Abstract
The classical tango is a dance characterized by a 2/4 or 4/4 rhythm in which the partners dance in a coordinated way, allowing dynamic contact. There is a surprising similarity between the tango and how KCNE β-subunits “dance” to the fast rhythm of the cell with their partners from the Kv channel family. The five KCNE β-subunits interact with several members of the Kv channels, thereby modifying channel gating via the interaction of their single transmembrane-spanning segment, the extracellular amino terminus, and/or the intracellular carboxy terminus with the Kv α-subunit. Best studied is the molecular basis of interactions between KCNE1 and Kv7.1, which, together, supposedly form the native cardiac IKs channel. Here we review the current knowledge about functional and molecular interactions of KCNE1 with Kv7.1 and try to summarize and interpret the tango of the KCNEs.
Introduction
Voltage-gated potassium channels (Kv) are ubiquitously expressed in human tissues. They enable the rapid, selective movement of potassium ions through cellular membranes, thereby regulating physiological processes such as transmembrane ion passage and hormone secretion, vesicle cycling, and cell excitability. Kv channels display a huge diversity due to the large number of different α-subunits, alternative splicing, post-transcriptional modifications, and their ability to form heteromeric channels with other pore-forming α-subunits. To complicate the situation even more, the number of functionally different Kv channels in native tissues is further increased by interaction with regulatory β-subunits. These accessory subunits modify subcellular localization as well as biophysical properties of Kv channels such as gating kinetics, ion selectivity, and pharmacology. Among the best studied modulations of Kv channels by regulatory β-subunits is the effect of KCNE1 on Kv7.1.
Kv7.1-Expression and Physiological Function
The KCNQ1 gene was first identified by Wang et al. (1996b) in a linkage study of patients with long QT syndrome (LQTS1). Its gene product, Kv7.1 (also termed KvLQT1 or KCNQ1), is a voltage-gated potassium channel α-subunit, and its expression was detected in several mammalian tissues, including heart, epithelia, and smooth muscle (Figure 1; Table A1 in Appendix). Kv7.1 can assemble with different members of the KCNE family of regulatory β-subunits to fulfill a variety of physiological functions.
Figure 1
In the heart, Kv7.1 is involved in the termination of the cardiac action potential. The repolarizing potassium current, IK, consists of two major components, the rapid delayed rectifier potassium current IKr and the slow delayed rectifier potassium current IKs (Sanguinetti and Jurkiewicz, 1990). Kv7.1 coassembles with the KCNE1 β-subunit to form the channel complex that mediates IKs (Barhanin et al., 1996; Sanguinetti et al., 1996). Although KCNE1 is the major accessory subunit assembling with Kv7.1 in the heart, other subunits of the KCNE family might be present (Bendahhou et al., 2005), serving as additional regulators of IKs (Wu et al., 2006). The significance of Kv7.1 and its accessory β-subunits for maintaining normal rhythmicity is further emphasized by the numerous KCNQ1 and KCNE mutations associated with cardiac arrhythmias (http://www.fsm.it/cardmoc/). Most of these mutations lead to loss of channel function causing LQTS, a disorder predisposing affected individuals to torsade de pointes arrhythmia and cardiac sudden death.
Besides its cardiac function, several lines of evidence suggest an important role of Kv7.1 and its accessory β-subunit KCNE1 in the hearing process. In patients suffering from Jervell and Lange-Nielsen syndrome – the recessive form of inherited LQTS – cardiac arrhythmia is accompanied by profound bilateral deafness. Mutations in both KCNQ1 and KCNE1 genes have been reported to cause this disorder (Jervell and Lange-Nielsen, 1957; Neyroud et al., 1997; Schulze-Bahr et al., 1997). In addition, targeted disruption of the KCNQ1 gene in mice leads to deafness caused by morphological abnormalities of the inner ear (Lee et al., 2000; Casimiro et al., 2001). Expression of Kv7.1 and KCNE1 has been detected in the marginal cells of the stria vascularis of the cochlea and the vestibular dark cells (Neyroud et al., 1997; Nicolas et al., 2001; Knipper et al., 2006; Hur et al., 2007). Both cell types are involved in the generation of the potassium-rich endolymph, and Kv7.1/KCNE1 channels have been suggested to be key mediators of this K+ secretion (Marcus and Shen, 1994; Shen et al., 1995; Wangemann, 1995; Wangemann et al., 1995; Sunose et al., 1997).
In addition to the inner ear epithelium, Kv7.1 has been detected in a variety of other epithelial cell types, where it participates in secretory transduction. In the kidney, Kv7.1/KCNE1 channels seem to be located in the proximal tubule of the nephron (Sugimoto et al., 1990; Vallon et al., 2001), conducting a K+ current to counterbalance membrane depolarization induced by electrogenic Na+-coupled transport of glucose or amino acids (Vallon et al., 2001, 2005). The relevance of Kv7.1/KCNE1 channels for renal function is further underlined by the observation that KCNE1 knockout mice suffer from hypokalemia, urinary and fecal salt wasting, and volume depletion (Arrighi et al., 2001; Warth and Barhanin, 2002). Kv7.1 expression has also been detected in the small intestine and the colon (Schroeder et al., 2000; Dedek and Waldegger, 2001; Demolombe et al., 2001; Kunzelmann et al., 2001; Horikawa et al., 2005). In colonic crypt cells Kv7.1 is believed to assemble with another accessory β-subunit, KCNE3, and to mediate a K+ conductance that provides the driving force for chloride secretion (Schroeder et al., 2000; Kunzelmann et al., 2001). Two further examples of Kv7.1 expression and function in chloride-secreting tissues are pancreatic acinar cells and airway epithelium (Kim and Greger, 1999; Kottgen et al., 1999; Mall et al., 2000; Demolombe et al., 2001; Grahammer et al., 2001b; Lee et al., 2004). In parietal cells of the stomach Kv7.1 coassembles with KCNE2 and participates in gastric acid secretion (Dedek and Waldegger, 2001; Demolombe et al., 2001; Grahammer et al., 2001a; Heitzmann et al., 2004). In KCNQ1 knockout mice gastric hyperplasia and profound hypochlorhydria have been observed, indicating the importance of Kv7.1 in normal stomach development and function (Lee et al., 2000). Kv7.1 expression has also been detected in the human thyroid gland, and it has been shown that mice lacking functional Kv7.1 develop hypothyroidism (Frohlich et al., 2011). Recently, Kv7.1 channels have been shown to relax systemic and pulmonary arteries upon pharmacological activation (Chadha et al., 2012).
Regulation of Kv7.1 by Accessory β-Subunits of the KCNE Gene Family
All five members of the KCNE family of regulatory β-subunits can functionally coassemble with Kv7.1 to subunit-specifically change its current characteristics (for an excellent review about KCNEs, see McCrossan and Abbott, 2004). The founding member KCNE1 (previously termed MinK or IsK) was identified in 1988 (Takumi et al., 1988). As its expression in Xenopus oocytes produced slowly activating potassium currents resembling the cardiac IKs, KCNE1 was originally believed to be the ion channel α-subunit forming the IKs channel. However, it is now well established that KCNE1 cannot form functional K+ channels alone, but can serve as a regulatory β-subunit for several voltage-gated cation channels, including Kv7.1 (Barhanin et al., 1996; Sanguinetti et al., 1996). The IKs-like potassium currents observed in oocytes upon heterologous expression of KCNE1 were actually caused by coassembly with endogenous Kv7.1. So far, four other members of the KCNE family, KCNE2-KCNE5 (also termed MiRP1-4) have been identified (Abbott et al., 1999; Piccini et al., 1999), and their influence on Kv7.1 channels has been studied extensively in heterologous expression systems. Kv7.1 expressed alone forms a classical delayed rectifier potassium-selective channel with fast activation, delayed partial inactivation, and relatively slow deactivation (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). The presence of KCNE1 drastically modifies Kv7.1 activity by increasing unitary conductance as well as macroscopic currents, slowing activation, right-shifting voltage dependence of activation, suppressing currents at low activating voltages, suppressing partial inactivation, increasing Q10-value, and modulating pharmacology (Figures 2A–C; Barhanin et al., 1996; Sanguinetti et al., 1996; Pusch, 1998; Pusch et al., 1998; Sesti and Goldstein, 1998; Tristani-Firouzi and Sanguinetti, 1998; Lerche et al., 2000; Seebohm et al., 2001a,b,c, 2003a,b,c, 2005; Morokuma et al., 2008). Coassembly of Kv7.1 and KCNE2 gives rise to currents with decreased amplitude, instantaneous activation, rapid partial deactivation, and a linear current-voltage relationship (Tinel et al., 2000). KCNE3 also converts Kv7.1 to a channel with nearly instantaneous activation and a linear current-voltage relationship (Schroeder et al., 2000; Seebohm et al., 2003c), but in contrast to Kv7.1/KCNE2, complexes containing KCNE3 show some time dependency of gating at positive potentials and increased current densities (Schroeder et al., 2000; Melman et al., 2001; Mazhari et al., 2002). KCNE4 completely suppresses Kv7.1 currents at physiologically relevant membrane potentials (Grunnet et al., 2002) and KCNE5 shifts the voltage dependence of Kv7.1 activation to more positive potentials toward an activation threshold of about +40 mV (Figure 2D; Angelo et al., 2002; Seebohm et al., 2003c).
Figure 2
Gating of Kv7.1 and Its Modulation by KCNE1
The biophysical properties of voltage-gated Kv7.1 channels change dramatically when they coassemble with KCNEs. Homotetrameric Kv7.1 channels activate with relatively fast kinetics with τactivation by about 80−100 ms at 60 mV (Figure 2; Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). The channels undergo partial inactivation, with about 60% of the channels being inactivated at 60 mV in steady-state (Figure 2). The kinetic behavior of Kv7.1 can be approximated by a linear gating scheme of the form (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998):
model 1:
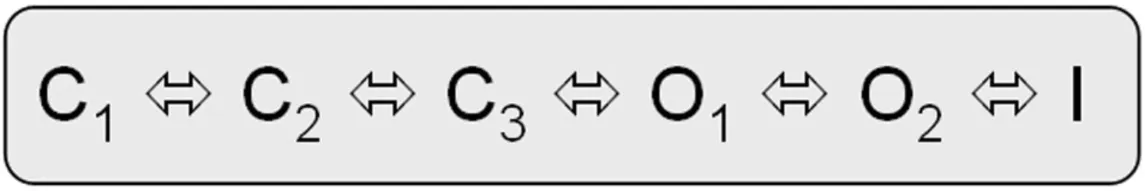
In this model C1 C2, C2 C3, and O1 O2 are the voltage-dependent gating steps. Recently, an alternative circular gating scheme was proposed by Ma et al. (2011).
C2, C2 C3, and O1 O2 are the voltage-dependent gating steps. Recently, an alternative circular gating scheme was proposed by Ma et al. (2011).
model 2:
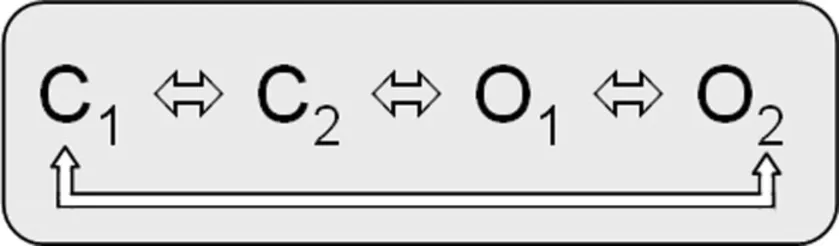
This novel gating scheme accounts for an open fraction that is determined by allosteric coupling of residues in the S4–S5 linker/pore domain (PD). C1 C2 and O1 O2 are the voltage-dependent gating steps.
Kv7.1/KCNE1 heteromeric channels activate far more slowly and at more positive potentials than homomeric Kv7.1 channels, do not partially inactivate, and have an increased single-channel conductance compared to homomeric Kv7.1 recorded in standard extracellular solutions (Barhanin et al., 1996; Sanguinetti et al., 1996; Pusch, 1998; Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998). It is believed that Kv7.1/KCNE1 channel currents resemble the cardiac repolarizing, slowly activating potassium current IKs and that LQTS1/5 mutations might directly or allosterically modify interactions of Kv7.1 and the β-subunit KCNE1 (e.g., Schmitt et al., 2000; Seebohm et al., 2001c; Osteen et al., 2010; Wang et al., 2011b). Cui et al. (1994) proposed two linear-branched gating schemes for Kv7.1/KCNE1 heteromers, which depend on varying KCNE1 amounts coexpressed with Kv7.1 and imply interactions among individual channel proteins during activation. Yet Tzounopoulos et al. reported on a crossover gating phenomenon after prepulsing to different voltages that cannot be explained by a classical Cole–Moore shift in a linear gating model of Kv7.1/KCNE1 channels and presented results that argue for a circular gating model similar to model 2 (Cole and Moore, 1960; Tzounopoulos et al., 1998). However, a branched model can account for this crossover gating phenomenon after prepulsing to different voltages as well (Strutz-Seebohm et al., 2011):
model 3:
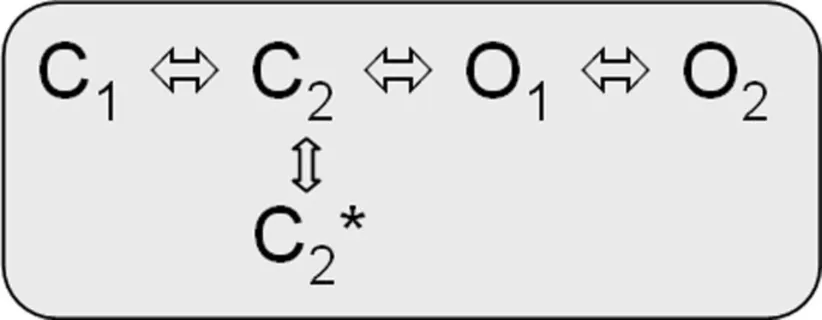
Further gating schemes have been proposed to describe Kv7.1/KCNE1 gating (Silva and Rudy, 2005, 2010; Osteen et al., 2010; Ghosh et al., 2011). In summary, the gating behaviors of Kv7.1 and Kv7.1/KCNE1 are relatively complex, and simple, linear gating models reach too short to approximate a realistic mathematical description.
Stoichiometry of the Kv7.1/KCNE1 Complex
After the discovery that the channel complex mediating the cardiac IKs is composed of Kv7.1 α-subunits and KCNE1 β-subunits (Barhanin et al., 1996; Sanguinetti et al., 1996), detailed investigations on the nature of this association started. Like all members of the Kv channel family, Kv7.1 α-subunits assemble into tetramers to form functional channels. Each α-subunit consists of six transmembrane segments, S1–S6, flanked by intracellular amino- and carboxy-terminal domains. The central pore domain (PD) of the channel, which contains the ion conduction pathway and the activation gate, is formed by segments S5 and S6 and the pore loop between them. The pore loop carries the K+ channel signature sequence TTIGYGD allowing rapid ion conduction and potassium selectivity. The PD is surrounded and controlled by four voltage-sensing domains (VSDs) formed by segments S1–S4 from each subunit (Figures 3A,B). S4 contains several positively charged amino acids and is a key element of voltage sensing. Further charged residues in S1–S3 may contribute additional charge to allow for voltage sensing. Tetrameric assembly of Kv7.1 α-subunits is mediated via their C-terminal domains (Schmitt et al., 2000; Schwake et al., 2003). Although Kv7.1 homotetramers are functional, the presence of the accessory β-subunit KCNE1 is needed to produce currents with characteristics of native IKs (Figure 2A). KCNE1 is a small, integral membrane protein with a single transmembrane-spanning segment flanked by an extracellular N-terminal and an intracellular C-terminal domain (Figure 3A). The number of KCNE1 β-subunits recruited by tetrameric Kv7.1 channels has been a matter of extensive debate (Figure 3B). Previous studies have either suggested a fixed stoichiometry of four Kv7.1 subunits and two KCNE1 subunits (Wang and Goldstein, 1995; Chen et al., 2003a; Kang et al., 2008; Morin and Kobertz, 2008) or a variable stoichiometry with up to four KCNE1 subunits assembling with a Kv7.1 tetramer (Cui et al., 1994; Wang et al., 1998, 2011a; Morokuma et al., 2008; Nakajo et al., 2010; Zheng et al., 2010; Strutz-Seebohm et al., 2011). A summary of the data obtained during the last two decades is provided in Table 1. The results reported leave the possibility open that KCNE1 subunits coassemble with Kv7.1 in different modes allowing up to four KCNE1 subunits to approach the Kv7.1 tetramer but permitting only formation of 4 Kv7.1 : 2 KCNE1 assemblies in specific regions of the heteromeric channels. Prerequisites for such an interaction would be variable and dynamic interactions. Indeed, several studies suggest interactions of Kv7.1 with KCNE1 at different protein regions in the various channel states (summarized in Table 2). However, further studies are required to support such a hypothesis.
Table 1
| Experimental evidence leading to conclusion of fixed stoichiometry (4:2) | Reference |
|---|---|
| Suppression of current induced by coexpression of wildtype and mutant KCNE1 indicates 4:2 stoichiometry | Wang and Goldstein (1995) |
| Kv7.1/Kv7.1/KCNE1 fusion proteins and naturally assembled Kv7.1/KCNE1 channels show similar characteristics of CTX inhibition; quantification of Kv7.1 and KCNE1 subunits using 3H-CTX and an antibody indicates a 4:2 stoichiometry | Chen et al. (2003a) |
| Chemical subunit counting experiments indicate that association of two KCNE1 subunits with the Kv7.1 tetramer is sufficient to induce KCNE1-typical modulation of channel properties | Morin and Kobertz (2008) |
| Computational model of Kv7.1/KCNE1 channels indicates that binding of more than two KCNE1 subunits to the Kv7.1 tetramer might be sterically hindered | Kang et al. (2008) |
| Experimental evidence leading to conclusion of variable stoichiometry | Reference |
| Current amplitude, activation kinetics, and voltage dependence of Kv7.1/KCNE1 channels vary with the amount of KCNE1 | Cui et al. (1994) |
| Both Kv7.1/KCNE1 fusion proteins and Kv7.1–Kv7.1 with additional KCNE1 produce currents with activation kinetics and voltage dependence similar to naturally assembled Kv7.1/KCNE1 channels | Wang et al. (1998) |
| Voltage dependence of activation of Kv7.1/KCNE1 channels varies with the amount of KCNE1 | Morokuma et al. (2008) |
| Single-molecule fluorescent bleaching studies indicate that up to four KCNE1 subunits associate with the Kv7.1 tetramer depending on the relative densities of the two subunits | Nakajo et al. (2010) |
| Effects of free KCNE1 C-terminals on voltage dependence of activation of Kv7.1 and Kv7.1/KCNE1 channels are complex, indicating multiple stoichiometries or saturation of possible binding sites | Zheng et al. (2010) |
| Overexpression of KCNE1 markedly changed activation kinetics and voltage dependence of native IKs, indicating assembly of additional KCNE1 subunits with endogenous channels | Wang et al. (2011a) |
| Modeling of transmembrane domain suggests that variable stoichiometry seems possible in this region | Strutz-Seebohm et al. (2011) |
Stoichiometry of Kv7.1/KCNE1 channels.
Table 2
| Approach | Conclusion | Reference |
|---|---|---|
| Deletion analysis, chimeric approach, and/or site-directed mutagenesis | TM segment and cytoplasmic portion immediately following TM segment of KCNE1 mediate KCNE1 function | Takumi et al. (1991) |
| TM segment and C-terminal domain of KCNE1 mediate KCNE1 function | Tapper and George (2000) | |
| Residues 57–59 of KCNE1 are important for KCNE1 function (“activation triplet”) | Melman et al. (2001) | |
| Residue L273 of Kv7.1 is important for normal modulation by KCNE1 | Seebohm et al. (2001c) | |
| Residue T58 is a key element of KCNE1 function | Melman et al. (2002) | |
| Requirements to interact with KCNE1 are located in regions C-terminal to S5; Residues S338, F339, and F340 in S6 are important for normal modulation by KCNE1 | Melman et al. (2004) | |
| Residues S338, F339, and F340 in S6 are possible interaction sites of KCNE1 | Panaghie et al. (2006) | |
| KCNE1 C-terminus is crucial for channel assembly, open state destabilization, kinetics of deactivation | Chen et al. (2009) | |
| Amino acids important for normal modulation by KCNE1 are located in S5 and S6 (G272, V324, V334) of Kv7.1 | Nakajo et al. (2011) | |
| Cysteine scanning mutagenesis combined with chemical modifications (Cd2+ coordination, MTS reagent binding, and spontaneous disulfide formation) | TM segment of KCNE1 lines the conduction pathway | Wang et al. (1996a) |
| TM segment of KCNE1 lines the conduction pathway | Tai and Goldstein (1998) | |
| KCNE1 is located outside the conduction pathway | Kurokawa et al. (2001) | |
| KCNE1 is located outside the conduction pathway but in very close proximity to S6 of Kv7.1 | Tapper and George (2001) | |
| E44 in KCNE1 is close to A226 in S4 of Kv7.1 in the open state; KCNE1 is close to or possibly interacts with the VSD | Nakajo and Kubo (2007) | |
| KCNE1 makes state-dependent contact with S1 of Kv7.1; KCNE1 is in close proximity to the VSD | Xu et al. (2008) | |
| KCNE1 is located close to S1 and S4 of two adjacent VSDs | Shamgar et al. (2008) | |
| C-terminal region directly following the KCNE1 TM segment interacts with activation gate of Kv7.1 | Lvov et al. (2010) | |
| Proof of direct physical interaction or close proximity (e.g., co-immunoprecipitation, FRET) | C-terminus of KCNE1 directly interacts with the pore region of Kv7.1 | Romey et al. (1997) |
| C-termini of Kv7.1 and KCNE1 move close to each other during channel activation; distal C-terminus of KCNE1 interacts with dimeric coiled coil helix C of Kv7.1 | Haitin et al. (2009) | |
| Physical interaction between C-termini of Kv7.1 and KCNE1; portion of A-helix and its linker to S6 bind KCNE1 | Zheng et al. (2010) | |
| Analysis of disease-causing mutations | Functional interaction of KCNE1 with S4/S4–S5 linker of Kv7.1 | Franqueza et al. (1999) |
| Functional interaction of KCNE1 with S4/S4–S5 linker of Kv7.1 | Chouabe et al. (2000) | |
| Functional interaction of KCNE1 with S1 of Kv7.1 | Chan et al. (2012) | |
| Computational model | KCNE1 is located in a cleft between the pore domain and the VSD of Kv7.1 | Kang et al. (2008) |
| KCNE1 binds to the outer face of the Kv7.1 channel pore, KCNE1 is located in a cleft between pore domain and VSD of Kv7.1 | Strutz-Seebohm et al. (2011) |
Structural basis of Kv7.1/KCNE1 interaction.
Figure 3
Structural Basis of Kv7.1/KCNE1 Interaction
The structural basis of the Kv7.1/KCNE1 interaction has been studied extensively over the last 20 years. Experimental data obtained from deletion analysis, chimeric approaches, and site-directed mutagenesis identified regions in both Kv7.1 and KCNE1 that might be crucial for association and modulation (summarized in Table 2). Based on these data it has been suggested that the transmembrane segment and the C-terminal domain of KCNE1 mediate its ability to modulate Kv7.1 (Takumi et al., 1991; Tapper and George, 2000). Molecular key elements of KCNE1 function seem to be three glycines, several bulky, aromatic phenylalanines, and the threonine 58 located at the center of the KCNE1 transmembrane segment (Melman et al., 2001, 2002; Strutz-Seebohm et al., 2011). A comparison with other KCNE subunits underscores the importance of the transmembrane segment for KCNE function: interactions of Kv7.1 with KCNE1 and Kv7.1 with KCNE3 show similar chemical modification rates within the transmembrane region (Rocheleau and Kobertz, 2008), and the KCNE4 transmembrane segment modulates voltage dependence of Kv7.1 activation (Ciampa et al., 2011). A recent study indicates that different KCNE proteins contact different regions in Kv7.1 all located within the transmembrane segments (Nakajo et al., 2011). In segment S6 of Kv7.1, a three amino acid motif (S338, F339, F340) has been identified that might constitute a site of specific interaction with KCNE1, especially with its residue T58 (Melman et al., 2004; Panaghie et al., 2006; Strutz-Seebohm et al., 2011). Further residues in the pore region of Kv7.1 (F270, G272, and L273 in S5, V307, V310, and T311 in the lower pore helix, and V324 and V334 in the upper S6) are crucial for an accurate modulation by KCNE1 (Seebohm et al., 2001c, 2003c; Nakajo et al., 2011).
Based on experimental evidence, localizations of KCNE1 in the channel complex have been suggested. A series of observations obtained from cysteine scanning mutagenesis combined with chemical modifications (Cd2+ coordination, MTS reagent binding, and spontaneous disulfide formation) indicate that KCNE1 lies in close proximity to both the PD and the VSD of the Kv7.1 tetramer (summarized in Table 2). First interpretations suggested that KCNE1 lines the conduction pathway (Wang et al., 1996a; Tai and Goldstein, 1998). However, subsequent studies indicate that the KCNE1 transmembrane segment is located outside the PD and interacts with the outer S5 and S6 segments of Kv7.1 (Lerche et al., 2000; Kurokawa et al., 2001; Tapper and George, 2001; Chung et al., 2009; Strutz-Seebohm et al., 2011). Its extracellular flank seems to face the extracellular ends of segments S1 and S6 of Kv7.1, with the residues interacting with segment S1 varying dependent on the specific channel state (Xu et al., 2008; Chung et al., 2009). In addition to segment S1 of the VSD and S5/S6 of the PD, the transmembrane domain of KCNE1 gets in close contact with the primary cationic voltage sensor S4 (Nakajo and Kubo, 2007; Shamgar et al., 2008; Silva et al., 2009; Strutz-Seebohm et al., 2011). A possible interaction of KCNE1 with the VSD of Kv7.1 is further underscored by the fact that the abnormal phenotypes of several disease-causing mutations in S1, S4, or the S4–S5 linker of Kv7.1 in vitro only manifest in the presence of KCNE1 (summarized in Table 2; Franqueza et al., 1999; Chouabe et al., 2000; Chen et al., 2003c; Chan et al., 2012). The cytoplasmic portion directly following the transmembrane segment of KCNE1 seems to interact with the gating machinery of Kv7.1 (Lvov et al., 2010). A hallmark of all Kv7 channels is their sensitivity to muscarinic signaling, which gave them the name M-channels. The Kv7.1 membrane-proximal region may link membrane lipid metabolism of phosphoinositide PI(4,5)P2 to the gating machinery to allow for integration of muscarinic signaling into gating alterations in Kv7.1 (Ikeda and Kammermeier, 2002; Loussouarn et al., 2003). The lower S6 is expected to be in close proximity to allow for electrostatic interaction with the PI(4,5)P2 head groups, and PI(4,5)P2 binding sites have been reported to be positioned close to the lower S6-helices bundle in the PD of Kv7.1 (Loussouarn et al., 2003; Thomas et al., 2011). However, it may be mentioned that further PI(4,5)P2 binding sites may exist in the TM domain. KCNE1 residues R67SKKLEH73 in the vicinity of the inner membrane leaflet increase the PI(4,5)P2 sensitivity of Kv7.1 by a factor of 100, possibly by electrostatic interaction with PI(4,5)P2 head groups and/or Kv7.1 residues interacting with PI(4,5)P2 (Li et al., 2011). Thus, PI(4,5)P2 may function as a molecular coupler of Kv7.1 and KCNE1 to modulate gating in complex ways.
Recent structural modeling approaches generated 3D models of the transmembrane region of Kv7.1/KCNE1 that are in large agreement with the various experimental data. Specifically, homology modeling of the Kv7.1 S1–S6 region and Rosetta-based docking of the KCNE1 transmembrane domain produced an open and a closed state model of the heteromeric channel complex (Smith et al., 2007; Kang et al., 2008). Very recently, homology modeling of Kv7.1 with manual docking of the KCNE1 transmembrane domain was used to generate a pre-open closed state model of the Kv7.1/KCNE1 channel (Strutz-Seebohm et al., 2011). All three models are relatively stable in molecular dynamics simulations, which suggests that they are close to native states.
The C-termini of Kv7.1 and KCNE1 are in close proximity and physically interact, as indicated by FRET measurements and co-immunoprecipitation, yet the structural basis of this interaction is unknown (Haitin et al., 2009; Zheng et al., 2010). The cytosolic domain of Kv7.1/KCNE1 channels represents a multi-modular structure serving multiple functions. It contains several amphipathic α-helices. Structural prediction algorithms place two α-helices, A and B, to the proximal C-terminus of Kv7.1 (Haitin and Attali, 2008). These α-helices may contain functionally relevant calmodulin interaction motifs. The residues Kv7.1586–618 form α-helix D that contain a leucine-zipper and a coiled coil fold (Kanki et al., 2004). This parallel, four-stranded coiled coil allows for specific tetrameric assembly of Kv7.1 channel subunits (Schmitt et al., 2000; Schwake et al., 2003; Nakajo and Kubo, 2008). Recently, Xu and Minor (2009) presented a high-resolution X-ray crystal structure (1.7 Å resolution) of Kv7.1583–611 confirming the predictions of the coiled coil assembly specificity domain. Interestingly, helix D is supposedly two helical turns longer in this crystal structure than helices D of the closely related Kv7.2–5 channels, which allows for specific homomeric assembly with other Kv7.1 subunits but not for heteromeric assemblies with other family members (Haitin and Attali, 2008; Xu and Minor, 2009). The S6 transmembrane segment and the A-domain are linked by the region Kv7.1350–582, which according to in silico prediction assumes an apo-chloroperoxidase fold and can thus be modeled using the structural coordinates of apo-chloroperoxidase (pdb: 1VNS; Macedo-Ribeiro et al., 1999). However, these predictions appear somewhat preliminary, and several assumptions have to be made to incorporate the modeled region into the rest of the Kv7.1–3D model. Further, how exactly the proposed helices A/B and linker region containing the calmodulin binding site can be integrated into this fold is speculative. The structural folds of the N-terminal region Kv7.11–115 and the most distal residues Kv7.1612–676 remain completely unknown.
PI(4,5)P2 modulates function of Kv7.1/KCNE1 channels, and functional PI(4,5)P2 interaction sites were reported not only for the lower S6 segment but also for the C-terminus, where a cluster of basic amino acids may form a PI(4,5)P2 interaction site allowing for electrostatic interactions with the PI(4,5)P2 head groups (Loussouarn et al., 2003; Zhang et al., 2003; Delmas and Brown, 2005). This PI(4,5)P2 interaction site may functionally and physically overlap with the calmodulin binding sites in the inter helix A/B linker region (Hernandez et al., 2008). The Kv7.1–calmodulin interaction can be modulated by coassembly with KCNE4. This modulation is dependent on a juxtamembrane tetra-leucine motif (L69–L72) in KCNE4. However, deletion of this motif leaves the kinetic effects on Kv7.1 mediated by the KCNE4 transmembrane domain intact (Ciampa et al., 2011). Possibly, PI(4,5)P2 and calmodulin “glue” these C-terminal regions to the transmembrane proximal regions mentioned above and allow for modulation of gating at the activation gate and the VSD. Clearly, further evidence is needed to prove this hypothesis (Li et al., 2011).
The C-termini of Kv7.1/KCNE1 channels physically interact with several proteins like the AKAP yotiao, epidermal growth factor receptor kinase, and NEDD4.2 (Marx et al., 2002; Kanki et al., 2004; Kurokawa et al., 2004; Jespersen et al., 2007; Dong et al., 2010). The protein kinase A phosphorylates Ser27 in the N-terminus of Kv7.1 and modulates Kv7.1/KCNE1 function (Marx et al., 2002). This modulation critically depends on the presence of KCNE1 (Marx et al., 2002; Kurokawa et al., 2004). This observation suggests that the Kv7.1 N-terminus contributes to Kv7.1/KCNE1 subunit interaction. The binding sites for these proteins have to be located at the surface of the C-terminal structure to be accessible for interaction. This information will be of help for further structural modeling. A 3D model of the Kv7.1/KCNE1 channel satisfying most of the aforementioned hypotheses is shown in Figure 4.
Figure 4
Toward a Dynamic View on Structural Rearrangements during Kv7.1/KCNE1 Gating
In the last 14 years a wealth of potassium channel crystal structures have been reported. Although central structural elements in Kv7.1 clearly differ from classical Shaker-type Kv channels, the available crystal structures are well-suited templates to model the Kv7.1 S1–S6 region in different states (Seebohm et al., 2006; Silva et al., 2009). Direct interactions of the transmembrane segments of Kv7.1 with KCNE1 have been proposed, but the specific amino acid interactions remain controversial (summarized in Table 2). CD spectra in a membrane-like environment and the NMR solution structure of KCNE1 transmembrane peptides show that this peptide adopts an α-helical structure (Aggeli et al., 1998; Strutz-Seebohm et al., 2011). Recently, two NMR structures of full-length KCNE1 confirmed that the transmembrane segment of KCNE1 folds an α-helix (Tian et al., 2007; Kang et al., 2008). An open and a closed state model of Kv7.1/KCNE1 channels were proposed based on the NMR-structural coordinates and Rosetta-dockings of KCNE1 to Kv7.1 that suggest binding of KCNE1 to the so-called “gain-of-function cleft”, a space formed between two adjacent voltage sensor domains and the outer PD (Smith et al., 2007; Kang et al., 2008).
Localized to the center of the α-helical KCNE1 transmembrane segment are the residues G52FFGFFTLG60. As predicted, this α-helical transmembrane domain allows for flexibility at glycine residues G52, G55, and G60 (Tian et al., 2007; Kang et al., 2008; Strutz-Seebohm et al., 2011). Key to functional modulation of Kv7.1 by KCNE1 is the residue T58 (Melman et al., 2002). The surrounding phenylalanine residues generate a bulky aromatic cuff. The combination of these structural elements may enable formation of a stable hydrogen bond with residue S338 in Kv7.1 and diverse van der Waals interactions with residues in S4 and the S4–S5 linker region as well as S5/S6 amino acids F270, F339, and F340 to stabilize a pre-open closed state. The combination of the Kv7.1/KCNE1 models, the closed state, the pre-open closed state and the open state model are in good agreement with published results of several experimental studies (Smith et al., 2007; Kang et al., 2008; Strutz-Seebohm et al., 2011). The modeled transmembrane domain structures can be used to assess a molecular view on the slow activation of IKs (Figure 5). The three states closed, pre-open, and open can be used for morphing calculations (Echols et al., 2003). The basic idea is that the channel undergoes conformational transitions from closed (C) to pre-open (C*), from pre-open (C*) to open (O), and from open (O) to closed (C) states. These seem to represent the major steps in the Kv7.1 and Kv7.1/KCNE1 gating (Silva et al., 2009). The position of the voltage sensor is “down” for the closed state and “up” for the pre-open and open states (Tian et al., 2007; Kang et al., 2008; Silva et al., 2009; Strutz-Seebohm et al., 2011). The interaction of KCNE1T58 with Kv7.1S338 and Kv7.1F339 may uncouple the gate at the lower S6 from the S4–S5 linker, which leads to S6 bundle crossing and closure of the gate (Strutz-Seebohm et al., 2011). The formation of the S6 bundle upon S4–S5 linker uncoupling may be supported by the lack of a central S6 gating hinge (Seebohm et al., 2006). The uncoupling of S4–S5 by KCNE1 during closed state inactivation shows similarities to an uncoupling event of the PD from the voltage sensor domain during closed state inactivation of some Kv channels as described by the group of Bähring et al. (2012). In the light of the ongoing discussion about fixed stoichiometry of two KCNE1 subunits per Kv7.1 tetramer vs. variable stoichiometry it is fair to study a channel complex with two KCNE1 subunits docked to four Kv7.1 subunits (Strutz-Seebohm et al., 2011). This stoichiometry is consistent with both hypotheses. Morphing experiments have been performed (Figure 5). As expected the gating is associated with large structural rearrangements in the voltage sensor domain. On the other hand the PD is rather rigid in the region around the selectivity filter. The KCNE1 transmembrane segment undergoes relatively small rearrangements during the gating steps C C* and O C but dramatic structural alterations during the C* O transition (Figure 5; the extent of structural rearrangement around key residue KCNE1 T58 is indicated by a yellow arrow). This dramatic rearrangement of KCNE1 associated with the C* O transition may generate an energetic barrier that can only slowly be overcome, resulting in dramatically slowed activation, the pivotal characteristic of Kv7.1/KCNE1 gating. Clearly, these models present a preliminary view on the dynamics of Kv7.1/KCNE1 channels. Silva et al. (2009) modeled further structural closed states of the Kv7.1 voltage sensor module. Incorporation of these data in future Kv7.1/KCNE1 morphing experiments may prove valuable. Increase in knowledge combined with the already available data, upcoming improvements in computational methodology, and decreasing computational costs will allow for much more facilitated dynamic modeling of Kv7.1/KCNE1 channel gating in the future.
Figure 5
The novel structural “snapshots” allow for a better understanding of Kv7.1/KCNE1 molecular pharmacology. Several Kv7.1/KCNE1 inhibitors and activators bind to distinct sites in state-dependent manner (Lerche et al., 2000, 2007; Seebohm et al., 2001b, 2003a; Chen et al., 2003b). Docking of various compounds to the different models and experimental verifications are now possible. An interesting observation is that the KCNE1 transmembrane segment induces large fenestrations in the Kv7.1 channel pre-open state that may allow for a novel access pathway not present in Kv7.1 homomeric channels (Strutz-Seebohm et al., 2011). Affected by these structural effects is the central cavity, the binding and blocking site of several Kv7.1/KCNE1 inhibitors (Lerche et al., 2000, 2007; Seebohm et al., 2003a). This altered structure could be used as a scaffold for the computer-aided design of specific Kv7.1/KCNE1 modulators not targeting homomeric Kv7.1 channels, which would be a prerequisite for tissue specific pharmacology (Seebohm et al., 2003b; Strutz-Seebohm et al., 2011). Insights into KCNE1-specific molecular pharmacology will be highly valuable for both drug development and safety pharmacology alike (Towart et al., 2009; Pugsley et al., 2011).
Upcoming physiological, pathophysiological, pharmacological, and structural analyses will fill the gaps in our current knowledge, and therefore the old Kv7.1/KCNE1 channel faces an exciting future. Let’s dance the KCNQ1 tango!
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AbbottG. W.SestiF.SplawskiI.BuckM. E.LehmannM. H.TimothyK. W.KeatingM. T.GoldsteinS. A. (1999). MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell97, 175–187.10.1016/S0092-8674(00)80728-X
2
AggeliA.BannisterM. L.BellM.BodenN.FindlayJ. B.HunterM.KnowlesP. F.YangJ. C. (1998). Conformation and ion-channeling activity of a 27-residue peptide modeled on the single-transmembrane segment of the IsK (minK) protein. Biochemistry37, 8121–8131.10.1021/bi972112h
3
AngeloK.JespersenT.GrunnetM.NielsenM. S.KlaerkeD. A.OlesenS. P. (2002). KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys. J.83, 1997–2006.10.1016/S0006-3495(02)73961-1
4
ArrighiI.Bloch-FaureM.GrahammerF.BleichM.WarthR.MengualR.DriciM. D.BarhaninJ.MenetonP. (2001). Altered potassium balance and aldosterone secretion in a mouse model of human congenital long QT syndrome. Proc. Natl. Acad. Sci. U.S.A.98, 8792–8797.10.1073/pnas.141233398
5
BähringR.BarghaanJ.WestermeierR.WollbergJ. (2012). Voltage sensor inactivation in potassium channels. Front. Pharmacol.3:100.10.3389/fphar.2012.00100
6
BarhaninJ.LesageF.GuillemareE.FinkM.LazdunskiM.RomeyG. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature384, 78–80.10.1038/384078a0
7
BendahhouS.MarionneauC.HaurogneK.LarroqueM. M.DerandR.SzutsV.EscandeD.DemolombeS.BarhaninJ. (2005). In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc. Res.67, 529–538.10.1016/j.cardiores.2005.02.014
8
BrahmajothiM. V.MoralesM. J.RasmussonR. L.CampbellD. L.StraussH. C. (1997). Heterogeneity in K+ channel transcript expression detected in isolated ferret cardiac myocytes. Pacing Clin. Electrophysiol.20, 388–396.10.1111/j.1540-8159.1997.tb06198.x
9
BrueggemannL. I.MoranC. J.BarakatJ. A.YehJ. Z.CribbsL. L.ByronK. L. (2007). Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol.292, H1352–H1363.10.1152/ajpheart.00065.2006
10
CasimiroM. C.KnollmannB. C.EbertS. N.VaryJ. C.Jr.GreeneA. E.FranzM. R.GrinbergA.HuangS. P.PfeiferK. (2001). Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc. Natl. Acad. Sci. U.S.A.98, 2526–2531.10.1073/pnas.041398998
11
ChadhaP. S.ZunkeF.DavisA. J.JeppsT. A.LindersJ. T.SchwakeM.TowartR.GreenwoodI. A. (2012). Pharmacological dissection of K(v) 7.1 channels in systemic and pulmonary arteries. Br. J. Pharmacol.166, 1377–1387.10.1111/j.1476-5381.2012.01863.x
12
ChanP. J.OsteenJ. D.XiongD.BohnenM. S.DoshiD.SampsonK. J.MarxS. O.KarlinA.KassR. S. (2012). Characterization of KCNQ1 atrial fibrillation mutations reveals distinct dependence on KCNE1. J. Gen. Physiol.139, 135–144.10.1085/jgp.201110672
13
ChenH.KimL. A.RajanS.XuS.GoldsteinS. A. (2003a). Charybdotoxin binding in the I(Ks) pore demonstrates two MinK subunits in each channel complex. Neuron40, 15–23.10.1016/S0896-6273(03)00570-1
14
ChenH.SestiF.GoldsteinS. A. (2003b). Pore- and state-dependent cadmium block of I(Ks) channels formed with MinK-55C and wild-type KCNQ1 subunits. Biophys. J.84, 3679–3689.10.1016/S0006-3495(03)74998-4
15
ChenY. H.XuS. J.BendahhouS.WangX. L.WangY.XuW. Y.JinH. W.SunH.SuX. Y.ZhuangQ. N.YangY. Q.LiY. B.LiuY.XuH. J.LiX. F.MaN.MouC. P.ChenZ.BarhaninJ.HuangW. (2003c). KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science299, 251–254.10.1126/science.1080819
16
ChenJ.ZhengR.MelmanY. F.McDonaldT. V. (2009). Functional interactions between KCNE1 C-terminus and the KCNQ1 channel. PLoS ONE4, e5143.10.1371/journal.pone.0005143
17
ChouabeC.NeyroudN.GuicheneyP.LazdunskiM.RomeyG.BarhaninJ. (1997). Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J.16, 5472–5479.10.1093/emboj/16.17.5472
18
ChouabeC.NeyroudN.RichardP.DenjoyI.HainqueB.RomeyG.DriciM. D.GuicheneyP.BarhaninJ. (2000). Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc. Res.45, 971–980.10.1016/S0008-6363(99)00411-3
19
ChungD. Y.ChanP. J.BankstonJ. R.YangL.LiuG.MarxS. O.KarlinA.KassR. S. (2009). Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc. Natl. Acad. Sci. U.S.A.106, 743–748.10.1073/pnas.0911436106
20
CiampaE. J.WelchR. C.VanoyeC. G.GeorgeA. L.Jr. (2011). KCNE4 juxtamembrane region is required for interaction with calmodulin and for functional suppression of KCNQ1. J. Biol. Chem.286, 4141–4149.10.1074/jbc.M110.158865
21
ColeK. S.MooreJ. W. (1960). Potassium ion current in the squid giant axon: dynamic characteristic. Biophys. J.1, 1–14.10.1016/S0006-3495(60)86871-3
22
CuiJ.KlineR. P.PennefatherP.CohenI. S. (1994). Gating of IsK expressed in Xenopus oocytes depends on the amount of mRNA injected. J. Gen. Physiol. 104, 87–105.10.1085/jgp.104.1.87
23
DedekK.WaldeggerS. (2001). Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflugers Arch.442, 896–902.10.1007/s004240100609
24
DelmasP.BrownD. A. (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci.6, 850–862.10.1038/nrm1746
25
DemolombeS.FrancoD.de BoerP.KuperschmidtS.RodenD.PereonY.JarryA.MoormanA. F.EscandeD. (2001). Differential expression of KvLQT1 and its regulator IsK in mouse epithelia. Am. J. Physiol., Cell Physiol.280, C359–C372.
26
DongM. Q.SunH. Y.TangQ.TseH. F.LauC. P.LiG. R. (2010). Regulation of human cardiac KCNQ1/KCNE1 channel by epidermal growth factor receptor kinase. Biochim. Biophys. Acta1798, 995–1001.10.1016/j.bbamem.2010.01.010
27
EcholsN.MilburnD.GersteinM. (2003). MolMovDB: analysis and visualization of conformational change and structural flexibility. Nucleic Acids Res.31, 478–482.10.1093/nar/gkg104
28
FinleyM. R.LiY.HuaF.LillichJ.MitchellK. E.GantaS.GilmourR. F.Jr.FreemanL. C. (2002). Expression and coassociation of ERG1, KCNQ1, and KCNE1 potassium channel proteins in horse heart. Am. J. Physiol. Heart Circ. Physiol.283, H126–H138.
29
FranquezaL.LinM.ShenJ.SplawskiI.KeatingM. T.SanguinettiM. C. (1999). Long QT syndrome-associated mutations in the S4–S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem.274, 21063–21070.10.1074/jbc.274.30.21063
30
FrohlichH.BoiniK. M.SeebohmG.Strutz-SeebohmN.UrecheO. N.FollerM.EichenmullerM.ShumilinaE.PathareG.SinghA. K.SeidlerU.PfeiferK. E.LangF. (2011). Hypothyroidism of gene-targeted mice lacking Kcnq1. Pflugers Arch.461, 45–52.10.1007/s00424-010-0890-5
31
GaboritN.Le BouterS.SzutsV.VarroA.EscandeD.NattelS.DemolombeS. (2007). Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. (Lond.)582, 675–693.10.1113/jphysiol.2006.126714
32
GhoshS.SilvaJ. N.CanhamR. M.BowmanT. M.ZhangJ.RheeE. K.WoodardP. K.RudyY. (2011). Electrophysiologic substrate and intraventricular left ventricular dyssynchrony in nonischemic heart failure patients undergoing cardiac resynchronization therapy. Heart Rhythm8, 692–699.10.1016/j.hrthm.2011.01.017
33
GrahammerF.HerlingA. W.LangH. J.Schmitt-GraffA.WittekindtO. H.NitschkeR.BleichM.BarhaninJ.WarthR. (2001a). The cardiac K+ channel KCNQ1 is essential for gastric acid secretion. Gastroenterology120, 1363–1371.10.1053/gast.2001.24053
34
GrahammerF.WarthR.BarhaninJ.BleichM.HugM. J. (2001b). The small conductance K+ channel, KCNQ1: expression, function, and subunit composition in murine trachea. J. Biol. Chem.276, 42268–42275.10.1074/jbc.M105014200
35
GrunnetM.JespersenT.RasmussenH. B.LjungstromT.JorgensenN. K.OlesenS. P.KlaerkeD. A. (2002). KCNE4 is an inhibitory subunit to the KCNQ1 channel. J. Physiol. (Lond.)542, 119–130.10.1113/jphysiol.2002.017301
36
HaitinY.AttaliB. (2008). The C-terminus of Kv7 channels: a multifunctional module. J. Physiol. (Lond.)586, 1803–1810.10.1113/jphysiol.2007.149187
37
HaitinY.WienerR.ShahamD.PeretzA.CohenE. B.ShamgarL.PongsO.HirschJ. A.AttaliB. (2009). Intracellular domains interactions and gated motions of I(KS) potassium channel subunits. EMBO J.28, 1994–2005.10.1038/emboj.2009.157
38
HanW.BaoW.WangZ.NattelS. (2002). Comparison of ion-channel subunit expression in canine cardiac Purkinje fibers and ventricular muscle. Circ. Res.91, 790–797.10.1161/01.RES.0000039534.18114.D9
39
HeitzmannD.GrahammerF.von HahnT.Schmitt-GraffA.RomeoE.NitschkeR.GerlachU.LangH. J.VerreyF.BarhaninJ.WarthR. (2004). Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J. Physiol. (Lond.)561, 547–557.10.1113/jphysiol.2004.075168
40
HeitzmannD.WarthR. (2007). No potassium, no acid: K+ channels and gastric acid secretion. Physiology (Bethesda)22, 335–341.10.1152/physiol.00016.2007
41
HernandezC. C.ZaikaO.ShapiroM. S. (2008). A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J. Gen. Physiol.132, 361–381.10.1085/jgp.200810007
42
HorikawaN.SuzukiT.UchiumiT.MinamimuraT.TsukadaK.TakeguchiN.SakaiH. (2005). Cyclic AMP-dependent Cl- secretion induced by thromboxane A2 in isolated human colon. J. Physiol. (Lond.)562, 885–897.10.1113/jphysiol.2004.077776
43
HurD. G.LeeJ. H.OhS. H.KimY. H.LeeJ. H.ShinD. H.ChangS. O.KimC. S. (2007). KCNQ1/KCNE1 K+ channel and P2Y4 receptor are co-expressed from the time of birth in the apical membrane of rat strial marginal cells. Acta Otolaryngol. Suppl.558, 30–35.10.1080/03655230701624830
44
IannottiF. A.PanzaE.BarreseV.ViggianoD.SoldovieriM. V.TaglialatelaM. (2010). Expression, localization, and pharmacological role of Kv7 potassium channels in skeletal muscle proliferation, differentiation, and survival after myotoxic insults. J. Pharmacol. Exp. Ther.332, 811–820.10.1124/jpet.109.162800
45
IkedaS. R.KammermeierP. J. (2002). M current mystery messenger revealed?Neuron35, 411–412.10.1016/S0896-6273(02)00792-4
46
JervellA.Lange-NielsenF. (1957). Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart J.54, 59–68.10.1016/0002-8703(57)90079-0
47
JespersenT.MembrezM.NicolasC. S.PitardB.StaubO.OlesenS. P.BaroI.AbrielH. (2007). The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc. Res.74, 64–74.10.1016/j.cardiores.2007.01.008
48
JoshiS.SedivyV.HodycD.HergetJ.GurneyA. M. (2009). KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J. Pharmacol. Exp. Ther.329, 368–376.10.1124/jpet.108.147785
49
KangC.TianC.SonnichsenF. D.SmithJ. A.MeilerJ.GeorgeA. L.Jr.VanoyeC. G.KimH. J.SandersC. R. (2008). Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry47, 7999–8006.10.1021/bi800875q
50
KankiH.KupershmidtS.YangT.WellsS.RodenD. M. (2004). A structural requirement for processing the cardiac K+ channel KCNQ1. J. Biol. Chem.279, 33976–33983.10.1074/jbc.M404539200
51
KimS. J.GregerR. (1999). Voltage-dependent, slowly activating K+ current (I(Ks)) and its augmentation by carbachol in rat pancreatic acini. Pflugers Arch.438, 604–611.10.1007/s004240051083
52
KnipperM.ClaussenC.RuttigerL.ZimmermannU.Lullmann-RauchR.EskelinenE. L.SchroderJ.SchwakeM.SaftigP. (2006). Deafness in LIMP2-deficient mice due to early loss of the potassium channel KCNQ1/KCNE1 in marginal cells of the stria vascularis. J. Physiol. (Lond.)576, 73–86.10.1113/jphysiol.2006.116889
53
KnollmannB. C.SirenkoS.RongQ.KatchmanA. N.CasimiroM.PfeiferK.EbertS. N. (2007). Kcnq1 contributes to an adrenergic-sensitive steady-state K+ current in mouse heart. Biochem. Biophys. Res. Commun.360, 212–218.10.1016/j.bbrc.2007.06.038
54
KottgenM.HoeferA.KimS. J.BeschornerU.SchreiberR.HugM. J.GregerR. (1999). Carbachol activates a K+ channel of very small conductance in the basolateral membrane of rat pancreatic acinar cells. Pflugers Arch.438, 597–603.10.1007/s004240051082
55
KunzelmannK.HubnerM.SchreiberR.Levy-HolzmanR.GartyH.BleichM.WarthR.SlavikM.von HahnT.GregerR. (2001). Cloning and function of the rat colonic epithelial K+ channel KVLQT1. J. Membr. Biol.179, 155–164.10.1007/s002320010045
56
KurokawaJ.MotoikeH. K.KassR. S. (2001). TEA(+)-sensitive KCNQ1 constructs reveal pore-independent access to KCNE1 in assembled I(Ks) channels. J. Gen. Physiol.117, 43–52.10.1085/jgp.117.1.43
57
KurokawaJ.MotoikeH. K.RaoJ.KassR. S. (2004). Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc. Natl. Acad. Sci. U.S.A.101, 16374–16378.10.1073/pnas.0405583101
58
LambrechtN. W.YakubovI.ScottD.SachsG. (2005). Identification of the K efflux channel coupled to the gastric H-K-ATPase during acid secretion. Physiol. Genomics21, 81–91.10.1152/physiolgenomics.00212.2004
59
LeeJ. E.ParkH. S.UhmD. Y.KimS. J. (2004). Effects of KCNQ1 channel blocker, 293B, on the acetylcholine-induced Cl- secretion of rat pancreatic acini. Pancreas28, 435–442.10.1097/00006676-200405000-00013
60
LeeM. P.RavenelJ. D.HuR. J.LustigL. R.TomaselliG.BergerR. D.BrandenburgS. A.LitziT. J.BuntonT. E.LimbC.FrancisH.GorelikowM.GuH.WashingtonK.ArganiP.GoldenringJ. R.CoffeyR. J.FeinbergA. P. (2000). Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J. Clin. Invest.106, 1447–1455.10.1172/JCI10897
61
LeeW. K.TorchalskiB.RoussaE.ThevenodF. (2008). Evidence for KCNQ1 K+ channel expression in rat zymogen granule membranes and involvement in cholecystokinin-induced pancreatic acinar secretion. Am. J. Physiol. Cell Physiol.294, C879–C892.10.1152/ajpcell.00403.2007
62
LercheC.BruhovaI.LercheH.SteinmeyerK.WeiA. D.Strutz-SeebohmN.LangF.BuschA. E.ZhorovB. S.SeebohmG. (2007). Chromanol 293B binding in KCNQ1 (Kv7.1) channels involves electrostatic interactions with a potassium ion in the selectivity filter. Mol. Pharmacol.71, 1503–1511.10.1124/mol.106.031682
63
LercheC.SeebohmG.WagnerC. I.SchererC. R.DehmeltL.AbitbolI.GerlachU.BrendelJ.AttaliB.BuschA. E. (2000). Molecular impact of MinK on the enantiospecific block of I(Ks) by chromanols. Br. J. Pharmacol.131, 1503–1506.10.1038/sj.bjp.0703734
64
LiY.ZaydmanM. A.WuD.ShiJ.GuanM.Virgin-DowneyB.CuiJ. (2011). KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc. Natl. Acad. Sci. U.S.A.108, 9095–9100.10.1073/pnas.1109773109
65
LiangG. H.JinZ.UlfendahlM.JarlebarkL. (2006). Molecular analyses of KCNQ1-5 potassium channel mRNAs in rat and guinea pig inner ears: expression, cloning, and alternative splicing. Acta Otolaryngol.126, 346–352.10.1080/00016480500416777
66
LoussouarnG.ParkK. H.BellocqC.BaroI.CharpentierF.EscandeD. (2003). Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J.22, 5412–5421.10.1093/emboj/cdg526
67
LvovA.GageS. D.BerriosV. M.KobertzW. R. (2010). Identification of a protein-protein interaction between KCNE1 and the activation gate machinery of KCNQ1. J. Gen. Physiol.135, 607–618.10.1085/jgp.200910386
68
MaL. J.OhmertI.VardanyanV. (2011). Allosteric features of KCNQ1 gating revealed by alanine scanning mutagenesis. Biophys. J.100, 885–894.10.1016/j.bpj.2010.12.2825
69
Macedo-RibeiroS.HemrikaW.RenirieR.WeverR.MesserschmidtA. (1999). X-ray crystal structures of active site mutants of the vanadium-containing chloroperoxidase from the fungus Curvularia inaequalis. J. Biol. Inorg. Chem.4, 209–219.10.1007/s007750050306
70
MackieA. R.BrueggemannL. I.HendersonK. K.ShielsA. J.CribbsL. L.ScroginK. E.ByronK. L. (2008). Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J. Pharmacol. Exp. Ther.325, 475–483.10.1124/jpet.107.135764
71
MallM.WissnerA.SchreiberR.KuehrJ.SeydewitzH. H.BrandisM.GregerR.KunzelmannK. (2000). Role of K(V)LQT1 in cyclic adenosine monophosphate-mediated Cl(-) secretion in human airway epithelia. Am. J. Respir. Cell Mol. Biol.23, 283–289.
72
MarcusD. C.ShenZ. (1994). Slowly activating voltage-dependent K+ conductance is apical pathway for K+ secretion in vestibular dark cells. Am. J. Physiol.267, C857–C864.
73
MarxS. O.KurokawaJ.ReikenS.MotoikeH.D’ArmientoJ.MarksA. R.KassR. S. (2002). Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science295, 496–499.10.1126/science.1066843
74
MazhariR.NussH. B.ArmoundasA. A.WinslowR. L.MarbanE. (2002). Ectopic expression of KCNE3 accelerates cardiac repolarization and abbreviates the QT interval. J. Clin. Invest.109, 1083–1090.10.1172/JCI15062
75
McCallumL. A.GreenwoodI. A.TribeR. M. (2009). Expression and function of K(v)7 channels in murine myometrium throughout oestrous cycle. Pflugers Arch.457, 1111–1120.10.1007/s00424-008-0567-5
76
McCrossanZ. A.AbbottG. W. (2004). The MinK-related peptides. Neuropharmacology47, 787–821.10.1016/j.neuropharm.2004.06.018
77
MelmanY. F.DomenechA.de la LunaS.McDonaldT. V. (2001). Structural determinants of KvLQT1 control by the KCNE family of proteins. J. Biol. Chem.276, 6439–6444.10.1074/jbc.M103717200
78
MelmanY. F.KrumermanA.McDonaldT. V. (2002). A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J. Biol. Chem.277, 25187–25194.10.1074/jbc.M200564200
79
MelmanY. F.UmS. Y.KrumermanA.KaganA.McDonaldT. V. (2004). KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron42, 927–937.10.1016/j.neuron.2004.06.001
80
MorinT. J.KobertzW. R. (2008). Counting membrane-embedded KCNE beta-subunits in functioning K+ channel complexes. Proc. Natl. Acad. Sci. U.S.A.105, 1478–1482.10.1073/pnas.0710366105
81
MorokumaJ.BlackistonD.AdamsD. S.SeebohmG.TrimmerB.LevinM. (2008). Modulation of potassium channel function confers a hyperproliferative invasive phenotype on embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A.105, 16608–16613.10.1073/pnas.0808328105
82
NakajoK.KuboY. (2007). KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J. Gen. Physiol.130, 269–281.10.1085/jgp.200709805
83
NakajoK.KuboY. (2008). Second coiled-coil domain of KCNQ channel controls current expression and subfamily specific heteromultimerization by salt bridge networks. J. Physiol. (Lond.)586, 2827–2840.10.1113/jphysiol.2007.148601
84
NakajoK.NishinoA.OkamuraY.KuboY. (2011). KCNQ1 subdomains involved in KCNE modulation revealed by an invertebrate KCNQ1 orthologue. J. Gen. Physiol.138, 521–535.10.1085/jgp.201110677
85
NakajoK.UlbrichM. H.KuboY.IsacoffE. Y. (2010). Stoichiometry of the KCNQ1 – KCNE1 ion channel complex. Proc. Natl. Acad. Sci. U.S.A.107, 18862–18867.10.1073/pnas.1010354107
86
NeyroudN.TessonF.DenjoyI.LeiboviciM.DongerC.BarhaninJ.FaureS.GaryF.CoumelP.PetitC.SchwartzK.GuicheneyP. (1997). A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet.15, 186–189.10.1038/nbt0297-186
87
NgF. L.DavisA. J.JeppsT. A.HarhunM. I.YeungS. Y.WanA.ReddyM.MelvilleD.NardiA.KhongT. K.GreenwoodI. A. (2011). Expression and function of the K+ channel KCNQ genes in human arteries. Br. J. Pharmacol.162, 42–53.10.1111/j.1476-5381.2010.01027.x
88
NicolasC. S.ParkK. H.El HarchiA.CamonisJ.KassR. S.EscandeD.MerotJ.LoussouarnG.Le BouffantF.BaroI. (2008). IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiovasc. Res.79, 427–435.10.1093/cvr/cvn085
89
NicolasM.DememesD.MartinA.KupershmidtS.BarhaninJ. (2001). KCNQ1/KCNE1 potassium channels in mammalian vestibular dark cells. Hear. Res.153, 132–145.10.1016/S0378-5955(00)00268-9
90
OhyaS.SergeantG. P.GreenwoodI. A.HorowitzB. (2003). Molecular variants of KCNQ channels expressed in murine portal vein myocytes: a role in delayed rectifier current. Circ. Res.92, 1016–1023.10.1161/01.RES.0000070880.20955.F4
91
OsteenJ. D.GonzalezC.SampsonK. J.IyerV.RebolledoS.LarssonH. P.KassR. S. (2010). KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc. Natl. Acad. Sci. U.S.A.107, 22710–22715.10.1073/pnas.1014150107
92
PanaghieG.TaiK. K.AbbottG. W. (2006). Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J. Physiol. (Lond.)570, 455–467.10.1113/jphysiol.2005.100644
93
ParkK. S.PangB.ParkS. J.LeeY. G.BaeJ. Y.ParkS.KimI.KimS. J. (2011). Identification and functional characterization of ion channels in CD34(+) hematopoietic stem cells from human peripheral blood. Mol. Cells32, 181–188.10.1007/s10059-011-0122-7
94
PicciniM.VitelliF.SeriM.GaliettaL. J.MoranO.BulfoneA.BanfiS.PoberB.RenieriA. (1999). KCNE1-like gene is deleted in AMME contiguous gene syndrome: identification and characterization of the human and mouse homologs. Genomics60, 251–257.10.1006/geno.1999.5904
95
PugsleyM. K.TowartR.AuthierS.GallacherD. J.CurtisM. J. (2011). Innovation in safety pharmacology testing. J. Pharmacol. Toxicol. Methods64, 1–6.10.1016/j.vascn.2011.03.008
96
PuschM. (1998). Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with minK. Pflugers Arch.437, 172–174.10.1007/s004240050765
97
PuschM.MagrassiR.WollnikB.ContiF. (1998). Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J.75, 785–792.10.1016/S0006-3495(98)77568-X
98
RasmussenH. B.MollerM.KnausH. G.JensenB. S.OlesenS. P.JorgensenN. K. (2004). Subcellular localization of the delayed rectifier K(+) channels KCNQ1 and ERG1 in the rat heart. Am. J. Physiol. Heart Circ. Physiol.286, H1300–H1309.10.1152/ajpheart.00344.2003
99
RocheleauJ. M.KobertzW. R. (2008). KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. J. Gen. Physiol.131, 59–68.10.1085/jgp.200709816
100
RomeyG.AttaliB.ChouabeC.AbitbolI.GuillemareE.BarhaninJ.LazdunskiM. (1997). Molecular mechanism and functional significance of the MinK control of the KvLQT1 channel activity. J. Biol. Chem.272, 16713–16716.10.1074/jbc.272.27.16713
101
Roura-FerrerM.SoleL.Martinez-MarmolR.VillalongaN.FelipeA. (2008). Skeletal muscle Kv7 (KCNQ) channels in myoblast differentiation and proliferation. Biochem. Biophys. Res. Commun.369, 1094–1097.10.1016/j.bbrc.2008.02.152
102
SanguinettiM. C.CurranM. E.ZouA.ShenJ.SpectorP. S.AtkinsonD. L.KeatingM. T. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature384, 80–83.10.1038/384080a0
103
SanguinettiM. C.JurkiewiczN. K. (1990). Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol.96, 195–215.10.1085/jgp.96.1.195
104
SchmittN.SchwarzM.PeretzA.AbitbolI.AttaliB.PongsO. (2000). A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO J.19, 332–340.10.1093/emboj/19.3.332
105
SchroederB. C.WaldeggerS.FehrS.BleichM.WarthR.GregerR.JentschT. J. (2000). A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature403, 196–199.10.1038/35003200
106
Schulze-BahrE.WangQ.WedekindH.HaverkampW.ChenQ.SunY.RubieC.HordtM.TowbinJ. A.BorggrefeM.AssmannG.QuX.SombergJ. C.BreithardtG.ObertiC.FunkeH. (1997). KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat. Genet.17, 267–268.10.1038/ng1197-267
107
SchwakeM.JentschT. J.FriedrichT. (2003). A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep.4, 76–81.10.1038/sj.embor.embor715
108
SeebohmG.ChenJ.StrutzN.CulbersonC.LercheC.SanguinettiM. C. (2003a). Molecular determinants of KCNQ1 channel block by a benzodiazepine. Mol. Pharmacol.64, 70–77.10.1124/mol.64.1.70
109
SeebohmG.PuschM.ChenJ.SanguinettiM. C. (2003b). Pharmacological activation of normal and arrhythmia-associated mutant KCNQ1 potassium channels. Circ. Res.93, 941–947.10.1161/01.RES.0000102866.67863.2B
110
SeebohmG.SanguinettiM. C.PuschM. (2003c). Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. (Lond.)552, 369–378.10.1113/jphysiol.2003.046490
111
SeebohmG.LercheC.BuschA. E.BachmannA. (2001a). Dependence of I(Ks) biophysical properties on the expression system. Pflugers Arch.442, 891–895.10.1007/s004240100608
112
SeebohmG.LercheC.PuschM.SteinmeyerK.BruggemannA.BuschA. E. (2001b). A kinetic study on the stereospecific inhibition of KCNQ1 and I(Ks) by the chromanol 293B. Br. J. Pharmacol.134, 1647–1654.10.1038/sj.bjp.0704421
113
SeebohmG.SchererC. R.BuschA. E.LercheC. (2001c). Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. J. Biol. Chem.276, 13600–13605.
114
SeebohmG.Strutz-SeebohmN.UrecheO. N.BaltaevR.LampertA.KornichukG.KamiyaK.WuttkeT. V.LercheH.SanguinettiM. C.LangF. (2006). Differential roles of S6 domain hinges in the gating of KCNQ potassium channels. Biophys. J.90, 2235–2244.10.1529/biophysj.105.067165
115
SeebohmG.WestenskowP.LangF.SanguinettiM. C. (2005). Mutation of colocalized residues of the pore helix and transmembrane segments S5 and S6 disrupt deactivation and modify inactivation of KCNQ1 K+ channels. J. Physiol. (Lond.)563, 359–368.10.1113/jphysiol.2004.080887
116
SestiF.GoldsteinS. A. (1998). Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol.112, 651–663.10.1085/jgp.112.6.651
117
ShamgarL.HaitinY.YisharelI.MalkaE.SchottelndreierH.PeretzA.PaasY.AttaliB. (2008). KCNE1 constrains the voltage sensor of Kv7.1 K+ channels. PLoS ONE3, e1943.10.1371/journal.pone.0001943
118
ShenZ.LiuJ.MarcusD. C.ShigaN.WangemannP. (1995). DIDS increases K+ secretion through an IsK channel in apical membrane of vestibular dark cell epithelium of gerbil. J. Membr. Biol.146, 283–291.
119
SilvaJ.RudyY. (2005). Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation112, 1384–1391.10.1161/CIRCULATIONAHA.105.543306
120
SilvaJ. R.PanH.WuD.NekouzadehA.DeckerK. F.CuiJ.BakerN. A.SeptD.RudyY. (2009). A multiscale model linking ion-channel molecular dynamics and electrostatics to the cardiac action potential. Proc. Natl. Acad. Sci. U.S.A.106, 11102–11106.10.1073/pnas.0904505106
121
SilvaJ. R.RudyY. (2010). Multi-scale electrophysiology modeling: from atom to organ. J. Gen. Physiol.135, 575–581.10.1085/jgp.200910358
122
SmithJ. A.VanoyeC. G.GeorgeA. L.Jr.MeilerJ.SandersC. R. (2007). Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry46, 14141–14152.10.1021/bi700637a
123
Strutz-SeebohmN.PuschM.WolfS.StollR.TapkenD.GerwertK.AttaliB.SeebohmG. (2011). Structural basis of slow activation gating in the cardiac I Ks channel complex. Cell. Physiol. Biochem.27, 443–452.10.1159/000329965
124
Strutz-SeebohmN.SeebohmG.FedorenkoO.BaltaevR.EngelJ.KnirschM.LangF. (2006). Functional coassembly of KCNQ4 with KCNE-beta- subunits in Xenopus oocytes. Cell. Physiol. Biochem.18, 57–66.10.1159/000097675
125
SugimotoT.TanabeY.ShigemotoR.IwaiM.TakumiT.OhkuboH.NakanishiS. (1990). Immunohistochemical study of a rat membrane protein which induces a selective potassium permeation: its localization in the apical membrane portion of epithelial cells. J. Membr. Biol.113, 39–47.10.1007/BF01869604
126
SunoseH.LiuJ.ShenZ.MarcusD. C. (1997). cAMP increases apical IsK channel current and K+ secretion in vestibular dark cells. J. Membr. Biol.156, 25–35.10.1007/s002329900184
127
TaiK. K.GoldsteinS. A. (1998). The conduction pore of a cardiac potassium channel. Nature391, 605–608.10.1038/35416
128
TakumiT.MoriyoshiK.AramoriI.IshiiT.OikiS.OkadaY.OhkuboH.NakanishiS. (1991). Alteration of channel activities and gating by mutations of slow ISK potassium channel. J. Biol. Chem.266, 22192–22198.
129
TakumiT.OhkuboH.NakanishiS. (1988). Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science242, 1042–1045.10.1126/science.3194754
130
TapperA. R.GeorgeA. L.Jr. (2000). MinK subdomains that mediate modulation of and association with KvLQT1. J. Gen. Physiol.116, 379–390.10.1085/jgp.116.3.379
131
TapperA. R.GeorgeA. L.Jr. (2001). Location and orientation of minK within the I(Ks) potassium channel complex. J. Biol. Chem.276, 38249–38254.
132
ThomasA. M.HarmerS. C.KhambraT.TinkerA. (2011). Characterization of a binding site for anionic phospholipids on KCNQ1. J. Biol. Chem.286, 2088–2100.10.1074/jbc.M110.153551
133
TianC.VanoyeC. G.KangC.WelchR. C.KimH. J.GeorgeA. L.Jr.SandersC. R. (2007). Preparation, functional characterization, and NMR studies of human KCNE1, a voltage-gated potassium channel accessory subunit associated with deafness and long QT syndrome. Biochemistry46, 11459–11472.10.1021/bi700705j
134
TinelN.DiochotS.BorsottoM.LazdunskiM.BarhaninJ. (2000). KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J.19, 6326–6330.10.1093/emboj/19.23.6326
135
TowartR.LindersJ. T.HermansA. N.RohrbacherJ.van der LindeH. J.ErckenM.CikM.RoevensP.TeismanA.GallacherD. J. (2009). Blockade of the I(Ks) potassium channel: an overlooked cardiovascular liability in drug safety screening?J. Pharmacol. Toxicol. Methods60, 1–10.10.1016/j.vascn.2009.04.197
136
Tristani-FirouziM.SanguinettiM. C. (1998). Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. (Lond.)510(Pt 1), 37–45.10.1111/j.1469-7793.1998.037bz.x
137
TzounopoulosT.MaylieJ.AdelmanJ. P. (1998). Gating of I(sK) channels expressed in Xenopus oocytes. Biophys. J.74, 2299–2305.10.1016/S0006-3495(98)77939-1
138
VallonV.GrahammerF.RichterK.BleichM.LangF.BarhaninJ.VolklH.WarthR. (2001). Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J. Am. Soc. Nephrol.12, 2003–2011.
139
VallonV.GrahammerF.VolklH.SanduC. D.RichterK.RexhepajR.GerlachU.RongQ.PfeiferK.LangF. (2005). KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc. Natl. Acad. Sci. U.S.A.102, 17864–17869.10.1073/pnas.0505860102
140
WangK.TerrenoireC.SampsonK. J.IyerV.OsteenJ. D.LuJ.KellerG.KottonD. N.KassR. S. (2011a). Biophysical properties of slow potassium channels in human embryonic stem cell derived cardiomyocytes implicate subunit stoichiometry. J. Physiol. (Lond.)589, 6093–6104.10.1113/jphysiol.2010.203463
141
WangY. H.JiangM.XuX. L.HsuK. L.ZhangM.TsengG. N. (2011b). Gating-related molecular motions in the extracellular domain of the IKs channel: implications for IKs channelopathy. J. Membr. Biol.239, 137–156.10.1007/s00232-010-9333-7
142
WangK. W.GoldsteinS. A. (1995). Subunit composition of minK potassium channels. Neuron14, 1303–1309.10.1016/0896-6273(95)90277-5
143
WangK. W.TaiK. K.GoldsteinS. A. (1996a). MinK residues line a potassium channel pore. Neuron16, 571–577.10.1016/S0896-6273(00)80076-8
144
WangQ.CurranM. E.SplawskiI.BurnT. C.MillhollandJ. M.VanRaayT. J.ShenJ.TimothyK. W.VincentG. M.de JagerT.SchwartzP. J.ToubinJ. A.MossA. J.AtkinsonD. L.LandesG. M.ConnorsT. D.KeatingM. T. (1996b). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet.12, 17–23.10.1038/ng0196-17
145
WangW.XiaJ.KassR. S. (1998). MinK-KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J. Biol. Chem.273, 34069–34074.10.1074/jbc.273.38.24867
146
WangemannP. (1995). Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hear. Res.90, 149–157.10.1016/0378-5955(95)00157-2
147
WangemannP.LiuJ.MarcusD. C. (1995). Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear. Res.84, 19–29.10.1016/0378-5955(95)00009-S
148
WarthR.BarhaninJ. (2002). The multifaceted phenotype of the knockout mouse for the KCNE1 potassium channel gene. Am. J. Physiol. Regul. Integr. Comp. Physiol.282, R639–R648.
149
WuD. M.JiangM.ZhangM.LiuX. S.KorolkovaY. V.TsengG. N. (2006). KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of I(Ks) current amplitude in the heart. Heart Rhythm3, 1469–1480.10.1016/j.hrthm.2006.08.019
150
XuQ.MinorD. L.Jr. (2009). Crystal structure of a trimeric form of the K(V)7.1 (KCNQ1) A-domain tail coiled-coil reveals structural plasticity and context dependent changes in a putative coiled-coil trimerization motif. Protein Sci.18, 2100–2114.10.1002/pro.105
151
XuX.JiangM.HsuK. L.ZhangM.TsengG. N. (2008). KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J. Gen. Physiol.131, 589–603.10.1085/jgp.200809976
152
YangW. P.LevesqueP. C.LittleW. A.ConderM. L.ShalabyF. Y.BlanarM. A. (1997). KvLQT1, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proc. Natl. Acad. Sci. U.S.A.94, 4017–4021.10.1073/pnas.94.26.14524
153
YeungS. Y.PucovskyV.MoffattJ. D.SaldanhaL.SchwakeM.OhyaS.GreenwoodI. A. (2007). Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br. J. Pharmacol.151, 758–770.10.1038/sj.bjp.0707284
154
ZhangH.CraciunL. C.MirshahiT.RohacsT.LopesC. M.JinT.LogothetisD. E. (2003). PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron37, 963–975.10.1016/S0896-6273(03)00125-9
155
ZhengR.ThompsonK.Obeng-GyimahE.AlessiD.ChenJ.ChengH.McDonaldT. V. (2010). Analysis of the interactions between the C-terminal cytoplasmic domains of KCNQ1 and KCNE1 channel subunits. Biochem. J.428, 75–84.10.1042/BJ20090977
156
ZhongX. Z.HarhunM. I.OlesenS. P.OhyaS.MoffattJ. D.ColeW. C.GreenwoodI. A. (2010). Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J. Physiol. (Lond.)588, 3277–3293.10.1113/jphysiol.2010.192823
157
ZichaS.MossI.AllenB.VarroA.PappJ.DumaineR.AntzelevichC.NattelS. (2003). Molecular basis of species-specific expression of repolarizing K+ currents in the heart. Am. J. Physiol. Heart Circ. Physiol.285, H1641–H1649.
Appendix
Table A1
| Specified tissue | Species | mRNA/protein | Reference |
|---|---|---|---|
| Brain | Mouse | mRNA | Lee et al. (2000) |
| Mouse | mRNA | Ohya et al. (2003) | |
| Mouse | mRNA | Yeung et al. (2007) | |
| Mouse | mRNA | Iannotti et al. (2010) | |
| Inner ear | Mouse | mRNA | Neyroud et al. (1997) |
| Mouse | mRNA/protein | Nicolas et al. (2001) | |
| Mouse | Protein | Knipper et al. (2006) | |
| Rat | mRNA | Liang et al. (2006) | |
| Rat | Protein | Hur et al. (2007) | |
| Guinea pig | mRNA | Liang et al. (2006) | |
| Trachea | Mouse | mRNA/protein | Grahammer et al. (2001b) |
| Thyroid gland | Mouse | mRNA/protein | Frohlich et al. (2011) |
| Human | mRNA | Yang et al. (1997) | |
| Human | mRNA | Frohlich et al. (2011) | |
| Thymus | Mouse | mRNA | Demolombe et al. (2001) |
| Human | mRNA | Chouabe et al. (1997) | |
| Lung | Mouse | mRNA | Demolombe et al. (2001) |
| Human | mRNA | Wang et al. (1996b) | |
| Human | mRNA | Sanguinetti et al. (1996) | |
| Human | mRNA | Chouabe et al. (1997) | |
| Human | mRNA | Yang et al. (1997) | |
| Heart | Mouse | mRNA | Barhanin et al. (1996) |
| Mouse | mRNA | Lee et al. (2000) | |
| Mouse | mRNA | Casimiro et al. (2001) | |
| Mouse | mRNA | Demolombe et al. (2001) | |
| Mouse | mRNA | Ohya et al. (2003) | |
| Mouse | mRNA | Yeung et al. (2007) | |
| Mouse | mRNA | Iannotti et al. (2010) | |
| Mouse | mRNA | Strutz-Seebohm et al. (2006) | |
| Mouse | Protein | Knollmann et al. (2007) | |
| Rat | Protein | Rasmussen et al. (2004) | |
| Guinea pig | mRNA/protein | Zicha et al. (2003) | |
| Guinea pig | Protein | Nicolas et al. (2008) | |
| Ferret | mRNA | Brahmajothi et al. (1997) | |
| Rabbit | mRNA/protein | Zicha et al. (2003) | |
| Horse | Protein | Finley et al. (2002) | |
| Canine | mRNA/protein | Han et al. (2002) | |
| Human | mRNA | Wang et al. (1996b) | |
| Human | mRNA | Sanguinetti et al. (1996) | |
| Human | mRNA | Chouabe et al. (1997) | |
| Human | mRNA | Yang et al. (1997) | |
| Human | mRNA | Bendahhou et al. (2005) | |
| Human | mRNA | Gaborit et al. (2007) | |
| Human | mRNA | Frohlich et al. (2011) | |
| Human | mRNA/protein | Zicha et al. (2003) | |
| Liver | Mouse | mRNA | Lee et al. (2000) |
| Mouse | mRNA | Demolombe et al. (2001) | |
| Spleen | Mouse | mRNA | Demolombe et al. (2001) |
| Human | mRNA | Chouabe et al. (1997) | |
| Pancreas | Mouse | mRNA | Demolombe et al. (2001) |
| Rat | Protein | Lee et al. (2008) | |
| Human | mRNA | Sanguinetti et al. (1996) | |
| Human | mRNA | Chouabe et al. (1997) | |
| Human | mRNA | Yang et al. (1997) | |
| Stomach | Mouse | mRNA | Lee et al. (2000) |
| Mouse | mRNA | Demolombe et al. (2001) | |
| Mouse | mRNA/protein | Dedek and Waldegger (2001) | |
| Mouse | Protein | Grahammer et al. (2001a) | |
| Mouse | Protein | Heitzmann et al. (2004) | |
| Mouse | Protein | Heitzmann and Warth (2007) | |
| Rat | mRNA/protein | Lambrecht et al. (2005) | |
| Human | mRNA | Yang et al. (1997) | |
| Human | mRNA | Frohlich et al. (2011) | |
| Human | Protein | Grahammer et al. (2001a) | |
| Colon | Mouse | mRNA/protein | Dedek and Waldegger (2001) |
| Rat | mRNA | Kunzelmann et al. (2001) | |
| Human | mRNA | Chouabe et al. (1997) | |
| Human | mRNA | Yang et al. (1997) | |
| Human | mRNA | Frohlich et al. (2011) | |
| Human | mRNA/protein | Horikawa et al. (2005) | |
| Small intestine | Mouse | mRNA | Demolombe et al. (2001) |
| Mouse | mRNA/protein | Dedek and Waldegger (2001) | |
| Human | mRNA | Chouabe et al. (1997) | |
| Human | mRNA | Yang et al. (1997) | |
| Kidney | Mouse | mRNA | Barhanin et al. (1996) |
| Mouse | mRNA | Lee et al. (2000) | |
| Mouse | mRNA | Demolombe et al. (2001) | |
| Mouse | Protein | Vallon et al. (2001) | |
| Human | mRNA | Wang et al. (1996b) | |
| Human | mRNA | Sanguinetti et al. (1996) | |
| Human | mRNA | Chouabe et al. (1997) | |
| Human | mRNA | Yang et al. (1997) | |
| Uterus | Mouse | mRNA/protein | McCallum et al. (2009) |
| Placenta | Human | mRNA | Wang et al. (1996b) |
| Human | mRNA | Sanguinetti et al. (1996) | |
| Human | mRNA | Chouabe et al. (1997) | |
| Human | mRNA | Yang et al. (1997) | |
| Ovary | Human | mRNA | Chouabe et al. (1997) |
| Prostate | Human | mRNA | Chouabe et al. (1997) |
| Human | mRNA | Yang et al. (1997) | |
| Testis | Human | mRNA | Chouabe et al. (1997) |
| Blood vessels | Mouse | mRNA/protein | Ohya et al. (2003) |
| Mouse | mRNA/protein | Yeung et al. (2007) | |
| Rat | mRNA | Brueggemann et al. (2007) | |
| Rat | mRNA | Mackie et al. (2008) | |
| Rat | mRNA | Joshi et al. (2009) | |
| Rat | mRNA/protein | Zhong et al. (2010) | |
| Human | mRNA/protein | Ng et al. (2011) | |
| Skeletal muscle | Mouse | mRNA | Demolombe et al. (2001) |
| Mouse | mRNA | Iannotti et al. (2010) | |
| Rat | mRNA | Roura-Ferrer et al. (2008) | |
| Peripheral blood leukocytes | Human | mRNA | Chouabe et al. (1997) |
| Hematopoietic stem cells | Human | mRNA | Park et al. (2011) |
Kv7.1 expression in mammalian tissues.
Summary
Keywords
Kv channel, Kv7.1, β-subunit, KCNE1
Citation
Wrobel E, Tapken D and Seebohm G (2012) The KCNE Tango – How KCNE1 Interacts with Kv7.1. Front. Pharmacol. 3:142. doi: 10.3389/fphar.2012.00142
Received
29 March 2012
Accepted
29 June 2012
Published
02 August 2012
Volume
3 - 2012
Edited by
Gildas Loussouarn, University of Nantes, France
Reviewed by
Robert Kass, Colombia University in the city of New York, USA; Tom V. McDonald, Albert Einstein College of Medicine, USA
Copyright
© 2012 Wrobel, Tapken and Seebohm.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Guiscard Seebohm, Myozelluläre Elektrophysiologie und Molekularbiologie, Westfälische Wilhelms-Universität Münster, Domagkstraße 3, 48149 Münster, Germany. e-mail: guiscard.seebohm@gmx.de
This article was submitted to Frontiers in Pharmacology of Ion Channels and Channelopathies, a specialty of Frontiers in Pharmacology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.