 Stefania Zappettini1
Stefania Zappettini1 Massimo Grilli1
Massimo Grilli1 Guendalina Olivero1
Guendalina Olivero1 Elisa Mura2
Elisa Mura2 Stefania Preda2
Stefania Preda2 Stefano Govoni2 Alessia Salamone1
Stefano Govoni2 Alessia Salamone1 Mario Marchi1,3*
Mario Marchi1,3*- 1 Section of Pharmacology and Toxicology, Department of Experimental Medicine, University of Genoa, Genoa, Italy
- 2 Department of Drug Sciences, Centre of Excellence in Applied Biology, University of Pavia, Pavia, Italy
- 3 Centre of Excellence for Biomedical Research, University of Genoa, Genoa, Italy
Using both in vitro (hippocampal synaptosomes in superfusion) and in vivo (microdialysis) approaches we investigated whether and to what extent β amyloid peptide 1–40 (Aβ 1–40) interferes with the cholinergic modulation of the release of glycine (GLY) in the rat hippocampus. The nicotine-evoked overflow of endogenous GLY in hippocampal synaptosomes in superfusion was significantly inhibited by Aβ 1–40 (10 nM) while increasing the concentration to 100 nM the inhibitory effect did not further increase. Both the Choline (Ch; α7 agonist; 1 mM) and the 5-Iodo-A-85380 dihydrochloride (5IA85380, α4β2 agonist; 10 nM)-evoked GLY overflow were inhibited by Aβ 1–40 at 100 nM but not at 10 nM concentrations. The KCl evoked [3H]GLY and [3H]Acetylcholine (ACh) overflow were strongly inhibited in presence of oxotremorine; however this inhibitory muscarinic effect was not affected by Aβ 1–40. The effects of Aβ 1–40 on the administration of nicotine, veratridine, 5IA85380, and PHA543613 hydrochloride (PHA543613; a selective agonist of α7 subtypes) on hippocampal endogenous GLY release in vivo were also studied. Aβ 1–40 significantly reduced (at 10 μM but not at 1 μM) the nicotine-evoked in vivo release of GLY. Aβ 1–40 (at 10 μM but not at 1 μM) significantly inhibited the PHA543613 (1 mM)-elicited GLY overflow while was ineffective on the GLY overflow evoked by 5IA85380 (1 mM). Aβ 40–1 (10 μM) did not produce any inhibitory effect on nicotine-evoked GLY overflow both in the in vitro and in vivo experiments. Our results indicate that (a) the cholinergic modulation of the release of GLY occurs by the activation of both α7 and α4β2 nicotinic ACh receptors (nAChRs) as well as by the activation of inhibitory muscarinic ACh receptors (mAChRs) and (b) Aβ 1–40 can modulate cholinergic evoked GLY release exclusively through the interaction with α7 and the α4β2 nAChR nicotinic receptors but not through mAChR subtypes.
Introduction
Nicotinic and muscarinic receptors are widely expressed in the brain and implicated in the pathophysiology of many neurological conditions, including Alzheimer’s disease (AD), where typical symptoms include the loss of cognitive function and dementia. The presence of extracellular neuritic plaques composed of β amyloid (Aβ) peptide is a characteristic feature of AD, however, although neurotoxicity is a prominent feature of Aβ, recent data emphasize the existence of synaptic functional roles of this peptide which can modulate the release of several neurotransmitters (see Mura et al., 2012 and references therein). Accordingly, Aβ isoforms and oligomers of increasing molecular dimensions may have different biological actions in a continuum from physiology to pathology, determining loss and gains of function along the course of the disease (Mura et al., 2010a). Indeed, it has been shown that non-neurotoxic Aβ 1–40 concentrations were able to modulate (predominantly, but not exclusively, to inhibit) the release of several neurotransmitters (dopamine, γ aminobutyric acid, aspartate, glutamate) elicited by the stimulation of cholinergic muscarinic and nicotinic receptor [muscarinic ACh receptors (mAChR); nicotinic ACh receptors (nAChR)] subtypes in different brain areas; (Preda et al., 2008; Puzzo et al., 2008; Grilli et al., 2009; Jürgensen and Ferreira, 2010;Mura et al., 2010a,b, 2012; Ondrejcak et al., 2010). The hippocampus, an area which is particularly vulnerable and early target of Alzheimer’s disease and in which the cholinergic pathways are critical for modulation of attention and memory (Parri et al., 2011), Aβ regulates the nicotine-evoked release of both excitatory (glutamate and aspartate) and inhibitory (γ aminobutyric acid) aminoacids (Mura et al., 2012).
Increasing evidence demonstrate that glycine (GLY) is an important aminoacid at hippocampal level which may have a dual role acting as an inhibitory neurotransmitter, when interacting with the strychnine-sensitive receptors, and playing a stimulatory role, when co-activating excitatory N-Methyl-D-aspartic acid receptors together with glutamate (Johnson and Ascher, 1987; Luccini et al., 2008; Romei et al., 2009, 2011; Zappettini et al., 2011). No data are available so far on the possible effects of Aβ on the cholinergic receptors which modulate GLY release at the hippocampal level.
In the present study using both in vitro (hippocampal synaptosomes in superfusion) and in vivo (microdialysis) approaches we investigated whether and to what extent Aβ interferes with the cholinergic modulation of the release of GLY in the rat hippocampus.
The results indicate that (a) the cholinergic modulation of the release of GLY occurs by the activation of both α7 and α4β2 (nAChRs) as previously reported (Zappettini et al., 2011) as well as by the activation of inhibitory mAChRs; (b) the nicotinic modulation of GLY release by α4β2, and α7 nAChRs is inhibited in vitro in presence of a nanomolar concentration of Aβ 1–40 which is on the contrary ineffective on the inhibitory mAChR receptor subtypes.
Materials and Methods
Animals
Adult male Wistar rats (200–250 g, Harlan, Udine) were used for both in vivo experiments and as brain tissue source for in vitro experiments. Animals were housed at constant temperature (22 ± 1°C) and relative humidity (50%) under a regular light–dark schedule (light 7 a.m. to 7 p.m.). The in vitro experimental procedures were approved by the Ethical Committee of the Pharmacology and Toxicology Section, Department of Experimental Medicine, in accordance with the European legislation (European Communities Council Directive of 24 November 1986, 86/609/EEC) and were approved by Italian legislation on animal experimentation (Decreto Ministeriale number 124/2003-A). The in vivo protocol was approved by Ethical Committee of Pavia’s University (registered as 2/2008) according to international regulations for the care and treatment of laboratory animals, to the Italian Act (DL n 116, GU, suppl 40, 18 February, 1992) and to EEC Council Directive (86/609, OJ L 358, 1, 12 December, 1987). All efforts were made to minimize animal suffering and to use the minimal number of animals necessary to produce reliable results.
In vitro Experiments
Experiments of release
Rats were killed by decapitation and the hippocampus rapidly removed at 0–4°C. Purified synaptosomes were prepared on Percoll® gradients (Sigma-Aldrich, St Louis, MO, USA) essentially according to (Nakamura et al., 1993), with only minor modifications. Briefly, the tissue was homogenized in six volumes of 0.32 M sucrose, buffered at pH 7.4 with Tris–HCl, using a glass-teflon tissue grinder (clearance 0.25 mm, 12 up–down strokes in about 1 min). The homogenate was centrifuged (5 min, 1000 × g at 4°C) to remove nuclei and debris; the supernatant was gently stratified on a discontinuous Percoll® gradient (2, 6, 10, and 20% v/v in Tris-buffered sucrose) and centrifuged at 33,500 × g for 5 min at 4°C. The layer between 10 and 20% Percoll® (synaptosomal fraction) was collected, washed by centrifugation, and resuspended in physiological HEPES-buffered medium having the following composition (mM): NaCl 128, KCl 2.4, CaCl2 3.2, KH2PO4 1.2, MgSO4 1.2, HEPES 25, pH 7.5, glucose 10, pH 7.2–7.4 (Lu et al., 1998). Synaptosomal protein content following purification was 10–15% of that in the supernatant stratified on the Percoll® gradient.
The synaptosomal suspension was layered on microporous filters at the bottom of a set of parallel superfusion chambers maintained at 37°C (Raiteri and Raiteri, 2000; Superfusion System, Ugo Basile, Comerio, Varese, Italy). Synaptosomes were superfused at 1 ml/min with standard physiological medium as previously described. The system was first equilibrated during 36.5 min of superfusion; subsequently, four consecutive 90 s fractions of superfusate were collected and the endogenous GLY content was measured by high performance liquid chromatography as below described. Synaptosomes were exposed to agonists for 90 s starting from the second fraction collected (t = 38 min), with antagonists being added 8 min before agonists. The evoked overflow was calculated by subtracting the corresponding basal release from each fraction and was expressed as pmol/mg of synaptosomal proteins. We previously demonstrated that in our superfusion system the indirect drug effects exerted by other mediators in the monolayer of synaptosomes in superfusion are absolutely minimized (Raiteri and Raiteri, 2000).
When studying the release of [3H]GLY or [3H]Acetylcholine (ACh) hippocampal synaptosomes were incubated for 20 min at 37°C with [3H]GLY (final concentration 0.1 μM) in the presence of the selective GLY transporter 1 transporter blocker N-[(3R)-3-([1,1′-biphenyl]-4-yloxy)-3-(4-fluorophenyl)propyl]-N-methylglycine hydrochloride (final concentration 0.3 μM) or with [3H]Choline (Ch, final concentration 0.08 μM). The K+-induced overflow from synaptosomes was estimated by subtracting the neurotransmitter content into the first and the third fractions collected (basal release, b1 and b3) from that in the 6-min fraction collected during and after the depolarization pulse (evoked release, b2). The amount of radioactivity released into each superfusate fraction was expressed as a percentage of the total synaptosomal tritium content at the start of the fraction collected (fractional efflux).
Endogenous GLY Determination
Endogenous GLY was measured by high performance liquid chromatography analysis following precolumn derivatization with o-phthalaldehyde and resolution through a C18-reverse phase chromatographic column (10 mm × 4.6 mm, 3 μm; Chrompack, Middleburg, The Netherlands) coupled with fluorometric detection (excitation wavelength 350 nm; emission wavelength 450 nm). Homoserine was used as internal standard. Buffers and gradient program were prepared and executed as follows: solvent A, 0.1 M sodium acetate (pH 5.8)/methanol, 80:20; solvent B, 0.1 M sodium acetate (pH 5.8)/methanol, 20:80; solvent C, sodium acetate (pH 6.0)/methanol, 80:20; gradient program, 100% C for 4 min from the initiation of the program; 90% A and 10% B in 1 min; 42% A and 58% B in 14 min; 100% B in 1 min; isocratic flow 2 min; 100% C in 3 min; flow rate 0.9 ml/min.
In vivo Experiments
Microdialysis probe implantation
Rats were anesthetized with Equithesin 3 ml/kg (pentobarbital 9.7 g, chloral hydrate 42.5 g, MgSO4 21.3 g for 1 l, 10% ethanol, 40% propylene glycol v/v) administered intraperitoneally and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). The skin was shaved, disinfected, and cut with a sterile scalpel to expose the skull. A hole was drilled to allow the implantation of the probe into the brain parenchyma. The probe was implanted in the hippocampus (CA1/CA2 regions; AP −5.8 mm, ML ± 5.0 mm from bregma, and DV −8.0 mm from dura) according to the Paxinos and Watson (1986) atlas, and secured to the skull with one stainless steel screw and dental cement. All in vivo experiments were performed using microdialysis probes, made in our laboratory according to the original method described by Di Chiara (1990; Emophan Bellco Artificial OR-internal diameter 200 μm, cutoff 40 kDa; Bellco, Mirandola, Modena, Italy), with a nominal active length of 5 mm. Finally, the skin was sutured, and the rats were allowed to recover from anesthesia for at least 24 h before the neurotransmitter release study.
Microdialysis samples collection
Microdialysis experiments were performed on conscious freely moving rats. On the day of the experiments (24 h after the surgical procedure), the probe was perfused with artificial CSF containing 145 mM NaCl, 3.0 mM KCl, 1.26 mM CaCl2, 1.0 mM MgCl2, 1.4 mM Na2HPO4, buffered at pH 7.2–7.4, and filtered through a Millipore 0.2 μm pore membrane. In all experiments, the microdialysis membrane was allowed to stabilize for 1 h at the flow rate of 4 μl/min, without collecting samples. At the end of the stabilization period, three samples were collected to evaluate baseline release of GLY and then the specific treatment started. All treatments were administered by manually switching syringes and tubing connections to allow drugs diluted in artificial CSF to flow through the probes. Tubing switches were performed taking care to maintain constant flow rates and collection volumes. Both basal and treatment samples were collected every 20 min in 100 μl Eppendorf tubes. The flow rate of 4 μl/min was maintained using a 1000-μl syringe (Hamilton) and a microinjection pump (CMA/100, CMA/Microdialysis AB). In vitro recovery of the probe for GLY was about 20%. Each rat was used for only one microdialysis session. At the end of each experiment animals were sacrificed by guillotine, rat brains were removed and the position of the microdialysis probe was verified by histological procedures, slicing the tissues by a cryostat microtome (LEICA CM 1510). Only data from rats in which probe tracks were exactly located in the target area were used for statistical analysis.
Immunohistochemical analysis
Immunohistochemical analysis was performed to verify the presence of Aβ in the perfused tissue and to confirm (according to HOECHST 33342 staining) the absence of neurotoxic-induced apoptotic phenomenon. Brain tissue samples were frozen and stored at −80°C. For immunodetection of infused Aβ peptide, 10 μm coronal sections (obtained on a cryostat Leica CM 1510) were incubated with a primary monoclonal antibody recognizing Aβ protein (clone 4G8; Chemicon International). Sections were then incubated with a mouse anti-IgG antibody RPE conjugated (Dako). After the fluorescent labeling procedures, sections were finally counterstained for DNA with HOECHST 33342 and mounted in a drop of Mowiol (Calbiochem, Inalco SpA, Milan, Italy). Fluorescent micrographs were acquired with a Leica TCS SP5 II confocal microscope. After acquisition of fluorescent micrographs, the slides were demounted and then the same sections were slightly counterstained with Mayer hematoxylin, dehydrated, and mounted in DPX for microanatomical analysis. The images were acquired with a BX51 Olympus microscope.
Statistical Analysis
In vitro experiments
Multiple comparisons were performed with one-way ANOVA followed by an appropriate post hoc test (Dunnett and Bonferroni). Data were considered significant for p < 0.05, at least.
In vivo experiments
Values were expressed either as amount of GLY measured in the dialyzate (pmol/80 μl) or as area under the curve (AUC), evaluating the cumulative release over time. AUC was used as a measure of treatment exposure and was calculated, for each animal, using GraphPad Prism (version 4.03 GraphPad Software, San Diego, CA, USA), defining as baseline of the area the basal value (average concentration of three consecutive samples immediately preceding the drug dose).
D’Agostino–Pearson Omnibus Test (GraphPad Prism, version 4.03, GraphPad Software, San Diego, CA, USA) and Grubb’s Test (GraphPad QuickCalcs, online calculator for scientists at http://www.graphpad.com/quickcalcs/, GraphPad Software, San Diego, CA, USA) were used as preliminary tests in order to evaluate whether data were sampled from a Gaussian distribution and to detect outliers respectively. All outliers were excluded from the analysis. Data were then analyzed by analysis of variance (one- or two-way ANOVA) followed, when significant, by an appropriate post hoc comparison test. Data were considered significant for p < 0.05. The reported data are expressed as means ± SEM. The number of animals used for each experiment is reported in the legend to figures.
Preparation of Beta Amyloid Solutions
In the case of both in vivo and in vitro experiments, synthetic human Aβ 1–40 (Sigma-Aldrich, Milan, Italy) was dissolved in aCSF at a concentration of 100 mM (stock solution). Then, this solution was filtered through a Millipore 0.2 μm pore membrane and stocked in small aliquots. Working solutions were freshly prepared by diluting an aliquot of Aβ 1–40 stock solution at the final concentrations (10 mM, 1 mM, or 100 nM Aβ 1–40 for in vivo experiments, 100 nM, 10 nM, 1 nM, or 100 pM for in vitro analysis).
Chemicals
Beta amyloid (1–40; 40–1), percoll®, choline, himbacine, dimethyl sulfoxide, veratridine, nicotine hydrogen tartrate salt (Sigma-Aldrich, St Louis, MO, USA); NFPS (ALX 5407), 5IA85380, PHA543613, AQRA741 (Tocris Bioscience, Bristol, UK); all salts used for the preparation of aCSF (NaCl, KCl, CaCl2, MgCl2, Na2HPO4) and for Equithesin (MgSO4) were purchased at Merck KGaA, Darmstadt, Germany; chloral hydrate, ethanol 96%, and propylene glycol were used for the preparation of Equithesin and were obtained at VWR BDH Prolabo, Belgium;[3H]Choline (specific activity: 60–90 Ci/mmol) and [3H]Glycine (specific activity: 15 Ci/mmol) were purchased from Perkin Elmer SpA.
Results
It is known that Aβ peptides structure and aggregation properties depend on several factors. It is therefore important when using Aβ peptides to specify both the concentration of the soluble Aβ but also the methods used to prepare Aβ solution and to verify the presence of Aβ aggregates (see Parri et al., 2011). This facilitates the interpretation of the results when reporting on Aβ effects and, which is even more important, would facilitate the comparison of data obtained in different laboratories. Hence, in our previous paper we characterized the Aβ peptide conformation we administered in vivo by Western Blot procedure (Mura et al., 2012) showing that we administered, at least predominantly, Aβ monomers. We cannot completely exclude by the adopted methods that small amounts of Aβ oligomers are present and may participate to produce the observed effects. In regard to the in vitro Aβ preparations, since we did not observe aggregation at the concentrations and the timing (up to 40 min) analyzed in vivo, we also do not expect to observe aggregation at the lower concentrations and shorter times used in vitro in light of the fact that aggregation is a concentration and time-dependent process.
Figure 1A shows that the nicotine-evoked overflow of endogenous GLY was significantly inhibited by Aβ 1–40 already at a concentration of 10 nM while increasing the concentration to 100 nM the inhibitory effect did not further increase. The 1-nM Aβ 1–40 concentration was ineffective. The reverse peptide Aβ 40–1 was ineffective even at high (100 nM) concentration. Aβ 1–40 (100 nM) did not affect the veratridine (10 nM)-evoked GLY overflow.
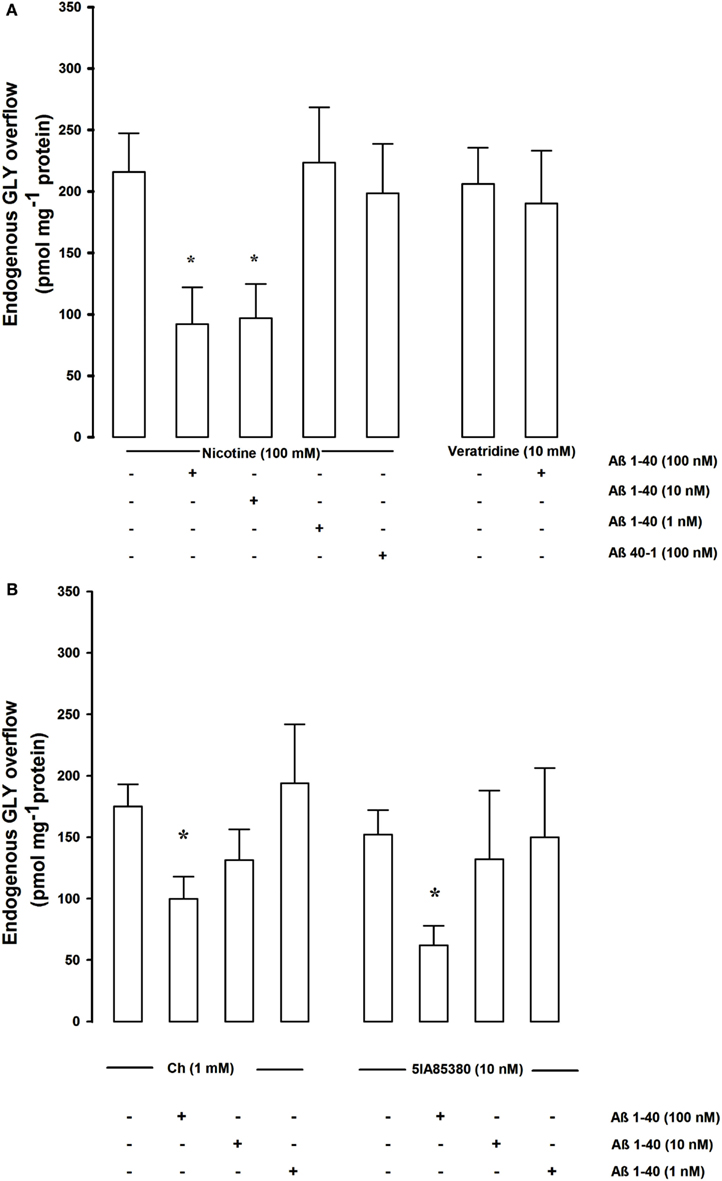
Figure 1. (A) Shows the concentration dependence effects of Aβ on nicotine- and veratridine-evoked endogenous GLY overflows from rat hippocampal synaptosomes. Data are mean ± SEM of three to six experiments for each concentration run in triplicate. *p < 0.05 versus nicotine-evoked GLY overflow (one-way ANOVA followed by Bonferroni post hoc Test). (B) Shows the concentration dependence effects of Aβ on Ch and 5IA85380 evoked endogenous GLY overflows from rat hippocampal synaptosomes. Data are mean ± SEM of three to six experiments for each concentration run in triplicate. *p < 0.05 versus Ch-evoked GLY overflow (one-way ANOVA followed by Dunnett’s Multiple Comparison Test).
Since it has been shown that two different nAChR subtypes, α7 and α4β2, modulate GLY release (Zappettini et al., 2011), the possibility that Aβ may differentially inhibit the nicotinic control of GLY release has been investigated. In order to verify this point we have studied the effects of two different agonists, Ch and 5-IA85380 hydrochloride (5IA85380), known to act selectively on the α7 and α4β2 nAChR subtypes respectively (Mukhin et al., 2000; Uteshev et al., 2003; Dickinson et al., 2007; Zappettini et al., 2010). Ch (1 mM) and the 5IA85380 (10 nM)-evoked a similar overflow of GLY confirming the involvement of the two receptor subtypes. Both the Ch (1 mM) and the 5IA85380 (10 nM)-evoked GLY overflow were significantly inhibited by Aβ 1–40 at 100 nM but not at 10–1 nM concentrations compared to controls (Figure 1B). Aβ 1–40 (100 nM) did not modify the basal release of endogenous GLY (data not shown).
It has been demonstrated that also different mAChR subtypes are involved in the modulation of both ACh and GLY release from brain hippocampal synaptosomes (Raiteri et al., 1984; Russo et al., 1993). We investigated whether Aβ was able to affect the muscarinic control of the release of these two transmitters as it was able to disrupt the nicotinic control of GLY release. Figure 2A shows that the KCl evoked [3H]GLY overflow was strongly inhibited in presence of oxotremorine. This inhibitory effect was totally antagonized by himbacine but was not affected by Aβ 1–40 (100 nM). In presence of oxotremorine also the KCl evoked release of [3H]ACh from hippocampal nerve endings was significantly inhibited (Figure 2B). This result was unexpected but demonstrates quite interestingly that the cholinergic muscarinic modulation of GLY release could be different according to the different brain areas or the different species studied since previous findings using human cortical nerve endings have shown a potentiating effect of oxotremorine on GLY release (Russo et al., 1993). Interestingly also the inhibitory effect of oxotremorine on [3H]ACh release was antagonized, as expected, by the specific M2 mAChR antagonist AQRA741 but was not affected by Aβ 1–40 (100 nM; Figure 2B).
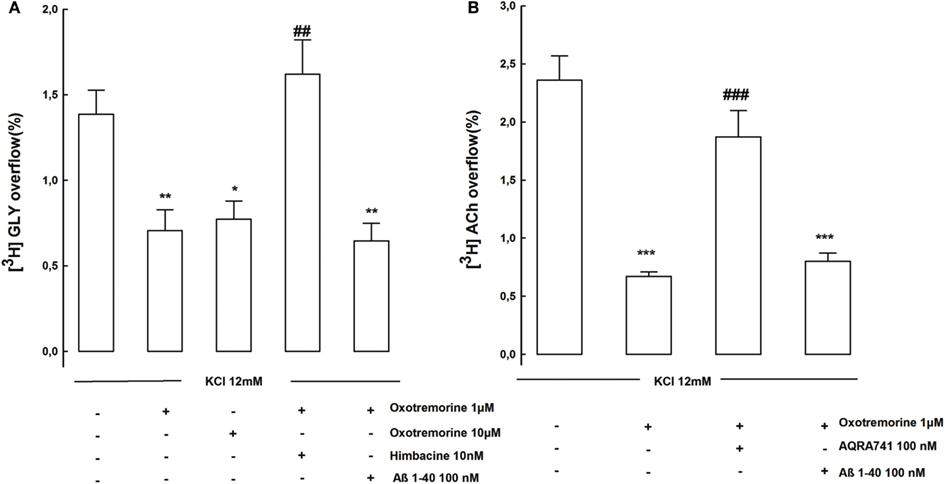
Figure 2. (A) Shows the effects of Aβ on muscarinic receptors controlling KCl evoked [3H]GLY overflow from rat hippocampal synaptosomes. Data are mean ± SEM of three to six experiments run in triplicate. *p < 0.05; **p < 0.01 versus KCl evoked [3H]GLY overflow; ##p < 0.01 versus KCl plus oxotremorine (1 μM)-evoked [3H]GLY overflow. (B) Shows the effects of Aβ on muscarinic receptors controlling KCl evoked [3H]ACh overflow from rat hippocampal synaptosomes. Data are mean ± SEM of three to six experiments run in triplicate. ***p < 0.001 versus KCl evoked [3H]ACh overflow; ##p < 0.001 versus KCl plus oxotremorine (1 μM) -evoked [3H]ACh overflow (one-way ANOVA followed by Bonferroni post hoc Test).
Based on the in vitro data we then analyzed the effects of Aβ 1–40 on the administration of nicotine, veratridine, 5IA85380, and PHA543613 hydrochloride (PHA543613, a selective agonist of α7 subtypes) on hippocampal GLY release in vivo. In order to test whether the administration of Aβ 1–40 through the dialysis probe allowed the delivery of the peptide to the tissue we performed an immunohistochemical analysis. Figure 3 shows the presence of the peptide for the two concentrations tested in vivo (1 and 10 μM) within the hippocampus. Despite the fact that we do not know the exact amounts of Aβ reaching the tissue, there was a visible positive correlation between the concentration administered and the signal of Aβ immunoreactivity in the tissue. Moreover, immunohistochemical analysis shows that no evident signs of apoptosis were observed within the area of amyloid diffusion as shown by Hoechst staining.
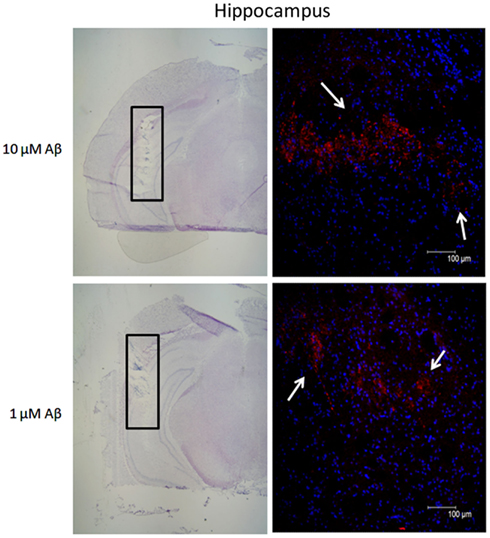
Figure 3. Immunohistochemical analysis showing the presence of beta-amyloid (Aβ) in hippocampal tissue after the perfusion of the peptide at two different concentrations. Coronal section indicating the location of the microdialysis probe (hippocampus, black rectangle) counterstained with Mayer Hematoxylin and relative fluorescence micrographs of the area within the black rectangle showing the presence of human Aβ protein. Aβ immunoreactivity (red-PE staining, white arrows) immediately after perfusion of 10 and 1 μM Aβ 1–40. Nuclear DNA was counterstained with Hoechst 33,342 (blue staining). Scale bars for Mayer hematoxylin sections: 200 μM. Scale bars for fluorescent micrographs: 100 μM.
The choice of the concentration of the nicotinic cholinergic agonists to be delivered in vivo was derived from previous data demonstrating that the administration by microdialysis of 50 mM nicotine was able to significantly increase the levels of GLY in hippocampal extracellular compartment (Toth, 1996; Fedele et al., 1998; Zappettini et al., 2011). As previously shown in our experimental conditions 40 min-long administration of 50 mM nicotine was able to greatly enhance GLY release from basal values. Figure 4A show that Aβ 1–40 significantly reduced (at 10 μM but not at 1 μM) the nicotine-evoked in vivo release of GLY. Aβ 40–1 did not produce any inhibitory effect used at 10 μM concentration. The GLY overflow stimulated by veratridine was unaltered in presence of Aβ 1–40 (Figure 4B). Then we compared the effects Aβ 1–40 after exposure to the selective nAChRs agonists, 5IA85380, and PHA543613. As shown in Figure 4C, Aβ 1–40 (at 10 μM but not at 1 μM) significantly inhibited the PHA543613 (1 mM)-elicited GLY overflow while was ineffective on the GLY overflow evoked by 5IA85380 (1 mM).
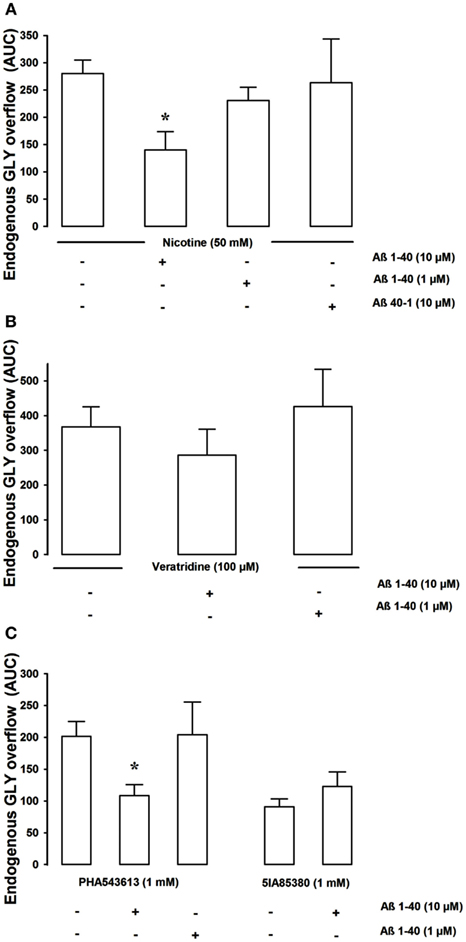
Figure 4. The figure shows the in vivo effect of Aβ 1–40 on the nicotine-, veratrine-, PHA543613-, and 5IA85380-induced overflow of GLY, in rat hippocampus [(A–C) respectively]. Aβ 1–40 (10 μM but not 1 μM) significantly inhibited the 50-mM nicotine-induced overflow of GLY *p < 0.01 versus nicotine (one-way ANOVA followed by Dunnett’s Multiple Comparison Test). Aβ 40–1 10 μM was ineffective (A). Aβ 1–40 (10 μM) which significantly inhibited the 1-mM PHA543613-induced overflow of GLY was unable to inhibit both the 100-μM veratridine- and the 5IA85380-induced overflow of GLY (B,C). Data are expressed as mean ± SEM of 4–15 individual rats for each experimental group. *p < 0.01 versus PHA543613 (one-way ANOVA followed by Dunnett’s Multiple Comparison Test.
Discussion
It is expected that cognitive deficits and memory impairments in AD patients could be related, at least in part, to Aβ mediated decrease of cholinergic function (Wang et al., 2009a,b, 2010; Parri et al., 2011). However it is still unclear whether these impairments are a consequence of a loss of cholinergic neurons and a decrease of nAChRs or of a direct molecular interaction of Aβ with nAChRs leading to a dysregulation of receptor function or of both mechanisms. To shed some light on these mechanisms we investigated in the present study as well as in previous researches whether Aβ, at concentrations not producing acutely neurotoxicity, is able to disrupt the cholinergic control of neurotransmitter release. To do this we took advantage of the fact that both α4β2, and α7 nAChRs receptors are known to be expressed in rat hippocampal interneurons (McQuiston and Madison, 1999; Sudweeks and Yakel, 2000; Yakel and Shao, 2004), and it is well-known that they have a positive role in regulating cognitive function (Picciotto et al., 1995; Levin and Simon, 1998). Being aware that several neurotransmitters that play important roles in cognitive functions could be affected either directly or indirectly by Aβ, we focused our attention on GLY. Indeed increasing evidence demonstrate that GLY is an important aminoacid at hippocampal level which has a dual role (a) acting as an inhibitory neurotransmitter when interacting with the strychnine-sensitive receptors and (b) playing a fundamental stimulatory role when co-activating excitatory N-Methyl-D-aspartic acid receptors together with glutamate (Johnson and Ascher, 1987; Hirai et al., 1996; Luccini et al., 2008; Kubota et al., 2010; Zappettini et al., 2011). The changes in the GLY release may therefore directly interfere with glutamate neurotransmission, which plays an important role in the processes of learning and memory in this brain area.
We here report that the cholinergic modulation of GLY release at the presynaptic level on hippocampal nerve endings was modulated not only by stimulatory α4β2 and α7 nAChRs as previously reported (Zappettini et al., 2011) but also by an inhibitory, mAChR subtype. We do not know whether both nicotinic and muscarinic receptors are present on all nerve endings or they are peculiar of a specific neuronal population and/or their physiological importance in the intact tissue. It is quite interesting however that the two modulatory mechanisms display a different sensitivity to Aβ. The stimulatory effects of both α4β2 and α7 nAChRs were partially blocked by nanomolar concentration of Aβ 1–40 which was on the contrary inactive on the mAChRs inhibiting GLY release. This observation allows to speculate that an excess of Aβ (the range of active concentrations was between 10 and 100 nM in vitro and 10 μM in vivo) as it may happen because of the disease may dysregulate the cholinergic modulation of hippocampal activity leading to a disproportionate inhibition since it leaves unaffected the muscarinic inhibitory control and impairs the nicotinic stimulatory one. All this occurs in absence of Aβ acutely induced neuronal damage.
The decreased release of GLY may have several functional consequences. Some of them may be relevant to the development of AD pathology. As an example, a decrease of GLY release may reduce the tonic inhibition exerted through the activation of GLY receptors (Mori et al., 2002; Petrini et al., 2004; Farrant and Nusser, 2005) normally providing neuroprotection under pathological conditions, when extracellular GLY levels are elevated (Baker et al., 1991; Saransaari et al., 1997; Zhao et al., 2005; Saransaari and Oja, 2008). A second relevant event caused by a reduced glycine release may consist in a decrease of N-Methyl-D-aspartic acid receptors co-activation. At this regard it is important to recall that intracerebroventricular injection of Aβ 1–40 significantly suppress high frequency stimulation-induced LTP (Chen et al., 2006; Wu et al., 2008).
The exact nature of the Aβ interaction with nAChR subtypes is so far not well understood.
Interestingly in our study both the α7 and the α4β2 nAChR subtypes which almost equally contribute to the stimulation of GLY release, were functionally inhibited in vitro apparently in a similar extent by Aβ concentrations in the nanomolar range (Figure 2B). However we do not know whether Aβ might have a similar mechanism of action on the two different nAChR subtypes. The inhibitory effect on both the α7 and the α4β2 nAChR was incomplete with a maximal inhibition of about 30–40%. A recently described allosteric binding pocket located within the trans membrane domain of the α7 and of non-α7 nAChRs might provide a potential structure-function mechanism to explain the inhibitory effects of Aβ (Young et al., 2008; Gill et al., 2011, 2012).
Indeed Aβ at concentration in the upper nanomolar range (10 nM) produced full effect while did not produced any effect at 1 nM concentration. Ten nanomolars Aβ beyond the estimated normal concentrations in human CSF but may be caused by the altered precursor protein processing as it occurs along with the disease (Reaume et al., 1996) or be related to defects in the removal of the peptide from the extracellular space as it may occur in the disease in association with the ApoE ε4 genotype (Cramer et al., 2012).
Of course talking about cholinergic modulation of GLY release we have also to consider the possibility that Aβ might for instance interfere directly with the mechanisms which modulate the release of ACh. However our data show that Aβ was ineffective also on the inhibitory muscarinic autoreceptors which inhibit ACh release (Figure 2). We can therefore foresee that in an integrated system, where cellular networks and their functional relationships are completely preserved and several direct and indirect processes are simultaneously taking place in neurons, the effect of Aβ on the cholinergic modulation of GLY function may mostly depend on the interaction of Aβ with the nAChRs.
Our findings in vivo largely support this view even if Aβ was unable to inhibit the release of GLY elicited by the specific α4β2 nAChRs agonist while it exerted an inhibition (over 40%) of both nicotine and PHA543613 stimulated GLY release. On the other hand in vivo important differences were noticed at the baseline in the ability of the two specific nAChRs agonists to elicit GLY release as compare to in vitro. Indeed in vitro both α7 and α4β2 nAChRs agonists were able to stimulate to the same extent GLY release; while in vivo the release elicited by 5IA85380 was less than a half that obtained in response to PHA543613. In vivo to many variables may take place, and in spite of the above described discrepancy, there is a general good agreement of the effects in vivo and in vitro of Aβ on the release of various neurotransmitters (present work and references) including the observation that so far Aβ has never been able to modify the depolarization (veratridine or potassium)-elicited release of neurotransmitters, but only the one modulated by presynaptic receptors.
In conclusion our findings show that Aβ can modulate cholinergic evoked GLY release exclusively through the interaction with α7 and the α4β2 nAChR nicotinic receptors, acting probably through different mechanisms of action, but not through mAChRs. The present study may therefore provide further insight into the mechanism by which AB impairs synaptic plasticity and cholinergic function in AD brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by from the Italian Ministero Università Ricerca to Prof. Mario Marchi (Prot. No 2009R7WCZS_003), by Compagnia di San Paolo, by University of Genoa “Progetto Ricerca Ateneo,” and by project AROMA (ALCOTRA 2007-2013) by the European Community Project PYRGI, project no. B51H10000000006, and by the project 7FP-KBBE BAMMBO and to Prof. Govoni (MIUR 2009). This work is supported by a grant of the Alzheimer’s Association (NIRG-11-205183) to Elisa Mura. We wish to thank Dr. Cristina Lanni for technical support in immunohistochemical analysis, Maura Agate for editorial assistance and Dr. Silvia E. Smith, Ph.D. (University of Utah) for reviewing the manuscript.
References
Baker, A. J., Zornow, M. H., Scheller, M. S., Yaksh, T. L., Skilling, S. R., Smullin, D. H., Larson, A. A., and Kuczenski, R. (1991). Changes in extracellular concentrations of glutamate, aspartate, glycine, dopamine, serotonin, and dopamine metabolites after transient global ischemia in the rabbit brain. J. Neurochem. 57, 1370–1379.
Chen, L., Yamada, K., Nabeshima, T., and Sokabe, M. (2006). alpha7 Nicotinic acetylcholine receptor as a target to rescue deficit in hippocampal LTP induction in beta-amyloid infused rats. Neuropharmacology 50, 254–268.
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., Casali, B. T., Restivo, J. L., Goebel, W. D., James, M. J., Brunden, K. R., Wilson, D. A., and Landreth, G. E. (2012). ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506.
Di Chiara, G. (1990). In-vivo brain dialysis of neurotransmitters. Trends Pharmacol. Sci. 11, 116–121.
Dickinson, J. A., Hanrott, K. E., Mok, M. H., Kew, J. N., and Wonnacott, S. (2007). Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC12 cells. J. Neurochem. 100, 1089–1096.
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229.
Fedele, E., Varnier, G., Ansaldo, M. A., and Raiteri, M. (1998). Nicotine administration stimulates the in vivo N-methyl-D-aspartate receptor/nitric oxide/cyclic GMP pathway in rat hippocampus through glutamate release. Br. J. Pharmacol. 125, 1042–1048.
Gill, J. K., Dhankher, P., Sheppard, T. D., Sher, E., and Millar, N. S. (2012). A series of α7 nicotinic acetylcholine receptor allosteric modulators with close chemical similarity but diverse pharmacological properties. Mol. Pharmacol. 81, 710–718.
Gill, J. K., Savolainen, M., Young, G. T., Zwart, R., Sher, E., and Millar, N. S. (2011). Agonist activation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 108, 5867–5872.
Grilli, M., Zappettini, S., Raiteri, L., and Marchi, M. (2009). Nicotinic and muscarinic cholinergic receptors coexist on GABAergic nerve endings in the mouse striatum and interact in modulating GABA release. Neuropharmacology 56, 610–614.
Hirai, H., Kirsch, J., Laube, B., Betz, H., and Kuhse, J. (1996). The glycine binding site of the N-methyl-D-aspartate receptor subunit NR1: identification of novel determinants of co-agonist potentiation in the extracellular M3-M4 loop region. Proc. Natl. Acad. Sci. U.S.A. 93, 6031–6036.
Johnson, J. W., and Ascher, P. (1987). Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–531.
Jürgensen, S., and Ferreira, S. T. (2010). Nicotinic receptors, amyloid-beta, and synaptic failure in Alzheimer’s disease. J. Mol. Neurosci. 40, 221–229.
Kubota, H., Alle, H., Betz, H., and Geiger, J. R. (2010). Presynaptic glycine receptors on hippocampal mossy fibers. Biochem. Biophys. Res. Commun. 393, 587–591.
Levin, E., and Simon, B. (1998). Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl.) 138, 217–230.
Lu, Y., Grady, S., Marks, M. J., Picciotto, M., Changeux, J. P., and Collins, A. C. (1998). Pharmacological characterization of nicotinic receptor-stimulated GABA release from mouse brain synaptosomes. J. Pharmacol. Exp. Ther. 287, 648.
Luccini, E., Romei, C., and Raiteri, L. (2008). Glycinergic nerve endings in hippocampus and spinal cord release glycine by different mechanisms in response to identical depolarizing stimuli. J. Neurochem. 105, 2179–2189.
McQuiston, A. R., and Madison, D. V. (1999). Nicotinic receptor activation excites distinct subtypes of interneurons in the rat hippocampus. J. Neurosci. 19, 2887–2896.
Mori, M., Gähwiler, B. H., and Gerber, U. (2002). Beta-alanine and taurine as endogenous agonists at glycine receptors in rat hippocampus in vitro. J. Physiol. (Lond.) 539, 191–200.
Mukhin, A. G., Gündisch, D., Horti, A. G., Koren, A. O., Tamagnan, G., Kimes, A. S., Chambers, J., Vaupel, D. B., King, S. L., Picciotto, M. R., Innis, R. B., and London, E. D. (2000). 5-Iodo-A-85380, an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol. Pharmacol. 57, 642–649.
Mura, E., Lanni, C., Preda, S., Pistoia, F., Sarà, M., Racchi, M., Schettini, G., Marchi, M., and Govoni, S. (2010a). Beta-amyloid: a disease target or a synaptic regulator affecting age-related neurotransmitter changes? Curr. Pharm. Des. 16, 672–683.
Mura, E., Preda, S., Govoni, S., Lanni, C., Trabace, L., Grilli, M., Lagomarsino, F., Pittaluga, A., and Marchi, M. (2010b). Specific neuromodulatory actions of amyloid-beta on dopamine release in rat nucleus accumbens and caudate putamen. J. Alzheimers Dis. 19, 1041–1053.
Mura, E., Zappettini, S., Preda, S., Biundo, F., Lanni, C., Grilli, M., Cavallero, A., Olivero, G., Salamone, A., Govoni, S., and Marchi, M. (2012). Dual effect of beta-amyloid on α7 and α4β2 nicotinic receptors controlling the release of glutamate, aspartate and GABA in rat hippocampus. PLoS ONE 7, e29661. doi:10.1371/journal.pone.0029661
Nakamura, Y., Iga, K., Shibata, T., Shudo, M., and Kataoka, K. (1993). Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia 9, 48–56.
Ondrejcak, T., Klyubin, I., Hu, N. W., Barry, A. E., Cullen, W. K., and Rowan, M. J. (2010). Alzheimer’s disease amyloid beta-protein and synaptic function. Neuromolecular Med. 12, 13–26.
Parri, H. R., Hernandez, C. M., and Dineley, K. T. (2011). Research update: alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem. Pharmacol. 82, 931–942.
Paxinos, G., and Watson, C. (1986). The Rat Brain in Stereotaxic Coordinates, 2nd Edn. New York: Academic Press.
Petrini, E. M., Marchionni, I., Zacchi, P., Sieghart, W., and Cherubini, E. (2004). Clustering of extrasynaptic GABA(A) receptors modulates tonic inhibition in cultured hippocampal neurons. J. Biol. Chem. 279, 45833–45843.
Picciotto, M., Zoli, M., Léna, C., Bessis, A., Lallemand, Y., Le Novère, N., Vincent, P., Pich, E., Brûlet, P., and Changeux, J. (1995). Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature 374, 65–67.
Preda, S., Govoni, S., Lanni, C., Racchi, M., Mura, E., Grilli, M., and Marchi, M. (2008). Acute beta-amyloid administration disrupts the cholinergic control of dopamine release in the nucleus accumbens. Neuropsychopharmacology 33, 1062–1070.
Puzzo, D., Privitera, L., Leznik, E., Fà, M., Staniszewski, A., Palmeri, A., and Arancio, O. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545.
Raiteri, L., and Raiteri, M. (2000). Synaptosomes still viable after 25 years of superfusion. Neurochem. Res. 25, 1265–1274.
Raiteri, M., Leardi, R., and Marchi, M. (1984). Heterogeneity of presynaptic muscarinic receptors regulating neurotransmitter release in the rat brain. J. Pharmacol. Exp. Ther. 228, 209.
Reaume, A. G., Howland, D. S., Trusko, S. P., Savage, M. J., Lang, D. M., Greenberg, B. D., Siman, R., and Scott, R. W. (1996). Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer’s disease mutations and a “humanized” Abeta sequence. J. Biol. Chem. 271, 23380–23388.
Romei, C., Di Prisco, S., Raiteri, M., and Raiteri, L. (2011). Glycine release provoked by disturbed Na+, Na+ and Ca2+ homeostasis in cerebellar nerve endings: roles of Ca2+ channels, Na+/Ca2+ exchangers and GlyT2 transporter reversal. J. Neurochem. 119, 50–63.
Romei, C., Luccini, E., Raiteri, M., and Raiteri, L. (2009). GABA(B) presynaptic receptors modulate glycine exocytosis from mouse spinal cord and hippocampus glycinergic nerve endings. Pharmacol. Res. 59, 154–159.
Russo, C., Marchi, M., Andrioli, G. C., Cavazzani, P., and Raiteri, M. (1993). Enhancement of glycine release from human brain cortex synaptosomes by acetylcholine acting at M4 muscarinic receptors. J. Pharmacol. Exp. Ther. 266, 142.
Saransaari, P., and Oja, S. S. (2008). Effects of inhibitory amino acids on adenosine release in the mouse hippocampus. Proc. West. Pharmacol. Soc. 51, 15–17.
Saransaari, P., Oja, S. S., Borkowska, H. D., Koistinaho, J., Hilgier, W., and Albrecht, J. (1997). Effects of thioacetamide-induced hepatic failure on the N-methyl-D-aspartate receptor complex in the rat cerebral cortex, striatum, and hippocampus. Binding of different ligands and expression of receptor subunit mRNAs. Mol. Chem. Neuropathol. 32, 179–193.
Sudweeks, S. N., and Yakel, J. L. (2000). Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J. Physiol. (Lond.) 527(Pt 3), 515–528.
Toth, E. (1996). Effect of nicotine on the level of extracellular amino acids in the hippocampus of rat. Neurochem. Res. 21, 903–907.
Uteshev, V., Meyer, E., and Papke, R. (2003). Regulation of neuronal function by choline and 4OH-GTS-21 through alpha 7 nicotinic receptors. J. Neurophysiol. 89, 1797–1806.
Wang, H. Y., Bakshi, K., Shen, C., Frankfurt, M., Trocmé-Thibierge, C., and Morain, P. (2010). S 24795 limits beta-amyloid-alpha7 nicotinic receptor interaction and reduces Alzheimer’s disease-like pathologies. Biol. Psychiatry 67, 522–530.
Wang, H. Y., Stucky, A., Liu, J., Shen, C., Trocme-Thibierge, C., and Morain, P. (2009a). Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s disease brain. J. Neurosci. 29, 10961–10973.
Wang, Y., Zhang, G., Zhou, H., Barakat, A., and Querfurth, H. (2009b). Opposite effects of low and high doses of Abeta42 on electrical network and neuronal excitability in the rat prefrontal cortex. PLoS ONE 4, e8366. doi:10.1371/journal.pone.0008366
Wu, M. N., He, Y. X., Guo, F., and Qi, J. S. (2008). Alpha4beta2 nicotinic acetylcholine receptors are required for the amyloid beta protein-induced suppression of long-term potentiation in rat hippocampal CA1 region in vivo. Brain Res. Bull. 77, 84–90.
Yakel, J., and Shao, Z. (2004). Functional and molecular characterization of neuronal nicotinic ACh receptors in rat hippocampal interneurons. Prog. Brain Res. 145, 95–107.
Young, G. T., Zwart, R., Walker, A. S., Sher, E., and Millar, N. S. (2008). Potentiation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 105, 14686–14691.
Zappettini, S., Grilli, M., Salamone, A., Fedele, E., and Marchi, M. (2010). Pre-synaptic nicotinic receptors evoke endogenous glutamate and aspartate release from hippocampal synaptosomes by way of distinct coupling mechanisms. Br. J. Pharmacol. 161, 1161–1171.
Zappettini, S., Mura, E., Grilli, M., Preda, S., Salamone, A., Olivero, G., Govoni, S., and Marchi, M. (2011). Different presynaptic nicotinic receptor subtypes modulate in vivo and in vitro the release of glycine in the rat hippocampus. Neurochem. Int. 59, 729–738.
Keywords: β amyloid, glycine release, nicotinic receptors, muscarinic receptors, microdialysis
Citation: Zappettini S, Grilli M, Olivero G, Mura E, Preda S, Govoni S, Salamone A and Marchi M (2012) Beta amyloid differently modulate nicotinic and muscarinic receptor subtypes which stimulate in vitro and in vivo the release of glycine in the rat hippocampus. Front. Pharmacol. 3:146. doi: 10.3389/fphar.2012.00146
Received: 16 May 2012; Accepted: 09 July 2012;
Published online: 27 July 2012.
Edited by:
Valentina Echeverria Moran, Bay Pines VA Medical Center, USAReviewed by:
Enrico Sanna, University of Cagliari, ItalyValentina Echeverria Moran, Bay Pines VA Medical Center, USA
Copyright: © 2012 Zappettini, Grilli, Olivero, Mura, Preda, Govoni, Salamone and Marchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Mario Marchi, Section of Pharmacology and Toxicology, Department of Experimental Medicine, University of Genoa, Viale Cembrano 4, 16148 Genoa, Italy. e-mail:bWFyY2hpQHBoYXJtYXRveC51bmlnZS5pdA==