 Yael Yaniv
Yael Yaniv Victor A. Maltsev
Victor A. Maltsev- 1Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging – National Institutes of Health, Baltimore, MD, USA
- 2Department of Biomedical Engineering, Technion – Israel Institute of Technology, Haifa, Israel
Sinoatrial node (SAN) is the primary heart pacemaker which initiates each heartbeat under normal conditions. Numerous experimental data have demonstrated that Ca2+- and CaMKII-dependent processes are crucially important for regulation of SAN cells. However, specific mechanisms of this regulation and their relative contribution to pacemaker function remain mainly unknown. Our review summarizes available data and existing numerical modeling approaches to understand Ca2+ and CaMKII effects on the SAN. Data interpretation and future directions to address the problem are given within the coupled-clock theory, i.e., a modern view on the cardiac pacemaker cell function generated by a system of sarcolemmal and intracellular proteins.
Introduction
Under normal conditions, SAN cells (SANC) generate spontaneous rhythmic action potentials (AP) that initiate the heartbeat. The evolution of thought regarding the cardiac pacemaker cell operation paradigm switched back and forth between intracellular origin [e.g., a “metabolic” intracellular clock (Bozler, 1943) or sarcoplasmic reticulum (SR)-based Ca2+-clock (Maltsev et al., 2006)] and cell membrane origin [voltage membrane clock or M-clock (Noble, 1960)]. A more recent paradigm shift has been the realization that both intracellular and sarcolemmal mechanisms are tightly, dynamically coupled to each other and are indispensable for normal pacemaker function. These ideas have been summarized within a “coupled-clock” theory of interacting M-clock and Ca2+-clock (Maltsev and Lakatta, 2009; Figure 1) that explained numerous experimental findings (Lakatta et al., 2010; Maltsev and Lakatta, 2012). The key processes of the coupled-clock system depend on Ca2+, calmodulin (CaM), and CaMKII signaling (Figure 1, red). Interactions within the system are extremely complex and their detailed investigation requires numerical model simulations. The CaMKII function in pacemaker cells has not been systematically studied using numerical simulations. Our review summarizes major principles of the coupled-clock theory, available data, and existing numerical modeling approaches that are important to delineate future numerical integration and exploration of CaMKII within the pacemaker cell system.
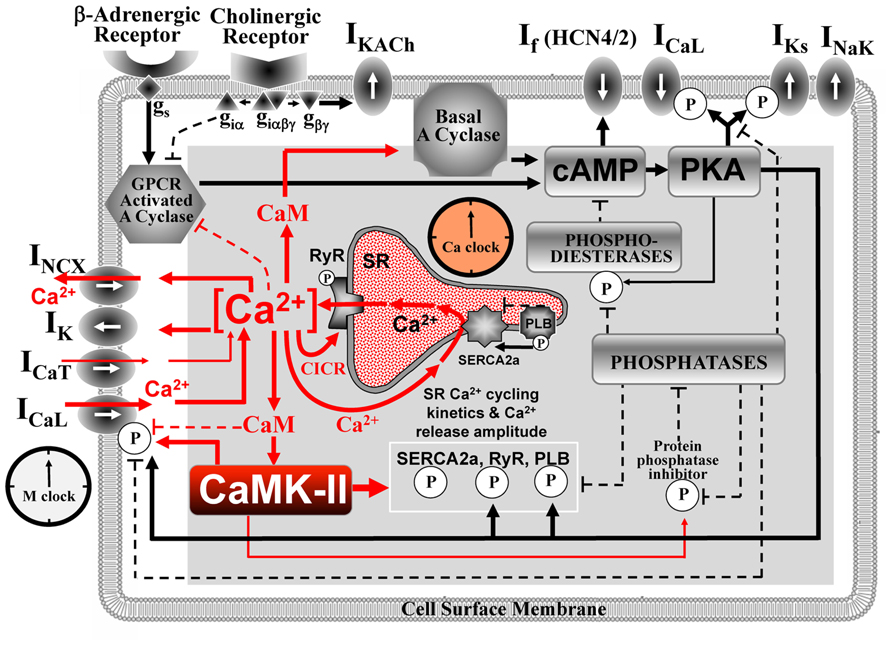
FIGURE 1. Schematic illustration of sinoatrial node cell as a coupled-clock system of voltage membrane clock (M-clock) and an intracellular, sarcoplasmic reticulum (SR)-based Ca2+-clock (gray intracellular area). Interactions of key molecules comprising the system with Ca2+ and CaMKII signaling are shown in red. Note that common regulatory factors govern the function of both clocks. These common factors, including CaMKII, act as nodes within the system to couple the function of both clocks activities. The system is balanced: signals accelerating action potential (AP) firing are balanced by signals suppressing AP firing. This balance determines a given steady-state level of net Ca2+, cAMP, and protein phosphorylation via PKA, and CaMKII. G protein-coupled receptors (top left corner) within the cell membrane modulate both the Ca2+-clock and M-clock function via the same crucial signaling nodes of the system. Modified from Lakatta et al. (2010).
Integration of Ca2+ and CaMKII Signaling within the Coupled-Clock System
Operation of the coupled-clock system has been explored in recent numerical model studies (Maltsev and Lakatta, 2009, 2013; Yaniv et al., 2013a, d), and experimental evidence for the coupled-clock theory has been summarized (Lakatta et al., 2010; Maltsev and Lakatta, 2012). The system generates spontaneous, rhythmic APs separated by a slow diastolic depolarization (DD) that starts each cycle from the maximum diastolic potential (MDP ~-60 mV) and brings the membrane potential (V m) to a cell excitation threshold of ~-40 mV. The coupled-clock theory postulates that the DD is generated by the two coupled oscillators, Ca2+-clock and M-clock, rather than just by M-clock alone (Figure 1).
The first numerical model of M-clock was developed by Noble (1960), by application of Hodgkin–Huxley (HH) theory to cardiac pacemaker cells. The M-clock-based models generate the DD via time-dependent kinetics of ion channels upon AP repolarization, e.g., by inactivation of a K+ current (Noble, 1960) or by activation of a non-selective, “funny” current (DiFrancesco and Noble, 2012). The SR, a major Ca2+ store in cardiac cells, can also generate spontaneous oscillations via rhythmic cycles of SR Ca2+ pumping (via SERCA) and release (via release channels, RyR; Figure 1). Ventricular muscle cells can spontaneously cycle Ca2+ (under conditions of high Ca2+ loading) via global Ca2+ waves via regenerative Ca2+-induced Ca2+ release (CICR) propagating by Ca2+ diffusion (Fabiato, 1983). However, cardiac pacemaker cells generate rhythmic, spontaneous Ca2+ releases during DD under normal Ca2+ conditions (in the absence of Ca2+ overload; Huser et al., 2000; Bogdanov et al., 2001). These releases occur in the form of abrogated waves, dubbed local Ca2+ releases or LCRs. The synchronous occurrence of the LCRs generates a powerful, diastolic, net Ca2+ signal, dubbed the late diastolic Ca2+ elevation or LDCaE (Figure 2). The rhythmic LCRs are generated in the absence of M-clock, e.g., under voltage clamp or in membrane-permeabilized SANC [when [Ca2+] is normal, ~100 nM, review (Lakatta et al., 2010)]. The Ca2+-clock in SANC is driven by Ca2+ cycling proteins (e.g., phospholamban and RyR, Figure 1), whose function is enhanced by phosphorylation via basal activity of PKA (Vinogradova et al., 2006) and CaMKII (Vinogradova et al., 2000). In turn, the PKA is activated by a high basal level of cAMP produced by Ca2+-activated adenylyl cyclases (ACs) which are highly expressed in SANC [particularly types 1 and 8, (Mattick et al., 2007; Younes et al., 2008)]. The high rate of cAMP production and protein phosphorylation is counterbalanced by activities of phosphatases and phosphodiesterases. Interestingly, a powerful Ca2+-clock generating rhythmic LCRs (similar to that in SANC) also emerges in ventricular myocytes when the phosphorylation of Ca2+ cycling protein increases (e.g., via inhibition of phosphatases and/or phosphodiesterases; Sirenko et al., 2014).
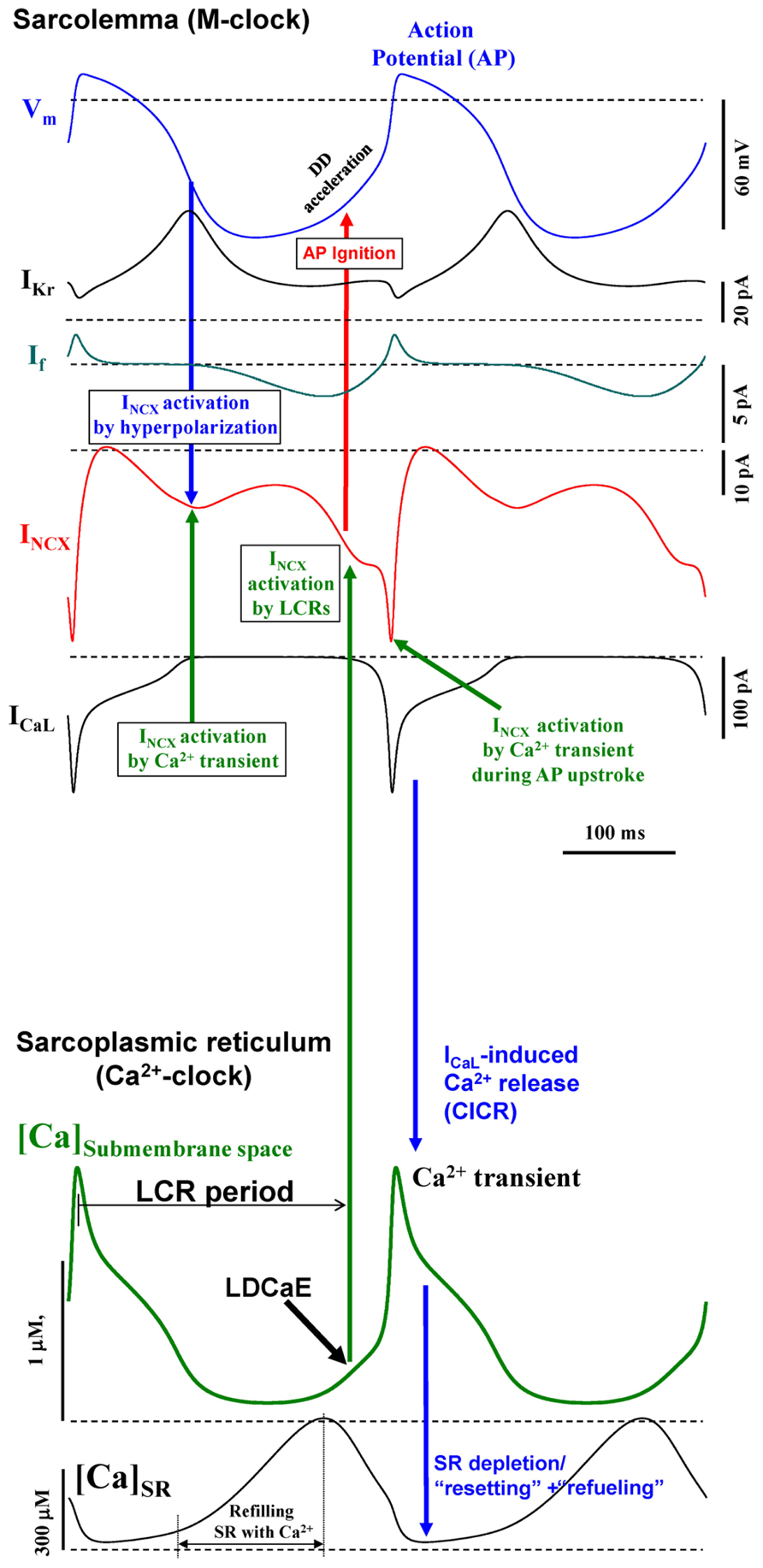
FIGURE 2. Coupled-clock Maltsev–Lakatta numerical model (Maltsev and Lakatta, 2009) predicts complex synergistic interactions between cell membrane and Ca2+ cycling proteins within SANC (see “Integration of Ca2+ and CaMKII Signaling within the Coupled-Clock System”). Modified from Maltsev and Lakatta (2009).
The Ca2+-clock and the M-clock are coupled in SANC via Na+/Ca2+ exchanger (NCX; Figure 2) that senses the LCR ensemble (i.e., LDCaE) and, operating in the forward mode, generates a substantial inward current (I NCX) during DD. M-clock, in turn, regulates Ca2+-clock via L-type Ca2+ current (I CaL) by (1) resetting phases of local Ca2+ oscillators that synchronizes LCR ensemble; (2) supplying Ca2+, i.e., the Ca2+-clock’s oscillatory substrate. Both clocks are coupled not only directly via V m and Ca2+, but indirectly, enzymatically, by coupling factors, such as PKA and CaMKII, affecting multiple targets within both clocks (Figure 1). PKA- and CaMKII-dependent phosphorylation enhances function of the proteins comprising the system and is required for normal pacemaker function and autonomic modulation. Because of these complex interactions (which define the Ca2+ balance and enzymatic activity balance), each component of the system contributes to the LCR spatiotemporal characteristics, especially the LCR period, i.e., the time when LCRs emerge and accelerate the DD (Figure 2). Thus, the LCR period is contributed not only directly by the Ca2+ release channels RyR, but also indirectly by L-type Ca2+ channels (LCCh), SERCA, and NCX regulating Ca2+ fluxes (Maltsev et al., 2013), and even by K+ channels or “funny” channels via respective V m changes, also regulating Ca2+ fluxes (Yaniv et al., 2013a).
Experimental Evidence for Importance of CaMKII Signaling in Pacemaker Cells
CaMKII indirectly senses [Ca2+] by binding Ca2+-CaM complex at the CaM region in its regulatory domain, which increases its activity (Anderson et al., 2011). (Of note, there are two predominant CaMKII isoforms in the heart: CaMKIIδ B localizes in nuclei and CaMKIIδ C in cytosol). While CaMKII does not regulate directly cAMP production, reduction in CaMKII activity is associated with reduction in [cAMP] in rabbit SANC (Yaniv et al., 2013b), indicating a complex interplay of the CaMKII, ACs, and PKA signaling (Figure 1). In this special issue Wu and Anderson discuss in detail experimental evidence for contribution of CaMKII activity to SAN function during health and heart disease (Wu and Anderson, 2014). Here we summarize the key facts with respect to the integration of CaMKII within the coupled-clock system of SANC (Figure 1) and its future numerical modeling.
Pharmacological inhibition of CaMKII signaling (using AIP or KN-93) depresses the basal rate and amplitude of spontaneous APs in SANC of rabbit (Vinogradova et al., 2000; Yaniv et al., 2013b) and guinea-pig (Rigg et al., 2003). Confocal imaging of immunolabeled proteins demonstrates that active CaMKII is highly localized beneath the surface membrane (Vinogradova et al., 2000). Thus, CaMKII activity is geographically associated with proteins of both M- and Ca2+-clocks. CaMKII modulates several membrane ion channels in the heart: LCCh, K+ channels, and Ca2+-clock proteins: SERCA (directly and indirectly via phospholamban) and RyR. Studies in isolated rabbit SANC suggested that CaMKII regulates the pacemaker activity via modulating I CaL inactivation and reactivation (Vinogradova et al., 2000) and LCR morphology (Vinogradova et al., 2011). I f is not affected directly by CaMKII inhibition (Rigg et al., 2003).
Thus, contribution of CaMKII to basal AP generation by SANC was demonstrated for rabbit and guinea pig [but remains controversial for mice (Zhang et al., 2005; Wu et al., 2009)]. Because CaMKII is sensitive to the frequency of the Ca2+ transients, CaMKII is ideally suited to respond to changes in SAN rhythm. For example, electrical stimulation alone increases CaMKII-dependent phosphorylation of phospholamban at CaMKII phosphorylation site in a frequency-dependent manner in ventricular myocytes (Hagemann et al., 2000). CaMKII also mediates SAN response to β-adrenergic receptor stimulation (Wu et al., 2009). Moreover, SANC and isolated hearts from mice with CaMKII inhibition (by transgenic expression of AC3-I) were insensitive to BayK, an LCCh agonist, which increases pacemaker rate in wild type mice (Gao et al., 2011). New evidence that CaMKII is a key part of the coupled-clocked system (Figure 1) has been obtained in studies of specific I f inhibitor ivabradine (Yaniv et al., 2013a; discussed below).
CaMKII activity can also be enhanced by pro-oxidant conditions (Erickson et al., 2008). Clinical studies show that right atrial tissue from patients with heart failure who also required artificial pacemakers have more Oxidize-CaMKII compared to patients with heart failure alone and patients without heart failure or severe SAN dysfunction (Swaminathan et al., 2011). Ang II infusion in mice increases Oxidize-CaMKII and elicits SAN dysfunction that is prevented by overexpression of a synthetic CaMKII inhibitory peptide (AC3-I) or by CaMKIIN, an endogenous CaMKII protein present in neurons, but absent in the heart (Swaminathan et al., 2011).
CaMKII activity appears to be increased in heart disease (e.g., arrhythmia, heart failure, atrial fibrillation; Anderson et al., 2011). Sinus sick syndrome prevails during heart failure and hypertension conditions (with both conditions exhibiting elevated angiotensin II levels). Because CaMKII inhibition is sufficient to protect against angiotensin II-induced sick sinus syndrome in aforementioned mouse model (Swaminathan et al., 2012), CaMKII inhibition may be a useful approach to prevent sinus sick syndrome.
It was demonstrated that basal AC-cAMP/PKA signaling directly, and Ca2+ indirectly, regulate mitochondrial ATP production (Yaniv et al., 2011, 2013c). As a crucial element of normal automaticity in rabbit SANC, CaMKII signaling is also involved in SANC bioenergetics. When ATP demand is reduced by interfering with CaMKII or CaM activity, SANC become depleted of ATP, indicating reduction in ATP generation with lower demand (Yaniv et al., 2013b).
Numerical Modeling Studies that Show Importance of CaMKII Signaling for San Function
Although CaMKII signaling, per se, has not been systematically studied in pacemaker cell models, at least two recent numerical model studies point to a key functional importance of CaMKII signaling in pacemaker cells and tissues.
Yaniv et al. (2013a) have recently demonstrated that CaMKII likely serves as a key functional integrator of M-clock and Ca2+-clock signals (Figure 1) by testing effects of specific perturbations of either clock in rabbit SANC. The M-clock was specifically perturbed by ivabradine that at low concentrations (<3 μM) specifically inhibits I f, i.e., it does not suppress I CaL (Yaniv et al., 2012a), other membrane ion currents (Bois et al., 1996), or Ca2+ cycling in permeabilized SANC (Yaniv et al., 2013a).
Numerical simulations (Yaniv et al., 2013a) using a modified coupled-clock Maltsev–Lakatta model (Yaniv et al., 2012b), provided new insights in ivabradine-induced bradycardia. An initial I f reduction slows AP rate that, in turn, reduces the number of I CaL activations/unit time, average Ca2+ influx, and Ca2+ available for SR pumping. This results in lower SR Ca2+ load and longer LCR period (both effects were also found experimentally). Later activation of diastolic I NCX by the LCRs (and I NCX-linked DD acceleration) leads to a delayed activation of I CaL, i.e., M-clock slowing. Thus, inhibition of the M-clock inhibits (indirectly) Ca2+-clock that further suppresses the M-clock, and so on, until the coupled-clock system attains a new steady-state.
Interestingly, model simulations show that the complex ivabradine effects extend further, beyond “biophysical” entrainment, and likely include an additional “biochemical” component. The aforementioned decrease in average Ca2+ influx produced by ivabradine not only decreases Ca2+ available for SR pumping, but also likely decreases protein phosphorylation signaling via Ca2+-activated-CaMKII and Ca2+-activated-ACs-cAMP/PKA pathways. This leads to further reductions in the average Ca2+ influx and, therefore, SR Ca2+ loading and AP firing rate. Simultaneously, reduction in cAMP shifts the I f activation curve (effecting further M-clock slowing). If the “biochemical” crosstalk is lacking, model simulations (Yaniv et al., 2013a) predict only about 50% of the experimentally measured bradycardia produced by ivabradine. Thus, the entire ivabradine effect is explained by a crosstalk of equally important biophysical and biochemical mechanisms (including CaMKII signaling).
According to the coupled-clock theory (Maltsev and Lakatta, 2009) any selective perturbation of either clock will inevitably affect the function of the other and the entire coupled-clock system. In line with this postulate, the bradycardic effect is symmetric: it does not depend on which clock was initially perturbed. Both the LCR period and AP cycle length become prolonged by either perturbations of M-clock (e.g., using ivabradine) or Ca2+-clock (e.g., using cyclopiazonic acid to selectively inhibit SERCA), with the LCR period reporting the resultant complex effect (Yaniv et al., 2013a).
Heart rate reductions produced by ivabradine or HCN4 mutations have been interpreted as a pure result of insufficient I f function. However, based on the results discussed above, these effects are likely complex, involving the secondary changes in Ca2+-clock and the entire coupled-clock system (that includes CaMKII signaling; Yaniv and Lakatta, 2013). Effects of mutations of Ca2+ cycling proteins on pacemaker function also likely include clocks coupling, i.e., secondary effect on I f (via Ca2+-activated-ACs and cAMP), rate-dependent effects on both clocks, ultimately resulting in mutual entrainment of the clocks (Yaniv and Lakatta, 2013; Yaniv et al., 2013a).
Luo et al. (2013) numerically modeled a further level of CaMKII effects related to cell death that is important to approach the mechanisms of insufficient pacemaker function in disease and aging. They developed a two-dimensional histologically reconstructed mathematical model that takes into account SAN cell death and fibrosis expressed in myocardial infarction by oxidizing CaMKII. Their simulations predict decreased conduction velocity and shift of the leading pacemaker site under these conditions. Thus, changes in CaMKII signaling can result in morphological changes of the SAN tissue which can affect cardiac impulse initiation.
Local Ca2+ and CaMKII Signaling in Pacemaker Cells
The local Ca2+ control theory (Stern, 1992) remains a key in understanding the mechanisms of cardiac excitation-contraction coupling. This theory explained graded CICR phenomenon via statistics of success and failure of an initiating event (such as LCCh opening) to recruit stochastic Ca2+ release units (CRUs) to fire. While partially periodic LCRs (comprising Ca2+ clocks) in cardiac pacemaker cells are generated by the CRUs, they are, in fact, a product of complex local interactions of proteins residing in both cell membrane and the SR, i.e., RyR, SERCA, LCCh, and NCX. These interactions, in turn, are regulated by PKA and CaMKII signaling (Figure 1).
During the last decade mathematical models have been developed in ventricular myocytes to describe the CaMKII effects via regulation of ionic currents (Hund and Rudy, 2004; Grandi et al., 2007). More recent models describe CaMKII activity as a function of subspace Ca2+, CaM, and phosphatase activity (Saucerman and Bers, 2008). These studies have demonstrated that the different affinities of CaM and CaMKII and calcineurin determine their sensitivity to local versus global Ca2+ signals that regulates excitation-contraction coupling. Hashambhoy et al. (2009) developed a stochastic model describing the dynamic interactions among CaMKII, LCCh, and phosphatases as a function of dyadic Ca2+ and CaM levels.
Local Ca2+ mechanisms have been recently modeled in pacemaker cells. The LCRs are generated via stochastic recruitment of the neighboring CRUs (Maltsev et al., 2011) regulated by local interactions of RyR, SERCA, and NCX (Maltsev et al., 2013). Some irregularity in RyR spatial distribution is not an imperfection, but rather a functional modality of the pacemaker cells [abstract (Maltsev et al., 2014)]. The irregularity decreases nearest neighbor-to-neighbor distances among the CRUs and thereby facilitates local CICR forming wavelet-like LCRs. This new local control mechanism regulates the balance between robustness and flexibility of pacemaker cell function.
The most advanced SANC model (Stern et al., 2014) features stochastic propagated spontaneous diastolic Ca2+ release in three dimensions. This model describes explicit gating of individual Ca2+ channels (both RyR and LCCh), without assuming either a discrete sub-membrane compartment or an inactivated state of the RyR. The model succeeded in reproducing observed propagating local Ca2+ releases and realistic pacemaker rates only when RyR locations were assigned taking into account irregular, hierarchical distribution of RyR clusters (small and large) observed in 3D confocal scan sections of immunofluorescence staining. When the RyR sensitivity is very high or the NCX density is low, synchronization is lost, causing sympathetic stimulation to reduce (rather than increase) beating rate, often exhibiting arrhythmias (Maltsev et al., 2013; Stern et al., 2014). This regime may be important for rhythm abnormalities caused by heart failure, RyR mutations, or pharmacological NCX blockade.
Compared to previous models, lacking local Ca2+ dynamics (i.e., “common pool” models [Kurata et al., 2002; Maltsev and Lakatta, 2009)], the new models provide mechanistic insights into local crosstalk of the key molecules of the system: recruitment of RyRs (generating diastolic LCRs), RyR-LCCh and RyR-NCX crosstalk, and efficient SERCA operation (Maltsev et al., 2013). Indeed, Ca2+ signals within LCRs exhibit much higher amplitudes vs. those predicted by “common pool” models (~tens of μM vs. ~1 μM). Thus, the “local” models, predicting the realistic scale of Ca2+ signals within the inhomogeneous signaling network of SANC, seem to be a better choice to explore CaMKII effects in future studies of pacemaker cells.
Summary
In this review we have summarized the present state of experimental and numerical modeling studies on Ca2+ and CaMKII roles in cardiac pacemaker cells. Taking into account emerging importance of local Ca2+ control in cardiac pacemaker cells and also importance of local CaMKII signaling (reported in ventricular myocytes), accurate interpretation of experimental data on CaMKII effects in pacemaker cells will likely require integration of local (Saucerman and Bers, 2008) and molecular (Hashambhoy et al., 2009) mechanisms into new pacemaker cell models. Another important aspect that needs numerical integration is CaMKII involvement in SANC bioenergetics (Yaniv et al., 2013b). The new experimental studies combined with new model simulations will explore CaMKII interactions (Figure 1, red) with key regulatory molecules (e.g., ACs, PDEs, phosphatases, PKA, phospholamban), effector molecules (RyR, SERCA, NCX, LCCh, NCX, etc), and energy production of the system. This knowledge will contribute greatly to our understanding of cardiac impulse initiation and specific role of CaMKII signaling in the pacemaker regulation.
Author Contributions
Both authors contributed to the conception of the work, drafted the paper, approved the version to be published, and are accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging.
References
Anderson, M. E., Brown, J. H., and Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Bogdanov, K. Y., Vinogradova, T. M., and Lakatta, E. G. (2001). Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ. Res. 88, 1254–1258. doi: 10.1161/hh1201.092095
Bois, P., Bescond, J., Renaudon, B., and Lenfant, J. (1996). Mode of action of bradycardic agent, S 16257, on ionic currents of rabbit sinoatrial node cells. Br. J. Pharmacol. 118, 1051–1057. doi: 10.1111/j.1476-5381.1996.tb15505.x
Bozler, E. (1943). Tonus changes in cardiac muscle and their significance for the initiation of impulses. Am. J. Physiol. 139, 477–480.
DiFrancesco, D., and Noble, D. (2012). The funny current has a major pacemaking role in the sinus node. Heart Rhythm 9, 299–301. doi: 10.1016/j.hrthm.2011.09.021
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Fabiato, A. (1983). Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245, C1–C14.
Gao, Z., Singh, M. V., Hall, D. D., Koval, O. M., Luczak, E. D., Joiner, M. L., et al. (2011). Catecholamine-independent heart rate increases require Ca2+/calmodulin-dependent protein kinase II. Circ. Arrhythm. Electrophysiol. 4, 379–387. doi: 10.1161/CIRCEP.110.961771
Grandi, E., Puglisi, J. L., Wagner, S., Maier, L. S., Severi, S., and Bers, D. M. (2007). Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys. J. 93, 3835–3847. doi: 10.1529/biophysj.107.114868
Hagemann, D., Kuschel, M., Kuramochi, T., Zhu, W., Cheng, H., and Xiao, R. P. (2000). Frequency-encoding Thr17 phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. J. Biol. Chem. 275, 22532–22536. doi: 10.1074/jbc.C000253200
Hashambhoy, Y. L., Winslow, R. L., and Greenstein, J. L. (2009). CaMKII-induced shift in modal gating explains L-type Ca2+ current facilitation: a modeling study. Biophys. J. 96, 1770–1785. doi: 10.1016/j.bpj.2008.11.055
Hund, T. J., and Rudy, Y. (2004). Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation 110, 3168–3174. doi: 10.1161/01.CIR.0000147231.69595.D3
Huser, J., Blatter, L. A., and Lipsius, S. L. (2000). Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J. Physiol. 524(Pt 2), 415–422. doi: 10.1111/j.1469-7793.2000.00415.x
Kurata, Y., Hisatome, I., Imanishi, S., and Shibamoto, T. (2002). Dynamical description of sinoatrial node pacemaking: improved mathematical model for primary pacemaker cell. Am. J. Physiol. 283, H2074–H2101.
Lakatta, E. G., Maltsev, V. A., and Vinogradova, T. M. (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 106, 659–673. doi: 10.1161/CIRCRESAHA.109.206078
Luo, M., Guan, X., Luczak, E. D., Lang, D., Kutschke, W., Gao, Z., et al. (2013). Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Invest. 123, 1262–1274. doi: 10.1172/JCI65268
Maltsev, A. V., Maltsev, V. A., Mikheev, M., Maltseva, L. A., Sirenko, S. G., Lakatta, E. G., et al. (2011). Synchronization of stochastic Ca2+ release units creates a rhythmic Ca2+ clock in cardiac pacemaker cells. Biophys. J. 100, 271–283. doi: 10.1016/j.bpj.2010.11.081
Maltsev, A. V., Yaniv, Y., Stern, M. D., Lakatta, E. G., and Maltsev, V. A. (2013). RyR-NCX-SERCA local crosstalk ensures pacemaker cell function at rest and during the fight-or-flight reflex. Circ. Res. 113, e94–e100. doi: 10.1161/CIRCRESAHA.113.302465
Maltsev, V. A., and Lakatta, E. G. (2009). Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am. J. Physiol. Heart Circ. Physiol. 296, H594–H615. doi: 10.1152/ajpheart.01118.2008
Maltsev, V. A., and Lakatta, E. G. (2012). The funny current in the context of the coupled clock pacemaker cell system. Heart Rhythm 9, 302–307. doi: 10.1016/j.hrthm.2011.09.022
Maltsev, V. A., and Lakatta, E. G. (2013). Numerical models based on a minimal set of sarcolemmal electrogenic proteins and an intracellular Ca clock generate robust, flexible, and energy-efficient cardiac pacemaking. J. Mol. Cell Cardiol. 59, 181–195. doi: 10.1016/j.yjmcc.2013.03.004
Maltsev, V. A., Maltsev, A. V., Lakatta, E. G., and Stern, M. D. (2014). Spatial imperfection encodes functional perfection: success and failure of calcium release to propagate regulate pacemaker cell function. Biophys. J. 106, 319a (Abstract).
Maltsev, V. A., Vinogradova, T. M., and Lakatta, E. G. (2006). The emergence of a general theory of the initiation and strength of the heartbeat. J. Pharmacol. Sci. 100, 338–369. doi: 10.1254/jphs.CR0060018
Mattick, P., Parrington, J., Odia, E., Simpson, A., Collins, T., and Terrar, D. (2007). Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I f pacemaker current. J. Physiol. 582, 1195–1203. doi: 10.1113/jphysiol.2007.133439
Noble, D. (1960). Cardiac action and pacemaker potentials based on the Hodgkin–Huxley equations. Nature 188, 495–497. doi: 10.1038/188495b0
Rigg, L., Mattick, P. A., Heath, B. M., and Terrar, D. A. (2003). Modulation of the hyperpolarization-activated current (If) by calcium and calmodulin in the guinea-pig sino-atrial node. Cardiovasc. Res. 57, 497–504. doi: 10.1016/S0008-6363(02)00668-5
Saucerman, J. J., and Bers, D. M. (2008). Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys. J. 95, 4597–4612. doi: 10.1529/biophysj.108.128728
Sirenko, S., Maltsev, V. A., Maltseva, L. A., Yang, D., Lukyanenko, Y., Vinogradova, T. M., et al. (2014). Sarcoplasmic reticulum Ca cycling protein phosphorylation in a physiologic Ca milieu unleashes a high-power, rhythmic Ca clock in ventricular myocytes: relevance to arrhythmias and bio-pacemaker design. J. Mol. Cell Cardiol. 66C, 106–115. doi: 10.1016/j.yjmcc.2013.11.011
Stern, M. D. (1992). Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63, 497–517. doi: 10.1016/S0006-3495(92)81615-6
Stern, M. D., Maltseva, L. A., Juhaszova, M., Sollott, S. J., Lakatta, E. G., and Maltsev, V. A. (2014). Hierarchical clustering of ryanodine receptors enables emergence of a Calcium Clock in SA node cells. J. General Physiol. (in press). doi: 10.1085/jgp.201311123
Swaminathan, P. D., Purohit, A., Hund, T. J., and Anderson, M. E. (2012). Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ. Res. 110, 1661–1677. doi: 10.1161/CIRCRESAHA.111.243956
Swaminathan, P. D., Purohit, A., Soni, S., Voigt, N., Singh, M. V., Glukhov, A. V., et al. (2011). Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Invest. 121, 3277–3288. doi: 10.1172/JCI57833
Vinogradova, T., Sirenko, S., Li, Y., Spurgeon, H. A., and Lakatta, E. G. (2011). CaMKII activity modulates basal sarcoplasmic reticulum Ca2+ cycling to drive normal automaticity of sinoatrial node cells. Circulation 124, A17100.
Vinogradova, T. M., Lyashkov, A. E., Zhu, W., Ruknudin, A. M., Sirenko, S., Yang, D., et al. (2006). High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 98, 505–514. doi: 10.1161/01.RES.0000204575.94040.d1
Vinogradova, T. M., Zhou, Y. Y., Bogdanov, K. Y., Yang, D., Kuschel, M., Cheng, H., et al. (2000). Sinoatrial node pacemaker activity requires Ca2+/calmodulin-dependent protein kinase II activation. Circ. Res. 87, 760–767. doi: 10.1161/01.RES.87.9.760
Wu, Y., and Anderson, M. E. (2014). CaMKII in sinoatrial node physiology and dysfuction. Front. Pharmacol. 5:48. doi: 10.3389/fphar.2014.00048
Wu, Y., Gao, Z., Chen, B., Koval, O. M., Singh, M. V., Guan, X., et al. (2009). Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc. Natl. Acad. Sci. U.S.A. 106, 5972–5977. doi: 10.1073/pnas.0806422106
Yaniv, Y., Juhaszova, M., Lyashkov, A. E., Spurgeon, H. A., Sollott, S. J., and Lakatta, E. G. (2011). Ca2+-regulated-cAMP/PKA signaling in cardiac pacemaker cells links ATP supply to demand. J. Mol. Cell Cardiol. 51, 740–748. doi: 10.1016/j.yjmcc.2011.07.018
Yaniv, Y., and Lakatta, E. G. (2013). Pacemaker gene mutations, bradycardia, arrhythmias and the coupled clock theory. J. Cardiovasc. Electrophysiol. 24, E28–E29. doi: 10.1111/jce.12236
Yaniv, Y., Maltsev, V. A., Ziman, B. D., Spurgeon, H. A., and Lakatta, E. G. (2012a). The “Funny” current (I f) inhibition by ivabradine at membrane potentials encompassing spontaneous depolarization in pacemaker cells. Molecules 17, 8241–8254. doi: 10.3390/molecules17078241
Yaniv, Y., Spurgeon, H. A., Lyashkov, A. E., Yang, D., Ziman, B. D., Maltsev, V. A., et al. (2012b). Crosstalk between mitochondrial and sarcoplasmic reticulum Ca2+ cycling modulates cardiac pacemaker cell automaticity. PLoS ONE 7:e37582. doi: 10.1371/journal.pone.0037582
Yaniv, Y., Sirenko, S., Ziman, B. D., Spurgeon, H. A., Maltsev, V. A., and Lakatta, E. G. (2013a). New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: bradycardic effects of ivabradine are linked to suppression of intracellular Ca cycling. J. Mol. Cell Cardiol. 62C, 80–89. doi: 10.1016/j.yjmcc.2013.04.026
Yaniv, Y., Spurgeon, H. A., Ziman, B. D., and Lakatta, E. G. (2013b). Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity and sinoatrial nodal pacemaker cell energetics. PLoS ONE 8:e57079. doi: 10.1371/journal.pone.0057079
Yaniv, Y., Spurgeon, H. A., Ziman, B. D., Lyashkov, A. E., and Lakatta, E. G. (2013c). Mechanisms that match ATP supply to demand in cardiac pacemaker cells during high ATP demand. Am. J. Physiol. Heart Circ. Physiol. 304, H1428–H1438. doi: 10.1152/ajpheart.00969.2012
Yaniv, Y., Stern, M. D., Lakatta, E. G., and Maltsev, V. A. (2013d). Mechanisms of beat-to-beat regulation of cardiac pacemaker cell function by Ca2+ cycling dynamics. Biophys. J. 105, 1551–1561. doi: 10.1016/j.bpj.2013.08.024
Younes, A., Lyashkov, A. E., Graham, D., Sheydina, A., Volkova, M. V., Mitsak, M., et al. (2008). Ca2+-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 283, 14461–14468. doi: 10.1074/jbc.M707540200
Keywords: cardiac pacemaker, sinoatrial node, numerical modeling, calcium, CaMKII, ion channels
Citation: Yaniv Y and Maltsev VA (2014) Numerical modeling calcium and CaMKII effects in the SA node. Front. Pharmacol. 5:58. doi: 10.3389/fphar.2014.00058
Received: 17 January 2014; Accepted: 16 March 2014;
Published online: 01 April 2014.
Edited by:
Eleonora Grandi, University of California, Davis, USAReviewed by:
Eric A. Sobie, Mount Sinai School of Medicine, USAYuanfang Xie, University of California, Davis, USA
Copyright © 2014 Yaniv and Maltsev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor A. Maltsev, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging – National Institutes of Health, 251 Bayview Boulevard, Baltimore, MD 21224, USA e-mail:bWFsdHNldnZpQGdyYy5uaWEubmloLmdvdg==