 Naghmeh H. Asvadi
Naghmeh H. Asvadi Michael Morgan
Michael Morgan Amitha K. Hewavitharana
Amitha K. Hewavitharana Peter J. Cabot
Peter J. CabotEndogenous opioid peptides have been aligned with a diverse array of effects. Their activity is not only attributable to action the three main opioid receptors, mu (MOR), delta (DOR), and kappa (KOR) opioid receptors but their impacts appear to extend to activities at sodium channels, cytokine receptors (Finley et al., 2008), calcium channels and non-specific and partially undefined pharmacological effects inconsistent with G-protein coupled opioid receptor activity.
Of the family of opioid peptides beta-endorphin (BE 1-31) is one of the most prominent and is the prototypical endogenous peptide for the MOR class of opioid receptors and is found within the CNS and the immune system (Cabot et al., 1997). BE 1-31 is derived from pro-opiomelanocortin (POMC) in the cytosol of cell bodies. BE has been shown to possess peripheral and central analgesic activity (Van Den Burg et al., 2001), producing a morphine-like effect by inhibiting the signals of C- and Aδ-fiber activation (Duggan and Fleetwood-Walker, 1993). In addition, BE 1-31 is a non-selective endogenous peptide with the highest affinities for MOR and DOR (Binder et al., 2004), suggesting that the endogenous system is not modulated by specific and selective opioid agonists in isolation.
This concept touches on a new theme evolving in novel therapeutic strategies in the pain field, i.e. the targeting of multiple channels with either one non-selective ligand or a combination of selective ligands to produce effects that are either synergistic or, at a minimum, differential in terms of side effects. This could seemingly point to a multitude of combinations of drugs of both G-protein receptor targeting ligands or extend to those targeting other receptor classes including sodium channels (Su et al., 2002), potassium channels (Welch and Dunlow, 1993) and calcium channels (Smart et al., 1995). The scope of the possible therapeutic targets is immense and, potentially of even greater complexity, is the dose determination for such combinations. Perhaps the answer in part lies in the endogenous opioid system, which is, in essence, the system designed to mediate noxious stimuli as well as interact with the immune system in disease (Figure 1).
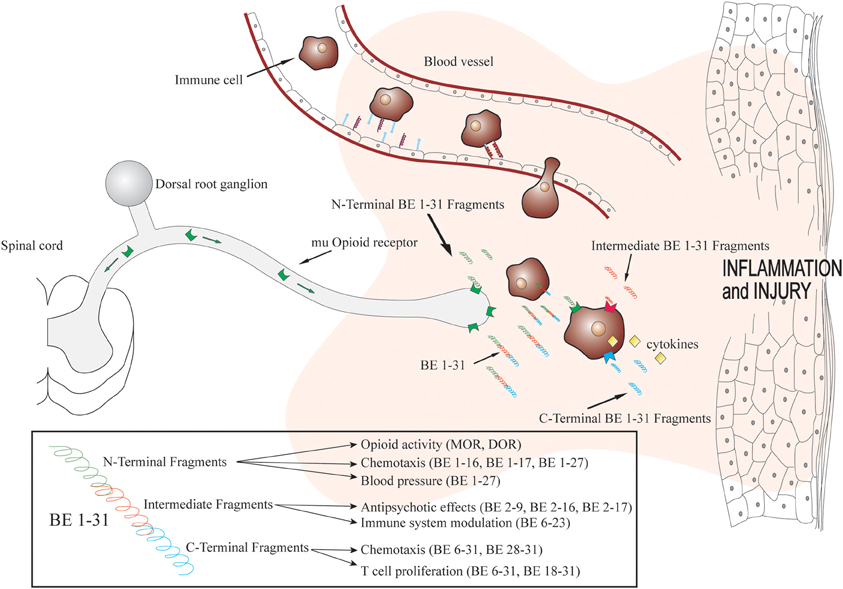
Figure 1. Biotransformation of beta-endorphin 1–31 within inflamed tissue and fragment actions. Immune cells containing beta-endorphin migrate to inflamed tissue in a site directed manner. Beta-endorphin is released within the inflammatory mileu and biotransformed rapidly producing fragments with various pharmacological actions. Adapted with permission from Kapitzke et al. (2005).
Enzymatic Processing of Beta-Endorphin
It is well known that peptides including opioid peptides are susceptible to rapid enzymatic degradation (McKnight et al., 1983). The major peptidases involved in the degradation of opioid peptides are aminopeptidases (Montiel et al., 1997), angiotensin-converting enzyme (ACE), insulin degrading enzyme (Reed et al., 2008), serine peptidases (Sandin et al., 1998), dipeptidyl peptidase III and IV (DPP III, DPP IV) (Sakurada et al., 2003). DPP IV is a serine protease (Shane et al., 1999) and has demonstrated a structural preference for the cleavage of opioid peptides at proline (Augustyns et al., 2005) and is a likely candidate responsible for the cleavage of BE 1–31 producing BE 1–13. Insulin degrading enzyme similarly has been shown to have selective cleaving properties, producing BE 1–17 and BE 1–18 from BE 1–31. In addition, BE 1–19 and BE 20–31 are the likely products of the enzymatic activity of metallo sensitive serine proteases (Sandin et al., 1998). ACE is however has broad peptide cleavage properties and is found widely distributed in many cells (Brownson et al., 1994). Undoubtedly, a major degraditive pathway to non-opioid metabolites will be via aminopeptidases, yet to be demonstrated for BE 1-31 but has been shown to be responsible for cleavage of dynorphin A 1–13 to 2–13 (Müller and Hochhaus, 1995), a conserved region within BE 1–31 and dynorphin A 1–13.
Inhibiting Beta-Endorphin Biotransformation as a Therapeutic Strategy
Peptidase inhibition has been investigated as a therapeutic strategy with some success. This is not necessarily a novel approach but it has received a recent resurgence with the development of more selective inhibitors (Schreiter et al., 2012). Certainly, peptidases have been blocked selectively and non-selectively by a number of strategies, e.g., di-isopropyl fluorophosphate and metal ions (Pb2+, Hg2+, Zn2+) are effective inhibitors for DPP IV, albeit di-isopropyl fluorophosphate has been shown to induce compensatory anticholinesterase activity producing tremors due to its irreversible nature. Spinorphin (Tien et al., 2004), which is endogenous factor derived from bovine spinal cord, and its truncated fragment, tynorphin, are both inhibitors of DPP III (Yamamoto et al., 2000). Leupeptin has been shown to block cysteine-containing enzymes and EDTA and phenanthroline inhibit metalloproteases generally (Mentlein, 1999), whilst aminopeptidases are inhibited non-selectively by bestatin without affecting DPP IV (Scornik and Botbol, 2001). A different approach undertaken is to modify the structure of endogenous peptides at specific labile or susceptible bonds. These include the routine modification at Gly2 with substitution by D or L Ala2 or N-methylation of the Tyrosine1, both increasing stability of the peptide by reducing the N-terminal degradation (Hiramatsu et al., 2001). These approaches have been shown to produce long lasting analgesia, thereby suggesting that peptide processing is simply a means to facilitate the degradation of bioactive peptides to their non-pharmacologically active forms. It is likely that this, however, may not be the complete story, opioid peptide fragment and in-situ biotransformation may be an integral part of the bodies efforts in addressing disease and pain.
Biotransformation Alteration in Disease
A recent study in our laboratory has identified biotransformation fragments of BE 1–31 in rat inflamed tissue (Herath et al., 2012). This study demonstrated that the hydrolytic metabolism of BE 1–31 in homogenized inflamed tissue was faster than in serum and trypsin incubation; similar results have been noted for the processing of dynorphin (the endogenous ligand for KOR) within inflamed tissue homogenates (Morgan et al., 2012). The rate of metabolism of BE 1–31 at pH 5.5 was also higher than the rate of metabolism of BE 1–31 at pH 7.4. These acidic pH values have been shown to be concordant with those found within inflamed tissue (Dray, 1995). In addition, the nature of the biotransformation hydrolysis was altered, BE 1–31 was shown in inflamed tissue homogenates to be most susceptible for hydrolytic degradation at specific amino acid bonds: (Tyr1-Gly2), (Lys9-Ser10), (Leu17-Phe18-Lys19-Asn20), (Lys24-Asn25), (Lys28-Lys29-Gly30-Gln31) (Herath et al., 2012). This is likely to be a consequence of the inflammatory conditions that affect the enzymes independently and specifically (Lin et al., 2001). These results highlight the presence of a unique panel of peptides which would be produced dependent upon the disease state, possessing potentially unique pharmacological properties.
Biotransformation and Opioid Activity
Many studies have investigated the pharmacological changes observed following opioid peptide modification and truncation. Deakin et al. showed that the removal of one, two, or four amino acids from the C-terminal of BE 1–31 reduced the analgesic effect of fragments and that the removal of eight amino acids from the N-terminal of BE 1–31 resulted in an absence of analgesic activity (Deakin et al., 1980). Many other studies have provided evidence for the structural necessity of a tyrosine residue at position 1 in BE 1–31 for the retention of analgesic activity. In agreement with this notion, N-acetyl derivatives of BE 1–31 naturally found in the pituitary do not produce opioid activity (Deakin et al., 1980). In addition, a number of studies have demonstrated the C-terminal sequence of BE 1–31determines the potency of opioid peptide in analgesia. Naturally occurring forms of BE 1–31, truncated at the C-terminal, BE 1–28, BE 1–27, and BE 1–26 are found in the pituitary (Zakarian and Smyth, 1982). These compounds are not only ineffective as analgesics but BE 1–27 intra-cerebroventricularly injected into mice has been shown to block the analgesia produced by BE 1–31, with a potency four times greater than that of naloxone - the non-selective opioid antagonist (Hammonds et al., 1984). However, further truncation to BE 1-26 decreased the antagonist effect whilst further reduction of the peptide chain resulted in the complete loss of inhibition of analgesic activity (Nicolas and Choh Hao, 1985). The analgesic potency of further abbreviated forms remains from peptide sequences of BE 1–31 right down to BE 1–4, the overwhelming consequence of the truncation to smaller N-terminal conserved sequences is decreased affinity for MOR, but increased activity at DOR and KOR (Jaba et al., 2007).
Biotransformation and Non-Opioid Activity
The presence of BE 1–31 in both the neuronal and immune systems indicates that the pharmacological effects of these peptides may extend past those of the management of nociceptive signals. A number of studies have examined potential immune-related mechanisms for BE 1–31 and a variety of truncated forms. Interestingly, effects on human monocyte chemotaxis showed both a lack of requirement for opioid receptor action and the presence of the N-terminal Tyrosine. These effects occurred for a range of truncated forms of BE 1–31 (namely: BE 1–16, BE 1–17, BE 1–27, BE 6–31, BE 28–31) (Sacerdote and Panerai, 1989). Similarly, T cell proliferation was modulated at non-opioid receptors by BE 1–31, BE 6–31, and BE 18–31 (Van Den Bergh et al., 1993). Separate to their immune system effects but aligned with the systemic availability of these peptides, the effects on blood pressure and heart rate in anesthetized rats have also been examined for BE 1–31 and truncated peptides. BE 1–27, shown in previous studies to possess opioid antagonist activity against BE 1–31, and reduced blood pressure to an extent which was similar to that of the effects of the parent molecule, BE 1–31 (Giersbergen et al., 1991). In neurological experiments BE 1–16 and BE 1–17 modulated avoidance behavior and this was not inhibited by naltrexone, an opioid receptor antagonist. The non-opioid peptide fragment BE 2–17 also displayed strong anti-psychotic effects in schizophrenic patients (De Wied, 1979). This non-opioid effect of truncated BE 1–31 was supported in a separate study that showed similar effects with BE 2–16 and BE 2–9 (Van Ree and De Wied, 1982). Furthermore, BE 1–31, when cultured with rat splenocytes, showed suppression of plaque-forming cells (PFC) in response to coculture with sheep red blood cells, not reversed by naloxone (Hemmick and Bidlack, 1989). BE 1–31 has also been shown to interact with protein S in a C-terminal specific manner, implicating BE 1–31 in anticoagulation through antithrombin III (Hildebrand et al., 1989).
Non-Opioid Site of Action
The search for the sites of action for the non-opioid effects of endogenous opioids has been largely focused on the immune system (Rittner et al., 2008). There is evidence of receptor binding sites for BE 1–31 on a number of immune cells that are not modulated by common analgesics or opioid selective antagonists. There is also a substantial body of evidence for opioids interacting with Toll-like receptors within the immune system (Franchi et al., 2012), with stereo selectivity for the plus isomers of common opioids such as morphine-3-glucuronide (Lewis et al., 2010), naloxone and naltrexone (Hutchinson et al., 2008). These effects have been correlated with modulation of cytokine expression or release, and result in changes that may effect cell proliferation and chemotaxis. Consistent with immune system modulation a non-opioid binding site for BE 1–31 has been demonstrated in immune cells, which would appear to exist in combination with classical opioid receptors and naloxone dependent effects. These sites have been proposed to be activated by restricted sequences of BE 1–31 to BE 6–23 and not modulated by naloxone or alkaloid agonists such as morphine (Kovalitskaya and Navolotskaya, 2011).
Concluding Remarks
Increasing our understanding of the role of beta-endorphin and its biotransformation fragments provides an insight into the complexity of the endogenous opioid system. The current analgesics are targeted at the modulation of analgesia by directly binding to one or more of the opioid receptors, with the analgesic being predominantly designed as a MOP agonist. The above observations would suggest that this is solely one aspect of opioid pharmacology, albeit one that has been explored widely and utilized in therapy. Biotransformation is a process that produces an array of compounds having a plethora of specific actions which contribute to the body's and its biological systems response to disease or injury. Future therapeutic strategies should consider such actions in designing better treatments or disease modulators.
Acknowledgments
Naghmeh Asvadi was funded off a University of Queensland International Postgraduate Research Scholarship.
References
Augustyns, K., Veken, P. V. D., Senten, K., and Haemers, A. (2005). The therapeutic potential of inhibitors of dipeptidyl peptidase IV (DPP IV) and related proline-specific dipeptidyl aminopeptidases. Curr. Med. Chem. 12, 971–998. doi: 10.2174/0929867053507298
Binder, W., Mousa, S. A., Sitte, N., Kaiser, M., Stein, C., and Schäfer, M. (2004). Sympathetic activation triggers endogenous opioid release and analgesia within peripheral inflamed tissue. Eur. J. Neurosci. 20, 92–100. doi: 10.1111/j.1460-9568.2004.03459.x
Brownson, E. A., Abbruscato, T. J., Gillespie, T. J., Hruby, V. J., and Davis, T. P. (1994). Effect of peptidases at the blood brain barrier on the permeability of enkephalin. J. Pharmacol. Exp. Ther. 270, 675–680.
Cabot, P. J., Carter, L., Gaiddon, C., Zhang, Q., Schäfer, M., Loeffler, J. P., et al. (1997). Immune cell-derived beta-endorphin. Production, release, and control of inflammatory pain in rats. J. Clin. Invest. 100, 142–148.
Deakin, J. F., Doströvsky, J. O., and Smyth, D. G. (1980). Influence of N-terminal acetylation and C-terminal proteolysis on the analgesic activity of beta-endorphin. Biochem. J. 189, 501–506.
De Wied, D. (1979). Schizophrenia as an inborn error in the degradation of β-endorphin a hypothesis. Trends Neurosci. 2, 79–82.
Dray, A. (1995). Inflammatory mediators of pain. Br. J. Anaesth. 75, 125–131. doi: 10.1093/bja/75.2.125
Duggan, A. W., and Fleetwood-Walker, S. M. (1993). “Opioids and sensory processing in the central nervous system,” in Opioids, eds A. Herz, H. Akil and E. Simon (Berlin; Heidelberg: Springer), 731–771.
Finley, M. J., Happel, C. M., Kaminsky, D. E., and Rogers, T. J. (2008). Opioid and nociceptin receptors regulate cytokine and cytokine receptor expression. Cell. Immunol. 252, 146–154. doi: 10.1016/j.cellimm.2007.09.008
Franchi, S., Moretti, S., Castelli, M., Lattuada, D., Scavullo, C., Panerai, A. E., et al. (2012). Mu opioid receptor activation modulates Toll like receptor 4 in murine macrophages. Brain Behav. Immun. 26, 480–488. doi: 10.1016/j.bbi.2011.12.010
Giersbergen, P. L. M. V., Lang, H. D., and Jong, W. D. (1991). Effects of dynorphin A(1 – 13) and of fragments of β-endorphin on blood pressure and heart rate of anesthetized rats. Can. J. Physiol. Pharmacol. 69, 327–333. doi: 10.1139/y91-050
Hammonds, R. G., Nicolas, P., and Li, C. H. (1984). beta-endorphin-(1-27) is an antagonist of beta-endorphin analgesia. Proc. Natl. Acad. Sci. U.S.A. 81, 1389–1390.
Hemmick, L. M., and Bidlack, J. M. (1989). Beta-endorphin suppresses rat plaque-forming cell response by a non-opioid mechanism. J. Neuroimmunol. 23, 67–71.
Herath, H., Cabot, P., Shaw, P., and Hewavitharana, A. (2012). Study of beta endorphin metabolism in inflamed tissue, serum and trypsin solution by liquid chromatography–tandem mass spectrometric analysis. Anal. Bioanal. Chem. 402, 2089–2100. doi: 10.1007/s00216-011-5686-8
Hildebrand, A., Preissner, K. T., Müller-Berghaus, G., and Teschemacher, H. (1989). A novel beta-endorphin binding protein. Complement S protein (= vitronectin) exhibits specific non-opioid binding sites for beta-endorphin upon interaction with heparin or surfaces. J. Biol. Chem. 264, 15429–15434.
Hiramatsu, M., Inoue, K., Ambo, A., Sasaki, Y., and Kameyama, T. (2001). Long-lasting antinociceptive effects of a novel dynorphin analogue, Tyr-D-Ala-Phe-Leu-Arg ψ (CH2NH) Arg-NH2, in mice. Br. J. Pharmacol. 132, 1948–1956. doi: 10.1038/sj.bjp.0703982
Hutchinson, M. R., Zhang, Y., Brown, K., Coats, B. D., Shridhar, M., Sholar, P. W., et al. (2008). Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 28, 20–29. doi: 10.1111/j.1460-9568.2008.06321.x
Jaba, I., Tamba, B., Manolidis, G., and Mungiu, O. (2007). Variation in the analgesic activity of opioid peptide fragments in correlation with the amino acidic sequence. Cent. Eur. J. Med. 2, 417–429. doi: 10.2478/s11536-007-0036-0
Kapitzke, D., Vetter, I., and Cabot, P. J. (2005). Endogenous opioid analgesia in peripheral tissues and the clinical implications for pain control. Ther Clin. Risk Manag. 1, 279–297.
Kovalitskaya, Y. A., and Navolotskaya, E. V. (2011). Nonopioid effect of β-endorphin. Biochemistry (Mosc.) 76, 379–393. doi: 10.1134/S0006297911040018
Lewis, S. S., Hutchinson, M. R., Rezvani, N., Loram, L. C., Zhang, Y., Maier, S. F., et al. (2010). Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1β. Neuroscience 165, 569–583. doi: 10.1016/j.neuroscience.2009.10.011
Lin, L., Sohar, I., Lackland, H., and Lobel, P. (2001). The human CLN2 Protein/Tripeptidyl-peptidase I is a serine protease that autoactivates at acidic pH. J. Biol. Chem. 276, 2249–2255. doi: 10.1074/jbc.M008562200
McKnight, A. T., Corbett, A. D., and Kosterlitz, H. W. (1983). Increase in potencies of opioid peptides after peptidase inhibition. Eur. J. Pharmacol. 86, 393–402. doi: 10.1016/0014-2999(83)90189-9
Mentlein, R. (1999). Dipeptidyl-peptidase IV (CD26)-role in the inactivation of regulatory peptides. Regul. Pept. 85, 9–24. doi: 10.1016/S0167-0115(99)00089-0
Montiel, J.-L., Cornille, F., Roques, B. P., and Noble, F. (1997). Nociceptin/Orphanin FQ metabolism: role of aminopeptidase and endopeptidase 24.15. J. Neurochem. 68, 354–361. doi: 10.1046/j.1471-4159.1997.68010354.x
Morgan, M., Herath, H. M. D. R., Cabot, P. J., Shaw, P. N., and Hewavitharana, A. K. (2012). Dynorphin A 1–17 biotransformation in inflamed tissue, serum and trypsin solution analysed by liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 404, 3111–3121. doi: 10.1007/s00216-012-6406-8.
Müller, S., and Hochhaus, G. (1995). Metabolism of dynorphin A 1-13 in human blood and plasma. Pharm. Res. 12, 1165–1170. doi: 10.1023/A:1016211910107
Nicolas, P., and Choh Hao, L. (1985). Inhibition of analgesia by C-terminal deletion analogs of human β-endorphin. Biochem. Biophys. Res. Commun. 127, 649–655. doi: 10.1016/s0006-291x(85)80210-2
Reed, B., Bidlack, J. M., Chait, B. T., and Kreek, M. J. (2008). Extracellular biotransformation of β-endorphin in rat striatum and cerebrospinal fluid. J. Neuroendocrinol. 20, 606–616. doi: 10.1111/j.1365-2826.2008.01705.x
Rittner, H. L., Brack, A., and Stein, C. (2008). Pain and the immune system. Br. J. Anaesth. 101, 40–44. doi: 10.1093/bja/aen078
Sacerdote, P., and Panerai, A. E. (1989). Analysis of the beta-endorphin structure-related activity on human monocyte chemotaxis: importance of the N- and C-terminal. Peptides 10, 565–569. doi: 10.1016/0196-9781(89)90143-5
Sakurada, C., Sakurada, S., Hayashi, T., Katsuyama, S., Tan-No, K., and Sakurada, T. (2003). Degradation of endomorphin-2 at the supraspinal level in mice is initiated by dipeptidyl peptidase IV: an in vitro and in vivo study. Biochem. Pharmacol. 66, 653–661. doi: 10.1016/S0006-2952(03)00391-5
Sandin, J., Nylander, I., and Silberring, J. (1998). Metabolism of β-endorphin in plasma studied by liquid chromatography–electrospray ionization mass spectrometry. Regul. Pept. 73, 67–72. doi: 10.1016/s0167-0115(97)01065-3
Schreiter, A., Gore, C., Labuz, D., Fournie-Zaluski, M.-C., Roques, B. P., Stein, C., et al. (2012). Pain inhibition by blocking leukocytic and neuronal opioid peptidases in peripheral inflamed tissue. FASEB J. 26, 5161–5171. doi: 10.1096/fj.12-208678
Scornik, O. A., and Botbol, V. (2001). Bestatin as an experimental tool in mammals. Curr. Drug Metab. 2, 67–85. doi: 10.2174/1389200013338748
Shane, R., Wilk, S., and Bodnar, R. J. (1999). Modulation of endomorphin-2-induced analgesia by dipeptidyl peptidase IV. Brain Res. 815, 278–286. doi: 10.1016/S0006-8993(98)01121-4
Smart, D., Smith, G., and Lambert, D. G. (1995). Mu-opioids activate phospholipase C in SH-SY5Y human neuroblastoma cells via calcium-channel opening. Biochem. J. 305, 577–581.
Su, X., Joshi, S. K., Kardos, S., and Gebhart, G. F. (2002). Sodium channel blocking actions of the κ-opioid receptor agonist U50,488 contribute to its visceral antinociceptive effects. J. Neurophysiol. 87, 1271–1279.
Tien, L.-T., Fan, L.-W., Ma, T., Loh, H. H., and Ho, I.-K. (2004). Increased diisopropylfluorophosphate-induced toxicity in μ-opioid receptor knockout mice. J. Neurosci. Res. 78, 259–267. doi: 10.1002/jnr.20259
Van Den Bergh, P., Rozing, J., and Nagelkerken, L. (1993). Identification of two moieties of beta-endorphin with opposing effects on rat T-cell proliferation. Immunology 79, 18–23.
Van Den Burg, E., Metz, J. R., Arends, R., Devreese, B., Vandenberghe, I., Van Beeumen, J., et al. (2001). Identification of beta-endorphins in the pituitary gland and blood plasma of the common carp (Cyprinus carpio). J. Endocrinol. 169, 271–280. doi: 10.1677/joe.0.1690271
Van Ree, J. M., and De Wied, D. (1982). Behavioral effects of the beta-endorphin fragment 2-9. Life Sci. 31, 2383–2386.
Welch, S. P., and Dunlow, L. D. (1993). Antinociceptive activity of intrathecally administered potassium channel openers and opioid agonists: a common mechanism of action? J. Pharmacol. Exp. Ther. 267, 390–399.
Yamamoto, Y., Hashimoto, J.-I., Shimamura, M., Yamaguchi, T., and Hazato, T. (2000). Characterization of tynorphin, a potent endogenous inhibitor of dipeptidyl peptidaseIII. Peptides 21, 503–508. doi: 10.1016/S0196-9781(00)00174-1
Keywords: peptide, biotransformation, inflammation, opioid, proteomics, pain
Citation: Asvadi NH, Morgan M, Hewavitharana AK, Shaw PN and Cabot PJ (2014) Biotransformation of beta-endorphin and possible therapeutic implications. Front. Pharmacol. 5:18. doi: 10.3389/fphar.2014.00018
Received: 13 December 2013; Accepted: 28 January 2014;
Published online: 19 February 2014.
Edited by:
Susan Hua, The University of Newcastle, AustraliaReviewed by:
James W. Grau, Texas A&M University, USAClaudia Bregonzio, IFEC-CONICET-UNC, Argentina
Copyright © 2014 Asvadi, Morgan, Hewavitharana, Shaw and Cabot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:cC5jYWJvdEB1cS5lZHUuYXU=