

- 1Department of Physiology, Faculty of Medicine, Maastricht University, Maastricht, Netherlands
- 2Department of Cardiovascular Medicine, Division of Electrophysiology, University of Muenster, Muenster, Germany
Cardiac conduction is mediated by gap junction channels that are formed by connexin (Cx) protein subunits. The connexin family of proteins consists of more than 20 members varying in their biophysical properties and ability to combine with other connexins into heteromeric gap junction channels. The mammalian heart shows regional differences both in connexin expression profile and in degree of electrical coupling. The latter reflects functional requirements for conduction velocity which needs to be low in the sinoatrial and atrioventricular nodes and high in the ventricular conduction system. Over the past 20 years knowledge of the biology of gap junction channels and their role in the genesis of cardiac arrhythmias has increased enormously. This review focuses on the insights gained from transgenic mouse models. The mouse heart expresses Cx30, 30.2, 37, 40, 43, 45, and 46. For these connexins a variety of knock-outs, heart-specific knock-outs, conditional knock-outs, double knock-outs, knock-ins and overexpressors has been studied. We discuss the cardiac phenotype in these models and compare Cx expression between mice and men. Mouse models have enhanced our understanding of (patho)-physiological implications of Cx diversity in the heart. In principle connexin-specific modulation of electrical coupling in the heart represents an interesting treatment strategy for cardiac arrhythmias and conduction disorders.
Introduction
Gap junction channels form continuous pores between the cytoplasms of closely apposed cells that are permeable to ions and molecules <1 kDa, thereby conferring electrical and metabolic coupling between neighboring cells. Gap junction channels consist of connexin (Cx) protein subunits. More than 20 Cx isoforms have been described in both humans and mice and mutations in Cx genes have been implicated in various inheritable diseases (Sohl and Willecke, 2004; Dobrowolski and Willecke, 2009). Complete gap junction channels are formed by the docking of two hemichannels, or connexons, contributed by closely apposed cells (Figure 1). Each connexon consists of 6 connexin subunits, and a homomeric gap junction channel consists of 12 Cx proteins of the same isoform. When two homomeric connexons consisting of different connexins are apposed, a heterotypic channel may form if those connexins are compatible. In heteromeric connexons, different isoforms are mixed, and a heteromeric channel may form. Some Cxs also form functional hemichannels, i.e., connexons that can open under certain conditions, causing depolarization of the plasma membrane (John et al., 1999; Bukauskas et al., 2006).
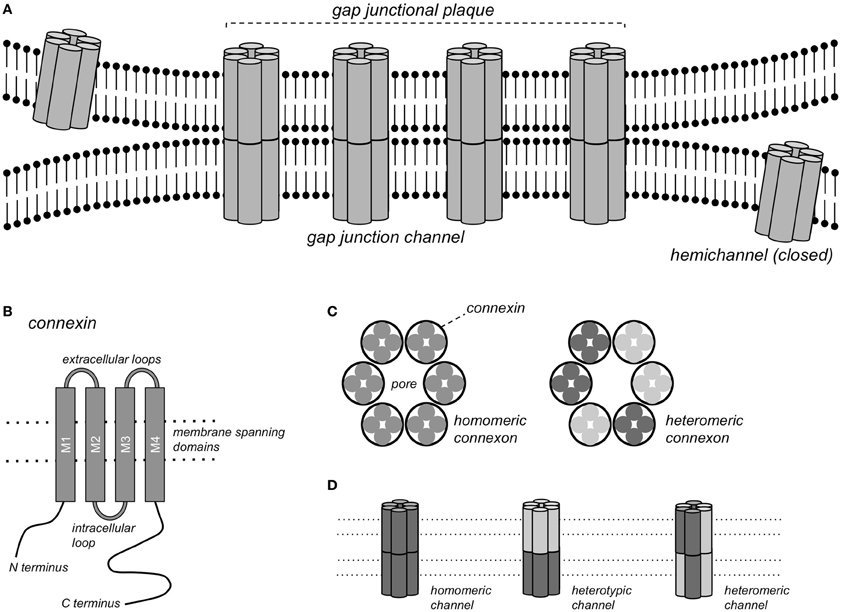
Figure 1. Gap junction channels. (A) Schematic representation of a gap junctional plaque, with each of the neighboring cells contributing one hemichannel to form the complete gap junction channel. (B) Structure of the pattern of a single connexin subunit with 4 domains traversing the membrane. (C) A hemichannel or connexon consists of a hexagonal arrangement of 6 Cx subunits, either of the same Cx isoform (homomeric connexon) or of different isoforms (heteromeric connexon). (D) Gap junction channels formed by homomeric connexons of the same isoform (homomeric channel), by homomeric connexons of different isoforms (heterotypic channel) or by heteromeric connexons (heteromeric channels).
Different regions of the heart show specific profiles of Cx expression, which in mouse and human hearts show many similarities, but also some important differences (Table 1). Different parts of the heart also have varying requirements for the degree of electrical coupling. To spread the activation wave rapidly over the ventricles, the large Purkinje myocytes of the specialized ventricular conduction system need to be strongly coupled. To create a delay between the atrial and ventricular contraction, a very low degree of coupling is required in the atrioventricular (AV) node. Similarly, to allow pacemaker function, pacemaker myocytes in the sinoatrial (SA) node need to be weakly coupled, otherwise the pacemaker (or ectopic focus) would in effect be silenced by the surrounding working myocardium (Joyner and van Capelle, 1986; Joyner et al., 2000).
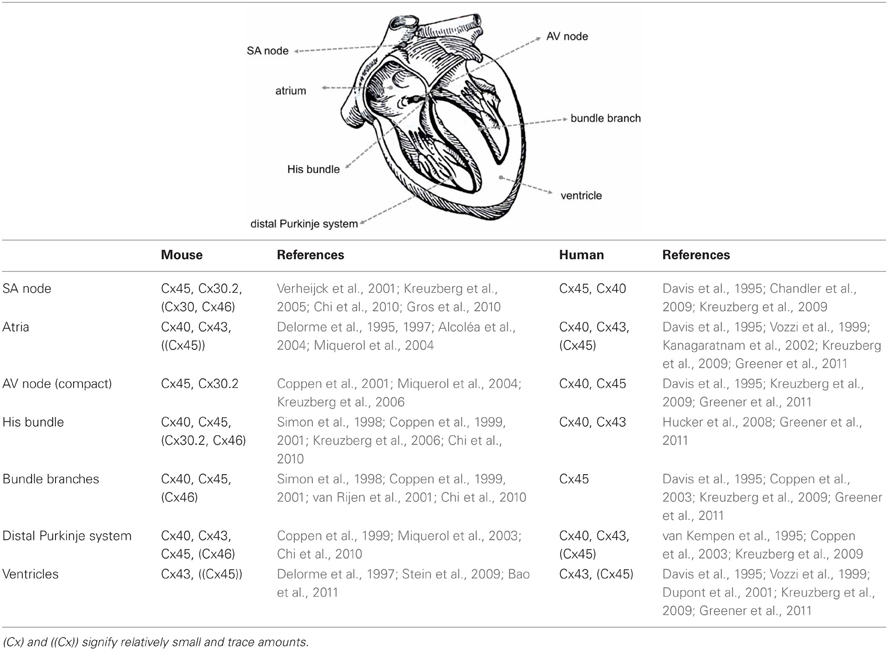
Table 1. Comparison of Cx regional expression in the mouse and human heart.
Cardiac gap junctions are highly dynamic structures, with a Cx43 protein half-life of a few hours and a comparably rapid redistribution in response to e.g., ischemia (Beardslee et al., 1998; Smyth et al., 2012). Connexins interact directly with numerous other cellular proteins, e.g., cytoskeletal components (Herve et al., 2007). Apart from their well-established roles in electrical and metabolic coupling, a number of other functions of Cx proteins are emerging, for example in mechanical adhesion and interactions with voltage-gated membrane channels (Agullo-Pascual and Delmar, 2012) and, in the case of Cx43, in mitochondrial metabolism (Ruiz-Meana et al., 2008). Conversely, electrical coupling may be mediated not only by gap junction channels, but also to some degree by ephaptic coupling, i.e., by electric field effects (Sperelakis, 2002; Lin and Keener, 2010).
Connexins differ in various biophysical properties, such as single channel conductance, permeability to larger (dye) molecules and sensitivity to the cytoplasmic pH and transjunctional potential difference, (see e.g., Elfgang et al., 1995; Gonzalez et al., 2007; Rackauskas et al., 2010). Concerning the latter, gap junction channels may close during prolonged, large voltage differences between cells, for example when one myocyte fires an action potential while its direct neighbor remains at rest (Lin and Veenstra, 2004; Lin et al., 2005). While such large voltage gradients can be imposed in voltage clamp experiments, they are unlikely to occur in the heart. Even in regions with a relatively low degree of electrical coupling, such as the SA node, transjunctional potential differences >10 mV last only a few milliseconds (Verheule et al., 2001), which would be too short for the slow process of voltage-dependent inactivation of gap junction channels.
Cx trafficking, degradation, channel gating and permeability can be regulated by phosphorylation of serine and tyrosine residues (Solan and Lampe, 2009). During ischemia, intracellular acidification, release of lipids and channel phosphorylation cause channel closure and redistribution (Dhein, 2006). This response to ischemia allows the confinement of diseased from healthy myocardium, thus enabling the myocardium to “heal-over” (de Mello et al., 1969). On the other hand, redistribution of connexins under pathological conditions has been implicated in arrhythmogenesis (Salameh and Dhein, 2011; Duffy, 2012).
Much of our current knowledge of cardiac connexins stems from studies on mouse models (Table 2). There are numerous similarities, but also some important differences between the hearts of mice and humans (Kaese and Verheule, 2012). In this review, we will discuss evidence from genetically engineered mouse models on the properties and roles of the various cardiac connexins, highlighting both their role during cardiac development and in the adult heart.
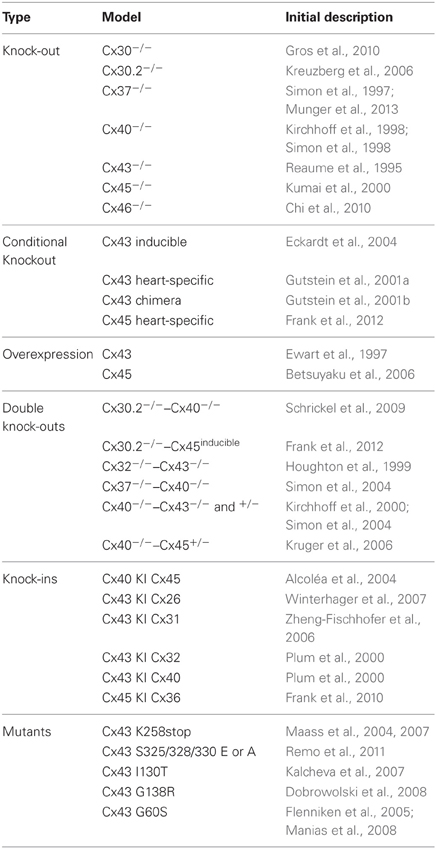
Table 2. Overview of genetically engineered mouse models.
Connexin 30
Cx30 forms channels with a large single channel conductance (γj) of 179 pS (in K-aspartate), a potential of half-maximal inactivation (V1/2) of 27 mV and a voltage-independent fraction (Gj, min) of 0.15 (Valiunas et al., 1999). Cx30 is expressed at low abundance in the murine SA node (Gros et al., 2010). Interestingly, Cx30-deficient mice have a 9% higher heart rate than wild type (Wt) mice and a lower beat-to-beat variability (Gros et al., 2010). This increased rate was still present under autonomic blockade, indicating that Cx30 affects the intrinsic pacemaker frequency. The presence of Cx30 could conceivably enhance electrical coupling between the SA node and the surrounding atrium and thereby decelerate impulse formation within the SA node (Dhein, 2010; Gros et al., 2010). However, it is not known whether Cx30 in the SA node forms heteromeric gap junction channels with the other connexins present (Cx30.2 and Cx45), and whether this would increase or decrease cell-to-cell electrical coupling.
Connexin 30.2
Cx30.2, the murine ortholog of human Cx31.9 (Belluardo et al., 2001), forms small conductance, weakly voltage-dependent channels [γj = 9 pS, V1/2 > 60 mV (Kreuzberg et al., 2005)]. Cx30.2 is expressed in the murine SA node, AV node and His bundle (Kreuzberg et al., 2005). In transfected HeLa cells, Cx30.2 can form both heterotypic (Kreuzberg et al., 2005) and heteromeric (Gemel et al., 2008) channels with Cx40, Cx43, and Cx45, the major myocardial connexins. Cx30.2 was colocalized with Cx45 in the SA and AV nodes, but not with Cx40 and Cx43 (Kreuzberg et al., 2005). Surprisingly, Cx30.2 deficient mice display accelerated AV nodal conduction, a decrease in AV Wenckebach period and a higher ventricular rate during atrial fibrillation (Kreuzberg et al., 2006). In addition, whereas deletion of Cx40 slows AV conduction, it is normal in mice deficient for both Cx30.2 and Cx40 (Schrickel et al., 2009), suggesting that the balance between Cx40 and Cx30.2 is an important determinant of AV conduction in mice. Cx30.2 is able to form functional hemichannels (Bukauskas et al., 2006), which in the open state would decrease the membrane resistance and thereby slow conduction, but the role of these hemichannels in the adult heart is uncertain at present. During development, Cx30.2 expression and proper development of the AV conduction system is determined by an enhancer region under the control of Tbx5 and GATA4. Accordingly, the PR interval is shortened in GATA4+/− mice (Munshi et al., 2009). Moreover, inhibition of Notch signaling during development leads to a loss of Cx30.2 expressing cells, hypoplasia of the AV node and accelerated AV conduction (Rentschler et al., 2011). It is important to note that Cx31.9, the human ortholog of murine Cx30.2, is not expressed in the human SA and AV nodes, ventricular conduction system or working myocardium (Kreuzberg et al., 2009). This indicates that the presence of Cx30.2 may reflect an adaptation in the small mouse heart to allow high activation frequencies and optimize A-V timing. In addition to this difference, AV conduction system in the mouse is also electrically connected to the ventricles at the basal part of the septum, leading to a baso-apical activation pattern within the septum (van Rijen et al., 2001).
Connexin 37
Cx37 forms large conductance channels that are moderately voltage sensitive [γj = 200–250 pS in K-glutamate, V1/2 = 28 mV, Gj, min = 0.27 (Reed et al., 1993)]. In the adult heart, Cx37 is expressed by endothelial cells in blood vessels and the endocardial lining of the chambers, although expression has also been observed in parts of the ventricular myocardium during embryonic development (Haefliger et al., 2000). Cx37−/− mice do not develop venous and lymphatic valves (Munger et al., 2013). In addition, mice lacking both Cx37 and Cx40 show a high incidence of atrial and ventricular septal defects at birth (Simon et al., 2004).
Connexin 40
Properties and Expression
Cx40 forms channels with a large single channel conductance and moderate voltage-sensitivity [γj = 150 pS in KCl, V1/2 = 44 mV, Gj, min = 0.5 (Traub et al., 1994)]. In the developing mouse heart, Cx40 is widely expressed in the ventricles and atria at embryonic day 11. From day 14 onwards, Cx40 becomes restricted to the conduction system in the ventricles, but it remains present in the atrial working myocardium (Delorme et al., 1995). The transcription factors Tbx2 and Tbx3 repress Cx40 expression (Hoogaars et al., 2004; Aanhaanen et al., 2011). Inactivation of Tbx2 leads to the formation of Cx40-expressing accessory pathways and ventricular preexcitation (Aanhaanen et al., 2011). On the other hand, Cx40 expression is increased by Nkx2-5 (Harris et al., 2006) and Tbx5 (Pizard et al., 2005; Arnolds et al., 2012) that delineate the conduction system. In fact, normal development of the ventricular conduction system requires the expression of Cx40 (Sankova et al., 2012).
In the adult mouse heart, Cx40 is expressed by atrial myocytes and ventricular conduction system (His bundle, left and right bundle branches and Purkinje network) (Simon et al., 1998; Miquerol et al., 2004; van Veen et al., 2005b). The selective expression of Cx40 by the conduction system within the ventricle has allowed sophisticated functional studies on conduction within the Purkinje network (Miquerol et al., 2004; Tallini et al., 2007).
Atria
Information on the effect of Cx40-deficiency on atrial conduction is somewhat contradictory. In the first studies, the P wave duration was significantly prolonged (Kirchhoff et al., 1998; Simon et al., 1998; Hagendorff et al., 1999; Verheule et al., 1999; Bagwe et al., 2005). Some other studies have not reproduced this observation (Bevilacqua et al., 2000; Tamaddon et al., 2000; Vanderbrink et al., 2000). Measurement of P-wave duration in mice is not straightforward, requiring different leads to accurately determine the end of the biphasic P-wave. In two studies that measured atrial conduction directly using direct contact mapping or optical mapping, a reduction in conduction velocity was observed (Verheule et al., 1999; Bagwe et al., 2005). However, a later study used optical mapping to show that deletion of Cx40 did not affect conduction velocity, but did abolish the difference in conduction velocity between the left and right atria (Leaf et al., 2008).
Atrial myocytes express both Cx40 and 43. Based on expression studies in Xenopus oocytes, Cx40 and 43 were originally thought to be incompatible (White and Bruzzone, 1996). However, later studies presented compelling evidence that Cx40 and 43 can form both heterotypic (Valiunas et al., 2000) and heteromeric (He et al., 1999; Cottrell and Burt, 2001) gap junction channels in mammalian cells, including adult atrial myocytes (Elenes et al., 1999). In cultured neonatal mouse atrial myocytes, Cx40 and 43 appear to make equal contributions to total gap junctional conductance (Lin et al., 2010). Intriguingly, a study on cultured strands of atrial myocytes that the Cx40/Cx43 ratio was an important determinant of propagation, with Cx43 increasing and Cx40 decreasing conduction velocity (Beauchamp et al., 2006).
In intact Cx40−/− mice, episodes of atrial tachyarrythmias/atrial fibrillation could be induced by transesophageal pacing (Hagendorff et al., 1999) and direct atrial pacing (Verheule et al., 1999; Bevilacqua et al., 2000). In perfused Cx40−/− mouse hearts studied with optical mapping, atrial ectopic beats and intraatrial conduction block during pacing at high rates were reported (Bagwe et al., 2005).
Sinoatrial Node
In contrast to larger species, such as rabbits (Verheule et al., 2001), pacemaker myocytes in the central SA node do not express Cx40 (Verheijck et al., 2001; Wiese et al., 2009) However, Cx40-deficiency does cause a modest increase in sinus cycle length (Hagendorff et al., 1999; Verheule et al., 1999; Bevilacqua et al., 2000; de Wit et al., 2003), and an increase in (corrected) SA node recovery time (Hagendorff et al., 1999; Verheule et al., 1999). The effects of Cx40-deficiency on SA node function may be caused either by altered conduction from the SA node to the atrium and/or by the phenomenon that the “atrial pacemaking complex” may be much larger than the area that is classically considered to be the sinus node (Glukhov et al., 2010).
Atrioventricular Conduction
AV conduction is also affected in Cx40−/− mice, reflected in a prolonged PR interval, increased QRS duration and a QRS morphology reminiscent of right bundle branch block in humans (Kirchhoff et al., 1998; Simon et al., 1998; Verheule et al., 1999; Bevilacqua et al., 2000; Vanderbrink et al., 2000). In addition, episodes of 2nd and 3rd degree AV block were observed in Cx40−/− mice (Kirchhoff et al., 1998; Hagendorff et al., 1999). With minor differences between studies, Cx40-deficiency prolongs the AV effective refractory period (ERP), the cycle lengths of A to V Wenckebach and 2:1 block, while the atrial ERP, ventricular ERP, cycle lengths of V to A Wenckebach and 2:1 block are not affected (Hagendorff et al., 1999; Verheule et al., 1999; Bevilacqua et al., 2000; Vanderbrink et al., 2000). Although Cx40 is not expressed in the central AV node (Delorme et al., 1995; Simon et al., 1998; Coppen et al., 1999), the AH interval is prolonged in intact mice (Bevilacqua et al., 2000; Vanderbrink et al., 2000), AV nodal conduction curves are shifted (Vanderbrink et al., 2000) and AV nodal facilitation is reversed (Zhu et al., 2005), indicative of an effect on the AV node itself. However, AV nodal conduction curves recorded in perfused hearts did not differ between Wt and Cx40−/− mice (van Rijen et al., 2001), suggesting a possible role of autonomic activity in the observations in intact mice.
Both the left and right bundle branch normally express high levels of Cx40 (Simon et al., 1998; van Rijen et al., 2001). High resolution mapping has revealed that deletion of Cx40 leads to slower conduction in the left bundle branch and conduction block in the thinner right bundle branch, causing a delayed activation of the right ventricle (Tamaddon et al., 2000; van Rijen et al., 2001). Interestingly, in mice the common bundle (expressing Cx40 and 45) is electrically connected to the base of the interventricular septum (expressing Cx43) through a small transitional zone expressing Cx43 and 45 (van Rijen et al., 2001; van Veen et al., 2005b) This arrangement results in an activation pattern within the septum from base to apex that is quite different from the pattern in humans (Durrer et al., 1970).
Ventricles
In the ventricular myocardium, which expresses Cx43 but not Cx40, conduction velocity is unaffected by Cx40-deficiency and the inducibility of ventricular arrhythmias under normal conditions was not increased (Verheule et al., 1999; Bevilacqua et al., 2000; Tamaddon et al., 2000), although one study reported an increased inducibility of ventricular tachycardia (VT) in Cx40−/− mice during infusion of isoproterenol (Bevilacqua et al., 2000), possibly involving the Purkinje system.
In none of the studies mentioned above did heterozygous Cx40+/− mice display a electrophysiological phenotype that differed from Cx40+/+ mice, indicating that even a substantial reduction in Cx40 protein does not greatly affect cardiac conduction in mice. Moreover, although it differs considerably in its biophysical properties, Cx45 can replace Cx40 almost completely, because cardiac activation in Cx40KICx45 mice is normal, except for a slower conduction velocity in the left atrium and right bundle branch (Alcoléa et al., 2004).
Two factors complicate the interpretation of electrophysiological data from Cx40−/− mice. First, although this was not noted in the earliest studies, Cx40 deficient mice display a high incidence of a variety of cardiac malformations, including ventricular septal defects, tetralogy of Fallot, double-outlet right ventricles, endocardial cushion and aortic arch defects (Kirchhoff et al., 2000; Gu et al., 2003). The occurrence of cardiac malformations is exacerbated in mice in which a homozygous deletion of Cx40 is combined with a heterozygous deletion of either Cx43 (Kirchhoff et al., 2000) or Cx45 (Kruger et al., 2006), leading to neonatal lethality in the former and additional atrial defects and further delayed AV conduction in the latter. Mice that are homozygously deficient for both Cx40 and 43 die much earlier than Cx43−/− mice, around embryonic day 12, with an abnormal rotation of the ventricles (Simon et al., 2004). Second, apart from expression by atrial myocytes and Purkinje cells, Cx40 is also expressed by endothelial cells (Dahl et al., 1995), together with Cx37. Interestingly, de Wit et al have reported that in Cx40−/− mice, arterioles show spontaneous, irregular vasomotion and that blood pressure is greatly increased from 90 to 120 mmHg (de Wit et al., 2003). To what extent hypertension in Cx40−/− mice is responsible for secondary changes in cardiac pump function, autonomic regulation and electrophysiology (e.g., decreased heart rate and slower AV nodal conduction) is uncertain at present.
Connexin 43
Properties and Expression
Cx43 forms channels with a single channel conductance of 120 pS (in CsCl) and a modest voltage dependence (V1/2 = 60 mV and Gj, min = 0.4) (Elenes et al., 2001).
In the developing mouse heart, Cx43 is specifically present in the trabeculated parts of the ventricle at embryonic day 12.5, but is abundantly expressed by the entire working myocardium later on (Coppen et al., 2003; Miquerol et al., 2003). Cx43 expression is suppressed by the transcription factors Tbx18 (specifying the SA node), Tbx2 and Tbx3 (specifying the AV conduction system) (Christoffels et al., 2004; Bakker et al., 2008; Kapoor et al., 2011; Sizarov et al., 2011).
Cx43−/− mice die shortly after birth from respiratory failure caused by a right ventricular outflow tract obstruction (Reaume et al., 1995). Abnormal cardiac development is already visible during the looping phase at embryonic day 10 (Ya et al., 1998). The outflow tract defect can be traced back to the neural crest, where abnormal p53 activation in Cx43−/− mice causes apoptosis of primordial germ cells that would otherwise have migrated to the heart (Lo et al., 1997; Francis and Lo, 2006). Both a deficiency and an excess of Cx43 can derail this process (Ewart et al., 1997; Huang et al., 1998), although it does not necessarily depend on Cx43 expression in the neural crest itself (Kretz et al., 2006). In addition to this defect, Cx43−/− (and Cx43+/−) mice also show abnormal patterning of the main coronary arteries (Li et al., 2002; Walker et al., 2005; Liu et al., 2006).
Ventricular Conduction
Using optical mapping in Cx43−/− mouse embryos, Vaidya et al reported that ventricular conduction velocity was unaffected at embryonic day 12.5, when Cx40 is still present abundantly in the ventricle. At embryonic day 17.5 however, ventricular conduction velocity was greatly reduced and ventricular arrhythmias were frequently observed (Vaidya et al., 2001). Accordingly, gap junctional coupling and conduction velocity are dramatically reduced in cultured neonatal myocytes from Cx43−/− mice, which do not show a compensatory increase in Cx40 and 45 (Beauchamp et al., 2004; Vink et al., 2004).
In adult mice, Cx43 is expressed in the atrial and ventricular working myocardium and in the distal Purkinje system (Gourdie et al., 1991; Gros and Jongsma, 1996; van Veen et al., 2005b). Within the ventricular wall, Cx43 expression is lower in the epicardium than in deeper regions (Yamada et al., 2004). Unlike Cx43−/− mice, Cx43+/− mice survive and age normally (Betsuyaku et al., 2002), although ventricular and atrial level of Cx43 protein are reduced by approximately 50% (Guerrero et al., 1997; Thomas et al., 1998). There has been a debate about to what extent this reduction in expression affects conduction, with one group reporting a 50% reduction in ventricular conduction velocity (Guerrero et al., 1997; Eloff et al., 2001), and another group reporting no change (Morley et al., 2000). The latter result is in better agreement with studies using mathematical models, which predict a weaker dependence of conduction velocity on gap junctional coupling (Shaw and Rudy, 1997; Wiegerinck et al., 2006) and a study on strands of cultured myocytes from Cx43+/+ and Cx43+/− mice, which did not show a difference in conduction velocity (Thomas et al., 2003). Moreover, two later studies in adult mice with conditional deletion of Cx43 showed a reduction of conduction velocity by approximately 40% while Cx43 protein level was decreased by 90% (Gutstein et al., 2001a; van Rijen et al., 2004). Conduction anisotropy and heterogeneity were increased, especially in the RV. VT was readily induced by pacing, often with a stable reentrant circuit in the RV and fibrillatory conduction in the LV (van Rijen et al., 2004). Telemetric recordings revealed that most mice die from arrhythmic sudden death within weeks of the start of Cx elimination (Gutstein et al., 2001a). By contrast, in a conditional knock-out strain with a 50% reduction in Cx43, no change in conduction velocity was observed and no arrhythmias were inducible (van Rijen et al., 2004).
Similar findings were reported in mice with a cardiac-specific deletion with a gradual postnatal decline in Cx43 expression. At birth, cardiac structure in these mice is normal (Eckardt et al., 2006), but Cx43 protein decreases to 59% of control level at 25 days (without a change in conduction velocity) and 18% at 45 days (with a 50% decrease in conduction velocity). At the latter stage, lethal VTs could be induced in 80% of the mice (Danik et al., 2004). Interestingly, optical mapping during sinus rhythm in this model revealed ectopic sites of ventricular activation caused by a paradoxical increase in conduction across Purkinje-ventricular junctions (Morley et al., 2005). In another approach, chimeric mice were produced with a patchy Cx43 expression pattern in the ventricles. In addition to a depressed pump function, these mice showed heterogeneous ventricular conduction and spontaneous non-sustained VT (Gutstein et al., 2001b, 2005).
Replacement of Cx43
Several studies have assessed whether the role of Cx43 can be taken over by other Cxs, using knock-in mouse models in which Cx43 was replaced by Cx40, 32, 31, or 26. Anatomically, Cx43KI40 hearts showed no abnormalities at birth (apart from mild hypertrophy in some mice), whereas Cx43KI32 hearts showed a mild form of the RV outflow tract abnormalities seen in Cx43−/− mice (Plum et al., 2000). ECG intervals in adult Cx43KI32 and Cx43KI40 mice did not differ from those in control mice. However, spontaneous ventricular extrasystoles were observed more frequently in Cx43KI40 mice than in Cx43KI32 and control mice. In Cx43KI31 mice, the RV outflow tract obstruction is severe, cardiac conduction is markedly slow and mice die within days after birth (Zheng-Fischhofer et al., 2006). In this context it is worth noting that Cx31 is highly restricted in its ability to form channels with other Cxs (Elfgang et al., 1995). Neonatal Cx43KI26 mice do not have structurally abnormal hearts, have only slightly decelerated cardiac conduction, but died within weeks because of deficient lactation of their heterozygous mothers (Winterhager et al., 2007). Adult Cx43KI26 mice (reared by foster mothers) showed a small prolongation in His-to-ventricle conduction time and QRS duration. Thus, several connexins are able to replace Cx43 in the developing and heatlhy adult heart to a large degree. However, the consequences of replacing Cx43 may be more pronounced under pathological conditions (see below for an example in Cx43KI32 mice).
Response to Ischemia
In the ventricles, the closing of gap junction channels in response to ischemia enables the myocardium to “heal-over” (de Mello et al., 1969), but it is also associated with (phase 1b) arrhythmias occurring 15–60 min after the onset of ischemia (De Groot and Coronel, 2004; Wit and Peters, 2012). During acute ischemia, spontaneous and induced VT occurred more frequently in Cx43+/− than in Cx43+/+ mice (Lerner et al., 2000). However, infarct size in the weeks after coronary occlusion was smaller in Cx43+/− mice (Kanno et al., 2003). At those later time points, the incidence of spontaneous and induced VT did not differ between Cx43+/− and Cx43+/+ mice, suggesting that effect of reduced coupling is offset by the smaller infarct size (Betsuyaku et al., 2004).
Cx43 hemichannels are present in the plasma membrane and can open as a non-selective pore in response to metabolic inhibition, which would accelerate cell death (John et al., 1999; Kondo et al., 2000). Gap 19, a peptide that prevents hemichannel opening, without affecting gap junction channels, reduces ischemia/reperfusion damage (Wang et al., 2013).
In addition, Cx43 is located in the nuclear membrane and in subsarcolemmal mitochondria (Rodriguez-Sinovas et al., 2007; Boengler et al., 2009). Ischemic preconditioning increases the levels of mitochondrial Cx43 (Boengler et al., 2005) and accordingly, ischemic preconditioning is lost in Cx43+/− mice (Schwanke et al., 2002; Li et al., 2004). Cx43 is involved in mitochondrial K+ uptake (Miro-Casas et al., 2009) and respiration (Boengler et al., 2012), forming hemichannels in the mitochondrial inner membrane that interact with other proteins (Rodriguez-Sinovas et al., 2007).
The carboxy terminus of the Cx43 protein is an important regulatory domain, involved in e.g., the response to intracellular acidification (Duffy et al., 2004). A mutant with a C terminal truncation shows a redistribution of Cx43 to the periphery of larger, sparser gap junctions (Maass et al., 2007). After ischemia/reperfusion, this mutant has larger infarcts and a higher incidence of induced VT (Maass et al., 2009). Similarly, increased gap junctional coupling by adenoviral transfection of the less pH-sensitive Cx32 increases infarct size in mice with a coronary occlusion (Prestia et al., 2011). Replacement of Cx43 with Cx32 has more complex effects (Rodriguez-Sinovas et al., 2010). Under normal conditions, ATP levels were decreased and lactate levels were increased in the myocardium of these Cx43KI32 mice. Although infarct size following ischemia/ reperfusion was smaller, protection by preconditioning was lost, indicating that Cx32 cannot replace Cx43 in its role in mitochondrial metabolism and preconditioning.
Cx43 Mutations
Several models of mutations in Cx genes have been developed that correspond to inherited diseases in humans, as reviewed in (Dobrowolski and Willecke, 2009) and (Delmar and Makita, 2012). The G60S missense mutation in Cx43 acts in a dominant negative way to cause oculodentodigital dysplasia, including various cardiac manifestations (patent foramen ovale, reduced cardiac function, a large decrease in Cx43 due to impaired trafficking to the intercalated disc, a small decrease in ventricular conduction velocity and a variety of ECG abnormalities, both brady- and tachyarrhythmias) (Flenniken et al., 2005; Kalcheva et al., 2007; Manias et al., 2008; Tuomi et al., 2011). The G138R (Dobrowolski et al., 2008) and I130T (Kalcheva et al., 2007) mutants show a similar phenotype of oculodentodigital dysplasia, including similar alterations in cardiac structure and function. However, in the case of G138R, Cx43 trafficking seems to be normal, but Cx43 (hemichannel) function is reduced due to a loss of phosphorylation. The importance of Cx43 phosphorylation is also underscored by a study on mice with “phosphatase resistant” Cx43, in which serine residues 325, 328, and 330 are replaced by either phosphomimetic glutamine (S3E) or non-phosphorylatable alanine (S3A) (Remo et al., 2011). S3E mice were resistant to gap junctional remodeling in response to transverse aortic constriction and showed a lower inducibility of VT than wildtype mice. Conversely, in S3A mice Cx43 was lost from the intercalated disc in response to transverse aortic constriction and inducibility of VT was higher than in wildtype mice.
Interactions with Other Proteins
Cx43 interacts with a number of other proteins. Whereas the organizations of adherens junctions is not disrupted in the absence of Cx43 (Gutstein et al., 2003), conversely a loss of E-cadherin, N-cadherin, desmin or desmoplakin does greatly decrease Cx43 in intercalated discs (Ferreira-Cornwell et al., 2002; Gard et al., 2005; Kostetskii et al., 2005; Li et al., 2005; Gomes et al., 2012). Among the many changes in gene expression (directly or indirectly) caused by the absence of Cx43 (Iacobas et al., 2005), potassium current also show regional alterations in Cx43−/− mice, leading to proarrhythmic shortening of the action potential duration (Danik et al., 2008), whereas sodium channel function may (Desplantez et al., 2012; Jansen et al., 2012a) or may not (Johnson et al., 1999) be affected. Short term (6 h) ventricular pacing causes a decrease in Cx43 gene expression (Kontogeorgis et al., 2008a), while the shift in potassium channel expression in response to pacing was altered in Cx43+/− mice (Kontogeorgis et al., 2008b).
Interestingly, inducible deletion of Cx43 leads to increased fibrosis during aging and pressure overload because of enhanced fibroblast activity, with a concomitant increase in conduction heterogeneity and vulnerability to VT (Jansen et al., 2012b). On the other hand, TGFβ—dependent profibrotic signaling in response to myocardial infarction is blunted in Cx43+/− mice compared to Cx43+/+ mice, leading to a decrease in post-infarct fibrosis (Zhang et al., 2010b), probably because of the difference in Cx43 expression in fibroblasts (Zhang et al., 2010a). With respect to arrhythmogenesis, these and some other studies highlight the clinically relevant conjunction of changes in excitability, electrical coupling and structural remodeling (van Veen et al., 2005a; Stein et al., 2009) that is poorly captured by monogenic alterations in transgenic mouse models.
Connexin 45
Properties and Expression
Cx45 forms channels with small conductance of 30 pS that are the most sensitive to transjunctional voltage of all cardiac Cxs (V1/2 = 13 mV, Gj, min = 0.12) (Moreno et al., 1995). Cx45 is the connexin expressed earliest in the developing heart, and the only one present before embryonic day 9. Cx45-deficient mice develop conduction block and a cushion defect caused by impairment of the epithelial-mesenchymal transformation of the cardiac endothelium (Kumai et al., 2000). Mice with cardiac myocyte-specific deletion of Cx45 do not have the cushion defect, but still develop conduction block and also die at around embryonic day 10 from pump failure (Nishii et al., 2003). The function of Cx45 in early cardiac development cannot be replaced by the neuronal connexin Cx36 (Frank et al., 2010).
Pacemaker and Conduction System
During later embryonic development, Cx45 becomes increasingly localized to pacemaker and conduction system Alcolea et al., 1999; Coppen et al., 1999. In adult mice, Cx45 is the main connexin in the central SA node, a region apposed by protrusions of Cx40 and 43 positive atrial cells (Verheijck et al., 2001). Nevertheless, the heart rate is unaffected in mice with a heart-specific inducible Cx45 deletion (Frank et al., 2012). Furthermore, Cx45 is expressed along the entire AV conduction system, including the AV node, His bundle, proximal bundle branches and Purkinje system (Coppen et al., 1999; van Veen et al., 2005b). In adult mice, the dependence of AV conduction on the 3 Cxs expressed in the AV node region is complex. As noted above, mice deficient in Cx30.2 and 40 show accelerated and decelerated AV conduction, respectively (Schrickel et al., 2009). Apparently, Cx45 is sufficient for AV conduction, because mice deficient in both Cx30.2 and 40 show normal AV conduction (Schrickel et al., 2009). In mice with a heart-specific, inducible deficiency in Cx45, AV nodal conduction is slower than normal (Frank et al., 2012), whereas atrial and ventricular conduction was not affected (Bao et al., 2011). Interestingly, these mice also displayed a posttranslational reduction of Cx30.2 expression in the conduction system. Combining the inducible deficiency in Cx45 with a deficiency in Cx30.2 further slowed AV nodal conduction (Frank et al., 2012). Although a heterozygous deletion of Cx45 does not prolong the PR interval by itself, it does further increase the observed PR prolongation in Cx40-deficient mice (Kruger et al., 2006).
Working Myocardium
Cx45 was initially thought to be widely expressed in the mammalian working myocardium (Davis et al., 1994; Verheule et al., 1997). Coppen et al later showed that this observation was mainly caused by cross-reactivity of a Cx45 antibody with Cx43 (Coppen et al., 1998). Low levels of Cx45 are expressed in the mouse ventricular working myocardium, amounting to 0.3% of total gap junction protein (Bao et al., 2011). Cx45 can form both heterotypic (Elenes et al., 2001) and heteromeric (Desplantez et al., 2004) channels with Cx43. However, co-expression of Cx45 with Cx43 appears to decrease the size of gap junctions (Grikscheit et al., 2008). This phenomenon could be more important under conditions with a higher Cx45/Cx43 ratio, for example in the ventricular epicardium, where Cx43 levels are relatively low (Yamada et al., 2004), or during heart failure, where the expression of Cx45 increases and Cx43 decreases (Yamada et al., 2003). Indeed, mice overexpressing Cx45 in the heart showed a reduction in gap junctional coupling and an increased inducibility of VT (Betsuyaku et al., 2006).
Connexin 46
Along with Cx50, Cx46 is the main connexin expressed in the lens, forming channels with a γj = 140 pS in CsCl, V1/2 = 48 mV, Gj, min = 0.11 (Hopperstad et al., 2000). mRNA for Cx46 (and Cx50) has been detected in the adult canine heart (Davis et al., 1994). The localization of Cx46 (and Cx50) protein in the dog heart is unknown, although one study detected minimal Cx46 immunoreactivity between some atrial and ventricular myocytes (Davis et al., 1995). However, in neonatal mice Cx46-positive myocytes were detected in the cardiac conduction system (or more precisely in the atrium, AV canal, intraventricular septum and ventricular subendocardium) (Chi et al., 2010). Cx46-deficient mice displayed a slightly lower heart rate, a prolonged QRS and QT duration and a QRS morphology consistent with bundle branch block (Chi et al., 2010). Cx46 has a propensity to form functional hemichannels (Trexler et al., 1996; Pfahnl and Dahl, 1999), but their role in the heart is unknown.
Conclusions
The diversity of connexins in the heart allows fine-tuning of electrical coupling depending on the region and on conditions. As therapeutic strategies, both increasing and decreasing gap junctional coupling could in theory be beneficial under certain circumstances. However, a non-specific treatment targeting gap junctions may have beneficial effects in one part of the heart but cause deleterious side effects in another. Several compounds have been developed that can modulate gap junctions (Herve and Dhein, 2010; De Vuyst et al., 2011). Some of these have been successfully tested in animal models. For example, in canine model of heart failure, enhancing gap junctional coupling with rotigaptide can reduce the vulnerability to atrial fibrillation (Guerra et al., 2006). For more selective pharmacological manipulation of gap junction coupling, it is desirable for a treatment to be (1) specific to the heart and/or (2) specific for a particular connexin. In this respect, gap junction blocking “peptidomimetics,” a class of small peptide molecules that have some Cx selectivity, are especially promising (Evans et al., 2012). An enormous amount of knowledge on cardiac connexins has been gathered from genetically engineered mouse models, as described above. There are some important differences in cardiac electrophysiology in general and connexin distribution in particular between murine and human hearts (Kaese and Verheule, 2012). Nevertheless, for the development of connexin-specific treatment strategies, knowledge derived from transgenic mouse models provides a wealth of valuable insights.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Sven Kaese received a scholarship from the Peter-Osypka-Foundation. Also supported by the European Union (FP7 Collaborative project EUTRAF, 261057) to Sander Verheule.
References
Aanhaanen, W. T., Boukens, B. J., Sizarov, A., Wakker, V., De Gier-De Vries, C., Van Ginneken, A. C., et al. (2011). Defective Tbx2-dependent patterning of the atrioventricular canal myocardium causes accessory pathway formation in mice. J. Clin. Invest. 121, 534–544. doi: 10.1172/JCI44350
Agullo-Pascual, E., and Delmar, M. (2012). The noncanonical functions of Cx43 in the heart. J. Membr. Biol. 245, 477–482. doi: 10.1007/s00232-012-9466-y
Alcoléa, S., Jarry-Guichard, T., De Bakker, J., Gonzalez, D., Lamers, W., Coppen, S., et al. (2004). Replacement of connexin40 by connexin45 in the mouse: impact on cardiac electrical conduction. Circ. Res. 94, 100–109. doi: 10.1161/01.RES.0000108261.67979.2A
Alcolea, S., Theveniau-Ruissy, M., Jarry-Guichard, T., Marics, I., Tzouanacou, E., Chauvin, J. P., et al. (1999). Downregulation of connexin 45 gene products during mouse heart development. Circ. Res. 84, 1365–1379. doi: 10.1161/01.RES.84.12.1365
Arnolds, D. E., Liu, F., Fahrenbach, J. P., Kim, G. H., Schillinger, K. J., Smemo, S., et al. (2012). TBX5 drives Scn5a expression to regulate cardiac conduction system function. J. Clin. Invest. 122, 2509–2518. doi: 10.1172/JCI62617
Bagwe, S., Berenfeld, O., Vaidya, D., Morley, G. E., and Jalife, J. (2005). Altered right atrial excitation and propagation in connexin40 knockout mice. Circulation 112, 2245–2253. doi: 10.1161/CIRCULATIONAHA.104.527325
Bakker, M. L., Boukens, B. J., Mommersteeg, M. T., Brons, J. F., Wakker, V., Moorman, A. F., et al. (2008). Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ. Res. 102, 1340–1349. doi: 10.1161/CIRCRESAHA.107.169565
Bao, M., Kanter, E. M., Huang, R. Y., Maxeiner, S., Frank, M., Zhang, Y., et al. (2011). Residual Cx45 and its relationship to Cx43 in murine ventricular myocardium. Channels (Austin) 5, 489–499. doi: 10.4161/chan.5.6.18523
Beardslee, M. A., Laing, J. G., Beyer, E. C., and Saffitz, J. E. (1998). Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 83, 629–635. doi: 10.1161/01.RES.83.6.629
Beauchamp, P., Choby, C., Desplantez, T., De Peyer, K., Green, K., Yamada, K. A., et al. (2004). Electrical propagation in synthetic ventricular myocyte strands from germline connexin43 knockout mice. Circ. Res. 95, 170–178. doi: 10.1161/01.RES.0000134923.05174.2f
Beauchamp, P., Yamada, K. A., Baertschi, A. J., Green, K., Kanter, E. M., Saffitz, J. E., et al. (2006). Relative contributions of connexins 40 and 43 to atrial impulse propagation in synthetic strands of neonatal and fetal murine cardiomyocytes. Circ. Res. 99, 1216–1224. doi: 10.1161/01.RES.0000250607.34498.b4
Belluardo, N., White, T. W., Srinivas, M., Trovato-Salinaro, A., Ripps, H., Mudo, G., et al. (2001). Identification and functional expression of HCx31.9, a novel gap junction gene. Cell Commun. Adhes. 8, 173–178. doi: 10.3109/15419060109080719
Betsuyaku, T., Kanno, S., Lerner, D. L., Schuessler, R. B., Saffitz, J. E., and Yamada, K. A. (2004). Spontaneous and inducible ventricular arrhythmias after myocardial infarction in mice. Cardiovasc. Pathol. 13, 156–164. doi: 10.1016/S1054-8807(03)00152-2
Betsuyaku, T., Kovacs, A., Saffitz, J. E., and Yamada, K. A. (2002). Cardiac structure and function in young and senescent mice heterozygous for a connexin43 null mutation. J. Mol. Cell. Cardiol. 34, 175–184. doi: 10.1006/jmcc.2001.1499
Betsuyaku, T., Nnebe, N. S., Sundset, R., Patibandla, S., Krueger, C. M., and Yamada, K. A. (2006). Overexpression of cardiac connexin45 increases susceptibility to ventricular tachyarrhythmias in vivo. Am. J. Physiol. Heart Circ. Physiol. 290, H163–H171. doi: 10.1152/ajpheart.01308.2004
Bevilacqua, L. M., Simon, A. M., Maguire, C. T., Gehrmann, J., Wakimoto, H., Paul, D. L., et al. (2000). A targeted disruption in connexin40 leads to distinct atrioventricular conduction defects. J. Interv. Card. Electrophysiol. 4, 459–467. doi: 10.1023/A:1009800328836
Boengler, K., Dodoni, G., Rodriguez-Sinovas, A., Cabestrero, A., Ruiz-Meana, M., Gres, P., et al. (2005). Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 67, 234–244. doi: 10.1016/j.cardiores.2005.04.014
Boengler, K., Ruiz-Meana, M., Gent, S., Ungefug, E., Soetkamp, D., Miro-Casas, E., et al. (2012). Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J. Cell. Mol. Med. 16, 1649–1655. doi: 10.1111/j.1582-4934.2011.01516.x
Boengler, K., Stahlhofen, S., Van De Sand, A., Gres, P., Ruiz-Meana, M., Garcia-Dorado, D., et al. (2009). Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 104, 141–147. doi: 10.1007/s00395-009-0007-5
Bukauskas, F. F., Kreuzberg, M. M., Rackauskas, M., Bukauskiene, A., Bennett, M. V., Verselis, V. K., et al. (2006). Properties of mouse connexin 30.2 and human connexin 31.9 hemichannels: implications for atrioventricular conduction in the heart. Proc. Natl. Acad. Sci. U.S.A. 103, 9726–9731. doi: 10.1073/pnas.0603372103
Chandler, N. J., Greener, I. D., Tellez, J. O., Inada, S., Musa, H., Molenaar, P., et al. (2009). Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation 119, 1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369
Chi, N. C., Bussen, M., Brand-Arzamendi, K., Ding, C., Olgin, J. E., Shaw, R. M., et al. (2010). Cardiac conduction is required to preserve cardiac chamber morphology. Proc. Natl. Acad. Sci. U.S.A. 107, 14662–14667. doi: 10.1073/pnas.0909432107
Christoffels, V. M., Hoogaars, W. M., Tessari, A., Clout, D. E., Moorman, A. F., and Campione, M. (2004). T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev. Dyn. 229, 763–770. doi: 10.1002/dvdy.10487
Coppen, S. R., Dupont, E., Rothery, S., and Severs, N. J. (1998). Connexin45 expression is preferentially associated with the ventricular conduction system in mouse and rat heart. Circ. Res. 82, 232–243. doi: 10.1161/01.RES.82.2.232
Coppen, S. R., Gourdie, R. G., and Severs, N. J. (2001). Connexin45 is the first connexin to be expressed in the central conduction system of the mouse heart. Exp. Clin. Cardiol. 6, 17–23.
Coppen, S. R., Kaba, R. A., Halliday, D., Dupont, E., Skepper, J. N., Elneil, S., et al. (2003). Comparison of connexin expression patterns in the developing mouse heart and human foetal heart. Mol. Cell. Biochem. 242, 121–127. doi: 10.1023/A:1021150014764
Coppen, S. R., Severs, N. J., and Gourdie, R. G. (1999). Connexin45 (alpha 6) expression delineates an extended conduction system in the embryonic and mature rodent heart. Dev. Genet. 24, 82–90.
Cottrell, G. T., and Burt, J. M. (2001). Heterotypic gap junction channel formation between heteromeric and homomeric Cx40 and Cx43 connexons. Am. J. Physiol. Cell Physiol. 281, C1559–C1567.
Dahl, E., Winterhager, E., Traub, O., and Willecke, K. (1995). Expression of gap junction genes, connexin40 and connexin43, during fetal mouse development. Anat. Embryol. 191, 267–278. doi: 10.1007/BF00187825
Danik, S. B., Liu, F., Zhang, J., Suk, H. J., Morley, G. E., Fishman, G. I., et al. (2004). Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 95, 1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a
Danik, S. B., Rosner, G., Lader, J., Gutstein, D. E., Fishman, G. I., and Morley, G. E. (2008). Electrical remodeling contributes to complex tachyarrhythmias in connexin43-deficient mouse hearts. FASEB J. 22, 1204–1212. doi: 10.1096/fj.07-8974com
Davis, L. M., Kanter, H. L., Beyer, E. C., and Saffitz, J. E. (1994). Distinct gap junction protein phenotypes in cardiac tissues with disparate conduction properties. J. Am. Coll. Cardiol. 24, 1124–1132. doi: 10.1016/0735-1097(94)90879-6
Davis, L. M., Rodefeld, M. E., Green, K., Beyer, E. C., and Saffitz, J. E. (1995). Gap junction protein phenotypes of the human heart and conduction system. J. Cardiovasc. Electrophysiol. 6, 813–822. doi: 10.1111/j.1540-8167.1995.tb00357.x
De Groot, J. R., and Coronel, R. (2004). Acute ischemia-induced gap junctional uncoupling and arrhythmogenesis. Cardiovasc. Res. 62, 323–334. doi: 10.1016/j.cardiores.2004.01.033
Delorme, B., Dahl, E., Jarry-Guichard, T., Briand, J. P., Willecke, K., Gros, D., et al. (1997). Expression pattern of connexin gene products at the early developmental stages of the mouse cardiovascular system. Circ. Res. 81, 423–437. doi: 10.1161/01.RES.81.3.423
Delorme, B., Dahl, E., Jarry-Guichard, T., Marics, I., Briand, J. P., Willecke, K., et al. (1995). Developmental regulation of connexin 40 gene expression in mouse heart correlates with the differentiation of the conduction system. Dev. Dyn. 204, 358–371. doi: 10.1002/aja.1002040403
Delmar, M., and Makita, N. (2012). Cardiac connexins, mutations and arrhythmias. Curr. Opin. Cardiol. 27, 236–241. doi: 10.1097/HCO.0b013e328352220e
de Mello, W. C., Motta, G. E., and Chapeau, M. (1969). A study on the healing-over of myocardial cells of toads. Circ. Res. 24, 475–487. doi: 10.1161/01.RES.24.3.475
Desplantez, T., Halliday, D., Dupont, E., and Weingart, R. (2004). Cardiac connexins Cx43 and Cx45: formation of diverse gap junction channels with diverse electrical properties. Pflugers Arch. 448, 363–375. doi: 10.1007/s00424-004-1250-0
Desplantez, T., McCain, M. L., Beauchamp, P., Rigoli, G., Rothen-Rutishauser, B., Parker, K. K., et al. (2012). Connexin43 ablation in foetal atrial myocytes decreases electrical coupling, partner connexins, and sodium current. Cardiovasc. Res. 94, 58–65. doi: 10.1093/cvr/cvs025
De Vuyst, E., Boengler, K., Antoons, G., Sipido, K. R., Schulz, R., and Leybaert, L. (2011). Pharmacological modulation of connexin-formed channels in cardiac pathophysiology. Br. J. Pharmacol. 163, 469–483. doi: 10.1111/j.1476-5381.2011.01244.x
de Wit, C., Roos, F., Bolz, S. S., and Pohl, U. (2003). Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol. Genomics 13, 169–177.
Dhein, S. (2006). Cardiac ischemia and uncoupling: gap junctions in ischemia and infarction. Adv. Cardiol. 42, 198–212. doi: 10.1159/000092570
Dhein, S. (2010). Cx30 in the sinus node of murine heart: just one connexin more, or more. Evidence for a construction principle? Cardiovasc. Res. 85, 7–8. doi: 10.1093/cvr/cvp351
Dobrowolski, R., Sasse, P., Schrickel, J. W., Watkins, M., Kim, J. S., Rackauskas, M., et al. (2008). The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 17, 539–554. doi: 10.1093/hmg/ddm329
Dobrowolski, R., and Willecke, K. (2009). Connexin-caused genetic diseases and corresponding mouse models. Antioxid. Redox Signal. 11, 283–295. doi: 10.1089/ars.2008.2128
Duffy, H. S. (2012). The molecular mechanisms of gap junction remodeling. Heart Rhythm 9, 1331–1334. doi: 10.1016/j.hrthm.2011.11.048
Duffy, H. S., Ashton, A. W., O'donnell, P., Coombs, W., Taffet, S. M., Delmar, M., et al. (2004). Regulation of connexin43 protein complexes by intracellular acidification. Circ. Res. 94, 215–222. doi: 10.1161/01.RES.0000113924.06926.11
Dupont, E., Matsushita, T., Kaba, R. A., Vozzi, C., Coppen, S. R., Khan, N., et al. (2001). Altered connexin expression in human congestive heart failure. J. Mol. Cell. Cardiol. 33, 359–371. doi: 10.1006/jmcc.2000.1308
Durrer, D., Van Dam, R. T., Freud, G. E., Janse, M. J., Meijler, F. L., and Arzbaecher, R. C. (1970). Total excitation of the isolated human heart. Circulation 41, 899–912. doi: 10.1161/01.CIR.41.6.899
Eckardt, D., Kirchhoff, S., Kim, J. S., Degen, J., Theis, M., Ott, T., et al. (2006). Cardiomyocyte-restricted deletion of connexin43 during mouse development. J. Mol. Cell. Cardiol. 41, 963–971. doi: 10.1016/j.yjmcc.2006.07.017
Eckardt, D., Theis, M., Degen, J., Ott, T., van Rijen, H. V., Kirchhoff, S., et al. (2004). Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J. Mol. Cell. Cardiol. 36, 101–110. doi: 10.1016/j.yjmcc.2003.10.006
Elenes, S., Martinez, A. D., Delmar, M., Beyer, E. C., and Moreno, A. P. (2001). Heterotypic docking of Cx43 and Cx45 connexons blocks fast voltage gating of Cx43. Biophys. J. 81, 1406–1418. doi: 10.1016/S0006-3495(01)75796-7
Elenes, S., Rubart, M., and Moreno, A. P. (1999). Junctional communication between isolated pairs of canine atrial cells is mediated by homogeneous and heterogeneous gap junction channels. J. Cardiovasc. Electrophysiol. 10, 990–1004. doi: 10.1111/j.1540-8167.1999.tb01270.x
Elfgang, C., Eckert, R., Lichtenberg-Frate, H., Butterweck, A., Traub, O., Klein, R. A., et al. (1995). Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J. Cell Biol. 129, 805–817. doi: 10.1083/jcb.129.3.805
Eloff, B. C., Lerner, D. L., Yamada, K. A., Schuessler, R. B., Saffitz, J. E., and Rosenbaum, D. S. (2001). High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc. Res. 51, 681–690. doi: 10.1016/S0008-6363(01)00341-8
Evans, W. H., Bultynck, G., and Leybaert, L. (2012). Manipulating connexin communication channels: use of peptidomimetics and the translational outputs. J. Membr. Biol. 245, 437–449. doi: 10.1007/s00232-012-9488-5
Ewart, J. L., Cohen, M. F., Meyer, R. A., Huang, G. Y., Wessels, A., Gourdie, R. G., et al. (1997). Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development 124, 1281–1292.
Ferreira-Cornwell, M. C., Luo, Y., Narula, N., Lenox, J. M., Lieberman, M., and Radice, G. L. (2002). Remodeling the intercalated disc leads to cardiomyopathy in mice misexpressing cadherins in the heart. J. Cell. Sci. 115, 1623–1634.
Flenniken, A. M., Osborne, L. R., Anderson, N., Ciliberti, N., Fleming, C., Gittens, J. E., et al. (2005). A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development 132, 4375–4386. doi: 10.1242/dev.02011
Francis, R. J., and Lo, C. W. (2006). Primordial germ cell deficiency in the connexin 43 knockout mouse arises from apoptosis associated with abnormal p53 activation. Development 133, 3451–3460. doi: 10.1242/dev.02506
Frank, M., Eiberger, B., Janssen-Bienhold, U., De Sevilla Muller, L. P., Tjarks, A., Kim, J. S., et al. (2010). Neuronal connexin-36 can functionally replace connexin-45 in mouse retina but not in the developing heart. J. Cell Sci. 123, 3605–3615. doi: 10.1242/jcs.068668
Frank, M., Wirth, A., Andrie, R. P., Kreuzberg, M. M., Dobrowolski, R., Seifert, G., et al. (2012). Connexin45 provides optimal atrioventricular nodal conduction in the adult mouse heart. Circ. Res. 111, 1528–1538. doi: 10.1161/CIRCRESAHA.112.270561
Gard, J. J., Yamada, K., Green, K. G., Eloff, B. C., Rosenbaum, D. S., Wang, X., et al. (2005). Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc. Res. 67, 539–547. doi: 10.1016/j.cardiores.2005.04.004
Gemel, J., Lin, X., Collins, R., Veenstra, R. D., and Beyer, E. C. (2008). Cx30.2 can form heteromeric gap junction channels with other cardiac connexins. Biochem. Biophys. Res. Commun. 369, 388–394. doi: 10.1016/j.bbrc.2008.02.040
Glukhov, A. V., Fedorov, V. V., Anderson, M. E., Mohler, P. J., and Efimov, I. R. (2010). Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. Am. J. Physiol. Heart Circ. Physiol. 299, H482–H491. doi: 10.1152/ajpheart.00756.2009
Gomes, J., Finlay, M., Ahmed, A. K., Ciaccio, E. J., Asimaki, A., Saffitz, J. E., et al. (2012). Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur. Heart J. 33, 1942–1953. doi: 10.1093/eurheartj/ehr472
Gonzalez, D., Gomez-Hernandez, J. M., and Barrio, L. C. (2007). Molecular basis of voltage dependence of connexin channels: an integrative appraisal. Prog. Biophys. Mol. Biol. 94, 66–106. doi: 10.1016/j.pbiomolbio.2007.03.007
Gourdie, R. G., Green, C. R., and Severs, N. J. (1991). Gap junction distribution in adult mammalian myocardium revealed by an anti-peptide antibody and laser scanning confocal microscopy. J. Cell Sci. 99, 41–55.
Greener, I. D., Monfredi, O., Inada, S., Chandler, N. J., Tellez, J. O., Atkinson, A., et al. (2011). Molecular architecture of the human specialised atrioventricular conduction axis. J. Mol. Cell. Cardiol. 50, 642–651. doi: 10.1016/j.yjmcc.2010.12.017
Grikscheit, K., Thomas, N., Bruce, A. F., Rothery, S., Chan, J., Severs, N. J., et al. (2008). Coexpression of connexin 45 with connexin 43 decreases gap junction size. Cell Commun. Adhes. 15, 185–193. doi: 10.1080/15419060802013943
Gros, D., Theveniau-Ruissy, M., Bernard, M., Calmels, T., Kober, F., Sohl, G., et al. (2010). Connexin 30 is expressed in the mouse sino-atrial node and modulates heart rate. Cardiovasc. Res. 85, 45–55. doi: 10.1093/cvr/cvp280
Gros, D. B., and Jongsma, H. J. (1996). Connexins in mammalian heart function. Bioessays 18, 719–730. doi: 10.1002/bies.950180907
Gu, H., Smith, F. C., Taffet, S. M., and Delmar, M. (2003). High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 93, 201–206. doi: 10.1161/01.RES.0000084852.65396.70
Guerra, J. M., Everett, T. H. T., Lee, K. W., Wilson, E., and Olgin, J. E. (2006). Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation 114, 110–118. doi: 10.1161/CIRCULATIONAHA.105.606251
Guerrero, P. A., Schuessler, R. B., Davis, L. M., Beyer, E. C., Johnson, C. M., Yamada, K. A., et al. (1997). Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J. Clin. Invest. 99, 1991–1998. doi: 10.1172/JCI119367
Gutstein, D. E., Danik, S. B., Lewitton, S., France, D., Liu, F., Chen, F. L., et al. (2005). Focal gap junction uncoupling and spontaneous ventricular ectopy. Am. J. Physiol. Heart Circ. Physiol. 289, H1091–H1098. doi: 10.1152/ajpheart.00095.2005
Gutstein, D. E., Liu, F. Y., Meyers, M. B., Choo, A., and Fishman, G. I. (2003). The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J. Cell. Sci. 116, 875–885. doi: 10.1242/jcs.00258
Gutstein, D. E., Morley, G. E., Tamaddon, H., Vaidya, D., Schneider, M. D., Chen, J., et al. (2001a). Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 88, 333–339. doi: 10.1161/01.RES.88.3.333
Gutstein, D. E., Morley, G. E., Vaidya, D., Liu, F., Chen, F. L., Stuhlmann, H., et al. (2001b). Heterogeneous expression of gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation 104, 1194–1199. doi: 10.1161/hc3601.093990
Haefliger, J. A., Polikar, R., Schnyder, G., Burdet, M., Sutter, E., Pexieder, T., et al. (2000). Connexin37 in normal and pathological development of mouse heart and great arteries. Dev. Dyn. 218, 331–344.
Hagendorff, A., Schumacher, B., Kirchhoff, S., Luderitz, B., and Willecke, K. (1999). Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation 99, 1508–1515. doi: 10.1161/01.CIR.99.11.1508
Harris, B. S., Spruill, L., Edmonson, A. M., Rackley, M. S., Benson, D. W., O'brien, T. X., et al. (2006). Differentiation of cardiac Purkinje fibers requires precise spatiotemporal regulation of Nkx2-5 expression. Dev. Dyn. 235, 38–49. doi: 10.1002/dvdy.20580
He, D. S., Jiang, J. X., Taffet, S. M., and Burt, J. M. (1999). Formation of heteromeric gap junction channels by connexins 40 and 43 in vascular smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 96, 6495–6500. doi: 10.1073/pnas.96.11.6495
Herve, J. C., Bourmeyster, N., Sarrouilhe, D., and Duffy, H. S. (2007). Gap junctional complexes: from partners to functions. Prog. Biophys. Mol. Biol. 94, 29–65. doi: 10.1016/j.pbiomolbio.2007.03.010
Herve, J. C., and Dhein, S. (2010). Peptides targeting gap junctional structures. Curr. Pharm. Des. 16, 3056–3070. doi: 10.2174/138161210793292528
Hoogaars, W. M., Tessari, A., Moorman, A. F., De Boer, P. A., Hagoort, J., Soufan, A. T., et al. (2004). The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc. Res. 62, 489–499. doi: 10.1016/j.cardiores.2004.01.030
Hopperstad, M. G., Srinivas, M., and Spray, D. C. (2000). Properties of gap junction channels formed by Cx46 alone and in combination with Cx50. Biophys. J. 79, 1954–1966. doi: 10.1016/S0006-3495(00)76444-7
Houghton, F. D., Thonnissen, E., Kidder, G. M., Naus, C. C., Willecke, K., and Winterhager, E. (1999). Doubly mutant mice, deficient in connexin32 and -43, show normal prenatal development of organs where the two gap junction proteins are expressed in the same cells. Dev. Genet. 24, 5–12.
Huang, G. Y., Wessels, A., Smith, B. R., Linask, K. K., Ewart, J. L., and Lo, C. W. (1998). Alteration in connexin 43 gap junction gene dosage impairs conotruncal heart development. Dev. Biol. 198, 32–44. doi: 10.1016/S0012-1606(98)80027-4
Hucker, W. J., McCain, M. L., Laughner, J. I., Iaizzo, P. A., and Efimov, I. R. (2008). Connexin 43 expression delineates two discrete pathways in the human atrioventricular junction. Anat. Rec. (Hoboken) 291, 204–215. doi: 10.1002/ar.20631
Iacobas, D. A., Iacobas, S., Li, W. E., Zoidl, G., Dermietzel, R., and Spray, D. C. (2005). Genes controlling multiple functional pathways are transcriptionally regulated in connexin43 null mouse heart. Physiol. Genomics 20, 211–223. doi: 10.1152/physiolgenomics.00229.2003
Jansen, J. A., Noorman, M., Musa, H., Stein, M., De Jong, S., van Der Nagel, R., et al. (2012a). Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 9, 600–607. doi: 10.1016/j.hrthm.2011.11.025
Jansen, J. A., van Veen, T. A., De Jong, S., van Der Nagel, R., van Stuijvenberg, L., Driessen, H., et al. (2012b). Reduced Cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ. Arrhythm. Electrophysiol. 5, 380–390. doi: 10.1161/CIRCEP.111.966580
John, S. A., Kondo, R., Wang, S. Y., Goldhaber, J. I., and Weiss, J. N. (1999). Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 274, 236–240. doi: 10.1074/jbc.274.1.236
Johnson, C. M., Green, K. G., Kanter, E. M., Bou-Abboud, E., Saffitz, J. E., and Yamada, K. A. (1999). Voltage-gated Na+ channel activity and connexin expression in Cx43-deficient cardiac myocytes. J. Cardiovasc. Electrophysiol. 10, 1390–1401. doi: 10.1111/j.1540-8167.1999.tb00195.x
Joyner, R. W., and van Capelle, F. J. (1986). Propagation through electrically coupled cells. How a small SA node drives a large atrium. Biophys. J. 50, 1157–1164. doi: 10.1016/S0006-3495(86)83559-7
Joyner, R. W., Wang, Y. G., Wilders, R., Golod, D. A., Wagner, M. B., Kumar, R., et al. (2000). A spontaneously active focus drives a model atrial sheet more easily than a model ventricular sheet. Am. J. Physiol. Heart Circ. Physiol. 279, H752–H763.
Kaese, S., and Verheule, S. (2012). Cardiac electrophysiology in mice: a matter of size. Front. Physiol. 3:345. doi: 10.3389/fphys.2012.00345
Kalcheva, N., Qu, J., Sandeep, N., Garcia, L., Zhang, J., Wang, Z., et al. (2007). Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc. Natl. Acad. Sci. U.S.A. 104, 20512–20516. doi: 10.1073/pnas.0705472105
Kanagaratnam, P., Rothery, S., Patel, P., Severs, N. J., and Peters, N. S. (2002). Relative expression of immunolocalized connexins 40 and 43 correlates with human atrial conduction properties. J. Am. Coll. Cardiol. 39, 116–123. doi: 10.1016/S0735-1097(01)01710-7
Kanno, S., Kovacs, A., Yamada, K. A., and Saffitz, J. E. (2003). Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J. Am. Coll. Cardiol. 41, 681–686. doi: 10.1016/S0735-1097(02)02893-0
Kapoor, N., Galang, G., Marban, E., and Cho, H. C. (2011). Transcriptional suppression of connexin43 by TBX18 undermines cell-cell electrical coupling in postnatal cardiomyocytes. J. Biol. Chem. 286, 14073–14079. doi: 10.1074/jbc.M110.185298
Kirchhoff, S., Kim, J. S., Hagendorff, A., Thonnissen, E., Kruger, O., Lamers, W. H., et al. (2000). Abnormal cardiac conduction and morphogenesis in connexin40 and connexin43 double-deficient mice. Circ. Res. 87, 399–405. doi: 10.1161/01.RES.87.5.399
Kirchhoff, S., Nelles, E., Hagendorff, A., Kruger, O., Traub, O., and Willecke, K. (1998). Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr. Biol. 8, 299–302. doi: 10.1016/S0960-9822(98)70114-9
Kondo, R. P., Wang, S. Y., John, S. A., Weiss, J. N., and Goldhaber, J. I. (2000). Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 32, 1859–1872. doi: 10.1006/jmcc.2000.1220
Kontogeorgis, A., Kaba, R. A., Kang, E., Feig, J. E., Gupta, P. P., Ponzio, M., et al. (2008a). Short-term pacing in the mouse alters cardiac expression of connexin43. BMC Physiol. 8:8. doi: 10.1186/1472-6793-8-8
Kontogeorgis, A., Li, X., Kang, E. Y., Feig, J. E., Ponzio, M., Kang, G., et al. (2008b). Decreased connexin43 expression in the mouse heart potentiates pacing-induced remodeling of repolarizing currents. Am. J. Physiol. Heart Circ. Physiol. 295, H1905–H1916. doi: 10.1152/ajpheart.590.2008
Kostetskii, I., Li, J., Xiong, Y., Zhou, R., Ferrari, V. A., Patel, V. V., et al. (2005). Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 96, 346–354. doi: 10.1161/01.RES.0000156274.72390.2c
Kretz, M., Eckardt, D., Kruger, O., Kim, J. S., Maurer, J., Theis, M., et al. (2006). Normal embryonic development and cardiac morphogenesis in mice with Wnt1-Cre-mediated deletion of connexin43. Genesis 44, 269–276. doi: 10.1002/dvg.20204
Kreuzberg, M. M., Liebermann, M., Segschneider, S., Dobrowolski, R., Dobrzynski, H., Kaba, R., et al. (2009). Human connexin31.9, unlike its orthologous protein connexin30.2 in the mouse, is not detectable in the human cardiac conduction system. J. Mol. Cell. Cardiol. 46, 553–559. doi: 10.1016/j.yjmcc.2008.12.007
Kreuzberg, M. M., Schrickel, J. W., Ghanem, A., Kim, J. S., Degen, J., Janssen-Bienhold, U., et al. (2006). Connexin30.2 containing gap junction channels decelerate impulse propagation through the atrioventricular node. Proc. Natl. Acad. Sci. U.S.A. 103, 5959–5964. doi: 10.1073/pnas.0508512103
Kreuzberg, M. M., Sohl, G., Kim, J. S., Verselis, V. K., Willecke, K., and Bukauskas, F. F. (2005). Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ. Res. 96, 1169–1177. doi: 10.1161/01.RES.0000169271.33675.05
Kruger, O., Maxeiner, S., Kim, J. S., van Rijen, H. V., De Bakker, J. M., Eckardt, D., et al. (2006). Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J. Mol. Cell. Cardiol. 41, 787–797. doi: 10.1016/j.yjmcc.2006.07.005
Kumai, M., Nishii, K., Nakamura, K., Takeda, N., Suzuki, M., and Shibata, Y. (2000). Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 127, 3501–3512.
Leaf, D. E., Feig, J. E., Vasquez, C., Riva, P. L., Yu, C., Lader, J. M., et al. (2008). Connexin40 imparts conduction heterogeneity to atrial tissue. Circ. Res. 103, 1001–1008. doi: 10.1161/CIRCRESAHA.107.168997
Lerner, D. L., Yamada, K. A., Schuessler, R. B., and Saffitz, J. E. (2000). Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 101, 547–552. doi: 10.1161/01.CIR.101.5.547
Li, J., Patel, V. V., Kostetskii, I., Xiong, Y., Chu, A. F., Jacobson, J. T., et al. (2005). Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ. Res. 97, 474–481. doi: 10.1161/01.RES.0000181132.11393.18
Li, W. E., Waldo, K., Linask, K. L., Chen, T., Wessels, A., Parmacek, M. S., et al. (2002). An essential role for connexin43 gap junctions in mouse coronary artery development. Development 129, 2031–2042.
Li, X., Heinzel, F. R., Boengler, K., Schulz, R., and Heusch, G. (2004). Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J. Mol. Cell. Cardiol. 36, 161–163. doi: 10.1016/j.yjmcc.2003.10.019
Lin, J., and Keener, J. P. (2010). Modeling electrical activity of myocardial cells incorporating the effects of ephaptic coupling. Proc. Natl. Acad. Sci. U.S.A. 107, 20935–20940. doi: 10.1073/pnas.1010154107
Lin, X., Gemel, J., Beyer, E. C., and Veenstra, R. D. (2005). Dynamic model for ventricular junctional conductance during the cardiac action potential. Am. J. Physiol. Heart Circ. Physiol. 288, H1113–H1123. doi: 10.1152/ajpheart.00882.2004
Lin, X., Gemel, J., Glass, A., Zemlin, C. W., Beyer, E. C., and Veenstra, R. D. (2010). Connexin40 and connexin43 determine gating properties of atrial gap junction channels. J. Mol. Cell. Cardiol. 48, 238–245. doi: 10.1016/j.yjmcc.2009.05.014
Lin, X., and Veenstra, R. D. (2004). Action potential modulation of connexin40 gap junctional conductance. Am. J. Physiol. Heart Circ. Physiol. 286, H1726–H1735. doi: 10.1152/ajpheart.00943.2003
Liu, S., Liu, F., Schneider, A. E., St Amand, T., Epstein, J. A., and Gutstein, D. E. (2006). Distinct cardiac malformations caused by absence of connexin 43 in the neural crest and in the non-crest neural tube. Development 133, 2063–2073. doi: 10.1242/dev.02374
Lo, C. W., Cohen, M. F., Huang, G. Y., Lazatin, B. O., Patel, N., Sullivan, R., et al. (1997). Cx43 gap junction gene expression and gap junctional communication in mouse neural crest cells. Dev. Genet. 20, 119–132.
Maass, K., Chase, S. E., Lin, X., and Delmar, M. (2009). Cx43 CT domain influences infarct size and susceptibility to ventricular tachyarrhythmias in acute myocardial infarction. Cardiovasc. Res. 84, 361–367. doi: 10.1093/cvr/cvp250
Maass, K., Ghanem, A., Kim, J. S., Saathoff, M., Urschel, S., Kirfel, G., et al. (2004). Defective epidermal barrier in neonatal mice lacking the C-terminal region of connexin43. Mol. Biol. Cell 15, 4597–4608. doi: 10.1091/mbc.E04-04-0324
Maass, K., Shibayama, J., Chase, S. E., Willecke, K., and Delmar, M. (2007). C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ. Res. 101, 1283–1291. doi: 10.1161/CIRCRESAHA.107.162818
Manias, J. L., Plante, I., Gong, X. Q., Shao, Q., Churko, J., Bai, D., et al. (2008). Fate of connexin43 in cardiac tissue harbouring a disease-linked connexin43 mutant. Cardiovasc. Res. 80, 385–395. doi: 10.1093/cvr/cvn203
Miquerol, L., Dupays, L., Theveniau-Ruissy, M., Alcolea, S., Jarry-Guichard, T., Abran, P., et al. (2003). Gap junctional connexins in the developing mouse cardiac conduction system. Novartis Found. Symp. 250, 80–98. discussion 98–109, 276–279.
Miquerol, L., Meysen, S., Mangoni, M., Bois, P., van Rijen, H. V., Abran, P., et al. (2004). Architectural and functional asymmetry of the His-Purkinje system of the murine heart. Cardiovasc. Res. 63, 77–86. doi: 10.1016/j.cardiores.2004.03.007
Miro-Casas, E., Ruiz-Meana, M., Agullo, E., Stahlhofen, S., Rodriguez-Sinovas, A., Cabestrero, A., et al. (2009). Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc. Res. 83, 747–756. doi: 10.1093/cvr/cvp157
Moreno, A. P., Laing, J. G., Beyer, E. C., and Spray, D. C. (1995). Properties of gap junction channels formed of connexin 45 endogenously expressed in human hepatoma (SKHep1) cells. Am. J. Physiol. 268, C356–C365.
Morley, G. E., Danik, S. B., Bernstein, S., Sun, Y., Rosner, G., Gutstein, D. E., et al. (2005). Reduced intercellular coupling leads to paradoxical propagation across the Purkinje-ventricular junction and aberrant myocardial activation. Proc. Natl. Acad. Sci. U.S.A. 102, 4126–4129. doi: 10.1073/pnas.0500881102
Morley, G. E., Vaidya, D., and Jalife, J. (2000). Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J. Cardiovasc. Electrophysiol. 11, 375–377. doi: 10.1111/j.1540-8167.2000.tb01811.x
Munger, S. J., Kanady, J. D., and Simon, A. M. (2013). Absence of venous valves in mice lacking Connexin37. Dev. Biol. 373, 338–348. doi: 10.1016/j.ydbio.2012.10.032
Munshi, N. V., McAnally, J., Bezprozvannaya, S., Berry, J. M., Richardson, J. A., Hill, J. A., et al. (2009). Cx30.2 enhancer analysis identifies Gata4 as a novel regulator of atrioventricular delay. Development 136, 2665–2674. doi: 10.1242/dev.038562
Nishii, K., Kumai, M., Egashira, K., Miwa, T., Hashizume, K., Miyano, Y., et al. (2003). Mice lacking connexin45 conditionally in cardiac myocytes display embryonic lethality similar to that of germline knockout mice without endocardial cushion defect. Cell Commun. Adhes. 10, 365–369.
Pfahnl, A., and Dahl, G. (1999). Gating of cx46 gap junction hemichannels by calcium and voltage. Pflugers Arch. 437, 345–353. doi: 10.1007/s004240050788
Pizard, A., Burgon, P. G., Paul, D. L., Bruneau, B. G., Seidman, C. E., and Seidman, J. G. (2005). Connexin 40, a target of transcription factor Tbx5, patterns wrist, digits, and sternum. Mol. Cell. Biol. 25, 5073–5083. doi: 10.1128/MCB.25.12.5073-5083.2005
Plum, A., Hallas, G., Magin, T., Dombrowski, F., Hagendorff, A., Schumacher, B., et al. (2000). Unique and shared functions of different connexins in mice. Curr. Biol. 10, 1083–1091. doi: 10.1016/S0960-9822(00)00690-4
Prestia, K. A., Sosunov, E. A., Anyukhovsky, E. P., Dolmatova, E., Kelly, C. W., Brink, P. R., et al. (2011). Increased cell-cell coupling increases infarct size and does not decrease incidence of ventricular tachycardia in mice. Front. Physiol. 2:1. doi: 10.3389/fphys.2011.00001
Rackauskas, M., Neverauskas, V., and Skeberdis, V. A. (2010). Diversity and properties of connexin gap junction channels. Medicina (Kaunas) 46, 1–12.
Reaume, A. G., De Sousa, P. A., Kulkarni, S., Langille, B. L., Zhu, D., Davies, T. C., et al. (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831–1834. doi: 10.1126/science.7892609
Reed, K. E., Westphale, E. M., Larson, D. M., Wang, H. Z., Veenstra, R. D., and Beyer, E. C. (1993). Molecular cloning and functional expression of human connexin37, an endothelial cell gap junction protein. J. Clin. Invest. 91, 997–1004. doi: 10.1172/JCI116321
Remo, B. F., Qu, J., Volpicelli, F. M., Giovannone, S., Shin, D., Lader, J., et al. (2011). Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ. Res. 108, 1459–1466. doi: 10.1161/CIRCRESAHA.111.244046
Rentschler, S., Harris, B. S., Kuznekoff, L., Jain, R., Manderfield, L., Lu, M. M., et al. (2011). Notch signaling regulates murine atrioventricular conduction and the formation of accessory pathways. J. Clin. Invest. 121, 525–533. doi: 10.1172/JCI44470
Rodriguez-Sinovas, A., Cabestrero, A., Lopez, D., Torre, I., Morente, M., Abellan, A., et al. (2007). The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog. Biophys. Mol. Biol. 94, 219–232. doi: 10.1016/j.pbiomolbio.2007.03.003
Rodriguez-Sinovas, A., Sanchez, J. A., Gonzalez-Loyola, A., Barba, I., Morente, M., Aguilar, R., et al. (2010). Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischaemia and preconditioning protection. J. Physiol. 588, 1139–1151. doi: 10.1113/jphysiol.2009.186577
Ruiz-Meana, M., Rodriguez-Sinovas, A., Cabestrero, A., Boengler, K., Heusch, G., and Garcia-Dorado, D. (2008). Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 77, 325–333. doi: 10.1093/cvr/cvm062
Salameh, A., and Dhein, S. (2011). Adrenergic control of cardiac gap junction function and expression. Naunyn Schmiedebergs Arch. Pharmacol. 383, 331–346. doi: 10.1007/s00210-011-0603-4
Sankova, B., Benes, J. Jr., Krejci, E., Dupays, L., Theveniau-Ruissy, M., Miquerol, L., et al. (2012). The effect of connexin40 deficiency on ventricular conduction system function during development. Cardiovasc. Res. 95, 469–479. doi: 10.1093/cvr/cvs210
Schrickel, J. W., Kreuzberg, M. M., Ghanem, A., Kim, J. S., Linhart, M., Andrie, R., et al. (2009). Normal impulse propagation in the atrioventricular conduction system of Cx30.2/Cx40 double deficient mice. J. Mol. Cell. Cardiol. 46, 644–652. doi: 10.1016/j.yjmcc.2009.02.012
Schwanke, U., Konietzka, I., Duschin, A., Li, X., Schulz, R., and Heusch, G. (2002). No ischemic preconditioning in heterozygous connexin43-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 283, H1740–H1742.
Shaw, R. M., and Rudy, Y. (1997). Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ. Res. 81, 727–741. doi: 10.1161/01.RES.81.5.727
Simon, A. M., Goodenough, D. A., Li, E., and Paul, D. L. (1997). Female infertility in mice lacking connexin 37. Nature 385, 525–529. doi: 10.1038/385525a0
Simon, A. M., Goodenough, D. A., and Paul, D. L. (1998). Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr. Biol. 8, 295–298. doi: 10.1016/S0960-9822(98)70113-7
Simon, A. M., McWhorter, A. R., Dones, J. A., Jackson, C. L., and Chen, H. (2004). Heart and head defects in mice lacking pairs of connexins. Dev. Biol. 265, 369–383. doi: 10.1016/j.ydbio.2003.09.036
Sizarov, A., Devalla, H. D., Anderson, R. H., Passier, R., Christoffels, V. M., and Moorman, A. F. (2011). Molecular analysis of patterning of conduction tissues in the developing human heart. Circ. Arrhythm. Electrophysiol. 4, 532–542. doi: 10.1161/CIRCEP.111.963421
Smyth, J. W., Vogan, J. M., Buch, P. J., Zhang, S. S., Fong, T. S., Hong, T. T., et al. (2012). Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circ. Res. 110, 978–989. doi: 10.1161/CIRCRESAHA.111.257964
Sohl, G., and Willecke, K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228–232. doi: 10.1016/j.cardiores.2003.11.013
Solan, J. L., and Lampe, P. D. (2009). Connexin43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261–272. doi: 10.1042/BJ20082319
Sperelakis, N. (2002). An electric field mechanism for transmission of excitation between myocardial cells. Circ. Res. 91, 985–987. doi: 10.1161/01.RES.0000045656.34731.6D
Stein, M., van Veen, T. A., Remme, C. A., Boulaksil, M., Noorman, M., van Stuijvenberg, L., et al. (2009). Combined reduction of intercellular coupling and membrane excitability differentially affects transverse and longitudinal cardiac conduction. Cardiovasc. Res. 83, 52–60. doi: 10.1093/cvr/cvp124
Tallini, Y. N., Brekke, J. F., Shui, B., Doran, R., Hwang, S. M., Nakai, J., et al. (2007). Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ. Res. 101, 1300–1309. doi: 10.1161/CIRCRESAHA.107.149484
Tamaddon, H. S., Vaidya, D., Simon, A. M., Paul, D. L., Jalife, J., and Morley, G. E. (2000). High-resolution optical mapping of the right bundle branch in connexin40 knockout mice reveals slow conduction in the specialized conduction system. Circ. Res. 87, 929–936. doi: 10.1161/01.RES.87.10.929
Thomas, S. A., Schuessler, R. B., Berul, C. I., Beardslee, M. A., Beyer, E. C., Mendelsohn, M. E., et al. (1998). Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction: evidence for chamber-specific molecular determinants of conduction. Circulation 97, 686–691. doi: 10.1161/01.CIR.97.7.686
Thomas, S. P., Kucera, J. P., Bircher-Lehmann, L., Rudy, Y., Saffitz, J. E., and Kleber, A. G. (2003). Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ. Res. 92, 1209–1216. doi: 10.1161/01.RES.0000074916.41221.EA
Traub, O., Eckert, R., Lichtenberg-Frate, H., Elfgang, C., Bastide, B., Scheidtmann, K. H., et al. (1994). Immunochemical and electrophysiological characterization of murine connexin40 and -43 in mouse tissues and transfected human cells. Eur. J. Cell Biol. 64, 101–112.
Trexler, E. B., Bennett, M. V., Bargiello, T. A., and Verselis, V. K. (1996). Voltage gating and permeation in a gap junction hemichannel. Proc. Natl. Acad. Sci. U.S.A. 93, 5836–5841. doi: 10.1073/pnas.93.12.5836
Tuomi, J. M., Tyml, K., and Jones, D. L. (2011). Atrial tachycardia/fibrillation in the connexin 43 G60S mutant (Oculodentodigital dysplasia) mouse. Am. J. Physiol. Heart Circ. Physiol. 300, H1402–H1411. doi: 10.1152/ajpheart.01094.2010
Vaidya, D., Tamaddon, H. S., Lo, C. W., Taffet, S. M., Delmar, M., Morley, G. E., et al. (2001). Null mutation of connexin43 causes slow propagation of ventricular activation in the late stages of mouse embryonic development. Circ. Res. 88, 1196–1202. doi: 10.1161/hh1101.091107
Valiunas, V., Manthey, D., Vogel, R., Willecke, K., and Weingart, R. (1999). Biophysical properties of mouse connexin30 gap junction channels studied in transfected human HeLa cells. J. Physiol. 3, 631–644. doi: 10.1111/j.1469-7793.1999.0631n.x
Valiunas, V., Weingart, R., and Brink, P. R. (2000). Formation of heterotypic gap junction channels by connexins 40 and 43. Circ. Res. 86, E42–E49. doi: 10.1161/01.RES.86.2.e42
Vanderbrink, B. A., Sellitto, C., Saba, S., Link, M. S., Zhu, W., Homoud, M. K., et al. (2000). Connexin40-deficient mice exhibit atrioventricular nodal and infra-Hisian conduction abnormalities. J. Cardiovasc. Electrophysiol. 11, 1270–1276. doi: 10.1046/j.1540-8167.2000.01270.x
van Kempen, M. J., Ten Velde, I., Wessels, A., Oosthoek, P. W., Gros, D., Jongsma, H. J., et al. (1995). Differential connexin distribution accommodates cardiac function in different species. Microsc. Res. Tech. 31, 420–436. doi: 10.1002/jemt.1070310511
van Rijen, H. V., Eckardt, D., Degen, J., Theis, M., Ott, T., Willecke, K., et al. (2004). Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 109, 1048–1055. doi: 10.1161/01.CIR.0000117402.70689.75
van Rijen, H. V., van Veen, T. A., Van Kempen, M. J., Wilms-Schopman, F. J., Potse, M., Krueger, O., et al. (2001). Impaired conduction in the bundle branches of mouse hearts lacking the gap junction protein connexin40. Circulation 103, 1591–1598. doi: 10.1161/01.CIR.103.11.1591
van Veen, T. A., Stein, M., Royer, A., Le Quang, K., Charpentier, F., Colledge, W. H., et al. (2005a). Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation 112, 1927–1935.
van Veen, T. A., van Rijen, H. V., Van Kempen, M. J., Miquerol, L., Opthof, T., Gros, D., et al. (2005b). Discontinuous conduction in mouse bundle branches is caused by bundle-branch architecture. Circulation 112, 2235–2244.
Verheijck, E. E., van Kempen, M. J., Veereschild, M., Lurvink, J., Jongsma, H. J., and Bouman, L. N. (2001). Electrophysiological features of the mouse sinoatrial node in relation to connexin distribution. Cardiovasc. Res. 52, 40–50. doi: 10.1016/S0008-6363(01)00364-9
Verheule, S., van Batenburg, C. A., Coenjaerts, F. E., Kirchhoff, S., Willecke, K., and Jongsma, H. J. (1999). Cardiac conduction abnormalities in mice lacking the gap junction protein connexin40. J. Cardiovasc. Electrophysiol. 10, 1380–1389. doi: 10.1111/j.1540-8167.1999.tb00194.x
Verheule, S., van Kempen, M. J., Postma, S., Rook, M. B., and Jongsma, H. J. (2001). Gap junctions in the rabbit sinoatrial node. Am. J. Physiol. Heart Circ. Physiol. 280, H2103–H2115.
Verheule, S., van Kempen, M. J., Te Welscher, P. H., Kwak, B. R., and Jongsma, H. J. (1997). Characterization of gap junction channels in adult rabbit atrial and ventricular myocardium. Circ. Res. 80, 673–681. doi: 10.1161/01.RES.80.5.673
Vink, M. J., Suadicani, S. O., Vieira, D. M., Urban-Maldonado, M., Gao, Y., Fishman, G. I., et al. (2004). Alterations of intercellular communication in neonatal cardiac myocytes from connexin43 null mice. Cardiovasc. Res. 62, 397–406. doi: 10.1016/j.cardiores.2004.01.015
Vozzi, C., Dupont, E., Coppen, S. R., Yeh, H. I., and Severs, N. J. (1999). Chamber-related differences in connexin expression in the human heart. J. Mol. Cell. Cardiol. 31, 991–1003. doi: 10.1006/jmcc.1999.0937
Walker, D. L., Vacha, S. J., Kirby, M. L., and Lo, C. W. (2005). Connexin43 deficiency causes dysregulation of coronary vasculogenesis. Dev. Biol. 284, 479–498. doi: 10.1016/j.ydbio.2005.06.004
Wang, N., De Vuyst, E., Ponsaerts, R., Boengler, K., Palacios-Prado, N., Wauman, J., et al. (2013). Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 108, 309. doi: 10.1007/s00395-012-0309-x
White, T. W., and Bruzzone, R. (1996). Multiple connexin proteins in single intercellular channels: connexin compatibility and functional consequences. J. Bioenerg. Biomembr. 28, 339–350. doi: 10.1007/BF02110110
Wiegerinck, R. F., Verkerk, A. O., Belterman, C. N., van Veen, T. A., Baartscheer, A., Opthof, T., et al. (2006). Larger cell size in rabbits with heart failure increases myocardial conduction velocity and QRS duration. Circulation 113, 806–813. doi: 10.1161/CIRCULATIONAHA.105.565804
Wiese, C., Grieskamp, T., Airik, R., Mommersteeg, M. T., Gardiwal, A., De Gier-De Vries, C., et al. (2009). Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ. Res. 104, 388–397. doi: 10.1161/CIRCRESAHA.108.187062
Winterhager, E., Pielensticker, N., Freyer, J., Ghanem, A., Schrickel, J. W., Kim, J. S., et al. (2007). Replacement of connexin43 by connexin26 in transgenic mice leads to dysfunctional reproductive organs and slowed ventricular conduction in the heart. BMC Dev. Biol. 7:26. doi: 10.1186/1471-213X-7-26
Wit, A. L., and Peters, N. S. (2012). The role of gap junctions in the arrhythmias of ischemia and infarction. Heart Rhythm 9, 308–311. doi: 10.1016/j.hrthm.2011.09.056
Ya, J., Erdtsieck-Ernste, E. B., De Boer, P. A., van Kempen, M. J., Jongsma, H., Gros, D., et al. (1998). Heart defects in connexin43-deficient mice. Circ. Res. 82, 360–366. doi: 10.1161/01.RES.82.3.360
Yamada, K. A., Kanter, E. M., Green, K. G., and Saffitz, J. E. (2004). Transmural distribution of connexins in rodent hearts. J. Cardiovasc. Electrophysiol. 15, 710–715. doi: 10.1046/j.1540-8167.2004.03514.x
Yamada, K. A., Rogers, J. G., Sundset, R., Steinberg, T. H., and Saffitz, J. (2003). Up-regulation of connexin45 in heart failure. J. Cardiovasc. Electrophysiol. 14, 1205–1212. doi: 10.1046/j.1540-8167.2003.03276.x
Zhang, Y., Kanter, E. M., and Yamada, K. A. (2010a). Remodeling of cardiac fibroblasts following myocardial infarction results in increased gap junction intercellular communication. Cardiovasc. Pathol. 19, e233–e240. doi: 10.1016/j.carpath.2009.12.002
Zhang, Y., Wang, H., Kovacs, A., Kanter, E. M., and Yamada, K. A. (2010b). Reduced expression of Cx43 attenuates ventricular remodeling after myocardial infarction via impaired TGF-beta signaling. Am. J. Physiol. Heart Circ. Physiol. 298, H477–H487. doi: 10.1152/ajpheart.00806.2009
Zheng-Fischhofer, Q., Ghanem, A., Kim, J. S., Kibschull, M., Schwarz, G., Schwab, J. O., et al. (2006). Connexin31 cannot functionally replace connexin43 during cardiac morphogenesis in mice. J. Cell. Sci. 119, 693–701. doi: 10.1242/jcs.02800
Keywords: gap junctions, connexins, mouse models, conduction, arrhythmias, cardiac
Citation: Verheule S and Kaese S (2013) Connexin diversity in the heart: insights from transgenic mouse models. Front. Pharmacol. 4:81. doi: 10.3389/fphar.2013.00081
Received: 15 April 2013; Paper pending published: 03 May 2013;
Accepted: 04 June 2013; Published online: 27 June 2013.
Edited by:
Aida Salameh, Heart Centre University of Leipzig, GermanyReviewed by:
Mirko Baruscotti, University of Milano, ItalyGregory E. Morley, New York University School of Medicine, USA
Copyright © 2013 Verheule and Kaese. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Sander Verheule, Department of Physiology, Universiteitssingel 50, PO Box 616, 6200 MD, Maastricht, Netherlands e-mail:cy52ZXJoZXVsZUBtYWFzdHJpY2h0dW5pdmVyc2l0eS5ubA==