 Yuejin Wu
Yuejin Wu Mark E. Anderson
Mark E. Anderson- 1Department of Internal Medicine, Carver College of Medicine, University of Iowa, Iowa City, IA, USA
- 2Department of Molecular Physiology and Biophysics, Carver College of Medicine, University of Iowa, Iowa City, IA, USA
The calcium and calmodulin-dependent protein kinase II (CaMKII) is present in sinoatrial node (SAN) pacemaker cells and is required for physiological “fight or flight” SAN beating rate responses. Inhibition of CaMKII in SAN does not affect baseline heart rate, but reduces heart rate increases in response to physiological stress. CaMKII senses intracellular calcium (Ca2+) changes, oxidation status, and hyperglycemia to phosphorylate substrates that regulate Ca2+-sensitive proteins, such as L-type Ca2+ channels, phospholamban, and cardiac ryanodine receptors (RyR2). All of these substrates are involved in the SAN pacemaking mechanism. Excessive CaMKII activity, as occurs under pathological conditions such as heart failure, ischemia, and diabetes, can promote intracellular Ca2+ overload and reactive oxygen species production. Oxidation of CaMKII (ox-CaMKII) locks CaMKII into a constitutively active configuration that contributes to SAN cell apoptosis and fibrosis. This ox-CaMKII-mediated loss of functional SAN cells contributes to SAN dysfunction (SND) and sudden death. Thus, CaMKII has emerged as a central regulator of physiological SAN responses and a key determinant of SND.
Introduction
The sinoatrial node (SAN) is a specialized region of heart tissue present at the junction of the right atrium and superior vena cava that extends along the cristae terminalis, where it initiates each normal heart beat. The pacemaking function of SAN cells is accomplished by generation of spontaneous action potentials. There appear to be redundant systems in SAN for generating spontaneous cell membrane potential depolarizations, which are ultimately necessary to sustain life by maintaining cardiac output. One of these systems comprises a set of cell membrane delimited ion channels. These ion channels include hyperpolarization-activated cyclic nucleotide-gated (HCN) channels that conduct an inward current, sometimes called a pacemaker current or funny current (If; DiFrancesco, 1991), L-type (CaV1.2/1.3; Christel et al., 2012) and T-type (CaV3.1/3.2) Ca2+ channels (Mangoni et al., 2006; Tanaka et al., 2008; Brahmajothi et al., 2010) and several K+ channels, including ERG (Brahmajothi et al., 1997, 2010) and KvLQT1 (Chandler et al., 2009; Brahmajothi et al., 2010 ). All of these ion channels have the potential to play a role in pacemaking under different conditions. The other system involves intracellular Ca2+ machinery that is used for excitation–contraction coupling in mechanically purposed myocardium, but that contributes to rhythmic intracellular Ca2+ oscillations in SAN. This system enables SAN fight or flight heart rate increases and contributes to SAN cell death under pathological stress. These components include the sarcoplasmic reticulum (SR; Rigg and Terrar, 1996), which contains the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a), the ryanodine receptor 2 (RYR2), a large Ca2+ channel that releases Ca2+ from the SR lumen to the cytoplasm and the cell membrane spanning Na+/Ca2+ exchanger (NCX1; Sanders et al., 2006). The components in both systems collaborate but are also capable of independent activity that ensures nonstop pacemaking activity (Lakatta and DiFrancesco, 2009; Lakatta et al., 2010).
We believe that the effects of the multifunctional Ca2+ and calmodulin-dependent protein kinase II (CaMKII) on SAN cell biology are related to actions on Ca2+ homeostasis. CaMKII is a multifunctional serine/threonine-specific protein kinase that is initially activated by the Ca2+/calmodulin complex (Schulman and Greengard, 1978). CaMKII is present in contracting myocardium and in SAN cells (Vinogradova et al., 2000). Details of CaMKII structure, function, activation, and inactivation are contained in another chapter in this compendium (XYZ). However, the CaMKII holomeric structure allows it to perform as a precisely regulated enzyme that activates and inactivates with Ca2+/calmodulin binding and unbinding but also to transition into a constitutively active conformation by post-translational modifications to the autoregulatory domain (Kuret and Schulman, 1985; Erickson et al., 2008, 2013; Chao et al., 2011; Gutierrez et al., 2013). Excessive levels of constitutively active CaMKII are linked to cardiovascular and pulmonary diseases, including SAN dysfunction (SND; Erickson et al., 2011; Sanders et al., 2013).
CaMKII in SAN Physiology
Activated CaMKII can catalyze phosphorylation of multiple Ca2+ homeostatic proteins, including L-type, e.g., CaV1.2 (Dzhura et al., 2000; Grueter et al., 2006) and T-type, e.g., CaV3.2 (Yao et al., 2006) Ca2+ channels, phospholamban (PLN; Lindemann et al., 1983), a protein that negatively regulates SERCA2a in the absence of CaMKII or protein kinase A catalyzed phosphorylation (Kranias and Hajjar, 2012), and RYR2 (Witcher et al., 1991; Wehrens et al., 2004). CaMKII catalyzed phosphorylation increases Ca2+ entry through Ca2+ channels, increases SERCA2a uptake of cytoplasmic Ca2+ into the SR lumen through phosphorylating PLN, which in turn increases the pool of SR releasable Ca2+, increases Ca2+ release from RYR2 by phosphorylation of RYR2 at several sites, including serine 2814. On one hand, these effects will increase intracellular Ca2+ flux through the SR and RYR2 to accelerate NCX1 to increase SAN cell action potential frequency and the physiological fight or flight heart rate response. On the other hand, excessive CaMKII activity will cause Ca2+ overload (Wagner et al., 2011), which can induce increased reactive oxygen species (ROS) production and cause SAN cell damage or death (Swaminathan et al., 2011; Luo et al., 2013).
The role of CaMKII in SAN function has been explored since 1989 (Hagiwara and Irisawa, 1989). The major focus of this study was on the effects of calmodulin or CaMKII on If currents by using calmidazolium, a calmodulin inhibitor with many off target actions (Klöckner and Isenberg, 1987). They found that If currents were sensitive to intracellular Ca2+ but no evidence that If was regulated by CaMKII. A more recent study (Rigg et al., 2003) confirmed that If currents are regulated by Ca2+ and calmodulin but not by the CaMKII pathway. They showed that If current amplitude was unaffected by the CaMKII inhibitor KN-93 (1 μM) although this CaMKII inhibition did reduce L-type Ca2+ current by 48 ± 19% at 0 mV voltage clamp command potential. However, a more recent study challenged the concept of calmodulin regulation of If (Chatelier et al., 2005) based on experiments in inside-out cell membrane macro-patches excised from rabbit SAN cells. They found that “intracellular” calmodulin perfusion had no effect on HCN activity and did not change the cAMP-induced If activation shift. This study suggested that another calmodulin inhibitor, W-7, with well documented off target actions had direct effects on If that were independent of Ca2+ and calmodulin. The myriad off target actions on ion channels represent major obstacles to the use of CaMKII inhibitors in functional studies ( Ledoux et al., 1999; Gao et al., 2006; Rezazadeh et al., 2006; Liao et al., 2011). CaMKII enhances CaV1.2 channel currents in ventricular myocytes (Anderson et al., 1994; Xiao et al., 1994; Yuan and Bers, 1994) and so could potentially affect SAN automaticity by actions on CaV1.2. A paper from the Xiao group showed that CaMKII was likely to play an important role in SAN pacemaker activity by actions at voltage-gated Ca2+ channels (Vinogradova et al., 2000). They were able to stop SAN cell automaticity by using CaMKII inhibitors KN-93 or myristoylated autocamtide-2-related inhibitory peptide (AIP) (a cell membrane permeant peptide inhibitor modeled after the CaMKII autoinhibitory region). The findings from the Xiao group supported an If-independent role for cardiac pacing. However, these studies were mostly performed using small molecule inhibitors with off target actions that complicate interpretation of the results. Taken together, these findings highlight some of the limitations of available small molecule calmodulin and CaMKII antagonists and suggest that If is not directly responsive to calmodulin or CaMKII but leave open the question whether CaMKII actions at CaV1.2 channels contribute to SAN automaticity. We developed a mouse with myocardial targeted transgenic expression of AC3-I, a highly selective CaMKII inhibitory peptide, under control of the α-myosin heavy chain promoter (Zhang et al., 2005). AC3-I expression was present in SAN cells and a study from our group using this mouse found that CaMKII inhibition did not affect baseline SAN pacemaking activity but selectively impaired the fight or flight response of SAN cells to isoproterenol (Wu et al., 2009). CaMKII was responsible for approximately half of the dynamic heart rate response range. We found that neither SAN cell Ca2+ channels nor If currents from AC3-I mice were different compared with their WT littermates nor control transgenic mice expressing AC3-C, an AC3-I like peptide without biological activity. We found that SR Ca2+ content responses to isoproterenol in those mice were reduced, potentially as a consequence of diminished CaMKII catalyzed phosphorylation of PLN. The reduced SR Ca2+ content likely contributed to reduced Ca2+ spark frequency as well as decreased Ca2+ release from SR (Figure 1). Our findings were later confirmed by studies from another group using a CaMKIIδ knock out mouse (Xu et al., 2010). Their study also showed that CaMKII is required for heart rate increases by isoproterenol stimulation or in response to a physiological fight or flight mechanism. A recent study from Terrar group (Collins and Terrar, 2012) suggested that the effect of CaMKII in atrial myocytes may be primarily on SR proteins due to different distribution of CaMKII in ventricular myocytes compare to atrial myocytes which lack of T-tube. The effects of CaMKII on atrial Ca2+ channels are indirectly through CaMKII enhanced SR Ca2+ releasee that stimulates adenylyl cyclases (ACs). Recently, one study from Lakatta group using KN-93, myristoylated AIP, and W-7 to inhibit CaMKII (Yaniv et al., 2013) suggest that CaMKII may affect SAN automaticity by actions on metabolism. In our opinion, these results are intriguing but inconclusive because of the documented off-target actions of these reagents (Ledoux et al., 1999; Chatelier et al., 2005; Gao et al., 2006; Rezazadeh et al., 2006; Liao et al., 2011). Taken together, these studies support a view that CaMKII is not required to maintain basal heart rates but plays a critical role in sustained heart rate increases during physiological stress. This selective role of CaMKII on heart rate suggests that CaMKII inhibition could protect against excessive heart rates without reducing baseline heart rate.
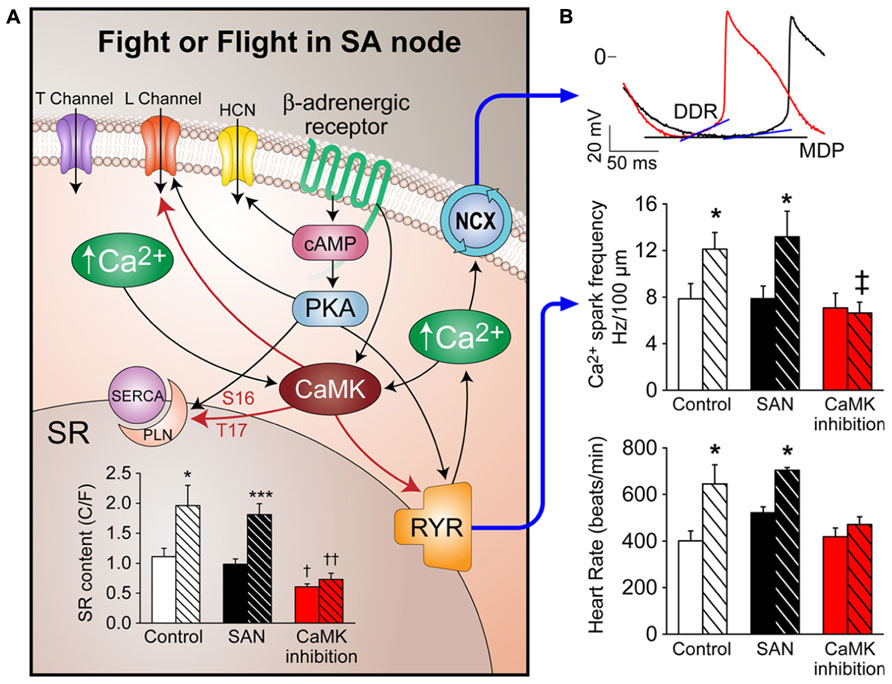
FIGURE 1. Mechanism of CaMKII effects on Fight or Flight in SA node. (A) Fight or flight stimulation (e.g., isoproterenol) activates PKA and CaMKII, which phosphorylate L-type Ca2+ channels and PLN to increase SR Ca2+ content. RYR2 phosphorylation increases Ca2+ release from SR. Increased Ca2+ release accelerates NCX1 activity which increases the diastolic depolarization rate (DDR) of SAN action potentials (B). SR Ca2+ content increases by isoproterenol are abolished by CaMKII inhibition. Upper panel shows DDR change with isoproterenol (red trace) compare to control DDR (black trace), MDP, maximal diastolic potential. Middle panel shows Ca2+ spark frequency increases after isoproterenol are abolished by CaMKII inhibition. The lower panel shows the heart rate increase by isoproterenol is abolished by CaMKII inhibition. All shaded bars in bar graphs represent data with isoproterenol effects. Both white bars (WT control) and black bars (transgenic control) represent data from control SAN. *p < 0.05, ***p < 0.001 before vs. after isoproterenol, † p < 0.05 compare to control SAN groups, †† or ‡ p < 0.01 compare to control SAN groups.
CaMKII in SND
Conditions that favor SND, such as heart failure, atrial fibrillation (AF), and advanced age are marked by heightened ROS (Cesselli et al., 2001; Kim et al., 2005; Dai et al., 2009). Because CaMKII is activated by ROS (Erickson et al., 2008) in the setting of increased angiotensin II (Ang II), a circulating neurohormone present at increased levels in heart failure, we tested if oxidized CaMKII (ox-CaMKII) could contribute to SND. We found Ang II increased atrial and SAN oxidation by activating NADPH oxidase, leading to increased ox-CaMKII, SAN cell apoptosis, and SND (Swaminathan et al., 2011). In order to test whether elevated ox-CaMKII could cause SND, mice were infused with Ang II. Ang II infusion for 3 weeks caused increased SAN ox-CaMKII, SAN cell apoptosis, fibrosis, slowed atrial impulse conduction velocity, and SND. Ang II-triggered SND was prevented by transgenic myocardial and SAN cell expression of AC3-I (Zhang et al., 2005) and by SAN-targeted gene therapy (Kikuchi et al., 2005) providing ectopic SAN expression of a CaMKII inhibitory peptide, CaMKIIN, that is endogenous to neurons but absent in heart (Chang et al., 1998). Neither transgenic nor gene-targeting approaches to SAN CaMKII inhibition affected the hypertensive response to Ang II, nor did they abrogate the increased SAN ROS due to Ang II infusion, indicating that CaMKII was a critical downstream signal for the pathological actions of ROS on SAN. The increase in SAN ox-CaMKII by Ang II required activation of NADPH oxidase, because it was absent in p47-/- mice (Huang et al., 2000) lacking functional NADPH oxidase. We developed a structural and computational model of the SAN that revealed a quantitative mechanism to explain how Ang II-induced SAN cell apoptosis resulted in SND by reducing SAN cell number and increasing electrotonic loading of surviving SAN cells to cause loss of high-fidelity impulse formation and propagation (Figure 2; Huke and Knollmann, 2011). We also found that right atrial tissue from patients with heart failure who required artificial pacemakers for SND or dogs with pacing-induced heart failure and SND had elevated ox-CaMKII compared with patients with heart failure alone or dogs with non-SND controls. These findings provide insights into how excessive activation of CaMKII in SAN cells causes SND, suggest ox-CaMKII is a biomarker for SND and identify what we believe to be a novel candidate approach to preventing SND in high risk settings by CaMKII inhibition.
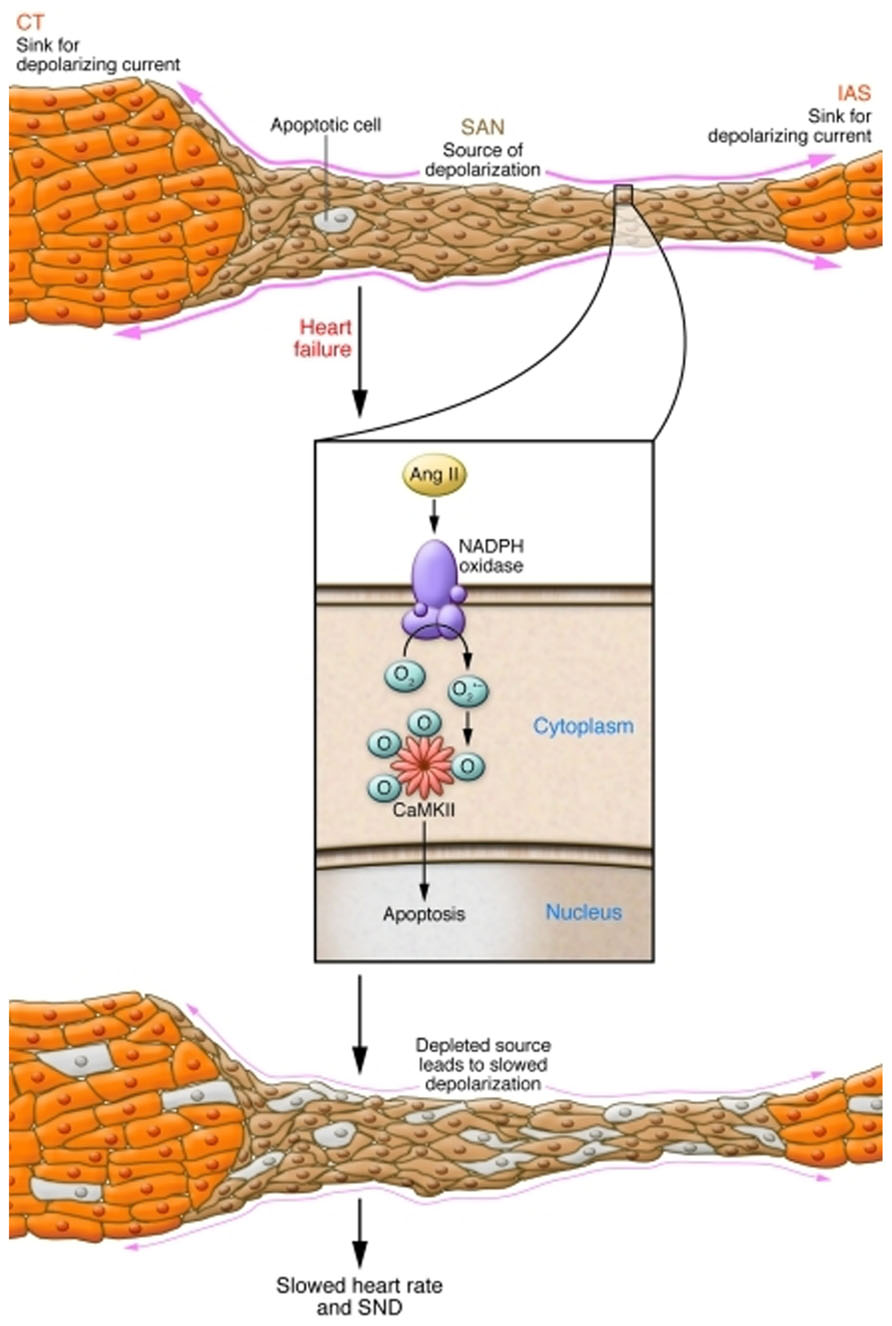
FIGURE 2. Mechanism of Ang II-induced SND. Normally, the small volume of excited tissue in the SAN (source) depolarizes the neighboring quiescent atrial tissue (sink). In conditions with increased Ang II, NADPH oxidase is activated, leading to oxidation of two methionine residues of CaMKII, rendering the enzyme autonomously active. Elevated activity of CaMKII leads to SAN cell death, reducing the threshold volume of automatic cells of the SAN and increasing non-excitable tissue in the form of fibrosis. This increased electrotonic loading produces a source-sink mismatch slows the beating rate, and causes SND. CT, crista terminalis; IAS, inferior atrial septum. Reproduced from Huke and Knollmann (2011), with permission from JCI.
Patients with AF are at increased risk for SND (Chang et al., 2013) and CaMKII activity and expression are increased in fibrillating human atria (Neef et al., 2010). We recently found that ox-CaMKII is increased in fibrillating compared to non-fibrillating human atria and that Ang II infusion increases AF induction in mice (Purohit et al., 2013). Mice with transgenic expression of AC3-I, mice with a knock-in mutation (MM-VV) in CaMKIIδ that prevents oxidative activation and mice with transgenic over-expression of methionine sulfoxide reductase A that reverses the first oxidation state (sulfoxide) of methionine were all resistant to Ang II-induced AF. We interpret these findings to suggest that ox-CaMKII is a unifying signal for SND and AF.
Diabetes is a risk factor for SND (Podlaha and Falk, 1992). We recently found significantly more ox-CaMKII in right atrium from patients with a history of diabetes and myocardial infarction (MI) compared with right atrial tissue from patients with MI but no diabetes, suggesting that ox-CaMKII could contribute to the increased mortality in diabetic patients after MI (Luo et al., 2013). Streptozotocin (STZ)-treated mice develop severe type I diabetes due to death of pancreatic β-cells. STZ-treated diabetic mice were twice as likely to die after MI surgery as vehicle-treated control mice, mimicking the increased mortality in diabetic patients compared with that in non-diabetic patients after MI. STZ-treated MM-VV mice and mice with transgenic myocardial and SAN expression of AC3-I (Zhang et al., 2005) were protected from increased mortality after MI, indicating that increased ox-CaMKII was essential for excess mortality after MI in STZ-treated mice. Death in STZ-treated mice after MI was due to severe bradycardia, consistent with known defects in cardiac pacemaker function in another diabetic animal model (Howarth et al., 2007). In contrast to our earlier studies with Ang II-triggered ROS by activation of NADPH oxidase (Swaminathan et al., 2011), we found that hyperglycemia-induced ROS were primarily from mitochondria. Excess mortality in STZ-treated diabetic mice after MI surgery was prevented by chronic infusion with a mitochondrial targeted antioxidant, Mito-TEMPO. Mito-TEMPO reduced ox-CaMKII, preserved heart rates, and improved survival after MI. These results provide new evidence that ox-CaMKII is a biomarker for SND and suggest that mitochondrial or CaMKII-targeted antioxidant therapies could benefit high-risk diabetic patients.
In summary, CaMKII appears to play important roles in tuning the fight or flight response and in promoting SND. It may be that the physiological benefits of CaMKII activation in early life are outweighed in later life by the tendency of CaMKII to become persistently active under conditions of high oxidative, neurohumoral and hyperglycemic stress. The tractability of CaMKII as a target for selectively controlling heart rate and preventing SND will depend upon the availability of clinically suitable CaMKII inhibitory drugs or gene therapy.
Conflict of Interest Statement
Mark E. Anderson is a cofounder of Allosteros Therapeutics, a biotech aiming to treat cardiovascular disease by enzyme inhibition.
Acknowledgments
Shawn Roach for graphic design. This work was funded by National Institutes of Health (NIH) Grants R01-HL 079031, R01-HL096652, and R01-HL070250, R01-HL071140 and the Fondation LeducqTransatlantic Alliance for CaMKII Signaling, 08CVD01.
References
Anderson, M. E., Braun, A. P., Schulman, H., and Premack, B. A. (1994). Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca2+-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ. Res. 75, 854–861. doi: 10.1161/01.RES.75.5.854
Brahmajothi, M. V., Morales, M. J., Campbell, D. L., Steenbergen, C., and Strauss, H. C. (2010). Expression and distribution of voltage-gated ion channels in ferret sinoatrial node. Physiol. Genomics. 42A, 131–40. doi: 10.1152/physiolgenomics.00049.2010
Brahmajothi, M. V., Morales, M. J., Reimer, K. A., and Strauss, H. C. (1997). Regional localization of ERG, the channel protein responsible for the rapid component of the delayed rectifier, K+ current in the ferret heart. Circ. Res. 81, 128–135. doi: 10.1161/01.RES.81.1.128
Cesselli, D., Jakoniuk, I., Barlucchi, L., Beltrami, A. P., Hintze, T. H., Nadal-Ginard, B., et al. (2001). Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 89, 279–286. doi: 10.1161/hh1501.094115
Chandler, N. J., Greener, I. D., Tellez, J. O., Inada, S., Musa, H., Molenaar, P., et al. (2009). Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation 119, 1562–1575. doi: 0.1161/CIRCULATIONAHA.108.804369
Chang, B. H., Mukherji, S., and Soderling, T. R. (1998). Characterization of a calmodulin kinase II inhibitor protein in brain. Proc. Natl. Acad. Sci. U.S.A. 95, 10890–10895. doi: 10.1073/pnas.95.18.10890
Chang, H. Y., Lin, Y. J., Lo, L. W., Chang, S. L., Hu, Y. F., Li, C. H., et al. (2013). Sinus node dysfunction in atrial fibrillation patients: the evidence of regional atrial substrate remodelling. Europace 15, 205–211. doi: 10.1093/europace/eus219
Chao, L. H., Stratton, M. M., Lee, I. H., Rosenberg, O. S., Levitz, J., Mandell, D. J., et al. (2011). A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell 146, 732–745. doi: 10.1016/j.cell.2011.07.038
Chatelier, A., Renaudon, B., Bescond, J., El Chemaly, A., Demion, M., and Bois, P. (2005). Calmodulin antagonist W7 directly inhibits f-type current in rabbit sino-atrial cells. Eur. J. Pharmacol. 521, 29–33. doi: 10.1016/j.ejphar.2005.08.024
Christel, C. J., Cardona, N., Mesirca, P., Herrmann, S., Hofmann, F., Striessnig, J., et al. (2012). Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J. Physiol. 590(Pt 24), 6327–6342. doi: 10.1113/jphysiol.2012.239954
Collins, T. P., and Terrar, D. A. (2012). Ca(2+)-stimulated adenylyl cyclases regulate the L-type Ca(2+) current in guinea-pig atrial myocytes. J. Physiol. 590(Pt 8), 1881–1893. doi: 10.1113/jphysiol.2011.227066
Dai, D. F., Santana, L. F., Vermulst, M., Tomazela, D. M., Emond, M. J., MacCoss, M. J., et al. (2009). Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119, 2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403
DiFrancesco, D. (1991). The contribution of the “pacemaker” current (if) to generation of spontaneous activity in rabbit sino-atrial node myocytes. J. Physiol. 434, 23–40.
Dzhura, I., Wu, Y., Colbran, R. J., Balser, J. R., and Anderson, M. E. (2000). Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat. Cell Biol. 2, 173–177. doi: 10.1038/35004052
Erickson, J. R., He, B. J., Grumbach, I. M., and Anderson, M. E. (2011). CaMKII in the cardiovascular system: sensing redox states. Physiol. Rev. 91, 889–915. doi: 10.1152/physrev.00018.2010
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Gao, L., Blair, L. A., and Marshall, J. (2006). CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem. Biophys. Res. Commun. 345, 1606–1610. doi: 10.1016/j.bbrc.2006.05.066
Grueter, C. E., Abiria, S. A., Dzhura, I., Wu, Y., Ham, A. J., Mohler, P. J., et al. (2006). L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol. Cell 23, 641–650 doi: 10.1016/j.molcel.2006.07.006
Gutierrez, D. A., Fernandez-Tenorio, M., Ogrodnik, J., and Niggli, E. (2013). NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 100, 392–401. doi: 10.1093/cvr/cvt201
Hagiwara, N., and Irisawa, H. (1989). Modulation by intracellular Ca2+ of the hyperpolarization-activated inward current in rabbit single sino-atrial node cells. J. Physiol. 409, 121–141.
Howarth, F. C., Al-Sharhan, R., Al-Hammadi, A., and Qureshi, M. A. (2007). Effects of streptozotocin-induced diabetes on action potentials in the sinoatrial node compared with other regions of the rat heart. Mol. Cell Biochem. 300, 39–46. doi: 10.1007/s11010-006-9366-5
Huang, C. K., Zhan, L., Hannigan, M. O., Ai, Y., and Leto, T. L. (2000). P47(phox)-deficient NADPH oxidase defect in neutrophils of diabetic mouse strains, C57BL/6J-m db/db and db/+. J. Leukoc. Biol. 67, 210–215.
Huke, S., and Knollmann, B. C. (2011). Oxidized CaMKII: a “heart stopper” for the sinus node? J. Clin. Invest. 121, 2975–2977. doi: 10.1172/JCI58389
Kikuchi, K., McDonald, A. D., Sasano, T., and Donahue, J. K. (2005). Targeted modification of atrial electrophysiology by homogeneous transmural atrial gene transfer. Circulation 111, 264–270. doi: 10.1161/01.CIR.0000153338.47507.83
Kim, Y. M., Guzik, T. J., Zhang, Y. H., Zhang, M. H., Kattach, H., Ratnatunga, C., et al. (2005). A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 97, 629–636. doi: 10.1161/01.RES.0000183735.09871.61
Klöckner, U., and Isenberg, G. (1987). Calmodulin antagonists depress calcium and potassium currents in ventricular and vascular myocytes. Am. J. Physiol. 253(6 Pt 2), H1601–H1611.
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/CIRCRESAHA.111.259754
Kuret, J., and Schulman, H. (1985). Mechanism of autophosphorylation of the multifunctional Ca2+/calmodulin-dependent protein kinase. J. Biol. Chem. 260, 6427–6433.
Lakatta, E. G., and DiFrancesco, D. (2009). What keeps us ticking: a funny current, a calcium clock, or both? J. Mol. Cell Cardiol. 47, 157–170. doi: 10.1016/j.yjmcc.2009.03.022
Lakatta, E. G., Maltsev, V. A., and Vinogradova, T. M. (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 106, 659–673. doi: 10.1161/CIRCRESAHA.109.206078
Ledoux, J., Chartier, D., and Leblanc, N. (1999). Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J. Pharmacol. Exp. Ther. 290, 1165–1174.
Liao, Z., St Clair, J. R., Larson, E. D., and Proenza, C. (2011). Myristoylated peptides potentiate the funny current (I(f)) in sinoatrial myocytes. Channels (Austin) 5, 115–119. doi: 10.4161/chan.5.2.14195
Lindemann, J. P., Jones, L. R., Hathaway, D. R., Henry, B. G., and Watanabe, A. M. (1983). beta-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J. Biol. Chem. 258, 464–471.
Luo, M., Guan, X., Luczak, E. D., Lang, D., Kutschke, W., Gao, Z., et al. (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 123, 1262–1274. doi: 10.1172/JCI65268
Mangoni, M. E., Traboulsie, A., Leoni, A. L., Couette, B., Marger, L., Le Quang, K., et al. (2006). Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ. Res. 98, 1422–1430. doi: 10.1161/01.RES.0000225862.14314.49
Neef, S., Dybkova, N., Sossalla, S., Ort, K. R., Fluschnik, N., Neumann, K., et al. (2010). CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 106, 1134–1144. doi: 10.1161/CIRCRESAHA.109.203836
Podlaha, R., and Falk, A. (1992). The prevalence of diabetes mellitus and other risk factors of atherosclerosis in bradycardia requiring pacemaker treatment. Horm. Metab. Res. Suppl. 26, 84–87.
Purohit, A., Rokita, A. G., Guan, X., Chen, B., Koval, O. M., Voigt, N., et al. (2013). Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 128, 1748–1757. doi: 10.1161/CIRCULATIONAHA.113.003313
Rezazadeh, S., Claydon, T. W., and Fedida, D. (2006). KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J. Pharmacol. Exp. Ther. 317, 292–299. doi: 10.1124/jpet.105.097618
Rigg, L., Mattick, P. A., Heath, B. M., and Terrar, D. A. (2003). Modulation of the hyperpolarization-activated current (I(f)) by calcium and calmodulin in the guinea-pig sino-atrial node. Cardiovasc. Res. 57, 497–504. doi: 10.1016/S0008-6363(02)00668-5
Rigg, L., and Terrar, D. A. (1996). Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 81, 877–880.
Sanders, L., Rakovic, S., Lowe, M., Mattick, P. A., and Terrar, D. A. (2006). Fundamental importance of Na+Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J. Physiol. 571(Pt 3), 639–649. doi: 10.1113/jphysiol.2005.100305
Sanders, P. N., Koval, O. M., Jaffer, O. A., Prasad, A. M., Businga, T. R., Scott, J. A., et al. (2013). CaMKII is essential for the proasthmatic effects of oxidation. Sci. Transl. Med. 5, 195ra97. doi: 10.1126/scitranslmed.3006135
Schulman, H., and Greengard, P. (1978). Stimulation of brain membrane protein phosphorylation by calcium and an endogenous heat-stable protein. Nature 271, 478–479. doi: 10.1038/271478a0
Swaminathan, P. D., Purohit, A., Soni, S., Voigt, N., Singh, M. V., Glukhov, A. V., et al. (2011). Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Invest. 121, 3277–3288. doi: 10.1172/JCI57833
Tanaka, H., Komikado, C., Namekata, I., Nakamura, H., Suzuki, M., Tsuneoka, Y., et al. (2008). Species difference in the contribution of T-type calcium current to cardiac pacemaking as revealed by r(-)-efonidipine. J. Pharmacol. Sci. 107, 99–102 doi: 10.1254/jphs.SC0070405
Vinogradova, T. M., Zhou, Y. Y., Bogdanov, K. Y., Yang, D., Kuschel, M., Cheng, H., et al. (2000). Sinoatrial node pacemaker activity requires Ca (2+)/calmodulin-dependent protein kinase II activation. Circ. Res. 87, 760–767. doi: 10.1161/01.RES.87.9.760
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108, 555–565. doi: 10.1161/CIRCRESAHA.110.221911
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004). Ca2+/calmodulindependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2
Witcher, D. R., Kovacs, R. J., Schulman, H., Cefali, D. C., and Jones, L. R. (1991). Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 266, 11144–11152.
Wu, Y., Gao, Z., Chen, B., Koval, O. M., Singh, M. V., Guan, X., et al. (2009). Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc. Natl. Acad. Sci. U. S. A. 106, 5972–5977. doi: 10.1073/pnas.0806422106
Xiao, R. P., Cheng, H., Lederer, W. J., Suzuki, T., and Lakatta, E. G. (1994). Dual regulation of Ca2+/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc. Natl. Acad. Sci. U. S. A. 91, 9659–9663. doi: 10.1073/pnas.91.20.9659
Xu, L., Lai, D., Cheng, J., Lim, H. J., Keskanokwong, T., Backs, J., et al. (2010). Alterations of L-type calcium current and cardiac function in CaMKII{delta} knockout mice. Circ. Res. 107, 398–407. doi: 10.1161/CIRCRESAHA.110.222562
Yaniv, Y., Spurgeon, H. A., Ziman, B. D., and Lakatta, E. G. (2013). Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity and sinoatrial nodal pacemaker cell energetics. PLoS ONE 8:e57079. doi: 10.1371/journal.pone.0057079
Yao, J., Davies, L. A., Howard, J. D., Adney, S. K., Welsby, P. J., Howell, N., et al. (2006). Molecular basis for the modulation of native T-type Ca2+ channels in vivo by Ca2+/calmodulin-dependent protein kinase II. J. Clin. Invest. 116, 2403–2412. doi: 10.1172/JCI27918
Yuan, W., and Bers, D. M. (1994). Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am. J. Physiol. 267(3 Pt 2), H982–H993.
Keywords: calcium/calmodulin-dependent protein kinase II, sinoatrial node, heart rate, sinoatrial node dysfunction, calcium
Citation: Wu Y and Anderson ME (2014) CaMKII in sinoatrial node physiology and dysfunction. Front. Pharmacol. 5:48. doi: 10.3389/fphar.2014.00048
Received: 16 January 2014; Accepted: 03 March 2014;
Published online: 18 March 2014.
Edited by:
Donald M. Bers, University of California, Davis, USAReviewed by:
Zhandi Liao, University of California, Davis, USASatoshi Matsuoka, University of Fukui, Japan
Copyright © 2014 Wu and Anderson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark E. Anderson, Department of Internal Medicine and Department of Molecular Physiology and Biophysics, Carver College of Medicine, University of Iowa, Iowa City, IA 52242, USA e-mail:bWFyay1lLWFuZGVyc29uQHVpb3dhLmVkdQ==