 Birce Onal1,2
Birce Onal1,2 Thomas J. Hund
Thomas J. Hund- 1The Dorothy M. Davis Heart and Lung Research Institute, Wexner Medical Center, The Ohio State University, Columbus, OH, USA
- 2Department of Biomedical Engineering, College of Engineering, The Ohio State University, Columbus, OH, USA
- 3Department of Internal Medicine, Wexner Medical Center, The Ohio State University, Columbus, OH, USA
Post-translational modification of membrane proteins (e.g., ion channels, receptors) by protein kinases is an essential mechanism for control of excitable cell function. Importantly, loss of temporal and/or spatial control of ion channel post-translational modification is common in congenital and acquired forms of cardiac disease and arrhythmia. The multifunctional Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates a number of diverse cellular functions in heart, including excitation-contraction coupling, gene transcription, and apoptosis. Dysregulation of CaMKII signaling has been implicated in human and animal models of disease. Understanding of CaMKII function has been advanced by mathematical modeling approaches well-suited to the study of complex biological systems. Early kinetic models of CaMKII function in the brain characterized this holoenzyme as a bistable molecular switch capable of storing information over a long period of time. Models of CaMKII activity have been incorporated into models of the cell and tissue (particularly in the heart) to predict the role of CaMKII in regulating organ function. Disease models that incorporate CaMKII overexpression clearly demonstrate a link between its excessive activity and arrhythmias associated with congenital and acquired heart disease. This review aims at discussing systems biology approaches that have been applied to analyze CaMKII signaling from the single molecule to intact cardiac tissue. In particular, efforts to use computational biology to provide new insight into cardiac disease mechanisms are emphasized.
Introduction
Signal transduction, whereby a cell receives and processes extracellular information to coordinate a cellular process, is critical for normal cell function. Signal-transduction systems are commonly perturbed in disease, making core constituents (e.g., kinases) attractive therapeutic targets (Levitzki, 2003). While we have learned a great deal about the components of key signaling pathways, the complex nature of these vast networks represents a significant obstacle to understand their dynamics, regulation, and function. Systems biology and computational modeling of biological systems have become increasingly valuable in enhancing our understanding of these complex protein interaction networks.
Systems biology involves the study of the complex interactions and associated dynamics found in biological systems. Systems biology approaches commonly involve translation of the system into a mathematical model for subsequent computer simulation and analysis. As systems-based approaches have gained favor in the study of human disease processes, so has mathematical modeling of biological systems with associated advancements in understanding complex biological phenomenon like circadian rhythms, apoptosis, synaptic plasticity, and cell communication (Herzel and Bluthgen, 2008; Kotaleski and Blackwell, 2010).
The multifunctional Ca2+/calmodulin-dependent protein kinase II (CaMKII) has emerged as an attractive target for systems-based approaches that aim to integrate large experimental data with mathematical modeling and computational approaches across spatial and temporal scales (Figure 1). CaMKII serves as a nodal point for a vast signaling network that regulates critical processes like learning and memory, cardiomyocyte contractility, T-cell selection, and expression and localization of class II MHC molecules in dendritic cells (Braun and Schulman, 1995; Maier and Bers, 2002; McGargill et al., 2005; Herrmann et al., 2007; Anderson et al., 2011; Swaminathan et al., 2012). For example, CaMKII regulates multiple important functions in neurons, including synthesis and release of neurotransmitters, modulation of ion channel activity, neurite extension, synaptic plasticity, learning, and gene expression (Braun and Schulman, 1995). Similarly, in heart, CaMKII phosphorylates ion channels, transcription factors, signaling molecules, and other membrane proteins that are critical to cardiac electrical activity and structure. Abnormal CaMKII activity has been observed in human and animal models of cardiovascular disease (e.g., heart failure, myocardial infarction, arrhythmia), and is thought to promote downstream dysfunction in excitation-contraction coupling, structural remodeling, cell death, and even transcriptional activation of inflammation factors (Maier and Bers, 2002; Swaminathan et al., 2012). Current research aims at elucidating how this large effector molecule acts as a pro-cardiac disease/arrhythmogenic molecule and whether it may be effectively targeted for therapy.
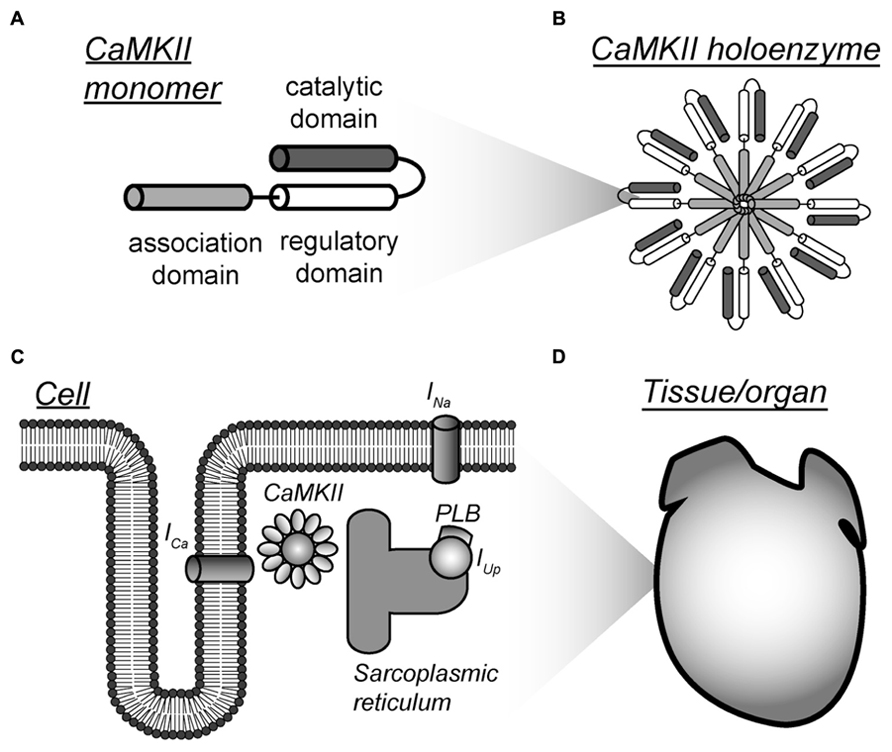
FIGURE 1. Multi-scalar mathematical modeling of CaMKII signaling. (A–D) Mathematical modeling has been applied to understand CaMKII function across scales from the molecular to the tissue level. Each scale presents unique challenges and opportunities for modeling efforts. Abbreviations are as follows: ICa, L-type Ca2+ current; INa, fast Na+ current; PLB, phospholamban; IUp, Ca2+ uptake into the sarcoplasmic reticulum.
Mathematical modeling studies over the past three decades have elucidated important aspects of CaMKII function and signaling mechanisms. Pioneering modeling studies focused on understanding CaMKII structure and function in the brain (Lisman, 1985; Lisman and Goldring, 1988; Hanson et al., 1994; Coomber, 1998; Kubota, 1999; Dupont et al., 2003). This early work motivated later studies that incorporated models of CaMKII activity into models of the whole cell and tissue (mostly cardiac) to understand the larger role of CaMKII signaling in cell/organ function (Figure 1; Hund and Rudy, 2004; Iribe et al., 2006; Grandi et al., 2007; Livshitz and Rudy, 2007; Hund et al., 2008; Saucerman and Bers, 2008; Christensen et al., 2009; Koivumaki et al., 2009; Hashambhoy et al., 2010; O’Hara et al., 2011). Recently, these efforts have been expanded to gain insight into the role of CaMKII in human disease (Hund et al., 2008; Christensen et al., 2009; Koivumaki et al., 2009; Hashambhoy et al., 2010; Swaminathan et al., 2011; Lascano et al., 2013; Luo et al., 2013; Zang et al., 2013). This review aims at describing the challenges, advances and opportunities for mathematical modeling of CaMKII signaling at each stage of development across scales from the molecular to the tissue level.
Modeling the Camkii Holoenzyme
The CaMKII holoenzyme possesses a number of distinguishing characteristics that pose unique challenges for modeling. Briefly (details may be found elsewhere (Couchonnal and Anderson, 2008; Anderson et al., 2011; Swaminathan et al., 2012), multiple CaMKII isoforms are expressed in cells with CaMKIIα and CaMKIIβ expressed predominantly in neurons, whereas CaMKIIγ and CaMKIIδ are more uniformly expressed in other tissues. Structurally, the CaMKII holoenzyme is organized as a hexamer of dimers arranged as two stacked rings. Each monomer is comprised of an N-terminal catalytic domain, a regulatory domain, and a C-terminal association domain. In its inactive conformation, the regulatory domain binds to the active site in catalytic domain, thereby inhibiting the activity of the enzyme. Association of Ca2+ bound calmodulin to the regulatory domain causes its release from the active site and exposes the active site in catalytic subunit, enabling the kinase to phosphorylate its substrates (Kolodziej et al., 2000; Rosenberg et al., 2005). Multiple residues within the regulatory domain are also exposed that may subsequently undergo post-translational regulation (e.g., phosphorylation, oxidation, glycosylation) that, in turn, alter kinase function (Lai et al., 1986; Braun and Schulman, 1995; Erickson et al., 2008; Swaminathan et al., 2012; Erickson et al., 2013). Enzyme regulation/activity depends heavily on the multimeric holoenzyme structure (Kolodziej et al., 2000; Hoelz et al., 2003; Rellos et al., 2010; Chao et al., 2011; Stratton et al., 2013). For example, a distinguishing characteristic is the ability of CaMKII to undergo autophosphorylation where an active (Ca2+/calmodulin bound) kinase subunit is phosphorylated at a specific residue (Thr286/287) by a neighboring active subunit (Lai et al., 1986; Braun and Schulman, 1995). The autophosphorylated kinase retains activity in the absence of bound Ca2+/calmodulin and is thought to contribute to synaptic plasticity and learning functions as well as myocyte excitation-contraction coupling (Silva et al., 1992a,b; Dupont et al., 2003).
One of the most obvious and compelling challenges for modeling of CaMKII is autoregulation. The simplest models consider the entire population of CaMKII subunits that are subject to autophosphorylation at a rate dependent on levels of Ca2+/calmodulin (Hanson et al., 1994; Dupont et al., 2003; Gaertner et al., 2004; Chiba et al., 2008). Detailed models have also been developed that incorporate structural information to account for the fact that CaMKII autophophosphorylation is constrained by physical proximity of active subunits (Lisman and Goldring, 1988; Michelson and Schulman, 1994; Zhabotinsky, 2000; Kubota and Bower, 2001; Miller et al., 2005; Lucic et al., 2008; Michalski, 2013). Recently, efforts have been made to also account for other kinase activation modes (e.g., oxidation; Christensen et al., 2009). Modeling studies at the molecular level have generated important insight into CaMKII function. In particular, models have been used to demonstrate that CaMKII activity is sensitive to changes in Ca2+ spike frequency and is capable of long-term storage of information at the post-synaptic density by acting as a bistable switch (Lisman, 1985; Lisman and Goldring, 1988; Hanson et al., 1994; Coomber, 1998; Kubota, 1999; Dupont et al., 2003). Furthermore, modeling studies have demonstrated the importance of autophosphorylation for bistability in CaMKII signaling, although there is some debate about the requisite conditions and physiological relevance (Zhabotinsky, 2000; Michalski, 2013). Together, these initial CaMKII modeling studies provided important insight into the link between holoenzyme structure, the ability of the kinase to encode Ca2+spike information, and behavior (e.g., long-term potentiation) in neurons. Moreover, this work laid the essential foundation for subsequent multi-scale studies in other systems (e.g., heart).
Modeling Camkii Signaling in the Intact Cell and Tissue
Much work has been done, particularly in the cardiac field, to incorporate models of the CaMKII signaling pathway into models of the intact cell (Figure 2). Modeling of CaMKII signaling at the cellular level poses a unique set of challenges in addition to those encountered at the molecular level (Table 1). First, the kinase is sensitive to intracellular Ca2+, whose temporal and spatial profile is tightly controlled. In the myocyte, for example, influx of Ca2+ through voltage-gated Ca2+ channels during the action potential (AP) triggers Ca2+ release from the sarcoplasmic reticulum (SR) that leads to a large increase in intracellular Ca2+(free and calmodulin-bound) levels. Thus, any cell model of the kinase pathway must address the dynamic nature of the input, namely Ca2+-bound calmodulin. Second, once activated, the multifunctional kinase targets a large number of substrates in the cell, from membrane ion channels, pumps and transporters to contractile proteins and even transcription factors. One must consider a priori which targets are likely important for the phenomenon of interest. Finally, CaMKII interacts with a vast and complex signaling web that includes other proteins directly regulated by Ca2+/calmodulin (e.g., ion channels, calcineurin), protein phosphatases that antagonize CaMKII phosphorylation (e.g., PP1), and other kinases that potentially synergize CaMKII effects (e.g., protein kinase A).
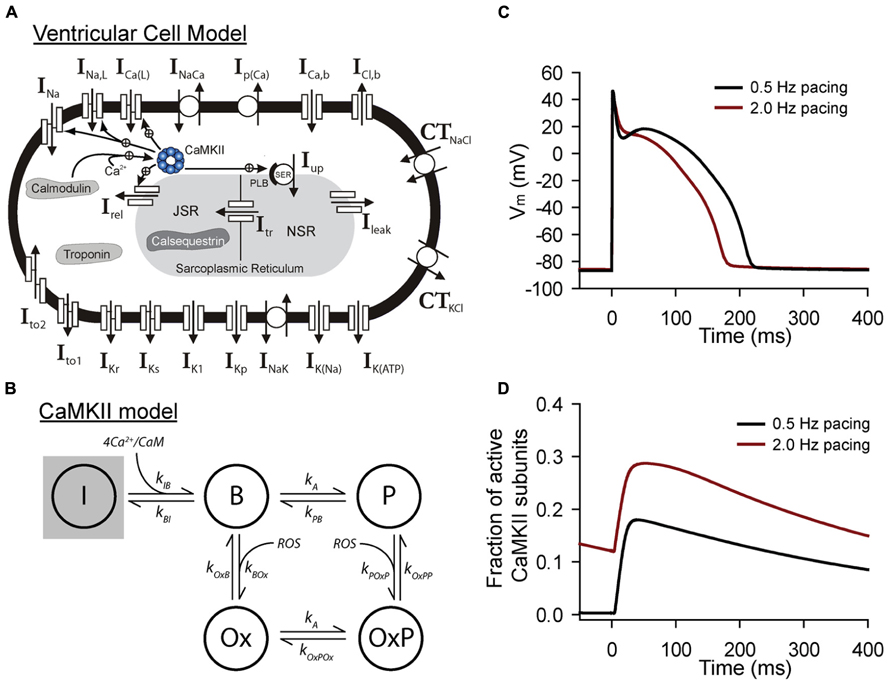
FIGURE 2. Mathematical modeling of CaMKII signaling in the intact cell. (A) Schematic of the Hund-Rudy model of the ventricular action potential that accounts for dynamic CaMKII signaling (Christensen et al., 2009). Abbreviations are as follows: INa, fast Na+ current; INa,L, late (persistent) Na+ current; ICa(L), L-type Ca2+ current; INaCa, Na+/Ca2+ exchanger; Ip(Ca), sarcolemmal Ca2+ pump; ICa,b, background Ca2+ current; ICl,b, background Cl- current; CTNaCl, Na+/Cl-cotransporter; CTKCl, K+/Cl-cotransporter; Ito1, transient outward K+ current; Ito2, Ca2+-dependent transient outward current; IKr, rapid delayed rectifier K+ current; IKs, slow delayed rectifier K+ current; IK1, inward rectifier K+ current; IKp, plateau K+ current; INaK, Na+/K+ ATPase; IK(Na), Na+-dependent K+ current; IK(ATP), ATP-sensitive K+ current; Irel, sarcoplasmic reticulum (SR) ryanodine receptor Ca2+ release channel; IUp, SR Ca2+ pump; PLB, phospholamban; Ileak, SR Ca2+ leak; NSR, network SR; JSR, junctional SR; Itr, Ca2+ transfer from NSR to JSR. (B) State diagram for integrated CaMKII model that includes inactive (I), Ca2+/calmodulin bound (B), autophosphorylated (P, OxP) and oxidized active states (Ox, OxP). Abbreviations are as follows: kIB, kBI, forward and reverse rate constants, respectively, for transition from inactive state to Ca2+/calmodulin bound state; kA, kPB, autophosphorylation and dephosphorylation rate constants, respectively, for transition from Ca2+/calmodulin bound state to autophosphorylated state; kA, kOxPOx, autophosphorylation and dephosphorylation rate constants, respectively, for transition from oxidized active state to oxidized and autophosphorylated state; kOxB, kBOx, forward and reverse rate constants, respectively, for transition fromCa2+/calmodulin bound state to oxidized active state; kPOxP, kOxPP, forward and reverse rate constants, respectively, for transition from autophosphorylated state to oxidized and autophosphorylated active state. Simulated (C) action potentials and (D) CaMKII activity from the model at two different pacing frequencies to demonstrate sensitivity of CaMKII to pacing frequency.

TABLE 1. Challenges for modeling of CaMKII activity across scales from molecule to tissue.
Despite these numerous obstacles, CaMKII signaling networks have been successfully incorporated with varying degrees of complexity into whole cell models of the myocyte (mostly ventricular) action potential and calcium transient (Hund and Rudy, 2004; Iribe et al., 2006; Grandi et al., 2007; Livshitz and Rudy, 2007; Hund et al., 2008; Saucerman and Bers, 2008; Christensen et al., 2009; Koivumaki et al., 2009; Hashambhoy et al., 2010; O’Hara et al., 2011), as well as other non-cardiac cell types (Dupont et al., 2010; Mironov, 2013). These models have employed different strategies to deal with challenges outlined above. The most common class of models incorporate a scheme where a single population of CaMKII responds to changes in bulk or subspace Ca2+/calmodulin (Hund and Rudy, 2004; Iribe et al., 2006; Livshitz and Rudy, 2007; Hund et al., 2008). In other cases, a static formalism is adopted where CaMKII-dependent effects on membrane substrates are implemented in the absence of dynamic changes in CaMKII activity (Grandi et al., 2007; Thiel et al., 2008; Koval et al., 2010; Koval et al., 2012). More recently, consideration has been given to compartmentalization of CaMKII signaling within the cell (Saucerman and Bers, 2008; Song et al., 2008; Soltis and Saucerman, 2010). In general, models account for CaMKII-dependent effects on membrane ion channels and transporters important for Ca2+ cycling, including the ryanodine receptor (RyR), SERCA 2a (SR Ca2+ ATPase), phospholamban (PLB), and L-type Ca2+ channels. As data have emerged regarding CaMKII-dependent effects on other channels important for the action potential (e.g., INa and Ito), these effects have also been incorporated (Grandi et al., 2007; Hund et al., 2008; Christensen et al., 2009; Hashambhoy et al., 2010; Koval et al., 2012). It is expected that as we learn more about the specific molecular targets for CaMKII within the cell, models will adapt to account for the new findings.
What have we learned from cellular models of CaMKII signaling? Several computational studies have demonstrated the ability of CaMKII to regulate myocyte action potential, Ca2+ transient, and even contractile force in a rate-dependent manner (Hund and Rudy, 2004; Iribe et al., 2006; Livshitz and Rudy, 2007; Soltis and Saucerman, 2010; O’Hara et al., 2011). Interestingly, a role for CaMKII has emerged not only in normal rate dependent behavior (e.g., AP duration adaptation and force-frequency relationships), but also in promoting cellular triggers for arrhythmias such as AP alternans and afterdepolarizations (Grandi et al., 2007; Livshitz and Rudy, 2007; Thiel et al., 2008; Hashambhoy et al., 2010; Koval et al., 2010; Soltis and Saucerman, 2010). Integrated myocyte models have also been applied to increase our understanding of spatial and temporal control of CaMKII signaling (Saucerman and Bers, 2008; Song et al., 2008; Soltis and Saucerman, 2010). Interestingly, studies in this area have demonstrated the importance of affinity for Ca2+/calmodulin in defining the differential response of CaMKII and the protein phosphatase calcineurin to the dynamic Ca2+ transient (Saucerman and Bers, 2008; Song et al., 2008). Furthermore, studies that incorporate both CaMKII and PKA signaling have shown how the two networks synergize for joint regulation of excitation-contraction coupling (Soltis and Saucerman, 2010). It will be interesting, going forward, to model how other factors such as interaction with scaffolding/anchoring proteins (e.g., βIV-spectrin) may contribute to spatial control of CaMKII signaling (Hund et al., 2010; Koval et al., 2010), similar to studies involving other signaling networks (Chan et al., 2012; Greenwald et al., 2014). Finally, although considerable less work has been done in this area compared to smaller scales, progress has been made to understand the role of CaMKII in coordinating function at the tissue/organ level (Livshitz and Rudy, 2007; Christensen et al., 2009; Swaminathan et al., 2011; Luo et al., 2013). These multicellular studies have identified roles for CaMKII in regulating AP heterogeneity and conduction, as well as cardiac pacemaking.
Modeling Camkii Signaling in Disease
CaMKII plays a critical role in regulating the substrate for both electrical and mechanical dysfunction in cardiovascular disease (Anderson et al., 2011; Swaminathan et al., 2012). Perhaps the greatest challenge for mathematical modeling of CaMKII signaling is how to ultimately link function at the molecular level to behavior at cell/tissue level in the setting of disease. Among the difficulties for modeling in this area involves distinguishing between acute and chronic effects of CaMKII activity. For example, while acute effects of CaMKII are mostly mediated by posttranslational modification of substrates, chronic CaMKII activation may facilitate large scale remodeling changes due to effects on transcription and gene expression (Koivumaki et al., 2009; Swaminathan et al., 2012). Mathematical modeling and computer simulation have been used to generate new insights into molecular mechanisms for arrhythmia in several disease states, including myocardial ischemia/infarction, heart failure, and diabetes (Hund et al., 2008; Christensen et al., 2009; Swaminathan et al., 2011; Lascano et al., 2013; Luo et al., 2013; Zang et al., 2013).
Arrhythmia mechanisms in the canine infarct border zone have been studied extensively using a mathematical modeling approach (Cabo and Boyden, 2003; Cabo et al., 2006; Hund et al., 2008; Christensen et al., 2009). The canine infarct border zone is particularly well suited to mathematical modeling approach due to the tremendous amount of available data at the molecular, cellular, and tissue level (Pinto and Boyden, 1999). Mathematical models have been used to link defects in CaMKII signaling with ion channel remodeling, abnormal Ca2+ handling, and arrhythmias in the infarct border zone. Specifically these studies have demonstrated that increased autophosphorylation and oxidation of the kinase results in increased activity that both increases Ca2+ leak from the sarcoplasmic reticulum and compromises availability of voltagegated Na+ channels to create a favorable substrate for arrhythmias (Hund et al., 2008; Christensen et al., 2009). More recently, mathematical models have been used to study the role of chronic CaMKII activation in sinus node dysfunction in the setting of heart failure and diabetes (Swaminathan et al., 2011; Luo et al., 2013). A two dimensional model of the intact sinus node has been applied to demonstrate that CaMKII-induced apoptosis and associated loss of sinoatrial node cells disrupts the source–sink balance between the sinoatrial node and surrounding atrial myocardium resulting in slowed pacemaking and even failure (Swaminathan et al., 2011; Luo et al., 2013). Other studies have used mathematical modeling to determine relative importance of direct CaMKII effects and compensatory changes in gene regulation in the setting of chronic CaMKII overexpression (Koivumaki et al., 2009). Finally, in addition to common forms of acquired disease (e.g., myocardial infarction, heart failure, diabetes), mathematical models have been used to better understand the role of CaMKII in congenital disease (Thiel et al., 2008; Koval et al., 2012). A recent study used mathematical modeling to demonstrate that human variants identified in the CaMKII phosphorylation motif of Nav1.5 confer arrhythmia susceptibility by mimicking the phosphorylated channel (Koval et al., 2012), while an earlier study examined the role of CaMKII regulation of SR Ca2+ release in increased incidence of afterdepolarizations in Timothy syndrome (Thiel et al., 2008). Together these studies demonstrate the potential for mathematical modeling and computer simulation in advancing our understanding of CaMKII biology and its role over a broad range of cardiovascular disease.
Future Directions
This review has outlined the many unique challenges and opportunities for multiscale mathematical modeling of CaMKII signaling. While great strides have been made in development and application of mathematical models of CaMKII signaling from molecule to tissue, clearly there are outstanding issues and unanswered questions to be addressed by future research in this area. At the molecular level, the recent discovery of the CaMKII crystal structure represents an exciting development with great potential for modeling (Chao et al., 2011). Similarly, it will be important for future modeling efforts to address novel pathways for regulation of CaMKII activity (e.g., glycosylation). At the cell level, a daunting challenge remains the sheer number of targets for CaMKII within the cell, with new substrates identified every year. Moreover, it remains to be understood the “tipping point” from the adaptive to the maladaptive aspects of CaMKII signaling. Finally, while most models have focused on the ventricular myocyte as a system, clearly CaMKII has important roles in other heart regions/cell types (e.g., atrial, sinoatrial node cells). Models of these different cell types that incorporate cell-specific CaMKII signaling will be of great use for studying CaMKII signaling at the organ level.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health (NIH) [grant number HL114893 to Thomas J. Hund] and James S. McDonnell Foundation [to Thomas J. Hund].
References
Anderson, M. E., Brown, J. H., and Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Braun, A. P., and Schulman, H. (1995). The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu. Rev. Physiol. 57, 417–445. doi: 10.1146/annurev.ph.57.030195.002221
Cabo, C., and Boyden, P. (2003). Electrical remodeling of the epicardial border zone in the canine infarcted heart: a computational analysis. Am. J. Physiol. Heart Circ. Physiol. 284, H372–H384.
Cabo, C., Yao, J., Boyden, P. A., Chen, S., Hussain, W., Duffy, H. S., et al. (2006). Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc. Res. 72, 241–249. doi: 10.1016/j.cardiores.2006.07.005
Chan, C., Liu, X., Wang, L., Bardwell, L., Nie, Q., and Enciso, G. (2012). Protein scaffolds can enhance the bistability of multisite phosphorylation systems. PLoS Comput. Biol. 8:e1002551. doi:10.1371/journal.pcbi.1002551
Chao, L. H., Stratton, M. M., Lee, I. H., Rosenberg, O. S., Levitz, J., Mandell, D. J., et al. (2011). A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell 146, 732–745. doi: 10.1016/j.cell.2011.07.038
Chiba, H., Schneider, N. S., Matsuoka, S., and Noma, A. (2008). A simulation study on the activation of cardiac CaMKII delta-isoform and its regulation by phosphatases. Biophys. J. 95, 2139–2149. doi: 10.1529/biophysj.107.118505
Christensen, M. D., Dun, W., Boyden, P. A., Anderson, M. E., Mohler, P. J., and Hund, T. J. (2009). Oxidized calmodulin kinase II regulates conduction following myocardial infarction: a computational analysis. PLoS Comput. Biol. 5:e1000583. doi:10.1371/journal.pcbi.1000583
Coomber, C. (1998). Current theories of neuronal information processing performed by Ca2+/calmodulin-dependent protein kinase II with support and insights from computer modelling and simulation. Comput. Chem. 22, 251–263. doi: 10.1016/S0097-8485(97)00002-8
Couchonnal, L. F., and Anderson, M. E. (2008). The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda) 23, 151–159. doi: 10.1152/physiol.00043.2007
Dupont, G., Heytens, E., and Leybaert, L. (2010). Oscillatory Ca2+ dynamics and cell cycle resumption at fertilization in mammals: a modelling approach. Int. J. Dev. Biol. 54, 655–665. doi: 10.1387/ijdb.082845gd
Dupont, G., Houart, G., and De Koninck, P. (2003). Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations: a simple model. Cell Calcium 34, 485–497. doi: 10.1016/S0143-4160(03)00152-0
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Gaertner, T. R., Kolodziej, S. J., Wang, D., Kobayashi, R., Koomen, J. M., Stoops, J. K., et al. (2004). Comparative analyses of the three–dimensional structures and enzymatic properties of α, β, γ, and δ isoforms of Ca2+–calmodulin–dependent protein kinase II. J. Biol. Chem. 279, 12484–12494. doi: 10.1074/jbc.M313597200
Grandi, E., Puglisi, J. L., Wagner, S., Maier, L. S., Severi, S., and Bers, D. M. (2007). Simulation of Ca–calmodulin–dependent protein kinase ii on rabbit ventricular myocyte ion currents and action potentials. Biophys. J. 93, 3835–3847. doi: 10.1529/biophysj.107.114868
Greenwald, E. C., Redden, J. M., Dodge–Kafka, K. L., and Saucerman, J. J. (2014). Scaffold stateswitching amplifies, accelerates and insulates PKC signaling. J. Biol. Chem. 289, 2353–2360. doi: 10.1074/jbc.M113.497941
Hanson, P. I., Meyer, T., Stryer, L., and Schulman, H. (1994). Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12, 943–956. doi: 10.1016/0896-6273(94)90306-9
Hashambhoy, Y. L., Greenstein, J. L., and Winslow, R. L. (2010). Role of CaMKII in RyR leak, EC coupling and action potential duration: a computational model. J. Mol. Cell Cardiol. 49, 617–624. doi: 10.1016/j.yjmcc.2010.07.011
Herrmann, T. L., Agrawal, R. S., Connolly, S. F., Mccaffrey, R. L., Schlomann, J., and Kusner, D. J. (2007). MHC Class II levels and intracellular localization in human dendritic cells are regulated by calmodulin kinase II. J. Leukoc. Biol. 82, 686–699. doi: 10.1189/jlb.0107045
Herzel, H., and Bluthgen, N. (2008). Mathematical models in mammalian cell biology. Genome Biol. 9, 316. doi: 10.1186/gb-2008-9-7-316
Hoelz, A., Nairn, A. C., and Kuriyan, J. (2003). Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin–dependent kinase II. Mol. Cell 11, 1241–1251. doi: 10.1016/S1097-2765(03)00171-0
Hund, T. J., Decker, K. F., Kanter, E., Mohler, P. J., Boyden, P. A., Schuessler, R. B., et al. (2008). Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: insights from mathematical modeling. J. Mol. Cell Cardiol. 45, 420–428. doi: 10.1016/j.yjmcc.2008.06.007
Hund, T. J., Koval, O. M., Li, J., Wright, P. J., Qian, L., Snyder, J. S., et al. (2010). A betaIV spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519. doi: 10.1172/JCI43621
Hund, T. J., and Rudy, Y. (2004). Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation 110, 3168–3174. doi: 10.1161/01.CIR.0000147231.69595.D3
Iribe, G., Kohl, P., and Noble, D. (2006). Modulatory effect of calmodulin–dependent kinase II (CaMKII) on sarcoplasmic reticulum Ca2+ handling and interval–force relations: a modelling study. Philos. Transact. A Math Phys. Eng. Sci. 364, 1107–1133. doi: 10.1098/rsta.2006.1758
Koivumaki, J. T., Korhonen, T., Takalo, J., Weckstrom, M., and Tavi, P. (2009). Regulation of excitation–contraction coupling in mouse cardiac myocytes: integrative analysis with mathematical modelling. BMC Physiol. 9:16. doi:10.1186/1472–6793–9–16
Kolodziej, S. J., Hudmon, A., Waxham, M. N., and Stoops, J. K. (2000). Three–dimensional reconstructions of calcium/calmodulin–dependent (CaM) kinase IIα and truncated CaM kinase IIα reveal a unique organization for its structural core and functional domains. J. Biol. Chem. 275, 14354–14359. doi: 10.1074/jbc.275.19.14354
Kotaleski, J. H., and Blackwell, K. T. (2010). Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat. Rev. Neurosci. 11, 239–251. doi: 10.1038/nrn2807
Koval, O. M., Guan, X., Wu, Y., Joiner, M. L., Gao, Z., Chen, B., et al. (2010). CaV1.2 beta–subunit coordinates CaMKII–triggered cardiomyocyte death and afterdepolarizations. Proc. Natl. Acad. Sci. U.S.A. 107, 4996–5000. doi: 10.1073/pnas.0913760107
Koval, O. M., Snyder, J. S., Wolf, R. M., Pavlovicz, R. E., Glynn, P., Curran, J., et al. (2012). Ca2+/calmodulin–dependent protein kinase II–based regulation of voltage–gated Na+ channel in cardiac disease. Circulation 126, 2084–2094. doi: 10.1161/CIRCULATIONAHA.112.105320
Kubota, Y. (1999). Decoding time–varying calcium signals by the postsynaptic biochemical network: computer simulations of molecular kinetics. Neurocomputing 26, 29–38. doi: 10.1016/S0925-2312(99)00085-5
Kubota, Y., and Bower, J. M. (2001). Transient versus asymptotic dynamics of CaM kinase II: possible roles of phosphatase. J. Comput. Neurosci. 11, 263–279. doi: 10.1023/A:1013727331979
Lai, Y., Nairn, A. C., and Greengard, P. (1986). Autophosphorylation reversibly regulates the Ca2+/calmodulin–dependence of Ca2+/calmodulin–dependent protein kinase II. Proc. Natl. Acad. Sci. U.S.A. 83, 4253–4257. doi: 10.1073/pnas.83.12.4253
Lascano, E. C., Said, M., Vittone, L., Mattiazzi, A., Mundina–Weilenmann, C., and Negroni, J. A. (2013). Role of CaMKII in post acidosis arrhythmias: a simulation study using a human myocyte model. J. Mol. Cell Cardiol. 60, 172–183. doi: 10.1016/j.yjmcc.2013.04.018
Levitzki, A. (2003). Protein kinase inhibitors as a therapeutic modality. Acc. Chem. Res. 36, 462–469. doi: 10.1021/ar0201207
Lisman, J. E. (1985). A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc. Natl. Acad. Sci. U.S.A. 82, 3055–3057. doi: 10.1073/pnas.82.9.3055
Lisman, J. E., and Goldring, M. A. (1988). Feasibility of long–term storage of graded information by the Ca2+/calmodulin–dependent protein kinase molecules of the postsynaptic density. Proc. Natl. Acad. Sci. U.S.A. 85, 5320–5324. doi: 10.1073/pnas.85.14.5320
Livshitz, L. M., and Rudy, Y. (2007). Regulation of Ca2+ and electrical alternans in cardiac myocytes: role of CaMKII and repolarizing currents. Am. J. Physiol. Heart Circ. Physiol. 292, H2854–H2866. doi: 10.1152/ajpheart.01347.2006
Lucic, V., Greif, G. J., and Kennedy, M. B. (2008). Detailed state model of CaMKII activation and autophosphorylation. Eur. Biophys. J. 38, 83–98. doi: 10.1007/s00249-008-0362-4
Luo, M., Guan, X., Di, L., Kutschke, W., Gao, Z., Yang, J., et al. (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 123, 1262–1274. doi: 10.1172/JCI65268
Maier, L. S., and Bers, D. M. (2002). Calcium, calmodulin, and calcium–calmodulin kinase II: heartbeat to heartbeat and beyond. J. Mol. Cell Cardiol. 34, 919–939. doi: 10.1006/jmcc.2002.2038
McGargill, M. A., Sharp, L. L., Bui, J. D., Hedrick, S. M., and Calbo, S. (2005). Active Ca2+/calmodulin–dependent protein kinase II gamma B impairs positive selection of T cells by modulating TCR signaling. J. Immunol. 175, 656–664.
Michalski, P. J. (2013). The delicate bistability of CaMKII. Biophys. J. 105, 794–806. doi: 10.1016/j.bpj.2013.06.038
Michelson, S., and Schulman, H. (1994). CaM Kinase: a model for its activation and dynamics. J. Theor. Biol. 171, 281–290. doi: 10.1006/jtbi.1994.1231
Miller, P., Zhabotinsky, A. M., Lisman, J. E., and Wang, X. J. (2005). The stability of a stochastic CaMKII switch: dependence on the number of enzyme molecules and protein turnover. PLoS Biol. 3:e107. doi: 10.1371/journal.pbio.0030107
Mironov, S. L. (2013). Calmodulin and calmodulin kinase II mediate emergent bursting activity in the brainstem respiratory network (preBotzinger complex). J. Physiol. 591, 1613–1630. doi: 10.1113/jphysiol.2012.237362
O’Hara, T., Virag, L., Varro, A., and Rudy, Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7:e1002061. doi:10.1371/journal.pcbi.1002061
Pinto, J. M., and Boyden, P. A. (1999). Electrical remodeling in ischemia and infarction. Cardiovasc. Res. 42, 284–297. doi: 10.1016/S0008-6363(99)00013-9
Rellos, P., Pike, A. C., Niesen, F. H., Salah, E., Lee, W. H., Von Delft, F., et al. (2010). Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 8:e1000426. doi: 10.1371/journal.pbio.1000426.
Rosenberg, O. S., Deindl, S., Sung, R. J., Nairn, A. C., and Kuriyan, J. (2005). Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 123, 849–860. doi: 10.1016/j.cell.2005.10.029
Saucerman, J. J., and Bers, D. M. (2008). Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys. J. 95, 4597–4612. doi: 10.1529/biophysj.108.128728
Silva, A. J., Paylor, R., Wehner, J. M., and Tonegawa, S. (1992a). Impaired spatial learning in α–calcium–calmodulin kinase II mutant mice. Science 257, 206–211. doi: 10.1126/science.1321493
Silva, A. J., Stevens, C. F., Tonegawa, S., and Wang, Y. (1992b). Deficient hippocampal long–term potentiation in α–calcium–calmodulin kinase II mutant mice. Science 257, 201–206. doi: 10.1126/science.1378648
Soltis, A. R., and Saucerman, J. J. (2010). Synergy between CaMKII substrates and beta–adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophys. J. 99, 2038–2047. doi: 10.1016/j.bpj.2010.08.016
Song, Q., Saucerman, J. J., Bossuyt, J., and Bers, D. M. (2008). Differential integration of Ca2+–calmodulin signal in intact ventricular myocytes at low and high affinity Ca2+–calmodulin targets. J. Biol. Chem. 283, 31531–31540. doi: 10.1074/jbc.M804902200
Stratton, M. M., Chao, L. H., Schulman, H., and Kuriyan, J. (2013). Structural studies on the regulation of Ca2+/calmodulin dependent protein kinase II. Curr. Opin. Struct. Biol. 23, 292–301. doi: 10.1016/j.sbi.2013.04.002
Swaminathan, P. D., Purohit, A., Hund, T. J., and Anderson, M. E. (2012). Calmodulin–dependent protein kinase II: linking heart failure and arrhythmias. Circ. Res. 110, 1661–1677. doi: 10.1161/CIRCRESAHA.111.243956
Swaminathan, P. D., Purohit, A., Soni, S., Voigt, N., Singh, M. V., Glukhov, A. V., et al. (2011). Oxidized CaMKII causes sinus node dysfunction in mice. J. Clin. Invest. 121, 3277–3288. doi: 10.1172/JCI57833
Thiel, W. H., Chen, B., Hund, T. J., Koval, O. M., Purohit, A., Song, L. S., et al. (2008). Proarrhythmic defects in Timothy syndrome require calmodulin kinase II. Circulation 118, 2225–2234. doi: 10.1161/CIRCULATIONAHA.108.788067
Zang, Y., Dai, L., Zhan, H., Dou, J., Xia, L., and Zhang, H. (2013). Theoretical investigation of the mechanism of heart failure using a canine ventricular cell model: especially the role of up–regulated CaMKII and SR Ca2+ leak. J. Mol. Cell Cardiol. 56, 34–43. doi: 10.1016/j.yjmcc.2012.11.020
Keywords: calmodulin kinase II, mathematical modeling, calcium, arrhythmias, heart failure
Citation: Onal B, Unudurthi SD and Hund TJ (2014) Modeling CaMKII in cardiac physiology: from molecule to tissue. Front. Pharmacol. 5:9. doi: 10.3389/fphar.2014.00009
Received: 15 December 2013; Accepted: 16 January 2014;
Published online: 04 February 2014.
Edited by:
Eleonora Grandi, University of California Davis, USAReviewed by:
Steven Alexander Niederer, King’s College London, UKJussi Tapani Koivumäki, Simula Research Laboratory, Norway
Copyright © 2014 Onal, Unudurthi and Hund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas J. Hund, The Dorothy M. Davis Heart and Lung Research Institute, Wexner Medical Center, The Ohio State University, 473 West 12th Avenue, Columbus, OH 43210, USA e-mail:dGhvbWFzLmh1bmRAb3N1bWMuZWR1