

 Nan Wang
Nan Wang
- Physiology Group, Department of Basic Medical Sciences, Ghent University, Ghent, Belgium
The coordination of tissue function is mediated by gap junctions (GJs) that enable direct cell–cell transfer of metabolic and electric signals. GJs are formed by connexins of which Cx43 is most widespread in the human body. In the brain, Cx43 GJs are mostly found in astroglia where they coordinate the propagation of Ca2+ waves, spatial K+ buffering, and distribution of glucose. Beyond its role in direct intercellular communication, Cx43 also forms unapposed, non-junctional hemichannels in the plasma membrane of glial cells. These allow the passage of several neuro- and gliotransmitters that may, combined with downstream paracrine signaling, complement direct GJ communication among glial cells and sustain glial-neuronal signaling. Mutations in the GJA1 gene encoding Cx43 have been identified in a rare, mostly autosomal dominant syndrome called oculodentodigital dysplasia (ODDD). ODDD patients display a pleiotropic phenotype reflected by eye, hand, teeth, and foot abnormalities, as well as craniofacial and bone malformations. Remarkably, neurological symptoms such as dysarthria, neurogenic bladder (manifested as urinary incontinence), spasticity or muscle weakness, ataxia, and epilepsy are other prominent features observed in ODDD patients. Over 10 mutations detected in patients diagnosed with neurological disorders are associated with altered functionality of Cx43 GJs/hemichannels, but the link between ODDD-related abnormal channel activities and neurologic phenotype is still elusive. Here, we present an overview on the nature of the mutants conveying structural and functional changes of Cx43 channels and discuss available evidence for aberrant Cx43 GJ and hemichannel function. In a final step, we examine the possibilities of how channel dysfunction may lead to some of the neurological manifestations of ODDD.
Introduction: ODDD Mutations, Clinical Manifestations and Review Focus
Oculodentodigital syndrome or oculodentodigital dysplasia (ODDD, OMIM #164200) is a mostly autosomal dominant disease caused by mutations in the GJA1 gene which is located on chromosome 6 (q21-q23.2). ODDD is a rare disease (prevalence < 1/1,000,0001) and symptoms have been mostly described in Caucasian families; it is uncertain though whether this is a matter of clustering or of inconsistent screening in other populations. In the affected families, male and female patients are found in equal numbers while in sporadic forms of ODDD, females seem to be more susceptible (reviewed in Paznekas et al., 2009; Avshalumova et al., 2013).
GJA1 encodes for one of the most abundant connexin (Cx) proteins, Cx43. Cxs are a family of transmembrane proteins with molecular weights (MW) varying from 26 to 60 kilodaltons (kDa) on which the current nomenclature is based (Cx43 has a MW of ~43 kDa). In vertebrates, Cxs are the building blocks of gap junction (GJ) channels, intercellular channels that connect the cytoplasm of two neighboring cells. A GJ channel consists of two hemichannels (HCs), each composed of six Cx proteins and delivered by each of the coupled cells. Cx43 is ubiquitously present in the human body in a large array of tissues and cells (reviewed in Laird, 2006). As a result, ODDD patients exhibit a pleiotropic phenotype with manifestations in a large variety of organ systems. Externally, mostly eyes, teeth, hands and feet are affected. Typical craniofacial dysmorphisms include a thin nose with hypoplastic alae nasi, small anteverted nares and prominent columnella. Some patients additionally have dysplastic ears. Ophthalmological anomalies include microphthalmia, microcornea, iris abnormalities, cataracts, glaucoma and optical neuropathy. Malformations of the extremities are another hallmark of ODDD and include syndactyly of fingers and toes. Additionally, campylodactyly (fixed flexion deformity of fingers and toes) and clinodactyly (lateral curvature of the fingers) are frequently observed. In the oral region, mandibular overgrowth, cleft lip, and cleft palate may be present. Abnormalities in primary and permanent teeth such as microdontia, partial anodontia, enamel hypoplasia, caries, and early tooth loss are observed in most patients. Brittle nails and hair abnormalities sometimes appear and skin diseases like palmoplantar keratoderma and subclinical wound healing defects are possible as well (Paznekas et al., 2003; Gong et al., 2006; Thibodeau et al., 2010; Churko et al., 2011; Amano et al., 2012).
In addition to this large variety of physically observable features, the disease is also characterized by cardiac and neurological malfunctioning. The cardiac phenotype is observed in 3% of the ODDD patients and includes endocardial cushion defects, atrial or ventricular septum defects, recurrent ventricular tachycardia, atrioventricular block, and idiopathic atrial fibrillation (Paznekas et al., 2003, 2009). Cardiac arrhythmia caused by a Cx43 mutation (E42K) has been associated with “sudden infant death syndrome” (Van Norstrand et al., 2012), making it plausible that some of the ODDD mutations result in a cardiac phenotype. Neurological symptoms are not universally seen, but about 30% of the patient population has been diagnosed with neurological problems that include conductive hearing loss, dysarthria (speech articulation problems), neurogenic bladder (voiding problems), ataxia (gait disturbance), muscle weakness, spasticity, and seizures. MRI imaging studies have brought up diffuse bilateral abnormalities in the subcortical cerebral white matter, possibly indicating a slow progressive leukodystrophy. Mental retardation may occur but is rare (Paznekas et al., 2003, 2009; Amador et al., 2008; Joss et al., 2008; Furuta et al., 2012). Of note, in most families, the appearance of neurological symptoms is unpredictable, but in 4 families genotyped for having the L90V, L113P, K134N, or G138R mutant, all ODDD patients exhibit neurological traits (Paznekas et al., 2009). Cardiac and neurological manifestations in ODDD have only started to emerge in the past decade and the list of “new” ODDD features still seems to expand continuously. Brice and co-workers have for instance recently described a case of lymphedema in ODDD (Brice et al., 2013). Given the low prevalence, the high pleiotropy and the still growing list of previously unnoticed ODDD features, currently, there are no clear data available about the life expectancy of the patients.
In this review, we focus on the neurological manifestations of ODDD and explore how Cx43 mutations and consequent channel aberrations may link to some of these manifestations (Table 1). We start with an overview of connexins and its channels (section Connexin Channels: Gap Junctions and Hemichannels), provide an in depth analysis of the consequence of ODDD mutations on channel function (section ODDD-Linked Mutations and Cx43 Channel Function) and end by examining how channel dysfunction may lead to alterations in neural tissue functioning (section Neurological Phenotype in ODDD—Link to Aberrant Cx43 Channels). Several excellent reviews have highlighted each of these aspects (Kielian, 2008; Laird, 2008, 2010; Chew et al., 2010; Orellana et al., 2011; Abrams and Scherer, 2012; Eugenin et al., 2012); here we aimed to correlate channel dysfunction with neurological manifestations and explore possible relations in the framework of currently available knowledge.
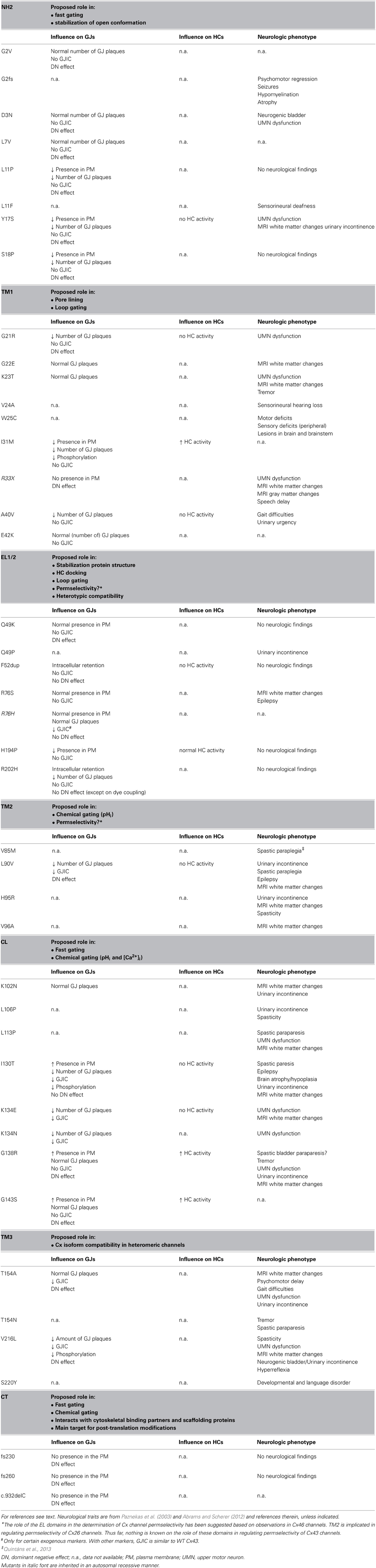
Table 1. Overview of ODDD-linked mutations in different Cx43 domains, their effect on Cx channel properties and the associated neurologic phenotypes.
Connexin Channels: Gap Junctions and Hemichannels
Cxs form two kinds of functional channels: GJs and HCs. GJs mediate the direct diffusion of ions and molecules with MWs up to 1.5 kDa, including inositol 1,4,5 trisphosphate (IP3), cyclic nucleotides, and energy molecules such as ATP (reviewed in Alexander and Goldberg, 2003), thereby contributing to the coordination of cell function in several organs and tissues. GJ channels are for instance implicated in the propagation of intercellular Ca2+ waves (ICWs) (reviewed by Leybaert and Sanderson, 2012), metabolic and electric coupling in astrocytes and cardiomyocytes (Rouach et al., 2008; Meme et al., 2009; Desplantez et al., 2012), exchange of bone modulating molecules (reviewed in Batra et al., 2012) and synchronization of smooth muscle cell contraction in the bladder and uterus (Miyoshi et al., 1996; Neuhaus et al., 2002). Moreover, GJs may also spread cell death signals to neighboring cells, thereby contributing to tissue/organ damage in pathology (Decrock et al., 2012). HCs can be present both as GJ precursors in the plasma membrane (PM) or as non-junctional channels, not incorporated into GJs. For a long time, it was thought that HCs remain closed until they form a GJ channel, since uncontrolled HC opening would lead to membrane depolarization and depletion of essential molecules from the cytoplasm, ultimately leading to cell dysfunction and possibly cell death. The first evidence of functional HCs, derived from in vitro work whereby Cx46 was expressed in Xenopus laevis oocytes, confirmed that HC opening resulted in uptake of Lucifer yellow, but also in depolarization and cell death (Paul et al., 1991). Research over the past decades has identified numerous scenarios in which HCs are activated (see section ODDD-Linked Mutations and Cx43 Channel Function). HCs have been shown to be involved in different forms of paracrine signaling through the release of ATP (Kang et al., 2008), glutamate (Ye et al., 2003), glutathione (Rana and Dringen, 2007), NAD+ (Goodenough and Paul, 2003), and prostaglandins (Jiang and Cherian, 2003; Orellana et al., 2011). HC-mediated ATP release functions as a paracrine signal in the propagation of ICWs (Leybaert and Sanderson, 2012) and evidence is accruing that HCs may additionally contribute to “center-surround” antagonism in the retina (Kamermans et al., 2001; Goodenough and Paul, 2003), osteogenesis (reviewed in Civitelli, 2008; Batra et al., 2012), regulation of vascular permeability (De Bock et al., 2011), central chemoreception (Huckstepp et al., 2010), atherosclerotic plaque formation (Wong et al., 2006), induction of astrogliosis (O'Carroll et al., 2008), ischemia-related cell death (Danesh-Meyer et al., 2008, 2012; Davidson et al., 2012; Wang et al., 2013a,b and reviewed in Contreras et al., 2004; Bargiotas et al., 2009) as well as in the propagation of apoptotic signals (Decrock et al., 2009). The role of HCs has been heavily debated since the discovery of pannexin channels, transmembrane channels that have similar tissue distribution and properties as HCs but are not likely to form GJ channels (Spray et al., 2006; Iglesias et al., 2009; Scemes, 2012). Much of the published data on the possible role of both HCs and pannexin channels is based on indirect measures that might be prone to misinterpretation and these issues are considered in detail in another review in this Frontiers Research Topic (Giaume et al., 2013).
Connexin Life Cycle and Channel Assembly
Because of the relatively short Cx half-life (1–6 h), there is a continuous synthesis and breakdown of the protein, enabling fast adaptation of GJ intercellular communication (GJIC) to the physiological needs of the tissue (reviewed in Herve et al., 2007; Rackauskas et al., 2010). This is for instance illustrated in the myometrium, where steroid hormones control the expression level of Cxs before and after parturition (Risek et al., 1990). Like most transmembrane proteins, Cxs are co-translationally integrated into the rough endoplasmic reticulum (ER) membrane where they adopt their native transmembrane configuration (Falk, 2000; Vanslyke et al., 2009). Hydropathy plots reveal that all Cx proteins share a common topology: four transmembrane alpha-helices (TM1-4) are connected through two extracellular loops (EL1-2). In each loop, three cysteine residues form intramolecular disulphide bonds that are required for appropriate folding of the protein (Dahl et al., 1992; Foote et al., 1998). The Cx protein further contains three intracellular domains: a cytoplasmic loop (CL), an amino-terminal domain (NT), and the carboxyl-terminal region (CT). The best conserved protein regions are the ELs and the TM domains, whereas the CT- and CL-regions show marked divergence (reviewed in Nicholson, 2003; Saez et al., 2003; Wei et al., 2004). The subsequent oligomerization of Cx proteins into HCs starts in the ER, progressing to the trans-Golgi network (Falk, 2000; Vanslyke et al., 2009). During this process, newly synthesized HCs remain closed in order to protect and maintain the integrity of the lumens of the intracellular compartments (Moreno, 2005; Laird, 2006).
After leaving the ER, Cxs first pass the ER-Golgi intermediate compartment and then transit through the cis- and trans-Golgi network before being shuttled to the PM. Pleomorphic vesicles and tubular extensions departing from the Golgi apparatus contribute to the delivery of HCs to the PM (Gaietta et al., 2002; Laird, 2006). It is not entirely clear to what extent post-translational modifications are required for Cx transport to the PM. Some data suggest that Cx43 is transiently phosphorylated early in the secretory pathway, but the vast majority of phosphorylation is thought to occur at the cell surface (Lampe et al., 2000; Solan and Lampe, 2007 and reviewed in Lampe and Lau, 2004), where it plays a complex role in Cx channel gating (see further). Notably, once inserted into the cell membrane, HCs undergo specific adhesions and dock with HCs of neighboring cells to form GJs. Hundreds to thousands of GJ channels cluster in plaques at the cell–cell interface with newly formed GJs located at the border of the plaque and older channels located centrally. These central channels are targeted for internalization and degradation of GJ channels (reviewed in Goodenough and Paul, 2003; Laird, 2006).
Internalization is mediated through large double-membrane bound vesicles that contain a complete GJ channel. These vesicles are called “annular junctions” or “connexosomes” (Gaietta et al., 2002; Laird, 2006) and also contain Cx-binding proteins and molecules that function as chaperones for internalization and intracellular degradation through the proteasomal or lysosomal pathway (Qin et al., 2003; Thevenin et al., 2013). Cxs have been shown to be a substrate for ubiquitination that is known to guide proteins to proteasomes. Connexosomes are identified in the majority of cell types, but are for instance difficult to find in hepatocytes. It therefore remains possible that Cxs are additionally internalized and degraded through the endosomal cascade. Furthermore, internalization of Cx43 has been suggested to occur via a clathrin-mediated, caveolae-dependent process. Clearly, more studies are required to investigate all possible pathways of internalization/degradation. The proteasomal pathway may be responsible for ER-associated degradation, while lysosomes degrade Cxs that are recycled to the PM trough endosomes (Laird, 2006).
ODDD-Linked Mutations and Cx43 Channel Function
Cx43 Mutations and Channel Gating
Collectively, there are over 62, mostly missense, mutations found throughout the expanse of the Cx43 protein that are associated with ODDD. All Cx domains have specific functions, be it in Cx trafficking, Cx assembly or channel gating. In contrast to “benign” polymorphisms that have no discernible effect on Cx43 channel function, a minority of mutants promote channel opening/function; however, most mutants carry loss-of-function mutations and may involve those leading to inappropriate membrane sorting, those leading to improper folding, interfering with the HC docking process, and those giving rise to altered permselectivity or gating. The latter implies a fast and reversible shift in the channel's conductive properties which can be due to conformational changes or which can be mediated by Cx-linked adaptor molecules and proteins (Bukauskas et al., 2000; Cottrell et al., 2003; Herve et al., 2007; Moreno and Lau, 2007).
Cx channels—both GJs and HCs—are sensitive to changes in voltage, intracellular pH and [Ca2+]i, allowing swift adaptations (faster than those brought about by assembly/disassembly) of GJIC and paracrine signaling to comply to the specific needs of the tissue. In terms of voltage-dependent channel gating, GJs are influenced mainly by the transjunctional voltage (Vj) which defines the difference in voltage measured between two coupled cells. In contrast, HCs are highly sensitive to Vm: they remain preferentially “silent” at inside negative potentials but are activated upon depolarization by a gating mechanism that resembles gating transitions associated with the docking of extracellular loop domains; therefore this gating is also referred to as loop gating (or slow gating) (Trexler et al., 1996; Bukauskas and Verselis, 2004; Gonzalez et al., 2006; Verselis et al., 2009). Loop gating represents transitions between the fully open and closed states whereas fast (<1 ms) HC gating involves transitions to long-lasting substates. Chemical gating, achieved by changes in the intra- or extracellular environment, much resembles gating transitions observed with slow voltage gating; therefore, it has been proposed that both phenomena are mediated by the same mechanisms.
Cx43 mutations are found in all protein domains, each of which has been associated with specific functions in terms of channel oligomerization, trafficking, gating and permeability. Importantly, many of the known mutations causing ODDD occur in residues that are highly conserved throughout the animal kingdom (Paznekas et al., 2003), hinting toward their importance in channel regulation. Below, we discuss the different Cx43 mutations that have been characterized in terms of trafficking, channel assembly and channel function. Unfortunately though, for most mutant channels there is not yet a clear structural-functional correlation that would allow a better understanding of what is going wrong with the channels. The effects of the Cx43 mutations are often not predictable from their location in the primary structure and mutations located at different domains may cause similar channel dysfunction phenotypes, ultimately leading to the ODDD phenotype.
N-Terminal Cx43 Mutations
Deletion of the first N-terminal amino acids (AAs 2–7) disables Cx43 trafficking and most of the Cx43 protein is retained in the intracellular compartment. Overall, the Y17S mutation has similar effects (Shao et al., 2012), although a few plaque-like structures remained observable at the PM (Shibayama et al., 2005; Lai et al., 2006). Nevertheless, given the problematic trafficking of this mutant, neither HC activity nor GJIC was detected (Lai et al., 2006). Unfortunately, fairly little is known on the role of the Cx43 N-terminal domain in terms of oligomerization or trafficking. Most of the other N-terminal missense mutations, including G2V, D3N, L7V, L11P, and S18P, do allow a normal Cx life cycle, but result in non-functional GJs, as evidenced by the lack of dye transfer and the absence of electrical coupling (Shibayama et al., 2005; Churko et al., 2011; Shao et al., 2012). In these mutant channels, the closed state is thus relatively more stable than the open one. Importantly, these mutant proteins exert a dominant negative effect on WT Cx43 in terms of coupling, indicating that the patients are likely experiencing more than 50% loss of normal Cx43 function, despite having one functional allele (Shao et al., 2012).
A hydrophobic core built around W4 has been put forward as a structural determinant of the Cx43 N-terminal domain, governing interactions with the TM1 domain. Replacement of G2 with a valine residue was suggested to introduce additional hydrophobic interactions, not present in the WT channel that would abolish NT-TM1 interactions (Shao et al., 2012). Based on electron crystallography of Cx26 channels and electrophysiological studies performed in Cx46 channels, interactions between the NT and the TM1 domains are suggested to keep the channel in the open conformation (Maeda et al., 2009; Kronengold et al., 2012). Preventing NT-TM1 interactions may thus be one mechanism by which the mutants result in non-functional GJ channels.
Cx43 Mutations in the Transmembrane Domains
The crystal structure of a C-terminally truncated Cx43 channel at 7.5 Å resolution indicates the presence of 24 transmembrane α-helices within each hemichannel, grouped in two concentric rings around the central pore (Unger et al., 1997, 1999); however, there has been controversy regarding the identities of the principal pore-lining segments. The substituted cysteine accessibility method (SCAM) that enables the identification of pore lining residues, has pointed out that TM3 might delineate the inner pore of Cx32 GJs (Skerrett et al., 2002); however, currently, most evidence points to the amphipathic TM1 as the major pore-lining domain in channels composed of Cx46 (Zhou et al., 1997; Kronengold et al., 2003a,b), Cx50 (Verselis et al., 2009) Cx32 (Oh et al., 1997; Tang et al., 2009) and Cx26 (Sanchez et al., 2010). The crystal structure of a Cx26 channel at 3.5 Å resolution confirms that TM1, but also TM2, are part of the inner lumen wall, whereas TM3 and TM4 face the hydrophobic environment (Maeda et al., 2009).
In TM1, mutants G21R, G22E, K23T, I31M, R33X, A40V, and E42K have all been identified in ODDD patients. Of these G21R, G22E, K23T, and E42K [the latter causing sudden infant death syndrome (Van Norstrand et al., 2012)], encounter normal trafficking to the PM and insertion into GJ plaques; yet cells are not electrically coupled (data on channel function of G22E and K23T mutant channels are still lacking) (Roscoe et al., 2005; Shibayama et al., 2005; Abrams and Scherer, 2012; Van Norstrand et al., 2012; Huang et al., 2013). When the Cx43-G21R mutant is co-expressed with endogenous WT Cx43, it negatively influences dye coupling (Shibayama et al., 2005). Both I31M and A40V mutants exhibit disturbed membrane insertion with just a fraction of the channels present in GJ plaques; these were, however, not sufficient to sustain GJIC (Shibayama et al., 2005; Dobrowolski et al., 2007). With respect to HC activity, G21R and A40V do not form functional HCs but I31M mutant channels release more ATP compared to the Cx43 WT. The half-life of the I31M mutant was not prolonged pointing to altered gating mechanisms or a shift in permselectivity (Lai et al., 2006; Dobrowolski et al., 2007). Additionally, Cx43-I31M presents with reduced phosphorylation that is, up to a certain degree, required for proper GJIC (Dobrowolski et al., 2007). This is quite surprising as Cx43 has thus far not been shown to be phosphorylated in transmembrane domains (reviewed in Johnstone et al., 2012; Marquez-Rosado et al., 2012). In addition, isoleucine residues are not generally direct targets for phosphorylation. Thus, it is more likely that a phosphorylation in TM1 is associated with conformational changes that somehow mask candidate phosphorylation sites in other protein domains.
Known mutations located in TM2 (L90V), TM3 (T154A), or TM4 (V216L) all form channels that are present as punctae in the PM, but the coupling capacity of each mutant channel is reduced. All mutants furthermore exert dominant negative effects on WT Cx43 when in a heteromeric or heterotypic conformation (McLachlan et al., 2005; Shibayama et al., 2005; Beahm et al., 2006; Churko et al., 2011). The H95R mutant (TM2) has additionally been described in an ODDD patient, but unfortunately, the manuscript provided no functional data (Honkaniemi et al., 2005). Earlier data indicate that this residue is involved in pH sensitivity of Cx43 channels (Ek et al., 1994): introducing a negative or positive charge at position 95 yields GJ channels that exhibit reduced or increased sensitivity to intracellular acidification, respectively. Finally, residues located in TM2 have been proposed to play a role in determining permselectivity. Permeation of the Ca2+ mobilizing messenger IP3 for instance, is abolished in GJ channels composed of the mutant Cx26-V84L which is associated with hereditary deafness. The mutant protein has, however, no effect on unitary conductance and permeability to Lucifer yellow, which remain indistinguishable from WT Cx26 (Beltramello et al., 2005). This suggests that subtle structural modifications in the channel pore may selectively hinder the passage of biologically relevant molecules, possibly by altering certain interactions between the permeant and the channel pore. As this valine is highly conserved (V85 in Cx43) it may well be involved in determining IP3 transfer through Cx43 channels. ODDD patients carrying a V85M mutant were recently identified (Fenwick et al., 2008; Quintáns et al., 2013), but again, the implications of this mutant on channel trafficking/gating/permselectivity were not studied.
Cx43 Mutations in the Extracellular Loops
The extracellular loops are characterized by conserved, cysteine-rich patterns (CX6CX3C in EL1 and CX4/5CX5C in EL2), and intramolecular disulphide bridges in and between the ELs stabilize the protein's tertiary structure (Dahl et al., 1992; Bruzzone et al., 1996; Foote et al., 1998). In Cx43 these involve C54, C61 and C65 in EL1 and C187, C192 and C198 in EL2. Mutations of each of these cysteines could result in distorted loops resulting in improper protein folding and retention in intracellular compartments. Although mutations of conserved cysteines have not been associated with ODDD, two mutations that lie in direct proximity to conserved cysteine residues (F52dup and Cx43-R202H) cannot be traced back to the PM (Shibayama et al., 2005; Lai et al., 2006). Other mutants (Q49K, R76S/H, H194P) are inserted into the lipid bilayer (Sokolova et al., 2002; McLachlan et al., 2005; Shibayama et al., 2005; Dobrowolski et al., 2007; Abrams and Scherer, 2012; Huang et al., 2013); yet, electrical coupling is abolished (Sokolova et al., 2002; McLachlan et al., 2005). Based on a model proposed by Foote et al. in which the interdigitation of apposed ELs is required for docking and thus GJ formation (Foote et al., 1998), even those mutants that are present in the PM, may not form GJs due to distorted loops. This is further supported by the fact that HC activity of Cys-less Cx43 mutant channels appears normal both in vitro and in vivo (Bao et al., 2004; Tong et al., 2007). Also for ODDD-linked mutants like H194P in which the insertion of a proline induces a kink in the EL2 domain, HC activity is normal (Dobrowolski et al., 2007). In addition, those mutant proteins that cannot traffic to the PM in a homomeric configuration, can do so when assembled in a heteromeric channel with WT Cx43 (Shibayama et al., 2005; Lai et al., 2006). Interestingly, main and subconductance states of these WT:R202H and WT:F52dup channels were not different from homomeric WT channels (Shibayama et al., 2005). The single channel conductance of Cx43-R76H channels was substantially reduced compared to WT Cx43 channels (Huang et al., 2013). Slow voltage gating of Cx channels generally involves the coordinated response of all Cx monomers in the channel (Bukauskas and Verselis, 2004; Kwon et al., 2012). This might explain why a remaining electrical coupling is observed in heteromeric WT:R202H channels. At the same time, however, the mutant proteins may narrow the pore as compared to homomeric WT channels, preventing the diffusion of larger molecules like Lucifer yellow while leaving its electrical conduction intact (Shibayama et al., 2005).
Cx43 Mutations Located in the Cytoplasmic Loop
It is now firmly established that the CL domain is of utmost importance for Cx channel gating. It has for instance been shown to contain the preferred interaction site for calmodulin (CaM; region 136–158) and as such, to mediate the closure of GJ channels in response to increasing [Ca2+]i (Zhou et al., 2007; Myllykoski et al., 2009). Indeed, Ca2+-induced closure of GJs is likely to be mediated by intracellular effector proteins since the uncoupled state may persist long after [Ca2+]i has been restored (Cotrina et al., 1998; Lurtz and Louis, 2003, 2007). Even more important, it is now widely accepted that the L2 region (second half of the CL; amino acids 119–144) serves as a receptor domain for the CT and that this CT-CL interaction, also known as the ball-and-chain mechanism, is implicated in both voltage gating and chemical gating of GJs by intracellular pH changes (Ek et al., 1994; Ek-Vitorin et al., 1996; Bukauskas et al., 2000; Anumonwo et al., 2001; Moreno et al., 2002; Shibayama et al., 2006). A CT-CL interaction is also important for the regulation of Cx43 HCs by extracellular Ca2+, since a conformational change induced by an increase of extracellular Ca2+ masks the CT from antibody binding, which is most likely to be the result of its association with the “receptor”-domain (Liu et al., 2006). The modulation of HCs in response to changes in [Ca2+]i has only been outlined over the last decade, demonstrating that HCs are differently influenced by [Ca2+]i as compared to GJs. While GJs generally close with a [Ca2+]i elevation, HCs display a bimodal response: a moderate increase in [Ca2+]i up to 500 nM strongly promotes HC opening while this effect disappears with [Ca2+]i > 500 nM. Further increases in [Ca2+]i to the micromolar level tend to close the HCs (Shintani-Ishida et al., 2007; De Vuyst et al., 2009; Wang et al., 2012a). Mechanistically, Ca2+-activation of Cx43 HCs is mediated by CaM-dependent signaling (De Vuyst et al., 2009) and necessitates a CT-CL interaction (Ponsaerts et al., 2010). Most notably, the necessity of CT-CL interaction to trigger opening of Cx43 HCs stands in stark contrast to the fact that such interaction results in closure of GJs (reviewed in Iyyathurai et al., 2013). Addition of a CT-mimicking peptide prevented HC closure at high [Ca2+]i and restored HC activity of CT truncated Cx43 (Ponsaerts et al., 2010), while not affecting HC activation by modest (<500 nM) [Ca2+]i.
Several ODDD-associated mutations have been identified in the CL domain, most of which are located in the L2 region. Cx43-I130T HCs are transported to the PM and assemble into GJ plaques. Moreover, mutant levels in the PM exceed those of WT Cx43 which might indicate a reduced degree of protein degradation. Despite of this, the mutant exhibits reduced dye and electrical coupling and HC activity is disrupted (Shibayama et al., 2005; Lai et al., 2006; Kalcheva et al., 2007). K134E/N mutants show a reduced degree of plaque formation and electrical coupling and a decrease in unitary conductance (Shibayama et al., 2005). Both I130 and K134, forming ionic bonds with C-terminal aspartate residues, are suggested to be involved in CT-CL interactions (Seki et al., 2004; Hirst-Jensen et al., 2007). As discussed higher, CT-CL interactions are necessary for HC opening while they result in closure of GJs. However, I130T and K134E mutant channels are closed in both the GJ and HC configuration, indicating that mechanisms different from altered CT-CL interactions may contribute to the closure of GJs.
Two CL mutations located further downstream in the L2 region (G138R and G143S), result in GJ channels that fail to sustain electrical or dye coupling and have a dominant negative effect on WT or endogenous (NRK—normal rat kidney—cells) Cx43. Interestingly, HC activity, measured by dye uptake and ATP release, is increased in both mutants (Roscoe et al., 2005; Dobrowolski et al., 2007, 2008). It is possible that the differential effects of these mutants on GJs and HCs mutants result from reinforced CT-CL interactions, caused by stronger electrostatic interactions between the CL equipped with an additional positively charged arginine residue (G138R), with negatively charged residues in the CT (Gong et al., 2007). Likewise, a shift from a non-polar glycine to a polar serine residue (G143S) may account for a higher number of hydrogen bonds that lock the CT to the CL. Further work is needed to substantiate these possibilities.
C-Terminal Cx43 Mutations
As highlighted above, the CT functions as a gating particle that alters its state in response to changes in the extra-or intracellular environment. The CT domain is additionally the primary target for post-translational modifications like S-nitrosylation (Retamal et al., 2006) and phosphorylation (reviewed by Johnstone et al., 2012). The majority of phosphorylation events occur on serine residues although tyrosine phosphorylation is abundant as well (Lampe and Lau, 2004; Solan and Lampe, 2007). Phosphorylation of Cx proteins seems to be intricately involved in Cx trafficking to the PM. Additionally, both under basal and stimulated conditions, Cx channel activity appears to be regulated by ongoing phosphorylation-dephosphorylation events. Substitution of C-terminal serines (S325, S328, S330) by alanines has indicated that phosphorylation of these serines is required for a fully open state of Cx43 GJs while phosphorylation of other residues (S255, S279, S282, S368, Y247, and Y265) favors channel closure (Ek-Vitorin and Burt, 2013). However, much of the details on how Cx phosphorylation can determine trafficking, turnover and the activity state of HCs and GJs still remains to be resolved (Johnstone et al., 2012). One detailed example of Cx channel regulation by phosphorylation is phosphorylation of the Cx43 CT tail by pH-dependent kinases which might introduce negative charges at this site, potentiating CT-CL interaction (Yahuaca et al., 2000).
Although the CT is the primary interaction domain of Cx-associated partner proteins like zonula occludens 1, tubulin, microtubules, and caveolins that may regulate protein trafficking and function (Giepmans et al., 2001; Langlois et al., 2008; Saidi Brikci-Nigassa et al., 2012); work with CT-truncated Cx43 mutants has repetitively shown that CT-truncated proteins are present at the PM of mammalian cells (Unger et al., 1999; Moreno et al., 2002; Kang et al., 2006; Maass et al., 2007). Oppositely, most of the ODDD-linked CT mutations are not inserted in the PM: the frame shift mutations fs230 and fs260 result in preliminary truncated Cx43 and present with reduced ability to form plaques while having a dominant negative effect on WT Cxs with respect to trafficking (Lai et al., 2006; Gong et al., 2007; Churko et al., 2011). In the atrial tissue of a patient with idiopathic atrial fibrillation, a frame shift mutation caused by a single nucleotide deletion (c.932delC) was identified. The mutation renders a Cx43 protein exhibiting aberrant CT amino acids starting from position 346 followed by a premature stop codon, leading to prompt truncation. The protein remains intracellular and exerts a dominant negative effect on wild type Cx43 (as well as Cx40) in the atrial tissue (Thibodeau et al., 2010). Importantly, all previously reported work with CT-truncated mutants was indeed based on exogenous expression systems and the c.932delC mutant was similarly observed at the PM when exogenously expressed in HeLa cells where it even sustained dye coupling, though to a smaller extent than WT Cx43 (Hong et al., 2010). Another major difference between exogenous CT-truncated mutants and the ODDD mutants is that in the former the CT is absent while with frame shifts and single nucleotide deletion mutants, a CT is still (partly) present, albeit with a wrong amino acid sequence which could give a different outcome on trafficking.
Neurological Phenotype in ODDD - Link to Aberrant Cx43 Channels
Apart from the physical appearances linked to ODDD, several of the above described mutations have been associated with seizures, spasticity, gait difficulties, tremor and incontinence. These symptoms are for the bigger part clinical manifestations of underlying neuronal damage in the central nervous system (CNS). Patients may also suffer from hearing loss and decreased visual acuity or blindness; however, the latter two phenotypes are infrequent and only observed in a small fraction of the patients presenting with neurological traits. Magnetic resonance imaging (MRI) and computed tomography (CT) scans of patients' brains revealed abnormalities in both white and gray matter (Loddenkemper et al., 2002; Amador et al., 2008; Joss et al., 2008; Paznekas et al., 2009; Alao et al., 2010; Abrams and Scherer, 2012). One patient was reported to exhibit central or sensorineural hearing loss which is associated with damaged cranial nerve VIII (cochlear part) that transmits signals from the cochlea to the inner ear. Loss of visual acuity (decreased sharp vision) or blindness result from atrophy of the optic nerve that relays visual information from the retina (via the thalamus) to the visual cortex. Similar loss of visual acuity follows optical nerve neuritis observed in the demyelinating disease multiple sclerosis (Florio and Maniscalco, 2011). Spasticity is a muscle control disorder characterized by stiff, uncontrollable muscles with hyperreactivity toward exogenous stimulation, and is generally associated with hyperactive, persisting reflexes (hyperreflexia). Tremor is characterized by involuntary rhythmic muscle contractions. According to the alpha/gamma-coactivation theory, muscle contraction is controlled by both alpha and gamma motor neurons. The alpha component delivers a feed forward command resulting in force delivery by activating extrafusal muscle fibers. The gamma motor component controls the length of muscle spindles (intrafusal muscle fibers), thereby delivering input to a servo-controlled neuronal circuit that receives feedback on the length status from the sensory output of muscle spindles. Spasticity may develop as a result of aberrantly increased gamma motor neuron activity, resulting in increased tension in the muscle spindles that become oversensitive to external stimulation. Increased gamma motor drive may occur as a consequence of decreased central (cerebral and cerebellar) inhibitory input on spinal gamma motor neurons (reviewed in Sheean and McGuire, 2009). Ataxia is a cerebellar phenomenon that manifests as a lack of voluntary muscle coordination, typically observed as disturbances in the gait pattern. Interestingly, one patient carrying the W25C (TM1) mutation presented with all characteristic craniofacial features and extremity anomalies, as well as with spastic tetraparesis, hyperreflexia and sensory disturbances (numbness in feet), indicating wide-spread sensori-motor neurological deficits (Furuta et al., 2012). In two siblings characterized for having an R33X mutation, myelination deficits were described (Joss et al., 2008). The other major neurological deficit, namely seizure activity, results from a repetitive and synchronized, excessive neuronal activity in the CNS. Epilepsy has long been viewed as a channelopathy and involves epileptogenic (genesis of the disease) and ictogenic (genesis of an epileptic ictus or seizure) components. Currently, our understanding of epileptogenesis is very incomplete, at least at a mechanistic level. Ictogenesis is related to an imbalance between glutamatergic excitation and (mostly) GABA-ergic inhibition, and has been associated with dysfunctional channels, mostly voltage-gated Na+, K+, Cl−, and Ca2+ channels that determine neuronal action potential firing. Moreover, the extracellular concentration of Na+, K+, Cl−, and Ca2+ may influence the glial cells and thereby exert control over neuronal activity. It is hypothesized that dysfunctional Na+, K+, Cl−, or Ca2+ channels alter the threshold for neuronal depolarization and action potential firing, thereby shifting the balance between excitation and inhibition. At the microscopic level, epileptic brain regions are characterized by injured neurons, gliosis, axonal sprouting, and the formation of new, aberrant, synaptic connections (reviewed in D'Ambrosio, 2004; Dichter, 2009; Reid et al., 2009).
Cx43 is abundant in astrocytes and can additionally be found in activated microglia, developing neurons, and endothelial cells (Orellana et al., 2009, 2011; Avila et al., 2011; Wang et al., 2012b) (Figure 1). Given its ubiquitous presence, it is not surprising that around 30% of the ODDD patients exhibit neurological symptoms. This number may further increase as neurological investigations of ODDD patients are becoming progressively more detailed.
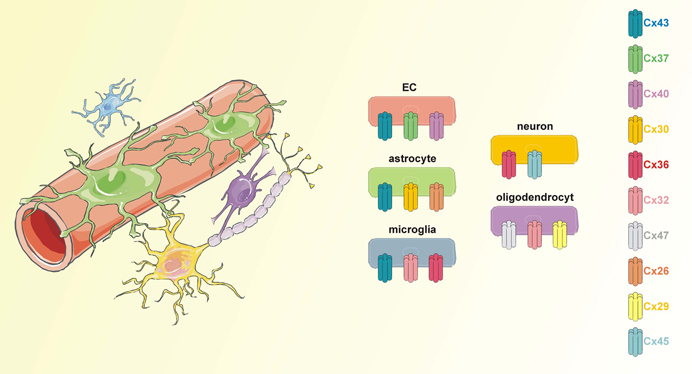
Figure 1. Connexins in the neurogliovascular unit. The neurogliovascular unit is a functional unit in which astrocytes are centrally involved. Astrocytic endfeet are tightly associated with endothelial cells that form the BBB while at their other site, astrocytes contact pre- and postsynaptic neuronal membranes. Additionally, astrocytes may contact other glial cells like microglia and oligodendrocytes. Each of the cell types in the neurogliovascular unit is endowed with a set of connexins proteins. Importantly, Cx43 is present in astrocytes as well as in BBB endothelial cells and microglial cells.
ODDD-Associated Mutations and Their Implications for Astrocyte Functioning in the Neurogliovascular Unit
Despite being originally described as “brain glue,” established functions of glia nowadays suggest an active role in brain function and information processing (reviewed in Allen and Barres, 2009). Together with neurons, glial cells work in concert with vascular and blood cells to establish the neurovascular unit, which should better be called the neurogliovascular unit (Figure 1). The concept of a neurogliovascular unit has been central in exploring improved strategies and approaches to treat stroke patients (reviewed in Berezowski et al., 2012; Kur et al., 2012).
Astrocytes are involved in key aspects of the nervous tissue including preservation of blood-brain barrier (BBB) integrity, modulation of blood supply, maintenance of brain homeostasis, myelination, and neurotransmission. The combined ablation of Cx30 and Cx43, which results in coupling-deficient astrocytes, lowers the threshold for epileptiform events (Theis et al., 2003; Wallraff et al., 2006), alters astrocyte energy metabolism (Rouach et al., 2008), gives rise to swollen astrocytic endfeet (Ezan et al., 2012) and leads to parenchymal vacuolation (Lutz et al., 2012). Astrocyte-targeted deletion of Cx43 severely reduces cell–cell coupling although it is not completely abolished, likely due to compensatory actions of Cx30 (Theis et al., 2003; Wallraff et al., 2006; Unger et al., 2012). Astrocyte-specific Cx43-ablated mice have been shown to exhibit reduced motor performance (Frisch et al., 2003), similar to ODDD patients suffering from cerebellar ataxia; yet, astrocyte-targeted deletion of Cx43 does not affect viability or astrocyte morphology nor does it cause neurodegeneration or astrogliosis (Theis et al., 2003). Conclusions from knock-out studies should be drawn with caution, as recent work in the heart has indicated that transgenic animals with a 5 amino acid deletion of the Cx43 CT have a much stronger cardiac phenotype than those with a complete deletion of the CT (Lubkemeier et al., 2013). In addition, mice missing one copy of the Cx43 allele do not mimic ODDD (Paznekas et al., 2009).
Below we describe how aberrant astrocytic signaling, due to defective Cx43, may contribute to ODDD-linked neurological symptoms.
Connexins and astrocytes contribute to blood-brain barrier function
Proper electrical signaling in the CNS requires a strict composition of the microenvironment around synapses and axons. The composition of the brain interstitial fluid is largely maintained by capillary endothelial cells that constitute the blood-brain barrier (BBB) and that survey solute diffusion in and out the brain (a highly comparable barrier is present between the capillaries and nervous tissue in the spinal cord). Astrocytic endfeet nearly completely enwrap these capillary endothelial cells, inducing and maintaining barrier properties. Cx43 may contribute to astrocyte maintenance of the endothelial barrier, as mice lacking Cx30 and astroglial Cx43 have a weakened BBB that is more vulnerable to hydrostatic vascular pressure and shear stress but that is otherwise intact in the absence of a pathological insult (Ezan et al., 2012; Lutz et al., 2012). It is not known whether this is mediated by the Cx protein itself or by an effect related to GJs or HCs. It is equally unknown whether a putative GJ involvement would be limited to GJs between astrocytic endfeet or between endfeet and endothelial cells. In vitro evidence has suggested astrocyte-endothelial communication via GJs (as well as HCs) (reviewed in Braet et al., 2001), but in vivo evidence argues against this possibility (Simard et al., 2003).
In a first approximation, it is conceivable that Cx43 mutations that result in a trafficking defect or loss of channel function may give similar manifestations as Cx43 silencing. BBB leakage enables the entry of potentially neurotoxic, circulating compounds that directly affect neuronal survival, but also gives rise to inflammation, edema and hypoxia, which will ultimately result in additional injury to the neural tissue. At the same time, the ionic homeostasis of the brain's interstitial compartment becomes disturbed, particularly the increased K+ concentration will interfere with action potential propagation and synaptic transmission. Additionally, influx of albumin and other circulating compounds can cause astrogliosis, characterized by the upregulation of GFAP (glial fibrillary acidic protein), astrocyte swelling and proliferation (David et al., 2009; Das et al., 2012), and promote extracellular K+ accumulation by indirect effects (Ivens et al., 2007; Janigro, 2012), favoring seizural activity by mechanisms described below.
Connexins and astrocytes control nutrient supply to the nervous tissue
While astrocytic endfeet contact the BBB endothelium and its associated basement membrane at one side, at the other end, astrocytic processes project toward pre- and postsynaptic neuronal membranes forming the “tripartite synapse” (Allen and Barres, 2009). Here, astrocytes exert homeostatic control over neural network excitability: they provide neurons with energy and substrates for neurotransmission while removing excessive neurotransmitters and K+ from the extracellular space by spatial buffering and siphoning. As such, neurons are protected from energy deprivation, large depolarisations and hyperexcitation that would eventually lead to neuronal death.
Astrocytes control blood flow and nutrient (oxygen and glucose) supply to those regions where neuronal activity is high by a process termed functional hyperemia or neurovascular coupling that acts at the level of cerebral arterioles and possibly also pericytes (Zonta et al., 2003 and reviewed in Iadecola and Nedergaard, 2007; Attwell et al., 2010; Petzold and Murthy, 2011). Unlike peripheral arterioles where the vasomotor response is propagated along smooth muscle cells, these do not seem to play a role in the regulation of blood flow in the CNS. Indeed, Xu et al. (2008) have shown that selective astrocyte death abolishes vasodilation despite the continued presence of viable smooth muscle cells (Xu et al., 2008). Although less documented, the vasomodulatory action of astrocytes can also imply a vasoconstriction (Mulligan and MacVicar, 2004; Metea and Newman, 2006; Filosa and Blanco, 2007; Gordon et al., 2008). Candidate astrocyte messengers mediating the vasomotor response include K+ as well as gliotransmitters (glutamate, ATP, adenosine, and NO) and arachidonic acid metabolites (reviewed in Mulligan and MacVicar, 2004; Koehler et al., 2006; Iadecola and Nedergaard, 2007; Attwell et al., 2010). A Cx43 mimetic peptide that prevents both HC opening and GJIC, has been shown to prevent neurovascular coupling (Xu et al., 2008); however, it remains to be established exactly how Cx channels contribute to neurovascular coupling. Likely, Cx channels mediate the propagation of ICWs between astrocytes, possibly spreading the vasomotor response. Increased blood flow in local parenchymal capillaries requires for example an upstream vasodilation in arterioles and pial vessels, which might be communicated by Cx-based astrocytic ICWs. ICWs rely on both a direct communication of Ca2+ mobilizing messengers mediated by GJs and an paracrine route involving the release of a Ca2+ mobilizing messenger in the extracellular space (Leybaert and Sanderson, 2012). Astrocytic Ca2+ changes play a key role in neurovascular coupling (Takano et al., 2006), with rises in [Ca2+]i along the path of the Ca2+ wave enabling the release of vasoactive messengers (Zonta et al., 2003; Mulligan and MacVicar, 2004). Additionally, HCs may provide a release pathway for vasoactive substances, yet this remains to be determined. Defective GJIC or HC responses are expected to disable ICWs and neurovascular coupling, resulting in a relative oxygen and glucose shortage during neuronal activity that may lead to convulsions and, on a longer term, to neuronal cell death. Chronically disturbed neurovascular coupling may lead to neurodegeneration (Zlokovic, 2011; Lasta et al., 2013) and this may be a possible mechanism at the basis of the motor deficiencies observed in ODDD patients.
Connexins and astrocyte modulation of synaptic transmission
Neuronal action potential firing is always accompanied by an increase of K+ levels in the extracellular space. Astrocytes buffer these excessive K+ ions through inward rectifier K+ channels (Kir4.1) aided by Na+/K+ pump activity and passive uptake with water through aquaporin 4 channels. Altered activity of both K+ channels and Na+ channels in the astrocyte PM are considered pro-epileptic (D'Ambrosio, 2004). Subsequently, K+ is redistributed over networks of GJ-connected astrocytes (reviewed in Carlen, 2012; Steinhauser and Seifert, 2012). Like K+, glutamate, the brain's major excitatory neurotransmitter, is taken up and diluted in those astrocytic networks during synaptic activity, keeping its levels in the synaptic cleft low and sheltering neurons from excitotoxic injury. Inside the astrocytes, glutamate is converted to glutamine that is shuttled back to presynaptic nerve terminals where it is reconverted to glutamate. Disturbed redistribution of glutamate and K+ in the astrocytic networks, via GJ loss or abnormal GJ channel gating and permselectivity, may lead to a local accumulation of both substances, paving the way for epileptic seizure activity (reviewed in Pannasch et al., 2011). The swelling of astrocytes may additionally result in a decreased extracellular space volume further increasing ambient concentrations of K+ and glutamate sensed by neurons. Reduced expression of Cx43 has been shown to increase expression of glutamate transporters (Unger et al., 2012), likely as a means to compensate the inadequate uptake of glutamate form the extracellular environment; its functional implications are, however, unknown. Also note that the clearance and redistribution of K+ is partially maintained in the hippocampus when Cx43 and Cx30 expression is silenced in astrocytes (Wallraff et al., 2006), indicating that mechanisms other than those related to astrocytic Cxs contribute to spatial buffering.
More than just exerting homeostatic control over the extracellular compartment, astrocytes are now considered to also more actively contribute to synaptic transmission, by responding to neuronal activity and releasing gliotransmitters. Individual astrocytes can contact up to 140,000 synapses (Bushong et al., 2002) and express a plethora of neurotransmitter receptors that in most cases trigger an increase in [Ca2+]i. In response to this, they release gliotransmitters that include glutamate, D-serine, ATP, adenosine, and GABA of which neurones on their turn express receptors (Perea and Araque, 2010; Orellana et al., 2011; Pannasch et al., 2012). HCs composed of Cx43 may well-contribute to gliotransmitter release as a [Ca2+]i-controlled diffusive pathway: HCs open with a [Ca2+]i increase up to 500 nM and close again at higher concentrations (see Cx43 Mutations Located in the Cytoplasmic Loop). Stehberg and co-workers have tested the hypothesis of gliotransmitter release via HC opening by making use of an inhibitory peptide (L2 peptide) that specifically targets Cx43 HCs without inhibiting Cx43 GJs (Ponsaerts et al., 2010). This work, performed in vivo, demonstrated that stereotactic injection of this peptide (coupled to a TAT-translocation motif) in the basolateral amygdala, blocked the consolidation of fear memory while addition of a cocktail of putative gliotransmitters rescued the L2-mediated inhibition of fear memory consolidation (Stehberg et al., 2012). Besides being activated by a moderate [Ca2+]i increase, HCs are also activated by a lowering of extracellular Ca2+. Extracellular Ca2+ can decrease as a result of neuronal activity and Nedergaard and co-workers have recently demonstrated that this may trigger astrocytic HC opening with consequent ATP release (Torres et al., 2012). The latter was demonstrated to activate inhibitory interneurons that put a brake on excitatory firing (Torres et al., 2012). Additionally, adenosine, derived from ATP degradation, has anticonvulsant effects as it inhibits presynaptic neurotransmitter release (Pascual et al., 2005). Overall, HC activation in astrocytes may thus contribute to gliotransmitter release, but with the evidence presently available, this may lead to excitatory as well as inhibitory signaling.
The contribution of Cx channels to ictogenesis has an ambiguous, two-faced aspect: on the one hand GJs have an anti-convulsive effect with respect to their K+ and glutamate buffering capacity, but on the other hand, GJs may act in a pro-convulsive way as well. In this respect, Cx43 gene ablation or pharmacological block of GJs reduced seizure activity while agents that potentiate cell–cell coupling enhanced neuronal bursting (Kohling et al., 2001; Wallraff et al., 2006; Yoon et al., 2010). Mechanistically, neuron-derived glutamate may trigger a [Ca2+]i increase in astrocytes that further stimulates glutamate release from these cells. The Ca2+ signal can propagate to neighboring astrocytes in the form of an intercellular Ca2+ wave, modulating groups of remotely located synapses and inducing a hyperexcited [Ca2+]i state over the entire astrocyte network (Giaume, 2010). Additionally, astrocyte GJs provide an intercellular route for the supply of energy substrates like glucose and lactate that are required for neuronal activity (Rouach et al., 2008). Once the astrocytic distribution of these substrates is compromised, energy delivery may become below demand, causing the accumulation of glutamate and K+ that may act in a pro-convulsive manner. Finally, both GJs and HCs have been implicated in the spread of apoptosis between astrocytes (Nodin et al., 2005; Eugenin and Berman, 2007; Decrock et al., 2009) which may well-break ground for epileptic seizures (Briellmann et al., 2002; Willoughby et al., 2003; Kang et al., 2006). With respect to HCs, Cx43 mutations giving a gain of HC function (I31M, G143S, and G138R) are, theoretically, expected to result in increased neuronal cell death, caused by excessively elevated neuronal [Ca2+]i (reviewed in Bennett et al., 2012). However, up to date, none of these gain-of-function mutants has been associated with epileptic seizures in ODDD patients (Abrams and Scherer, 2012). On the other hand, some mutants that result in a loss of HC function (L90V and I130T) are also associated with epileptic seizure activity which may result from a decrease in ATP release and consequent inhibitory signaling. At this stage, it is not clear what the net effect of an astrocytic HC contribution would be on excitability and ictogenesis but it emerges that the involvement of HCs in ictogenesis is, like for GJs, a double-edged sword. The use of novel tools such as the Gap19 peptide (Wang et al., 2013a) and the L2 peptide (Ponsaerts et al., 2010; Stehberg et al., 2012) may shed more light on the role of HCs in the healthy or pathological brain. In comparison to non-specific Cx channel blockers and peptides like Gap26/Gap27, L2/Gap19 peptides specifically block HCs composed of Cx43 (but not those composed of Cx40 and Panx1) while not blocking GJs.
When looking over the currently available evidence for ODDD-associated epilepsy, seizures have been reported in patients exhibiting mutants that give both dysfunctional HCs and GJs (Table 2); the loss of efficient K+ and glutamate buffering and inadequate nutrient supply may thus play a primary role in the appearance of an epileptic phenotype in ODDD, although this requires further research. Epilepsy has thus far not been reported for mutants with a gain of HC function.

Table 2. List of ODDD-linked mutations associated with epileptic seizures and their effect on Cx43 HCs and GJs.
Connexins and astrocyte-oligodendrocyte interactions involved in axonal myelination
As a last example of the astrocytic contribution to normal CNS function we highlight the role of astrocytes in axonal myelination. Astrocytes themselves do not produce myelin, but they are connected to oligodendrocytes, the brain's myelinating cells, via Cx43/Cx47 GJs (Orthmann-Murphy et al., 2007). Loss-of-function mutations in the Cx47 gene (GJA12/GJC2) lie at the basis of Pelizaeus-Merzbacher-like disease (PMLD1), an early onset, progressive dysmyelinating disorder affecting the CNS, and are also known to cause a distinct form of late onset hereditary spastic paraplegia (SPG44). In both cases, Cx47 mutations prevent the formation of functional Cx47/Cx47 and Cx43/Cx47 GJs causing hypomyelinating leukoencephalopathy (Orthmann-Murphy et al., 2007, 2009). The role of astrocyte-oligodendrocyte GJs is unclear but possibly relates to K+ shunting between both cell types. Although astrocytes and oligodendrocytes are also connected through Cx30/Cx32 GJs, these do not seem to compensate for the loss of Cx43/Cx47 coupling, probably as a consequence of differences in conductance, gating, and permeability (Orthmann-Murphy et al., 2007). We expect that spasticity observed with ODDD is similarly related to non-functional Cx43/Cx47 GJs caused by mutations in the GJA1 gene; however, there are, to our knowledge, no records that address astrocyte-oligodendrocyte coupling with ODDD-linked Cx43 mutants. The observation of a severe downregulation of astrocytic Cx43 in demyelinating lesions of patients suffering from Baló's disease, a disorder characterized by astrocytopathy and demyelination (Masaki et al., 2012), may give credit to the hypothesis that aberrant Cx43 function in astrocytes results in anomalous axonal myelination.
Finally, the retention of processed or unprocessed Cx43 protein can cause glial cell death through a process known as the “unfolded protein response” or through ER stress (reviewed by Roussel et al., 2013). The unfolded protein response and ER stress have been implicated in different neurodegenerative diseases, including those with progressive motor dysfunction (Huntington's disease, amyotrophic lateral sclerosis) as observed in ODDD as well.
Implications of ODDD-Linked Mutations in CNS Cells Other than Astrocytes
BBB-ECs
The role of Cxs, including Cx43, in BBB endothelial cells is only starting to emerge. Endothelial GJs have been implicated in maintenance of the interendothelial junctional complex (Nagasawa et al., 2006) and more recently, we have shown that Ca2+ dynamics triggered by low extracellular Ca2+ or inflammatory substances were sustained by GJs and HCs and contributed to disturbed BBB function and increased permeability (De Bock et al., 2011, 2012, 2013). Additionally, work from others has indicated that Cx43 HCs contribute to endothelial cell loss during pathologic insults (Danesh-Meyer et al., 2012). The effect of a Cx43 loss-of-function has not been studied at the level of the BBB, although one might expect a dysfunctional barrier due to a disorganisation of the junctions. On the other hand, following the observation that HCs contribute to increasing permeability, those mutants exhibiting increased HC activity are expected to render the BBB leaky to circulating compounds. The implications of such a leaky BBB have been outlined above.
Neurons
Cx43 is not expressed in adult neurons but it is prominently present in neuroblast cells where its role in embryonic development of the cortex becomes more and more acknowledged. Radial glial cells, the neuronal stem cells of the embryonic cerebral cortex, originate in the ventricular zone and differentiate into neuronal cells as the neocortex forms. These neuronal cells subsequently migrate to their final destination in the cortical plate. Additionally, radial glia give rise to radial fibers that form a guidance scaffold for neuronal migration. Cx43 can be found in the neuronal progenitors (or neuroblasts) as well as in the glial scaffold. Expression of the Cx43-T154A mutant that is able to make adhesions but is unable to form functional GJ channels in the developing cortex did not abolish the migration of new born neurons, but expression of the Cx43-C61S mutant that lacks docking ability (with maintained HC function) did prevent migration of new neurons. These observations suggested that channel activity does not play a role but Cxs are involved as adhesive contact points between the scaffold and the migrating neurones (Elias et al., 2007). More recently, it was shown that conditional Cx43 knock-out in radial glia disrupts neuronal migration, and this could be rescued by expression of full-length Cx43 but not by expression of CT-truncated Cx43 (removal of the last 125 residues) (Cina et al., 2009). These findings indicate that the C-terminal tail, which is known to interact with scaffolding proteins like ZO-1 and cytoskeletal tubulin (Giepmans et al., 2001), crucially links adhesive Cx43 properties to the cytoskeleton. Cx43 additionally co-localizes with proteins specialized in cell adhesion to neighboring cells or to the extracellular matrix (Nagasawa et al., 2006; Li et al., 2010; Sato et al., 2011). Although both studies suggest that the Cx43 channel pore has no function in neuronal migration, other studies have indicated that Cx43 HCs and GJs play a role in the initiation and propagation of Ca2+ signals in and between neuronal precursor cells respectively. These Ca2+ signals are likely to play a role in neuronal proliferation. Blocking HCs/GJs reduced neuronal motility, likely by interfering with Rho-GTPase activity, and gave rise to defective neurogenesis (Weissman et al., 2004; Liu et al., 2008, 2010). Cx43 knock-out mice generally do not present with a severely distorted cortex which might be explained by the fact that other Cxs like Cx30 or Cx26 contribute to neuronal migration (Elias et al., 2007), compensating for the loss of Cx43. One mouse strain that lacks Cx43 in embryonic radial glia, termed Shuffler, mimics part of the neuronal phenotype observed in ODDD (gait disturbance and ataxia). The mice exhibit structural abnormalities in the cerebellum, hippocampus and cortex which can explain the neuronal deficits. Importantly, the phenotype of these mice differs from other Cx43 knock-out mice strains in which no neurological phenotype was observed, suggesting that the genetic background is an important determinant of the occurrence of neurological symptoms (Wiencken-Barger et al., 2007). Such effect may apply for humans as well and might account for the rather low prevalence of neurological manifestations in ODDD patients.
Microglia
Microglial cells are the resident innate immune effector cells of the CNS and are essential in the primary defense against pathologic insults. Resting amoeboid microglia continuously scan the nervous tissue and rapidly respond to inflammatory molecules, pathogens or tissue injury by transforming into ramified cells that exhibit a high rate of proliferation and migration (reviewed in Kettenmann et al., 2011). Their activation involves the release of pro-inflammatory cytokines, free radicals and glutamate (Candelario-Jalil et al., 2007; Sumi et al., 2010; Takeuchi et al., 2011) which are all generally believed to be neurotoxic. Indeed, microglial activation is observed in many neurodegenerative diseases including Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis (Orellana et al., 2009) as well as in epilepsy (reviewed in Mirrione and Tsirka, 2011; Vezzani et al., 2012). In addition to direct neurotoxic effects, cytokines and other molecules released by microglia may inhibit GJIC between astrocytes (Meme et al., 2006) while stimulating HCs (Retamal et al., 2007), or activate BBB endothelial cells (Orellana et al., 2009), further contributing to neurodegeneration and epilepsy via mechanisms described above. However, in certain scenarios, microglia may also contribute to CNS repair through scavenging of reactive oxygen species, the release of neurotrophic factors and the removal of neurotoxic substances (Rouach et al., 2004, reviewed in Mika and Prochnow, 2012; Aguzzi et al., 2013). The role of Cx43 in the CNS' immune cells is uncertain. Whereas some groups find no indication for the presence of microglial Cx43 in the resting or ramified state (Dobrenis et al., 2005; Theodoric et al., 2012), others do report an increase in microglial Cx43 in inflammatory conditions (Eugenin et al., 2001; Garg et al., 2005; Shaikh et al., 2012). HCs have been implicated in glutamate release which could contribute to neuronal/astrocyte cell death and epilepsy (Ye et al., 2003; Takeuchi et al., 2008). In addition, dye coupling between microglial cells has been observed upon exposure to inflammatory conditions (Eugenin et al., 2001; Martinez et al., 2002). The functional implications of this activation-dependent microglial coupling remain unclear. Similar as in peripheral immune cells where Cx43 levels increase during inflammation, GJs could be involved in the cell–cell transfer of immunogens, rendering antigen cross-presentation more efficient (Neijssen et al., 2005). Altogether, altered Cx43 expression/channel function could disturb the proper microglial response to inflammation, failing in the defense against neurodegenerative mediators but contributing to neuronal bursting activity.
Conclusions
ODDD manifests as a pleiotropic disease with patients exhibiting both morphological and functional deficiencies caused by mutations in the widespread GJA1 gene. The GJA1 gene product Cx43 plays a leading role in CNS physiology and it thus comes with no surprise that neurological symptoms are included in the still expanding list of ODDD features. Analysis of the different mutants demonstrates that they can either alter insertion of Cx43 in the PM, GJ channel formation or HC/GJ channel gating. All CNS cells expressing aberrant Cx43 are potential contributors to nervous tissue dysfunction or damage, with a major involvement of astrocytes as the prime cell type expressing Cx43. In this paper we gave an overview of those mutants associated with nervous tissue dysfunctioning along with their outcome on Cx43 function. The information currently available on the possible effects of ODDD-linked mutations on Cx43 expression/channel function is impressive, as can be appreciated from Table 1. However, there is still a paucity of reports that document and analyze the mechanism by which the mutants give the neurological phenotype in a detailed manner. Thus, it remains difficult to present a clear genotype/phenotype correlation. None of the mutants identified thus far have been characterized for their specific effect on the function of astrocytes or other cells of the neurogliovascular unit, and therefore, genotype/phenotype linkage still remains in the realm of speculation. ODDD is, with a few exceptions, subject to an autosomal dominant inheritance pattern but unfortunately, thus far, there is no clear hypothesis that explains this pattern. Having one dysfunctional allele may cause a CNS phenotype in ODDD while in homozygous astrocyte-specific Cx43 knock-out animals no severe alterations are observed (Theis et al., 2003). This observation may plead in favor for a dominant negative effect of mutant Cx43 on co-expressed WT Cx43 protein. Most of the Cx43 mutants exhibit dominant negative effects, but a comparison of dominant negative mutants and neurological phenotypes is unfortunately inconclusive. The dominant negative effect of Cx43 mutants does not guarantee neurological findings (see for instance L11P and S18P) while mutants that do not exert a dominant effect may give a severe neurological phenotype (I130T). Trans-dominant negative effects of the mutants on other co-expressed Cxs are another likely explanation. Such effects have for instance been used to explain the dominant expression pattern of syndromic deafness caused by GJB2 (Cx26) mutations (reviewed in Laird, 2008). On the other hand, loss-of-function mutations of Cx26 and Cx30 have thus far not been associated with neuropathology (Abrams and Scherer, 2012). In the heart, Cx43-R33X mutants exert trans-dominant effects on Cx37 and Cx40 (Huang et al., 2013), whereas G138R does not (Dobrowolski et al., 2008). Unfortunately, we are not aware of any studies tackling this question with regards to the CNS. The (trans-)dominant effect of Cx43 mutants may additionally be cell type specific as the mouse G60S mutant has dominant negative effects on WT Cx43 in ovarian granulosa cells (Flenniken et al., 2005) but not in astrocytes (Wasseff et al., 2010). Finally, as illustrated in Table 2, epilepsy seems to be associated with loss of function ODDD mutants. We carefully checked this for motor deficiencies (Table 3) and found that a motor phenotype is always associated with mutants that give a loss or reduction of GJs, while the mutant's effects on HC function are variable. This generates a number of interesting working hypotheses that are open to be tested, hopefully strengthening our understanding of how ODDD mutants lead to channelopathy, CNS cell dysfunction and neurodegeneration.
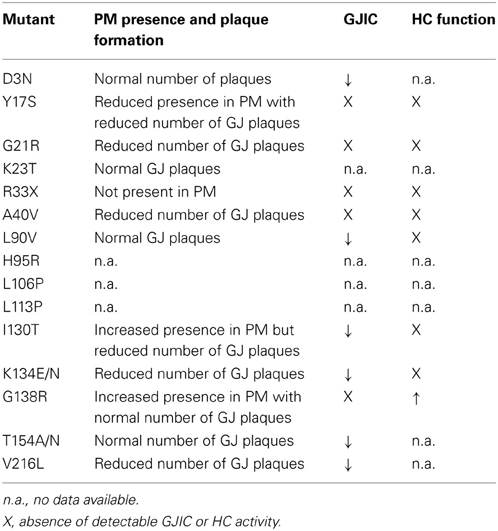
Table 3. List of ODDD mutations linked to upper motor neuron dysfunction, tremor, gait disturbances, and spasticity, indicating neurodegeneration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Fund for Scientific Research Flanders (FWO—grant numbers G.0354.07, G.0140.08 and 3G.0134.09 to Luc Leybaert) and the Interuniversity Attraction Poles Program (Belgian Science Policy Projects P6/31 and P7/10 to Luc Leybaert). Figure 1 was produced using Servier medical art.
Footnotes
References
Abrams, C. K., and Scherer, S. S. (2012). Gap junctions in inherited human disorders of the central nervous system. Biochim. Biophys. Acta 1818, 2030–2047. doi: 10.1016/j.bbamem.2011.08.015
Aguzzi, A., Barres, B. A., and Bennett, M. L. (2013). Microglia: scapegoat, saboteur, or something else? Science 339, 156–161. doi: 10.1126/science.1227901
Alao, M. J., Bonneau, D., Holder-Espinasse, M., Goizet, C., Manouvrier-Hanu, S., Mezel, A., et al. (2010). Oculo-dento-digital dysplasia: lack of genotype-phenotype correlation for GJA1 mutations and usefulness of neuro-imaging. Eur. J. Med. Genet. 53, 19–22. doi: 10.1016/j.ejmg.2009.08.007
Alexander, D. B., and Goldberg, G. S. (2003). Transfer of biologically important molecules between cells through gap junction channels. Curr. Med. Chem. 10, 2045–2058. doi: 10.2174/0929867033456927
Allen, N. J., and Barres, B. A. (2009). Neuroscience: glia - more than just brain glue. Nature 457, 675–677. doi: 10.1038/457675a
Amador, C., Mathews, A. M., Del Carmen Montoya, M., Laughridge, M. E., Everman, D. B., and Holden, K. R. (2008). Expanding the neurologic phenotype of oculodentodigital dysplasia in a 4-generation Hispanic family. J. Child Neurol. 23, 901–905. doi: 10.1177/0883073808317730
Amano, K., Ishiguchi, M., Aikawa, T., Kimata, M., Kishi, N., Fujimaki, T., et al. (2012). Cleft lip in oculodentodigital dysplasia suggests novel roles for connexin43. J. Dent. Res. 91, 38S-44S. doi: 10.1177/0022034512447952
Anumonwo, J. M., Taffet, S. M., Gu, H., Chanson, M., Moreno, A. P., and Delmar, M. (2001). The carboxyl terminal domain regulates the unitary conductance and voltage dependence of connexin40 gap junction channels. Circ. Res. 88, 666–673. doi: 10.1161/hh0701.088833
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., Macvicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi: 10.1038/nature09613
Avila, M. A., Sell, S. L., Hawkins, B. E., Hellmich, H. L., Boone, D. R., Crookshanks, J. M., et al. (2011). Cerebrovascular connexin expression: effects of traumatic brain injury. J. Neurotrauma 28, 1803–1811. doi: 10.1089/neu.2011.1900
Avshalumova, L., Fabrikant, J., and Koriakos, A. (2013). Overview of skin diseases linked to connexin gene mutations. Int. J. Dermatol. doi: 10.1111/ijd.12062. [Epub ahead of print].
Bao, X., Chen, Y., Reuss, L., and Altenberg, G. A. (2004). Functional expression in Xenopus oocytes of gap-junctional hemichannels formed by a cysteine-less connexin 43. J. Biol. Chem. 279, 9689–9692. doi: 10.1074/jbc.M311438200
Bargiotas, P., Monyer, H., and Schwaninger, M. (2009). Hemichannels in cerebral ischemia. Curr. Mol. Med. 9, 186–194. doi: 10.2174/156652409787581646
Batra, N., Kar, R., and Jiang, J. X. (2012). Gap junctions and hemichannels in signal transmission, function and development of bone. Biochim. Biophys. Acta 1818, 1909–1918. doi: 10.1016/j.bbamem.2011.09.018
Beahm, D. L., Oshima, A., Gaietta, G. M., Hand, G. M., Smock, A. E., Zucker, S. N., et al. (2006). Mutation of a conserved threonine in the third transmembrane helix of alpha- and beta-connexins creates a dominant-negative closed gap junction channel. J. Biol. Chem. 281, 7994–8009. doi: 10.1074/jbc.M506533200
Beltramello, M., Piazza, V., Bukauskas, F. F., Pozzan, T., and Mammano, F. (2005). Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat. Cell Biol. 7, 63–69. doi: 10.1038/ncb1205
Bennett, M. V., Garre, J. M., Orellana, J. A., Bukauskas, F. F., Nedergaard, M., and Saez, J. C. (2012). Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. 1487, 3–15. doi: 10.1016/j.brainres.2012.08.042
Berezowski, V., Fukuda, A. M., Cecchelli, R., and Badaut, J. (2012). Endothelial cells and astrocytes: a concerto en duo in ischemic pathophysiology. Int. J. Cell Biol. 2012, 176287. doi: 10.1155/2012/176287
Braet, K., Paemeleire, K., D'Herde, K., Sanderson, M. J., and Leybaert, L. (2001). Astrocyte-endothelial cell calcium signals conveyed by two signalling pathways. Eur. J. Neurosci. 13, 79–91. doi: 10.1046/j.1460-9568.2001.01372.x
Brice, G., Ostergaard, P., Jeffery, S., Gordon, K., Mortimer, P., and Mansour, S. (2013). A novel mutation in GJA1 causing oculodentodigital syndrome and primary lymphoedema in a three generation family. Clin. Genet. doi: 10.1111/cge.12158. [Epub ahead of print].
Briellmann, R. S., Kalnins, R. M., Berkovic, S. F., and Jackson, G. D. (2002). Hippocampal pathology in refractory temporal lobe epilepsy: T2-weighted signal change reflects dentate gliosis. Neurology 58, 265–271. doi: 10.1212/WNL.58.2.265
Bruzzone, R., White, T. W., and Paul, D. L. (1996). Connections with connexins: the molecular basis of direct intercellular signaling. Eur. J. Biochem. 238, 1–27. doi: 10.1111/j.1432-1033.1996.0001q.x
Bukauskas, F. F., Jordan, K., Bukauskiene, A., Bennett, M. V., Lampe, P. D., Laird, D. W., et al. (2000). Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Sci. U.S.A. 97, 2556–2561. doi: 10.1073/pnas.050588497
Bukauskas, F. F., and Verselis, V. K. (2004). Gap junction channel gating. Biochim. Biophys. Acta 1662, 42–60. doi: 10.1016/j.bbamem.2004.01.008
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192.
Candelario-Jalil, E., de Oliveira, A. C., Graf, S., Bhatia, H. S., Hull, M., Munoz, E., et al. (2007). Resveratrol potently reduces prostaglandin E2 production and free radical formation in lipopolysaccharide-activated primary rat microglia. J. Neuroinflammation 4, 25. doi: 10.1186/1742-2094-4-25
Carlen, P. L. (2012). Curious and contradictory roles of glial connexins and pannexins in epilepsy. Brain Res. 1487, 54–60. doi: 10.1016/j.brainres.2012.06.059
Chew, S. S., Johnson, C. S., Green, C. R., and Danesh-Meyer, H. V. (2010). Role of connexin43 in central nervous system injury. Exp. Neurol. 225, 250–261. doi: 10.1016/j.expneurol.2010.07.014
Churko, J. M., Shao, Q., Gong, X. Q., Swoboda, K. J., Bai, D., Sampson, J., et al. (2011). Human dermal fibroblasts derived from oculodentodigital dysplasia patients suggest that patients may have wound-healing defects. Hum. Mutat. 32, 456–466. doi: 10.1002/humu.21472
Cina, C., Maass, K., Theis, M., Willecke, K., Bechberger, J. F., and Naus, C. C. (2009). Involvement of the cytoplasmic C-terminal domain of connexin43 in neuronal migration. J. Neurosci. 29, 2009–2021. doi: 10.1523/JNEUROSCI.5025-08.2009
Civitelli, R. (2008). Cell-cell communication in the osteoblast/osteocyte lineage. Arch. Biochem. Biophys. 473, 188–192. doi: 10.1016/j.abb.2008.04.005
Contreras, J. E., Sanchez, H. A., Veliz, L. P., Bukauskas, F. F., Bennett, M. V., and Saez, J. C. (2004). Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res. Brain Res. Rev. 47, 290–303. doi: 10.1016/j.brainresrev.2004.08.002
Cotrina, M. L., Kang, J., Lin, J. H., Bueno, E., Hansen, T. W., He, L., et al. (1998). Astrocytic gap junctions remain open during ischemic conditions. J. Neurosci. 18, 2520–2537.
Cottrell, G. T., Lin, R., Warn-Cramer, B. J., Lau, A. F., and Burt, J. M. (2003). Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am. J. Physiol. Cell Physiol. 284, C511–C520. doi: 10.1152/ajpcell.00214.2002
D'Ambrosio, R. (2004). The role of glial membrane ion channels in seizures and epileptogenesis. Pharmacol. Ther. 103, 95–108. doi: 10.1016/j.pharmthera.2004.05.004
Dahl, G., Werner, R., Levine, E., and Rabadan-Diehl, C. (1992). Mutational analysis of gap junction formation. Biophys. J. 62, 172–180. discussion: 180–172. doi: 10.1016/S0006-3495(92)81803-9
Danesh-Meyer, H. V., Huang, R., Nicholson, L. F., and Green, C. R. (2008). Connexin43 antisense oligodeoxynucleotide treatment down-regulates the inflammatory response in an in vitro interphase organotypic culture model of optic nerve ischaemia. J. Clin. Neurosci. 15, 1253–1263. doi: 10.1016/j.jocn.2008.08.002
Danesh-Meyer, H. V., Kerr, N. M., Zhang, J., Eady, E. K., O'Carroll, S. J., Nicholson, L. F., et al. (2012). Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 135, 506–520. doi: 10.1093/brain/awr338
Das, A., Wallace, G. C. 4th., Holmes, C., McDowell, M. L., Smith, J. A., Marshall, J. D., et al. (2012). Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220, 237–246. doi: 10.1016/j.neuroscience.2012.06.002
David, Y., Cacheaux, L. P., Ivens, S., Lapilover, E., Heinemann, U., Kaufer, D., et al. (2009). Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J. Neurosci. 29, 10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009
Davidson, J. O., Green, C. R., Nicholson, L. F., O'Carroll, S. J., Fraser, M., Bennet, L., et al. (2012). Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol. 71, 121–132. doi: 10.1002/ana.22654
De Bock, M., Culot, M., Wang, N., Bol, M., Decrock, E., De Vuyst, E., et al. (2011). Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 31, 1942–1957. doi: 10.1038/jcbfm.2011.86
De Bock, M., Culot, M., Wang, N., da Costa, A., Decrock, E., Bol, M., et al. (2012). Low extracellular Ca2+ conditions induce an increase in brain endothelial permeability that involves intercellular Ca2+ waves. Brain Res. 1487, 78–87. doi: 10.1016/j.brainres.2012.06.046
De Bock, M., Wang, N., Decrock, E., Bol, M., Gadicherla, A., Culot, M., et al. (2013). Endothelial calcium dynamics: connexin channels and blood brain barrier function. Prog. Neurobiol. 108, 1–20. doi: 10.1016/j.pneurobio.2013.06.001
Decrock, E., De Vuyst, E., Vinken, M., Van Moorhem, M., Vranckx, K., Wang, N., et al. (2009). Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 16, 151–163. doi: 10.1038/cdd.2008.138
Decrock, E., Krysko, D. V., Vinken, M., Kaczmarek, A., Crispino, G., Bol, M., et al. (2012). Transfer of IP(3) through gap junctions is critical, but not sufficient, for the spread of apoptosis. Cell Death Differ. 19, 947–957. doi: 10.1038/cdd.2011.176
Desplantez, T., McCain, M. L., Beauchamp, P., Rigoli, G., Rothen-Rutishauser, B., Parker, K. K., et al. (2012). Connexin43 ablation in foetal atrial myocytes decreases electrical coupling, partner connexins, and sodium current. Cardiovasc. Res. 94, 58–65. doi: 10.1093/cvr/cvs025
De Vuyst, E., Wang, N., Decrock, E., De Bock, M., Vinken, M., Van Moorhem, M., et al. (2009). Ca(2+) regulation of connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium 46, 176–187. doi: 10.1016/j.ceca.2009.07.002
Dichter, M. A. (2009). Emerging concepts in the pathogenesis of epilepsy and epileptogenesis. Arch. Neurol. 66, 443–447. doi: 10.1001/archneurol.2009.10
Dobrenis, K., Chang, H. Y., Pina-Benabou, M. H., Woodroffe, A., Lee, S. C., Rozental, R., et al. (2005). Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. J. Neurosci. Res. 82, 306–315. doi: 10.1002/jnr.20650
Dobrowolski, R., Sasse, P., Schrickel, J. W., Watkins, M., Kim, J. S., Rackauskas, M., et al. (2008). The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 17, 539–554. doi: 10.1093/hmg/ddm329
Dobrowolski, R., Sommershof, A., and Willecke, K. (2007). Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J. Membr. Biol. 219, 9–17. doi: 10.1007/s00232-007-9055-7
Ek, J. F., Delmar, M., Perzova, R., and Taffet, S. M. (1994). Role of histidine 95 on pH gating of the cardiac gap junction protein connexin43. Circ. Res. 74, 1058–1064. doi: 10.1161/01.RES.74.6.1058
Ek-Vitorin, J. F., and Burt, J. M. (2013). Structural basis for the selective permeability of channels made of communicating junction proteins. Biochim. Biophys. Acta 1828, 51–68. doi: 10.1016/j.bbamem.2012.02.003
Ek-Vitorin, J. F., Calero, G., Morley, G. E., Coombs, W., Taffet, S. M., and Delmar, M. (1996). PH regulation of connexin43: molecular analysis of the gating particle. Biophys. J. 71, 1273–1284. doi: 10.1016/S0006-3495(96)79328-1
Elias, L. A., Wang, D. D., and Kriegstein, A. R. (2007). Gap junction adhesion is necessary for radial migration in the neocortex. Nature 448, 901–907. doi: 10.1038/nature06063
Eugenin, E. A., Basilio, D., Saez, J. C., Orellana, J. A., Raine, C. S., Bukauskas, F., et al. (2012). The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J. Neuroimmune Pharmacol. 7, 499–518. doi: 10.1007/s11481-012-9352-5
Eugenin, E. A., and Berman, J. W. (2007). Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J. Neurosci. 27, 12844–12850. doi: 10.1523/JNEUROSCI.4154-07.2007
Eugenin, E. A., Eckardt, D., Theis, M., Willecke, K., Bennett, M. V., and Saez, J. C. (2001). Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-gamma and tumor necrosis factor-alpha. Proc. Natl. Acad. Sci. U.S.A. 98, 4190–4195. doi: 10.1073/pnas.051634298
Ezan, P., Andre, P., Cisternino, S., Saubamea, B., Boulay, A. C., Doutremer, S., et al. (2012). Deletion of astroglial connexins weakens the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 1457–1467. doi: 10.1038/jcbfm.2012.45
Falk, M. M. (2000). Cell-free synthesis for analyzing the membrane integration, oligomerization, and assembly characteristics of gap junction connexins. Methods 20, 165–179. doi: 10.1006/meth.1999.0934
Fenwick, A., Richardson, R. J., Butterworth, J., Barron, M. J., and Dixon, M. J. (2008). Novel mutations in GJA1 cause oculodentodigital syndrome. J. Dent. Res. 87, 1021–1026. doi: 10.1177/154405910808701108
Filosa, J. A., and Blanco, V. M. (2007). Neurovascular coupling in the mammalian brain. Exp. Physiol. 92, 641–646. doi: 10.1113/expphysiol.2006.036368
Flenniken, A. M., Osborne, L. R., Anderson, N., Ciliberti, N., Fleming, C., Gittens, J. E., et al. (2005). A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development 132, 4375–4386. doi: 10.1242/dev.02011
Florio, C., and Maniscalco, G. T. (2011). Improvement of visual acuity in patients with severe visual loss affected by multiple sclerosis treated with natalizumab: a case report. Neurol. Sci. 31(Suppl. 3), 325–327. doi: 10.1007/s10072-010-0349-7
Foote, C. I., Zhou, L., Zhu, X., and Nicholson, B. J. (1998). The pattern of disulfide linkages in the extracellular loop regions of connexin 32 suggests a model for the docking interface of gap junctions. J. Cell Biol. 140, 1187–1197. doi: 10.1083/jcb.140.5.1187
Frisch, C., Theis, M., De Souza Silva, M. A., Dere, E., Sohl, G., Teubner, B., et al. (2003). Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur. J. Neurosci. 18, 2313–2318. doi: 10.1046/j.1460-9568.2003.02971.x
Furuta, N., Ikeda, M., Hirayanagi, K., Fujita, Y., Amanuma, M., and Okamoto, K. (2012). A novel GJA1 mutation in oculodentodigital dysplasia with progressive spastic paraplegia and sensory deficits. Intern. Med. 51, 93–98. doi: 10.2169/internalmedicine.51.5770
Gaietta, G., Deerinck, T. J., Adams, S. R., Bouwer, J., Tour, O., Laird, D. W., et al. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503–507. doi: 10.1126/science.1068793
Garg, S., Md Syed, M., and Kielian, T. (2005). Staphylococcus aureus-derived peptidoglycan induces Cx43 expression and functional gap junction intercellular communication in microglia. J. Neurochem. 95, 475–483. doi: 10.1111/j.1471-4159.2005.03384.x
Giaume, C. (2010). Astroglial wiring is adding complexity to neuroglial networking. Front. Neuroenerg. 2:129. doi: 10.3389/fnene.2010.00129
Giaume, C., Leybaert, L., Naus, C. C., and Sáez, J. C. (2013). Connexin and pannexin hemichannels in brain glial cells: properties, pharmacology, and roles. Front. Pharmacol. 4:88. doi: 10.3389/fphar.2013.00088
Giepmans, B. N., Verlaan, I., and Moolenaar, W. H. (2001). Connexin-43 interactions with ZO-1 and alpha- and beta-tubulin. Cell Commun. Adhes. 8, 219–223. doi: 10.3109/15419060109080727
Gong, X. Q., Shao, Q., Langlois, S., Bai, D., and Laird, D. W. (2007). Differential potency of dominant negative connexin43 mutants in oculodentodigital dysplasia. J. Biol. Chem. 282, 19190–19202. doi: 10.1074/jbc.M609653200
Gong, X. Q., Shao, Q., Lounsbury, C. S., Bai, D., and Laird, D. W. (2006). Functional characterization of a GJA1 frameshift mutation causing oculodentodigital dysplasia and palmoplantar keratoderma. J. Biol. Chem. 281, 31801–31811. doi: 10.1074/jbc.M605961200
Gonzalez, D., Gomez-Hernandez, J. M., and Barrio, L. C. (2006). Species specificity of mammalian connexin-26 to form open voltage-gated hemichannels. FASEB J. 20, 2329–2338. doi: 10.1096/fj.06-5828com
Goodenough, D. A., and Paul, D. L. (2003). Beyond the gap: functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 4, 285–294. doi: 10.1038/nrm1072
Gordon, G. R., Choi, H. B., Rungta, R. L., Ellis-Davies, G. C., and MacVicar, B. A. (2008). Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749. doi: 10.1038/nature07525
Herve, J. C., Derangeon, M., Bahbouhi, B., Mesnil, M., and Sarrouilhe, D. (2007). The connexin turnover, an important modulating factor of the level of cell-to-cell junctional communication: comparison with other integral membrane proteins. J. Membr. Biol. 217, 21–33. doi: 10.1007/s00232-007-9054-8
Hirst-Jensen, B. J., Sahoo, P., Kieken, F., Delmar, M., and Sorgen, P. L. (2007). Characterization of the pH-dependent interaction between the gap junction protein connexin43 carboxyl terminus and cytoplasmic loop domains. J. Biol. Chem. 282, 5801–5813. doi: 10.1074/jbc.M605233200
Hong, H. M., Yang, J. J., Shieh, J. C., Lin, M. L., and Li, S. Y. (2010). Novel mutations in the connexin43 (GJA1) and GJA1 pseudogene may contribute to nonsyndromic hearing loss. Hum. Genet. 127, 545–551. doi: 10.1007/s00439-010-0791-x
Honkaniemi, J., Kalkkila, J. P., Koivisto, P., Kahara, V., Latvala, T., and Simola, K. (2005). Letter to the editor: Novel GJA1 mutation in oculodentodigital dysplasia. Am. J. Med. Genet. A 139, 48–49.
Huang, T., Shao, Q., Macdonald, A., Xin, L., Lorentz, R., Bai, D., et al. (2013). Autosomal recessive GJA1 (Cx43) gene mutations cause oculodentodigital dysplasia by distinct mechanisms. J. Cell Sci. 126, 2857–2866. doi: 10.1242/jcs.123315
Huckstepp, R. T., id Bihi, R., Eason, R., Spyer, K. M., Dicke, N., Willecke, K., et al. (2010). Connexin hemichannel-mediated CO2-dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J. Physiol. 588, 3901–3920. doi: 10.1113/jphysiol.2010.192088
Iadecola, C., and Nedergaard, M. (2007). Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376. doi: 10.1038/nn2003
Iglesias, R., Dahl, G., Qiu, F., Spray, D. C., and Scemes, E. (2009). Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J. Neurosci. 29, 7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009
Ivens, S., Kaufer, D., Flores, L. P., Bechmann, I., Zumsteg, D., Tomkins, O., et al. (2007). TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547. doi: 10.1093/brain/awl317
Iyyathurai, J., D'Hondt, C., Wang, N., De Bock, M., Himpens, B., Retamal, M. A., et al. (2013). Peptides and peptide-derived molecules targeting the intracellular domains of Cx43: gap junctions versus hemichannels. Neuropharmacology. doi: 10.1016/j.neuropharm.2013.04.050. [Epub ahead of print].
Janigro, D. (2012). Are you in or out? Leukocyte, ion, and neurotransmitter permeability across the epileptic blood-brain barrier. Epilepsia 53(Suppl. 1), 26–34. doi: 10.1111/j.1528-1167.2012.03472.x
Jiang, J. X., and Cherian, P. P. (2003). Hemichannels formed by connexin 43 play an important role in the release of prostaglandin E(2) by osteocytes in response to mechanical strain. Cell Commun. Adhes. 10, 259–264.
Johnstone, S. R., Billaud, M., Lohman, A. W., Taddeo, E. P., and Isakson, B. E. (2012). Posttranslational modifications in connexins and pannexins. J. Membr. Biol. 245, 319–332. doi: 10.1007/s00232-012-9453-3
Joss, S. K., Ghazawy, S., Tomkins, S., Ahmed, M., Bradbury, J., and Sheridan, E. (2008). Variable expression of neurological phenotype in autosomal recessive oculodentodigital dysplasia of two sibs and review of the literature. Eur. J. Pediatr. 167, 341–345. doi: 10.1007/s00431-007-0468-1
Kalcheva, N., Qu, J., Sandeep, N., Garcia, L., Zhang, J., Wang, Z., et al. (2007). Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc. Natl. Acad. Sci. U.S.A. 104, 20512–20516. doi: 10.1073/pnas.0705472105
Kamermans, M., Fahrenfort, I., Schultz, K., Janssen-Bienhold, U., Sjoerdsma, T., and Weiler, R. (2001). Hemichannel-mediated inhibition in the outer retina. Science 292, 1178–1180. doi: 10.1126/science.1060101
Kang, J., Kang, N., Lovatt, D., Torres, A., Zhao, Z., Lin, J., et al. (2008). Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 28, 4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008
Kang, T. C., Kim, D. S., Kwak, S. E., Kim, J. E., Won, M. H., Kim, D. W., et al. (2006). Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 54, 258–271. doi: 10.1002/glia.20380
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kielian, T. (2008). Glial connexins and gap junctions in CNS inflammation and disease. J. Neurochem. 106, 1000–1016. doi: 10.1111/j.1471-4159.2008.05405.x
Koehler, R. C., Gebremedhin, D., and Harder, D. R. (2006). Role of astrocytes in cerebrovascular regulation. J. Appl. Physiol. 100, 307–317. doi: 10.1152/japplphysiol.00938.2005
Kohling, R., Gladwell, S. J., Bracci, E., Vreugdenhil, M., and Jefferys, J. G. (2001). Prolonged epileptiform bursting induced by 0-Mg(2+) in rat hippocampal slices depends on gap junctional coupling. Neuroscience 105, 579–587. doi: 10.1016/S0306-4522(01)00222-6
Kronengold, J., Srinivas, M., and Verselis, V. K. (2012). The N-terminal half of the connexin protein contains the core elements of the pore and voltage gates. J. Membr. Biol. 245, 453–463. doi: 10.1007/s00232-012-9457-z
Kronengold, J., Trexler, E. B., Bukauskas, F. F., Bargiello, T. A., and Verselis, V. K. (2003a). Pore-lining residues identified by single channel SCAM studies in Cx46 hemichannels. Cell Commun. Adhes. 10, 193–199.
Kronengold, J., Trexler, E. B., Bukauskas, F. F., Bargiello, T. A., and Verselis, V. K. (2003b). Single-channel SCAM identifies pore-lining residues in the first extracellular loop and first transmembrane domains of Cx46 hemichannels. J. Gen. Physiol. 122, 389–405. doi: 10.1085/jgp.200308861
Kur, J., Newman, E. A., and Chan-Ling, T. (2012). Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 31, 377–406. doi: 10.1016/j.preteyeres.2012.04.004
Kwon, T., Roux, B., Jo, S., Klauda, J. B., Harris, A. L., and Bargiello, T. A. (2012). Molecular dynamics simulations of the Cx26 hemichannel: insights into voltage-dependent loop-gating. Biophys. J. 102, 1341–1351. doi: 10.1016/j.bpj.2012.02.009
Lai, A., Le, D. N., Paznekas, W. A., Gifford, W. D., Jabs, E. W., and Charles, A. C. (2006). Oculodentodigital dysplasia connexin43 mutations result in non-functional connexin hemichannels and gap junctions in C6 glioma cells. J. Cell Sci. 119, 532–541. doi: 10.1242/jcs.02770
Laird, D. W. (2006). Life cycle of connexins in health and disease. Biochem. J. 394, 527–543. doi: 10.1042/BJ20051922
Laird, D. W. (2008). Closing the gap on autosomal dominant connexin-26 and connexin-43 mutants linked to human disease. J. Biol. Chem. 283, 2997–3001. doi: 10.1074/jbc.R700041200
Laird, D. W. (2010). The gap junction proteome and its relationship to disease. Trends Cell Biol. 20, 92–101. doi: 10.1016/j.tcb.2009.11.001
Lampe, P. D., and Lau, A. F. (2004). The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 36, 1171–1186. doi: 10.1016/S1357-2725(03)00264-4
Lampe, P. D., TenBroek, E. M., Burt, J. M., Kurata, W. E., Johnson, R. G., and Lau, A. F. (2000). Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 149, 1503–1512. doi: 10.1083/jcb.149.7.1503
Langlois, S., Cowan, K. N., Shao, Q., Cowan, B. J., and Laird, D. W. (2008). Caveolin-1 and -2 interact with connexin43 and regulate gap junctional intercellular communication in keratinocytes. Mol. Biol. Cell 19, 912–928. doi: 10.1091/mbc.E07-06-0596
Lasta, M., Pemp, B., Schmidl, D., Boltz, A., Kaya, S., Palkovits, S., et al. (2013). Neurovascular dysfunction precedes neural dysfunction in the retina of patients with type 1 diabetes. Invest. Ophthalmol. Vis. Sci. 54, 842–847. doi: 10.1167/iovs.12-10873
Leybaert, L., and Sanderson, M. J. (2012). Intercellular Ca2+ waves: mechanisms and function. Physiol. Rev. 92, 1359–1392. doi: 10.1152/physrev.00029.2011
Li, J., Cheng, L., Wang, L. J., Liu, H. C., Li, L., Wang, X. L., et al. (2010). Cell surface sialic acid inhibits Cx43 gap junction functions in constructed Hela cancer cells involving in sialylated N-cadherin. Mol. Cell. Biochem. 344, 241–251. doi: 10.1007/s11010-010-0548-9
Liu, F., Arce, F. T., Ramachandran, S., and Lal, R. (2006). Nanomechanics of hemichannel conformations: connexin flexibility underlying channel opening and closing. J. Biol. Chem. 281, 23207–23217. doi: 10.1074/jbc.M605048200
Liu, X., Hashimoto-Torii, K., Torii, M., Ding, C., and Rakic, P. (2010). Gap junctions/hemichannels modulate interkinetic nuclear migration in the forebrain precursors. J. Neurosci. 30, 4197–4209. doi: 10.1523/JNEUROSCI.4187-09.2010
Liu, X., Hashimoto-Torii, K., Torii, M., Haydar, T. F., and Rakic, P. (2008). The role of ATP signaling in the migration of intermediate neuronal progenitors to the neocortical subventricular zone. Proc. Natl. Acad. Sci. U.S.A. 105, 11802–11807. doi: 10.1073/pnas.0805180105
Loddenkemper, T., Grote, K., Evers, S., Oelerich, M., and Stogbauer, F. (2002). Neurological manifestations of the oculodentodigital dysplasia syndrome. J. Neurol. 249, 584–595. doi: 10.1007/s004150200068
Lubkemeier, I., Requardt, R. P., Lin, X., Sasse, P., Andrie, R., Schrickel, J. W., et al. (2013). Deletion of the last five C-terminal amino acid residues of connexin43 leads to lethal ventricular arrhythmias in mice without affecting coupling via gap junction channels. Basic Res. Cardiol. 108, 348. doi: 10.1007/s00395-013-0348-y
Lurtz, M. M., and Louis, C. F. (2003). Calmodulin and protein kinase C regulate gap junctional coupling in lens epithelial cells. Am. J. Physiol. Cell Physiol. 285, C1475–C1482. doi: 10.1152/ajpcell.00361.2002
Lurtz, M. M., and Louis, C. F. (2007). Intracellular calcium regulation of connexin43. Am. J. Physiol. Cell Physiol. 293, C1806–C1813. doi: 10.1152/ajpcell.00630.2006
Lutz, S. E., Raine, C. S., and Brosnan, C. F. (2012). Loss of astrocyte connexins 43 and 30 does not significantly alter susceptibility or severity of acute experimental autoimmune encephalomyelitis in mice. J. Neuroimmunol. 245, 8–14. doi: 10.1016/j.jneuroim.2012.01.007
Maass, K., Shibayama, J., Chase, S. E., Willecke, K., and Delmar, M. (2007). C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ. Res. 101, 1283–1291. doi: 10.1161/CIRCRESAHA.107.162818
Maeda, S., Nakagawa, S., Suga, M., Yamashita, E., Oshima, A., Fujiyoshi, Y., et al. (2009). Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 458, 597–602. doi: 10.1038/nature07869
Marquez-Rosado, L., Solan, J. L., Dunn, C. A., Norris, R. P., and Lampe, P. D. (2012). Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim. Biophys. Acta 1818, 1985–1992. doi: 10.1016/j.bbamem.2011.07.028
Martinez, A. D., Eugenin, E. A., Branes, M. C., Bennett, M. V., and Saez, J. C. (2002). Identification of second messengers that induce expression of functional gap junctions in microglia cultured from newborn rats. Brain Res. 943, 191–201. doi: 10.1016/S0006-8993(02)02621-5
Masaki, K., Suzuki, S. O., Matsushita, T., Yonekawa, T., Matsuoka, T., Isobe, N., et al. (2012). Extensive loss of connexins in Balo's disease: evidence for an auto-antibody-independent astrocytopathy via impaired astrocyte-oligodendrocyte/myelin interaction. Acta Neuropathol. 123, 887–900. doi: 10.1007/s00401-012-0972-x
McLachlan, E., Manias, J. L., Gong, X. Q., Lounsbury, C. S., Shao, Q., Bernier, S. M., et al. (2005). Functional characterization of oculodentodigital dysplasia-associated Cx43 mutants. Cell Commun. Adhes. 12, 279–292. doi: 10.1080/15419060500514143
Meme, W., Calvo, C. F., Froger, N., Ezan, P., Amigou, E., Koulakoff, A., et al. (2006). Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by beta-amyloid. FASEB J. 20, 494–496.
Meme, W., Vandecasteele, M., Giaume, C., and Venance, L. (2009). Electrical coupling between hippocampal astrocytes in rat brain slices. Neurosci. Res. 63, 236–243. doi: 10.1016/j.neures.2008.12.008
Metea, M. R., and Newman, E. A. (2006). Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J. Neurosci. 26, 2862–2870. doi: 10.1523/JNEUROSCI.4048-05.2006
Mika, T., and Prochnow, N. (2012). Functions of connexins and large pore channels on microglial cells: the gates to environment. Brain Res. 1487, 16–24. doi: 10.1016/j.brainres.2012.07.020
Mirrione, M. M., and Tsirka, S. E. (2011). “A functional role for microglia in epilepsy,” in Clinical and Genetic Aspects of Epilepsy, ed Z. Afawi (New York, NY: InTech), 3–22.
Miyoshi, H., Boyle, M. B., MacKay, L. B., and Garfield, R. E. (1996). Voltage-clamp studies of gap junctions between uterine muscle cells during term and preterm labor. Biophys. J. 71, 1324–1334. doi: 10.1016/S0006-3495(96)79332-3
Moreno, A. P. (2005). Connexin phosphorylation as a regulatory event linked to channel gating. Biochim. Biophys. Acta 1711, 164–171. doi: 10.1016/j.bbamem.2005.02.016
Moreno, A. P., Chanson, M., Elenes, S., Anumonwo, J., Scerri, I., Gu, H., et al. (2002). Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ. Res. 90, 450–457. doi: 10.1161/hh0402.105667
Moreno, A. P., and Lau, A. F. (2007). Gap junction channel gating modulated through protein phosphorylation. Prog. Biophys. Mol. Biol. 94, 107–119. doi: 10.1016/j.pbiomolbio.2007.03.004
Mulligan, S. J., and MacVicar, B. A. (2004). Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199. doi: 10.1038/nature02827
Myllykoski, M., Kuczera, K., and Kursula, P. (2009). Complex formation between calmodulin and a peptide from the intracellular loop of the gap junction protein connexin43: Molecular conformation and energetics of binding. Biophys. Chem. 144, 130–135. doi: 10.1016/j.bpc.2009.08.001
Nagasawa, K., Chiba, H., Fujita, H., Kojima, T., Saito, T., Endo, T., et al. (2006). Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell. Physiol. 208, 123–132. doi: 10.1002/jcp.20647
Neijssen, J., Herberts, C., Drijfhout, J. W., Reits, E., Janssen, L., and Neefjes, J. (2005). Cross-presentation by intercellular peptide transfer through gap junctions. Nature 434, 83–88. doi: 10.1038/nature03290
Neuhaus, J., Weimann, A., Stolzenburg, J. U., Wolburg, H., Horn, L. C., and Dorschner, W. (2002). Smooth muscle cells from human urinary bladder express connexin 43 in vivo and in vitro. World J. Urol. 20, 250–254.
Nicholson, B. J. (2003). Gap junctions - from cell to molecule. J. Cell. Sci. 116, 4479–4481. doi: 10.1242/jcs.00821
Nodin, C., Nilsson, M., and Blomstrand, F. (2005). Gap junction blockage limits intercellular spreading of astrocytic apoptosis induced by metabolic depression. J. Neurochem. 94, 1111–1123. doi: 10.1111/j.1471-4159.2005.03241.x
O'Carroll, S. J., Alkadhi, M., Nicholson, L. F., and Green, C. R. (2008). Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 15, 27–42. doi: 10.1080/15419060802014164
Oh, S., Ri, Y., Bennett, M. V., Trexler, E. B., Verselis, V. K., and Bargiello, T. A. (1997). Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron 19, 927–938. doi: 10.1016/S0896-6273(00)80973-3
Orellana, J. A., Figueroa, X. F., Sanchez, H. A., Contreras-Duarte, S., Velarde, V., and Saez, J. C. (2011). Hemichannels in the neurovascular unit and white matter under normal and inflamed conditions. CNS Neurol. Disord. Drug Targets 10, 404–414. doi: 10.2174/187152711794653869
Orellana, J. A., Saez, P. J., Shoji, K. F., Schalper, K. A., Palacios-Prado, N., Velarde, V., et al. (2009). Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid. Redox Signal. 11, 369–399. doi: 10.1089/ars.2008.2130
Orthmann-Murphy, J. L., Freidin, M., Fischer, E., Scherer, S. S., and Abrams, C. K. (2007). Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 27, 13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007
Orthmann-Murphy, J. L., Salsano, E., Abrams, C. K., Bizzi, A., Uziel, G., Freidin, M. M., et al. (2009). Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 132, 426–438. doi: 10.1093/brain/awn328
Pannasch, U., Derangeon, M., Chever, O., and Rouach, N. (2012). Astroglial gap junctions shape neuronal network activity. Commun. Integr. Biol. 5, 248–254. doi: 10.4161/cib.19410
Pannasch, U., Vargova, L., Reingruber, J., Ezan, P., Holcman, D., Giaume, C., et al. (2011). Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 8467–8472. doi: 10.1073/pnas.1016650108
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J. Y., et al. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. doi: 10.1126/science.1116916
Paul, D. L., Ebihara, L., Takemoto, L. J., Swenson, K. I., and Goodenough, D. A. (1991). Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 115, 1077–1089. doi: 10.1083/jcb.115.4.1077
Paznekas, W. A., Boyadjiev, S. A., Shapiro, R. E., Daniels, O., Wollnik, B., Keegan, C. E., et al. (2003). Connexin 43 (GJA1). mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 72, 408–418. doi: 10.1086/346090
Paznekas, W. A., Karczeski, B., Vermeer, S., Lowry, R. B., Delatycki, M., Laurence, F., et al. (2009). GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 30, 724–733. doi: 10.1002/humu.20958
Perea, G., and Araque, A. (2010). GLIA modulates synaptic transmission. Brain Res. Rev. 63, 93–102. doi: 10.1016/j.brainresrev.2009.10.005
Petzold, G. C., and Murthy, V. N. (2011). Role of astrocytes in neurovascular coupling. Neuron 71, 782–797. doi: 10.1016/j.neuron.2011.08.009
Ponsaerts, R., De Vuyst, E., Retamal, M., D'Hondt, C., Vermeire, D., Wang, N., et al. (2010). Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 24, 4378–4395. doi: 10.1096/fj.09-153007
Qin, H., Shao, Q., Igdoura, S. A., Alaoui-Jamali, M. A., and Laird, D. W. (2003). Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J. Biol. Chem. 278, 30005–30014. doi: 10.1074/jbc.M300614200
Quintáns, B., Martínez-Montero, P., Ordoñez-Ugalde, A., Esteban-Pérez, J., Sanz, I., Escudero, A., et al. (2013). “Oculodentodigital dysplasia: variable neurological phenotype associated with the p.V85M mutation in connexin 43,” in International Conference on Spinocerebellar Degenerations (Paris), 39
Rackauskas, M., Neverauskas, V., and Skeberdis, V. A. (2010). Diversity and properties of connexin gap junction channels. Medicina 46, 1–12.
Rana, S., and Dringen, R. (2007). Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 415, 45–48. doi: 10.1016/j.neulet.2006.12.043
Reid, C. A., Berkovic, S. F., and Petrou, S. (2009). Mechanisms of human inherited epilepsies. Prog. Neurobiol. 87, 41–57. doi: 10.1016/j.pneurobio.2008.09.016
Retamal, M. A., Cortes, C. J., Reuss, L., Bennett, M. V., and Saez, J. C. (2006). S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. U.S.A. 103, 4475–4480. doi: 10.1073/pnas.0511118103
Retamal, M. A., Froger, N., Palacios-Prado, N., Ezan, P., Saez, P. J., Saez, J. C., et al. (2007). Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 27, 13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007
Risek, B., Guthrie, S., Kumar, N., and Gilula, N. B. (1990). Modulation of gap junction transcript and protein expression during pregnancy in the rat. J. Cell Biol. 110, 269–282. doi: 10.1083/jcb.110.2.269
Roscoe, W., Veitch, G. I., Gong, X. Q., Pellegrino, E., Bai, D., McLachlan, E., et al. (2005). Oculodentodigital dysplasia-causing connexin43 mutants are non-functional and exhibit dominant effects on wild-type connexin43. J. Biol. Chem. 280, 11458–11466. doi: 10.1074/jbc.M409564200
Rouach, N., Calvo, C. F., Duquennoy, H., Glowinski, J., and Giaume, C. (2004). Hydrogen peroxide increases gap junctional communication and induces astrocyte toxicity: regulation by brain macrophages. Glia 45, 28–38. doi: 10.1002/glia.10300
Rouach, N., Koulakoff, A., Abudara, V., Willecke, K., and Giaume, C. (2008). Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322, 1551–1555. doi: 10.1126/science.1164022
Roussel, B. D., Kruppa, A. J., Miranda, E., Crowther, D. C., Lomas, D. A., and Marciniak, S. J. (2013). Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 12, 105–118. doi: 10.1016/S1474-4422(12)70238-7
Saez, J. C., Berthoud, V. M., Branes, M. C., Martinez, A. D., and Beyer, E. C. (2003). Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400.
Saidi Brikci-Nigassa, A., Clement, M. J., Ha-Duong, T., Adjadj, E., Ziani, L., Pastre, D., et al. (2012). Phosphorylation controls the interaction of the connexin43 C-terminal domain with tubulin and microtubules. Biochemistry 51, 4331–4342. doi: 10.1021/bi201806j
Sanchez, H. A., Mese, G., Srinivas, M., White, T. W., and Verselis, V. K. (2010). Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J. Gen. Physiol. 136, 47–62. doi: 10.1085/jgp.201010433
Sato, P. Y., Coombs, W., Lin, X., Nekrasova, O., Green, K. J., Isom, L. L., et al. (2011). Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ. Res. 109, 193–201. doi: 10.1161/CIRCRESAHA.111.247023
Scemes, E. (2012). Nature of plasmalemmal functional “hemichannels”. Biochim. Biophys. Acta 1818, 1880–1883. doi: 10.1016/j.bbamem.2011.06.005
Seki, A., Coombs, W., Taffet, S. M., and Delmar, M. (2004). Loss of electrical communication, but not plaque formation, after mutations in the cytoplasmic loop of connexin43. Heart Rhythm 1, 227–233. doi: 10.1016/j.hrthm.2004.03.066
Shaikh, S. B., Uy, B., Perera, A., and Nicholson, L. F. (2012). AGEs-RAGE mediated up-regulation of connexin43 in activated human microglial CHME-5 cells. Neurochem. Int. 60, 640–651. doi: 10.1016/j.neuint.2012.02.023
Shao, Q., Liu, Q., Lorentz, R., Gong, X. Q., Bai, D., Shaw, G. S., et al. (2012). Structure and functional studies of N-terminal Cx43 mutants linked to oculodentodigital dysplasia. Mol. Biol. Cell 23, 3312–3321. doi: 10.1091/mbc.E12-02-0128
Sheean, G., and McGuire, J. R. (2009). Spastic hypertonia and movement disorders: pathophysiology, clinical presentation, and quantification. PM R 1, 827–833. doi: 10.1016/j.pmrj.2009.08.002
Shibayama, J., Gutierrez, C., Gonzalez, D., Kieken, F., Seki, A., Carrion, J. R., et al. (2006). Effect of charge substitutions at residue his-142 on voltage gating of connexin43 channels. Biophys. J. 91, 4054–4063. doi: 10.1529/biophysj.106.085787
Shibayama, J., Paznekas, W., Seki, A., Taffet, S., Jabs, E. W., Delmar, M., et al. (2005). Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ. Res. 96, e83–e91. doi: 10.1161/01.RES.0000168369.79972.d2
Shintani-Ishida, K., Uemura, K., and Yoshida, K. (2007). Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 293, H1714–H1720. doi: 10.1152/ajpheart.00022.2007
Simard, M., Arcuino, G., Takano, T., Liu, Q. S., and Nedergaard, M. (2003). Signaling at the gliovascular interface. J. Neurosci. 23, 9254–9262.
Skerrett, I. M., Aronowitz, J., Shin, J. H., Cymes, G., Kasperek, E., Cao, F. L., et al. (2002). Identification of amino acid residues lining the pore of a gap junction channel. J. Cell Biol. 159, 349–360. doi: 10.1083/jcb.200207060
Sokolova, I. V., Martinez, A. M., and Fletcher, W. H. (2002). The highly conserved Gln49 and Ser50 of mammalian connexin43 are present in chick connexin43 and essential for functional gap junction channels. Cell Commun. Adhes. 9, 75–86. doi: 10.1080/15419060214149
Solan, J. L., and Lampe, P. D. (2007). Key connexin 43 phosphorylation events regulate the gap junction life cycle. J. Membr. Biol. 217, 35–41. doi: 10.1007/s00232-007-9035-y
Spray, D. C., Ye, Z. C., and Ransom, B. R. (2006). Functional connexin “hemichannels”: a critical appraisal. Glia 54, 758–773. doi: 10.1002/glia.20429
Stehberg, J., Moraga-Amaro, R., Salazar, C., Becerra, A., Echeverria, C., Orellana, J. A., et al. (2012). Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 26, 3649–3657. doi: 10.1096/fj.11-198416
Steinhauser, C., and Seifert, G. (2012). “Astrocyte dysfunction in epilepsy,” in: Jasper's Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: Oxford University Press), 606–617. doi: 10.1093/med/9780199746545.003.0047
Sumi, N., Nishioku, T., Takata, F., Matsumoto, J., Watanabe, T., Shuto, H., et al. (2010). Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell. Mol. Neurobiol. 30, 247–253. doi: 10.1007/s10571-009-9446-7
Takano, T., Tian, G. F., Peng, W., Lou, N., Libionka, W., Han, X., et al. (2006). Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267. doi: 10.1038/nn1623
Takeuchi, H., Jin, S., Suzuki, H., Doi, Y., Liang, J., Kawanokuchi, J., et al. (2008). Blockade of microglial glutamate release protects against ischemic brain injury. Exp. Neurol. 214, 144–146. doi: 10.1016/j.expneurol.2008.08.001
Takeuchi, H., Mizoguchi, H., Doi, Y., Jin, S., Noda, M., Liang, J., et al. (2011). Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer's disease. PLoS ONE 6:e21108. doi: 10.1371/journal.pone.0021108
Tang, Q., Dowd, T. L., Verselis, V. K., and Bargiello, T. A. (2009). Conformational changes in a pore-forming region underlie voltage-dependent “loop gating” of an unapposed connexin hemichannel. J. Gen. Physiol. 133, 555–570. doi: 10.1085/jgp.200910207
Theis, M., Jauch, R., Zhuo, L., Speidel, D., Wallraff, A., Doring, B., et al. (2003). Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J. Neurosci. 23, 766–776.
Theodoric, N., Bechberger, J. F., Naus, C. C., and Sin, W. C. (2012). Role of gap junction protein connexin43 in astrogliosis induced by brain injury. PLoS ONE 7:e47311. doi: 10.1371/journal.pone.0047311
Thevenin, A. F., Kowal, T. J., Fong, J. T., Kells, R. M., Fisher, C. G., and Falk, M. M. (2013). Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology (Bethesda) 28, 93–116. doi: 10.1152/physiol.00038.2012
Thibodeau, I. L., Xu, J., Li, Q., Liu, G., Lam, K., Veinot, J. P., et al. (2010). Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation 122, 236–244. doi: 10.1161/CIRCULATIONAHA.110.961227
Tong, D., Li, T. Y., Naus, K. E., Bai, D., and Kidder, G. M. (2007). In vivo analysis of undocked connexin43 gap junction hemichannels in ovarian granulosa cells. J. Cell Sci. 120, 4016–4024. doi: 10.1242/jcs.011775
Torres, A., Wang, F., Xu, Q., Fujita, T., Dobrowolski, R., Willecke, K., et al. (2012). Extracellular Ca(2). acts as a mediator of communication from neurons to glia. Sci. Signal. 5, ra8. doi: 10.1126/scisignal.2002160
Trexler, E. B., Bennett, M. V., Bargiello, T. A., and Verselis, V. K. (1996). Voltage gating and permeation in a gap junction hemichannel. Proc. Natl. Acad. Sci. U.S.A. 93, 5836–5841. doi: 10.1073/pnas.93.12.5836
Unger, T., Bette, S., Zhang, J., Theis, M., and Engele, J. (2012). Connexin-deficiency affects expression levels of glial glutamate transporters within the cerebrum. Neurosci. Lett. 506, 12–16. doi: 10.1016/j.neulet.2011.10.032
Unger, V. M., Kumar, N. M., Gilula, N. B., and Yeager, M. (1997). Projection structure of a gap junction membrane channel at 7 A resolution. Nat. Struct. Biol. 4, 39–43. doi: 10.1038/nsb0197-39
Unger, V. M., Kumar, N. M., Gilula, N. B., and Yeager, M. (1999). Three-dimensional structure of a recombinant gap junction membrane channel. Science 283, 1176–1180. doi: 10.1126/science.283.5405.1176
Van Norstrand, D. W., Asimaki, A., Rubinos, C., Dolmatova, E., Srinivas, M., Tester, D. J., et al. (2012). Connexin43 mutation causes heterogeneous gap junction loss and sudden infant death. Circulation 125, 474–481. doi: 10.1161/CIRCULATIONAHA.111.057224
Vanslyke, J. K., Naus, C. C., and Musil, L. S. (2009). Conformational maturation and post-ER multisubunit assembly of gap junction proteins. Mol. Biol. Cell 20, 2451–2463. doi: 10.1091/mbc.E09-01-0062
Verselis, V. K., Trelles, M. P., Rubinos, C., Bargiello, T. A., and Srinivas, M. (2009). Loop gating of connexin hemichannels involves movement of pore-lining residues in the first extracellular loop domain. J. Biol. Chem. 284, 4484–4493. doi: 10.1074/jbc.M807430200
Vezzani, A., Auvin, S., Ravizza, T., and Aronica, E. (2012). “Glia-neuronal interactions in ictogenesis and epileptogenesis: role of inflammatory mediators,” in Jasper's Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: Oxford University Press), 618–634. doi: 10.1093/med/9780199746545.003.0048
Wallraff, A., Kohling, R., Heinemann, U., Theis, M., Willecke, K., and Steinhauser, C. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26, 5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006
Wang, N., De Bock, M., Antoons, G., Gadicherla, A. K., Bol, M., Decrock, E., et al. (2012a). Connexin mimetic peptides inhibit Cx43 hemichannel opening triggered by voltage and intracellular Ca2+ elevation. Basic Res. Cardiol. 107, 304. doi: 10.1007/s00395-012-0304-2
Wang, X., Bukoreshtliev, N. V., and Gerdes, H. H. (2012b). Developing neurons form transient nanotubes facilitating electrical coupling and calcium signaling with distant astrocytes. PLoS ONE 7:e47429. doi: 10.1371/journal.pone.0047429
Wang, N., De Vuyst, E., Ponsaerts, R., Boengler, K., Palacios-Prado, N., Wauman, J., et al. (2013a). Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 108, 309. doi: 10.1007/s00395-012-0309-x
Wang, X., Ma, A., Zhu, W., Zhu, L., Zhao, Y., Xi, J., et al. (2013b). The role of connexin 43 and hemichannels correlated with the astrocytic death following ischemia/reperfusion insult. Cell. Mol. Neurobiol. 33, 401–410. doi: 10.1007/s10571-013-9906-y
Wasseff, S., Abrams, C. K., and Scherer, S. S. (2010). A dominant connexin43 mutant does not have dominant effects on gap junction coupling in astrocytes. Neuron Glia Biol. 6, 213–223. doi: 10.1017/S1740925X11000019
Wei, C. J., Xu, X., and Lo, C. W. (2004). Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 20, 811–838. doi: 10.1146/annurev.cellbio.19.111301.144309
Weissman, T. A., Riquelme, P. A., Ivic, L., Flint, A. C., and Kriegstein, A. R. (2004). Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43, 647–661. doi: 10.1016/j.neuron.2004.08.015
Wiencken-Barger, A. E., Djukic, B., Casper, K. B., and McCarthy, K. D. (2007). A role for Connexin43 during neurodevelopment. Glia 55, 675–686. doi: 10.1002/glia.20484
Willoughby, J. O., Mackenzie, L., Broberg, M., Thoren, A. E., Medvedev, A., Sims, N. R., et al. (2003). Fluorocitrate-mediated astroglial dysfunction causes seizures. J. Neurosci. Res. 74, 160–166. doi: 10.1002/jnr.10743
Wong, C. W., Christen, T., Roth, I., Chadjichristos, C. E., Derouette, J. P., Foglia, B. F., et al. (2006). Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 12, 950–954. doi: 10.1038/nm1441
Xu, H. L., Mao, L., Ye, S., Paisansathan, C., Vetri, F., and Pelligrino, D. A. (2008). Astrocytes are a key conduit for upstream signaling of vasodilation during cerebral cortical neuronal activation in vivo. Am. J. Physiol. Heart Circ. Physiol. 294, H622–H632. doi: 10.1152/ajpheart.00530.2007
Yahuaca, P., Ek-Vitorin, J. F., Rush, P., Delmar, M., and Taffet, S. M. (2000). Identification of a protein kinase activity that phosphorylates connexin43 in a pH-dependent manner. Braz. J. Med. Biol. Res. 33, 399–406. doi: 10.1590/S0100-879X2000000400005
Ye, Z. C., Wyeth, M. S., Baltan-Tekkok, S., and Ransom, B. R. (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 23, 3588–3596.
Yoon, J. J., Green, C. R., O'Carroll, S. J., and Nicholson, L. F. (2010). Dose-dependent protective effect of connexin43 mimetic peptide against neurodegeneration in an ex vivo model of epileptiform lesion. Epilepsy Res. 92, 153–162. doi: 10.1016/j.eplepsyres.2010.08.014
Zhou, X. W., Pfahnl, A., Werner, R., Hudder, A., Llanes, A., Luebke, A., et al. (1997). Identification of a pore lining segment in gap junction hemichannels. Biophys. J. 72, 1946–1953. doi: 10.1016/S0006-3495(97)78840-4
Zhou, Y., Yang, W., Lurtz, M. M., Ye, Y., Huang, Y., Lee, H. W., et al. (2007). Identification of the calmodulin binding domain of connexin 43. J. Biol. Chem. 282, 35005–35017. doi: 10.1074/jbc.M707728200
Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat. Rev. Neurosci. 12, 723–738.
Keywords: oculodentodigital dysplasia, Cx43, gap junction, hemichannel, astrocyte
Citation: De Bock M, Kerrebrouck M, Wang N and Leybaert L (2013) Neurological manifestations of oculodentodigital dysplasia: a Cx43 channelopathy of the central nervous system? Front. Pharmacol. 4:120. doi: 10.3389/fphar.2013.00120
Received: 13 May 2013; Paper pending published: 04 June 2013;
Accepted: 02 September 2013; Published online: 26 September 2013.
Edited by:
Aida Salameh, Heart Centre University of Leipzig, GermanyReviewed by:
Heike Wulff, University of California, Davis, USACharles K. Abrams, SUNY Downstate, USA
Copyright © 2013 De Bock, Kerrebrouck, Wang and Leybaert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luc Leybaert, Physiology Group, Department of Basic Medical Sciences, Faculty of Medicine and Health Sciences, Ghent University, De Pintelaan 185, Building B (Rm 310), 9000 Ghent, Belgium e-mail:bHVjLmxleWJhZXJ0QHVnZW50LmJl
†These authors have contributed equally to this work.