). GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J. Neurosci. 26, 5370–5382.
Shelton, M. K., and McCarthy, K. D. (2000). Hippocampal astrocytes exhibit Ca2+-elevating muscarinic cholinergic and histaminergic receptors in situ. J. Neurochem. 74, 555–563.
Shigetomi, E., Tong, X., Kwan, K. Y., Corey, D. P., and Khakh, B. S. (2011). TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 15, 70–80.
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R., and Coyle, A. J. (2010). HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388.
Skucas, V. A., Mathews, I. B., Yang, J., Cheng, Q., Treister, A., Duffy, A. M., Verkman, A. S., Hempstead, B. L., Wood, M. A., Binder, D. K., and Scharfman, H. E. (2011). Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J. Neurosci. 31, 6392–6397.
Takano, T., Tian, G.-F., Peng, W., Lou, N., Libionka, W., Han, X., and Nedergaard, M. (2006). Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267.
Takumi, T., Ishii, T., Horio, Y., Morishige, K., Takahashi, N., Yamada, M., Yamashita, T., Kiyama, H., Sohmiya, K., Nakanishi, S., and Kurachi, Y. (1995). A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J. Biol. Chem. 270, 16339–16346.
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., Iwama, H., Nishikawa, T., Ichihara, N., Kikuchi, T., Okuyama, S., Kawashima, N., Hori, S., Takimoto, M., and Wada, K. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702.
Tessler, S., Danbolt, N. C., Faull, R. L., Storm-Mathisen, J., and Emson, P. C. (1999). Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience 88, 1083–1091.
Theofilas, P., Brar, S., Stewart, K. A., Shen, H. Y., Sandau, U. S., Poulsen, D., and Boison, D. (2011). Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 52, 589–601.
Thrane, A. S., Rappold, P. M., Fujita, T., Torres, A., Bekar, L. K., Takano, T., Peng, W., Wang, F., Thrane, V. R., Enger, R., Haj-Yasein, N. N., Skare, O., Holen, T., Klungland, A., Ottersen, O. P., Nedergaard, M., and Nagelhus, E. A. (2011). Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc. Natl. Acad. Sci. U.S.A. 108, 846–851.
Thurman, D. J., Beghi, E., Begley, C. E., Berg, A. T., Buchhalter, J. R., Ding, D., Hesdorffer, D. C., Hauser, W. A., Kazis, L., Kobau, R., Kroner, B., Labiner, D., Liow, K., Logroscino, G., Medina, M. T., Newton, C. R., Parko, K., Paschal, A., Preux, P. M., Sander, J. W., Selassie, A., Theodore, W., Tomson, T., and Wiebe, S. (2011). Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 52(Suppl. 7), 2–26.
Tian, G.-F., Azmi, H., Takano, T., Xu, Q., Peng, W., Lin, J., Oberheim, N., Lou, N., Wang, X., Zielke, H. R., Kang, J., and Nedergaard, M. (2005). An astrocytic basis of epilepsy. Nat. Med. 11, 973–981.
Tomkins, O., Friedman, O., Ivens, S., Reiffurth, C., Major, S., Dreier, J. P., Heinemann, U., and Friedman, A. (2007). Blood-brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol. Dis. 25, 367–377.
Torres, A., Wang, F., Xu, Q., Fujita, T., Dobrowolski, R., Willecke, K., Takano, T., and Nedergaard, M. (2012). Extracellular Ca2+ acts as a mediator of communication from neurons to glia. Sci. Signal. 5, ra8.
Turrin, N. P., and Rivest, S. (2004). Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol. Dis. 16, 321–334.
Ulas, J., Satou, T., Ivins, K. J., Kesslak, J. P., Cotman, C. W., and Balazs, R. (2000). Expression of metabotropic glutamate receptor 5 is increased in astrocytes after kainate-induced epileptic seizures. Glia 30, 352–361.
van der Hel, W. S., Notenboom, R. G., Bos, I. W., Van Rijen, P. C., Van Veelen, C. W., and De Graan, P. N. (2005). Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64, 326–333.
Vezzani, A., Balosso, S., and Ravizza, T. (2008). The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 22, 797–803.
Vezzani, A., and Granata, T. (2005). Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46, 1724–1743.
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., Binaglia, M., Corsini, E., Di Luca, M., Galli, C. L., and Marinovich, M. (2003). Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700.
Wang, Y., Zaveri, H. P., Lee, T. S., and Eid, T. (2009). The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp. Neurol. 220, 293–302.
Wetherington, J., Serrano, G., and Dingledine, R. (2008). Astrocytes in the epileptic brain. Neuron 58, 168–178.
Ye, Z. C., and Sontheimer, H. (1996). Cytokine modulation of glial glutamate uptake: a possible involvement of nitric oxide. Neuroreport 7, 2181–2185.
Zhang, J.-M., Wang, H.-K., Ye, C.-Q., Ge, W., Chen, Y., Jiang, Z.-L., Wu, C.-P., Poo, M.-M., and Duan, S. (2003). ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40, 971–982.
Zonta, M., Angulo, M. C., Gobbo, S., Rosengarten, B., Hossmann, K.-A., Pozzan, T., and Carmignoto, G. (2003). Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 6, 43–50.
Keywords: astrocyte, epilepsy, seizures, glial-neuronal interactions, ictal event
Citation: Losi G, Cammarota M and Carmignoto G (2012) The role of astroglia in the epileptic brain. Front. Pharmacol. 3:132. doi: 10.3389/fphar.2012.00132
Received: 12 April 2012; Accepted: 23 June 2012;
Published online: 12 July 2012.
Edited by:
Roberto C. Sotero, McGill University, CanadaReviewed by:
Brenda Bartnik-Olson, Loma Linda University, USARoberto C. Sotero, McGill University, Canada
Copyright: © 2012 Losi, Cammarota and Carmignoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Giorgio Carmignoto, Institute of Neuroscience of the National Research Council and Department of Biomedical Sciences, University of Padova, Viale G. Colombo 3, 35121 Padova, Italy. e-mail:Z2lvcmdpby5jYXJtaWdub3RvQGJpby51bmlwZC5pdA==