 Tereza Cindrova-Davies
Tereza Cindrova-Davies- Centre for Trophoblast Research and Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, UK
Preeclampsia is a complex multifactorial disease. Placental oxidative stress, a result of deficient spiral artery remodeling, plays an important role in the pathophysiology of preeclampsia. Antiangiogenic factors secreted from malperfused placenta are instrumental in mediating maternal endothelial dysfunction and consequent symptoms of preeclampsia; the mechanism is likely to involve increased ET-1 secretion and reduced NO bioavailability. Therapeutic interventions so far remain only experimental and there is no established remedy for the treatment of preeclampsia. This review concentrates on the evidence for the therapeutic potential of antioxidants, ER chaperones, NO and H2S donors, and statins. These compounds display pleitropic antioxidant, anti-inflammatory, and pro-angiogenic effects in animal and in vitro studies. Although clinical trials on the use of antioxidant vitamins in pregnancy proved largely unsuccessful, the scope for their use still exists given the beneficial cardioprotective effects of antioxidant-rich Mediterranean diet, periconceptual vitamin use and the synergistic effect of vitamin C and L-arginine. Encouraging clinical evidence exists for the use of NO donors, and a clinical trial is underway testing the effect of statins in treatment of preeclampsia. H2S recently emerged as a novel therapeutic agent for cardiovascular disease, and its beneficial effects were also tested in animal models of preeclampsia. It is risky to prescribe any medication to pregnant women on a large scale, and any future therapeutic intervention has to be well tested and safe. Many of the compounds discussed could be potential candidates.
The Syndrome of Preeclampsia
Preeclampsia affects 3–5% of all pregnancies. The traditional definition of preeclampsia as de novo onset of hypertension and proteinuria after 20 weeks of gestation has been recently modified by the American College of Obstetricians and Gynecologists in recognition of the syndromic nature of preeclampsia. In the absence of proteinuria, preeclampsia is diagnosed as hypertension in association with thrombocytopenia, impaired liver function, new development of renal insufficiency, pulmonary edema, or new-onset cerebral or visual disturbances (American College of Obstetricians Gynecologists [ACOG], 2013). Hypertension, proteinuria, edema, and other systemic manifestations of the syndrome of preeclampsia are the direct consequences of maternal endothelial dysfunction. The syndrome is a major cause of maternal and fetal morbidity and mortality, and results in mild to severe microangiopathy of target organs, including the brain, liver, kidney, and placenta (Sibai et al., 2005). Severe maternal complications include seizures (eclampsia), stroke, renal failure, liver failure, and/or rupture, HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets), and death (Young et al., 2010). Fetal complications include prematurity due to preterm delivery, fetal growth restriction, fetal/neonatal hypoxic neurological injury, perinatal death, and long-term cardiovascular morbidity associated with low birth weight (Sibai et al., 2005).
Preeclampsia (PE) is a complex multifactorial disease; many factors, including genetic predisposition, immunological interactions, maternal endothelial function and environmental factors interact and culminate in the disease manifestation. In recent years, PE is commonly subdivided into early and late-onset, as the two conditions seem to have different underlying etiologies. Both early and late onset diseases present with abnormal placental perfusion and oxidative stress, however, poor placentation and deficient conversion of maternal spiral arteries only underlie the pathology of the early onset disease, whilst the late onset condition is associated with normal uterine spiral artery conversion and normally grown baby (Huppertz, 2008). Redman recently proposed a novel hypothesis for the pathology of late-onset PE, suggesting an intrinsic cause due to microvillous overcrowding, as placental growth reaches its functional limit. In both conditions, oxidatively stressed syncytiotrophoblast over-secrets proteins that perturb maternal angiogenic balance (Redman et al., 2014). Abnormal placentation is recognized as a main prerequisite for the pathogenesis of early onset preeclampsia. The disease seems to progress in two stages; in stage I (preclinical), poor development of the early placenta leads to deficient remodeling of maternal spiral arteries and consequent ischemia-reperfusion type injury of the placenta and oxidative stress. In stage II (clinical), the ischemic/malperfused placenta triggers the secretion of a mixture of placental factors, including antiangiogenic factors, proinflammatory cytokines and apoptotic debris that culminate in an enhanced maternal inflammatory response and in turn induce systemic endothelial dysfunction and the maternal syndrome of preeclampsia (Roberts and Cooper, 2001; Redman and Sargent, 2005).
In conditions of PE and IUGR which result in fetal hypoxia, there is a tendency to refer to fetoplacental hypoxia as the underlying cause. This concept has been challenged by several groups who advocate caution when interpreting results from pregnancies complicated by PE and IUGR (Kingdom and Kaufmann, 1997; Mayhew et al., 2004; Huppertz et al., 2014). These studies suggest that late-onset PE and late onset IUGR with present end diastolic flow (EDF) are associated with ischemic uteroplacental hypoxia in which the delivery of blood to the intervillous space is compromised due to deficient trophoblast invasion. This placental malperfusion results in ischemia-reperfusion injury affecting trophoblast and vascular endothelium, however fetal extraction of O2 is not compromised and there is evidence of increased placental branching angiogenesis (Mayhew et al., 2004). In contrast, pregnancies complicated by early onset PE, which is usually accompanied with early onset IUGR with absent/reversed EDF are often referred to as ‘placental hyperoxia’ due to compromised fetoplacental blood flow which fails to extract oxygen from the intervillous space, leading to poor fetal oxygenation and higher than normal intervillous oxygen levels (postplacental hypoxia; Kingdom and Kaufmann, 1997; Mayhew et al., 2004).
Medical intervention in preeclampsia remains limited, centering on management of maternal hypertension and systemic complications, seizure prevention with magnesium sulfate, and delivering the fetus before term as a measure of treating maternal effects and preventing further growth restriction. However, any benefits this confers are offset by the complications of prematurity. Hence, identifying interventions that would modulate the pathological processes involved in the disease pathogenesis are key to intervening effectively in preeclampsia to improve maternal and fetal prognosis. This review will focus on the role of angiogenic and antiangiogenic factors in mediating maternal endothelial dysfunction and explore some therapeutic strategies aimed at targeting these factors in order to reverse/ameliorate the disease pathology.
Evidence for the Role of Angiogenic Factors in Preeclampsia
Placental vasculature is plastic and changes constantly throughout pregnancy to facilitate the rapid growth, and supply the increasing metabolic needs of the growing fetus (Charnock-Jones et al., 2004). Normal placental development is regulated by proangiogenic and antiangiogenic factors. The processes of vasculogenesis, de novo formation of blood vessels from precursor cells, and angiogenesis, creation of vessels from pre-existing vessels, are critical in the fetal and placental development for effective transport of oxygen, nutrients and fetal growth and development (Charnock-Jones et al., 2004). In contrast, angiogenesis is a rare event in adult vascular networks under physiological conditions, being limited to organs of the female reproductive tract (endometrium, ovary, placenta). Appearance of new blood vessel growth in other organs is regarded as pathological, and can be involved in conditions such as wound healing, tumor growth, and retinopathy.
Placental abnormalities lead to secretion of a mixture of proteins that stimulate maternal endothelium and result in the disease presentation. Recently, circulating antiangiogenic proteins, soluble vascular endothelial growth factor receptor 1 [VEGF-R1, also referred to as soluble fms-like tyrosine kinase (sFlt1)] and soluble TGF-β co-receptor endoglin (sEng), have been implicated in the pathogenesis of many of the maternal features of preeclampsia. We will first consider the role of proangiogenic factors, VEGF and transforming growth factor (TGF-β), and their receptors in vascular homeostasis (Table 1). VEGFs are a family of secreted dimeric glycoproteins involved in vasculogenesis and angiogenesis. In humans and other mammals, VEGF-A (referred here as VEGF) and its placental homologue placental growth factor (PlGF) are the most important members with proangiogenic activity (Keck et al., 1989; Leung et al., 1989; Maglione et al., 1991). VEGF promotes survival and proliferation of endothelial cells and induces vascular permeability (Keck et al., 1989; Leung et al., 1989). Two VEGF receptors, VEGF-R1 (Flt-1) and VEGF-R2 (Flk-1/KDR) are present on endothelial cells. VEGF binds to both receptors whilst PlGF homodimers bind exclusively to VEGF-R1. VEGF-R2 it thought to primarily mediate the actions of VEGF on endothelial cells. The function of VEGF-R1 is less well defined, although it is thought to modulate VEGF-R2 signaling. Another function of VEGF-R1 is to act as a decoy receptor (sFlt-1), sequestering VEGF from VEGF-R2 binding (Table 1; Park et al., 1994; Waltenberger et al., 1994; Keyt et al., 1996). TGF-β is another important molecule involved in angiogenesis, it has been implicated in the development of the vasculature (Oshima et al., 1996). TGF-β signals via binding to both type I (ALK1 and ALK5) and type II receptors. In addition, TGF-β signaling in the vasculature involves coreceptors that act to modulate its effects. Endoglin (CD105) is a TGF-β coreceptor expressed by endothelial cells and placental syncytiotrophoblast. TGF-β effects in vitro seem concentration-dependent, inducing proliferation and migration of endothelial cells at low doses (ALK1 mediated), and inhibiting proliferation and migration of endothelial cells at high doses (via ALK5; Goumans et al., 2002). In addition, TGF-β is an important modulator of VEGF signaling, further linking TGF-β to angiogenesis (Table 1; Shao et al., 2009).
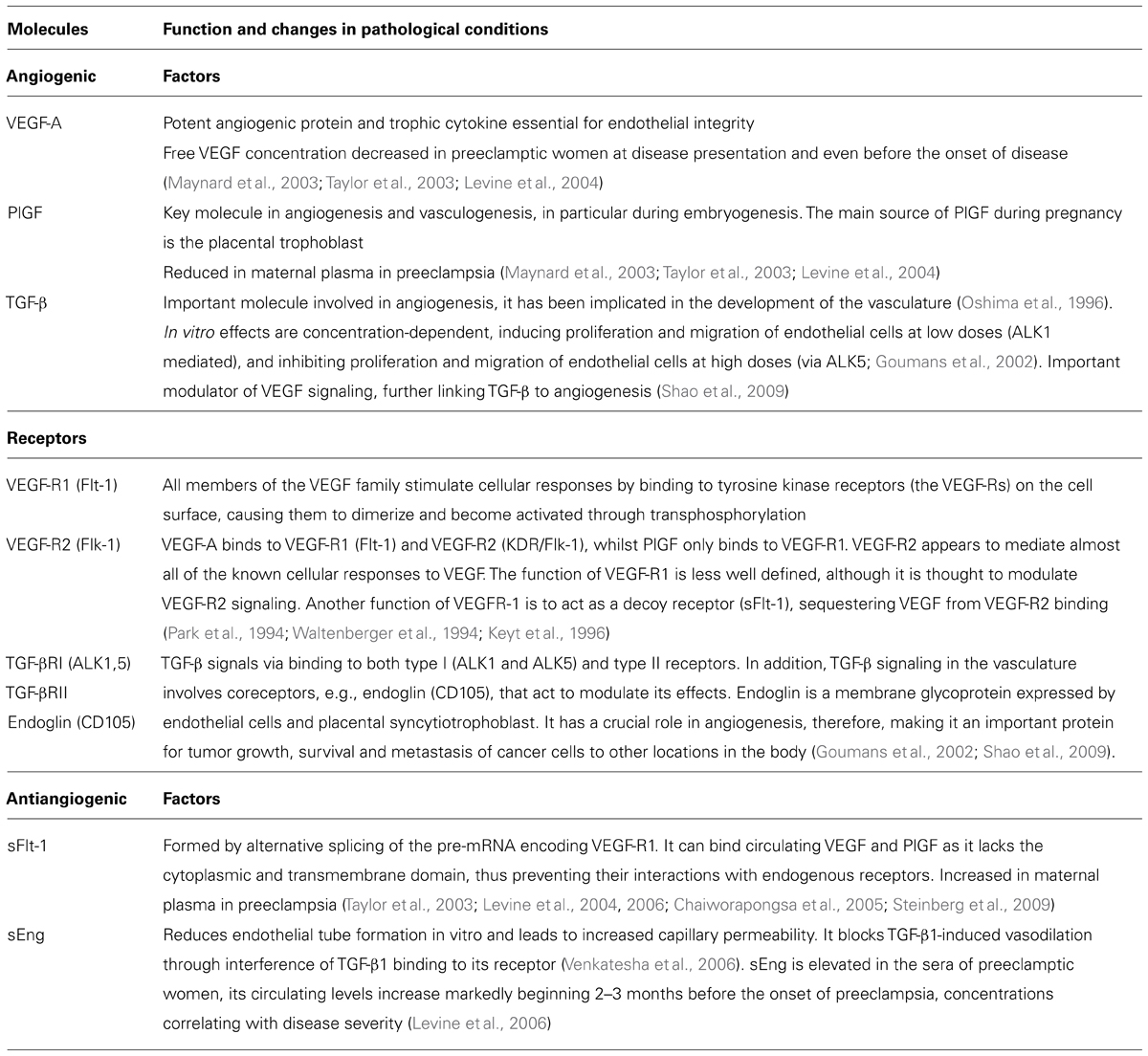
TABLE 1. A brief description of function of angiogenic and antiangiogenic growth factors and their receptors in pregnancy.
The vascular endothelium relies on proangiogenic factors. Release of placentally derived antiangiogenic factors into maternal circulation is thus likely to cause angiogenic factor imbalance, leading to endothelial dysfunction that manifests as the symptoms of preeclampsia. Two such factors, sFlt-1 and sEng, have been implicated in the pathophysiology of preeclampsia (Table 1). Soluble Flt-1 is formed by alternative splicing of the pre-mRNA encoding VEGF-R1. It can bind circulating VEGF and PlGF as it lacks the cytoplasmic and transmembrane domain, thus preventing their interactions with endogenous receptors. Soluble endoglin receptor sEng reduces endothelial tube formation in vitro and leads to increased capillary permeability. It blocks TGF-β1-induced vasodilation through interference of TGF-β1 binding to its receptor, this effect is likely to involve NO (Venkatesha et al., 2006).
Let us now consider the clinical and experimental evidence for the role of angiogenic factor imbalance in the pathophysiology of preeclampsia. The circulating levels of two placental-derived antiangiogenic factors, sFlt-1 and sEng, are elevated in the circulation of women with preeclampsia, and may begin to rise even before the onset of clinical symptoms, whereas the circulating concentrations of VEGF and PlGF are reduced in preeclamptic women at disease presentation and even before the onset of clinical symptoms (Taylor et al., 2003; Levine et al., 2004, 2006; Chaiworapongsa et al., 2005; Steinberg et al., 2009). The levels of circulating sFlt-1 increase and PlGF decrease during the last two months of pregnancy in normotensive women, however, these changes are significantly more pronounced in women who later develop preeclampsia and occur on average about 5 weeks before the onset of the disease (Levine et al., 2004). sEng is elevated in the sera of preeclamptic women, its circulating levels increase markedly beginning 2–3 months before the onset of preeclampsia, concentrations correlating with disease severity (Levine et al., 2006). However, it should be noted that despite the strong evidence linking angiogenic factor imbalance with PE, not all preeclamptic patients present with increased sFlt-1 and decreased VEGF/PlGF levels, and other mechanisms such as inflammation, oxidative stress, immunological interactions, maternal endothelial function, etc. also play an important role in the pathophysiology of this multifactorial disease.
Animal studies provide strong evidence linking sFlt-1 to the pathogenesis of preeclampsia. Although animal models do not develop preeclampsia, exogenous administration of sFlt-1 to pregnant rats induces some of the symptoms similar to the preeclamptic phenotype, including hypertension, proteinuria, and glomerular endotheliosis (Maynard et al., 2003). Similarly, glomerular endotheliosis and proteinuria can be induced in non-pregnant mice using antibodies against VEGF (Sugimoto et al., 2003), or by reducing glomerular VEGF levels by 50% (Eremina et al., 2003). In addition, administration of sFlt-1 can block VEGF and PlGF-induced microvascular relaxation of rat renal arterioles in vitro (Maynard et al., 2003). These studies all demonstrate that abnormal inhibition of endogenous VEGF activity can induce maternal preeclampsia-like symptoms. Similarly, administration of a therapeutic VEGF neutralizing antibody leads to renal pathology which can be replicated in adult animals by conditional gene targeting (Eremina et al., 2008). sFlt-treated animals do not develop additional symptoms of PE such as hemolysis and thrombocytopenia, seen in the HELLP syndrome. However, co-administration of sFlt-1 with sEng amplifies the symptoms, resulting in severe PE and/or HELLP syndrome, suggesting that these two placenta-derived factors could act in concert to induce severe preeclampsia (Venkatesha et al., 2006). We have shown similar synergistic effects between sFlt-1 and TNF-α. sFlt-1 sensitized endothelial cells to pro-inflammatory factors, increasing endothelial cell activation, as measured by increases in endothelial intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1, endothelin 1 (ET-1), von Willebrand factor and leukocyte adhesion, and led to reduction of AKT Ser473 and endothelial nitric oxide synthase (eNOS) Ser1177 phosphorylation (Cindrova-Davies et al., 2011). The survival of endothelial cells in vivo under non-pathological conditions is critically dependent on autocrine VEGF signaling. Endothelial specific ablation of VEGF results in progressive endothelial degeneration and sudden death of mutant animals (Lee et al., 2007), whilst autocrine VEGF is necessary for the survival of hematopoietic stem cells (Gerber et al., 2002), demonstrating that paracrine actions of VEGF are not sufficient to maintain the target cells. Thus, in preeclampsia the elevated circulating sFlt-1 is able to act in a dominant-negative fashion at the endothelial cell surface, blocking these autocrine signals (Cindrova-Davies et al., 2011).
Linking Antiangiogenic Factors with Endothelial Dysfunction
Maternal symptoms of preeclampsia present as a consequence of maternal endothelial cell dysfunction, not due to dysregulation of maternal angiogenesis. Secretion of antiangiogenic factors from the placenta can adversely affect maternal endothelial function. The higher relative concentrations of the antiangiogenic factors are thought to potentiate vascular endothelial cell injury in the liver, kidney, brain, and the placenta itself, and thus trigger the symptoms of preeclampsia. Placental oxidative stress due to malperfusion is now commonly considered as the underlying source for antiangiogenic factors which are at the root of the symptomatic phase of preeclampsia. However, the mechanism linking the angiogenic imbalance with endothelial dysfunction is still under investigation. Endothelin-1 (ET-1), a potent vasoconstrictor and pressor agent involved in the regulation of blood pressure, has been shown to play a crucial role in the development of hypertension in experimental animal models of placental hypoxia/ischemia (George and Granger, 2011). Long-term infusion of ET-1 into sheep also induced preeclampsia-like symptoms, i.e., hypertension, proteinuria, and decreased uteroplacental blood flow (Greenberg et al., 1997). In addition, several studies have reported elevated levels of plasma ET-1 in preeclamptic pregnant women, compared to controls (Taylor et al., 1990; Nova et al., 1991; Baksu et al., 2005).
The expression of VEGF and sFlt-1 can be regulated by the hypoxia-inducible factor-1 (HIF-1), a key component of a widely operative transcriptional response activated by hypoxia. Hypoxia increases HIF-1α stability, but it can also be up-regulated under non-hypoxic conditions by inflammatory cytokines or microtubule-depolymerising agents involving the NF-κB pathway (Jung et al., 2003). HIF-1-mediated sFlt-1 secretion can by induced in placental explants by hypoxia or hypoxia-reoxygenation (HR) in vitro (Achmad et al., 1997; Nagamatsu et al., 2004; Cindrova-Davies et al., 2007; Cudmore et al., 2007; Cindrova-Davies, 2009). These in vitro effects can be blocked by anti-oxidant vitamins, inhibitors of the p38 MAPK and NF-κB pathways, and by overexpression of the heme oxygenase (HO-1)/CO, suggesting the involvement of oxidative stress, NF-κB, p38 signaling and HO-1 in sFlt-1 and HIF-1α regulation (Cindrova-Davies, 2009). sEng inhibits TGF-β signaling in the vasculature, which includes effects on activation of eNOS and vasodilation. It has been postulated that increased circulating levels of sFlt-1 lead to reduction of VEGF and NOS, thus reducing NO production. Administration of sFlt-1 to pregnant rats induced hypertension and significantly increased the production of prepro-ET message levels in the renal cortex. The hypertensive response could be abolished with coadministration of an ETA antagonist (Murphy et al., 2010). Administration of sFlt-1 to pregnant animals thus impairs VEGF-R2-mediated NO production, reducing NO bioavailability, thereby increasing systemic peripheral resistance and inducing hypertension. NO bioavailability may be further reduced by increased oxidative stress, and leads to enhanced ET-1 production and increased blood pressure (George and Granger, 2011).
Differences have been reported in the oxygen sensing ability of early vs. late onset PE. Rolfo et al. (2010) reported a disruption of oxygen sensing in early onset disease but not in the late-onset, and they speculate this contributes to decreased HIF-1α hydroxylation and breakdown, leading to its accumulation in early onset PE (Rolfo et al., 2010). As mentioned previously, hypoxia is disputed by many as the leading cause of placental pathologies. Instead, ischemia-reperfusion (Hung et al., 2002; Burton et al., 2009) or hyperoxia (Kingdom and Kaufmann, 1997; Huppertz et al., 2014) have been proposed as the underlying etiological factors in PE. Nevertheless, all these conditions lead to generation of reactive oxygen species (ROS), which can increase HIF-1α levels and stability. In addition, HIF-1α can be stabilized under normoxic conditions by other mechanisms such as components of the immune system, especially inflammatory cytokines, and hormones (Zhou and Brüne, 2006). Preeclampsia is a condition of excessive inflammatory responses mediated by syncytiotrophoblast microparticles, pathogens and DAMPs (damage-associated molecular pattern), which activate toll-like receptors (TLR) and through binding to immune cells promote persistent inflammatory conditions in this syndrome (Laresgoiti-Servitje, 2010). TNF-α can promote the release of sFlt-1 (Parrish et al., 2010), and similarly, AT1 autoantibodies can promote the secretion of sEng and sFlt-1 through TNF-α-mediated mechanisms (Zhou et al., 2008; Irani et al., 2010).
Oxidative Stress and Antioxidants
Strong evidence exists that generation of placental oxidative stress is a key intermediary event in the pathology of preeclampsia (Hubel, 1999; Redman and Sargent, 2000; Burton and Jauniaux, 2004). It is thought that deficient conversion of the uterine spiral arteries and subsequent impaired perfusion of the placenta provides the initiating insult (Brosens et al., 2002). In early gestation of normal pregnancy, the spiral arteries undergo substantial remodeling by invasive extravillous trophoblasts, which penetrate into the myometrium and convert muscular spiral arteries into flaccid tubes with no muscularis or elastic lamina, capable of supplying the hugely expanded blood flow of the third trimester placenta. In contrast, the remodeling is minimal in PE, only affecting the decidual segments of the spiral arteries, which retain their high-resistance (Brosens et al., 2002). Maternal blood thus enters the intervillous space at a higher pressure and a faster rate, in a pulsatile jet-like manner, exposing placental villi to fluctuating oxygen concentrations, which contributes to the ischemia-reperfusion (I/R) type injury of the placenta (Jauniaux et al., 1994, 1995; Burton and Hung, 2003). The resulting oxidative stress is thought to induce the placenta to release a mixture of factors, including inflammatory cytokines, antiangiogenic factors, and apoptotic debris, which culminates in an enhanced maternal inflammatory response and endothelial dysfunction (Roberts, 1998; Redman and Sargent, 2005).
The role of oxidative stress in mediating PE pathophysiology is supported by reports of significantly decreased levels of antioxidant vitamins C, A, E, β-carotene, glutathione levels, and iron-binding capacity in the maternal circulation of women with preeclampsia. There is also evidence of diminished superoxide dismutase (SOD) levels and activity in the maternal and placental compartments of preeclamptic women, indicative of decreased total antioxidant protective capacity in preeclampsia (Walsh, 1998). In the experimental model of reduced uterine perfusion pressure (RUPP) of placental ischemia-induced hypertension, treatment with vitamins C and E did not decrease blood pressure, while the SOD mimetic Tempol attenuated RUPP hypertension. Interestingly, treatment with an NADP(H) oxidase inhibitor attenuated but did not normalize hypertension, suggesting other ROS-generating pathways (Gilbert et al., 2008; Sedeek et al., 2008). Challenging placental explants with hypoxia-reoxygenation (HR) or hypoxia in vitro induced sFlt-1 expression via HIF-1α upregulation (Ahmad and Ahmed, 2004; Nagamatsu et al., 2004; Cindrova-Davies et al., 2007). We showed that administration of vitamins C and E can block HR-mediated sFlt-1 secretion, via inhibition of p38 and NF-κB signaling pathways (Cindrova-Davies et al., 2007; Cindrova-Davies, 2009). Similarly, the in vivo challenge of labor results in increased oxidative stress, and increased expression of HIF-1 α, sFlt-1 and VEGF (Cindrova-Davies et al., 2007).
The regulatory role of oxidative stress in the I/R-type injury in preeclampsia and cardiovascular disease, together with encouraging in vitro and ex vivo data introduced the hypothesis that antioxidant vitamins might play an important role in the treatment of the ischemic disease pathology. However, despite encouraging in vitro data and cohort trials, clinical trials aimed to treat women at risk of preeclampsia with vitamins C and E (Poston et al., 2006; Rumbold et al., 2006) showed no reduction in the incidence of the disease. These results are disappointing. Periconceptional use of vitamins in women is associated with lower rates of severe preterm births and extreme SGA, which seems to suggest that vitamin use might be effective during conception and early pregnancy (Catov et al., 2007), and that the clinical trial administration of vitamins occurred too late in pregnancy to reverse already established disease pathology. The outcome could also be affected by the composition of natural vs. synthetic vitamins, as evidence exists that healthy Mediterranean diet lowers the incidence of preterm birth (Haugen et al., 2008) and coronary heart disease (Aravanis et al., 1970). Synergy between different mediators may also be important in disease treatment. Recent clinical trial found that whilst antioxidant vitamins alone did not have a protective effect for prevention of PE, there was a synergistic effect between L-arginine and vitamin C, reducing the incidence of PE in these patients (Vadillo-Ortega et al., 2011). Endogenous NADPH oxidase and manganese SOD are required to maintain VEGF signaling and vascular homeostasis (Abid et al., 2001, 2007). Interestingly, despite no effect of the vitamin trials on lowering the incidence of preeclampsia, antioxidant supplementation led to a significant decrease in the plasma concentration of sFlt-1 and an increase in the plasma concentration of PlGF (Poston et al., 2011), matching our in vitro findings and confirming the beneficial effect of antioxidants on VEGF signaling. Antioxidant vitamins could interfere with the normal physiological roles of VEGF and there seems to be a scope for potential use of antioxidants in preventing or ameliorating preeclampsia.
No Supplementation
Endothelium plays a crucial role in regulating vascular tone. In preeclamptic patients, endothelial activation is manifested by increased expression of markers of endothelial activation, including VCAM, endothelin, von Willebrand Factor, and thrombomodulin (Mutter and Karumanchi, 2008). Nitric oxide (NO), hydrogen sulfide (H2S), and carbon monoxide (CO) are three gaseous vasodilators that maintain the vascular tone. NO release from endothelial cells counter-balances the vasoconstriction produced by the sympathetic nervous system and the renin–angiotensin system. In addition, NO exerts many vasoprotective and anti-atherosclerotic properties, including protection from thrombosis, reduction of adhesion molecule expression and leukocyte adhesion. NO is produced by nitric oxide synthase (NOS), using L-arginine as a substrate. In addition, endogenously produced methylated amino acids such as asymmetric dimethylarginine (ADMA) can act as competitive inhibitors, and their secretion is increased in patients with cardiovascular disease and renal failure (Schnabel et al., 2005), as well as in women with high resistance placental circulation at risk of preeclampsia, IUGR, or both (Savvidou et al., 2003).
NO formation was found to be impaired in women with preeclampsia and gestational hypertension, compared to healthy pregnant controls. A negative correlation exists between plasma nitrite levels and sFlt-1 and sEng, suggesting an inhibitory effect of the angiogenic factors on NO formation (Sandrim et al., 2008). Given the important role of NO in the pathophysiology of preeclampsia, much research has concentrated on targeting the NO pathway as a potential therapy for preeclampsia. These include organic nitrates and S-nitrosothiols (e.g., S-nitrosoglutathione), L-arginine, inhibitors of cGMP breakdown (e.g., sildenafil), and other novel inhibitors of NO donor metabolism. This topic has been reviewed extensively by Johal et al. (2013). S-nitroglutathione (GSNO), endogenous S-nitrosothiol found ubiquitously in tissue, has been infused in women with preeclampsia and it has been shown to target not only the endothelial dysfunction, but also reduce platelet aggregation and activation, reduce sEng, and improve utero-placental perfusion with no adverse side effects, making it a good potential candidate for PE therapy (de Belder et al., 1995; Lees et al., 1996; Johal et al., 2013). The effects of supplementation with NO precursor, L-arginine, have been studied in several clinical trials. A recent randomized control trial of high risk pregnant women showed that dietary supplementation with a combination of L-arginine and antioxidants was associated with a significant reduction in the incidence of preeclampsia, compared to antioxidants alone or placebo prevention of preeclampsia (Vadillo-Ortega et al., 2011). These are encouraging results although they have to be interpreted with caution, given that the effects of L-arginine alone were not studied, and as such it is difficult to dissect out the relative contributions of L-arginine and antioxidants in reducing the incidence of the disease. These encouraging results were echoed in a recent systematic review of randomized trials focused on the role of L-arginine in the prevention of preeclampsia (Dorniak-Wall et al., 2014). The authors analyzed 7 randomized controlled trials testing the effects of L-arginine in pregnant women, and they reported L-arginine supplementation was associated with a significant reduction in the risk of preeclampsia in pregnant women with either established hypertension or who were considered at risk of preeclampsia (Dorniak-Wall et al., 2014).
The mechanism of the beneficial effect of L-arginine supplementation has been studied in pregnant rats in whom preeclampsia-like phenotype was induced with sFlt-1 administration (Murphy et al., 2011). sFlt-1 infusion into pregnant rats induced hypertension associated with reductions in circulating levels of VEGF, significant proteinuria, and endothelial dysfunction, as marked by a 3.5% increase in renal cortical pro-ET mRNA expression (Maynard et al., 2005; Gilbert et al., 2007; Murphy et al., 2010, 2011). Administration of L-arginine decreased sFlt-1 hypertension but had no effect on the blood pressure response in non-pregnant rats. In addition, L-arginine abolished the sFlt-mediated expression of renal cortical prepro-ET, suggesting that a reduction in NO synthesis may play an important role in the enhanced ET-1 production in response to sFlt-1 hypertension in pregnant rats (Murphy et al., 2011). These data are supported by previous studies, which showed a link between reduced NO production and enhanced ET-1 production (Kourembanas et al., 1993; Edwards et al., 1996). It has been postulated that increased circulating levels of sFlt-1 lead to reduction of VEGF and NOS, thus reducing NO production (George and Granger, 2011).
ER Stress and ER Stress Inhibitors
Endoplasmic reticulum (ER) processes all secreted proteins, facilitating folding and post-translational modifications. Accumulation of misfolded proteins leads to activation of ER stress response pathways, collectively known as the Unfolded Protein Response (UPR). The aim of the UPR is to restore homeostasis by inhibiting translation of non-essential proteins, increasing the capacity of the chaperone and folding machinery, and stimulating degradation of misfolded proteins. Failure of the mechanism results in activation of apoptotic pathways. ER stress is a major regulator of cell homeostasis (Xu et al., 2005; Cullinan and Diehl, 2006; Yoshida, 2007). The UPR is initiated by three transmembrane sensors normally held inactive by binding of the principal ER chaperone, GRP78. These sensors are PERK (PRKR-like ER kinase) that phosphorylates eukaryotic initiation factor 2α (eIF2α) to inhibit translation, ATF6 (activating transcription factor 6) and IRE1 (inositol-requiring enzyme 1). ATF6 is cleaved to produce an active transcription factor, while IRE1 splices the mRNA encoding XBP-1 (X-box binding protein-1) to produce a second transcription factor. These factors have overlapping functions, and both upregulate ER chaperone proteins, including GRP78, and degradation pathways (Xu et al., 2005; Cullinan and Diehl, 2006; Yoshida, 2007). In addition, ER stress can generate and accumulate ROS, thus promoting oxidative stress. Activation of the NF-κB pathway links increased oxidative stress, ER stress and inflammation. NF-κB can be activated by an increase in ER stress via PERK-eIF2α activation or IRE1α autophosphorylation. This effect can be blocked by both calcium chelators and antioxidants, suggesting NF-κB activation could be a result of the oxidative stress arising from excessive protein folding and/or ER-stress mediated Ca2+ leakage (Zhang and Kaufman, 2008).
Disorders of ER function are recognized to underlie many diverse pathologies, including diabetes and neurodegenerative diseases, and therapeutic interventions targeting it are being devised (Hetz et al., 2013). ER stress also seems instrumental in the pathophysiology of preeclampsia and IUGR (Yung et al., 2008, 2012). Use of ER chaperones proved a beneficial therapy for treatment of type 2 diabetes as it restored systemic insulin sensitivity, normalized hyperglycemia, resolved fatty liver disease, and enhanced insulin action in various tissues (Ozcan et al., 2006). Similarities exist between preeclampsia and metabolic syndrome, including dyslipidemia, inflammation, insulin resistance, oxidative stress and ER stress. We have demonstrated an increased expression of ET-1 and sFlt-1 in HR-treated placental explants (Cindrova-Davies et al., 2007; Cindrova-Davies, 2009). Conditioned medium from HR explants induced ER stress in JEG-3 cells, which was abolished by an ET-1 neutralizing antibody (Jain et al., 2012). Administration of ER chaperone TUDCA to placental explants challenged with HR reduced the levels of endothelin-1 (ET-1) in these explants, and it reduced the protein levels of sFlt-1 (unpublished observations). These data suggest that restoring ER function could also offer a therapeutic benefit for the treatment of preeclampsia. The ER chaperones are endogenously produced bile salts ursodeoxycholic acid (UDCA, TUDCA). The chaperones have excellent in vivo safety profiles and their use has been approved in clinical trials for the treatment of urea-cycle disorders, thalassemia and cystic fibrosis (Ozcan et al., 2006), and they are currently used to treat intrahepatic cholestasis of pregnancy (Geenes and Williamson, 2009).
Statins
Preeclampsia has been associated with dyslipidemia, and there is evidence of increased antibodies for oxidized form of LDL in patients with preeclampsia, which is consistent with oxidative stress, and it is analogous to changes described in atherosclerosis (Branch et al., 1994). Although preeclampsia is unique to pregnancy, there are many biological and pathological similarities, as well as risk factors (e.g., hypertension, obesity, dyslipidemia, diabetes, etc.) with adult cardiovascular disease. The mechanism underlying both atherosclerosis (Hansson, 2005) and preeclampsia (Roberts and Cooper, 2001; Redman and Sargent, 2005) is initiated by underlying endothelial dysfunction and inflammation, which lead to disease progression and manifestation. Pregnancy is often viewed as a stress test, and preeclampsia an early manifestation of CV disease. In fact, women who develop preeclampsia have a two- to threefold risk of hypertension, ischemic stroke and heart disease in later life (van Pampus and Aarnoudse, 2005; McDonald et al., 2008), and the risk is further increased with the severity of the disease, and early manifestation before 34 weeks gestation (Mongraw-Chaffin et al., 2010). Given the analogies between the two diseases, some remedies and treatments used to prevent and treat cardiovascular disease have been tested in preeclampsia.
Natural and synthetic statins (or 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors) are the most commonly prescribed classes of medication worldwide, used in primary and secondary prevention of cardiovascular mortality and other cardiovascular events through lipid-lowering remedy, as well as many pleiotropic effects, including endothelial protection, antioxidant properties, anti-inflammatory, antithrombotic and proangiogenic effects (Costantine and Cleary, 2013; Girardi, 2014). The beneficial effects of statins in the treatment of cardiovascular disease have been confirmed by recent meta-analysis of individual data from 90,000 individuals in 14 randomized trials of statin therapy vs. placebo, called the Cholesterol Treatment Trialists’ (CTT) Collaboration (Baigent et al., 2005). The CTT concluded that statin therapy can safely reduce the 5-year incidence of major coronary events, coronary revascularization, and stroke by about one fifth per mmol/L reduction in LDL cholesterol, largely irrespective of the initial lipid profile or other presenting characteristics (Baigent et al., 2005).
The effect of pravastatin administration in pregnancy has been tested in many rodent models of preeclampsia. Treatment with pravastatin significantly reduced maternal sFlt-1 levels, lowered blood pressure, improved the vascular profile, and prevented kidney injury (Ahmed et al., 2010; Costantine et al., 2010; Fox et al., 2011; Kumasawa et al., 2011; Saad et al., 2014). In addition, pravastatin also prevented the incidence of intrauterine growth restriction, without any adverse effects on pregnancy. In a mouse model of preeclampsia using placenta-specific lentiviral vector expression of sFlt-1, Kumasawa et al. elegantly demonstrated that pravastatin administration decreased sFlt-1 but importantly increased PlGF, thus restoring the angiogenic balance (Kumasawa et al., 2011). Pravastatin effects could be reproduced by administration of PlGF, which reduced sFlt-1 levels, ameliorated hypertension, glomerular endotheliosis, and proteinuria in the mice, suggesting that the beneficial effect of pravastatin on improving preeclampsia-like symptoms is mediated by increasing PlGF levels, which counteracts sFlt-1-mediated effects (Kumasawa et al., 2011). In addition, statins also exert protective effects on the endothelium and ameliorate preeclampsia symptoms by increasing the release of vasodilators NO (Redecha et al., 2009; Fox et al., 2011) and CO (Cudmore et al., 2007; Muchova et al., 2007). Pravastatin treatment of mice destined to develop preeclampsia using sFlt-1 overexpression increased eNOS protein expression in the vasculature (Fox et al., 2011), whilst pravastatin improved pregnancy outcome by increasing plasma NO levels in CBAxDBA/2 mice destined to develop recurrent miscarriage and IUGR (Redecha et al., 2009). Treatment of mice with statins resulted in increased HO-1 activity and increased CO release from tissues, as well as increased levels of plasma antioxidants (Muchova et al., 2007). Statins also induced the expression of HO-1 and inhibited sFlt-1 secretion in placental explants (Cudmore et al., 2007). These studies provide further evidence of a pleiotropic role of pravastatin in preventing the vascular dysfunction.
Given the encouraging evidence of the beneficial effects of statins on the prevention of cardiovascular disease in humans and of preeclampsia in animal models, the ability of pravastatin to restore angiogenic balance is currently being tested in the first randomized placebo controlled StAmP trial (Statins to ameliorate early onset preeclampsia) in the UK. The aim of the trial is to establish if pravastatin can lead to a significant reduction in circulating antiangiogenic factors and alleviate the severity of early onset preeclampsia. The pleiotropic anti-inflammatory, anti-thrombotic, antioxidant and vascular protective effects suggest that statins might be a good therapeutic option to prevent preeclampsia.
Future Directions – H2S Donors
Endogenous H2S plays an important role in regulating physiological processes such as blood flow, vasodilation, arterial diameter, and leukocyte adhesion (Wang, 2009). Endogenous production is catalyzed by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE). H2S is produced in the cardiovascular system by CSE, using cysteine as a substrate. It acts as a vasodilator, being able to hyperpolarize and relax SMCs by opening KATP channels (Zhao et al., 2001). Unlike NO and CO, which mediate vasorelaxation largely by cGMP pathway activation, the vasorelaxant effect of H2S is independent of cGMP pathway (Zhao et al., 2001). In addition, NO effects depend largely on the endothelial function integrity, whereas H2S is an endothelium-independent vasodilator and exerts its direct effect on SMCs. CSE knockout mice have markedly reduced serum H2S levels, which results in pronounced hypertension and diminished vasodilation, providing direct evidence that H2S is a potent vasodilator that can influence blood pressure (Yang et al., 2008). Similarly, genetic deficiency of CBS leads to homocysteinemia, which is associated with endothelial dysfunction and hypertension (Miles and Kraus, 2004). We reported reduced CSE expression in IUGR cases, as well as PE cases that were accompanied by abnormal UA Doppler profiles, and this could be recapitulated by subjecting placental explants to HR. In addition, we demonstrated a potent vasodilatory effect of an H2S donor in perfused placentas, capable of KATP dependent-reduction of placental vascular resistance. Reduced bioavailability of H2S may thus be implicated in placental vasoconstriction (Cindrova-Davies et al., 2013). In addition to being a potent vasodilator, many animal studies also showed a protective role of H2S donors against ischemia-reperfusion injury and inflammation (Bos et al., 2009, 2013; Sodha et al., 2009). The pro-survival pathways AKT, PKC, and ERK1/2 have been identified as H2S targets conferring its anti-apoptotic effects during reperfusion injury (Hu et al., 2008; Yong et al., 2008). Additionally, H2S can promote direct anti-apoptotic signaling during ischemia-reperfusion by activating eNOS and thus inducing NO (Yong et al., 2008), and H2S promotes DNA binding and transcriptional activation of anti-apoptotic genes regulated by NF-κB (Sen et al., 2012). The antioxidant properties of H2S can be mediated via two distinct mechanisms; by acting as a direct scavenger of ROS and by up-regulating antioxidant defenses. H2S may up-regulate endogenous antioxidants through a nuclear factor E2-related factor-2 (Nrf2; Calvert et al., 2009). Nrf2 binds the antioxidant responsive element (ARE) found in the promoter region of antioxidant genes, including heme oxygenase-1 (HO-1), thioredoxin, thioredoxin reductase, glutathione reductase, glutathione peroxidase (GPx), glutathione S-transferase (GSS), and catalase and is thus an important factor in controlling cardiac cellular susceptibility to reactive oxygen and nitrogen species-induced cytotoxicity (Zhu et al., 2005). Garlic oil can also promote Nrf-2 activation, further supporting the role for Nrf2 as the mediator of H2S-induced antioxidant effects (Fisher et al., 2007). Additionally, CSE expression appears to be a critical component of the cytoprotective ATF4 transcriptional response; CSE deficiency increases sensitivity to apoptosis induced by ER stressors and homocysteine, indicating the importance of GSH up-regulation through the transsulfuration pathway to promote survival (Dickhout et al., 2012). H2S also exerts important proangiogenic effects (Cai et al., 2007; Wang et al., 2010; Bir et al., 2012; Holwerda et al., 2014). H2S promotes proliferation, adhesion, migration and tube-like structure formation of endothelial cells in vitro via AKT pathway phosphorylation. It also stimulates angiogenesis in vivo at physiologically relevant doses (Cai et al., 2007). NO-H2S cross-talk seems critical in mediating these effects (Bir et al., 2012; Coletta et al., 2012). H2S therapy restores blood flow to ischemic tissues in a NO-dependent manner, by stimulating NOS expression and HIF-1α and VEGF expression and activity (Bir et al., 2012).
Consistent with these roles of H2S, the consumption of garlic is negatively correlated with the progression of CV disease, and causes a lower incidence of hypertension, enhances antioxidants and inhibits platelet aggregation (Banerjee and Maulik, 2002). It has been shown that the biological production of H2S from garlic-derived organic polysulfides mediates the major beneficial effects of garlic-rich diets, specifically on CV disease and more broadly on overall health (Benavides et al., 2007). In animal models of hypoxic pulmonary hypertension, H2S could reverse structural remodeling changes in pulmonary vessels of rats exposed to chronic HR (Hongfang et al., 2006). H2S therapy also protected against acute myocardial ischemia/reperfusion (Calvert et al., 2009), and attenuated cardiac dysfunction following heart failure (Kondo et al., 2013; Polhemus et al., 2013). Recent animal studies have focused on the use of “medical food,” i.e., drugs derived from natural sources such as garlic, as a cardioprotective therapy for heart failure. These include diallyl trisulfide (DATS; Polhemus et al., 2013), a long acting H2S donor derived from garlic, and sulfur-releasing “medical food” SG-1002 (Kondo et al., 2013), which is currently in clinical trials. Similar results have been reported in the placental field recently. Holwerda et al. (2014) evaluated the therapeutic effect of an H2S donor in an animal model of sFlt-induced hypertension. They reported a significant reduction in blood pressure, and proteinuria, as well as concomitant reduction in sFlt-1 levels and increase in free maternal VEGF concentrations in the treated group of animals (Holwerda et al., 2014). Wang et al. (2013) demonstrated that CSE enzyme inhibition in pregnant mice induced hypertension, increased sFlt-1 and sEng levels, caused placental abnormalities and compromised fetal growth. H2S donor therapy reduced the levels of the antiangiogenic factors and restored fetal growth, adding further weight to the evidence that a dysfunctional CSE/H2S pathway may contribute to the pathogenesis of preeclampsia (Wang et al., 2013), and that H2S donor therapy could have beneficial effects on prevention of preeclampsia.
Concluding Remarks
Therapeutic interventions to treat preeclampsia remain largely experimental and there is no established remedy for the treatment of this multifactorial disease. This review concentrated on the evidence for the therapeutic potential of antioxidants, ER chaperones, NO and H2S donors, and statins. Mechanistic effects of these compounds on the signaling pathways involved in the pathophysiology of preeclampsia, are summarized in Figure 1. Antioxidants, ER chaperones, NO donors, statins and H2S donors display pleitropic antioxidant, anti-inflammatory, and pro-angiogenic effects in animal and in vitro studies. Encouraging clinical evidence exists for the use of NO donors, and a clinical trial is currently underway testing the effect of statins in treatment of preeclampsia. Although clinical trials on the use of antioxidant vitamins in pregnancy proved largely unsuccessful, the scope for their use still exists given the beneficial cardioprotective effects of antioxidant-rich Mediterranean diet, periconceptual vitamin use and the synergistic effect of vitamin C and L-arginine. H2S recently emerged as a novel therapeutic agent for cardiovascular disease and its effects are being tested.
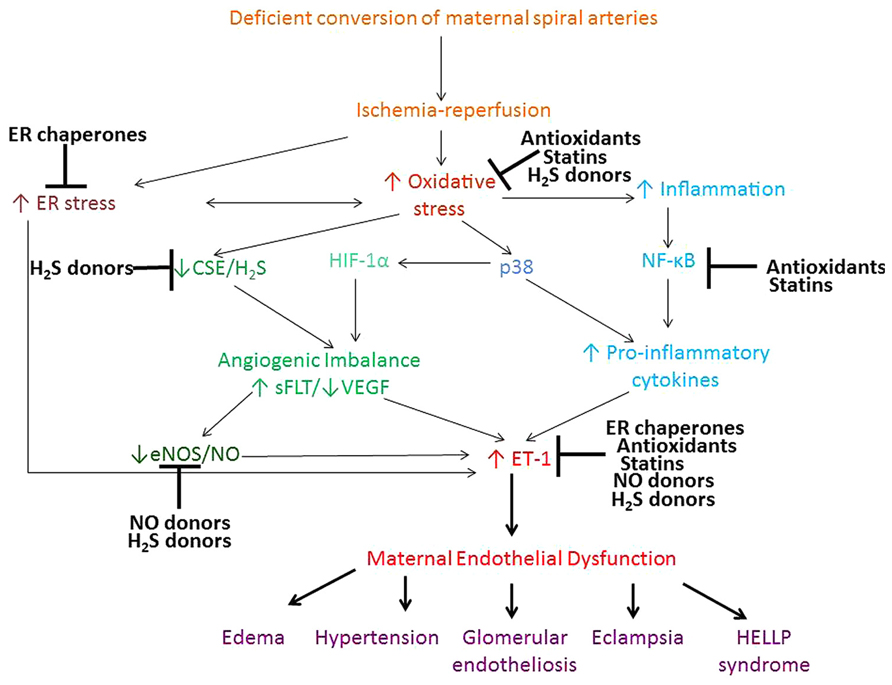
FIGURE 1. Proposed pathways contributing to the maternal syndrome of preeclampsia and the mechanism of action of antioxidants, ER chaperones, NO, and H2S donors and statins. Deficient conversion of spiral arteries leads to fluctuations in O2 concentration and triggers ischemia-reperfusion type injury of the placenta and consequent oxidative stress and ER stress. ROS activate various stress pathways, including proapoptotic p38 and inflammatory NF-κB pathway. These pathways promote increased shedding of microparticles, anti-angionenic factors and inflammatory cytokines, which lead to development of the peripheral maternal symptoms. Antioxidants, ER chaperones, NO and H2S donors and statins all display pleitrophic effects, targeting inflammatory, stress, antioxidant pathways, as well as VEGF and NO signaling.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to acknowledge the support of the Wellcome Trust (084804/2/08/Z) and the guidance and mentoring of Professors Graham J. Burton and D. Stephen Charnock-Jones.
Abbreviations
ADMA, asymmetric dimethylarginine; AKT, serine/threonine-specific protein kinase; ALK1 and ALK5, TGF-β receptors type I; ARE, antioxidant responsive element; ATF6, activating transcription factor 6; CBS, cystathionine β-synthase; CO, carbon monoxide; CSE, cystathionine γ-lyase; CTT, cholesterol treatment trialists; CV, cardiovascular; DAMPs, damage-associated molecular pattern; EDF, end diastolic flow; eIF2α, eukaryotic initiation factor 2α; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; ET-1, endothelin-1; GPx, glutathione peroxidase; GRP78, 78 kDa glucose-regulated protein, also known as binding immunoglobulin protein (BiP) or heat shock 70 kDa protein 5 (HSPA5); GSS, glutathione S-transferase; H2S, hydrogen sulfide; HELLP syndrome, hemolysis, elevated liver enzymes and low platelets; HO-1, heme oxygenase; HIF-1α, hypoxia-induced factor; HR, hypoxia-reoxygenation; ICAM, intercellular adhesion molecule; I/R, ischemia-reperfusion; IRE1, inositol-requiring enzyme 1; IUGR, intrauterine growth restriction; LDL, low density lipoprotein; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; Nrf2, nuclear factor E2-related factor-2; p38 MAPK, p38 mitogen-activated protein kinase; PE, preeclampsia; PERK, PRKR-like ER kinase; PlGF, placental growth factor; ROS, reactive oxygen species; RUPP, reduced uterine perfusion pressure; sEng, soluble TGF-β co-receptor endoglin; sFlt1, soluble VEGF receptor 1, also referred to as soluble fms-like tyrosine kinase; SGA, small for gestational age; SOD, superoxide dismutase; StAmP trial, Statins to ameliorate early onset preeclampsia; TLR, Toll-like receptors; TUDCA, Tauroursodeoxycholic acid, bile salt; UDCA, ursodeoxycholic acid, bile salt; UPR, Unfolded Protein Response; VCAM-1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor; XBP-1, X-box binding protein-1.
References
Abid, M. R., Spokes, K. C., Shih, S. C., and Aird, W. C. (2007). NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J. Biol. Chem. 282, 35373–35385. doi: 10.1074/jbc.M702175200
Abid, M. R., Tsai, J. C., Spokes, K. C., Deshpande, S. S., Irani, K., and Aird, W. C. (2001). Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J. 15, 2548–2550.
Achmad, T., Winterscheidt, A., Lindemann, C., and Rao, G. S. (1997). Oxidized low density lipoprotein acts on endothelial cells in culture to enhance endothelin secretion and monocyte migration. Methods Find. Exp. Clin. Pharmacol. 19, 153–159.
Ahmad, S., and Ahmed, A. (2004). Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ. Res. 95, 884–891.
Ahmed, A., Singh, J., Khan, Y., Seshan, S. V., and Girard, G. (2010). A new mouse model to explore therapies for preeclampsia. PLoS ONE 5:e13663. doi: 10.1371/journal.pone.0013663
American College of Obstetricians and Gynecologists (2013). Hypertension in pregnancy. DNLM: 1. Hypertension, Pregnancy-Induced-Practice Guideline. Washington: LoCCiP Data.
Aravanis, C., Corcondilas, A., Dontas, A. S., Lekos, D., and Keys, A. (1970). Coronary heart disease in seven countries. IX. The Greek islands of Crete and Corfu. Circulation 41(Suppl. 4), I88–I100.
Baigent, C., Keech, A., Kearney, P. M., Blackwell, L., Buck, G., Pollicino, C., et al. (2005). Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 366, 1267–1278. doi: 10.1016/S0140-6736(05)67394-1
Baksu, B., Davas, I., Baksu, A., Akyol, A., and Gulbaba, G. (2005). Plasma nitric oxide, endothelin-1 and urinary nitric oxide and cyclic guanosine monophosphate levels in hypertensive pregnant women. Int. J. Gynaecol. Obstet. 90, 112–117. doi: 10.1016/j.ijgo.2005.04.018
Banerjee, S. K., and Maulik, S. K. (2002). Effect of garlic on cardiovascular disorders: a review. Nutr. J. 1, 4. doi: 10.1186/1475-2891-1-4
Benavides, G. A., Squadrito, G. L., Mills, R. W., Patel, H. D., Isbell, T. S., Patel, R. P., et al. (2007). Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. U.S.A. 104, 17977–17982. doi: 10.1073/pnas.0705710104
Bir, S. C., Kolluru, G. K., McCarthy, P., Shen, X., Pardue, S., Pattillo, C. B., et al. (2012). Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart Assoc. 1, e004093. doi: 10.1161/JAHA.112.004093
Bos, E. M., Leuvenink, H. G., Snijder, P. M., Kloosterhuis, N. J., Hillebrands, J. L., Leemans, J. C., et al. (2009). Hydrogen sulfide-induced hypometabolism prevents renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 20, 1901–1905. doi: 10.1681/ASN.2008121269
Bos, E. M., Wang, R., Snijder, P. M., Boersema, M., Damman, J., Fu, M., et al. (2013). Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 24, 759–770. doi: 10.1681/ASN.2012030268
Branch, D. W., Mitchell, M. D., Miller, E., Palinski, W., and Witztum, J. L. (1994). Pre-eclampsia and serum antibodies to oxidised low-density lipoprotein. Lancet 343, 645–646. doi: 10.1016/S0140-6736(94)92639-5
Brosens, J. J., Pijnenborg, R., and Brosens, I. A. (2002). The myometrial junctional zone spiral arteries in normal and abnormal pregnancies: a review of the literature. Am. J. Obstet. Gynecol. 187, 1416–1423. doi: 10.1067/mob.2002.127305
Burton, G. J., and Hung, T.-H. (2003). Hypoxia-reoxygenation: a potential source of placental oxidative stress in normal pregnancy and preeclampsia. Fetal Maternal Med. Rev. 14, 97–117. doi: 10.1017/S0965539503001049
Burton, G. J., and Jauniaux, E. (2004). Placental oxidative stress: from miscarriage to preeclampsia. J. Soc. Gynecol. Investig. 11, 342–352. doi: 10.1016/j.jsgi.2004.03.003
Burton, G. J., Woods, A. W., Jauniaux, E., and Kingdom, J. C. (2009). Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 30, 473–482. doi: 10.1016/j.placenta.2009.02.009
Cai, W. J., Woods, A. W., Jauniaux, E., and Kingdom, J. C. (2007). The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 76, 29–40. doi: 10.1016/j.cardiores.2007.05.026
Calvert, J. W., Jha, S., Gundewar, S., Elrod, J. W., Ramachandran, A., Pattillo, C. B., et al. (2009). Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 105, 365–374. doi: 10.1161/CIRCRESAHA.109.199919
Catov, J. M., Bodnar, L. M., Ness, R. B., Markovic, N., and Roberts, J. M. (2007). Association of periconceptional multivitamin use and risk of preterm or small-for-gestational-age births. Am. J. Epidemiol. 166, 296–303. doi: 10.1093/aje/kwm071
Chaiworapongsa, T., Romero, R., Kim, Y. M., Kim, G. J., Kim, M. R., Espinoza, J., et al. (2005). Plasma soluble vascular endothelial growth factor receptor-1 concentration is elevated prior to the clinical diagnosis of pre-eclampsia. J. Matern. Fetal Neonatal. Med. 17, 3–18. doi: 10.1080/14767050400028816
Charnock-Jones, D. S., Kaufmann, P., and Mayhew, T. M. (2004). Aspects of human fetoplacental vasculogenesis and angiogenesis. I. Molecular regulation. Placenta 25, 103–113. doi: 10.1016/j.placenta.2003.10.004
Cindrova-Davies, T., Herrera, E. A., Niu, Y., Kingdom, J., Giussani, D. A., and Burton, G. J. (2013). Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am. J. Pathol. 182, 1448–1458. doi: 10.1016/j.ajpath.2013.01.001
Cindrova-Davies, T. (2009). Gabor Than Award Lecture 2008: pre-eclampsia - from placental oxidative stress to maternal endothelial dysfunction. Placenta 30(Suppl. A), S55–S65. doi: 10.1016/j.placenta.2008.11.020
Cindrova-Davies, T., Sanders, D. A., Burton, G. J., and Charnock-Jones, D. S. (2011). Soluble, F. L.T1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor-mediated signalling. Cardiovasc. Res. 89, 671–679. doi: 10.1093/cvr/cvq346
Cindrova-Davies, T., Spasic-Boskovic, O., Jauniaux, E., Charnock-Jones, D. S., and Burton, G. J. (2007). Nuclear factor-kappa B, p38, and stress-activated protein kinase mitogen-activated protein kinase signaling pathways regulate proinflammatory cytokines and apoptosis in human placental explants in response to oxidative stress: effects of antioxidant vitamins. Am. J. Pathol. 170, 1511–1520. doi: 10.2353/ajpath.2007.061035
Cindrova-Davies, T., Yung, H. W., Johns, J., Spasic-Boskovic, O., Korolchuk, S., Jauniaux, E., et al. (2007). Oxidative stress, gene expression, and protein changes induced in the human placenta during labor. Am. J. Pathol. 171, 1168–1179. doi: 10.2353/ajpath.2007.070528
Coletta, C., Papapetropoulos, A., Erdelyi, K., Olah, G., Módis, K., Panopoulos, P., et al. (2012). Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. U.S.A. 109, 9161–9166. doi: 10.1073/pnas.1202916109
Costantine, M. M., and Cleary, K. (2013). Pravastatin for the prevention of preeclampsia in high-risk pregnant women. Obstet. Gynecol. 121(Pt 1), 349–353. doi: 10.1097/AOG.0b013e31827d8ad5
Costantine, M. M., Tamayo, E., Lu, F., Bytautiene, E., Longo, M., Hankins, G. D., et al. (2010). Using pravastatin to improve the vascular reactivity in a mouse model of soluble fms-like tyrosine kinase-1-induced preeclampsia. Obstet. Gynecol. 116, 114–120. doi: 10.1097/AOG.0b013e3181e10ebd
Cudmore, M., Ahmad, S., Al-Ani, B., Fujisawa, T., Coxall, H., Chudasama, K., et al. (2007). Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 115, 1789–1797. doi: 10.1161/CIRCULATIONAHA.106.660134
Cullinan, S. B., and Diehl, J. A. (2006). Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 38, 317–332. doi: 10.1016/j.biocel.2005.09.018
de Belder, A., Lees, C., Martin, J., Moncada, S., and Campbell, S. (1995). Treatment of HELLP syndrome with nitric oxide donor. Lancet 345, 124–125. doi: 10.1016/S0140-6736(95)90088-8
Dickhout, J. G., Carlisle, R. E., Jerome, D. E., Mohammed-Ali, Z., Jiang, H., Yang, G. (2012). Integrated stress response modulates cellular redox state via induction of cystathionine gamma-lyase: cross-talk between integrated stress response and thiol metabolism. J. Biol. Chem. 287, 7603–7614. doi: 10.1074/jbc.M111.304576
Dorniak-Wall, T., Grivell, R. M., Dekker, G. A., Hague, W., and Dodd, J. M. (2014). The role of L-arginine in the prevention and treatment of pre-eclampsia: a systematic review of randomised trials. J. Hum. Hypertens. 28, 230–235. doi: 10.1038/jhh.2013.100
Edwards, D. L., Arora, C. P., Bui, D. T., and Castro, L. C. (1996). Long-term nitric oxide blockade in the pregnant rat: effects on blood pressure and plasma levels of endothelin-1. Am. J. Obstet. Gynecol. 175, 484–488. doi: 10.1016/S0002-9378(96)70166-7
Eremina, V., Jefferson, J. A., Kowalewska, J., Hochster, H., Haas, M., Weisstuch, J., et al. (2008). VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 358, 1129–1136. doi: 10.1056/NEJMoa0707330
Eremina, V., Sood, M., Haigh, J., Nagy, A., Lajoie, G., Ferrara, N., et al. (2003). Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Invest. 111, 707–716. doi: 10.1172/JCI17423
Fisher, C. D., Augustine, L. M., Maher, J. M., Nelson, D. M., Slitt, A. L., Klaassen, C. D., et al. (2007). Induction of drug-metabolizing enzymes by garlic and allyl sulfide compounds via activation of constitutive androstane receptor and nuclear factor E2-related factor 2. Drug. Metab. Dispos. 35, 995–1000. doi: 10.1124/dmd.106.014340
Fox, K. A., Longo, M., Tamayo, E., Kechichian, T., Bytautiene, E., Hankins, G. D., et al. (2011). Effects of pravastatin on mediators of vascular function in a mouse model of soluble Fms-like tyrosine kinase-1-induced preeclampsia. Am. J. Obstet. Gynecol. 205, 366e1–366e5. doi: 10.1016/j.ajog.2011.06.083
Geenes, V., and Williamson, C. (2009). Intrahepatic cholestasis of pregnancy. World J. Gastroenterol. 15, 2049–2066. doi: 10.3748/wjg.15.2049
George, E. M., and Granger, J. P. (2011). Endothelin: key mediator of hypertension in preeclampsia. Am. J. Hypertens. 24, 964–969. doi: 10.1038/ajh.2011.99
Gerber, H. P., Malik, A. K., Solar, G. P., Sherman, D., Liang, X. H., Meng, G., et al. (2002). VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 417, 954–958. doi: 10.1038/nature00821
Gilbert, J. S., Babcock, S. A., and Granger, J. P. (2007). Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 50, 1142–1147. doi: 10.1161/HYPERTENSIONAHA.107.096594
Gilbert, J. S., Ryan, M. J., LaMarca, B. B., Sedeek, M., Murphy, S. R., and Granger, J. P. (2008). Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 294, H541–H550. doi: 10.1152/ajpheart.01113.2007
Girardi, G. (2014). Can statins prevent pregnancy complications? J. Reprod. Immunol. 101–102, 161–167. doi:10.1016/j.jri.2013.07.005
Goumans, M. J., Valdimarsdottir, G., Itoh, S., Rosendahl, A., Sideras, P., and Ten Dijke, P. (2002). Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21, 1743–1753. doi: 10.1093/emboj/21.7.1743
Greenberg, S. G., Baker, R. S., Yang, D., and Clark, K. E. (1997). Effects of continuous infusion of endothelin-1 in pregnant sheep. Hypertension 30, 1585–1590. doi: 10.1161/01.HYP.30.6.1585
Hansson, G. K. (2005). Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695. doi: 10.1056/NEJMra043430
Haugen, M., Meltzer, H. M., Brantsaeter, A. L., Mikkelsen, T., Osterdal, M. L., Alexander, J., et al. (2008). Mediterranean-type diet and risk of preterm birth among women in the Norwegian, Mother and Child Cohort Study (MoBa): a prospective cohort study. Acta Obstet. Gynecol. Scand. 87, 319–324. doi: 10.1080/00016340801899123
Hetz, C., Chevet, E., and Harding, H. P. (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. doi: 10.1038/nrd3976
Holwerda, K. M., Burke, S. D., Faas, M. M., Zsengeller, Z., Stillman, I. E., Kang, P. M., et al. (2014). Hydrogen sulfide attenuates sFlt1-induced hypertension and renal damage by upregulating vascular endothelial growth factor. J. Am. Soc. Nephrol. 25, 717–725. doi: 10.1681/ASN.2013030291
Hongfang, J., Bailin, C., Bin, Z., Chunyu, Z., Xinmin, L., Weijin, Z., et al. (2006). Effects of hydrogen sulfide on hypoxic pulmonary vascular structural remodeling. Life Sci. 78, 1299–1309. doi: 10.1016/j.lfs.2005.07.009
Hu, Y., Chen, X., Pan, T. T., Neo, K. L., Lee, S. W., Khin, E. S., et al. (2008). Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch. 455, 607–616. doi: 10.1007/s00424-007-0321-4
Hubel, C. A. (1999). Oxidative stress in the pathogenesis of preeclampsia. Proc. Soc. Exp. Biol. Med. 222, 222–235. doi: 10.1046/j.1525-1373.1999.d01-139.x
Hung, T. H., Skepper, J. N., Charnock-Jones, D. S., and Burton, G. J. (2002). Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ. Res. 90, 1274–1281. doi: 10.1161/01.RES.0000024411.22110.AA
Huppertz, B. (2008). Placental origins of preeclampsia: challenging the current hypothesis. Hypertension 51, 970–975. doi: 10.1161/HYPERTENSIONAHA.107.107607
Huppertz, B., Weiss, G., and Moser, G. (2014). Trophoblast invasion and oxygenation of the placenta: measurements versus presumptions. J. Reprod. Immunol. 101–102, 74–79. doi: 10.1016/j.jri.2013.04.003
Irani, R., Zhang, Y., Zhou, C. C., Blackwell, S. C., Hicks, M. J., Ramin, S. M., et al. (2010). Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-alpha signaling. Hypertension 55, 1246–1253. doi: 10.1161/HYPERTENSIONAHA.110.150540
Jain, A., Olovsson, M., Burton, G. J., and Yung, H. W. (2012). Endothelin-1 induces endoplasmic reticulum stress by activating the PLC-IP(3) pathway: implications for placental pathophysiology in preeclampsia. Am. J. Pathol. 180, 2309–2320. doi: 10.1016/j.ajpath.2012.03.005
Jauniaux, E., Jurkovic, D., and Campbell, S. (1995). Current topic: in vivo investigation of the placental circulations by Doppler echography. Placenta 16, 323–331. doi: 10.1016/0143-4004(95)90089-6
Jauniaux, E., Ramsay, B., and Campbell, S. (1994). Ultrasonographic investigation of placental morphologic characteristics and size during the second trimester of pregnancy. Am. J. Obstet. Gynecol. 170(Pt 1), 130–137. doi: 10.1016/S0002-9378(94)70397-3
Johal, T., Lees, C. C., Everett, T. R., and Wilkinson, I. B. (2013). The nitric oxide pathway and possible therapeutic options in pre-eclampsia. Br. J. Clin. Pharmacol. doi: 10.1111/bcp.12301 [Epub ahead of print].
Jung, Y.-J., Isaacs, J. S., Lee, S., Trepel, J., Liu, Z. G., and Neckers, L. (2003). Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappaB activation. Biochem. J. 370, 1011–1017. doi: 10.1042/BJ20021279
Jung, Y.-J., Isaacs, J. S., Lee, S., Trepel, J., and Neckers, L. (2003). Microtubule disruption utilizes an NFkappa B-dependent pathway to stabilize HIF-1alpha protein. J. Biol. Chem. 278, 7445–7452. doi: 10.1074/jbc.M209804200
Jung, Y.-J., Jennifer, S., Sunmin, I., Jane, L., Trepel, L., and Neckers, N. (2003). IL-1b mediated up-regulation of HIF-1a via an NFkB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 17, 2115–2117.
Keck, P. J., Hauser, S. D., Krivi, G., Sanzo, K., Warren, T., Feder, J., et al. (1989). Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246, 1309–1312. doi: 10.1126/science.2479987
Keyt, B. A., Nguyen, H. V., Berleau, L. T., Duarte, C. M., Park, J., Chen, H., et al. (1996). Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors. Generation of receptor-selective VEGF variants by site-directed mutagenesis. J. Biol. Chem. 271, 5638–5646. doi: 10.1074/jbc.271.10.5638
Kingdom, J. C., and Kaufmann, P. (1997). Oxygen and placental villous development: origins of fetal hypoxia. Placenta 18, 613–621; discussion 623–626. doi: 10.1016/S0143-4004(97)90000-X
Kondo, K., Bhushan, S., King, A. L., Prabhu, S. D., Hamid, T., Koenig, S., et al. (2013). H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127, 1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855
Kourembanas, S., McQuillan, L. P., Leung, G. K., and Faller, D. V. (1993). Nitric oxide regulates the expression of vasoconstrictors and growth factors by vascular endothelium under both normoxia and hypoxia. J. Clin. Invest. 92, 99–104. doi: 10.1172/JCI116604
Kumasawa, K., Ikawa, M., Kidoya, H., Hasuwa, H., Saito-Fujita, T., Morioka, Y., et al. (2011). Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc. Natl. Acad. Sci. U.S.A. 108, 1451–1455. doi: 10.1073/pnas.1011293108
Laresgoiti-Servitje, E. (2010). A leading role for the immune system in the pathophysiology of preeclampsia. J. Leukoc. Biol. 94, 247–257. doi: 10.1189/jlb.1112603
Lee, S., Chen, T. T., Barber, C. L., Jordan, M. C., Murdock, J., Desai, S., et al. (2007). Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703. doi: 10.1016/j.cell.2007.06.054
Lees, C., Langford, E., Brown, A. S., de Belder, A., Pickles, A., Martin, J. F., et al. (1996). The effects of S-nitrosoglutathione on platelet activation, hypertension, and uterine and fetal Doppler in severe preeclampsia. Obstet. Gynecol. 88, 14–19. doi: 10.1016/0029-7844(96)00070-1
Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V., and Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309. doi: 10.1126/science.2479986
Levine, R. J., Lam, C., Qian, C., Yu, K. F., Maynard, S. E., Sachs, B. P., et al. (2006). Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 355, 992–1005. doi: 10.1056/NEJMoa055352
Levine, R. J., Maynard, S. E., Qian, C., Lim, K.-H, England, L. J., Yu, K. F., et al. (2004). Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 350, 672–683. doi: 10.1056/NEJMoa031884
Maglione, D., Guerriero, V., Viglietto, G., Delli-Bovi, P., and Persico, M. G. (1991). Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. U.S.A. 88, 9267–9271. doi: 10.1073/pnas.88.20.9267
Mayhew, T. M., Charnock-Jones, D. S., and Kaufmann, P. (2004). Aspects of human fetoplacental vasculogenesis and angiogenesis. III. Changes in complicated pregnancies. Placenta 25, 127–139. doi: 10.1016/j.placenta.2003.10.010
Maynard, S. E., Min, J. Y., Merchan, J., Lim, K. H., Li, J., Mondal, S., et al. (2003). Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 111, 649–658. doi: 10.1172/JCI17189
Maynard, S. E., Venkatesha, S., Thadhani, R., and Karumanchi, S. A. (2005). Soluble Fms-like tyrosine kinase 1 and endothelial dysfunction in the pathogenesis of preeclampsia. Pediatr. Res. 57(Pt 2), 1R–7R. doi: 10.1203/01.PDR.0000159567.85157.B7
McDonald, S. D., Malinowski, A., Zhou, Q., Yusuf, S., and Devereaux, P. J. (2008). Cardiovascular sequelae of preeclampsia/eclampsia: a systematic review and meta-analyses. Am. Heart J. 156, 918–930. doi: 10.1016/j.ahj.2008.06.042
Miles, E. W., and Kraus, J. P. (2004). Cystathionine beta-synthase: structure, function, regulation, and location of homocystinuria-causing mutations. J. Biol. Chem. 279, 29871–29874. doi: 10.1074/jbc.R400005200
Mongraw-Chaffin, M. L., Cirillo, P. M., and Cohn, B. A. (2010). Preeclampsia and cardiovascular disease death: prospective evidence from the child health and development studies cohort. Hypertension 56, 166–171. doi: 10.1161/HYPERTENSIONAHA.110.150078
Muchova, L., Wong, R. J., Hsu, M., Morioka, I., Vitek, L., Zelenka, J., et al. (2007). Statin treatment increases formation of carbon monoxide and bilirubin in mice: a novel mechanism of in vivo antioxidant protection. Can. J. Physiol. Pharmacol. 85, 800–810. doi: 10.1139/Y07-077
Murphy, S. R., Babbette, B., LaMarca D., Cockrell, K., and Granger, J. P. (2010). Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension 55, 394–398. doi: 10.1161/HYPERTENSIONAHA.109.141473
Murphy, S. R., LaMarca, B., Cockrell, K., Arany, M., and Granger, J. P. (2011). L-arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R259–R263. doi: 10.1152/ajpregu.00319.2011
Mutter, W. P., and Karumanchi, S. A. (2008). Molecular mechanisms of preeclampsia. Microvasc. Res. 75, 1–8. doi: 10.1016/j.mvr.2007.04.009
Nagamatsu, T., Fujii, T., Kusumi, M., Zou, L., Yamashita, T., Osuga, Y., et al. (2004). Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: an implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology 145, 4838–4845. doi: 10.1210/en.2004-0533
Nova, A., Sibai, B. M., Barton, J. R., Mercer, B. M., and Mitchell, M. D. (1991). Maternal plasma level of endothelin is increased in preeclampsia. Am. J. Obstet. Gynecol. 165, 724–727. doi: 10.1016/0002-9378(91)90317-K
Oshima, M., Oshima, H., and Taketo, M. M. (1996). TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 179, 297–302. doi: 10.1006/dbio.1996.0259
Ozcan, U., Yilmaz, E., Ozcan, L., Furuhashi, M., Vaillancourt, E., Smith, R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140. doi: 10.1126/science.1128294
Park, J. E., Chen, H. H., Winer, J., Houck, K. A., and Ferrara, N. (1994). Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 269, 25646–25654.
Parrish, M., Murphy, S. R., Rutland, S., Wallace, K., Wenzel, K., Wallukat, G., et al. (2010). The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am. J. Hypertens. 23, 911–916. doi: 10.1038/ajh.2010.70
Polhemus, D., Kondo, K., Bhushan, S., Bir, S. C., Kevil, C. G., Murohara, T., et al. (2013). Hydrogen sulfide attenuates cardiac dysfunction following heart failure via induction of angiogenesis. Circ. Heart Fail. 6, 1077–1086. doi: 10.1161/CIRCHEARTFAILURE.113.000299
Poston, L., Briley, A. L., Seed, P. T., Kelly, F. J., and Shennan, A. H. (2006). Vitamin, C. and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): randomised placebo-controlled trial. Lancet 367, 1145–1154. doi: 10.1016/S0140-6736(06)68433-X
Poston, L., Igosheva, N., Mistry, H. D., Seed, P. T., Shennan, A. H., Rana, S., et al. (2011). Role of oxidative stress and antioxidant supplementation in pregnancy disorders. Am. J. Clin. Nutr. 94(Suppl.), 1980S–1985S. doi: 10.3945/ajcn.110.001156
Redecha, P., van Rooijen, N., Torry, D., and Girardi, G. (2009). Pravastatin prevents miscarriages in mice: role of tissue factor in placental and fetal injury. Blood 113, 4101–4109. doi: 10.1182/blood-2008-12-194258
Redman, C. W. G., and Sargent, I. L. (2000). Placental debris, oxidative stress and pre-eclampsia. Placenta 21, 597–602. doi: 10.1053/plac.2000.0560
Redman, C. W., and Sargent, I. L. (2005). Latest advances in understanding preeclampsia. Science 308, 1592–1594. doi: 10.1126/science.1111726
Redman, C. W., Sargent, I. L., and Staff, A. C. (2014). IFPA Senior Award Lecture: making sense of pre-eclampsia - two placental causes of preeclampsia? Placenta 35(Suppl.), S20–S25. doi: 10.1016/j.placenta.2013.12.008
Roberts, J. M. (1998). Endothelial dysfunction in preeclampsia. Semin. Reprod. Endocrinol. 16, 5–15. doi: 10.1055/s-2007-1016248
Roberts, J. M., and Cooper, D. W. (2001). Pathogenesis and genetics of pre-eclampsia. Lancet 357, 53–56. doi: 10.1016/S0140-6736(00)03577-7
Rolfo, A., Many, A., Racano, A., Tal, R., Tagliaferro, A., Letta, F., et al. (2010). Abnormalities in oxygen sensing define early and late onset preeclampsia as distinct pathologies. PLoS ONE 5:e13288. doi: 10.1371/journal.pone.0013288
Rumbold, A. R., Crowther, C. A., Haslam, R. R., Dekker, G. A., and Robinson, J. S. (2006). Vitamins, C. and E and the risks of preeclampsia and perinatal complications. N. Engl. J. Med. 354, 1796–1806. doi: 10.1056/NEJMoa054186
Saad, A. F., Kechichian, T., Yin, H., Sbrana, E., Longo, M., Wen, M., et al. (2014). Effects of pravastatin on angiogenic and placental hypoxic imbalance in a mouse model of preeclampsia. Reprod. Sci. 21, 138–145. doi: 10.1177/1933719113492207
Sandrim, V. C., Palei, A. C., Metzger, I. F., Gomes, V. A., Cavalli, R. C., Tanus-Santos, J. E., et al. (2008). Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble fms-like tyrosine kinase-1 and soluble endogline in preeclampsia. Hypertension 52, 402–407. doi: 10.1161/HYPERTENSIONAHA.108.115006
Savvidou, M. D., Hingorani, A. D., Tsikas, D., Frölich, J. C., Vallance, P., Nicolaides, K. H., et al. (2003). Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre-eclampsia. Lancet 361, 1511–1517. doi: 10.1016/S0140-6736(03)13177-7
Schnabel, R., Blankenberg, S., Lubos, E., Lackner, K. J., Rupprecht, H. J., Espinola-Klein, C., et al. (2005). Asymmetric dimethylarginine and the risk of cardiovascular events and death in patients with coronary artery disease: results from the AtheroGene Study. Circ. Res. 97, e53–e59. doi: 10.1161/01.RES.0000181286.44222.61
Sedeek, M., Gilbert, J. S., LaMarca, B. B., Sholook, M., Chandler, D. L., et al. (2008). Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. Am. J. Hypertens. 21, 1152–1156. doi: 10.1038/ajh.2008.239
Sen, N., Paul, B. D., Gadalla, M. M., Mustafa, A. K., Sen, T., Xu, R., et al. (2012). Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell 45, 13–24. doi: 10.1016/j.molcel.2011.10.021
Shao, E. S., Lin, L., Yao, Y., and Boström, K. I. (2009). Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood 114, 2197–2206. doi: 10.1182/blood-2009-01-199166
Sibai, B., Dekker, G., and Kupferminc, M. (2005). Pre-eclampsia. Lancet 365, 785–799. doi: 10.1016/S0140-6736(05)17987-2
Sodha, N. R., Clements, R. T., Feng, J., Liu, Y., Bianchi, C., Horvath, E. M., et al. (2009). Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J. Thorac. Cardiovasc. Surg. 138, 977–984. doi: 10.1016/j.jtcvs.2008.08.074
Steinberg, G., Khankin, E. V., and Karumanchi, S. A. (2009). Angiogenic factors and preeclampsia. Thromb. Res. 123(Suppl. 2), S93–S99. doi: 10.1016/S0049-3848(09)70020-9
Sugimoto, H., Hamano, Y., Charytan, D., Cosgrove, D., Kieran, M., Sudhakar, A., et al. (2003). Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J. Biol. Chem. 278, 12605–12608. doi: 10.1074/jbc.C300012200
Taylor, R. N., Grimwood, J., Taylor, R. S., McMaster, M. T., Fisher, S. J., North, R. A., et al. (2003). Longitudinal serum concentrations of placental growth factor: evidence for abnormal placental angiogenesis in pathologic pregnancies. Am. J. Obstet. Gynecol. 188, 177–182. doi: 10.1067/mob.2003.111
Taylor, R. N., Varma, M., Teng, N. N., and Roberts, J. M. (1990). Women with preeclampsia have higher plasma endothelin levels than women with normal pregnancies. J. Clin. Endocrinol. Metab. 71, 1675–1677. doi: 10.1210/jcem-71-6-1675
Vadillo-Ortega, F., Perichart-Perera, O., Espino, S., Avila-Vergara, M. A., Ibarra, I., Ahued, R., et al. (2011). Effect of supplementation during pregnancy with L-arginine and antioxidant vitamins in medical food on pre-eclampsia in high risk population: randomised controlled trial. BMJ 342, d2901. doi: 10.1136/bmj.d2901
van Pampus, M. G., and Aarnoudse, J. G. (2005). Long-term outcomes after preeclampsia. Clin. Obstet. Gynecol. 48, 489–494. doi: 10.1097/01.grf.0000160316.67359.3d
Venkatesha, S., Toporsian, M., Lam, C., Hanai, J., Mammoto, T., Kim, Y. M., et al. (2006). Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 12, 642–649. doi: 10.1038/nm1429
Walsh, S. W. (1998). Maternal-placental interactions of oxidative stress and antioxidants in preeclampsia. Semin. Reprod. Endocrinol. 16, 93–104. doi: 10.1055/s-2007-1016256
Waltenberger, J., Claesson-Welsh, L., Siegbahn, A., Shibuya, M., and Heldin, C. H. (1994). Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 269, 26988–26995.
Wang, K., Ahmad, S., Cai, M., Rennie, J., Fujisawa, T., Crispi, F., et al. (2013). Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 127, 2514–2522. doi: 10.1161/CIRCULATIONAHA.113.001631
Wang, M. J., Cai, W. J., Li, N., Ding, Y. J., Chen, Y., Zhu, Y. C., et al. (2010). The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid. Redox Signal. 12, 1065–1077. doi: 10.1089/ars.2009.2945
Wang, R. (2009). Is H2S a stinky remedy for atherosclerosis? Arterioscler. Thromb. Vasc. Biol. 29, 156–157. doi: 10.1161/ATVBAHA.108.180190
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi: 10.1172/JCI26373
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590. doi: 10.1126/science.1162667
Yong, Q. C., Tang, Z. H., Xie, W., Shen, X. T., Liu, M. H., Peng, X. P., et al. (2008). Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. Am. J. Physiol. Heart Circ. Physiol. 295, H1330–H1340. doi: 10.1152/ajpheart.00244.2008
Yoshida, H. (2007). ER stress and diseases. FEBS J. 274, 630–658. doi: 10.1111/j.1742-4658.2007.05639.x
Young, B. C., Levine, R. J., and Karumanchi, S. A. (2010). Pathogenesis of preeclampsia. Annu. Rev. Pathol. 5, 173–192. doi: 10.1146/annurev-pathol-121808-102149
Yung, H. W., Calabrese, S., Hynx, D., Hemmings, B. A., Cetin, I., Charnock-Jones, D. S., et al. (2008). Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 173, 451–462. doi: 10.2353/ajpath.2008.071193
Yung, H. W., Hemberger, M., Watson, E. D., Senner, C. E., Jones, C. P., Kaufman, R. J., et al. (2012). Endoplasmic reticulum stress disrupts placental morphogenesis: implications for human intrauterine growth restriction. J. Pathol. 228, 554–564. doi: 10.1002/path.4068
Zhang, K., and Kaufman, R. J. (2008). From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462. doi: 10.1038/nature07203
Zhao, W., Zhang, J., Lu, Y., and Wang, R. (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 20, 6008–6016. doi: 10.1093/emboj/20.21.6008
Zhou, C., Ahmad, S., Mi, T., Abbasi, S., Xia, L., Day, M. C. (2008). Autoantibody from women with preeclampsia induces soluble Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated T-cells signaling. Hypertension 51, 1010–1019. doi: 10.1161/HYPERTENSIONAHA.107.097790
Zhou, J., and Brüne, B. (2006). Cytokines and hormones in the regulation of hypoxia inducible factor-1alpha (HIF-1alpha). Cardiovasc. Hematol. Agents Med. Chem. 4, 189–197. doi: 10.2174/187152506777698344
Zhu, H., Itoh, K., Yamamoto, M., Zweier, J. L., and Li, Y. (2005). Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett. 579, 3029–3036. doi: 10.1016/j.febslet.2005.04.058
Keywords: preeclampsia, angiogenesis, antioxidants, ER chaperones, NO, H2S, statins
Citation: Cindrova-Davies T (2014) The therapeutic potential of antioxidants, ER chaperones, NO and H2S donors, and statins for treatment of preeclampsia. Front. Pharmacol. 5:119. doi: 10.3389/fphar.2014.00119
Received: 31 March 2014; Accepted: 02 May 2014;
Published online: 27 May 2014.
Edited by:
Carlos Alonso Escudero, Universidad del Bío-Bío, ChileReviewed by:
Carmen Martínez, Université d’Angers, FranceBrett Mitchell, Texas A&M Health Science Center, USA
Copyright © 2014 Cindrova-Davies. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tereza Cindrova-Davies, Centre for Trophoblast Research and Department of Physiology, Development and Neuroscience, University of Cambridge, Downing Street, Cambridge, CB2 3EG, UK e-mail:dGMyNjlAY2FtLmFjLnVr