 Laura Silvestri
Laura Silvestri Antonella Nai
Antonella Nai Clara Camaschella
Clara Camaschella- Division of Genetics and Cell Biology, San Raffaele Scientific Institute, Università Vita-Salute San Raffaele, Milan, Italy
Transferrin receptor 2 (TFR2), a protein homologous to the cell iron importer TFR1, is expressed in the liver and erythroid cells and is reported to bind diferric transferrin, although at lower affinity than TFR1. TFR2 gene is mutated in type 3 hemochromatosis, a disorder characterized by iron overload and inability to upregulate hepcidin in response to iron. Liver TFR2 is considered a sensor of diferric transferrin, possibly in a complex with hemochromatosis protein. In erythroid cells TFR2 is a partner of erythropoietin receptor (EPOR) and stabilizes the receptor on the cell surface. However, Tfr2 null mice as well as TFR2 hemochromatosis patients do not show defective erythropoiesis and tolerate repeated phlebotomy. The iron deficient Tfr2-Tmprss6 double knock out mice have higher red cells count and more severe microcytosis than the liver-specific Tfr2 and Tmprss6 double knock out mice. TFR2 in the bone marrow might be a sensor of iron deficiency that protects against excessive microcytosis in a way that involves EPOR, although the mechanisms remain to be worked out.
The Second Transferrin Receptor
Transferrin receptor 2 (TFR2) is a type II transmembrane glycoprotein, member of the TFR family and homologous to TFR1, which provides iron to the cell by internalization of the transferrin-iron complex through receptor-mediated endocytosis. TFR2 was cloned during a project aimed at isolating genes encoding new transcription factors (Kawabata et al., 1999). The TFR2 gene comprises 18 exons and maps to chromosome 7q22 in close proximity to the EPO gene. Two TFR2 isoforms have been described: the alpha isoform, corresponding to all 18 exons, encodes a protein of about 89 kDa in its unglycosylated form. The beta form, which results from an alternative splicing, lacks exons 1–3, and has 142 additional nucleotides in its first exon (exon 4 of the alpha form). The resulting protein lacks the cytoplasmic and the transmembrane domain (Kawabata et al., 1999) and its function remains unknown. The alpha protein encompasses 801 amino acids and, as TFR1, has a short cytoplasmic tail that contains a consensus sequence for endocytosis, a transmembrane domain and a large extracellular region that comprises a protease-associated domain and two RGD motifs (only one is present in TFR1), that bind diferric transferrin. TFR2 differs from TFR1 for several aspects. First, TFR1 is ubiquitously expressed, whereas the expression of TFR2 is restricted to the hepatocytes and erythroid precursors. Second, TFR1 is post-transcriptionally regulated by iron through the iron regulatory proteins–iron responsive elements (IRP–IRE) interaction, while TFR2 is not regulated by IRPs. TFR2 5′ and 3′ untranslated regions do not contain IRE elements (Fleming et al., 2000); rather TFR2 promoter shows GATA-1 as well as c-EBP-alpha consensus sequences. Furthermore, although in transfected cells TFR2 may uptake iron loaded transferrin (holo-TF) in transfected cells, in vivo it does not rescue the embryo-lethality of Tfr1-/- mice (Levy et al., 1999), suggesting a function distinct from TFR1 and unrelated to iron transport. Moreover, the affinity of TFR2 for holo-TF is significantly lower than that of TFR1 (Kd 30 nM vs. 1 nM, respectively; Kawabata et al., 2000; West et al., 2000).
It has been reported that both TFRs bind hemochromatosis protein (HFE) in vitro. However, crystallographic studies have shown that HFE binds TFR1 (Bennett et al., 2000) at the same consensus sequences of diferric transferrin, implying a competition between the two ligands. On the contrary, based on in vitro data, binding of HFE to TFR2 and holo-Tf would occur simultaneously at two different TFR2 sequence motifs (Gao et al., 2009).
In the liver Tfr2 expression increases during mouse development, at variance with Tfr1, and in adult liver Tfr2 is much more expressed than Tfr1 (Kawabata et al., 2001). Our knowledge of the TFR2 hepatic function is still incomplete. In hepatoma cell lines TFR2 is stabilized on cell surface by the addition of holo-transferrin to the culture media, an effect due to the increased protein half-life (Enns, 2001). The divergent iron-mediated regulation of the two TFRs is confirmed also in vivo: while in iron loaded mice Tfr1 is downregulated by the loss of IRP-mediated mRNA stabilization, Tfr2 protein level is increased. In agreement with a ligand-mediated stabilization, levels of Tfr2 protein are decreased in the liver of hypotransferrinemic (hpx) mice (Robb and Wessling-Resnick, 2004). Thus the major regulation of TFR2 occurs at the protein rather than at RNA level.
In cell culture models, TFR2 localizes in caveolar microdomains (Calzolari et al., 2006), membrane structures involved in the recruitment of receptors that can be activated by ligand binding (Simons and Toomre, 2000). In the absence of holo-transferrin, both TFR1 and TFR2 are internalized by clathrin-mediated endocytosis (Chen et al., 2009), whereas in the presence of the ligand only TFR2, and not TFR1, activates ERK1/2 and p38 MAPK. This has been observed in hepatoma derived and in erythroid cells, supporting the hypothesis that TFR2 may function as a signaling receptor (Calzolari et al., 2006; Poli et al., 2010).
The stabilization of TFR2 by holo-transferrin and its ability to bind HFE led to the current model in which liver TFR2, in conjunction with HFE, represents a sensor of circulating iron and activates hepcidin in response to elevated transferrin saturation (Goswami and Andrews, 2006). In addition it has been shown that the TFR2-HFE interaction on the hepatocyte surface occurs within a multiprotein complex, that in vitro includes also the BMP-coreceptor hemojuvelin (D’Alessio et al., 2012). If this complex activates the intracellular signaling to upregulate hepcidin expression in vivo remains to be demonstrated. In addition, the binding of TFR2 to HFE has recently been questioned (Rishi et al., 2013) and some evidences are in favor of a distinct function for the two HFEs. Mice with inactivation of both Tfr2 and Hfe have a more severe phenotype compared to single mutant animals (Wallace et al., 2009). This occurs also in humans: patients with mutation in TFR2 and HFE were reported to develop a severe form of juvenile-like hemochromatosis (Pietrangelo et al., 2005). Moreover, patients with TFR2 mutations do not upregulate hepcidin upon oral iron administration, whereas the iron response is partially preserved in HFE patients (Girelli et al., 2011), thus strengthening the distinct and non-overlapping role of HFE and TFR2.
TFR2: The Gene of Hemochromatosis Type 3
In humans inactivating mutations of TFR2 lead to hemochromatosis type 3 (Camaschella et al., 2000), a rare recessive disorder characterized by iron overload, low hepcidin levels (Nemeth et al., 2005) and inability to properly regulate hepcidin after an oral iron challenge (Girelli et al., 2011). The disorder is quite rare among Caucasians and occasionally reported in Japanese, with single families identified in France, Portugal Spain and Taiwan. Currently, less than 30 pathogenic mutations have been described (Camaschella and Roetto, 2011). They are all rare, often private. Some insertions cause frameshift and premature stop codon, others are nonsense and small deletions. All the mutations are loss of function; missense mutations prevalently affect the protein C-terminus, especially the peptidase-like and the dimerization domains, suggesting that these regions have important functional roles (Camaschella and Roetto, 2011). The affected residues, usually highly conserved, might be essential for the proper folding and protein localization, or relevant for interaction with other proteins or for the regulatory function of TFR2.
The type of iron overload caused by mutations in TFR2 differs from the classic type 1 HFE-hemochromatosis, because of an earlier onset and more severe presentation (Camaschella, 2005). A study in patients affected by type 1 (HFE-related) or type 3 (TFR2-related) hemochromatosis showed a different role for TFR2 and HFE in hepcidin activation in response to a single oral iron challenge able to increase transferrin saturation: HFE patients showed a blunted hepcidin response, whereas TFR2 patients showed no response (Girelli et al., 2011). A similar difference in the hepcidin response after an acute iron loading has been observed in Tfr2Y245X/Y245X and Hfe-/- mice (Corradini et al., 2011; Ramos et al., 2011). These results led to the conclusion that TFR2 is important to up-regulate hepcidin in response to transferrin saturation.
TFR2 in Erythroid Cells
In favor of the relevance of TFR2 for the erythroid differentiation are two genetic observations: first, the close proximity of TFR2 and EPO genes on chromosome 7q22 that may suggest a common regulation. Second, the results of different genome-wide association studies: TFR2 single nucleotide polymorphisms (SNPs) have been identified associated with erythroid quantitative traits, such as red cell numbers, indexes and hematocrit (Ganesh et al., 2009; Soranzo et al., 2009; Ding et al., 2012). Although this observation could be due to an indirect effect mediated by serum iron levels, that were not measured in the original studies (Ganesh et al., 2009; Soranzo et al., 2009; Ding et al., 2012), a direct effect of TFR2 on erythropoiesis cannot be excluded.
More recently it was shown that in erythroid cells TFR2 is a partner of erythropoietin receptor (EPOR) that stabilizes the receptor on cell surface. TFR2 is co-expressed with EPOR during erythroid differentiation and its maximal expression precedes that of TFR1 (Kawabata et al., 2001). TFR2 protein, that associates with EPOR in the endoplasmic reticulum, is needed for the efficient transport of the receptor to the cell membrane (Forejtnikova et al., 2010). The interaction facilitates the stabilization of EPOR, likely contributing to EPO sensitivity and erythroid cell differentiation, both in vitro and in vivo. However the interaction does not seem to influence EPO binding to EPOR. In addition direct binding of TFR2 to EPO was excluded (Forejtnikova et al., 2010). Interestingly TFR2 silencing in human erythroid precursors delays their terminal differentiation in vitro (Forejtnikova et al., 2010). However, which signaling pathway is activated by TFR2 is still unclear. Moreover, although TFR2 is required for efficient erythropoiesis, Tfr2 null mice as well as TFR2 hemochromatosis patients do not show defective erythropoiesis and patients tolerate repeated courses of phlebotomy without developing anemia.
Animal Models of Tfr2 Inactivation Develop Iron Overload
The first animal model of hemochromatosis type 3 was generated by targeted mutagenesis, introducing a premature stop codon (Y245X; Fleming et al., 2002) in the murine Tfr2 coding sequence. This mutation is orthologous to the mutation (Y250X) originally detected in humans (Camaschella et al., 2000). Young (4 week-old) homozygous Y245X mutant mice had high liver iron concentration, even if maintained on a standard diet, in agreement with the observation of early iron overload in patients. The histological distribution of iron recapitulates features of hemochromatosis, with the typical liver periportal accumulation and low spleen iron stores. As in humans, heterozygous animals were normal. Later on, several murine models of Tfr2 inactivation were developed (Fleming et al., 2002; Wallace et al., 2007), including, among others, Tfr2 total (Tfr2-/-) and liver-specific (Tfr2 LCKO) knock-out (Wallace et al., 2007; Roetto et al., 2010) and Tfr2-Hfe double knock-out. All these models are characterized by low hepcidin expression and liver iron overload of variable severity (Table 1). However, when generated in the same genetic background, Tfr2 total knock-out was shown to have iron overload more severe than Hfe-/- although less severe than Hfe/Tfr2 double knock out. These observations are in agreement with the suggested distinct function of the two proteins.
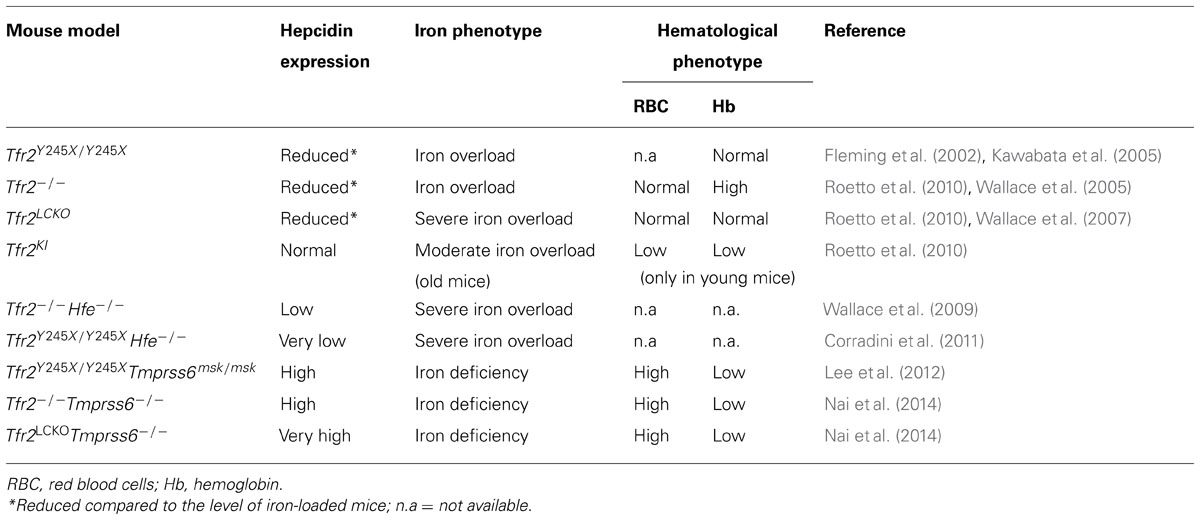
TABLE 1. Hepcidin levels, iron and hematological phenotype in the available murine models of Tfr2 inactivation.
A last model was generated with the M167K substitution in the Tfr2 protein (Roetto et al., 2010): this mutation destroys the methionine, putative start codon of the beta-isoform of the protein. β-Tfr2 is mostly expressed in the spleen (Kawabata et al., 1999; Roetto et al., 2010). Interestingly, the knock-in model Tfr2KI, specifically lacking the beta-isoform, is characterized by normal transferrin saturation, liver iron concentration, hepcidin and Bmp6 levels but show a transient anemia at young age. In addition adult animals accumulate iron in the spleen due to strong reduction of ferroportin mRNA, thus suggesting a possible regulatory effect of β-Tfr2 on splenic ferroportin expression.
Animal Models of Tfr2 Inactivation in Iron Deficiency
Tfr2-/- mice have slightly less severe iron overload than liver-specific (Tfr2LCKO) knock-out (Wallace et al., 2007; Roetto et al., 2010), slightly higher Hb levels (Roetto et al., 2010; Nai et al., 2014) and moderate macrocytosis. The Tmprss6-/- mice, which have a deletion of the hepcidin inhibitor, the serine protease Tmprss6, is a well established model of iron deficiency anemia with high hepcidin (Du et al., 2008; Folgueras et al., 2008). Tmprss6-/-Tfr2 -/- double knock out animals develop iron deficiency with high hepcidin, a phenotype similar to Tmprss6-/- mice (Lee et al., 2012; Nai et al., 2014) and to Tmprss6-/-Hfe-/- animals (Finberg et al., 2011; Lee et al., 2012). In a single study some degree of erythrocytosis were observed both in Tfr2-/- and in Hfe-/- knock-out with deletion of Tmprss6 (Lee et al., 2012), although results for Hfe-/-Tmprss6-/- are not unequivocal (Finberg et al., 2011).
Deleting Tmprss6 in the two hemochromatosis type 3 models, Tfr2-/- and Tfr2LCKO mice, revealed similarities but also differences in the hematological phenotype of the resulting double knock-out animals (Nai et al., 2014). Both models have the same degree of anemia, low transferrin saturation and low liver iron content (LIC), a phenotype quite similar to that of the iron deficient Tmprss6-/- (Table 1).
The modification of the phenotype of Tfr2-/- mice with deletion of Tmprss6 has important implications. First it indicates that hepatic TFR2 is genetically upstream TMPRSS6 in the BMP-SMAD signaling pathway, as previously shown for HFE (Finberg et al., 2011; Lee et al., 2012). Second, it excludes that TFR2 is a substrate of TMPRSS6, as previously observed in our in vitro studies (Pagani, unpublished observation, 2014). Further, it suggests that Tmprss6 is likely hyperactive in Tfr2-/- mice with iron overload and thus its inhibition might be effective in up-regulating hepcidin production and reducing iron overload, as shown by the use of small interference RNA (siRNA) or allele specific oligonucleotide (ASO) against Tmprss6 (Guo et al., 2013; Schmidt et al., 2013) in Hfe-/- animals.
We observed that the phenotype of Tfr2-Tmprss6 double knock-out mice is not exactly the same of the double knock-out for Tfr2LCKO and Tmprss6 or of Tmprss6-/- knock out (Table 1). An increased number of red cells are observed only in Tfr2-Tmprss6 double knock-out mice. Also, while hepcidin levels are increased in all models as compared with wild-type animals, they are less elevated in Tfr2-/-Tmprss6-/- mice compared with the other models. It seems that some inhibitory signal lowers hepcidin in Tfr2-/-Tmprss6-/- mice. In principle this may derive from the increased red cell production, exclusively present in Tfr2-/-Tmprss6-/- mice. It is of interest that the observed erythrocytosis is not due to enhanced erythropoietin (Epo) stimulation of erythropoiesis, since Epo levels are similar and consistent with similar degrees of anemia in all models. They are even decreased in Tmprss6-/-Tfr2-/- (Nai et al., 2014). We speculate that this positive modulation of erythropoiesis might result from the lack of Tfr2 expression in the erythroid compartment (Figure 1).
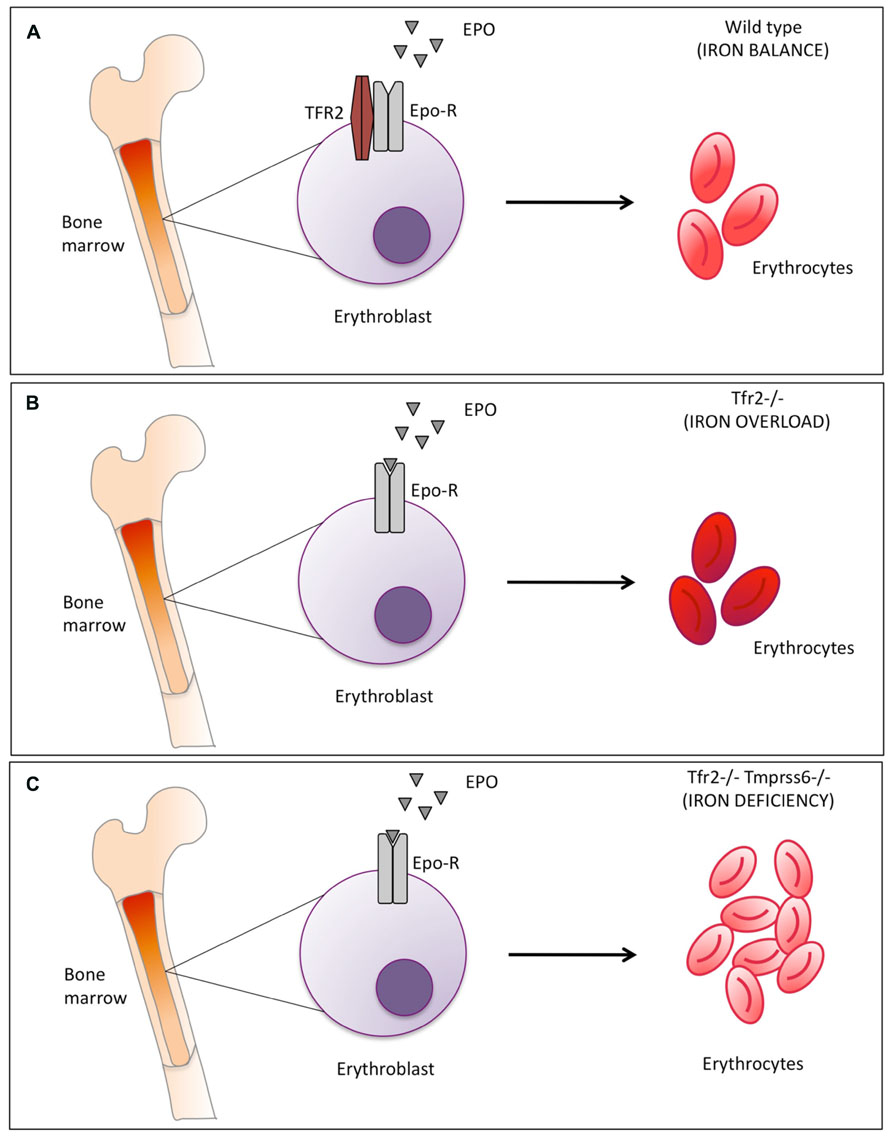
FIGURE 1. Schematic representation of Tfr2-mediated erythropoiesis modulation. (A) in normal conditions Tfr2 and erythropoietin receptor (EpoR) interact and localize on the cell surface and control red blood cells production (B) Tfr2 inactivation in mice causes iron overload: mean corpuscular hemoglobin (MCH) is enhanced (indicated by the darker red color of the erythrocytes) due to the increased iron availability. (C) Inactivation of Tmprss6 in Tfr2 KO mice increases the number of microcytic erythrocytes, as indicated by the light red color of the erythrocytes. Epo-EpoR interaction occurs in all conditions but is increased in the absence of Tfr2 (panel B and C vs. A).
Recently an erythroid function for Tfr2 was independently reported by Wallace et al. (2013) who noticed that the triple knock-out (Tfr2-/-, Hfe-/-, Tmprss6-/-) mice have more severe iron deficiency than Tmprss6-/- mice with deletion of either Hfe or Tfr2. However, the mechanism underlining this difference remains to be worked out.
From all the data available we concluded that Tfr2 in the erythroid compartment might serve to block excessive erythropoietic expansion. If this occurs in normal conditions (Figure 1 panel A) is difficult to verify because Tfr2 deletion leads to iron overload. In iron overload indeed the Tfr2 erythroid function is likely masked by the excessive iron availability that increases Hb and also Hb content per single cell (Figure 1 panel B vs. A; Roetto et al., 2010; (Nai et al., 2014). Tfr2 function becomes more evident in iron deficiency, as exemplified by Tmprss6-/-Tfr2-/- double knock out mice (Figure 1 panel C).
Conclusion
From its identification and cloning more than 10 years ago (Kawabata et al., 1999), the correct function of TFR2 in iron metabolism has remained mysterious. Several different roles have been proposed for this receptor: originally published as a second iron importer, after the identification of TFR2 mutations in hemochromatosis patients its proposed function became that of potential sensor of circulating iron-loaded transferrin and then of hepcidin (co)-activator. Although counteracting iron excess in the circulation remains its major function in the liver, an as well important erythropoietic function is emerging from our and other studies From the few available data it seems that TFR2 might serve as a brake to avoid iron consumption in excessive erythrocyte production in conditions of iron deficiency, likely within the perspective of global body iron economy. A prevalent role of erythroid TFR2 in iron deficiency might explain why this role is not evident in mice nor in patients with type 3 hemochromatosis, who are iron loaded and never experience iron deficiency. Further studies are needed to clarify the molecular mechanisms that mediate the TFR2 function in iron deficiency. However from now on the erythropoiesis status should be considered when interpreting the effect of TFR2 in iron metabolism and homeostasis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partially supported by MIUR PRIN 2010–2011 (Rome, Italy) and the Italian Ministry of Health (Grant RF-2010-2312048) to Clara Camaschella.
References
Bennett, M. J., Lebron, J. A., and Bjorkman, P. J. (2000). Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature 403, 46–53. doi: 10.1038/47417
Calzolari, A., Raggi, C., Deaglio, S., Sposi, N. M., Stafsnes, M., Fecchi, K., et al. (2006). TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J. Cell Sci. 119, 4486–4498. doi: 10.1242/jcs.03228
Camaschella, C. (2005). Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood 106, 3710–3717. doi: 10.1182/blood-2005-05-1857
Camaschella, C., and Roetto, A. (2011). “TFR2-related hereditary hemochromatosis,” in GeneReviews, eds R. A. Pagon, M. P. Adam, T. D. Bird, C. R. Dolan, C. T. Fong, and K. Stephens (Seattle, WA).
Camaschella, C., Roetto, A., Cali, A., De Gobbi, M., Garozzo, G., Carella, M., et al. (2000). The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25, 14–15. doi: 10.1038/75534
Chen, J., Wang, J., Meyers, K. R., and Enns, C. A. (2009). Transferrin-directed internalization and cycling of transferrin receptor 2. Traffic 10, 1488–1501. doi: 10.1111/j.1600-0854.2009.00961.x
Corradini, E., Rozier, M., Meynard, D., Odhiambo, A., Lin, H. Y., Feng, Q., et al. (2011). Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology 141, 1907–1914. doi: 10.1053/j.gastro.2011.06.077
D’Alessio, F., Hentze, M. W., and Muckenthaler, M. U. (2012). The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 57, 1052–1060. doi: 10.1016/j.jhep.2012.06.015
Ding, K., Shameer, K., Jouni, H., Masys, D. R., Jarvik, G. P., Kho, A. N., et al. (2012). Genetic Loci implicated in erythroid differentiation and cell cycle regulation are associated with red blood cell traits. Mayo Clin. Proc. 87, 461–474. doi: 10.1016/j.mayocp.2012.01.016
Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., et al. (2008). The serine protease TMPRSS6 is required to sense iron deficiency. Science 320, 1088–1092. doi: 10.1126/science.1157121
Enns, C. A. (2001). Pumping iron: the strange partnership of the hemochromatosis protein, a class I MHC homolog, with the transferrin receptor. Traffic 2, 167–174. doi: 10.1034/j.1600-0854.2001.020303.x
Finberg, K. E., Whittlesey, R. L., and Andrews, N. C. (2011). Tmprss6 is a genetic modifier of the Hfe-hemochromatosis phenotype in mice. Blood 117, 4590–4599. doi: 10.1182/blood-2010-10-315507
Fleming, R. E., Ahmann, J. R., Migas, M. C., Waheed, A., Koeffler, H. P., Kawabata, H., et al. (2002). Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. U.S.A. 99, 10653–10658. doi: 10.1073/pnas.162360699
Fleming, R. E., Migas, M. C., Holden, C. C., Waheed, A., Britton, R. S., Tomatsu, S., et al. (2000). Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc. Natl. Acad. Sci. U.S.A. 97, 2214–2219. doi: 10.1073/pnas.040548097
Folgueras, A. R., de Lara, F. M., Pendas, A. M., Garabaya, C., Rodriguez, F., Astudillo, A., et al. (2008). Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 112, 2539–2545. doi: 10.1182/blood-2008-04-149773
Forejtnikova, H., Vieillevoye, M., Zermati, Y., Lambert, M., Pellegrino, R. M., Guihard, S., et al. (2010). Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood 116, 5357–5367. doi: 10.1182/blood-2010-04-281360
Ganesh, S. K., Zakai, N. A., van Rooij, F. J., Soranzo, N., Smith, A. V., Nalls, M. A., et al. (2009). Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat. Genet. 41, 1191–1198. doi: 10.1038/ng.466
Gao, J., Chen, J., Kramer, M., Tsukamoto, H., Zhang, A. S., and Enns, C. A. (2009). Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 9, 217–227. doi: 10.1016/j.cmet.2009.01.010
Girelli, D., Trombini, P., Busti, F., Campostrini, N., Sandri, M., Pelucchi, S., et al. (2011). A time course of hepcidin response to iron challenge in patients with HFE and TFR2 hemochromatosis. Haematologica 96, 500–506. doi: 10.3324/haematol.2010.033449
Goswami, T., and Andrews, N. C. (2006). Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem. 281, 28494–28498. doi: 10.1074/jbc.C600197200
Guo, S., Casu, C., Gardenghi, S., Booten, S., Aghajan, M., Peralta, R., et al. (2013). Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J. Clin. Invest. 123, 1531–1541. doi: 10.1172/JCI66969
Kawabata, H., Fleming, R. E., Gui, D., Moon, S. Y., Saitoh, T., O’Kelly, J., et al. (2005). Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 105, 376–381. doi: 10.1182/blood-2004-04-1416
Kawabata, H., Germain, R. S., Ikezoe, T., Tong, X., Green, E. M., Gombart, A. F., et al. (2001). Regulation of expression of murine transferrin receptor 2. Blood 98, 1949–1954. doi: 10.1182/blood.V98.6.1949
Kawabata, H., Germain, R. S., Vuong, P. T., Nakamaki, T., Said, J. W., and Koeffler, H. P. (2000). Transferrin receptor 2-alpha supports cell growth both in iron-chelated cultured cells and in vivo. J. Biol. Chem. 275, 16618–16625. doi: 10.1074/jbc.M908846199
Kawabata, H., Yang, R., Hirama, T., Vuong, P. T., Kawano, S., Gombart, A. F., et al. (1999). Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem. 274, 20826–20832. doi: 10.1074/jbc.274.30.20826
Lee, P., Hsu, M. H., Welser-Alves, J., and Peng, H. (2012). Severe microcytic anemia but increased erythropoiesis in mice lacking Hfe or Tfr2 and Tmprss6. Blood Cells Mol. Dis. 48, 173–178. doi: 10.1016/j.bcmd.2011.12.005
Levy, J. E., Jin, O., Fujiwara, Y., Kuo, F., and Andrews, N. C. (1999). Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 21, 396–399. doi: 10.1038/7727
Nai, A., Pellegrino, R. M., Rausa, M., Pagani, A., Boero, M., Silvestri, L., et al. (2014). The erythroid function of Transferrin Receptor 2 revealed by Tmprss6 inactivation in different models of Transferrin Receptor 2 knock out mice. Haematologica doi: 10.3324/haematol.2013.103143 [Epub ahead of print].
Nemeth, E., Roetto, A., Garozzo, G., Ganz, T., and Camaschella, C. (2005). Hepcidin is decreased in TFR2 hemochromatosis. Blood 105, 1803–1806. doi: 10.1182/blood-2004-08-3042
Pietrangelo, A., Caleffi, A., Henrion, J., Ferrara, F., Corradini, E., Kulaksiz, H., et al. (2005). Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology 128, 470–479. doi: 10.1053/j.gastro.2004.11.057
Poli, M., Luscieti, S., Gandini, V., Maccarinelli, F., Finazzi, D., Silvestri, L., et al. (2010). Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling. Implications for transferrin-dependent hepcidin regulation. Haematologica 95, 1832–1840. doi: 10.3324/haematol.2010.027003
Ramos, E., Kautz, L., Rodriguez, R., Hansen, M., Gabayan, V., Ginzburg, Y., et al. (2011). Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice. Hepatology 53, 1333–1341. doi: 10.1002/hep.24178
Rishi, G., Crampton, E. M., Wallace, D. F., and Subramaniam, V. N. (2013). In situ proximity ligation assays indicate that hemochromatosis proteins Hfe and transferrin receptor 2 (Tfr2) do not interact. PLoS ONE 8:e77267. doi: 10.1371/journal.pone.0077267
Robb, A., and Wessling-Resnick, M. (2004). Regulation of transferrin receptor 2 protein levels by transferrin. Blood 104, 4294–4299. doi: 10.1182/blood-2004-06-2481
Roetto, A., Di Cunto, F., Pellegrino, R. M., Hirsch, E., Azzolino, O., Bondi, A., et al. (2010). Comparison of 3 Tfr2-deficient murine models suggests distinct functions for Tfr2-alpha and Tfr2-beta isoforms in different tissues. Blood 115, 3382–3389. doi: 10.1182/blood-2009-09-240960
Schmidt, P. J., Toudjarska, I., Sendamarai, A. K., Racie, T., Milstein, S., Bettencourt, B. R., et al. (2013). An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 121, 1200–1208. doi: 10.1182/blood-2012-09-453977
Simons, K., and Toomre, D. (2000). Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39. doi: 10.1038/35036052
Soranzo, N., Spector, T. D., Mangino, M., Kuhnel, B., Rendon, A., Teumer, A., et al. (2009). A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. 41, 1182–1190. doi: 10.1038/ng.467
Wallace, D. F., Summerville, L., Lusby, P. E., and Subramaniam, V. N. (2005). First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut 54, 980–986. doi: 10.1136/gut.2004.062018
Wallace, D. F., Summerville, L., and Subramaniam, V. N. (2007). Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 132, 301–310. doi: 10.1053/j.gastro.2006.11.028
Wallace, D. F., Trinder, D., and Subramaniam, V. N. (2009). Hepcidin regulation by HFE and TFR2: is it enough to give a hepatocyte a complex? Gastroenterology 137, 1173–1175; discussion 1175. doi: 10.1053/j.gastro.2009.07.009
Wallace, D., McDonald, C. J., Secondes, E. S., Ostini, L., Rishi, G., Hooper, J. D., et al. (2013). “An essential role for transferrin receptor 2 in erythropoiesis during iron restriction,” in Proceedings of the 2013 ASH Annual Meeting and Exposition, New Orleans, LA.
West, A. P. Jr., Bennett, M. J., Sellers, V. M., Andrews, N. C., Enns, C. A., and Bjorkman, P. J. (2000). Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J. Biol. Chem. 275, 38135–38138. doi: 10.1074/jbc.C000664200
Keywords: iron metabolism, transferrin, transferrin receptors, hepcidin, iron deficiency, hemochromatosis
Citation: Silvestri L, Nai A, Pagani A and Camaschella C (2014) The extrahepatic role of TFR2 in iron homeostasis. Front. Pharmacol. 5:93. doi: 10.3389/fphar.2014.00093
Received: 13 March 2014; Accepted: 14 April 2014;
Published online: 07 May 2014.
Edited by:
Paolo Arosio, University of Brescia, ItalyReviewed by:
Surjit Kaila Singh Srai, University College London, UKFrancesca Vinchi, Heidelberg University, Germany
Copyright © 2014 Silvestri, Nai, Pagani and Camaschella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clara Camaschella and Laura Silvestri, Division of Genetics and Cell Biology, San Raffaele Scientific Institute, Università Vita-Salute San Raffaele, Via Olgettina 60, 20132 Milan, Italy e-mail:Y2FtYXNjaGVsbGEuY2xhcmFAaHNyLml0;c2lsdmVzdHJpLmxhdXJhQGhzci5pdA==