
 Min-Yu Sun2,3
Min-Yu Sun2,3- 1UFR Biomédicale, CNRS UMR 8154, Université Paris Descartes, Paris, France
- 2Department of Cell Biology and Neuroscience, Center for Glial-Neuronal Interactions, University of California Riverside, Riverside, CA, USA
- 3Program in Cellular, Molecular, and Developmental Biology, University of California Riverside, Riverside, CA, USA
A prominent area of neuroscience research over the past 20 years has been the acute modulation of neuronal synaptic activity by Ca2+-dependent release of the transmitters ATP, D-serine, and glutamate (called gliotransmitters) by astrocytes. Although the physiological relevance of this mechanism is under debate, emerging evidence suggests that there are critical factors in addition to Ca2+ that are required for gliotransmitters to be released from astrocytes. Interestingly, these factors include activated microglia and the proinflammatory cytokine Tumor Necrosis Factor α (TNFα), chemotactic cytokine Stromal cell-Derived Factor-1α (SDF-1α), and inflammatory mediator prostaglandin E2 (PGE2). Of note, microglial activation and release of inflammatory molecules from activated microglia and reactive astrocytes can occur within minutes of a triggering stimulus. Therefore, activation of astrocytes by inflammatory molecules combined with Ca2+ elevations may lead to gliotransmitter release, and be an important step in the early sequence of events contributing to hyperexcitability, excitotoxicity, and neurodegeneration in the damaged or diseased brain. In this review, we will first examine evidence questioning Ca2+-dependent gliotransmitter release from astrocytes in healthy brain tissue, followed by a close examination of recent work suggesting that Ca2+-dependent gliotransmitter release occurs as an early event in the development of neurological disorders and neuroinflammatory and neurodegenerative diseases.
Introduction
Gliotransmission – defined as the rapid and regulated exocytosis of gliotransmitters by astrocytes in an activity and Gq G-protein-coupled receptor (GPCR), Ca2+-dependent manner – has continued to be an exciting area of neuroscience research ever since its initial discovery in 1994 (Parpura et al., 1994). Because the tight regulation provided by Ca2+ elevations and release by quantal-like vesicular exocytosis are typical properties of a physiological process (Bezzi et al., 2001), astrocytes have been touted as active, not only supportive, partners in the control of rapid synaptic communication in the healthy brain (Haydon, 2001; Volterra and Meldolesi, 2005; Perea et al., 2009; Santello and Volterra, 2009).
However, studies using novel techniques to selectively evoke or block Gq GPCR-mediated Ca2+ elevations in astrocytes have failed to observe changes in neuronal excitatory synaptic activity indicative of gliotransmission (Fiacco et al., 2007; Petravicz et al., 2008; Agulhon et al., 2010). This has raised controversy as to whether gliotransmission actually occurs in healthy brain tissue. Emerging evidence is providing a possible explanation for this discrepancy: Ca2+ elevations alone are not sufficient for gliotransmitter release by astrocytes. Additional factors such as inflammatory molecules appear to be required. These molecules may not be present in adequate quantities in healthy brain tissue to elicit sufficient gliotransmitter release to affect synaptic transmission. However, accumulating evidence suggests that in the early steps of inflammatory processes and in the diseased or damaged brain, activated microglia, and astrocytes engage in a partnership that transforms astrocytes, within seconds to minutes, into competent gliotransmitter releasing cells. As astrocytic gliotransmitters include ATP, D-serine, and glutamate, reactive astrocytes could increase neuronal excitability and contribute to excitotoxicity, synaptic damage, and pathogenesis of disease. Therefore, inflammatory transduction pathways that regulate Ca2+-dependent release of gliotransmitters from astrocytes are potential targets for therapy of neurological disorders as well as neuroinflammatory and neurodegenerative disease.
In this review, we will first examine evidence questioning Ca2+-dependent gliotransmitter release by astrocytes in the healthy brain, including the mechanisms driving astrocyte Ca2+ elevations, differences in basal (resting) Ca2+ vs. activity-induced Ca2+ elevations, and the temporal relationship between activation of postsynaptic receptors and astrocyte Ca2+ elevations. We will then review evidence suggesting that activated microglia and release of inflammatory mediators transform astrocytes into cells capable of releasing gliotransmitters in a Ca2+-dependent manner in the damaged or diseased brain.
Evidence Questioning Gliotransmission in Healthy Brain Tissue
What is Gliotransmission?
The process of gliotransmission has been defined as analogous to neurotransmission, except that the source of the transmitters is glia (i.e., astrocytes) rather than neurons (Volterra and Meldolesi, 2005). The other properties are essentially the same: a fast, Ca2+-dependent exocytosis of transmitters resulting in stimulation of synaptic receptors and acute synaptic modulation.
Mechanisms of Astrocyte Ca2+ Elevations
It is first important to point out that there is no firm definition of an astrocyte. If the definition is exclusively morphological, i.e., highly ramified star-shaped cells, then the so-called “complex” glia could be included as an astrocyte subtype (see Nishiyama et al., 2005). This broad classification results in considerable heterogeneity in the astrocyte population and can be problematic when considering sources of Ca2+ elevations in astrocytes. Complex glia, so named because of their complex current pattern evoked using voltage-step protocols, have also been referred to as GluR cells due to their expression of functional ionotropic glutamate receptors (iGluRs; Matthias et al., 2003; Jabs et al., 2005). Evidence suggests that the AMPA iGluRs expressed by complex glia and Bergmann glia of the cerebellum are of the Ca2+-permeable variety, thereby providing a source for fast focal Ca2+ elevations in these cells (Lin and Bergles, 2004; Lin et al., 2005; Piet and Jahr, 2007; Hamilton et al., 2010). In developing brain tissue, complex glia also include immature passive astrocytes (Walz, 2000; Zhou et al., 2006), but in the mature brain they appear to be a unique cell population called NG2+ glia due to their surface expression of NG2 proteoglycan. NG2+ glial cells have a star-shaped morphology and do not fire action potentials, but otherwise differ from the subpopulation of mature passive astrocytes in nearly every other way (reviewed in Lin and Bergles, 2002). Importantly, there is little convincing evidence to date that passive astrocytes in intact tissue express neurotransmitter-gated ionotropic receptor types that could provide a source for fast local Ca2+ elevations. Because of these differences, inclusion of complex glia or specialized astrocytes such as Bergmann glia in the sampled population of “astrocytes” could lead to very different conclusions about astrocyte signaling properties. In this review, for clarity our definition of astrocyte includes the protoplasmic passive subtype only, which are the glial fibrillary acidic protein (GFAP)-expressing glial cells that express glutamate transporters, are coupled by gap junctions, and whose processes envelop synapses and form endfeet on the cerebrovasculature.
In addition to lacking Ca2+-permeable ligand-gated ion channels, there is also evidence that astrocytes do not express voltage-gated calcium channels (VGCCs; Carmignoto et al., 1998), although the expression of VGCCs in passive astrocytes may still be a matter of debate. We have assayed for functional expression of astrocytic VGCCs as well by applying strong trains of depolarizing stimuli via whole-cell patch pipette (Figure 1). The depolarizing stimuli did not evoke Ca2+ elevations anywhere in the astrocyte, which nevertheless exhibited spontaneous Ca2+ elevations and responded to bath application of a cocktail of agonists to endogenous Gq GPCRs. In summary, it does not appear that astrocytes possess a fast source of Ca2+ elevations capable of responding to neuronal inputs on a millisecond timescale. This also distinguishes gliotransmission from neurotransmission, which is triggered by the rapid gating of presynaptic VGCCs located adjacent to a readily releasable pool of synaptic vesicles.
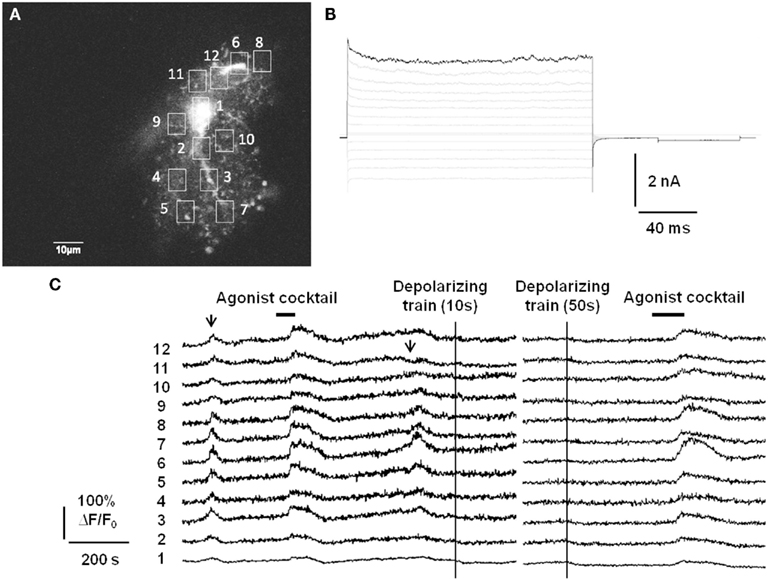
Figure 1. Passive astrocytes do not express functional voltage-gated calcium channels. (A) Astrocytes in stratum radiatum of acute hippocampal slices were patch-clamped with a small pipette tip (7–9 MΩ) in order to limit dialysis of the pipette contents during the recording. Included in the pipette was 150 μM of the cell impermeant Ca2+ indicator Oregon Green BAPTA-1 (OGB-1). (B) Passive mature astrocytes were characterized by the absence of voltage-gated currents evoked by a voltage-step protocol (−180 to +80 mV in 20 mV increments). (C) In 6/10 astrocytes from 10 slices, despite whole-cell voltage clamp the astrocytes exhibited spontaneous Ca2+ elevations (arrows) and evoked Ca2+ responses to an agonist cocktail consisting of 10 μM each of the Gq GPCR agonists histamine, carbachol, and ATP. However, strong depolarizing trains (−90 to 0 mV at 1 Hz for 10 or 50 s) through the patch pipette did not produce any increases in fluorescence, indicating a lack of voltage-gated Ca2+ channels in astrocytes. The remaining 4/10 astrocytes did not respond to depolarization or agonist cocktail with Ca2+ elevations, but did respond to agonist cocktail after pipette removal. This is presumably an effect of dialysis of the cell by the whole-cell recording pipette.
The source of astrocyte Ca2+ elevations appears to be almost exclusively Gq GPCR activated, inositol 1,4,5-triphosphate receptor (IP3R)-sensitive intracellular stores, involving the canonical phospholipase C (PLC)/IP3 pathway. Upon Gq GPCR activation, PLC hydrolyzes the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to generate diacylglycerol (DAG) and IP3, leading to IP3R activation and Ca2+ release from the endoplasmic reticulum (ER). The lines of evidence for Gq GPCR-mediated Ca2+ sources in astrocytes are multiple. First, there is strong evidence that astrocytes express Gq GPCRs for endogenous neurotransmitters (Porter and McCarthy, 1997) that can be activated by bath application of various agonists (e.g., Shelton and McCarthy, 2000). Second, Ca2+ elevations evoked by neuronal afferent stimulation in situ or by sensory stimulation in vivo are significantly inhibited by application of specific Gq GPCR antagonists (Porter and McCarthy, 1996; Perea and Araque, 2005; Wang et al., 2006). Third, spontaneous astrocytic Ca2+ transients are inhibited by the G-protein inhibitor GDP-βs, the IP3R antagonist heparin, or by depletion of internal Ca2+ stores using thapsigargin (Nett et al., 2002; Di Castro et al., 2011). Last, spontaneous and evoked astrocyte Ca2+ elevations are almost completely abolished by removal of the astrocyte-specific IP3R, the IP3R type 2 (IP3R2; Petravicz et al., 2008; Agulhon et al., 2010; Di Castro et al., 2011; Takata et al., 2011; Navarrete et al., 2012). Interestingly, one study has reported small residual and infrequent Ca2+ elevations remaining in astrocytic fine processes in IP3R2 knockout mice (Di Castro et al., 2011). The source of these Ca2+ elevations remains to be determined, but they may be Gq GPCR-independent and play a role in setting basal or resting Ca2+ levels in astrocytes (see below). Nevertheless, taken together the above evidence indicates that both evoked and spontaneous Ca2+ elevations in astrocytes are driven almost exclusively by Gq GPCR-coupled release from intracellular stores.
Are Astrocytic Gq GPCR-Mediated Ca2+ Elevations Sufficient to Induce Gliotransmitter Release?
Early work demonstrating gliotransmission in astrocyte-neuron co-cultures provided evidence that Ca2+ was “necessary and sufficient” for this process (Araque et al., 1998, 1999; Parpura and Haydon, 2000). Around this same period of time the emerging evidence that astrocytes in situ expressed numerous Gq-coupled metabotropic receptor types that could be activated by neuronal activity set the stage for the concept of gliotransmission; i.e., that spillover of synaptically released neurotransmitter stimulates astrocytic Gq GPCR signaling cascades, resulting in astrocytic Ca2+ elevations that reciprocally and acutely modulate synaptic transmission through release of gliotransmitters (Volterra and Meldolesi, 2005; Halassa et al., 2007). More recent work has called into question the Ca2+-dependency of gliotransmission. To test the hypothesis that physiologically relevant astrocyte Ca2+ elevations results in gliotransmission required that astrocytic Gq GPCRs actually be stimulated. This was problematic in intact tissue since bath application of Gq GPCR agonists such as the group I mGluR agonist DHPG directly stimulates metabotropic glutamate receptors (mGluRs) on both astrocytes and neurons as well as other cell types including microglia (Pocock and Kettenmann, 2007; Farso et al., 2009). Therefore, transgenic tools were developed to selectively stimulate or eliminate astrocytic Gq GPCR-mediated Ca2+ elevations (Fiacco et al., 2007; Petravicz et al., 2008; Agulhon et al., 2010). Surprisingly, selective stimulation or removal of astrocyte Gq GPCR-mediated Ca2+ elevations had no effect on CA1 pyramidal neuron excitatory synaptic transmission and hippocampal short- and long-term plasticity (LTP). These findings suggested that astrocyte Ca2+ elevations are not sufficient for gliotransmission in acute hippocampal slices. The results of these studies have already been well-documented as they have led to considerable debate as to whether the mechanisms of gliotransmission, as they have been conceived, are physiologically relevant (Kimelberg, 2007; Hamilton and Attwell, 2010; Kirchhoff, 2010; Smith, 2010).
What could be the source of activity-induced astrocyte Ca2+ elevations responsible for gliotransmission then, if it is not from iGluRs, VGCCs, or Gq GPCRs? It has been recently pointed out that, due to the tiny size of fine astrocyte processes (20–30 nm) surrounding synapses, the relevant Ca2+ elevations involved in gliotransmission may be too small, too fast, and too local to detect using available Ca2+ indicators and two-photon imaging methods (Rusakov et al., 2011). However, these putative Ca2+ sources would not likely include Gq GPCRs/IP3Rs, as the 20- to 30-nm, thin sheet-like processes of astrocytes frequently surrounding synapses are most often devoid of ER (Peters et al., 1991). Furthermore, numerous reports have provided evidence to suggest that only very large astrocyte Ca2+ elevations evoked using strong afferent stimulation of multiple inputs or uncaging of IP3 or Ca2+ may be sufficient to release sufficient gliotransmitter from diffusely scattered synaptic-like microvesicles to overcome reuptake by astrocytic and neuronal transporters to stimulate neuronal receptors (Fiacco and McCarthy, 2004; Takata et al., 2011; Navarrete et al., 2012). A more intriguing possibility is that gliotransmitter release is dependent on other signaling molecules produced by activated microglia and reactive astrocytes in addition to astrocyte Ca2+ (e.g., Santello et al., 2011), as will be discussed further below.
Additional evidence questioning Ca2+-dependent release of gliotransmitters by astrocytes in healthy brain tissue is the lack of propagating intercellular astrocyte Ca2+ waves. The original report of intercellular astrocyte Ca2+ waves in cultured astrocytes created quite a bit of excitement as it was postulated as a mechanism for long-distance signaling in the brain by an otherwise electrically non-excitable cell type (Cornell-Bell et al., 1990). Later work revealed that the mechanism underlying this phenomenon is release of the gliotransmitter ATP, which binds P2Y purinergic Gq GPCRs on adjacent astrocytes resulting in Ca2+ liberation from internal stores, ATP release, and thus wave regeneration and propagation (Cotrina et al., 2000; Arcuino et al., 2002; Bowser and Khakh, 2007). Propagating glial Ca2+ waves have also been observed in the white matter of neonatal brain tissue (Schipke et al., 2002) and in the retina (Newman, 2001), but not in cortical gray matter astrocytes in situ or in vivo (Fiacco and McCarthy, 2006; Kuchibhotla et al., 2009). We have performed additional experiments to assay for Ca2+ wave propagation between astrocytes using acute hippocampal slices from transgenic mice in which a large percentage of astrocytes selectively express the MrgA1 Gq GPCR (Fiacco et al., 2007; Figure 2). The design of these experiments is very simple: astrocytes were bulk-loaded with Ca2+ indicator and the MrgA1 receptor agonist FMRF and the group I mGluR agonist DHPG were applied in succession. Most astrocytes respond with characteristic long duration Ca2+ elevations to both agonists, but occasionally there are astrocytes that respond only to one agonist but not the other. Response to one agonist serves as a positive control that the astrocytes are viable. This is evidence against Ca2+-dependent release of ATP by astrocytes, because it is expected that astrocytes not expressing the MrgA1R or group I mGluRs would also have Ca2+ elevations due to the high amounts of ATP being released by the surrounding astrocytes. These data suggest that the only astrocytes capable of producing Ca2+ elevations are those cells that express the Gq GPCR being stimulated by the bath-applied agonist. It should be pointed out that cells expressing MrgA1R Ca2+ responses have been electrophysiologically confirmed as passive astrocytes in 100% of cases (n > 50 cells; unpublished observations). In summary, the propagating intercellular Ca2+ wave is clear evidence for astrocytic release of gliotransmitters, evidence which is lacking in astrocytes from healthy tissue in situ or in vivo. However, as will be discussed below, in the diseased or damaged brain propagating astrocytic Ca2+ waves are evident, suggesting that changes occurring in reactive astrocytes are permissive for gliotransmitter release.
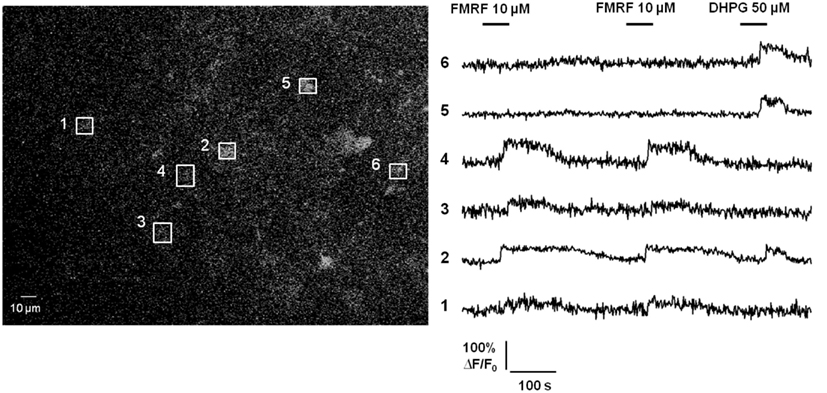
Figure 2. Propagating intercellular Ca2+ waves are unlikely to occur between astrocytes in acute hippocampal slices from healthy brain. Astrocytes in stratum radiatum of hippocampal slices from MrgA1 Gq GPCR transgenic mice were bulk-loaded with Ca2+ Green 1-AM indicator dye as described previously (Fiacco et al., 2007; left panel). Boxed regions of interest over individual astrocyte somata match numbered Ca2+ traces (right panel). Stimulation with either 10 μM FMRF or 50 μM DHPG evoked Ca2+ elevations in 56/75 (75%) or 67/75 (89%) of astrocytes, respectively, through activation of MrgA1Rs or group I mGluRs (n = 9 slices). In total, 50/75 (67%) of astrocytes responded to both agonists. However, 24/75 (32%) of astrocytes responded to one, but not the other, agonist. These results suggest that despite strong Ca2+ elevations in many astrocytes, the gliotransmitter ATP is not released in sufficient quantity to stimulate purinergic P2Y Gq GPCRs on adjacent astrocytes.
Basal (Resting) Ca2+ vs. Activity-Evoked Ca2+ Elevations
Many studies concluding a role for gliotransmission have used the Ca2+ chelators BAPTA and/or EGTA in an effort to block astrocytic Gq GPCR Ca2+ elevations. These manipulations have produced clear effects on neuronal activity, suggesting that activity-induced astrocyte Ca2+ elevations modulate neuronal activity by gliotransmission. It has been difficult to reconcile these findings with those using IP3R2 knockout mice, in which astrocytic Gq GPCR Ca2+ elevations are also abolished but without a corresponding effect on hippocampal excitatory synaptic activity or NMDA receptor-dependent LTP (for example, compare Agulhon et al., 2010 with Henneberger et al., 2010). Exciting new work has provided a potential explanation for this discrepancy (Shigetomi et al., 2012). Using a membrane-tethered genetically encoded Ca2+ indicator (Lck-GCaMP3), spotty Ca2+ signals were observed in cultured astrocytes that were not dependent on Ca2+ release from internal stores. Rather, these signals seemed to be dependent on extracellular influx of Ca2+ through transient receptor potential A1 (TRPA1) channels on the astrocyte membrane. These fast, focal Ca2+ elevations were shown to contribute to, and set resting Ca2+ levels in astrocytes. Could these channels represent the Ca2+ source required for gliotransmission? It cannot be ruled out; however, they seemed to open in an activity-independent manner and affect synaptic activity in hippocampal interneurons by a mechanism other than gliotransmission, i.e., by regulating the functional expression of astrocytic GABA transporters at the plasma membrane. It will be interesting in future work to determine if activity-independent Ca2+ influx setting basal Ca2+ levels is important for tonic release or uptake of gliotransmitters to set ambient concentrations of ATP/adenosine, D-serine, or glutamate (Jabaudon et al., 1999; Cavelier and Attwell, 2005; Cavelier et al., 2005; Le Meur et al., 2007). Together, these findings suggest that caution should be exercised in the interpretation of experiments dialyzing the astrocytic syncytium with Ca2+ chelators such as BAPTA and/or EGTA, which will not only block activity-induced Gq GPCR Ca2+ elevations but also clamp resting Ca2+ levels.
Questionable Timing for Gliotransmission to Acutely Modulate Synaptic Transmission: Astrocytes as Gliomodulators or “Re-Suppliers”
In order for gliotransmission to reciprocally affect synaptic events occurring on a millisecond timescale, it needs to occur along a similar timescale. As discussed above, evidence to date indicates that metabotropic Gq GPCRs are the prevailing mechanism behind activity-induced astrocytic Ca2+ elevations. Because the pathway to Ca2+ elevations involves a metabotropic signaling cascade, there is a considerable delay between neurotransmitter binding the receptor and the ensuing Ca2+ response. In our own recordings at room temperature in acute hippocampal slices, the time interval – on average – between a 50-Hz train of Schaffer collateral stimulation and the first responsive astrocytic compartment (almost always a small to medium-sized process) is 8 s (Figures 3A,C,E). This of course, is an average, and there may be some small compartments and/or a small subset (5%) of astrocytes capable of faster responses (Winship et al., 2007; Panatier et al., 2011). The remaining astrocytic compartments (encompassing the vast majority of the cell volume) take longer to respond due to the slow intracellular wave propagation rate of astrocyte Ca2+ (5–15 μm/s; Yagodin et al., 1994; Fiacco and McCarthy, 2006; Fiacco et al., 2007). Some of the delay to the initial astrocyte Ca2+ response can be attributed to the kinetics of the Ca2+ indicator dye converting the binding of Ca2+ to a fluorescence signal and to the spatial and temporal resolution of optical detection, but this can be taken into account by recording neuronal Ca2+ signals using the same fluorescence indicator and microscope settings. In contrast to astrocytic Ca2+ elevations, neuronal Ca2+ elevations using the same settings appear to occur instantaneously (Figures 3B,D,E). In the same preparation and in the same conditions, the delay to evoke excitatory field potentials (fEPSPs) from a single depolarizing pulse to the Schaffer collaterals is ∼5 ms, while whole-cell NMDA receptor-mediated eEPSCs are evoked after ∼4 ms (Figure 3F). Calcium elevations in the dendritic spines due to NMDA receptor activation are detected a little over 10 ms after photolysis of extracellular caged glutamate (Bloodgood and Sabatini, 2007), while elevations in the proximal dendrites generated by the opening of VGCCs gated by back-propagating action potentials occur within 100–200 ms (Fedirko et al., 2007). Therefore, detection issues cannot account for slow Ca2+ response times of astrocytes. Recording at room temperature will also slow down astrocyte Ca2+ response times, since the activity rates of effector proteins and enzymes in the Gq GPCR signaling cascade will be reduced compared to physiological temperature. However, even in vivo, astrocyte cell bodies respond to sensory or motor stimulation after a delay of ∼2–10 s, with Ca2+ increases in the processes occurring ∼1 s earlier than those in the soma (Wang et al., 2006; Dombeck et al., 2007; Schummers et al., 2008; Navarrete et al., 2012). Overall, the bulk of the evidence indicates that activity-induced astrocytic Gq GPCR Ca2+ responses in processes occur on a timescale of seconds. This is a 1000-fold slower than synaptic responses.
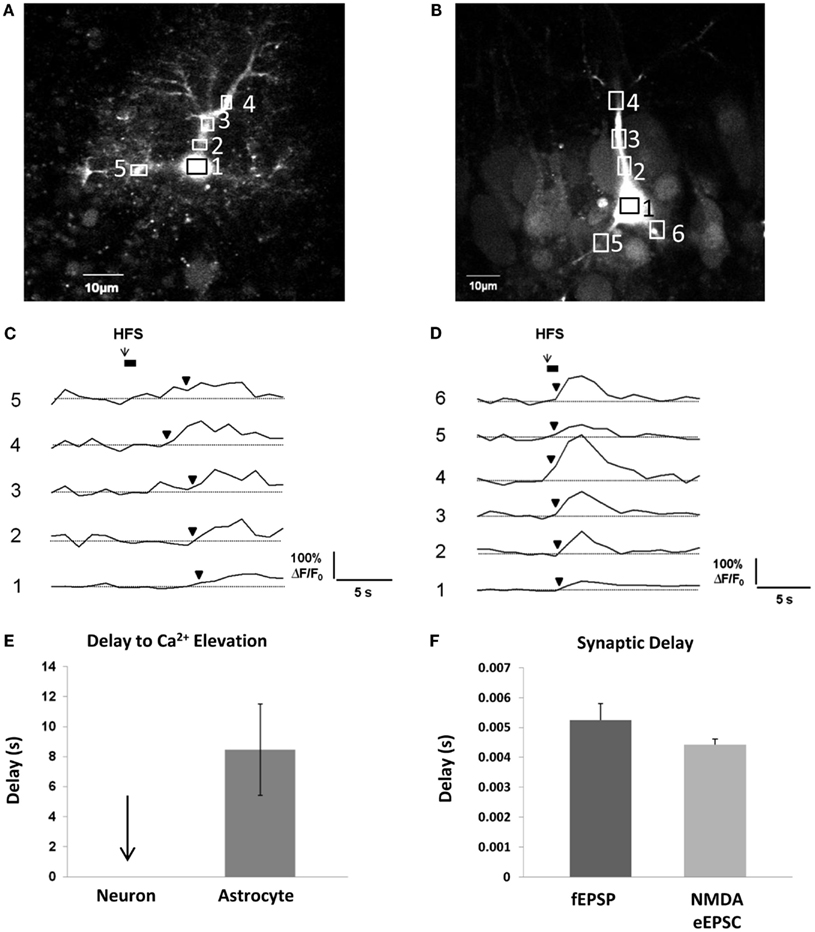
Figure 3. Delay between Schaffer collateral stimulation and evoked astrocyte or neuronal Ca2+ responses and AMPA and NMDA receptor currents. (A,C) Astrocyte in stratum radiatum (A) and CA1 pyramidal neuron (B) each filled with 150 μM OGB-1 Ca2+ indicator dye. Boxed regions of interest in subcellular compartments correspond to the fluorescence traces in (C,D). Boxes were placed all over the visible astrocyte but only a few are shown for clarity. Cells were given 10 min to recover after removal of the whole-cell patch clamp pipette prior to Schaffer collateral stimulation. The stimulating electrode was placed 75 μm from the recorded cells. (C,D) Schaffer collateral stimulation at 50 Hz for 1s produced astrocytic Ca2+ elevations after ∼3 s in the first responding process of this specific astrocyte (C), while neuronal Ca2+ elevations occurred almost instantaneously in proximal neuronal compartments in this example as well as all other recorded neurons (n = 4) (D). (E) Astrocyte Ca2+ elevations occurred on average after ∼8 s in the first responding compartment (n = 4 cells). Response initiation times were defined as the first data point that preceded two successive data points that were ≥3 S.D. above the mean baseline noise. We have observed that the astrocytic Ca2+ responses to the first stimulation are faster and more reliable than subsequent stimulations, so only the responses to the first stimulation were calculated. (F) In the same conditions, fEPSPs (n = 5 slices) and whole-cell CA1 neuronal NMDA eEPSCs (n = 16 cells, 16 slices) were evoked after ∼5 and 4 ms, respectively, in response to single depolarizing pulses (0.05 Hz) to the Schaffer collaterals.
Now, as an example, we will consider this timing in the context of release of the gliotransmitter D-serine by astrocytes. D-serine is a required co-agonist of the NMDA receptor – without D-serine bound the NMDA receptor ion channel cannot open (Johnson and Ascher, 1987; Mothet et al., 2000). The complete cycle of D-serine synthesis, release, uptake, and enzymatic breakdown is not yet fully understood, but most evidence suggests that there are high – although not completely saturating – concentrations of extracellular D-serine in most forebrain areas (Miller, 2004; Verrall et al., 2010). This leaves room for activity-dependent modulation of NMDA receptor function by regulated release of D-serine. For the sake of this discussion we must first assume that astrocytes are capable of producing and secreting D-serine, which has recently come under question. D-serine was originally thought to be produced and secreted only by astrocytes due to astrocyte-specific expression of the synthesizing enzyme serine racemase (SR; Wolosker et al., 1999). More recent data using different antibodies to SR and selective knockout studies suggest that SR is a neuronal enzyme (Kartvelishvily et al., 2006; Miya et al., 2008; Benneyworth et al., 2012). The recent study by Benneyworth et al. (2012) does suggest that a small portion of extracellular D-serine might be supplied by astrocytes.
Activity-dependent release of D-serine by astrocytes has been reported to be required for hippocampal LTP as well as LTP of local field potentials in the somatosensory cortex (Henneberger et al., 2010; Takata et al., 2011). The idea is that spillover of glutamate or glutamate + acetylcholine (ACh) from repetitive stimulation of neuronal afferents activates astrocytic mGluRs or mGluRs + metabotropic ACh receptors (mAChRs) to trigger Ca2+-dependent D-serine release from astrocytes, which is then necessary for postsynaptic NMDA receptor activation and LTP. However, if we walk through the steps in this process it becomes evident that the timing is problematic (Figure 4 – for simplicity only hippocampal NMDA receptor-mediated LTP is depicted). First, presynaptically released glutamate during the high-frequency stimulus (HFS) train crosses the narrow (20 nm) synaptic cleft and binds postsynaptic NMDA and AMPA receptors. In approximately 4–5 ms these channels open and pass postsynaptic current, and postsynaptic Ca2+ increases via entry through the NMDA receptor channel are observed after 10 ms (Bloodgood and Sabatini, 2007). Within 100–200 ms, Ca2+ is already elevating in the proximal apical dendrites due to back-propagating action potentials (Fedirko et al., 2007). During the HFS train the concentration of glutamate/ACh builds up and spills out of the synapse, reaching sufficient levels to overcome glutamate clearance or ACh breakdown to bind and activate astrocytic mGluRs or mAChRs. The activated Gq GPCRs exchange GDP for GTP and the α and βγ subunits dissociate. The α subunit then activates the enzyme phospholipase C (PLC) in the astrocyte membrane. Next, PLC mediates the conversion of inositol bisphosphate (PIP2) to IP3 and diacylglycerol (DAG). IP3 builds up in the astrocytic cytosol and binds its target receptor, IP3R2, on the ER. Activation of the IP3R2 results in channel opening and Ca2+ efflux from the ER into the cytosol, detected by the fluorescence indicator a few seconds after neuronal afferent stimulation. The locally elevated Ca2+ propagates into adjacent astrocytic compartments including the 20- to 30-nm sheet-like processes frequently surrounding synapses at a rate of 5–15 μm/s, where it encounters along the way small and diffuse synaptic-like microvesicles which are triggered to exocytose and release D-serine into the extrasynaptic space. The extracellular D-serine presumably reaches a sufficient concentration to percolate back into the synapse to bind synaptic NMDA receptors, permitting NMDA receptor channels to open to pass current and elevate postsynaptic Ca2+.
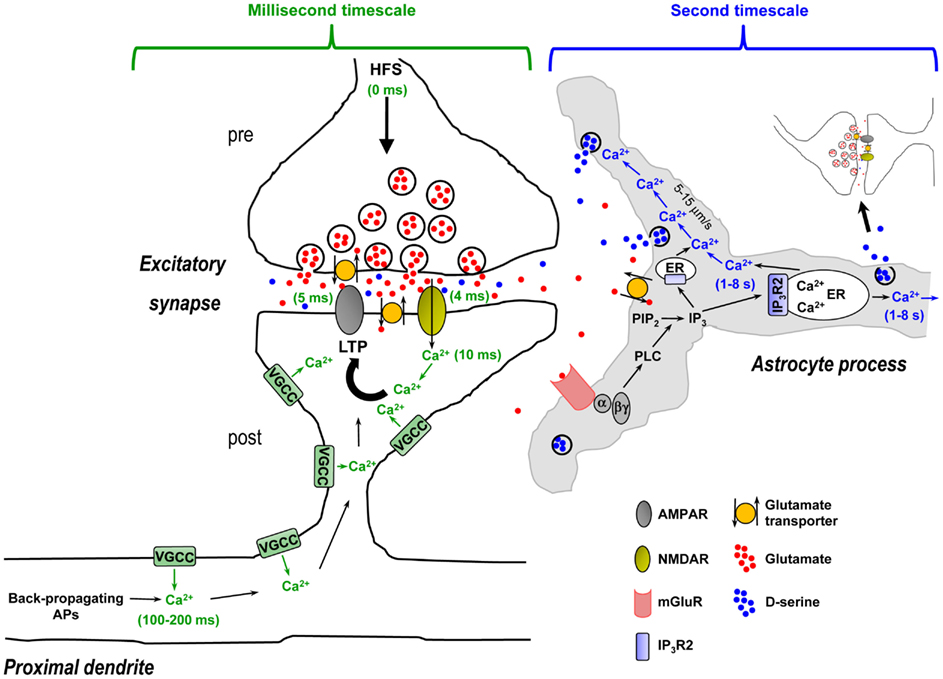
Figure 4. Time course of synaptic mechanisms leading to the induction of NMDA receptor-mediated LTP vs. time course of activity-evoked Gq GPCR Ca2+-mediated D-serine release from astrocytes in CA1 hippocampus: millisecond timescale mechanisms vs. second timescale mechanisms. Left panel, Induction of LTP requires: (i) High-frequency presynaptic stimulation (HFS) to release the neurotransmitter glutamate – beginning of HFS is noted as 0 ms time point; (ii) Depolarization of the postsynaptic compartment by glutamate-mediated AMPA receptor activation allowing Mg2+ block relief of the NMDA receptors, and activation of NMDA receptors by glutamate and necessary co-agonist D-serine (4–5 ms time points); (iii) Ca2+ entry through NMDA receptor channel (10 ms time point). This resulting rise in Ca2+ is crucially important to the induction of LTP and may be amplified by concurrent activation of VGCCs. At 100–200 ms time point, Ca2+ is elevating in the proximal apical dendrite due to back-propagating APs that might act as a retrograde signal to modulate LTP to some extent. All of these steps happen over a timescale of milliseconds and are represented in green. Right panel, HFS-evoked release of D-serine from astrocytes requires: (i) Spillover of presynaptically released glutamate from the synapse, reaching sufficient levels to overcome glutamate clearance by neuronal and astrocytic glutamate transporters; (ii) Glutamate binding and activation of astrocytic mGluRs on astrocyte processes enveloping synapses, which leads to PLC-mediated formation of IP3; (iii) Ca2+ release from ER upon IP3R2 receptor channel activation by IP3 binding (1–8 s time point – represented in blue). Note that in the astrocyte sheet-like processes surrounding synapses that are frequently 20–30 nm thick, the presence of ER might be limited and the IP3 must diffuse into thicker astrocyte processes where ER components are more likely present; (iiii) Intercellular Ca2+ wave propagation (5–15 μm/s) that triggers exocytosis of the sparse D-serine containing synaptic-like vesicles encountered along the way. In this context, activity-evoked D-serine release from astrocytes occurs in a second timescale after the beginning of HFS and therefore cannot be the source of the required co-agonist necessary for NMDA receptor activation, which occurs within 4–5 ms. Activity-induced and Gq GPCR Ca2+-mediated release of D-serine from astrocytes seems to be more optimally suited to modulate subsequent neuronal activity (i.e., involving gliomodulation as opposed to acute “gliotransmission”) or plays a role to “re-supply” ambient levels of extracellular D-serine, which is partially depleted through HFS. Abbreviations: AP, action potential; α, Gq α subunit; βγ, Gq βγ subunits; ER, endoplasmic reticulum; IP3, inositol 1,4,5-triphosphate; IP3R2, astrocyte-specific IP3 receptor type 2; LTP, long-term potentiation; PIP2, inositol bisphosphate; PLC, phospholipase C; VGCC, voltage-gated Ca2+ channel.
The obvious problem with this scenario is that the requirement for astrocytic D-serine as a necessary co-agonist at the NMDA receptor occurred a 1000 times earlier in relative synaptic time (Figure 4). This scenario does not take into account the rise time of astrocyte Ca2+ elevations, which is often seconds long (Wang et al., 2006; Henneberger et al., 2010; Navarrete et al., 2012). The very observation that NMDA receptor currents are evoked in ∼4 ms and postsynaptic Ca2+ elevates shortly thereafter indicates that the D-serine required for NMDA channel opening was already available, as suggested by the relatively high ambient D-serine levels in the synapse (Mothet et al., 2000). The remaining activity-dependent release of D-serine may come from the presynaptic or postsynaptic neuronal compartments, as reported in a recent study (Rosenberg et al., 2010). The advantage provided by this mechanism is that ambient and activity-induced release of D-serine will be available as a required co-agonist at the same time that presynaptically released glutamate is binding the NMDA receptor. Astrocytes, known to maintain ionic and neurotransmitter homeostasis (Kimelberg, 2007), might instead participate in setting ambient D-serine levels in a manner dependent on resting Ca2+. In this context, it seems conceivable that reducing or clamping resting Ca2+ levels in astrocytes using intracellular Ca2+ chelators minutes before HFS could decrease overall D-serine levels and LTP. Such a hypothesis, if validated, could reconcile discrepancies between studies (Agulhon et al., 2010; Henneberger et al., 2010).
Because of the slow timing of metabotropic receptor-evoked astrocyte Ca2+ elevations, astrocytes seem to be more optimally suited to modulate subsequent neuronal activity or play a role as “re-suppliers” as opposed to acute modulators. Is this gliotransmission? Arguably not, based on the current definition of gliotransmission. The term “gliomodulation” may be more appropriate and better distinguish this process from neurotransmission. Interestingly, Rosenberg et al. (2010) found that extracellular D-serine is reduced minutes after its initial increase following neuronal depolarization. Astrocyte Ca2+ may then participate in re-setting ambient levels of D-serine following depletion through repetitive neuronal activity. Astrocytes as re-suppliers can also be considered in the case of cerebrovascular coupling. As this process is also thought to be regulated by activity-induced astrocyte Gq GPCR Ca2+ (Simard et al., 2003; Zonta et al., 2003; Mulligan and MacVicar, 2004), astrocytes are well-positioned to re-supply neurons with metabolites and oxygen after these substrates are depleted during repetitive neuronal activity.
Evidence Supporting Ca2+-Dependent Gliotransmitter Release in the Early Steps of the Inflammatory Process and in the Diseased or Damaged Brain
Reactive Astrocytes
In the previous section we discussed evidence questioning the existence of gliotransmission in healthy brain tissue. In this section we will review data suggesting that activated microglia and reactive astrocytes engage in a partnership for Ca2+-dependent gliotransmitter release. First, what is a reactive astrocyte? Reactive astrocytes or reactive astrogliosis can be defined as a graded progression of molecular, cellular, and functional changes that range from subtle alterations in gene expression to glial scar formation (Hamby and Sofroniew, 2010). Reactive astrocytes are a hallmark of nearly all brain pathologies, including traumatic brain injury, stroke, ischemia, infectious disease, neuroinflammatory and neurodegenerative disease, epilepsy, brain tumors, and possibly even more subtle disorders such as schizophrenia, migraine, and depression. The list of changes that have been documented to occur in reactive astrocytes is extensive, and includes hypertrophy (Sofroniew, 2009), upregulated expression of GFAP (Pekny and Nilsson, 2005), altered expression of: mGluRs (Aronica et al., 2003), glutamate transporters and glutamine synthetase (Pardo et al., 2006; Ortinski et al., 2010), ephrins and their receptors (Goldshmit et al., 2006), aquaporin-4 (AQP4) and potassium channels (Binder and Steinhauser, 2006), and production and secretion of inflammatory molecules (e.g., TNFα, SDF-1α, PGE2) and oxygen free radicals (Lucas et al., 2006; Peng et al., 2006; Farina et al., 2007; Brambilla et al., 2009; Steele and Robinson, 2012).
One of the persistent problems in understanding astrocytic involvement in progression of changes occurring in the diseased or damaged brain is establishing cause-and-effect relationships. It seems that research is beginning to move past cataloging changes that are present in damaged tissue to carefully examining the initial sequence of events occurring in the progression of disease before overt neuroanatomical changes or onset of behavioral symptoms (Colangelo et al., 2012). Accumulating evidence is suggesting that microglia, the brain’s resident immune cells, are activated first, and through release of ATP and inflammatory mediators including interleukin-1 (IL-1), TNFα, or PGE2, subsequently trigger astrocytic activation (Bezzi et al., 2001; Ikeda-Matsuo et al., 2005; Liu et al., 2011; Machado et al., 2011; Colangelo et al., 2012; Pascual et al., 2012). Activated (reactive) astrocytes then increase their production and secretion of SDF-1α (Bajetto et al., 1999; Peng et al., 2006), TNFα, and/or PGE2 (Bezzi et al., 2001; Alvarez et al., 2010) which, in conjunction with Ca2+, promotes gliotransmitter (ATP, glutamate) release. All of these molecules may act in concert through complex autocrine/paracrine or astrocyte-microglial signaling pathways to generate and amplify propagating intercellular astrocyte Ca2+ waves and increase neuronal excitability (Bajetto et al., 1999; Cotrina et al., 2000; Bezzi et al., 2001; Han et al., 2001; Arcuino et al., 2002; Domercq et al., 2006; Bowser and Khakh, 2007; Santello et al., 2011; Pascual et al., 2012). These events are initiated rapidly, within seconds to minutes, suggesting that they may be among the early events underlying excitotoxic damage in disorders and diseases of the central nervous system.
Propagating Intercellular Ca2+ Waves
As discussed above, evidence for intercellularly propagating Ca2+ waves among cortical gray matter astrocytes in healthy brain tissue in situ or in vivo is lacking (Fiacco and McCarthy, 2006; Kuchibhotla et al., 2009). On the contrary, dramatic changes in astrocytic Ca2+ homeostasis have been observed under pathological conditions (e.g., epilepsy or Alzheimer’s disease) in situ and in vivo, affecting the frequency, duration, and amplitude of astrocytic Ca2+ transients (Aguado et al., 2002; Hirase et al., 2004; Kuchibhotla et al., 2009). Powerful astrocytic intercellular Ca2+ waves were found to travel across the cortex in a mouse model of Alzheimer’s disease in vivo but not in wild type littermates (Kuchibhotla et al., 2009), suggesting that Ca2+ waves signal the existence of a pathological insult, as previously postulated (Fiacco and McCarthy, 2006; Scemes and Giaume, 2006). Long-distance and tetrodotoxin-insensitive astrocytic intercellular Ca2+ waves have also been described in spreading depression (Basarsky et al., 1998; Kunkler and Kraig, 1998; Peters et al., 2003; Chuquet et al., 2007), which is believed to occur in several neurological disorders including migraine (Hadjikhani et al., 2001; James et al., 2001), trauma (Fabricius et al., 2006), and stroke (Strong et al., 2002; Dreier et al., 2006; Fabricius et al., 2006), contributing to the death of compromised tissue (Busch et al., 1996; Nedergaard, 1996). In summary, in the diseased or damaged brain propagating astrocytic Ca2+ waves are evident, suggesting that changes occurring in reactive astrocytes may be permissive for Ca2+-dependent release of gliotransmitters (ATP).
Requirement of Activated Microglia, Reactive Astrocytes and Inflammatory Molecules, and Potential for Therapeutic Intervention
An intriguing possibility conferring pathological relevance to astrocytic release of gliotransmitters is that this process is actually dependent on other signaling molecules produced by activated microglia and reactive astrocytes in addition to Ca2+. A new report supports this hypothesis by showing that activated microglia recruit astrocytes to modulate neuronal activity early in the inflammatory process (Pascual et al., 2012). Activation of microglia using a proinflammatory toll-like receptor 4 ligand (TLR4), lipopolysaccharide (LPS), induced a rapid (within minutes) and transient (∼10 min-long) increase in the frequency of excitatory synaptic events in acute hippocampal slices. The mechanism for this effect involved the release of ATP by microglia to activate metabotropic P2Y1 receptors (P2Y1Rs) on astrocytes, triggering glutamate release from astrocytes to modulate synaptic mGluRs. Because activation of microglia and alteration of neurotransmission are two early symptoms of most brain diseases, these findings support the idea that activated microglia are an upstream partner of astrocytes that transforms astrocytes into glutamate releasing cells, which may contribute to the initiation of bursting neuronal activity in the epileptic brain (Pascual et al., 2012) or other neurological disorders. Remarkably, the inflammatory mediators TNFα and PGE2 can also be co-released downstream of astrocytic ATP-induced P2Y1R activation, dramatically enhancing glutamate release from astrocytes, with TNFα action being a requirement for this glutamate response to occur (Domercq et al., 2006; Santello et al., 2011); although the requirement of TNFα is debated (Pascual et al., 2012). Even more striking is the observation that inflammatory molecules themselves can elicit a rapid (seconds) astrocytic release of glutamate without involvement of any Gq GPCR stimulation. SDF-1α, TNFα, or PGE2 by themselves are sufficient to induce glutamate release from astrocytes through selective activation of their respective receptors: Gi/o GPCRs (CXCR4), trimeric TNF receptors (TNFR), or Gi/Gs GPCRs [prostaglandin E (EP) receptors; Bezzi et al., 1998, 2001; Sanzgiri et al., 1999; Cali et al., 2008]. Although understanding of the pathways downstream of CXCR4, TNFR, or EPs remains largely incomplete in astrocytes, it has been assumed that selective activation of one of these receptor types is sufficient to elicit a Ca2+-dependent exocytosis of glutamate from astrocytes. This assumption is based mainly on a study suggesting that CXCR4, TNFR, and EPs participate in the same sequence of events that ultimately leads to Ca2+-dependent exocytosis of glutamate, a response otherwise blocked by intracellular Ca2+ chelators or inhibitors of exocytosis (Bezzi et al., 2001). CXCR4 activation induces a complex ensuing signaling cascade involving extracellular release of TNFα from astrocytes, autocrine/paracrine TNFα-dependent signaling, PGE2 formation and secretion from astrocytes, autocrine/paracrine PGE2-dependent signaling, and finally glutamate release from astrocytes (Bezzi et al., 2001). In this context, Ca2+ is mobilized from internal stores following activation of CXCR4 (Zheng et al., 1999; Bezzi et al., 2001; Cali et al., 2008) and EP receptors (Sanzgiri et al., 1999), including Gi GPCR EP3 (Takemiya et al., 2011) and Gs GPCR EP2 receptors (Di Cesare et al., 2006; Hsiao et al., 2007). CXCR4- or EP-evoked glutamate release from astrocytes induces presynaptic NMDA receptor-dependent synaptic potentiation (Santello et al., 2011) or postsynaptic/extrasynaptic NMDA receptor-dependant slow inward currents (Sanzgiri et al., 1999). Interestingly, EP-mediated Ca2+ elevations spread through neighboring astrocytes as intercellular Ca2+ waves (Sanzgiri et al., 1999), strongly suggesting that not only glutamate, but also ATP is released (Cotrina et al., 2000; Arcuino et al., 2002; Bowser and Khakh, 2007) following EP activation. Notably, SDF-1α-induced CXCR4-mediated glutamate release is dramatically enhanced in the presence of reactive microglia, leading to pro-apoptotic neurotoxicity, a mechanism potentially involved in AIDS dementia complex (Bezzi et al., 2001; Rossi and Volterra, 2009). Not only astrocytes, but also microglia and neurons express CXCR4 (Tanabe et al., 1997; Asensio and Campbell, 1999; Kaul and Lipton, 1999), and activation of this receptor in microglia causes significant TNFα release. Microglial TNFα would then add to astrocytically released TNFα to amplify the release of glutamate from astrocytes.
The synergistic initiation of signaling transduction pathways involving microglia and astrocytes suggests that both cell types and their signaling pathways may be required, in addition to astrocytic Ca2+, to trigger the release of glutamate from astrocytes (Figure 5). The observation that activated microglia or inflammatory mediators cannot only induce, but also substantially amplify glutamate release from astrocytes confers pathological relevance to this process. Furthermore, the rapid (seconds to minutes) induction of these different pathways suggests that they may be critical early events contributing to initiation of neuronal signaling cascades leading to hyperexcitability, excitotoxicity, and cell death. Therefore these transduction pathways could be potential targets for therapy of multiple neurological, neuroinflammatory, and neurodegenerative disorders, including stroke, trauma, epilepsy, Alzheimer’s disease, Huntington’s disease, AIDS dementia complex, and amyotrophic lateral sclerosis. These and a growing list of other neurological disorders are now understood to share the final common destructive metabolic pathway of excitotoxicity, which is an excessive activation of neuronal glutamatergic receptors and associated signaling molecules (Shaw, 1993; Lau and Tymianski, 2010). Additionally, activated/reactive microglia and astrocytes, and production of inflammatory mediators is also another common element in most, if not all, neurological, neuroinflammatory, and neurodegenerative disorders (Lucas et al., 2006). Glial inflammatory transduction pathways leading to astrocytic Ca2+-dependent glutamate release could contribute to excitotoxicity, providing a new framework for elucidating the mechanistic basis of excitotoxicity and potentially improving upon existing medications.
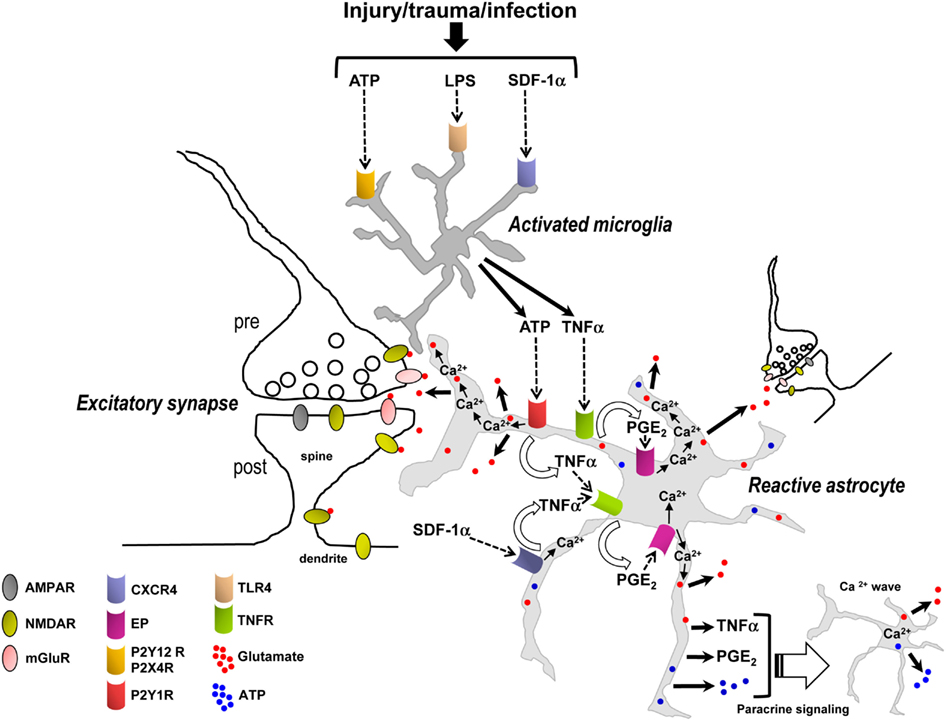
Figure 5. Synergistic signaling transduction pathways involving activated microglia and reactive astrocytes and their inflammatory mediators that, in addition to astrocytic Ca2+, may induce the release of gliotransmitters (glutamate, ATP) in the early steps of the inflammatory process and in the diseased or damaged brain. ATP, LPS, and/or SDF-1α are produced following acute and chronic pathological insults to the central nervous system, e.g., hypoxia, ischemia, epilepsy-associated seizures, mechanical injury and trauma, or infection (Bajetto et al., 2001; Bodin and Burnstock, 2001; Cook and McCleskey, 2002; Rivest, 2003; Qin et al., 2005). These mediators activate microglial purinergic receptors (P2Y12R, P2X4R; Haynes et al., 2006; Ohsawa and Kohsaka, 2011), TLR4 or CXCR4 as well as astrocytic CXCR4. Activated microglia then release ATP and/or TNFα to recruit astrocytes. In astrocytes, binding of ATP and SDF-1α – alone or in concert – to their Gq and Gi/o protein-coupled receptors (P2Y1R and CXCR4) stimulates Ca2+ mobilization from internal stores. P2Y1R and/or CXCR4-mediated intracellular Ca2+ elevations trigger an ensuing cascade of pathways involving the production and secretion of TNFα and PGE2, TNFR and EP activation, EP-induced Ca2+ elevations, and ultimately followed by the release of glutamate from astrocytes. For simplicity TNFα and PGE2 have been depicted as acting on the same astrocyte from which they are released; i.e., in an autocrine manner, although they can also act on neighboring astrocytes to propagate signals in a paracrine way and induce glutamate release. While astrocytic P2Y1R activation may also induce Ca2+-dependent release of glutamate without implication of TNFα, TNFα (from microglial and astrocytic origin), and PGE2 dramatically enhance the astrocyte glutamate response. The locally elevated Ca2+ due to activation of P2Y1R, CXCR4, and EPs, working alone or in concert, propagates some distance into astrocytic compartments including the sheet-like processes surrounding synapses where it encounters small and diffuse synaptic-like microvesicles which are triggered to exocytose glutamate into the extracellular space. Additionally, EP-mediated Ca2+ increases in astrocytes can also induce the release of ATP, which through autocrine/paracrine activation of astrocytic P2YRs evokes regenerative intercellular Ca2+ waves. Definitive proof for Ca2+-dependent astrocyte glutamate release to occur via exocytosis awaits further evidence. The overall glutamate release from astrocytes can occur within seconds to minutes of a triggering stimulus to increase ongoing pre- and postsynaptic mGluR- and/or NMDA receptor-mediated synaptic transmission, which may be a critical early step contributing to the initiation of neuronal signaling cascades leading to hyperexcitability, excitotoxicity, and cell death. Therefore the transduction pathways depicted in this figure could be potential targets for therapy of multiple neurological, neuroinflammatory, and neurodegenerative disorders.
Interestingly, in the inflammatory process involving activated microglia and reactive astrocytes, glutamate release from astrocytes may be mediated by Ca2+ elevations downstream of not only Gq (P2Y1R) GPCRs, but also Gi/o (CXCR4) or Gi/Gs (EP) GPCRs. Additionally, the ligands of these receptors (ATP, SDF-1α, PGE2) and the ligand for TNFR (TNFα) appear to be mainly of microglial and astrocytic origin (Asensio and Campbell, 1999; Bajetto et al., 1999; Sanzgiri et al., 1999; Bezzi et al., 2001; Rostasy et al., 2003; Domercq et al., 2006; Peng et al., 2006; Rossi and Volterra, 2009; Santello et al., 2011; Pascual et al., 2012) and not of presynaptic origin; although, a contribution of ATP co-released with glutamate from neuronal afferents (Khakh, 2001) or of neuronal SDF-1α (Rostasy et al., 2003) cannot be excluded. The seconds to minutes timescale for the astrocytic glutamate response to occur fits with the timing expected of a metabotropic signaling pathway and also indicates that microglia can become activated, and astrocytes reactive, very quickly in response to a triggering stimulus (Figure 5). The data suggest that inflammation – in response to infection or injury, rather than synaptically induced stimulation of astrocytic GPCRs – is essential for triggering glutamate release from astrocytes to subsequently increase ongoing excitatory synaptic transmission (Figure 5). These emerging findings also need to be taken into consideration in the interpretation of studies that have used tetanus toxin to block glutamate exocytosis from astrocytes (Bezzi et al., 2001). Transport of cell surface connexin hemichannels or volume-activated Cl− channels to the membrane via exosome trafficking may also be impaired by incubation in tetanus toxin (Proux-Gillardeaux et al., 2005), which could alter efflux of glutamate into the extracellular space by these alternate pathways (Malarkey and Parpura, 2008). Tetanus toxin would also presumably affect: (i) the transport of multidrug resistance protein 4 (MRP4) – a PGE2 export pump – to the membrane (Reid et al., 2003; Proux-Gillardeaux et al., 2005; Ronaldson et al., 2008) which could alter PGE2 release; or (ii) exocytosis of cytokine vesicular carriers which could decrease the secretion of inflammatory mediators (e.g., TNFα or other unidentified factors; Stow et al., 2009; Duitman et al., 2011). Thus, caution needs to be exercised in the interpretation of experiments using blockers of exocytosis or Ca2+ chelators in astrocytes (see discussion above) in combination with neuronal electrophysiological readouts, which may lead to misinterpretation of how astrocytes actually control extracellular glutamate levels in a Ca2+-dependent manner.
Glial inflammatory mediators such as SDF-1α or TNFα are expressed in the healthy brain, albeit at much lower levels than during inflammatory reactions and are thought to be implicated in regulating homeostatic brain functions (Vitkovic et al., 2000; Bajetto et al., 2001; Beattie et al., 2002; Stellwagen et al., 2005; Stellwagen and Malenka, 2006; Kaneko et al., 2008; Boulanger, 2009). Furthermore, Santello et al. (2011) reported that knockout of TNFα prevented P2Y1R Ca2+-dependent glutamate release from astrocytes in acute hippocampal slices from otherwise healthy mice. Therefore, constitutive levels of inflammatory mediators could also regulate to some extent astrocytic glutamate release in brain tissue from healthy mice. However, it is worth noting that astrocytic inflammatory mediator-dependent glutamate release has been obtained in cultured astrocytes, mixed microglia/astrocyte co-cultures or acute hippocampal slices (Bezzi et al., 1998, 2001; Sanzgiri et al., 1999; Domercq et al., 2006; Cali et al., 2008; Santello et al., 2011; Pascual et al., 2012). Microglia and astrocytes, by virtue of culturing conditions become activated (Cahoy et al., 2008; Zamanian et al., 2012), and their activity would therefore resemble more closely that which occurs in the late stages of inflammation. Acute slices, while offering the advantage of maintaining relationships between cell types and limiting reactive changes induced by culturing, are nevertheless also compromised to varying degrees. The slicing process invariably causes cellular and tissue damage including axotomy of afferent fibers (Coltman and Ide, 1996), accompanied by immediate release of ATP via cytolysis due to cell membrane damage, which may only be partially recovered during the subsequent incubation process (Bodin and Burnstock, 2001; Cook and McCleskey, 2002; Fiala et al., 2003). Central nervous system tissue can exhibit elevated ATP release for several hours after trauma (Wang et al., 2004). This form of lesion/trauma-induced injury is likely to cause activation of microglia (Nimmerjahn et al., 2005; Haynes et al., 2006; Ohsawa and Kohsaka, 2011) and astrocytes (Fiala et al., 2003; Neary et al., 2003) within the first minute of slicing. It is therefore reasonable to suggest that microglia and astrocytes in acute slices may be releasing higher levels of inflammatory mediators than they would do otherwise in vivo. This may make astrocytes more sensitive to stimulation by neurotransmitters or bath application of ATP, SDF-1α, TNFα, or PGE2. Astrocytic Ca2+-dependent glutamate release in these conditions may therefore most likely reflect responses of activated microglia and reactive astrocytes at the early stages of inflammation rather than physiological roles of presumed resting glial cells. In any case, differences in levels of TNFα and other inflammatory mediators between slice preparations could explain the variability between groups to produce or observe Ca2+-dependent glutamate release by astrocytes. Carefully controlled future studies in vivo may partially circumvent pathology associated with cell culture and acutely isolated brain slices, and help resolve whether astrocytic GPCR Ca2+-dependent glutamate release is relevant in physiological and/or pathological situations.
Summary and Perspectives
A “calcium-centric” view has pervaded astrocyte research ever since the discovery of propagating astrocyte intercellular Ca2+ waves in vitro in the early 1990s (e.g., Cornell-Bell et al., 1990). While astrocyte Ca2+ increases undoubtedly play a role in certain aspects of astrocyte function, accumulating evidence suggests that there is more to astrocyte physiology than Ca2+ elevations. One of the most documented areas of astrocyte research over the past 20 years has been Gq GPCR Ca2+-dependent gliotransmitter exocytosis to acutely modulate the excitability of adjacent astrocytes and neurons. However, gliotransmission in healthy tissue has been called into question based on numerous findings. First, some studies have reported that selective stimulation or removal of Gq GPCR-dependent astrocyte Ca2+ elevations does not result in changes in neuronal synaptic transmission and plasticity, suggesting that astrocyte Ca2+ elevations are not sufficient to trigger the release of gliotransmitters (ATP, D-serine, glutamate). Second, new research suggests that basal or resting astrocyte Ca2+ levels need to be carefully considered when Ca2+ chelators are infused into the astrocyte syncytium, which will block not only synaptically induced astrocyte Gq GPCR Ca2+ elevations, but also resting astrocyte Ca2+ levels. Manipulation of resting astrocyte Ca2+ may affect neuronal activity via mechanisms including changes in neurotransmitter uptake or ambient neurotransmitter concentrations. Third, propagating intercellular Ca2+ waves between astrocytes indicating release of the gliotransmitter ATP have not been substantiated in intact brain tissue from healthy subjects, arguing against gliotransmitter release in normal physiology. Last, because the endogenous means by which synaptically induced astrocyte Ca2+ elevations occur is through activation of Gq GPCRs, the kinetics of astrocyte Ca2+ elevations seem too slow to modulate neuronal synaptic activity occurring on a millisecond timescale (Figure 4). It only seems possible that astrocytes could modulate neuronal activity in a Gq GPCR-dependent manner seconds after the initiating neuronal stimulus, supporting the concept that astrocytes act as “re-suppliers” to support synaptic transmission through recovery of neuronal metabolism or re-establishment of extracellular transmitter homeostasis. The fact that neuronally evoked Gq GPCR Ca2+ elevations in astrocytes are not sufficient to trigger gliotransmitter release to acutely modulate neurotransmission in the healthy brain, but instead would have a role in maintaining the homeostatic levels of ambient transmitters or ions, has profound implications in our understanding of synaptic transmission. Indeed, the concept of astrocytes being the third functional element of the synapse, in addition to the pre- and postsynaptic compartments, by releasing gliotransmitters to acutely (millisecond timescale) influence synaptic transmission affects the interpretation of a broad range of findings in neurophysiology. Thus, we propose that synaptically induced Gq GPCR Ca2+-dependent release of gliotransmitters (i.e., gliotransmission) be rebranded as gliomodulation.
Emerging research is suggesting that Ca2+-dependent release of gliotransmitters (glutamate and ATP) is also dependent on activated microglia, reactive astrocytes, and inflammatory molecules (Figure 5). These molecules include ATP, SDF-1α, TNFα, and PGE2, mediators that are rapidly upregulated and secreted by activated microglia and reactive astrocytes. This new information is exciting for the following reasons: First, it provides a possible explanation for the disparate findings among research groups regarding the Ca2+-dependence of gliotransmitter release from astrocytes (Agulhon et al., 2008; Hamilton and Attwell, 2010). Second, because it appears that activated microglia and increased release of inflammatory mediators and glutamate can occur rapidly, within seconds to minutes, glutamate release by reactive astrocytes may be a key early event in the progression of changes leading to neuronal hyperexcitability and excitotoxicity in neurological disorders and neuroinflammatory and neurodegenerative diseases. The observation that Ca2+-dependent release of glutamate by astrocytes depends on glial mediators, as well as the second-to-minute time course of this response, argues that gliomodulation but not gliotransmission is involved. Definitive proof for Ca2+-dependent astrocyte glutamate release to occur via exocytosis awaits further evidence, and mechanisms other than exocytosis might also be at work including release through volume-regulated anion channels, pore-forming P2X7 channels, connexin hemichannels, or reversal of glutamate uptake (Haskew-Layton et al., 2008; Malarkey and Parpura, 2008;Zhang et al., 2008, 2011; Li et al., 2012). It will be important in future studies assessing the impact of astrocyte transmitter release in pathological conditions to dissociate between these alternative release pathways. The IP3R2 KO mice will be particularly useful for dissociating astrocyte Ca2+ specifically from other astrocyte transmitter release mechanisms. Future experiments that can measure and manipulate secretion and levels of inflammatory mediators vivo may also provide more compelling information regarding the conditions required for inflammatory mediator/Ca2+-dependent release of gliotransmitters by astrocytes. Overall, these findings provide promising new glial targets for therapeutical intervention to treat a wide range of central nervous system pathologies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant (Chair of Excellence) from the Paris School of Neuroscience (Ecole des Neurosciences de Paris) to Cendra Agulhon, and an Epilepsy Foundation grant and a Hellman Foundation fellowship to Todd A. Fiacco.
References
Aguado, F., Espinosa-Parrilla, J. F., Carmona, M. A., and Soriano, E. (2002). Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J. Neurosci. 22, 9430–9444.
Agulhon, C., Fiacco, T. A., and McCarthy, K. D. (2010). Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science 327, 1250–1254.
Agulhon, C., Petravicz, J., McMullen, A. B., Sweger, E. J., Minton, S. K., Taves, S. R., Casper, K. B., Fiacco, T. A., and McCarthy, K. D. (2008). What is the role of astrocyte calcium in neurophysiology? Neuron 59, 932–946.
Alvarez, S., Blanco, A., Fresno, M., and Munoz-Fernandez, M. A. (2010). Nuclear factor-kappaB activation regulates cyclooxygenase-2 induction in human astrocytes in response to CXCL12: role in neuronal toxicity. J. Neurochem. 113, 772–783.
Araque, A., Sanzgiri, R. P., Parpura, V., and Haydon, P. G. (1998). Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J. Neurosci. 18, 6822–6829.
Araque, A., Sanzgiri, R. P., Parpura, V., and Haydon, P. G. (1999). Astrocyte-induced modulation of synaptic transmission. Can. J. Physiol. Pharmacol. 77, 699–706.
Arcuino, G., Lin, J. H., Takano, T., Liu, C., Jiang, L., Gao, Q., Kang, J., and Nedergaard, M. (2002). Intercellular calcium signaling mediated by point-source burst release of ATP. Proc. Natl. Acad. Sci. U.S.A. 99, 9840–9845.
Aronica, E., Gorter, J. A., Jansen, G. H., van Veelen, C. W., van Rijen, P. C., Ramkema, M., and Troost, D. (2003). Expression and cell distribution of group I and group II metabotropic glutamate receptor subtypes in taylor-type focal cortical dysplasia. Epilepsia 44, 785–795.
Asensio, V. C., and Campbell, I. L. (1999). Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 22, 504–512.
Bajetto, A., Bonavia, R., Barbero, S., Florio, T., and Schettini, G. (2001). Chemokines and their receptors in the central nervous system. Front. Neuroendocrinol. 22, 147–184.
Bajetto, A., Bonavia, R., Barbero, S., Piccioli, P., Costa, A., Florio, T., and Schettini, G. (1999). Glial and neuronal cells express functional chemokine receptor CXCR4 and its natural ligand stromal cell-derived factor 1. J. Neurochem. 73, 2348–2357.
Basarsky, T. A., Duffy, S. N., Andrew, R. D., and MacVicar, B. A. (1998). Imaging spreading depression and associated intracellular calcium waves in brain slices. J. Neurosci. 18, 7189–7199.
Beattie, E. C., Stellwagen, D., Morishita, W., Bresnahan, J. C., Ha, B. K., Von Zastrow, M., Beattie, M. S., and Malenka, R. C. (2002). Control of synaptic strength by glial TNFalpha. Science 295, 2282–2285.
Benneyworth, M. A., Li, Y., Basu, A. C., Bolshakov, V. Y., and Coyle, J. T. (2012). Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell. Mol. Neurobiol. 32, 613–624.
Bezzi, P., Carmignoto, G., Pasti, L., Vesce, S., Rossi, D., Rizzini, B. L., Pozzan, T., and Volterra, A. (1998). Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391, 281–285.
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq, E., Vescovi, A., Bagetta, G., Kollias, G., Meldolesi, J., and Volterra, A. (2001). CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710.
Binder, D. K., and Steinhauser, C. (2006). Functional changes in astroglial cells in epilepsy. Glia 54, 358–368.
Bloodgood, B. L., and Sabatini, B. L. (2007). Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53, 249–260.
Bodin, P., and Burnstock, G. (2001). Purinergic signalling: ATP release. Neurochem. Res. 26, 959–969.
Boulanger, L. M. (2009). Immune proteins in brain development and synaptic plasticity. Neuron 64, 93–109.
Bowser, D. N., and Khakh, B. S. (2007). Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J. Gen. Physiol. 129, 485–491.
Brambilla, R., Persaud, T., Hu, X., Karmally, S., Shestopalov, V. I., Dvoriantchikova, G., Ivanov, D., Nathanson, L., Barnum, S. R., and Bethea, J. R. (2009). Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol. 182, 2628–2640.
Busch, E., Gyngell, M. L., Eis, M., Hoehn-Berlage, M., and Hossmann, K. A. (1996). Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J. Cereb. Blood Flow Metab. 16, 1090–1099.
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., Xing, Y., Lubischer, J. L., Krieg, P. A., Krupenko, S. A., Thompson, W. J., and Barres, B. A. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278.
Cali, C., Marchaland, J., Regazzi, R., and Bezzi, P. (2008). SDF 1-alpha (CXCL12) triggers glutamate exocytosis from astrocytes on a millisecond time scale: imaging analysis at the single-vesicle level with TIRF microscopy. J. Neuroimmunol. 198, 82–91.
Carmignoto, G., Pasti, L., and Pozzan, T. (1998). On the role of voltage-dependent calcium channels in calcium signaling of astrocytes in situ. J. Neurosci. 18, 4637–4645.
Cavelier, P., and Attwell, D. (2005). Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J. Physiol. (Lond.) 564, 397–410.
Cavelier, P., Hamann, M., Rossi, D., Mobbs, P., and Attwell, D. (2005). Tonic excitation and inhibition of neurons: ambient transmitter sources and computational consequences. Prog. Biophys. Mol. Biol. 87, 3–16.
Chuquet, J., Hollender, L., and Nimchinsky, E. A. (2007). High-resolution in vivo imaging of the neurovascular unit during spreading depression. J. Neurosci. 27, 4036–4044.
Colangelo, A. M., Cirillo, G., Lavitrano, M. L., Alberghina, L., and Papa, M. (2012). Targeting reactive astrogliosis by novel biotechnological strategies. Biotechnol. Adv. 30, 261–271.
Coltman, B. W., and Ide, C. F. (1996). Temporal characterization of microglia, IL-1 beta-like immunoreactivity and astrocytes in the dentate gyrus of hippocampal organotypic slice cultures. Int. J. Dev. Neurosci. 14, 707–719.
Cook, S. P., and McCleskey, E. W. (2002). Cell damage excites nociceptors through release of cytosolic ATP. Pain 95, 41–47.
Cornell-Bell, A. H., Finkbeiner, S. M., Cooper, M. S., and Smith, S. J. (1990). Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247, 470–473.
Cotrina, M. L., Lin, J. H., Lopez-Garcia, J. C., Naus, C. C., and Nedergaard, M. (2000). ATP-mediated glia signaling. J. Neurosci. 20, 2835–2844.
Di Castro, M. A., Chuquet, J., Liaudet, N., Bhaukaurally, K., Santello, M., Bouvier, D., Tiret, P., and Volterra, A. (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 14, 1276–1284.
Di Cesare, A., Del Piccolo, P., Zacchetti, D., and Grohovaz, F. (2006). EP2 receptor stimulation promotes calcium responses in astrocytes via activation of the adenylyl cyclase pathway. Cell. Mol. Life Sci. 63, 2546–2553.
Dombeck, D. A., Khabbaz, A. N., Collman, F., Adelman, T. L., and Tank, D. W. (2007). Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron 56, 43–57.
Domercq, M., Brambilla, L., Pilati, E., Marchaland, J., Volterra, A., and Bezzi, P. (2006). P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J. Biol. Chem. 281, 30684–30696.
Dreier, J. P., Woitzik, J., Fabricius, M., Bhatia, R., Major, S., Drenckhahn, C., Lehmann, T. N., Sarrafzadeh, A., Willumsen, L., Hartings, J. A., Sakowitz, O. W., Seemann, J. H., Thieme, A., Lauritzen, M., and Strong, A. J. (2006). Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 129, 3224–3237.
Duitman, E. H., Orinska, Z., and Bulfone-Paus, S. (2011). Mechanisms of cytokine secretion: a portfolio of distinct pathways allows flexibility in cytokine activity. Eur. J. Cell Biol. 90, 476–483.
Fabricius, M., Fuhr, S., Bhatia, R., Boutelle, M., Hashemi, P., Strong, A. J., and Lauritzen, M. (2006). Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 129, 778–790.
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145.
Farso, M. C., O’Shea, R. D., and Beart, P. M. (2009). Evidence group I mGluR drugs modulate the activation profile of lipopolysaccharide-exposed microglia in culture. Neurochem. Res. 34, 1721–1728.
Fedirko, N., Avshalumov, M., Rice, M. E., and Chesler, M. (2007). Regulation of postsynaptic Ca2+ influx in hippocampal CA1 pyramidal neurons via extracellular carbonic anhydrase. J. Neurosci. 27, 1167–1175.
Fiacco, T. A., Agulhon, C., Taves, S. R., Petravicz, J., Casper, K. B., Dong, X., Chen, J., and McCarthy, K. D. (2007). Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626.
Fiacco, T. A., and McCarthy, K. D. (2004). Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. 24, 722–732.
Fiacco, T. A., and McCarthy, K. D. (2006). Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia 54, 676–690.
Fiala, J. C., Kirov, S. A., Feinberg, M. D., Petrak, L. J., George, P., Goddard, C. A., and Harris, K. M. (2003). Timing of neuronal and glial ultrastructure disruption during brain slice preparation and recovery in vitro. J. Comp. Neurol. 465, 90–103.
Goldshmit, Y., McLenachan, S., and Turnley, A. (2006). Roles of Eph receptors and ephrins in the normal and damaged adult CNS. Brain Res. Rev. 52, 327–345.
Hadjikhani, N., Sanchez Del Rio, M., Wu, O., Schwartz, D., Bakker, D., Fischl, B., Kwong, K. K., Cutrer, F. M., Rosen, B. R., Tootell, R. B., Sorensen, A. G., and Moskowitz, M. A. (2001). Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. U.S.A. 98, 4687–4692.
Halassa, M. M., Fellin, T., and Haydon, P. G. (2007). The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol. Med. 13, 54–63.
Hamby, M. E., and Sofroniew, M. V. (2010). Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7, 494–506.
Hamilton, N., Vayro, S., Wigley, R., and Butt, A. M. (2010). Axons and astrocytes release ATP and glutamate to evoke calcium signals in NG2-glia. Glia 58, 66–79.
Hamilton, N. B., and Attwell, D. (2010). Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 11, 227–238.
Han, Y., He, T., Huang, D. R., Pardo, C. A., and Ransohoff, R. M. (2001). TNF-alpha mediates SDF-1 alpha-induced NF-kappa B activation and cytotoxic effects in primary astrocytes. J. Clin. Invest. 108, 425–435.
Haskew-Layton, R. E., Rudkouskaya, A., Jin, Y., Feustel, P. J., Kimelberg, H. K., and Mongin, A. A. (2008). Two distinct modes of hypoosmotic medium-induced release of excitatory amino acids and taurine in the rat brain in vivo. PLoS ONE 3, e3543. doi:10.1371/journal.pone.0003543
Haynes, S. E., Hollopeter, G., Yang, G., Kurpius, D., Dailey, M. E., Gan, W. B., and Julius, D. (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519.
Henneberger, C., Papouin, T., Oliet, S. H., and Rusakov, D. A. (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–236.
Hirase, H., Qian, L., Bartho, P., and Buzsaki, G. (2004). Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2, e96. doi:10.1371/journal.pbio.0020096
Hsiao, H. Y., Mak, O. T., Yang, C. S., Liu, Y. P., Fang, K. M., and Tzeng, S. F. (2007). TNF-alpha/IFN-gamma-induced iNOS expression increased by prostaglandin E2 in rat primary astrocytes via EP2-evoked cAMP/PKA and intracellular calcium signaling. Glia 55, 214–223.
Ikeda-Matsuo, Y., Ikegaya, Y., Matsuki, N., Uematsu, S., Akira, S., and Sasaki, Y. (2005). Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J. Neurochem. 94, 1546–1558.
Jabaudon, D., Shimamoto, K., Yasuda-Kamatani, Y., Scanziani, M., Gahwiler, B. H., and Gerber, U. (1999). Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc. Natl. Acad. Sci. U.S.A. 96, 8733–8738.
Jabs, R., Pivneva, T., Huttmann, K., Wyczynski, A., Nolte, C., Kettenmann, H., and Steinhauser, C. (2005). Synaptic transmission onto hippocampal glial cells with hGFAP promoter activity. J. Cell. Sci. 118, 3791–3803.
James, M. F., Smith, J. M., Boniface, S. J., Huang, C. L., and Leslie, R. A. (2001). Cortical spreading depression and migraine: new insights from imaging? Trends Neurosci. 24, 266–271.
Johnson, J. W., and Ascher, P. (1987). Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–531.
Kaneko, M., Stellwagen, D., Malenka, R. C., and Stryker, M. P. (2008). Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 58, 673–680.
Kartvelishvily, E., Shleper, M., Balan, L., Dumin, E., and Wolosker, H. (2006). Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. J. Biol. Chem. 281, 14151–14162.
Kaul, M., and Lipton, S. A. (1999). Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. U.S.A. 96, 8212–8216.
Khakh, B. S. (2001). Molecular physiology of P2X receptors and ATP signalling at synapses. Nat. Rev. Neurosci. 2, 165–174.
Kimelberg, H. K. (2007). Supportive or information-processing functions of the mature protoplasmic astrocyte in the mammalian CNS? A critical appraisal. Neuron Glia Biol. 3, 181–189.
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., and Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215.
Kunkler, P. E., and Kraig, R. P. (1998). Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. J. Neurosci. 18, 3416–3425.
Lau, A., and Tymianski, M. (2010). Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 460, 525–542.
Le Meur, K., Galante, M., Angulo, M. C., and Audinat, E. (2007). Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J. Physiol. (Lond.) 580, 373–383.
Li, D., Herault, K., Isacoff, E. Y., Oheim, M., and Ropert, N. (2012). Optogenetic activation of LiGluR-expressing astrocytes evokes anion channel-mediated glutamate release. J. Physiol. (Lond.) 590, 855–873.
Lin, S. C., and Bergles, D. E. (2002). Physiological characteristics of NG2-expressing glial cells. J. Neurocytol. 31, 537–549.
Lin, S. C., and Bergles, D. E. (2004). Synaptic signaling between neurons and glia. Glia 47, 290–298.
Lin, S. C., Huck, J. H., Roberts, J. D., Macklin, W. B., Somogyi, P., and Bergles, D. E. (2005). Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron 46, 773–785.
Liu, W., Tang, Y., and Feng, J. (2011). Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 89, 141–146.
Lucas, S. M., Rothwell, N. J., and Gibson, R. M. (2006). The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147(Suppl. 1), S232–S240.
Machado, A., Herrera, A. J., Venero, J. L., Santiago, M., De Pablos, R. M., Villaran, R. F., Espinosa-Oliva, A. M., Arguelles, S., Sarmiento, M., Delgado-Cortes, M. J., Maurino, R., and Cano, J. (2011). Peripheral inflammation increases the damage in animal models of nigrostriatal dopaminergic neurodegeneration: possible implication in Parkinson’s disease incidence. Parkinsons Dis. 2011, 393769.
Malarkey, E. B., and Parpura, V. (2008). Mechanisms of glutamate release from astrocytes. Neurochem. Int. 52, 142–154.
Matthias, K., Kirchhoff, F., Seifert, G., Huttmann, K., Matyash, M., Kettenmann, H., and Steinhauser, C. (2003). Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J. Neurosci. 23, 1750–1758.
Miya, K., Inoue, R., Takata, Y., Abe, M., Natsume, R., Sakimura, K., Hongou, K., Miyawaki, T., and Mori, H. (2008). Serine racemase is predominantly localized in neurons in mouse brain. J. Comp. Neurol. 510, 641–654.
Mothet, J. P., Parent, A. T., Wolosker, H., Brady, R. O. Jr., Linden, D. J., Ferris, C. D., Rogawski, M. A., and Snyder, S. H. (2000). D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 4926–4931.
Mulligan, S. J., and MacVicar, B. A. (2004). Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199.
Navarrete, M., Perea, G., Fernandez de Sevilla, D., Gomez-Gonzalo, M., Nunez, A., Martin, E. D., and Araque, A. (2012). Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10, e1001259. doi:10.1371/journal.pbio.1001259
Neary, J. T., Kang, Y., Willoughby, K. A., and Ellis, E. F. (2003). Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. J. Neurosci. 23, 2348–2356.
Nedergaard, M. (1996). Spreading depression as a contributor to ischemic brain damage. Adv. Neurol. 71, 75–83; discussion 83–74.
Nett, W. J., Oloff, S. H., and McCarthy, K. D. (2002). Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J. Neurophysiol. 87, 528–537.
Newman, E. A. (2001). Propagation of intercellular calcium waves in retinal astrocytes and Muller cells. J. Neurosci. 21, 2215–2223.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Nishiyama, A., Yang, Z., and Butt, A. (2005). Astrocytes and NG2-glia: what’s in a name? J. Anat. 207, 687–693.
Ohsawa, K., and Kohsaka, S. (2011). Dynamic motility of microglia: purinergic modulation of microglial movement in the normal and pathological brain. Glia 59, 1793–1799.
Ortinski, P. I., Dong, J., Mungenast, A., Yue, C., Takano, H., Watson, D. J., Haydon, P. G., and Coulter, D. A. (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591.
Panatier, A., Vallee, J., Haber, M., Murai, K. K., Lacaille, J. C., and Robitaille, R. (2011). Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146, 785–798.
Pardo, A. C., Wong, V., Benson, L. M., Dykes, M., Tanaka, K., Rothstein, J. D., and Maragakis, N. J. (2006). Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp. Neurol. 201, 120–130.
Parpura, V., Basarsky, T. A., Liu, F., Jeftinija, K., Jeftinija, S., and Haydon, P. G. (1994). Glutamate-mediated astrocyte-neuron signalling. Nature 369, 744–747.
Parpura, V., and Haydon, P. G. (2000). Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc. Natl. Acad. Sci. U.S.A. 97, 8629–8634.
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A., and Bessis, A. (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 109, E197–E205.
Peng, H., Erdmann, N., Whitney, N., Dou, H., Gorantla, S., Gendelman, H. E., Ghorpade, A., and Zheng, J. (2006). HIV-1-infected and/or immune activated macrophages regulate astrocyte SDF-1 production through IL-1beta. Glia 54, 619–629.
Perea, G., and Araque, A. (2005). Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J. Neurosci. 25, 2192–2203.
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431.
Peters, A., Palay, S. L., and Webster, H. deF. (1991). The Fine Structure of the Nervous System: Neurons and Their Supporting Cells, 3rd Edn. New York: Oxford University Press, 282.
Peters, O., Schipke, C. G., Hashimoto, Y., and Kettenmann, H. (2003). Different mechanisms promote astrocyte Ca2+ waves and spreading depression in the mouse neocortex. J. Neurosci. 23, 9888–9896.
Petravicz, J., Fiacco, T. A., and McCarthy, K. D. (2008). Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 28, 4967–4973.
Piet, R., and Jahr, C. E. (2007). Glutamatergic and purinergic receptor-mediated calcium transients in Bergmann glial cells. J. Neurosci. 27, 4027–4035.
Pocock, J. M., and Kettenmann, H. (2007). Neurotransmitter receptors on microglia. Trends Neurosci. 30, 527–535.
Porter, J. T., and McCarthy, K. D. (1996). Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci. 16, 5073–5081.
Porter, J. T., and McCarthy, K. D. (1997). Astrocytic neurotransmitter receptors in situ and in vivo. Prog. Neurobiol. 51, 439–455.
Proux-Gillardeaux, V., Rudge, R., and Galli, T. (2005). The tetanus neurotoxin-sensitive and insensitive routes to and from the plasma membrane: fast and slow pathways? Traffic 6, 366–373.
Qin, L., Li, G., Qian, X., Liu, Y., Wu, X., Liu, B., Hong, J. S., and Block, M. L. (2005). Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 52, 78–84.
Reid, G., Wielinga, P., Zelcer, N., van der Heijden, I., Kuil, A., de Haas, M., Wijnholds, J., and Borst, P. (2003). The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 100, 9244–9249.
Rivest, S. (2003). Molecular insights on the cerebral innate immune system. Brain Behav. Immun. 17, 13–19.
Ronaldson, P. T., Persidsky, Y., and Bendayan, R. (2008). Regulation of ABC membrane transporters in glial cells: relevance to the pharmacotherapy of brain HIV-1 infection. Glia 56, 1711–1735.
Rosenberg, D., Kartvelishvily, E., Shleper, M., Klinker, C. M., Bowser, M. T., and Wolosker, H. (2010). Neuronal release of D-serine: a physiological pathway controlling extracellular D-serine concentration. FASEB J. 24, 2951–2961.
Rossi, D., and Volterra, A. (2009). Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res. Bull. 80, 224–232.
Rostasy, K., Egles, C., Chauhan, A., Kneissl, M., Bahrani, P., Yiannoutsos, C., Hunter, D. D., Nath, A., Hedreen, J. C., and Navia, B. A. (2003). SDF-1alpha is expressed in astrocytes and neurons in the AIDS dementia complex: an in vivo and in vitro study. J. Neuropathol. Exp. Neurol. 62, 617–626.
Rusakov, D. A., Zheng, K., and Henneberger, C. (2011). Astrocytes as regulators of synaptic function: a quest for the Ca2+ master key. Neuroscientist 17, 513–523.
Santello, M., Bezzi, P., and Volterra, A. (2011). TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69, 988–1001.
Santello, M., and Volterra, A. (2009). Synaptic modulation by astrocytes via Ca2+-dependent glutamate release. Neuroscience 158, 253–259.
Sanzgiri, R. P., Araque, A., and Haydon, P. G. (1999). Prostaglandin E(2) stimulates glutamate receptor-dependent astrocyte neuromodulation in cultured hippocampal cells. J. Neurobiol. 41, 221–229.
Scemes, E., and Giaume, C. (2006). Astrocyte calcium waves: what they are and what they do. Glia 54, 716–725.
Schipke, C. G., Boucsein, C., Ohlemeyer, C., Kirchhoff, F., and Kettenmann, H. (2002). Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J. 16, 255–257.
Schummers, J., Yu, H., and Sur, M. (2008). Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320, 1638–1643.
Shaw, P. J. (1993). Excitatory amino acid receptors, excitotoxicity, and the human nervous system. Curr. Opin. Neurol. Neurosurg. 6, 414–422.
Shelton, M. K., and McCarthy, K. D. (2000). Hippocampal astrocytes exhibit Ca2+-elevating muscarinic cholinergic and histaminergic receptors in situ. J. Neurochem. 74, 555–563.
Shigetomi, E., Tong, X., Kwan, K. Y., Corey, D. P., and Khakh, B. S. (2012). TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 15, 70–80.
Simard, M., Arcuino, G., Takano, T., Liu, Q. S., and Nedergaard, M. (2003). Signaling at the gliovascular interface. J. Neurosci. 23, 9254–9262.
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647.
Steele, M. L., and Robinson, S. R. (2012). Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol. Aging 33, 423 e421–e413.
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228.
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059.
Stow, J. L., Low, P. C., Offenhauser, C., and Sangermani, D. (2009). Cytokine secretion in macrophages and other cells: pathways and mediators. Immunobiology 214, 601–612.
Strong, A. J., Fabricius, M., Boutelle, M. G., Hibbins, S. J., Hopwood, S. E., Jones, R., Parkin, M. C., and Lauritzen, M. (2002). Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 33, 2738–2743.
Takata, N., Mishima, T., Hisatsune, C., Nagai, T., Ebisui, E., Mikoshiba, K., and Hirase, H. (2011). Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 31, 18155–18165.
Takemiya, T., Matsumura, K., Sugiura, H., Yasuda, S., Uematsu, S., Akira, S., and Yamagata, K. (2011). Endothelial microsomal prostaglandin E synthase-1 facilitates neurotoxicity by elevating astrocytic Ca2+ levels. Neurochem. Int. 58, 489–496.
Tanabe, S., Heesen, M., Yoshizawa, I., Berman, M. A., Luo, Y., Bleul, C. C., Springer, T. A., Okuda, K., Gerard, N., and Dorf, M. E. (1997). Functional expression of the CXC-chemokine receptor-4/fusin on mouse microglial cells and astrocytes. J. Immunol. 159, 905–911.
Verrall, L., Burnet, P. W., Betts, J. F., and Harrison, P. J. (2010). The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol. Psychiatry 15, 122–137.
Vitkovic, L., Bockaert, J., and Jacque, C. (2000). “Inflammatory” cytokines: neuromodulators in normal brain? J. Neurochem. 74, 457–471.
Volterra, A., and Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640.
Walz, W. (2000). Controversy surrounding the existence of discrete functional classes of astrocytes in adult gray matter. Glia 31, 95–103.
Wang, X., Arcuino, G., Takano, T., Lin, J., Peng, W. G., Wan, P., Li, P., Xu, Q., Liu, Q. S., Goldman, S. A., and Nedergaard, M. (2004). P2X7 receptor inhibition improves recovery after spinal cord injury. Nat. Med. 10, 821–827.
Wang, X., Lou, N., Xu, Q., Tian, G. F., Peng, W. G., Han, X., Kang, J., Takano, T., and Nedergaard, M. (2006). Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 9, 816–823.
Winship, I. R., Plaa, N., and Murphy, T. H. (2007). Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J. Neurosci. 27, 6268–6272.
Wolosker, H., Blackshaw, S., and Snyder, S. H. (1999). Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 96, 13409–13414.
Yagodin, S. V., Holtzclaw, L., Sheppard, C. A., and Russell, J. T. (1994). Nonlinear propagation of agonist-induced cytoplasmic calcium waves in single astrocytes. J. Neurobiol. 25, 265–280.
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., and Barres, B. A. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410.
Zhang, H., Cao, H. J., Kimelberg, H. K., and Zhou, M. (2011). Volume regulated anion channel currents of rat hippocampal neurons and their contribution to oxygen-and-glucose deprivation induced neuronal death. PLoS ONE 6, e16803. doi:10.1371/journal.pone.0016803
Zhang, Y., Zhang, H., Feustel, P. J., and Kimelberg, H. K. (2008). DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp. Neurol. 210, 514–520.
Zheng, J., Ghorpade, A., Niemann, D., Cotter, R. L., Thylin, M. R., Epstein, L., Swartz, J. M., Shepard, R. B., Liu, X., Nukuna, A., and Gendelman, H. E. (1999). Lymphotropic virions affect chemokine receptor-mediated neural signaling and apoptosis: implications for human immunodeficiency virus type 1-associated dementia. J. Virol. 73, 8256–8267.
Zhou, M., Schools, G. P., and Kimelberg, H. K. (2006). Development of GLAST(+) astrocytes and NG2(+) glia in rat hippocampus CA1: mature astrocytes are electrophysiologically passive. J. Neurophysiol. 95, 134–143.
Keywords: glia, IP3R, microglia, neurological disorder, neurodegenerative disease, TNFα, Gq GPCR, inflammation
Citation: Agulhon C, Sun M-Y, Murphy T, Myers T, Lauderdale K and Fiacco TA (2012) Calcium signaling and gliotransmission in normal vs. reactive astrocytes. Front. Pharmacol. 3:139. doi: 10.3389/fphar.2012.00139
Received: 15 May 2012; Accepted: 26 June 2012;
Published online: 13 July 2012.
Edited by:
Ramón Martínez-Cancino, Instituto Superior de Tecnologías y Ciencias Aplicadas, CubaReviewed by:
Li Zhang, National Institutes of Health, USARamón Martínez-Cancino, Instituto Superior de Tecnologías y Ciencias Aplicadas, Cuba
Copyright: © 2012 Agulhon, Sun, Murphy, Myers, Lauderdale and Fiacco. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Cendra Agulhon, UFR Biomédicale, CNRS UMR 8154, Université Paris Descartes, 45 rue des Saints Pères, 75006 Paris, France. e-mail:Y2VuZHJhLmFndWxob25AcGFyaXNkZXNjYXJ0ZXMuZnI=; Todd A. Fiacco, Department of Cell Biology and Neuroscience, University of California Riverside, 1109 Biological Sciences Building, Riverside, CA 92521, USA. e-mail:dG9kZC5maWFjY29AdWNyLmVkdQ==