 Dagmar Kubitza
Dagmar Kubitza Elisabeth Perzborn
Elisabeth Perzborn Scott D. Berkowitz
Scott D. Berkowitz- 1Clinical Pharmacology, Bayer HealthCare AG, Wuppertal, Germany
- 2Formerly Global Drug Discovery, Bayer Pharma AG, Wuppertal, Germany
- 3Bayer HealthCare Pharmaceuticals, Whippany, NJ, USA
Direct oral anticoagulants that target a single coagulation factor (such as factor Xa or thrombin) have been developed in recent years in an attempt to address some of the limitations of traditional anticoagulants. Rivaroxaban is an oral, direct factor Xa inhibitor that inhibits free and clot-bound factor Xa and factor Xa in the prothrombinase complex. Preclinical studies demonstrated a potent anticoagulant effect of rivaroxaban in plasma as well as the ability of this agent to prevent and treat venous and arterial thrombosis in animal models. These studies led to an extensive phase I clinical development program that investigated the pharmacological properties of rivaroxaban in humans. In these studies, rivaroxaban was shown to exhibit predictable pharmacokinetics and pharmacodynamics and to have no clinically relevant interactions with many commonly prescribed co-medications. The pharmacodynamic effects of rivaroxaban (for example, inhibition of factor Xa and prolongation of prothrombin time) were closely correlated with rivaroxaban concentrations in plasma. The encouraging findings from preclinical and early clinical studies were expanded upon in large, randomized phase III studies, which demonstrated the clinical efficacy and safety of rivaroxaban in a broad spectrum of patients. This article provides an overview of the discovery and development of rivaroxaban, describing the pharmacodynamic profile established in preclinical studies and the optimal translation to clinical studies in healthy subjects and patient populations.
Introduction
Anticoagulant drugs are routinely used for the prevention and treatment of thromboembolic disorders. Although effective, traditional anticoagulant agents (which have a broad effect on multiple coagulation factors) are associated with several limitations. For example, heparins require a parenteral route of administration, and unfractionated heparin and vitamin K antagonists have significant variability in their pharmacodynamic responses, thus requiring routine coagulation monitoring and dose adjustments (McRae and Ginsberg, 2004; Ageno et al., 2012; Garcia et al., 2012). These shortcomings have spurred the search for novel agents that specifically target a single clotting factor within the coagulation cascade, such as factor Xa or thrombin.
Factor X has long been known to have a key role in hemostasis, and its activated form, factor Xa, has a significant function in the blood coagulation pathway because it catalyzes the production of thrombin, which leads to clot formation (Koller, 1960; Leadley, 2001). Activation of factor X to factor Xa occurs through both the intrinsic and extrinsic pathways of the coagulation cascade. Factor Xa initiates the final, common pathway that results in thrombin activation via the prothrombinase complex (Figure 1). It has been estimated that one molecule of factor Xa can catalyze the production of ~1000 molecules of thrombin because of the amplification inherent in the coagulation cascade (Rand et al., 1996; Mann et al., 2003; Kubitza and Haas, 2006; Perzborn et al., 2011); therefore, effective thrombin inhibition requires sustained high levels of free inhibitor under conditions of thrombin “burst.” These requirements are expected to be less stringent for factor Xa, because factor X is present in plasma at much lower concentrations compared with prothrombin (Brummel-Ziedins et al., 2012). Factor Xa is, therefore, an attractive target for anticoagulant agents.
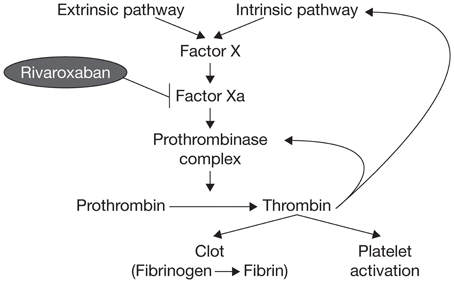
Figure 1. The blood coagulation pathway.
Early studies of naturally occurring factor Xa inhibitors indicated that targeting factor Xa could provide effective anticoagulation (Dunwiddie et al., 1989; Nicolini et al., 1996). Further studies with the synthetic indirect factor Xa inhibitor fondaparinux provided the proof of principle that selective inhibition of factor Xa could provide clinically effective anticoagulation (Turpie et al., 2002). Selective inhibition of factor Xa produces antithrombotic effects by decreasing the generation of thrombin, thus diminishing thrombin-mediated activation of both coagulation and platelets without affecting the activity of existing thrombin (Ieko et al., 2004).
Among the novel, direct, oral factor Xa inhibitors that have been developed in recent years, rivaroxaban was the first to gain regulatory approval for clinical use (which occurred in 2008). The pharmacodynamic effects of rivaroxaban were first shown in preclinical studies, and were subsequently demonstrated in early clinical studies in humans. The promising results from the early phase of development were confirmed in large-scale phase III studies, in which the efficacy and safety of rivaroxaban were demonstrated. Rivaroxaban is now approved for clinical use for the prevention and treatment of venous and arterial thromboembolic disorders (Bayer Pharma AG, 2013). This article will provide an overview of the discovery and development of rivaroxaban, describing the pharmacodynamic profile established in preclinical studies and the optimal translation to clinical studies in healthy subjects and patient populations.
Preclinical Pharmacodynamic Profile
High-throughput screening of approximately 200,000 compounds revealed several hits that selectively inhibited the cleavage of a chromogenic substrate by human factor Xa. The subsequent optimization program led to the identification of rivaroxaban as the lead compound for further development because of its high binding affinity to factor Xa, potency in vitro, and in vivo antithrombotic activity, as well as its favorable oral bioavailability (Perzborn et al., 2011).
The mode of action of rivaroxaban is the direct and specific competitive inhibition of factor Xa (inhibition constant [Ki] 0.4 ± 0.02 nM), with >10,000-fold selectivity for factor Xa over other serine proteases (Perzborn et al., 2005). Unlike the indirect factor Xa inhibitor fondaparinux, or low molecular weight heparins and unfractionated heparin, which exert their actions via antithrombin, rivaroxaban directly inhibits prothrombinase (half maximal inhibitory concentration [IC50] 2.1 ± 0.40 nM). In human plasma, rivaroxaban also inhibits the activity of clot-bound factor Xa (IC50 75 nM) in a concentration-dependent manner (Depasse et al., 2005).
In whole blood and platelet-rich human plasma, nanomolar concentrations of rivaroxaban significantly prolonged the initiation phase of thrombin generation (TG), significantly reduced the rate of the propagation phase of coagulation after tissue factor (TF) activation, and led to a reduction in endogenous thrombin potential (ETP)—a measure of the activity of thrombin multiplied by the time for which it remains active (Gerotziafas et al., 2007). Rivaroxaban reduced the ETP by 50% at a concentration of 35 nM; TG was almost completely inhibited in platelet-rich plasma at physiologically relevant concentrations (80–100 nM) of rivaroxaban (Gerotziafas et al., 2007). In platelet-poor human plasma, rivaroxaban prolonged prothrombin time (PT; assessed using Neoplastin Plus® from Roche Diagnostics, Mannheim, Germany) in a concentration-dependent manner; the assay concentration required to double the PT was ~691 nM (Perzborn et al., 2005).
The promising data on the pharmacodynamic effects of rivaroxaban obtained in these coagulation tests were supported by studies in animal models of venous and arterial thrombosis. In a rat venous stasis model, PT and factor Xa activity were affected slightly at the half maximal effective dose (ED50) of rivaroxaban (1.8-fold increase and 32% inhibition, respectively) and, at a dose leading to almost complete inhibition of thrombus formation (0.3 mg/kg), rivaroxaban prolonged PT moderately (3.2 ± 0.5-fold) and inhibited factor Xa activity (65 ± 3%) (Perzborn et al., 2005). In a rat arteriovenous shunt model, oral rivaroxaban inhibited factor Xa activity by 74% and prolonged PT by 3.2-fold; in a rabbit arteriovenous shunt model, factor Xa activity was almost completely inhibited (92%) by oral rivaroxaban, but PT was prolonged only slightly (1.2-fold), at the respective ED50 values (Perzborn et al., 2005). These pharmacodynamic effects of rivaroxaban were mirrored by its antithrombotic activity. Rivaroxaban reduced thrombus formation in a dose-dependent manner after intravenous administration in the rat venous stasis model, and after oral administration in the rat and rabbit arteriovenous shunt models (Figure 2) (ED50 0.1, 5.0, and 0.6 mg/kg, respectively), providing the first demonstration that rivaroxaban is effective in both arterial and venous thrombosis (Perzborn et al., 2005). In addition to prophylactic use, results obtained in an experimental rabbit jugular vein thrombosis model found that oral rivaroxaban (3.0 mg/kg) significantly reduced thrombus growth compared with oral control (36.9 ± 1.7% vs. 46.3 ± 1.3%; p < 0.05), demonstrating the potential of rivaroxaban to treat established thrombi (Biemond et al., 2007). No significant increase in bleeding time was observed at antithrombotic-effective doses (Perzborn et al., 2005; Biemond et al., 2007).
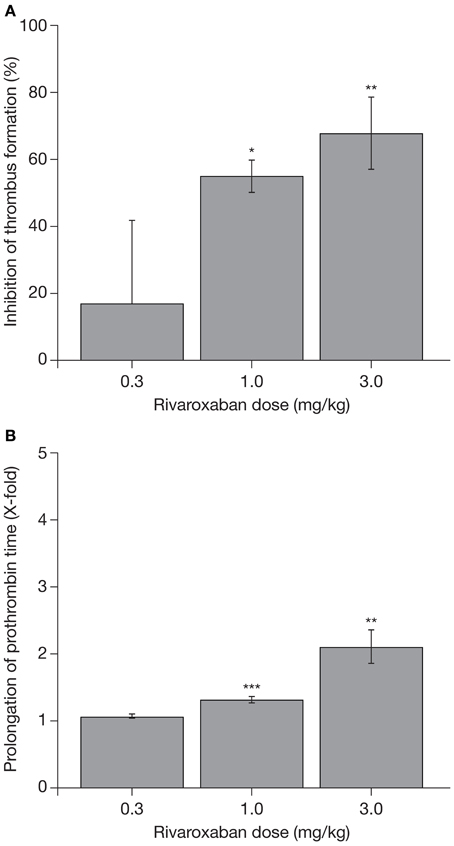
Figure 2. (A) Reduction in thrombus formation and (B) prolongation of prothrombin time with rivaroxaban in a rabbit arteriovenous shunt model. Each value represents the mean ± SEM of six animals. *p < 0.05; **p < 0.01; ***p < 0.001. Data published previously in the Journal of Thrombosis and Haemostasis (Perzborn et al., 2005).
Pharmacodynamic Effect in Clinical Studies
Phase I Studies
Results of single- and multiple-dose escalation studies in healthy male subjects were consistent with those obtained in preclinical studies, confirming the anticoagulation effects of rivaroxaban in humans (Kubitza et al., 2005a,b). In the single-dose study, factor Xa activity was inhibited in a dose-dependent manner, and up to 75% inhibition was achieved with a single 80 mg dose of rivaroxaban (Kubitza et al., 2005a). Maximal factor Xa inhibition with suspension and tablet formulations was achieved after 45 min and 1–4 h, respectively (Kubitza et al., 2005a). Factor Xa activity was inhibited even at 24 h after administration at doses >5 mg (Kubitza et al., 2005a). Rivaroxaban prolonged the PT in a dose-dependent manner (assessed using Neoplastin Plus® from Roche Diagnostics, as in the preclinical studies). The inhibition of factor Xa activity and prolongation of PT both correlated strongly with plasma concentrations of rivaroxaban (r = 0.949 and r = 0.935, respectively) (Kubitza et al., 2005a).
After multiple dosing, maximal inhibition of factor Xa and prolongation of PT did not show cumulative effects, illustrating predictable pharmacodynamics with repeated dosing (Kubitza et al., 2005b).
In healthy male subjects, rivaroxaban (5 or 30 mg) dose-dependently inhibited TG in response to TF or collagen stimulation in platelet-rich and platelet-poor plasma, suggesting effective inhibition of TG induced by the extrinsic and intrinsic coagulation pathways (Graff et al., 2007, 2008). All parameters of ETP [area under the plasma concentration–time curve (AUC), peak, lag time] were significantly affected by rivaroxaban 5 and 30 mg doses, with the maximal effect achieved at 2 h (Graff et al., 2007, 2008). ETP peak (activated by collagen) remained reduced by 40% at 24 h after administration of a 30 mg dose (Graff et al., 2007, 2008); this was the first evidence to support the concept of once-daily administration (Graff et al., 2007, 2008).
Phase II and III Studies
In phase II studies investigating rivaroxaban compared with enoxaparin for the prevention of venous thromboembolism (VTE) after hip or knee replacement surgery, rivaroxaban exhibited predictable pharmacodynamics with both once- and twice-daily dosing (Turpie et al., 2006; Eriksson et al., 2007a). As was observed in phase I studies in healthy subjects, factor Xa inhibition and PT prolongation correlated closely with rivaroxaban plasma concentrations (Turpie et al., 2006; Eriksson et al., 2007b). The correlation of PT prolongation with rivaroxaban plasma concentration was also demonstrated in a phase II study, ODIXa-DVT, which investigated the optimal dose of rivaroxaban for the treatment of acute, proximal deep vein thrombosis (Agnelli et al., 2007; Mueck et al., 2007). These data demonstrated that the pharmacodynamic effects of rivaroxaban, as shown for PT prolongation, were consistent between the preclinical and clinical studies performed in healthy volunteers and patients, when the same assay methods were used (e.g., Neoplastin Plus® from Roche Diagnostics for PT) (Table 1). The consistent response among different patient populations and the low-to-moderate inter-individual variability in the exposure–PT relationship confirmed the predictable pharmacodynamics of rivaroxaban (Mueck et al., 2008, 2011). Furthermore, the observed pharmacodynamic effects in these early studies were successfully translated into clinical efficacy, as demonstrated in a number of large phase III studies in several thromboembolic indications, including the prevention of VTE after elective hip or knee replacement surgery (Eriksson et al., 2008; Kakkar et al., 2008; Lassen et al., 2008; Turpie et al., 2009), treatment of deep vein thrombosis and pulmonary embolism and prevention of recurrent VTE (The EINSTEIN Investigators, 2010; The EINSTEIN–PE Investigators, 2012), stroke prevention in patients with non-valvular atrial fibrillation (Patel et al., 2011), and secondary prevention in patients with acute coronary syndrome (Mega et al., 2012). In these phase III studies, rivaroxaban was shown to be superior or non-inferior to standard of care therapies (Table 2) with a similar or improved safety profile (including major bleeding events). The favorable benefit–risk profile of rivaroxaban was also evident across patient subgroups.
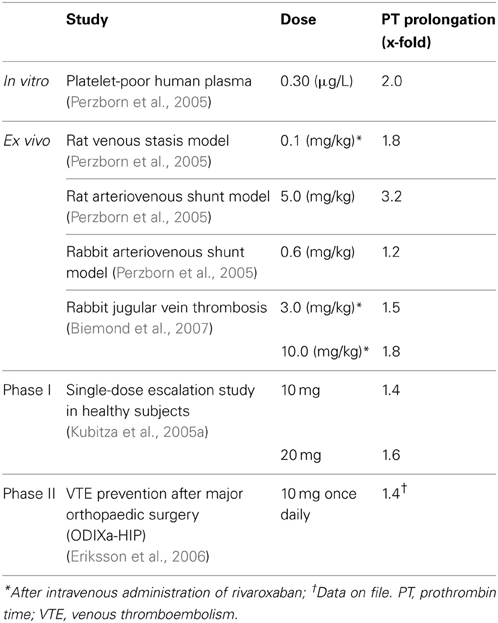
Table 1. Prolongation of PT in preclinical and clinical studies of rivaroxaban.
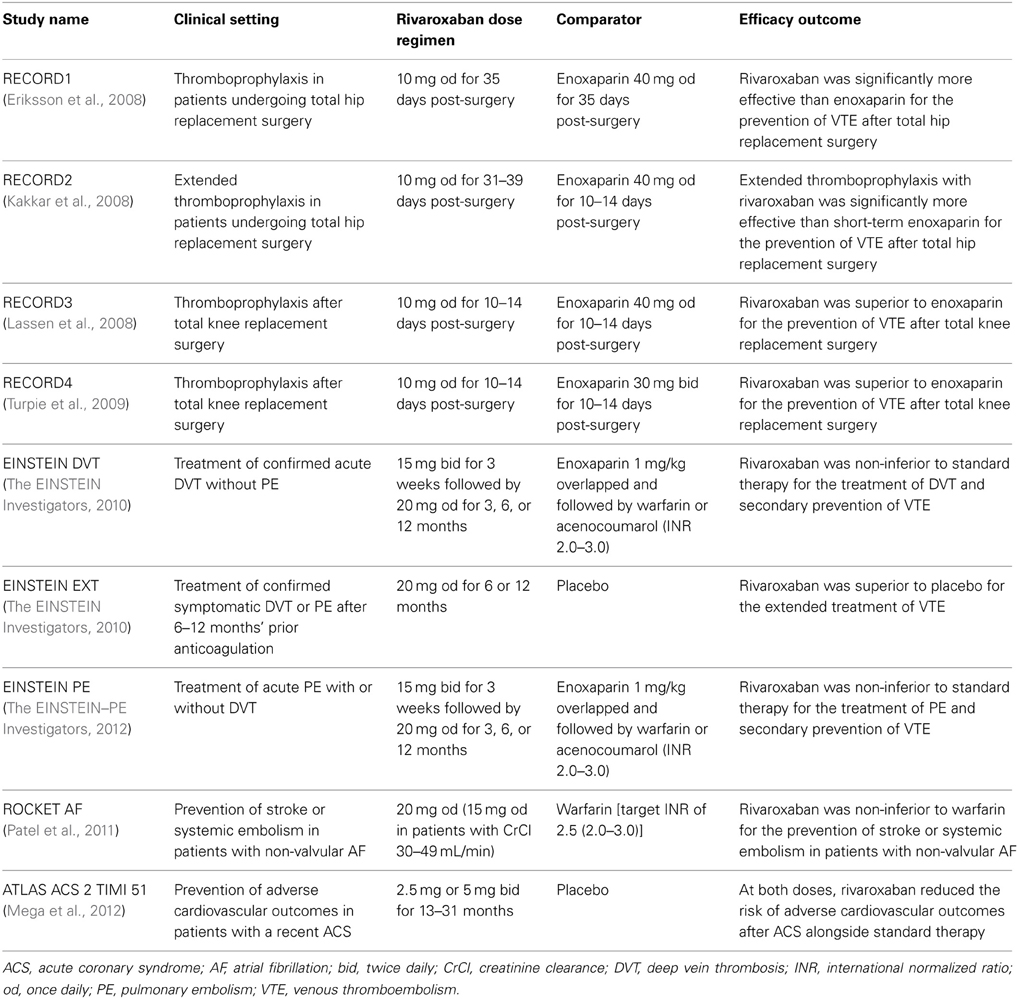
Table 2. Clinical evaluation of rivaroxaban in phase III studies.
Pharmacokinetic Profile and Clinical Relevance
Preclinical animal studies have shown rivaroxaban to have predictable pharmacokinetic properties. Rivaroxaban had a rapid absorption, a moderate or high absolute bioavailability in rats (57–66%) and dogs (60–86%), and dose-proportional plasma concentrations with small variability between animals for the tested dose-interval (1.0–10.0 mg/kg in rats, 0.3–3.0 mg/kg in dogs) (Weinz et al., 2005). Rivaroxaban also showed a low plasma clearance rate, a moderate volume of distribution, and a short elimination half-life (Weinz et al., 2005). The tissue affinity of rivaroxaban was moderate, with only small or no retention observed; however, a species-dependent and fully reversible high plasma protein binding was observed (Weinz et al., 2005).
Rivaroxaban has a dual route of excretion through the biliary/fecal and urinary pathways, with a recovery of total radioactivity >91% being observed in all species tested. Urinary excretion of radioactivity was 25 and 52%, and fecal excretion was 67 and 43% of the dose in rats and dogs, respectively (Weinz et al., 2005). In humans, 36% of the administered dose was excreted renally as unchanged drug, whereas 28% of the dose was excreted in the feces (Weinz et al., 2009). Unchanged rivaroxaban was the main component in human plasma, with no major or active circulating metabolites present (Weinz et al., 2005, 2009; Bayer Pharma AG, 2013).
In phase I studies in healthy male subjects, rivaroxaban showed rapid absorption after oral administration and reached maximum plasma concentration (Cmax) within 4 h (Kubitza et al., 2005a,b). Exposure to rivaroxaban, in terms of the AUC and Cmax, was dose proportional for doses up to 10 mg irrespective of food intake, and dose proportional at all doses up to 30 mg twice-daily if administered with food (Kubitza et al., 2005a,b). No substantial accumulation of rivaroxaban at steady state was detected in the multiple-dose study (Kubitza et al., 2005b). The oral bioavailability of rivaroxaban was high (80–100%) for the 10 mg tablet dose, regardless of food intake. Under fasting conditions, absorption of a 20 mg tablet decreased to 66%, but approached completeness when the tablet was administered with food (Stampfuss et al., 2013). The terminal half-life of rivaroxaban ranged between 5 and 9 h in healthy young subjects and between ~11 and 13 h in elderly subjects (Kubitza et al., 2005a,b, 2008).
Interactions with Drugs Affecting the Cytochrome P450 3A4 and P-Glycoprotein Pathways
Rivaroxaban is metabolized via cytochrome P450 (CYP) 3A4, CYP2J2, and CYP-independent mechanisms (Bayer Pharma AG, 2013; Mueck et al., 2013). Based on in vitro investigations, it was shown that rivaroxaban is a substrate of the transporter proteins P-glycoprotein (P-gp) and breast cancer resistance protein. Consequently, drugs that affect CYP3A4, P-gp, or breast cancer resistance protein are expected to influence the pharmacokinetics of rivaroxaban (Gnoth et al., 2011; Mueck et al., 2013).
In phase I studies in healthy subjects, substrates of CYP3A4 or P-gp (such as the cardiac glycoside digoxin, the statin atorvastatin or the benzodiazepine midazolam) had no clinically relevant effect on the plasma pharmacokinetics of rivaroxaban (Kubitza et al., 2012b; Bayer Pharma AG, 2013; Mueck et al., 2013). Substances strongly inhibiting only one pathway of elimination (such as the antibiotics clarithromycin and erythromycin and the antifungal agent fluconazole) caused a modest increase in rivaroxaban exposure (54, 34, and 42% increases, respectively), but this did not result in clinically relevant increases in exposure that would necessitate dose adaptations (Bayer Pharma AG, 2013; Mueck et al., 2013). However, rivaroxaban exposure was found to be significantly increased when it was co-administered with strong inhibitors of both P-gp and CYP3A4, such as ketoconazole (158% increase in rivaroxaban exposure) and ritonavir (153% increase in rivaroxaban exposure) (Bayer Pharma AG, 2013; Mueck et al., 2013). The use of rivaroxaban is, therefore, not recommended in patients receiving systemic azole-antimycotics (such as ketoconazole, itraconazole, voriconazole, and posaconazole) or HIV protease inhibitors (such as ritonavir) (Bayer Pharma AG, 2013). Co-administration of the antibiotic rifampicin, a strong CYP3A4 inducer, led to an approximately 50% decrease in the AUC of rivaroxaban. It is likely that other strong CYP3A4 inducers (such as the anticonvulsants phenytoin, carbamazepine, and phenobarbital, and the herbal supplement St John's wort) may lead to reduced plasma concentrations of rivaroxaban (Bayer Pharma AG, 2013). These agents should, therefore, be co-administered with rivaroxaban with caution (Bayer Pharma AG, 2013).
Effect of Age, Sex, Body Weight, and Renal Impairment on Pharmacokinetic Parameters
Phase I clinical studies have investigated the effect of age and sex on rivaroxaban pharmacokinetics. In healthy subjects aged >75 years, there was an increase in rivaroxaban exposure after the administration of a single 10 mg dose, as indicated by higher AUC values, compared with those observed in younger subjects (18–45 years), without a significant alteration in Cmax (Kubitza et al., 2013). The AUC values were, on average, 41% greater than those observed in younger individuals (Kubitza et al., 2013). These changes were the result of reduced rivaroxaban clearance in elderly subjects, mainly owing to decreased renal function (Kubitza et al., 2013). Sex had no significant influence on the pharmacokinetics of rivaroxaban (Kubitza et al., 2013).
The influence of body weight on the pharmacokinetics of rivaroxaban was investigated in healthy subjects weighing ≤50 and >120 kg, compared with those with a normal body weight (70–80 kg) (Kubitza et al., 2007b). The Cmax of rivaroxaban was unaffected in subjects >120 kg but was increased by 24% in subjects weighing ≤50 kg, with no effect on AUC. This was not considered clinically significant, suggesting that rivaroxaban is unlikely to require dose adjustment for body weight in adults (Kubitza et al., 2007b). Pharmacokinetic modeling has suggested that dose adjustment may be required in pediatric patients (Willmann et al., 2013); however, although studies are underway (clinicaltrials.gov, NCT01145859), no clinical data are yet available on the use of rivaroxaban in this population, and rivaroxaban is currently not recommended in patients aged <18 years (Bayer Pharma AG, 2013).
Rivaroxaban clearance is decreased with increasing renal impairment, leading to increased exposure. In subjects with mild (creatinine clearance [CrCl], 50–79 mL/min), moderate (CrCl, 30–49 mL/min), or severe (CrCl <30 mL/min) impairment of renal function, the AUCs of rivaroxaban were 44, 52, and 64% higher, respectively, compared with healthy control subjects after administration of a 10 mg rivaroxaban dose (Kubitza et al., 2010). The increase in AUC correlated inversely with CrCl (r = −0.45) (Kubitza et al., 2010). Rivaroxaban Cmax was relatively unaffected, indicating that the increase in AUC was caused by reduced clearance and not by increased absorption (Kubitza et al., 2010). The influence of renal function on rivaroxaban clearance is considered to be moderate (Kubitza et al., 2010).
Clinical Relevance of Pharmacokinetic Parameters
The consistent, predictable pharmacokinetics and pharmacodynamics of rivaroxaban translated into predictable effects in the clinical programs (Table 2). Analysis of data obtained across pre-specified subgroups demonstrated consistent efficacy and safety outcomes irrespective of age or renal function (Eriksson et al., 2008; Kakkar et al., 2008; Lassen et al., 2008; Turpie et al., 2009; The EINSTEIN Investigators, 2010; Patel et al., 2011; Mega et al., 2012; The EINSTEIN–PE Investigators, 2012), including in patients with atrial fibrillation with renal impairment who received a reduced dose of rivaroxaban (15 mg instead of 20 mg) for prevention of stroke and systemic embolism (Patel et al., 2011). It should be noted that patients with CrCl <30 mL/min were excluded from all phase III studies; therefore, because limited data are available, rivaroxaban should be used with caution in patients with severe renal impairment (CrCl 15–29 mL/min) (Bayer Pharma AG, 2013). The use of rivaroxaban is not recommended in patients with CrCl <15 mL/min (Bayer Pharma AG, 2013).
Data from the subgroup analyses further confirmed that rivaroxaban dose adjustment is not required for age, sex, or body weight (Eriksson et al., 2008; Kakkar et al., 2008; Lassen et al., 2008; Turpie et al., 2009; The EINSTEIN Investigators, 2010; Patel et al., 2011; Mega et al., 2012; The EINSTEIN–PE Investigators, 2012).
Pharmacodynamic Interactions with Drugs Affecting Hemostasis
Phase I studies in healthy male subjects evaluated the potential for interactions with commonly prescribed drugs that influence hemostasis, including non-steroidal anti-inflammatory drugs such as naproxen, platelet aggregation inhibitors such as clopidogrel, and the antiplatelet drug acetylsalicylic acid (ASA) (Kubitza et al., 2006, 2007a, 2012a; Bayer Pharma AG, 2013).
The maximum inhibition of factor Xa and prolongation of PT by rivaroxaban (administered as a single 15 mg dose) were unaffected in the presence of naproxen (500 mg) (Kubitza et al., 2007a). Rivaroxaban and naproxen given together significantly increased bleeding time compared with rivaroxaban alone (p = 0.017) (Kubitza et al., 2007a). However, this difference was small compared with the effect of naproxen given alone (Kubitza et al., 2007a).
Co-administration of clopidogrel (300 mg loading dose followed by 75 mg maintenance dose) and rivaroxaban (15 mg, given on day 2) had no effect on rivaroxaban-mediated inhibition of factor Xa or prolongation of PT (Kubitza et al., 2012a). Inhibition of adenosine 5′-diphosphate-stimulated platelet aggregation by clopidogrel was not affected by rivaroxaban (Kubitza et al., 2012a). Bleeding time was increased by clopidogrel, and co-administration of rivaroxaban and clopidogrel further increased bleeding time in a subset of subjects (Kubitza et al., 2012a).
Acetylsalicylic acid (500 mg loading dose followed by 100 mg maintenance dose) did not alter the effects of rivaroxaban (15 mg) on factor Xa activity or prolongation of PT (Kubitza et al., 2006). Bleeding time was significantly prolonged with ASA; the combination of rivaroxaban and ASA prolonged bleeding time slightly more than ASA alone (Kubitza et al., 2006).
These findings suggested a low potential for clinically relevant interactions of rivaroxaban with these commonly prescribed drugs; their use alongside rivaroxaban was investigated further in phase III studies.
In the phase III studies for the prevention of VTE after elective hip or knee replacement surgery (the RECORD program; Table 2), the concomitant use of ASA (no limit on dosage) and other platelet inhibitors with rivaroxaban (10 mg) was permitted (Eriksson et al., 2008, 2012; Kakkar et al., 2008; Lassen et al., 2008; Turpie et al., 2009). The relative rate ratios for any bleeding events, or major or non-major clinically relevant bleeding events, with different platelet inhibitors or ASA co-medication use vs. non-use remained relatively constant and were similar between the rivaroxaban and enoxaparin groups (Eriksson et al., 2012). In the EINSTEIN program (Table 2), ASA (up to 100 mg), and clopidogrel (75 mg per day), or both, were allowed if indicated (The EINSTEIN Investigators, 2010; The EINSTEIN–PE Investigators, 2012). Results obtained in a pooled analysis of patients in the EINSTEIN DVT and EINSTEIN PE studies indicated that co-administration of ASA with rivaroxaban increased the risk of bleeding, with no statistically significant increase in major bleeding, whereas use of non-steroidal anti-inflammatory drugs increased the overall risk of bleeding and the risk of major bleeding (Davidson et al., 2013). However, in general, an increased risk of bleeding complications is expected when combining an anticoagulant and antiplatelet agents, or other agents that affect hemostasis. In the ATLAS ACS 2 TIMI 51 study, increases in major bleeding (not related to coronary artery bypass grafting) and intracranial hemorrhage were observed when a low dose of rivaroxaban (2.5 or 5 mg twice daily) was co-administered with ASA and a thienopyridine (either clopidogrel or ticlopidine), but no increases in fatal bleeding were observed (Mega et al., 2012).
Summary
Rivaroxaban is an oral, direct factor Xa inhibitor with high selectivity for factor Xa, and which has been shown in clotting assays in human plasma to effectively prolong PT, inhibit TG, and reduce both collagen- and TF-induced ETP. The antithrombotic potential of rivaroxaban was first demonstrated in in vivo preclinical studies in both arterial and venous thrombosis models, in which the potential for the treatment of established thrombi was also shown. These, together with data from initial (or early) clinical studies, provided further support that factor Xa is a viable target for anticoagulant therapy.
The pharmacodynamic and pharmacokinetic profiles of rivaroxaban seen in preclinical studies were further demonstrated in phase I and II studies in humans. Here, the rapid onset of action, short half-life, and plasma concentration-dependent effects on coagulation were confirmed. Data obtained during phase I and II studies also confirmed the predictability of rivaroxaban effects across a range of patient populations. Alongside the low potential for interacting with food or other drugs and the wide therapeutic window, the predictable pharmacokinetic and pharmacodynamic profile of rivaroxaban irrespective of age, sex, or body weight obviates the need for routine coagulation monitoring (in certain clinical situations laboratory testing may be helpful, such as prior to urgent surgery), which opens the possibility of offering fixed dosing to a broad range of patient populations.
The promising preclinical data, first confirmed by results of phase I studies, have been successfully translated into clinically meaningful results in patients. The efficacy and safety of rivaroxaban have been demonstrated in large-scale phase III studies in multiple indications, with rivaroxaban having non-inferior or superior efficacy to standards of care. In light of these data, rivaroxaban has been approved for the prevention or treatment of several thromboembolic indications. In order to ensure responsible use in patients, prescribers should adhere to the prescribing information for the respective indications. There are patient populations in which rivaroxaban is contraindicated or not recommended, including patients with severe renal impairment with CrCl <15 mL/min. With effective and safe anticoagulation coupled with oral, fixed dosing, rivaroxaban provides an attractive alternative to traditional anticoagulants and has the potential to simplify the management of thromboembolic disorders.
Author Contributions
Dagmar Kubitza designed, evaluated, and reported the clinical pharmacology studies referenced in this paper. She gave input by providing these data. In addition, she conceptualized the content of the manuscript. Elisabeth Perzborn was involved in the discovery of rivaroxaban and the characterization of the in vitro and in vivo pharmacological profile of rivaroxaban. She provided input by describing the preclinical pharmacodynamic profile. Scott D. Berkowitz contributed to the writing, revisions, and finalization of the manuscript. All authors revised the manuscript and approved it for submission.
Conflict of Interest Statement
Dagmar Kubitza and Scott D. Berkowitz are employees of Bayer HealthCare Pharmaceuticals, Inc. Elisabeth Perzborn is a former employee of and consultant for Bayer HealthCare Pharmaceuticals, Inc., and has stock ownership in Bayer HealthCare Pharmaceuticals, Inc.
Acknowledgments
The authors wish to acknowledge Nicola Hamil and Yong-Ling Liu, who provided editorial support with funding from Bayer HealthCare Pharmaceuticals and Janssen Scientific Affairs, LLC.
References
Ageno, W., Gallus, A. S., Wittkowsky, A., Crowther, M., Hylek, E. M., and Palareti, G. (2012). Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 141, e44S–e88S. doi: 10.1378/chest.11-2292
Agnelli, G., Gallus, A., Goldhaber, S. Z., Haas, S., Huisman, M. V., Hull, R. D., et al. (2007). Treatment of proximal deep-vein thrombosis with the oral direct factor Xa inhibitor rivaroxaban (BAY 59-7939): the ODIXa-DVT (oral direct factor Xa inhibitor BAY 59-7939 in patients with acute symptomatic deep-vein thrombosis) study. Circulation 116, 180–187. doi: 10.1161/CIRCULATIONAHA.106.668020
Bayer Pharma AG. (2013). Xarelto® (rivaroxaban) Summary of Product Characteristics. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000944/WC500057108.pdf
Biemond, B. J., Perzborn, E., Friederich, P. W., Levi, M., Buetehorn, U., and Büller, H. R. (2007). Prevention and treatment of experimental thrombosis in rabbits with rivaroxaban (BAY 59-7939) - an oral, direct factor Xa inhibitor. Thromb. Haemost. 97, 471–477. doi: 10.1160/TH06-11-0620
Brummel-Ziedins, K. E., Orfeo, T., Gissel, M., Mann, K. G., and Rosendaal, F. R. (2012). Factor Xa generation by computational modeling: an additional discriminator to thrombin generation evaluation. PLoS ONE 7:e29178. doi: 10.1371/journal.pone.0029178
Davidson, B. L., Verheijen, S., Lensing, A. W. A., and Gebel, M. (2013). Risk of bleeding in patients with acute venous thromboembolism treated with rivaroxaban or enoxaparin/VKA and concomitant ASA therapy or NSAIDs: subanalysis from EINSTEIN DVT and PE studies. J. Thromb. Haemost. 11(Suppl. s2), Abstract AS 17.1. doi: 10.1111/jth.12284
Depasse, F., Busson, J., Mnich, J., Le Flem, L., Gerotziafas, G. T., and Samama, M. M. (2005). Effect of BAY 59-7939 - a novel, oral, direct factor Xa inhibitor - on clot-bound factor Xa activity in vitro. J. Thromb. Haemost. 3(Suppl. s1), Abstract P1104. doi: 10.1111/j.1538-7836.2005.0300c.x
Dunwiddie, C., Thornberry, N. A., Bull, H. G., Sardana, M., Friedman, P. A., Jacobs, J. W., et al. (1989). Antistasin, a leech-derived inhibitor of factor Xa. Kinetic analysis of enzyme inhibition and identification of the reactive site. J. Biol. Chem. 264, 16694–16699.
Eriksson, B. I., Borris, L. C., Dahl, O. E., Haas, S., Huisman, M. V., Kakkar, A. K., et al. (2006). A once-daily, oral, direct factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement. Circulation 114, 2374–2381. doi: 10.1161/CIRCULATIONAHA.106.642074
Eriksson, B. I., Borris, L. C., Dahl, O. E., Haas, S., Huisman, M. V., Kakkar, A. K., et al. (2007a). Dose-escalation study of rivaroxaban (BAY 59-7939) – an oral, direct factor Xa inhibitor – for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb. Res. 120, 685–693. doi: 10.1016/j.thromres.2006.12.025
Eriksson, B. I., Misselwitz, F., and Mueck, W. (2007b). Pharmacokinetics (PK) and pharmacodynamics (PD) of rivaroxaban: a comparison of once-and twice-daily dosing in patients undergoing total hip replacement (THR). J. Thromb. Haemost. 5(Suppl. 1), Abstract P-M-659.
Eriksson, B. I., Borris, L. C., Friedman, R. J., Haas, S., Huisman, M. V., Kakkar, A. K., et al. (2008). Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N. Engl. J. Med. 358, 2765–2775. doi: 10.1056/NEJMoa0800374
Eriksson, B. I., Rosencher, N., Friedman, R. J., Homering, M., and Dahl, O. E. (2012). Concomitant use of medication with antiplatelet effects in patients receiving either rivaroxaban or enoxaparin after total hip or knee arthroplasty. Thromb. Res. 130, 147–151. doi: 10.1016/j.thromres.2011.12.005
Garcia, D. A., Baglin, T. P., Weitz, J. I., and Samama, M. M. (2012). Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 141, e24S–e43S. doi: 10.1378/chest.11-2291
Gerotziafas, G. T., Elalamy, I., Depasse, F., Perzborn, E., and Samama, M. M. (2007). In vitro inhibition of thrombin generation, after tissue factor pathway activation, by the oral, direct factor Xa inhibitor rivaroxaban. J. Thromb. Haemost. 5, 886–888. doi: 10.1111/j.1538-7836.2007.02429.x
Gnoth, M. J., Buetehorn, U., Muenster, U., Schwarz, T., and Sandmann, S. (2011). In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J. Pharmacol. Exp. Ther. 338, 372–380. doi: 10.1124/jpet.111.180240
Graff, J., von Hentig, N., Misselwitz, F., Kubitza, D., Becka, M., Breddin, H. K., et al. (2007). Effects of the oral, direct factor Xa inhibitor rivaroxaban on platelet-induced thrombin generation and prothrombinase activity. J. Clin. Pharmacol. 47, 1398–1407. doi: 10.1177/0091270007302952
Graff, J., von Hentig, N., Misselwitz, F., Kubitza, D., Becka, M., Breddin, H. K., et al. (2008). Effects of the oral, direct factor Xa inhibitor rivaroxaban on platelet-induced thrombin generation and prothrombinase activity: errata. J. Clin. Pharmacol. 48, 1368. doi: 10.1177/0091270008326304
Ieko, M., Tarumi, T., Takeda, M., Naito, S., Nakabayashi, T., and Koike, T. (2004). Synthetic selective inhibitors of coagulation factor Xa strongly inhibit thrombin generation without affecting initial thrombin forming time necessary for platelet activation in hemostasis. J. Thromb. Haemost. 2, 612–618. doi: 10.1111/j.1538-7933.2004.00649.x
Kakkar, A. K., Brenner, B., Dahl, O. E., Eriksson, B. I., Mouret, P., Muntz, J., et al. (2008). Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 372, 31–39. doi: 10.1016/S0140-6736(08)60880-6
Kubitza, D., Becka, M., Mueck, W., Halabi, A., Maatouk, H., Klause, N., et al. (2010). Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct factor Xa inhibitor. Br. J. Clin. Pharmacol. 70, 703–712. doi: 10.1111/j.1365-2125.2010.03753.x
Kubitza, D., Becka, M., Mueck, W., and Schwers, S. (2012a). Effect of co-administration of rivaroxaban and clopidogrel on bleeding time, pharmacodynamics and pharmacokinetics: a phase I study. Pharmaceuticals 5, 279–296. doi: 10.3390/ph5030279
Kubitza, D., Becka, M., Roth, A., and Mueck, W. (2012b). Absence of clinically relevant interactions between rivaroxaban - an oral, direct factor Xa inhibitor - and digoxin or atorvastatin in healthy subjects. J. Int. Med. Res. 40, 1688–1707. doi: 10.1177/030006051204000508
Kubitza, D., Becka, M., Mueck, W., and Zuehlsdorf, M. (2006). Safety, tolerability, pharmacodynamics, and pharmacokinetics of rivaroxaban – an oral, direct factor Xa inhibitor – are not affected by aspirin. J. Clin. Pharmacol. 46, 981–990. doi: 10.1177/0091270006292127
Kubitza, D., Becka, M., Mueck, W., and Zuehlsdorf, M. (2007a). Rivaroxaban (BAY 59-7939) - an oral, direct factor Xa inhibitor - has no clinically relevant interaction with naproxen. Br. J. Clin. Pharmacol. 63, 469–476. doi: 10.1111/j.1365-2125.2006.02776.x
Kubitza, D., Becka, M., Zuehlsdorf, M., and Mueck, W. (2007b). Body weight has limited influence on the safety, tolerability, pharmacokinetics, or pharmacodynamics of rivaroxaban (BAY 59-7939) in healthy subjects. J. Clin. Pharmacol. 47, 218–226. doi: 10.1177/0091270006296058
Kubitza, D., Becka, M., Roth, A., and Mueck, W. (2008). Dose-escalation study of the pharmacokinetics and pharmacodynamics of rivaroxaban in healthy elderly subjects. Curr. Med. Res. Opin. 24, 2757–2765. doi: 10.1185/03007990802361499
Kubitza, D., Becka, M., Roth, A., and Mueck, W. (2013). The influence of age and gender on the pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct factor Xa inhibitor. J. Clin. Pharmacol. 53, 249–255. doi: 10.1002/jcph.5
Kubitza, D., Becka, M., Voith, B., Zuehlsdorf, M., and Wensing, G. (2005a). Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin. Pharmacol. Ther. 78, 412–421. doi: 10.1016/j.clpt.2005.06.011
Kubitza, D., Becka, M., Wensing, G., Voith, B., and Zuehlsdorf, M. (2005b). Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939 – an oral, direct factor Xa inhibitor – after multiple dosing in healthy male subjects. Eur. J. Clin. Pharmacol. 61, 873–880. doi: 10.1007/s00228-005-0043-5
Kubitza, D., and Haas, S. (2006). Novel factor Xa inhibitors for prevention and treatment of thromboembolic diseases. Expert Opin. Investig. Drugs 15, 843–855. doi: 10.1517/13543784.15.8.843
Lassen, M. R., Ageno, W., Borris, L. C., Lieberman, J. R., Rosencher, N., Bandel, T. J., et al. (2008). Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N. Engl. J. Med. 358, 2776–2786. doi: 10.1056/NEJMoa076016
Leadley, R. J. Jr. (2001). Coagulation factor Xa inhibition: biological background and rationale. Curr. Top. Med. Chem. 1, 151–159. doi: 10.2174/1568026013395380
Mann, K. G., Brummel, K., and Butenas, S. (2003). What is all that thrombin for? J. Thromb. Haemost. 1, 1504–1514. doi: 10.1046/j.1538-7836.2003.00298.x
McRae, S. J., and Ginsberg, J. S. (2004). Initial treatment of venous thromboembolism. Circulation 110, I3–I9. doi: 10.1161/01.CIR.0000140904.52752.0c
Mega, J. L., Braunwald, E., Wiviott, S. D., Bassand, J. P., Bhatt, D. L., Bode, C., et al. (2012). Rivaroxaban in patients with a recent acute coronary syndrome. N. Engl. J. Med. 366, 9–19. doi: 10.1056/NEJMoa1112277
Mueck, W., Agnelli, G., and Büller, H. (2007). Rivaroxaban has predictable pharmacokinetics (PK) and pharmacodynamics (PD) when given once or twice daily for the treatment of acute, proximal deep vein thrombosis (DVT). Blood (ASH Annual Meeting Abstracts) 110, Abstract 1880.
Mueck, W., Borris, L. C., Dahl, O. E., Haas, S., Huisman, M. V., Kakkar, A. K., et al. (2008). Population pharmacokinetics and pharmacodynamics of once- and twice-daily rivaroxaban for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb. Haemost. 100, 453–461. doi: 10.1160/TH07-12-0714
Mueck, W., Kubitza, D., and Becka, M. (2013). Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br. J. Clin. Pharmacol. 76, 455–466. doi: 10.1111/bcp.12075
Mueck, W., Lensing, A. W., Agnelli, G., Decousus, H., Prandoni, P., and Misselwitz, F. (2011). Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep-vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin. Pharmacokinet. 50, 675–686. doi: 10.2165/11595320-000000000-00000
Nicolini, F. A., Lee, P., Malycky, J. L., Lefkovits, J., Kottke-Marchant, K., Plow, E. F., et al. (1996). Selective inhibition of factor Xa during thrombolytic therapy markedly improves coronary artery patency in a canine model of coronary thrombosis. Blood Coagul. Fibrinolysis 7, 39–48. doi: 10.1097/00001721-199601000-00005
Patel, M. R., Mahaffey, K. W., Garg, J., Pan, G., Singer, D. E., Hacke, W., et al. (2011). Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N. Engl. J. Med. 365, 883–891. doi: 10.1056/NEJMoa1009638
Perzborn, E., Roehrig, S., Straub, A., Kubitza, D., and Misselwitz, F. (2011). The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discov. 10, 61–75. doi: 10.1038/nrd3185
Perzborn, E., Strassburger, J., Wilmen, A., Pohlmann, J., Roehrig, S., Schlemmer, K. H., et al. (2005). In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939 - an oral, direct factor Xa inhibitor. J. Thromb. Haemost. 3, 514–521. doi: 10.1111/j.1538-7836.2005.01166.x
Rand, M. D., Lock, J. B., van't Veer, C., Gaffney, D. P., and Mann, K. G. (1996). Blood clotting in minimally altered whole blood. Blood 88, 3432–3445.
Stampfuss, J., Kubitza, D., Becka, M., and Mueck, W. (2013). The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int. J. Clin. Pharmacol. Ther. 51, 549–561. doi: 10.5414/CP201812
The EINSTEIN Investigators. (2010). Oral rivaroxaban for symptomatic venous thromboembolism. N. Engl. J. Med. 363, 2499–2510. doi: 10.1056/NEJMoa1007903
The EINSTEIN–PE Investigators. (2012). Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N. Engl. J. Med. 366, 1287–1297. doi: 10.1056/NEJMoa1113572
Turpie, A. G. G., Bauer, K. A., Eriksson, B. I., Lassen, M. R., and Steering Committees of the Pentasaccharide Orthopedic Prophylaxis Studies. (2002). Fondaparinux vs enoxaparin for the prevention of venous thromboembolism in major orthopedic surgery: a meta-analysis of 4 randomized double-blind studies. Arch. Intern. Med. 162, 1833–1840. doi: 10.1001/archinte.162.16.1833
Turpie, A. G. G., Eriksson, B. I., Mueck, W., Bauer, K. A., Borris, L. C., Dahl, O. E., et al. (2006). Pharmacokinetic and pharmacodynamic analyses of rivaroxaban in patients undergoing orthopaedic surgery. Pathophysiol. Haemost. Thromb. 35, Abstract 1182. doi: 10.1159/000093567
Turpie, A. G. G., Lassen, M. R., Davidson, B. L., Bauer, K. A., Gent, M., Kwong, L. M., et al. (2009). Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial. Lancet 373, 1673–1680. doi: 10.1016/S0140-6736(09)60734-0
Weinz, C., Buetehorn, U., Daehler, H. P., Kohlsdorfer, C., Pleiss, U., Sandmann, S., et al. (2005). Pharmacokinetics of BAY 59-7939 – an oral, direct factor Xa inhibitor – in rats and dogs. Xenobiotica 35, 891–910. doi: 10.1080/00498250500250493
Weinz, C., Schwarz, T., Kubitza, D., Mueck, W., and Lang, D. (2009). Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs and humans. Drug Metab. Dispos. 37, 1056–1064. doi: 10.1124/dmd.108.025569
Keywords: anticoagulation, factor Xa inhibition, pharmacodynamics, prothrombin time, rivaroxaban, thrombin generation
Citation: Kubitza D, Perzborn E and Berkowitz SD (2013) The discovery of rivaroxaban: translating preclinical assessments into clinical practice. Front. Pharmacol. 4:145. doi: 10.3389/fphar.2013.00145
Received: 30 August 2013; Paper pending published: 17 September 2013;
Accepted: 04 November 2013; Published online: 25 November 2013.
Edited by:
Eliot Ohlstein, Drexel Med School, USAReviewed by:
Juan Badimon, The Mount Sinai School of Medicine, USAColin H. Macphee, GlaxoSmithKline, USA
Copyright © 2013 Kubitza, Perzborn and Berkowitz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dagmar Kubitza, Clinical Pharmacology, Bayer HealthCare AG, Aprather Weg 18a, 42096 Wuppertal, Germany e-mail:ZGFnbWFyLmt1Yml0emFAYmF5ZXJoZWFsdGhjYXJlLmNvbQ==