 Marina Kawaguchi-Suzuki
Marina Kawaguchi-Suzuki Reginald F. Frye
Reginald F. Frye- Department of Pharmacotherapy and Translational Research, Center for Pharmacogenomics, College of Pharmacy, University of Florida, Gainesville, FL, USA
Pioglitazone is the most widely used thiazolidinedione and acts as an insulin-sensitizer through activation of the Peroxisome Proliferator-Activated Receptor-γ (PPARγ). Pioglitazone is approved for use in the management of type 2 diabetes mellitus (T2DM), but its use in other therapeutic areas is increasing due to pleiotropic effects. In this hypothesis article, the current clinical evidence on pioglitazone pharmacogenomics is summarized and related to variability in pioglitazone response. How genetic variation in the human genome affects the pharmacokinetics and pharmacodynamics of pioglitazone was examined. For pharmacodynamic effects, hypoglycemic and anti-atherosclerotic effects, risks of fracture or edema, and the increase in body mass index in response to pioglitazone based on genotype were examined. The genes CYP2C8 and PPARG are the most extensively studied to date and selected polymorphisms contribute to respective variability in pioglitazone pharmacokinetics and pharmacodynamics. We hypothesized that genetic variation in pioglitazone pathway genes contributes meaningfully to the clinically observed variability in drug response. To test the hypothesis that genetic variation in PPARG associates with variability in pioglitazone response, we conducted a meta-analysis to synthesize the currently available data on the PPARG p.Pro12Ala polymorphism. The results showed that PPARG 12Ala carriers had a more favorable change in fasting blood glucose from baseline as compared to patients with the wild-type Pro12Pro genotype (p = 0.018). Unfortunately, findings for many other genes lack replication in independent cohorts to confirm association; further studies are needed. Also, the biological functionality of these polymorphisms is unknown. Based on current evidence, we propose that pharmacogenomics may provide an important tool to individualize pioglitazone therapy and better optimize therapy in patients with T2DM or other conditions for which pioglitazone is being used.
Introduction
Pioglitazone (PIO) is the most widely used thiazolidinedione (TZD) anti-diabetic drug. The first TZD, troglitazone, was approved for clinical use by the United States Food and Drug Administration (FDA) in 1997 (Kung and Henry, 2012). However, troglitazone was withdrawn from the market due to hepatotoxicity and is not currently available (Kung and Henry, 2012). PIO and rosiglitazone (ROSI) were approved by the FDA in 1999 (Kung and Henry, 2012). Unfortunately, due to an apparent increase in risk of myocardial infarction, ROSI was withdrawn from the European market in 2010; the FDA restricted the use of ROSI in the United States in 2011 (Kung and Henry, 2012). Therefore, PIO is currently the only TZD available without regulatory restrictions.
The exact mechanism of action of TZDs remains unclear, but the current consensus is that TZDs target the transcription factor peroxisome proliferator-activated receptor-γ (PPARγ) to improve insulin sensitivity (Cariou et al., 2012; Yau et al., 2013). Ligand-dependent transactivation of PPARγ causes heterodimerization with the retinoid-X receptor followed by recognition of peroxisome proliferator response elements (PPREs) that stimulate transcription of genes involved in metabolic homeostasis (Cariou et al., 2012). Additionally, PPARγ represses transcription of genes related to inflammation via transrepression, which does not require binding to PPREs but antagonizes other transcription factors that activate inflammatory pathways, such as NF-κ B and AP-1 (Cariou et al., 2012).
PIO was approved for the management of type 2 diabetes mellitus (T2DM) (Cariou et al., 2012; Yau et al., 2013). The efficacy of PIO as an anti-hyperglycemic agent was demonstrated in randomized controlled trials (Cariou et al., 2012). PIO monotherapy was shown to lower A1C by ~1% in T2DM patients (Yau et al., 2013). An advantage of PIO over sulfonylureas and insulin is that comparable A1C reduction is achieved without significant hypoglycemia (Yau et al., 2013). TZDs also improve insulin sensitivity, which may reduce insulin dose or eliminate the need for insulin in some patients (Yau et al., 2013). In addition to T2DM, PIO use has increased in other therapeutic areas such as non-alcoholic fatty liver disease, atherosclerosis, inflammation, infertility, and cancer due to the wide spectrum of effects secondary to PPARγ activation (Cariou et al., 2012; Yau et al., 2013). At the same time, more undesirable off-target effects have been identified. The use of PIO is typically limited by adverse drug reactions (ADRs) including bone fractures, peripheral edema, congestive heart failure (CHF), weight gain, and possible risk of bladder cancer (Cariou et al., 2012; Yau et al., 2013).
As more benefits and risks of PIO have been reported, clinically relevant variability in response has also been noted (Umpierrez and Dagogo-Jack, 2006; Yau et al., 2013). However, the sources of variability in response to PIO are not fully understood. In this article, we will address the question whether pharmacogenomics contributes to the observed variability in pioglitazone response. First, the current evidence on genetic variation in pioglitazone pathway genes will be summarized. Then, current evidence on genetic variation in the gene encoding the PPARγ receptor (PPARG) will be evaluated; because the data with PPARG are inconsistent, we conducted a meta-analysis to estimate the association between the best studied PPARG polymorphism and pioglitazone response. Finally, future research directions and methods to incorporate current pharmacogenomic findings in clinical practice are proposed.
Methods
Literature Search
Two databases were used to identify pharmacogenomics evidence: PubMed and Web of Science. The searches were conducted through April 30, 2013. Two searches were performed with PubMed by using the following terms: (1) (“thiazolidinediones”[MeSH Terms] OR “thiazolidinediones”[All Fields]) AND (“pharmacogenetics”[MeSH Terms] OR “pharmacogenetics”[All Fields]) and (2) (“thiazolidinediones”[MeSH Terms] OR “thiazolidinediones”[All Fields]) AND (“polymorphism, genetic”[MeSH Terms] OR (“polymorphism”[All Fields] AND “genetic”[All Fields]) OR “genetic polymorphism”[All Fields] OR “polymorphism”[All Fields]). The numbers of articles found from the two searches were 33 and 91, respectively without any restrictions. The following searches were conducted with Web of Science: Topic = (pioglitazone) AND [Topic = (pharmacogenomics) OR Topic = (pharmacogenetics) OR Topic = (polymorphism)]. The original hit number was 84. The titles and abstracts were screened to include only clinical studies that investigated differences in PK or PD of PIO based on genetic polymorphisms in human participants. Articles only written in English were examined and meeting abstracts were not included in the summary. Sixteen full-text original research articles meeting our criteria and published in peer-reviewed journals were identified by our literature search.
Meta-Analysis
A meta-analysis was conducted with five identified studies examining the PPARG p.Pro12Ala polymorphism. The change in fasting plasma glucose (FG) from baseline was used as the primary outcome of interest because the definition of the responder phenotype was not consistent between studies. The data presented in the original articles were used. For the study by Bluher et al. (2003), the values were not available in the published article, so they were obtained directly from the authors [p.Pro12Pro −3.01 ± 3.53 mmol/L vs. p.Pro12Ala −2.98 ± 3.16 mmol/L (mean ± standard deviation)]. Units were converted from mmol/L to mg/dL to analyze changes in FG consistently among studies. Standard differences in means were used to compare the outcome of interest between the PPARG p.Pro12Pro genotype and 12Ala carriers by using the Comprehensive Meta-Analysis V2 program (Biostat, Englewood, NJ, USA). A test for heterogeneity (I2 = 48.7; p = 0.10) was conducted, and a fixed-effects model was used. The publication bias was checked (Supplemental figure 1).
Results/Discussions
Effects of Genetic Variation on PIO Pharmacokinetics
Table 1 summarizes clinical studies that investigated the effect of genetic polymorphisms on PIO pharmacokinetics. The primary metabolites of PIO are designated as M-I, M-II, M-IV, M-V, and M-VI (Eckland and Danhof, 2000) (Figure 1). M-IV is further metabolized to M-III and M-VI may also be formed from M-V (Eckland and Danhof, 2000). M-IV and M-III are known to be the major active metabolites and are responsible for the extended hypoglycemic effect (Eckland and Danhof, 2000). Multiple cytochrome P450 (CYP) enzymes are involved in the metabolism of PIO. However, CYP2C8 and CYP3A4 are the most important enzymes and contribute to ~60% and less than 20% of total PIO metabolism, respectively (Eckland and Danhof, 2000; Scheen, 2007; Vandenbrink et al., 2011).
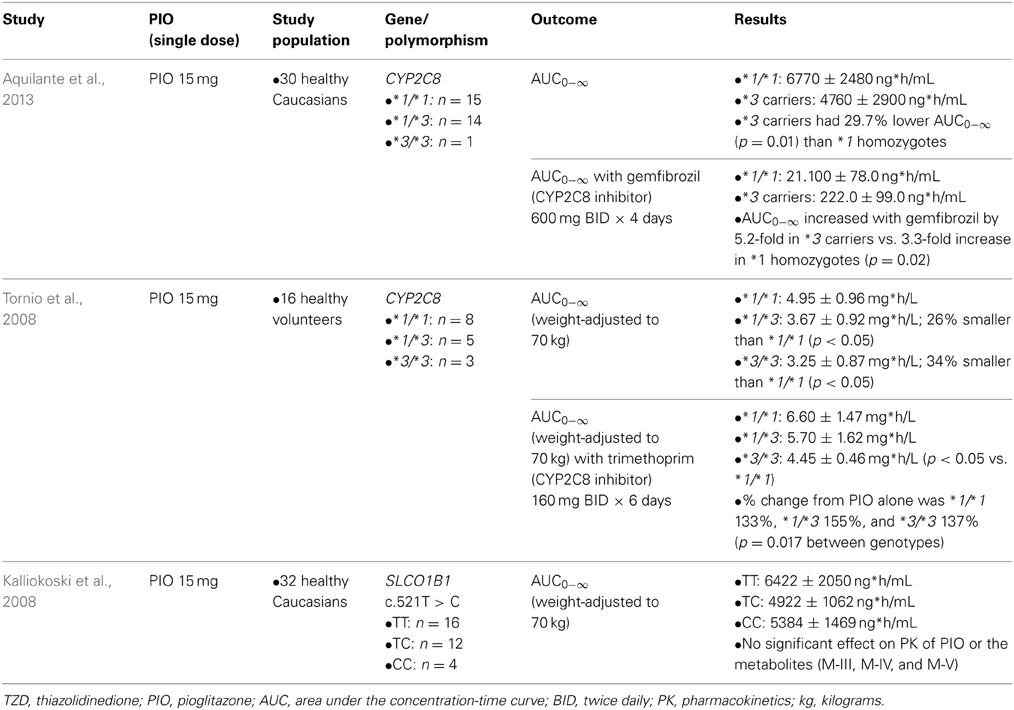
Table 1. Effect of genetic polymorphisms on pioglitazone pharmacokinetics.
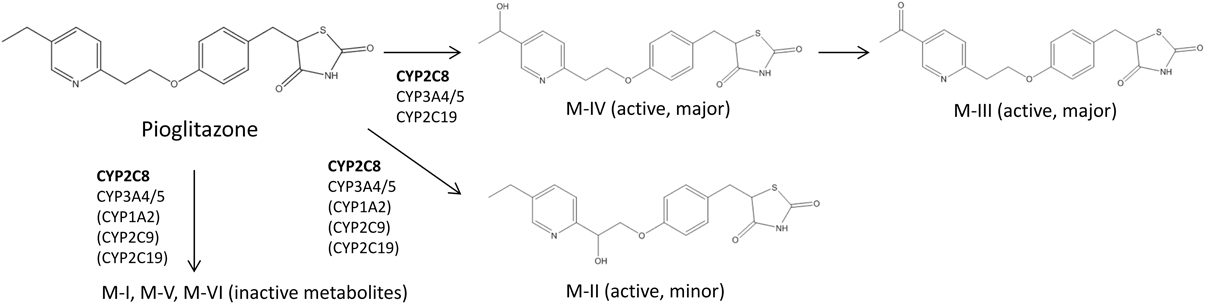
Figure 1. Abbreviated Pioglitazone Metabolism Pathway. CYP2C8 is the major enzyme metabolizing pioglitazone and is shown in bold. Enzymes in the parenthesis are suggested to be involved in pioglitazone metabolism, but their roles in the formation of particular metabolites are not clear according to currently available data (Eckland and Danhof, 2000; Jaakkola et al., 2006; Lai et al., 2009).
Two clinical studies examined genotypes of CYP2C8, the gene coding for the CYP2C8 enzyme. Both studies showed that compared to *1 homozygotes, the *3 variant allele carriers had ~30% lower total systemic exposure to PIO measured as the area under the plasma concentration-time curve (AUC) (Tornio et al., 2008; Aquilante et al., 2013). CYP2C8*3 is the best studied functional polymorphism in this gene. The *3 allele is designated by the presence of two non-synonymous polymorphisms, rs11572080:G>A in exon 3 (p.Arg139Lys) and rs10509681:A>G in exon 8 (p.Lys399Arg) (Aquilante et al., 2013). The functional relevance of these polymorphisms appears to be substrate-dependent. For example, metabolism of PIO, ROSI, and repaglinide was increased, but metabolism of R-ibuprofen was decreased in carriers of the CYP2C8*3 allele (Martinez et al., 2005; Daily and Aquilante, 2009; Aquilante et al., 2013). The substrate selectivity of the enzyme is likely due to the amino acid changes in the protein structure caused by the polymorphisms. According to a recent population pharmacokinetic model, the oral clearance of PIO was ~52% higher in CYP2C8*3 carriers (Kadam et al., 2013). Considering that 10–23% of Caucasians carry the *3 allele and that PIO exposure is decreased in carriers (Aquilante et al., 2013), this genotype is expected to explain some of the variability in response to PIO therapy. CYP2C8*3 is most frequently observed among Caucasians, followed by Hispanics, and the *3 variant is rare among people with African and Asian ancestry (Aquilante et al., 2013; Martis et al., 2013).
The clinical studies also examined the influence of CYP2C8*3 on the magnitude of drug-drug interactions. Gemfibrozil was administered as a CYP2C8 inhibitor and demonstrated that the resulting increase in AUC was significantly greater in *3 carriers (5.2-fold) compared with *1 homozygotes (3.3-fold) (Aquilante et al., 2013). This resulted in PIO exposure after inhibition being similar in the two genotype groups (Aquilante et al., 2013). When trimethoprim was administered as a CYP2C8 inhibitor, the increase in AUC was larger in *3 carriers, leading to a non-significant difference in the PIO exposure between *1 homozygotes and *1/*3 heterozygotes after the inhibition (Tornio et al., 2008). A similar genotype-dependent effect on the magnitude of drug-drug interaction has been reported with other CYP enzymes, such as CYP2C9, whereby the magnitude of interaction is greater in the individuals having the genotype associated with higher activity or clearance (Castellan et al., 2013). If a therapy known to inhibit CYP2C8 is initiated in patients with the *3 allele taking PIO, close monitoring of PIO therapy is warranted because the inhibition may be more pronounced in this patient population.
The effect of genetic variation in the gene that encodes the organic anion-transporting poly-peptide OATP1B1 (SLCO1B1, Solute carrier organic anion transporter family member 1B1) has also been studied. OATP1B1 is an uptake transporter located at the basolateral membrane of hepatocytes that facilitates drug entry into the hepatocyte (Zamek-Gliszczynski et al., 2012). PIO is a substrate and potent competitive inhibitor of OATP1B1 (Kalliokoski et al., 2008). However, no significant effect of SLCO1B1 polymorphisms on the PK of PIO and its metabolites (M-III, M-IV, and M-V) was observed (Kalliokoski et al., 2008). Because the results have not yet been replicated, transporter polymorphisms should be further studied with PIO.
Effects of Genetic Variation on PIO Pharmacodynamics
Table 2 summarizes studies that examined the extent to which genetic polymorphisms affect PIO pharmacodynamics.
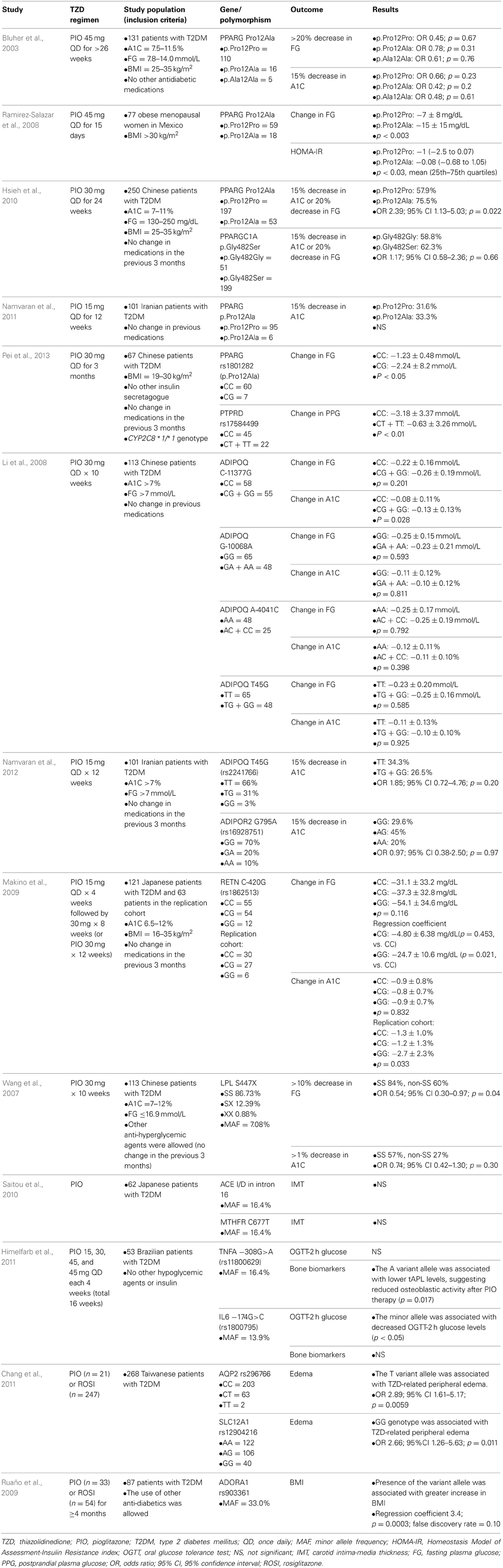
Table 2. Effect of genetic polymorphisms on pioglitazone pharmacodynamics.
Hypoglycemic effect
The most well-studied polymorphism with respect to PIO response is PPARG p.Pro12Ala (rs1801282). PPARG is the gene encoding PPARγ, a ligand-activated transcription factor that regulates glucose homeostasis (Auwerx, 1999). The minor allele frequency for this polymorphism has been reported as 2–18% in healthy humans (Saraf et al., 2012). The 12Ala allele appears to improve insulin sensitivity due to an alteration in the transcriptional activity of PPARγ (Deeb et al., 1998; Saraf et al., 2012). This variant has been consistently associated with decreased risk of T2DM in previous studies (Altshuler et al., 2000). According to a meta-analysis, the odds ratio of T2DM risk was 0.86 (95% confidence interval 0.81–0.90) with the presence of the 12Ala allele (Gouda et al., 2010).
PPARγ is the drug target of TZDs, thus it would be biologically plausible for the functional p.Pro12Ala polymorphism in the PPARG gene to be associated with response to TZDs. Previously, in response to ROSI, PPARG p.Pro12Ala allele carriers achieved greater reduction in FG (50.6 ± 27.8 vs. 24.3 ± 41.9 mg/dL, p = 0.026) and in A1C (1.41 ± 1.47% vs. 0.57 ± 1.16%, p = 0.015) than patients without the variant allele (Kang et al., 2005).
Our search identified five pharmacogenetic studies in which the effect of PPARG p.Pro12Ala polymorphism on PIO pharmacodynamic response was examined. The results were not entirely consistent across studies. Three out of five studies demonstrated a better response in terms of improvements in FG and A1C in carriers of the 12Ala allele (Ramirez-Salazar et al., 2008; Hsieh et al., 2010; Pei et al., 2013). However, two studies reported a non-significant response to PIO (Bluher et al., 2003; Namvaran et al., 2011). It is difficult to explain exactly why the results were disparate, but possible reasons include the heterogeneity of the studies and small sample sizes of patients with the minor allele. The studies were quite heterogeneous with respect to the patient population, the inclusion/exclusion criteria, PIO treatment regimen, and the presence of other anti-diabetic treatments. In addition, the two negative studies had a relatively small number of patients with the 12Ala minor allele, suggesting power may not have been adequate to detect a difference. Although the results are conflicting, the PPARG p.Pro12Ala polymorphism may contribute to the variability observed in response to PIO as shown in the three favorable studies. Indeed, according to our meta-analysis of the five studies identified, PPARG 12Ala allele carriers had a more favorable change in FG compared to people with the p.Pro12Pro genotype (p = 0.018, Figure 2). However, this polymorphism alone does not fully explain why some patients failed to respond to PIO. Other genetic and environmental factors are likely to be involved and may have contributed to the negative results in the two studies. Thus, consideration of other genes appears important to better explain the variability in PIO treatment response.
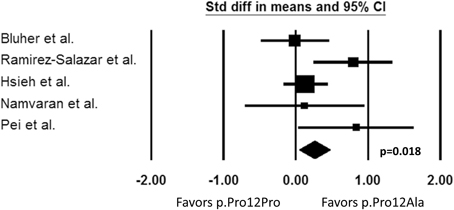
Figure 2. Change in fasting plasma glucose from baseline. Comparison of PPARG p.Pro12Pro genotype vs. p.Pro12Ala carriers (Std diff in means, standard difference in means; 95% CI, 95% confidence interval).
In addition to the PPARG gene, protein tyrosine phosphatase receptor type D (PTPRD) gene was investigated. PTPRD polymorphisms associated with T2DM in a genome wide association study in a Chinese Han population (Tsai et al., 2010). Rs17584499, located in intron 10, was the single nucleotide polymorphism (SNP) with the strongest association (Tsai et al., 2010). The functionality of this polymorphism has not yet been identified. Considering that it is located in an intron, the polymorphism may affect mRNA splicing or may be in linkage disequilibrium with other functional polymorphisms. One study found that patients with rs17584499 wild-type CC genotype had better postprandial plasma glucose concentrations after being treated with PIO for 3 months compared with T minor allele carriers (Pei et al., 2013). Confirmation of these results is needed in an independent cohort. However, it is clear that more polymorphisms remain to be uncovered to better explain the variability in PIO response. Such polymorphisms may be identified through populations that have been previously under-represented.
The adiponectin (ADIPOQ) gene has been studied in patients receiving PIO therapy. Adiponectin is an adipokine, a peptide hormone secreted from adipocytes, known to play an important role in insulin sensitization and fat β-oxidation (Li et al., 2008; Ruscica et al., 2012). T2DM and insulin resistance have been associated with low serum adiponectin levels (Li et al., 2008). Multiple polymorphisms in ADIPOQ associated with T2DM in various ethnic populations, including Asians and Caucasians with European ancestry (Hara et al., 2002; Menzaghi et al., 2002; Stumvoll et al., 2002; Vasseur et al., 2002, 2003; Vozarova De Courten et al., 2005; Woo et al., 2006; Li et al., 2008; Jing et al., 2012).
Two studies investigating response to PIO with respect to ADIPOQ genotypes were identified in the literature search. These studies were conducted in Chinese and Iranian populations (Li et al., 2008; Namvaran et al., 2012). However, in the two studies, the only significant change in FG or A1C in response to PIO was found with ADIPOQ C-11377G genotype in the study of the Chinese population (Li et al., 2008; Namvaran et al., 2012). The −11377 CC genotype patients had smaller reduction in A1C than the minor G allele carriers (Li et al., 2008). Response to ROSI was also previously studied with regard to the ADIPOQ polymorphism. In Chinese T2DM patients treated with ROSI, the wild-type −11377 CC genotype was associated with greater reduction in FG, compared to the CG and GG genotypes (−34.2 vs. −9.7 mmol/L, p = 0.001) (Sun et al., 2008). A definitive conclusion on this ADIPOQ polymorphism cannot be made due to the lack of replication and because the direction of the association was opposite with the ADIPOQ C-11377G genotype between the ROSI and PIO studies. Studies have shown that exercise affects adiponectin concentrations (Gueugnon et al., 2012; Lee et al., 2013). The Korean study reported that exercise increased adiponectin levels irrespective of ADIPOQ polymorphisms (Lee et al., 2013). Therefore, it may be possible that environmental factors, such as exercise level, have a large impact and make it difficult to differentiate the effect of ADIPOQ polymorphisms on response to PIO.
Adiponectin receptor 2 (ADIPOR2) is another gene studied along with ADIPOQ polymorphism to explain PIO response variability. The ADIPOR2 gene is highly expressed at critical sites involved in glucose metabolism and activation of this receptor is known to increase fatty acid oxidation and adiponectin-mediated glucose utilization (Kadowaki et al., 2006; Namvaran et al., 2012). However, in an Iranian population, ADIPOR2 G795A genotype did not have any significant effect on FG and A1C in response to PIO (Namvaran et al., 2012). No definitive conclusions can be made from one study and the functionality of ADIPOR2 polymorphisms needs to be investigated further to understand the involvement in PIO response. However, if any polymorphism in ADIPOR2 can affect the functionality of the receptor, it may still be possible that the polymorphism could have an influence on response to PIO therapy.
The involvement of the resistin (RETN) gene in response to PIO was studied in a Japanese population. Resistin is another adipokine known to antagonize insulin (Osawa et al., 2004). The role of RETN polymorphisms in T2DM or obese patients has not been fully elucidated, but the RETN C-420G (rs1862513) GG genotype was previously associated with T2DM susceptibility (Osawa et al., 2004; Ochi et al., 2007; Makino et al., 2009). Sp1 and Sp3 are transcription factors that bind to the DNA element, if the −420 G allele is present, to enhance serum resistin concentrations via increased RETN promoter activity (Osawa et al., 2004). The binding of Sp1/3 does not occur with the −420 C allele (Osawa et al., 2004). One report suggested that TZD-mediated PPARγ activation represses the expression of the resistin gene by modulating Sp1 activity (Chung et al., 2005; Makino et al., 2009). Makino et al. studied RETN C-420G polymorphism in Japanese T2DM patients and found that the GG genotype was associated with better FG and A1C reduction (Makino et al., 2009). However, the reduction in A1C was observed only in the replication cohort, and the significance with the FG reduction did not remain after adjustment for multiple testing. The authors commented on differences in the patient characteristics between the original and replication cohorts and that the sample size might not have been adequate. It is reasonable to hypothesize that patients with the GG genotype can recruit more Sp1 at baseline and therefore have more pronounced reduction in serum resistin levels with PIO treatment. However, the effect of this RETN C-420G SNP on PIO response needs to be further investigated with the involvement of Sp1/3 signaling pathway.
The lipoprotein lipase (LPL) S447X polymorphism has been studied with respect to PIO response. Lipoprotein lipase is the rate-limiting enzyme for the breakdown of lipoproteins rich in triglycerides (Groenemeijer et al., 1997). Lipoprotein lipase is synthesized by and secreted from various parenchyma cells including adipocytes, muscle cells, and macrophages and expression was increased by PIO in a previous in vivo study (Bogacka et al., 2004; Wang et al., 2007). Among more than 40 polymorphisms in LPL gene, S447X is the most extensively studied polymorphism; it causes a premature stop codon and the elimination of serine and glycine at the C-terminal (Wang et al., 2007). Previous independent studies reported an association of the S447X variant with dyslipoproteinemia and coronary artery disease (Kuivenhoven et al., 1997; Wang et al., 2007). This variant was investigated in T2DM patients who received PIO and found that the response rate (defined as >10% reduction in FG) to PIO was 0.54-fold smaller in patients with the LPL S447X genotype, compared to patients with the S447S genotype (Wang et al., 2007). Although independent confirmation of this result is needed, LPL is another candidate gene to be studied with response to PIO. In addition, more functional studies are necessary to understand how this polymorphism relates to glycemic control in humans.
Anti-atherosclerotic effect
The effect of genetic polymorphism on carotid atherosclerosis progression has been studied in patients who received PIO. TZDs were shown to improve lipid profiles, inflammation, coagulation, and endothelial cell function, and PIO was shown to significantly slow progression of carotid intima-media thickness (IMT) (Saitou et al., 2010). In contrast to ROSI, which was associated with an increased risk of MI, meta-analyses showed that PIO did not increase the risk of MI and might have a benefit of decreasing revascularization events (Nagajothi et al., 2008; Tannen et al., 2013). A study investigated 99 candidate gene polymorphisms, which were previously associated with atherosclerosis, diabetes mellitus, hypertension, or dyslipidemia, among T2DM patients treated with PIO (Saitou et al., 2010). Results for the angiotensin-converting enzyme (ACE) gene and methylene-tetrahydrofolate reductase (MTHFR) gene were reported, but the IMT was not significantly affected by polymorphisms in these two genes (Saitou et al., 2010). However, they compared the results with patients treated with diet alone. The IMT significantly increased in ACE D allele carriers (average IMT from 0.802 ± 0.017 to 0.831 ± 0.017 mm, p = 0.006; max IMT from 0.969 ± 0.023 to 0.998 ± 0.021 mm, p = 0.05) and the MTHER 677T allele carriers (average IMT from 0.804 ± 0.012 to 0.839 ± 0.013 mm, p = 0.001; max IMT from 0.969 ± 0.023 to 1.010 ± 0.018 mm, p = 0.007) (Saitou et al., 2010). Thus, the study demonstrated that PIO attenuated the increase in IMT irrespective of the genotypes.
Fracture risk
There is much concern with bone fractures as an ADR with TZDs. Both ROSI and PIO are known to provoke bone loss, and increased risks of fractures are reported, especially among women with T2DM taking a TZD (Betteridge, 2011). The most common sites of fractures are at distal limbs rather than at hips and spines even though the reason for this localization of fractures is still unclear (Betteridge, 2011; Cariou et al., 2012). PPARγ-mediated actions on bone homeostasis are believed to be responsible for these fractures. The activation of PPARγ was shown to promote osteoclastogenesis and suppress osteoblastogenesis, leading to bone loss as the net effect (Wan, 2010). Tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) are inflammatory cytokines, and their release is suppressed by PPARγ activation (Cariou et al., 2012). TNFα differentiates human peripheral monocytes into activated osteoclasts, so the reduced TNFα level can result in inhibition of osteoclastogenesis (Hounoki et al., 2008).
TNFα and IL-6 are encoded by TNFA and IL6 genes respectively, and the effect of TNFA and IL6 genotypes was studied in T2DM patients who received PIO (Himelfarb et al., 2011). They showed that TNFA –308A allele carriers had decreased mRNA expression compared with those with the wild GG genotype (Himelfarb et al., 2011). They also found that alkaline phosphatase, a biomarker for osteoblastic activity, was reduced in response to PIO in TNFA −308A allele carriers with T2DM (Himelfarb et al., 2011). IL6 −174C allele was associated with decreased OGTT-2h glucose but not with any bone metabolic markers (Himelfarb et al., 2011). IL6 −174C allele was previously associated with higher plasma levels of IL-6, but in this study, mRNA expression did not differ by the IL6 genotype (Himelfarb et al., 2011). Since replication is lacking and studies relating the genotypes directly to the occurrence of fractures are missing, no definitive conclusion can be made at this point.
Edema risk
Edema is a potential ADR from ROSI and PIO, and both TZDs carry a FDA black box warning for CHF (Kung and Henry, 2012). The safety of TZDs for patients with New York Heart Association class III or IV CHF is not established, and the use of TZDs is contraindicated in this patient population (Kung and Henry, 2012; Yau et al., 2013). The underlying mechanism of both edema and CHF appears to be fluid retention and expansion of plasma volume (Kung and Henry, 2012). Two studies that analyzed the TZD-related edema risk were identified in the literature search.
Twenty eight SNPs were genotyped from genes related to sodium and water reabsorption (Chang et al., 2011). They found that the AQP2 rs296766 T allele and SLC12A1 rs12904216 GG genotype were associated with edema in T2DM patients treated with a TZD (Chang et al., 2011). AQP2 gene codes aquaporins, which function as a water channel on the apical membrane, predominantly in the collecting duct of the kidney (Knepper et al., 1996). The AQP2 rs296766 SNP is located at the 3' untranslated region and may regulate mRNA stability (Chang et al., 2011). SLC12A1 encodes apical Na-K-2CL cotransporter, NKCC2, which plays an essential role in concentrating urine with the uptake of CL− and K+ driven by the Na+ influx (Ji et al., 2008; Chang et al., 2011). This transporter, coded by SLC12A1, is a target of thiazides and furosemide, and genetic polymorphisms in this gene were associated with blood pressure variation, Bartter syndrome, and response to loop diuretics (Ji et al., 2008; Chang et al., 2011). The SLC12A1 rs12904216 SNP is located in intron 7, and the protein expression may be altered by this SNP (Chang et al., 2011). Further studies are necessary to replicate the association, to describe how AQP2 rs296766 and SLC12A1 rs12904216 SNPs affect the coded transporter function, and to clarify a possible existence of other functional polymorphisms in linkage disequilibrium.
The other study genotyped 384 SNPs in genes involved in cardiovascular and metabolic pathways (Ruaño et al., 2009). The top five polymorphisms possibly associated with edema were: neuropeptide Y (NPY) rs1468271 (p = 0.006), glycogen synthase 1 (GYS1) rs2287754 (p = 0.013), chemokine C-C motif ligand 2 (CCL2) rs3760396 (p = 0.015), oxidized low density lipoprotein receptor 1 (OLR1) rs2742115 (p = 0.015), and growth hormone releasing hormone (GHRH) rs6032470 (p = 0.023) (Ruaño et al., 2009). However, none of the polymorphisms associated with edema after accounting for multiple comparisons.
The other factors related to edema were women sex and older age (Chang et al., 2011). According to these findings and the pharmacogenetic data, the investigators developed a simple point system to predict the likelihood of TZD-related edema based on age, sex, and AQP2 rs296766 and SLC12A1 rs12904216 genotypes (Chang et al., 2011). Although this point system needs to be validated in an independent cohort, the development of a simple scoring system is a good example of making pharmacogenetic information more useful and meaningful in clinical practice.
Increase in body mass index (BMI)
Besides the occurrence of edema, Ruaño et al. (2009) investigated possible associations of the 384 SNPs from cardiovascular and metabolic pathways with change in BMI. Weight gain is one of the common ADRs of TZDs. In a PIO monotherapy trial, patients gained 2.82 kg after the treatment of 45 mg PIO for 26 weeks while weight loss was observed in a placebo group (Kung and Henry, 2012). The weight gain appears to be dose-dependent, and the mechanism is not only related to the fluid retention but also increase in subcutaneous fat depots (Kung and Henry, 2012). The top five polymorphisms possibly associated with change in BMI were: adenosine A1 receptor (ADORA1) rs903361 (0.0003), pyruvate kinase (PKM2) rs2856929 (p = 0.002), ADIPOR2 rs7975375 (p = 0.007), uncoupling protein 2 (UCP2) rs660339 (p = 0.008), and apolipoprotein H (APOH) rs8178847 (p = 0.010) (Ruaño et al., 2009). After adjustment for multiple comparisons, rs903361, a SNP located in intron 2 of the ADORA1 gene, was significantly associated with increase in BMI (Ruaño et al., 2009). The adenosine A1 receptor is a G-protein-coupled receptor highly expressed in adipose tissue and activation of this receptor leads to inhibition of lipolysis (Dhalla et al., 2009). How the rs903361 SNP affects the functionality of the receptor is unknown and the association of this SNP with BMI must still be replicated in an independent cohort.
Conclusions
Clinically relevant variability in pioglitazone response has been demonstrated, but the underlying factors contributing to variability are not well-understood. This hypothesis article summarizes current evidence on pioglitazone pharmacogenomics; current data suggest that genetic variation is indeed an important factor contributing to pioglitazone response. Thus, we posit a strategy incorporating pharmacogenomic information may provide a more rational approach to achieve optimal outcomes. According to PharmGKB (https://www.pharmgkb.org), CYP2C8*3 is the only genetic variant annotated for PIO treatment as level 3 evidence, which was defined as “annotation for a variant-drug combination based on a single significant (not yet replicated) or annotation for a variant-drug combination evaluated in multiple studies but lacking clear evidence of an association”(Whirl-Carrillo et al., 2012). However, more studies investigating genetic effect on PIO therapy are now available. In this article, the PIO pharmacogenetic evidence to date is summarized. The genes investigated for sources of variability in PIO PK are CYP2C8 and SLCO1B1. The variant allele CYP2C8*3 is associated with higher PIO clearance, which results in reduced exposure to PIO in carriers of this allele. The alteration in PIO PK has clinical implications, and the relevant consequences with drug interaction are a concern. Thus, if an inhibitor, inducer or substrate of CYP2C8 is to be administered with PIO in known CYP2C8*3 carriers, closer monitoring may be warranted. For future studies of PIO, CYP2C8 genotyping is recommended to better understand the variability in response to PIO and to interpret data more clearly when CYP2C8*3 carriers are present in the study. This may be especially important because of racial differences in CYP2C8*3 allele frequency. The *3 variant has been the focus of clinical PK studies with PIO, but the functionality of other CYP2C8 variant alleles need to be further determined preferably in vivo because the discrepancy between in vitro and in vivo data have been shown with CYP2C8 activity. In contrast to CYP2C8, there is currently no evidence to suggest that genetic variation in drug transporters affects PIO disposition.
The genetic polymorphism most widely studied to better understand variability in PIO PD is PPARG p.Pro12Ala. T2DM patients with PPARG p.Pro12Ala variant are likely to have better glycemic response with PIO. Thus, the PPARG p.Pro12Ala is a promising polymorphism and one that should be included in clinical models developed to predict a patient's response based on genotype. However, this polymorphism alone is unlikely to explain all of the variability seen among patients. Consequently, other genetic variants as well as clinical factors must be considered. Indeed, several polymorphisms were identified in other genes related to PIO pathways. However, it is difficult to make any firm conclusions, based on single studies reporting a significant association, especially if it is not known how the polymorphism affects the functionality of the coded protein. Similarly, the identified polymorphism may merely be in linkage disequilibrium with the true functional variant. Therefore, the data must be interpreted with caution. Validation or replication of the finding is necessary before the pharmacogenetic knowledge can be applied to patient care. To better explain the variability in response, gene-gene and gene-environment interactions should also be considered.
Based on current evidence, we propose that pharmacogenomics may provide an important tool to individualize pioglitazone therapy. For example, developing a scoring system that incorporates both clinical and genetic factors may provide more clinical utility and make the pharmacogenetic evidence more accessible to clinicians who are unfamiliar with this field. This is important because the data from several genes suggest PIO pharmacogenomics has the potential to be an important means to better optimize therapy in patients with T2DM.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fphar.2013.00147/abstract
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACE, angiotensin-converting enzyme; ADIPOQ, adiponectin; ADIPOR, Adiponectin receptor; ADORA1, adenosine A1 receptor; ADR, adverse drug reaction; APOH, apolipoprotein H; AUC, area under the plasma concentration-time curve; BMI, body mass index; CCL2, chemokine C-C motif ligand 2; CHF, congestive heart failure; CYP, cytochrome P450; FDA, Food and Drug Administration; FG, fasting plasma glucose; GHRH, growth hormone releasing hormone; GYS1, glycogen synthase 1; IL-6, interleukin-6; IMT, intima-media thickness; LPL, lipoprotein lipase; MTHFR, methylene-tetrahydrofolate reductase; NPY, neuropeptide Y; OATP, organic anion transporter family member; OLR1, oxidized low density lipoprotein receptor 1; PD, pharmacodynamics; PIO, pioglitazone; PK, pharmacokinetics; PKM2, pyruvate kinase; PPARγ, peroxisome proliferator-activated receptor γ; PPRE, peroxisome proliferator response elements; PTPRD, protein tyrosine phosphatase receptor type D; RETN, resistin; ROSI, rosiglitazone; SLCO, solute carrier organic anion transporter family member; SNP, single nucleotide polymorphism; TNFα, Tumor necrosis factor α; TZD, thiazolidinedione; T2DM, type 2 diabetes mellitus; UCP2, uncoupling protein 2.
References
Altshuler, D., Hirschhorn, J. N., Klannemark, M., Lindgren, C. M., Vohl, M. C., Nemesh, J., et al. (2000). The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 26, 76–80. doi: 10.1038/79216
Aquilante, C. L., Kosmiski, L. A., Bourne, D. W., Bushman, L. R., Daily, E. B., Hammond, K. P., et al. (2013). Impact of the CYP2C8 *3 polymorphism on the drug-drug interaction between gemfibrozil and pioglitazone. Br. J. Clin. Pharmacol. 75, 217–226. doi: 10.1111/j.1365-2125.2012.04343.x
Auwerx, J. (1999). PPARgamma, the ultimate thrifty gene. Diabetologia 42, 1033–1049. doi: 10.1007/s001250051268
Betteridge, D. J. (2011). Thiazolidinediones and fracture risk in patients with Type 2 diabetes. Diabet. Med. 28, 759–771. doi: 10.1111/j.1464-5491.2010.03187.x
Bluher, M., Lubben, G., and Paschke, R. (2003). Analysis of the relationship between the Pro12Ala variant in the PPAR-gamma2 gene and the response rate to therapy with pioglitazone in patients with type 2 diabetes. Diabetes Care 26, 825–831. doi: 10.2337/diacare.26.3.825
Bogacka, I., Xie, H., Bray, G. A., and Smith, S. R. (2004). The effect of pioglitazone on peroxisome proliferator-activated receptor-gamma target genes related to lipid storage in vivo. Diabetes Care 27, 1660–1667. doi: 10.2337/diacare.27.7.1660
Cariou, B., Charbonnel, B., and Staels, B. (2012). Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol. Metab. 23, 205–215. doi: 10.1016/j.tem.2012.03.001
Castellan, A. C., Tod, M., Gueyffier, F., Audars, M., Cambriels, F., Kassai, B., et al. (2013). Quantitative prediction of the impact of drug interactions and genetic polymorphisms on cytochrome P450 2C9 substrate exposure. Clin. Pharmacokinet. 52, 199–209. doi: 10.1007/s40262-013-0031-3
Chang, T. J., Liu, P. H., Liang, Y. C., Chang, Y. C., Jiang, Y. D., Li, H. Y., et al. (2011). Genetic predisposition and nongenetic risk factors of thiazolidinedione-related edema in patients with type 2 diabetes. Pharmacogenet. Genomics 21, 829–836. doi: 10.1097/FPC.0b013e32834bfff1
Chung, S. S., Choi, H. H., Kim, K. W., Cho, Y. M., Lee, H. K., and Park, K. S. (2005). Regulation of human resistin gene expression in cell systems: an important role of stimulatory protein 1 interaction with a common promoter polymorphic site. Diabetologia 48, 1150–1158. doi: 10.1007/s00125-005-1762-y
Daily, E. B., and Aquilante, C. L. (2009). Cytochrome P450 2C8 pharmacogenetics: a review of clinical studies. Pharmacogenomics 10, 1489–1510. doi: 10.2217/pgs.09.82
Deeb, S. S., Fajas, L., Nemoto, M., Pihlajamaki, J., Mykkanen, L., Kuusisto, J., et al. (1998). A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 20, 284–287. doi: 10.1038/3099
Dhalla, A. K., Chisholm, J. W., Reaven, G. M., and Belardinelli, L. (2009). A1 adenosine receptor: role in diabetes and obesity. Handb. Exp. Pharmacol. 193, 271–295. doi: 10.1007/978-3-540-89615-9_9
Eckland, D. A., and Danhof, M. (2000). Clinical pharmacokinetics of pioglitazone. Exp. Clin. Endocrinol. Diabetes 108, S234–S242. doi: 10.1055/s-2000-8525
Gouda, H. N., Sagoo, G. S., Harding, A. H., Yates, J., Sandhu, M. S., and Higgins, J. P. (2010). The association between the peroxisome proliferator-activated receptor-gamma2 (PPARG2) Pro12Ala gene variant and type 2 diabetes mellitus: a HuGE review and meta-analysis. Am. J. Epidemiol. 171, 645–655. doi: 10.1093/aje/kwp450
Groenemeijer, B. E., Hallman, M. D., Reymer, P. W., Gagné, E., Kuivenhoven, J. A., Bruin, T., et al. (1997). Genetic variant showing a positive interaction with beta-blocking agents with a beneficial influence on lipoprotein lipase activity, HDL cholesterol, and triglyceride levels in coronary artery disease patients. The Ser447-stop substitution in the lipoprotein lipase gene. REGRESS Study Group. Circulation 95, 2628–2635. doi: 10.1161/01.CIR.95.12.2628
Gueugnon, C., Mougin, F., Simon-Rigaud, M. L., Regnard, J., Negre, V., and Dumoulin, G. (2012). Effects of an in-patient treatment program based on regular exercise and a balanced diet on high molecular weight adiponectin, resistin levels, and insulin resistance in adolescents with severe obesity. Appl. Physiol. Nutr. Metab. 37, 672–679. doi: 10.1139/h2012-045
Hara, K., Boutin, P., Mori, Y., Tobe, K., Dina, C., Yasuda, K., et al. (2002). Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes 51, 536–540. doi: 10.2337/diabetes.51.2.536
Himelfarb, S. T., Silva, F. A., Arazi, S. S., Farjado, C. M., Garofalo, A., Bertolami, M. C., et al. (2011). Tumor necrosis factor-α and interleukin-6 expression in leukocytes and their association with polymorphisms and bone markers in diabetic individuals treated with pioglitazone. Drug Metabol. Drug Interact. 26, 37–40. doi: 10.1515/dmdi.2011.100
Hounoki, H., Sugiyama, E., Mohamed, S. G., Shinoda, K., Taki, H., Abdel-Aziz, H. O., et al. (2008). Activation of peroxisome proliferator-activated receptor gamma inhibits TNF-alpha-mediated osteoclast differentiation in human peripheral monocytes in part via suppression of monocyte chemoattractant protein-1 expression. Bone 42, 765–774. doi: 10.1016/j.bone.2007.11.016
Hsieh, M. C., Lin, K. D., Tien, K. J., Tu, S. T., Hsiao, J. Y., Chang, S. J., et al. (2010). Common polymorphisms of the peroxisome proliferator-activated receptor-gamma (Pro12Ala) and peroxisome proliferator-activated receptor-gamma coactivator-1 (Gly482Ser) and the response to pioglitazone in Chinese patients with type 2 diabetes mellitus. Metabolism 59, 1139–1144. doi: 10.1016/j.metabol.2009.10.030
Jaakkola, T., Laitila, J., Neuvonen, P. J., and Backman, J. T. (2006). Pioglitazone is metabolised by CYP2C8 and CYP3A4 in vitro: potential for interactions with CYP2C8 inhibitors. Basic Clin. Pharmacol. Toxicol. 99, 44–51. doi: 10.1111/j.1742-7843.2006.pto_437.x
Ji, W., Foo, J. N., O'Roak, B. J., Zhao, H., Larson, M. G., Simon, D. B., et al. (2008). Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 40, 592–599. doi: 10.1038/ng.118
Jing, C., Xueyao, H., and Linong, J. (2012). Meta-analysis of association studies between five candidate genes and type 2 diabetes in Chinese Han population. Endocrine 42, 307–320. doi: 10.1007/s12020-012-9643-x
Kadam, R., Bourne, D., Kompella, U., and Aquilante, C. (2013). Effect of cytochrome P450 2C8*3 on the population pharmacokinetics of pioglitazone in healthy caucasian volunteers. Biol. Pharm. Bull. 36, 245–251. doi: 10.1248/bpb.b12-00657
Kadowaki, T., Yamauchi, T., Kubota, N., Hara, K., Ueki, K., and Tobe, K. (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116, 1784–1792. doi: 10.1172/JCI29126
Kalliokoski, A., Neuvonen, M., Neuvonen, P. J., and Niemi, M. (2008). No significant effect of SLCO1B1 polymorphism on the pharmacokinetics of rosiglitazone and pioglitazone. Br. J. Clin. Pharmacol. 65, 78–86. doi: 10.1111/j.1365-2125.2007.02986.x
Kang, E. S., Park, S. Y., Kim, H. J., Kim, C. S., Ahn, C. W., Cha, B. S., et al. (2005). Effects of Pro12Ala polymorphism of peroxisome proliferator-activated receptor gamma2 gene on rosiglitazone response in type 2 diabetes. Clin. Pharmacol. Ther. 78, 202–208. doi: 10.1016/j.clpt.2005.04.013
Knepper, M. A., Wade, J. B., Terris, J., Ecelbarger, C. A., Marples, D., Mandon, B., et al. (1996). Renal aquaporins. Kidney Int. 49, 1712–1717. doi: 10.1038/ki.1996.253
Kuivenhoven, J. A., Groenemeyer, B. E., Boer, J. M., Reymer, P. W., Berghuis, R., Bruin, T., et al. (1997). Ser447stop mutation in lipoprotein lipase is associated with elevated HDL cholesterol levels in normolipidemic males. Arterioscler. Thromb. Vasc. Biol. 17, 595–599. doi: 10.1161/01.ATV.17.3.595
Kung, J., and Henry, R. R. (2012). Thiazolidinedione safety. Expert Opin. Drug Saf. 11, 565–579. doi: 10.1517/14740338.2012.691963
Lai, X. S., Yang, L. P., Li, X. T., Liu, J. P., Zhou, Z. W., and Zhou, S. F. (2009). Human CYP2C8: structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr. Drug Metab. 10, 1009–1047. doi: 10.2174/138920009790711832
Lee, K. Y., Kang, H. S., and Shin, Y. A. (2013). Exercise improves adiponectin concentrations irrespective of the adiponectin gene polymorphisms SNP45 and the SNP276 in obese Korean women. Gene 516, 271–276. doi: 10.1016/j.gene.2012.12.028
Li, Z., Peng, X., Wu, Y., Xia, Y., Liu, X., and Zhang, Q. (2008). The influence of adiponectin gene polymorphism on the pioglitazone response in the Chinese with type 2 diabetes. Diabetes Obes. Metab. 10, 794–802. doi: 10.1111/j.1463-1326.2008.00905.x
Makino, H., Shimizu, I., Murao, S., Kondo, S., Tabara, Y., Fujiyama, M., et al. (2009). A pilot study suggests that the G/G genotype of resistin single nucleotide polymorphism at -420 may be an independent predictor of a reduction in fasting plasma glucose and insulin resistance by pioglitazone in type 2 diabetes. Endocr. J. 56, 1049–1058. doi: 10.1507/endocrj.K08E-320
Martinez, C., Garcia-Martin, E., Blanco, G., Gamito, F. J., Ladero, J. M., and Agundez, J. A. (2005). The effect of the cytochrome P450 CYP2C8 polymorphism on the disposition of (R)-ibuprofen enantiomer in healthy subjects. Br. J. Clin. Pharmacol. 59, 62–69. doi: 10.1111/j.1365-2125.2004.02183.x
Martis, S., Peter, I., Hulot, J. S., Kornreich, R., Desnick, R. J., and Scott, S. A. (2013). Multi-ethnic distribution of clinically relevant CYP2C genotypes and haplotypes. Pharmacogenomics J. 13, 369–377. doi: 10.1038/tpj.2012.10
Menzaghi, C., Ercolino, T., Di Paola, R., Berg, A. H., Warram, J. H., Scherer, P. E., et al. (2002). A haplotype at the adiponectin locus is associated with obesity and other features of the insulin resistance syndrome. Diabetes 51, 2306–2312. doi: 10.2337/diabetes.51.7.2306
Nagajothi, N., Adigopula, S., Balamuthusamy, S., Velazquez-Cecena, J. L., Raghunathan, K., Khraisat, A., et al. (2008). Pioglitazone and the risk of myocardial infarction and other major adverse cardiac events: a meta-analysis of randomized, controlled trials. Am. J. Ther. 15, 506–511. doi: 10.1097/MJT.0b013e318167180c
Namvaran, F., Azarpira, N., Rahimi-Moghaddam, P., and Dabbaghmanesh, M. H. (2011). Polymorphism of peroxisome proliferator-activated receptor gamma (PPARgamma) Pro12Ala in the Iranian population: relation with insulin resistance and response to treatment with pioglitazone in type 2 diabetes. Eur. J. Pharmacol. 671, 1–6. doi: 10.1016/j.ejphar.2011.09.158
Namvaran, F., Rahimi-Moghaddam, P., Azarpira, N., and Dabbaghmanesh, M. H. (2012). Polymorphism of adiponectin (45T/G) and adiponectin receptor-2 (795G/A) in an Iranian population: relation with insulin resistance and response to treatment with pioglitazone in patients with type 2 diabetes mellitus. Mol. Biol. Rep. 39, 5511–5518. doi: 10.1007/s11033-011-1354-5
Ochi, M., Osawa, H., Hirota, Y., Hara, K., Tabara, Y., Tokuyama, Y., et al. (2007). Frequency of the G/G genotype of resistin single nucleotide polymorphism at -420 appears to be increased in younger-onset type 2 diabetes. Diabetes 56, 2834–2838. doi: 10.2337/db06-1157
Osawa, H., Yamada, K., Onuma, H., Murakami, A., Ochi, M., Kawata, H., et al. (2004). The G/G genotype of a resistin single-nucleotide polymorphism at -420 increases type 2 diabetes mellitus susceptibility by inducing promoter activity through specific binding of Sp1/3. Am. J. Hum. Genet. 75, 678–686. doi: 10.1086/424761
Pei, Q., Huang, Q., Yang, G. P., Zhao, Y. C., Yin, J. Y., Song, M., et al. (2013). PPAR-gamma2 and PTPRD gene polymorphisms influence type 2 diabetes patients' response to pioglitazone in China. Acta Pharmacol. Sin. 34, 255–261. doi: 10.1038/aps.2012.144
Ramirez-Salazar, M., Perez-Luque, E., Fajardo-Araujo, M., Garza, S. M., and Malacara, J. M. (2008). Effect of the Pro12Ala polymorphism of the PPAR gamma 2 gene on response to pioglitazone treatment in menopausal women. Menopause 15, 1151–1156. doi: 10.1097/gme.0b013e31816d5b2d
Ruaño, G., Bernene, J., Windemuth, A., Bower, B., Wencker, D., Seip, R. L., et al. (2009). Physiogenomic comparison of edema and BMI in patients receiving rosiglitazone or pioglitazone. Clin. Chim. Acta 400, 48–55. doi: 10.1016/j.cca.2008.10.009
Ruscica, M., Steffani, L., and Magni, P. (2012). Adiponectin interactions in bone and cartilage biology and disease. Vitam. Horm. 90, 321–339. doi: 10.1016/B978-0-12-398313-8.00012-9
Saitou, M., Osonoi, T., Kawamori, R., Katakami, N., Kaneto, H., Matsuhisa, M., et al. (2010). Genetic risk factors and the anti-atherosclerotic effect of pioglitazone on carotid atherosclerosis of subjects with type 2 diabetes–a retrospective study. J. Atheroscler. Thromb. 17, 386–394. doi: 10.5551/jat.2527
Saraf, N., Sharma, P. K., Mondal, S. C., Garg, V. K., and Singh, A. K. (2012). Role of PPARg2 transcription factor in thiazolidinedione-induced insulin sensitization. J. Pharm. Pharmacol. 64, 161–171. doi: 10.1111/j.2042-7158.2011.01366.x
Scheen, A. J. (2007). Pharmacokinetic interactions with thiazolidinediones. Clin. Pharmacokinet. 46, 1–12. doi: 10.2165/00003088-200746010-00001
Stumvoll, M., Tschritter, O., Fritsche, A., Staiger, H., Renn, W., Weisser, M., et al. (2002). Association of the T-G polymorphism in adiponectin (exon 2) with obesity and insulin sensitivity: interaction with family history of type 2 diabetes. Diabetes 51, 37–41. doi: 10.2337/diabetes.51.1.37
Sun, H., Gong, Z. C., Yin, J. Y., Liu, H. L., Liu, Y. Z., Guo, Z. W., et al. (2008). The association of adiponectin allele 45T/G and -11377C/G polymorphisms with Type 2 diabetes and rosiglitazone response in Chinese patients. Br. J. Clin. Pharmacol. 65, 917–926. doi: 10.1111/j.1365-2125.2008.03145.x
Tannen, R., Xie, D., Wang, X., Yu, M., and Weiner, M. G. (2013). A new “Comparative Effectiveness” assessment strategy using the THIN database: comparison of the cardiac complications of pioglitazone and rosiglitazone. Pharmacoepidemiol. Drug Saf. 22, 86–97. doi: 10.1002/pds.3360
Tornio, A., Niemi, M., Neuvonen, P. J., and Backman, J. T. (2008). Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab. Dispos. 36, 73–80. doi: 10.1124/dmd.107.018010
Tsai, F. J., Yang, C. F., Chen, C. C., Chuang, L. M., Lu, C. H., Chang, C. T., et al. (2010). A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet. 6:e1000847. doi: 10.1371/journal.pgen.1000847
Umpierrez, G., and Dagogo-Jack, S. (2006). Role of thiazolidinediones in the management of type 2 diabetes: focus on ethnic minority populations. Ethn. Dis. 16, 51–57.
Vandenbrink, B. M., Foti, R. S., Rock, D. A., Wienkers, L. C., and Wahlstrom, J. L. (2011). Evaluation of CYP2C8 inhibition in vitro: utility of montelukast as a selective CYP2C8 probe substrate. Drug Metab. Dispos. 39, 1546–1554. doi: 10.1124/dmd.111.039065
Vasseur, F., Helbecque, N., Dina, C., Lobbens, S., Delannoy, V., Gaget, S., et al. (2002). Single-nucleotide polymorphism haplotypes in the both proximal promoter and exon 3 of the APM1 gene modulate adipocyte-secreted adiponectin hormone levels and contribute to the genetic risk for type 2 diabetes in French Caucasians. Hum. Mol. Genet. 11, 2607–2614. doi: 10.1093/hmg/11.21.2607
Vasseur, F., Leprêtre, F., Lacquemant, C., and Froguel, P. (2003). The genetics of adiponectin. Curr. Diab. Rep. 3, 151–158. doi: 10.1007/s11892-003-0039-4
Vozarova De Courten, B., Hanson, R. L., Funahashi, T., Lindsay, R. S., Matsuzawa, Y., Tanaka, S., et al. (2005). Common polymorphisms in the adiponectin gene ACDC are not associated with diabetes in Pima Indians. Diabetes 54, 284–289. doi: 10.2337/diabetes.54.1.284
Wan, Y. (2010). PPARγ in bone homeostasis. Trends Endocrinol. Metab. 21, 722–728. doi: 10.1016/j.tem.2010.08.006
Wang, G., Wang, X., Zhang, Q., and Ma, Z. (2007). Response to pioglitazone treatment is associated with the lipoprotein lipase S447X variant in subjects with type 2 diabetes mellitus. Int. J. Clin. Pract. 61, 552–557. doi: 10.1111/j.1742-1241.2006.01242.x
Whirl-Carrillo, M., McDonagh, E. M., Hebert, J. M., Gong, L., Sangkuhl, K., Thorn, C. F., et al. (2012). Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92, 414–417. doi: 10.1038/clpt.2012.96
Woo, J. G., Dolan, L. M., Deka, R., Kaushal, R. D., Shen, Y., Pal, P., et al. (2006). Interactions between noncontiguous haplotypes in the adiponectin gene ACDC are associated with plasma adiponectin. Diabetes 55, 523–529. doi: 10.2337/diabetes.55.02.06.db05-0446
Yau, H., Rivera, K., Lomonaco, R., and Cusi, K. (2013). The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr. Diab. Rep. 13, 329–341. doi: 10.1007/s11892-013-0378-8
Keywords: pioglitazone, thiazolidinedione, CYP2C8, cytochrome P450, PPAR, pharmacokinetics, pharmacodynamics
Citation: Kawaguchi-Suzuki M and Frye RF (2013) Current clinical evidence on pioglitazone pharmacogenomics. Front. Pharmacol. 4:147. doi: 10.3389/fphar.2013.00147
Received: 30 July 2013; Accepted: 07 November 2013;
Published online: 26 November 2013.
Edited by:
Andrea Gaedigk, The Children's Mercy Hospital and Clinics, USAReviewed by:
Louise Warnich, Stellenbosch University, South AfricaStuart Scott, Mount Sinai School of Medicine, USA
Copyright © 2013 Kawaguchi-Suzuki and Frye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reginald F. Frye, Department of Pharmacotherapy and Translational Research, Center for Pharmacogenomics, College of Pharmacy, University of Florida, 1225 Center Drive, HPNP 3308, PO Box 100486, Gainesville, FL 32610-0486, USA e-mail:ZnJ5ZUBjb3AudWZsLmVkdQ==