 Simone L. Cree
Simone L. Cree Martin A. Kennedy
Martin A. Kennedy- Department of Pathology, Carney Centre for Pharmacogenomics, University of Otago, Christchurch, New Zealand
G-quadruplexes are non-canonical secondary structures formed within nucleic acids that are involved in modulating cellular processes such as replication, gene regulation, recombination and epigenetics. Within genes, there is mounting evidence of G-quadruplex involvement in transcriptional and post transcriptional regulation. We report the presence of potential G-quadruplex motifs within relevant sites of some important pharmacogenes and discuss the possible implications of this on the function and expression of these genes. Appreciating the location and potential functions of these motifs may be of value when considering the impacts of some pharmacogenetic variants. G-quadruplexes are also the focus of drug development efforts in oncology and we highlight the broader pharmacological implications of treatment strategies that may target G-quadruplexes.
Introduction
The desire to implement pharmacogenetic testing as a means to improve drug safety and treatment efficacy has led to intense scrutiny of the functional and clinical relevance of variation in pertinent genes (Sim et al., 2013). Over 300 human genes are recognized to be involved in the absorption, distribution, metabolism, and excretion of drugs, as well as encoding protein targets for therapeutics (Sangkuhl et al., 2008; Madian et al., 2012). Extensive cataloging of variants within these genes has been carried out (Nelson et al., 2012), with the 1000 Genome Project providing a global view of such variants (Durbin et al., 2010). In addition, genome wide association studies (GWAS) and high throughput gene expression analyses are implicating new genes in drug response phenotypes (Daly, 2012; Madian et al., 2012; Wheeler and Dolan, 2012). Our ability to interpret the clinical significance of this growing catalog of gene variants depends on a fuller appreciation of functional genomic features (Sadee et al., 2011). Here we seek to highlight the importance of a genomic feature called the G-quadruplex that has been implicated in many critical cellular functions, and which may be of considerable interest to those working in pharmacogenomics.
G-quadruplexes (G4s) are stable secondary structures found in G rich nucleic acids wherein guanine bases associate via Hoogsteen hydrogen bonds to form tetrads (Figure 1A) that stack in a planar arrangement with a stabilizing monovalent cation occupying a central position of the cavity (Sen and Gilbert, 1988; Sundquist and Klug, 1989). G4s can form in one strand (Figure 1B) or from multiple strands where sequences intervening G tracts of two or more guanines exist as loops of various sizes (Burge et al., 2006). The stability and topology of G4 are influenced by several factors including length and sequence composition, strand stoichiometry, and orientation, nature of the binding cation and loop size between G tracts with longer tracts and small sized loops contributing to more stable structures (Bugaut and Balasubramanian, 2008; Guedin et al., 2010). The existence and topology of G4s can be studied in vitro by methods such as circular dichroism, nuclear magnetic resonance, and X-ray crystallography whereas fluorescence imaging, small molecule and protein pull downs, ChIP-Seq and ChIP-chip experiments can be designed to detect G4s in cellular DNA (Di Antonio et al., 2012b).
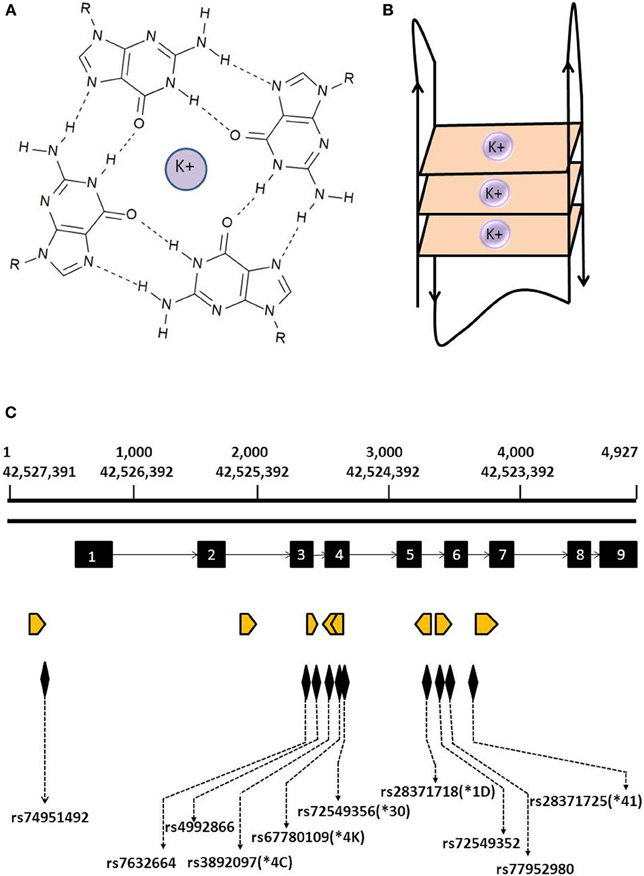
Figure 1. Structure of G4 and location of predicted G4s within the CYP2D6 gene. (A) Guanine tetrad formed by the association of four guanine bases via Hoogsten hydrogen bonds in a coplanar arrangement and stabilized by a potassium ion. (B) Schematic representation of a G-quadruplex formed by the stacking of the three guanine tetrads from a single strand of DNA. (C) Location of predicted G4s in the CYP2D6 gene. Orange arrowheads indicate location and orientation of predicted G4s; exons are depicted by black blocks. Also indicated are the locations of SNPs and CYP2D6* alleles found within predicted G4s.
The human genome contains more than 375,000 predicted G4 forming sequences (Huppert and Balasubramanian, 2005) with clusters of them occurring in biologically important regions such as telomeres, promoters, 5′-untranslated regions (UTRs), 3′UTRs, replication origins, exons and introns. G4s are implicated in maintenance of chromosomal ends, transcription, translation, DNA replication, alternative splicing, recombination, and epigenetic stability (Maizels and Gray, 2013). It is not yet clear how often putative G4 motifs actually give rise to G4 structures in vivo, although this process is expected to be tightly regulated within different cell types. Evidence for G4 formation in vivo has come from the identification of G4 interacting cellular proteins and the ability of small molecules that bind G4 to affect transcription, translation and replication (Rodriguez, 2012; Biffi et al., 2013; Valton et al., 2014). Furthermore, the location and composition of G4s are conserved in human populations, implying that they play an important role in cellular processes (Eddy and Maizels, 2008).
G4 formation in the promoters of proto-oncogenes has been linked with repression of transcription; most likely due to polymerase pausing at the site of G4 formation. Addition of small molecule ligands was found to stabilize G4s in the promoters of the proto-oncogene RET (Shin et al., 2014) and the gene encoding tumor angiogenesis factor VEGF (Salvati et al., 2014) resulting in altered protein expression. Likewise, RNA G4s in the 5′UTR of the matrix metalloproteinase (MT3-MMP), estrogen receptor (ESR1), apoptotic regulator (BCL2), telomere shelterin protein (TRF2) and several other proteins have been reported to negatively modulate translation in gene-reporter expression studies (Morris and Basu, 2009; Bugaut and Balasubramanian, 2012). RNA G4s have also been shown to play a role in alternative splicing and expression patterns of genes Bcl-xL (Hai et al., 2008) and FMR1(Didiot et al., 2008) while modulation of G4 formation in the third intron of the tumor supressor gene TP53 was recently shown to result in varied expression of the p53 transcript (Marcel et al., 2011).
Owing to the abundance of predicted G4s in the telomeric regions of the genome and in promoters of many oncogenes, including MYC, G4 represent an appealing and novel target for small-molecule chemotherapeutic development (Verma et al., 2009; Neidle, 2010). Inhibition of MYC transcription was achieved by exposing HeLa S3 cells to the small molecule TMPyP4, which binds to its promoter G4, providing proof of principle for the targeting of regulatory G4 structures of oncogenes as a novel therapeutic strategy (Siddiqui-Jain et al., 2002). Likewise, ongoing research is directed toward promoter G4 mediated transcriptional repression of proto-oncogenes such as the receptor tyrosine kinase KIT, the small GTPase KRAS, and more recently of an RNA G4 in the 5′ UTR region of the small GTPase gene NRAS (Heinrich et al., 2003; Bugaut et al., 2010).
Given the apparent significance of G4s, we have analyzed the occurrence of potential G-quadruplex forming sequence within “Very Important Pharmacogenes” (VIP) (Whirl-Carrillo et al., 2012) using web based G4 prediction algorithms Quadparser (Wong et al., 2010) and QGRSH (Kikin et al., 2006). The VIP is a manually curated list of genes with well described pharmacogenetic relationships which includes genes that encode drug targets, metabolic enzymes as well as drug transporters. It is by no means an exclusive listing of all known pharmacogenes, but it includes many of the best understood and widely studied pharmacogenes. Our analysis indicates that predicted G4 are found in the promoters, 5′UTRs, 3′UTRs, exons, and introns of pharmacogenes. The position and location of predicted G4 within the VIP suggests the potential for G4 structures to impact on pharmacogene expression (Supplementary Table 1).
G4s and the Regulation of Pharmacogenes
Promoter G4s Function as Transcriptional Regulatory Elements
Studies focused on interaction of promoter G4s within the cell have revealed a role in transcriptional regulation. The proto-oncogene MYC which is overexpressed in more than 80% of solid tumors contains a G4 forming region in the nuclease hypersensitive element of its promoter (Siddiqui-Jain et al., 2002). The nuclear protein nucleolin promotes G4 formation leading to transcriptional repression (Gonzalez et al., 2009). Conversely, transcriptional activation of this gene is induced by the G4-unwinding activity of the nucleoside diphosphate kinase NM23H2 protein (Postel et al., 1993) and poly ADP-ribose polymerase (Fekete et al., 2012). Palumbo et al. (2008) found that multiple GGA repeats within the C-MYB promoter form a G4 that acts as a transcriptional repressor by interacting with Myc-associated Zn finger proteins. Likewise, transcriptional repression of the human PDGFB gene was observed by ligand-mediated stabilization of its promoter G4 (Qin et al., 2010). In addition, it appears that the transcriptional regulatory protein SP1 can bind to G4 structures with high affinity, implying a role for transcription factor binding sites to form G4 as a key determinant for regulation of some genes (Raiber et al., 2012).
More than 40% of human gene promoters contain at least one predicted G4 (Huppert and Balasubramanian, 2007); in line with this our analysis of the VIP list (Whirl-Carrillo et al., 2012) indicates the occurrence of at least one promoter predicted G4 in 38% of these genes (Supplementary Table 1). Among these are genes encoding important metabolic enzymes (CYP2D6, CYP2J2, G6PD, TPMT), receptor proteins (ADRB1, ADRB2, DRD2, VDR), folate transporter (SLC19A1) and proteins involved in potassium voltage-gated and sodium channels (KCNH2, SCN5A). Presence of predicted G4 in the promoters of these pharmacogenes is suggestive of transcriptional regulation at the promoter G4 site. Mutations or polymorphisms affecting these predicted G4 could alter stability of secondary structures impacting gene expression and activity, and this may be important when considering the regulatory potential of genetic variants. This type of an effect was demonstrated by the recent observation at rs2255888 of the lipoxygenase gene ALOX15 promoter implicated in pathogenesis of atherosclerosis. Using biophysical and in vitro studies the G but not the A allele at this position was found to be involved in the DNA secondary structure formation (although this did not appear to be a G4), affecting binding of the cytoskeleton protein vimentin and resulting in altered transcriptional regulation (Samanta et al., 2012).
Role of RNA G4s in Post Transcriptional Regulation
Evidence has emerged for the role of G4s not only in transcription but also in post transcriptional regulation, as G4s can readily form in RNA molecules (Biffi et al., 2014). RNA G4s occur within the 5′UTR, exons, introns and 3′UTRs where they regulate translation, alternative splicing and expression patterns (Marcel et al., 2011; Bugaut and Balasubramanian, 2012; Endoh et al., 2013; Murat et al., 2014). G4s formed in the 5′UTR were found to play a role in translational down-regulation of genes with cap-dependent translation (Morris and Basu, 2009; Bugaut and Balasubramanian, 2012) whereas they have been shown to mediate internal ribosome entry site translation initiation in cap-independent translation (Bonnal et al., 2003; Morris et al., 2010).
Our analysis revealed G4 motifs in the 5′UTRs of the pharmacogenes G6PD, GSTP1, KCNH2, and PTGIS (Supplementary Table 1), suggesting these motifs have the potential to affect translational efficiencies and modulate protein expression levels. Detailed in silico, in vitro and cellular analysis of SNPs among 5′UTR predicted RNA G4s provide further evidence for G4 involvement in translation (Beaudoin and Perreault, 2010). Beaudoin and Perreault (2010) demonstrated that a G to C substitution within the 5′UTR RNA G4 of AASDHPPT (L-aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl transferase) restricted G4 formation by favoring stem loop structures, which led to increased translation efficiency of a reporter gene. This example also illustrates the potential significance of SNPs within G4 of 5′UTRs in causing differential gene expression among individuals.
Our investigation of G4 motifs in the VIP gene set also reveals that 20% of the genes queried contain a predicted G4 in their 3′UTR. This includes ACE, ADRB1, CYP2A6, G6PD, MTHFR, SLC19A1, and VDR (Supplementary Table 1). The presence of predicted G4s in the 3′UTR implies a role for RNA G4s in other stages of RNA metabolism. A 3′UTR in the PIM1 proto-oncogene was reported to reduce translation efficiency (Arora and Suess, 2011). Recent findings indicate that 3′UTR G4s can also regulate gene expression by increasing the efficiencies of alternate polyadenylation sites leading to expression of shorter transcripts, and eluding of the miRNA regulatory network (Beaudoin and Perreault, 2013).
Potential G4s in the CYP2D6 Gene
The highly polymorphic CYP2D6 gene is of particular pharmacogenetic interest owing to its implications for metabolism of a variety of drugs (Becker and Leeder, 2010). We detected a total of eight predicted G4s in this gene (Supplementary Table 1; Figure 1C) with one present 437 bp upstream from the transcriptional start site in the promoter region which could potentially influence transcriptional regulation. We also observed predicted G4 within introns 2, 3, 5, and 6 in an orientation such that they would occur in nuclear pre-mRNA transcripts (Figure 1C), with possible relevance to RNA processing. Notably, some of the CYP2D6* alleles involved in poor and rapid metabolizer phenotypes are situated within predicted G4s. Among these are CYP2D6*1D (rs28371718), CYP2D6*4K (rs67780109), CYP2D6*4C (rs3892097), CYP2D6*30 (rs72549356) and CYP2D6*41 (rs28371725). Furthermore, dbSNPs rs76326664 (A/G), rs77952980 (A/G), rs4992866 (C/T) are located within predicted G4 in CYP2D6 introns where they may affect G4 formation and stability. In addition to SNPs, mutations such as insertions, deletions, and gene rearrangements all have the ability to exert effects on G4 stability and hence perturb their role in transcriptional or translational regulation.
G4s as Drug Targets
G4 Interacting Ligands in Cancer Therapy
The large amount of research into G4 ligand induced gene regulation over the last decade has led to several G4 targeting compounds that are currently under investigation as potential chemotherapeutic drugs. G4 targeting anticancer drugs evaluated for some well-studied proto-oncogenes alongside their advantage and disadvantages are listed in Table 1. The first line of ligands such as the tri-substituted acridines targeted the terminal quartet present at the top and bottom of the G4 structure (Mergny et al., 2002). Platinum-derived complexes have been shown to preferentially target the MYC G4 structure over duplex DNA (Wang et al., 2013). The fluoroquinone derivative quarfloxin was the first G4-interacting compound to reach Phase II clinical trials, with potential for the treatment of neuroendocrine tumors (Drygin et al., 2009). Its ability to strongly interact with parallel G4 structures has been studied in detail within the cellular context. Although treatment with quarfloxin revealed the absence of organ or genotoxicity, it led to the disruption of interaction between nucleolin and ribosomal RNA G4 with consequent inhibition of Pol I driven transcription and redistribution of nucleolin within the cell, limiting its bioavailability (Balasubramanian et al., 2011).
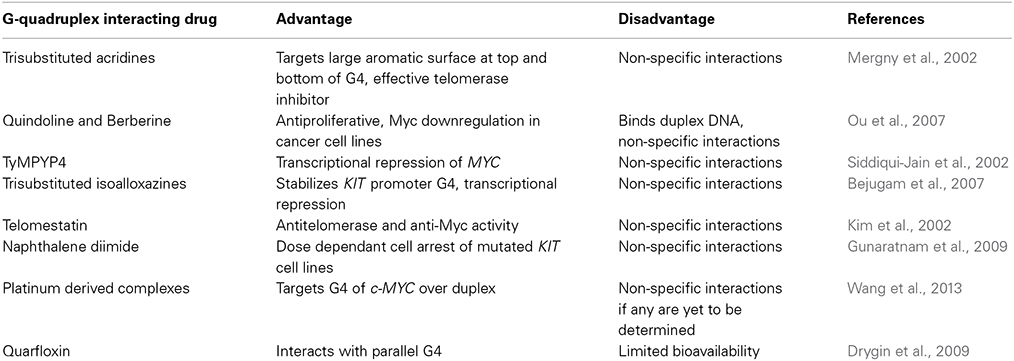
Table 1. List of G4 binding anticancer drugs tested.
As chemotherapeutic drugs that target regulatory G4 enter and move through clinical trials it may be prudent to consider whether binding of drugs to non-target G4s could impact on expression of genes that are important in metabolism of other chemotherapeutic drugs and xenobiotics. We have identified predicted G4 in a number of pharmacogenes relevant to the treatment of cancer (Supplementary Table 1) which could conceivably be impacted by such off target effects of G4 drugs. For example, if a G4 targeting drug also targets the predicted G4 in the promoter of CYP2D6 gene that is involved in transcriptional repression, it may result in reduced efficacy of other drugs that are metabolized by CYP2D6, like tamoxifen (Province et al., 2014). Similar situations could arise if the drug targeted G4s within the TPMT or UGT1A1 gene whose products are involved in metabolism of thiopurine drugs (Weinshilboum, 2001), and irinotecan (Wang et al., 2011) respectively.
More Widespread Opportunities for G4 Based Therapeutics?
Although much effort has been directed toward understanding G4s in proto-oncogenes and more recently cardiovascular genes (Zhou et al., 2013), there has been little consideration of the potential significance of G4s in many of the genes that impact on responses to treatment of these and other diseases. Among these are the G-protein coupled receptors, ADRB1 and ADRB2 involved in mediating heart rate, contractibility, bronchodilator response and cardiomyopathy (Sandilands and O'shaughnessy, 2005; Lymperopoulos and Bathgate, 2012). It is worth considering whether G4 formation in these and other important pharmacogenes could represent valid targets for modification of drug metabolism using targeted therapeutics.
We detected putative G4 in the promoter and 5′UTR of the TPMT gene (Supplementary Table 1), and it may be of value to investigate the formation of G4s and the role they may play in the regulation of this gene product as it is involved in the treatment of autoimmune diseases, inflammatory bowel disease, lupus, transplantation, and acute lymphoblastic leukemia (Lennard, 2002). We also described variant trinucleotide repeats within the promoter of TPMT from two patients with ultra-high activity of this enzyme, which showed increased transcriptional activity in reporter gene assays (Roberts et al., 2008). These GCC repeat arrays may be capable of forming G4 structures, and different forms of these could conceivably contribute to different levels of transcriptional activity.
In addition to CYP2D6, discussed above, other predicted G4 containing pharmacogenes include UGT1A1, involved in bilirubin metabolism and degradation and removal of xenobiotic waste (Tukey and Strassburg, 2000); CYP2A6, best known for its role in conversion of nicotine to cotinine (Benowitz et al., 2006); and MTHFR, important in folate and methionine biosynthesis (Marini et al., 2008) as well as de novo purine biosynthesis (Jongbloet et al., 2008) and DNA methylation. Determining the formation of G4 structures at these sites and establishing whether this is of pharmacogenetic significance is important as there may be merit in exploring novel strategies for targeting G4 mediated regulation in these and other genes.
Future of G4 Drug Targeting
The quest to identify and devise G4 binding ligands has garnered much attention in the last few years. Development of small molecules with high specificity and affinity for a particular G4 could also be achieved by using structure-based design methods and screening against virtual compound libraries (Ma et al., 2011). Investigation of the alkyl derivative of TMPyP4 (TMpyP4-C14), led to the observation that TMpyP4-C14 could efficiently enter cells and preferentially localize into the cytoplasm with binding to G4 structures in the 5′UTR of KRAS mRNA, resulting in 90% down regulation of KRAS protein expression in pancreatic cancer cells (Xodo et al., 2008). Small molecules like the pyridine-2,6-bis-quinolinodicarboxamide derivative and its variant have been shown to specifically decrease translation efficiency by stabilizing a G4 in the 5′UTR of the NRAS proto-oncogene (Bugaut and Balasubramanian, 2012). In situ click chemistry is another promising approach aimed at enhancing the interaction specificity of a small molecule G4 ligand to its corresponding nucleic acid structure (Di Antonio et al., 2012a). Virtual screening approaches on the other hand have identified novel G4 ligands (Alcaro et al., 2013; Gonzalez et al., 2013). Another approach to molecules with selective affinity for specific G4 is to generate specific antibodies (Fernando et al., 2009). Huppert (2007) presented the idea of adding functional specificity to the G4 binding ligand by tagging it with sequence specific complementarity to its neighboring DNA. The biological activity of these compounds depends on the structural interaction of G4 small molecule ligand complexes to disrupt normal regulatory processes that may involve the targeted G4 structure.
G4 aptamers that bind to and block clinically relevant proteins are the converse situation, and the prototypical molecule of this class is the thrombin binding G4 aptamer which inhibits thrombin catalyzed clot formation (Nagatoishi et al., 2011). Similarly, HIV DNA integration into the genome was shown to be inhibited by binding of G4 aptamers to the HIV integrase protein (Do et al., 2011). Finally, G4 decoys could be explored in treating patients that harbor G4 alleviating SNPs in clinically relevant genes. This strategy was shown to successfully mediate apoptosis in HeLa and T24 urinary bladder cancer cells expressing the hyper activated HRAS protein. G4 decoys designed to mimic the promoter G4s of the HRAS proto-oncogene functioned as transcriptional repressors (Membrino et al., 2011). Introduction of G4 decoys also resulted in effective inhibition of KRAS and tumor growth arrest in pancreatic cancer cells (Cogoi et al., 2013).
Conclusion
Among the many DNA and RNA structures that have been described, G4s have gained increasing prominence because of their implication in so many fundamental cellular, evolutionary and genomic processes (Wu and Brosh, 2010; Maizels and Gray, 2013). Although there is an increased understanding of the role played by G4s in a number of cellular processes the structural intricacies of G4 motifs associated with specific function remain to be explored. Small molecule based G4 binding ligands have proved successful in regulating transcription and expression of proto-oncogenes, providing a novel approach to the treatment of cancerous cells (Siddiqui-Jain et al., 2002; Gunaratnam et al., 2009). Our survey revealed several important pharmacogenes that harbor one or more predicted G4 in relevant sites that may contribute to regulation and expression. Investigating the role of these predicted G4s may also provide evidence for alternative targets through which specific regulation of pharmacogenes could be achieved in situations where this may confer clinical benefit. While G4s located in regulatory regions of pharmacogenes may provide opportunities for pharmacokinetic modulation by small molecules that target G4, they also represent a risk for off-target regulatory effects of chemotherapeutics designed to target regulatory G4 in oncogenes. For example, tamoxifen and codeine are both prodrugs that are activated by CYP2D6. If a small molecule designed to target an oncogene G4 also acted on the G4 sites in CYP2D6, reducing expression of this metabolic enzyme, unexpected effects on the efficacy of tamoxifen or codeine could occur. Alternatively, if the chemotherapeutic drug itself was a substrate of CYP2D6, then concurrent modulation of CYP2D6 via off-target G4 binding could markedly affect the pharmacokinetic behavior of the drug.
Finally, G4 structures are also vulnerable to modulation by mutation or polymorphism, so our analysis has the potential to assign regulatory significance to newly discovered pharmacogenetic variants that may affect function by impacting on such structural features. Because pharmacogenes, like oncogenes and other classes of genes, have the potential to be regulated by formation of G4s within either genomic DNA or RNA transcripts, it is our hope that the knowledge of G4 structures and consideration of their potential to be modified may provide useful insights in the field of pharmacogenomics.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by the Carney Centre for Pharmacogenomics, and the Marsden Fund Council (New Zealand) administered by the Royal Society of New Zealand.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fphar.2014.00160/abstract
References
Alcaro, S., Musetti, C., Distinto, S., Casatti, M., Zagotto, G., Artese, A., et al. (2013). Identification and characterization of new DNA G-quadruplex binders selected by a combination of ligand and structure based virtual screening approaches. J. Med. Chem. 56, 843–855. doi: 10.1021/jm3013486
Arora, A., and Suess, B. (2011). An RNA G-quadruplex in the 3′ UTR of the proto-oncogene PIM1 represses translation. RNA Biol. 8, 802–805. doi: 10.4161/rna.8.5.16038
Balasubramanian, S., Hurley, L. H., and Neidle, S. (2011). Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug discov. 10, 261–275. doi: 10.1038/nrd3428
Beaudoin, J. D., and Perreault, J. P. (2010). 5′-UTR G-quadruplex structures acting as translational repressors. Nucleic Acids Res. 38, 7022–7036. doi: 10.1093/nar/gkq557
Beaudoin, J. D., and Perreault, J. P. (2013). Exploring mRNA 3′-UTR G-quadruplexes: evidence of roles in both alternative polyadenylation and mRNA shortening. Nucleic Acids Res. 41, 5898–5911. doi: 10.1093/nar/gkt265
Becker, M. L., and Leeder, J. S. (2010). Identifying genomic and developmental causes of adverse drug reactions in children. Pharmacogenomics 11, 1591–1602. doi: 10.2217/pgs.10.146
Bejugam, M., Sewitz, S., Shirude, P. S., Rodriguez, R., Shahid, R., and Balasubramanian, S. (2007). Trisubstituted isoalloxazines as a new class of G-quadruplex binding ligands: small molecule regulation of c-kit oncogene expression. J. Am. Chem. Soc. 129, 12926–12927. doi: 10.1021/ja075881p
Benowitz, N. L., Swan, G. E., Jacob, P. 3rd., Lessov-Schlaggar, C. N., and Tyndale, R. F. (2006). CYP2A6 genotype and the metabolism and disposition kinetics of nicotine. Clin. Pharmacol. Ther. 80, 457–467. doi: 10.1016/j.clpt.2006.08.011
Biffi, G., Di Antonio, M., Tannahill, D., and Balasubramanian, S. (2014). Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat. Chem. 6, 75–80. doi: 10.1038/nchem.1805
Biffi, G., Tannahill, D., McCafferty, J., and Balasubramanian, S. (2013). Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 5, 182–186. doi: 10.1038/nchem.1548
Bonnal, S., Schaeffer, C., Creancier, L., Clamens, S., Moine, H., Prats, A. C., et al. (2003). A single internal ribosome entry site containing a G quartet RNA structure drives fibroblast growth factor 2 gene expression at four alternative translation initiation codons. J. Biol. Chem. 278, 39330–39336. doi: 10.1074/jbc.M305580200
Bugaut, A., and Balasubramanian, S. (2008). A sequence-independent study of the influence of short loop lengths on the stability and topology of intramolecular DNA G-quadruplexes. Biochemistry 47, 689–697. doi: 10.1021/bi701873c
Bugaut, A., and Balasubramanian, S. (2012). 5′-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res. 40, 4727–4741. doi: 10.1093/nar/gks068
Bugaut, A., Rodriguez, R., Kumari, S., Hsu, S. T., and Balasubramanian, S. (2010). Small molecule-mediated inhibition of translation by targeting a native RNA G-quadruplex. Org. Biomol. Chem. 8, 2771–2776. doi: 10.1039/c002418j
Burge, S., Parkinson, G. N., Hazel, P., Todd, A. K., and Neidle, S. (2006). Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 34, 5402–5415. doi: 10.1093/nar/gkl655
Cogoi, S., Zorzet, S., Rapozzi, V., Geci, I., Pedersen, E. B., and Xodo, L. E. (2013). MAZ-binding G4-decoy with locked nucleic acid and twisted intercalating nucleic acid modifications suppresses KRAS in pancreatic cancer cells and delays tumor growth in mice. Nucleic Acids Res. 41, 4049–4064. doi: 10.1093/nar/gkt127
Daly, A. K. (2012). Genetic polymorphisms affecting drug metabolism: recent advances and clinical aspects. Adv. Pharmacol. 63, 137–167. doi: 10.1016/B978-0-12-398339-8.00004-5
Di Antonio, M., Biffi, G., Mariani, A., Raiber, E. A., Rodriguez, R., and Balasubramanian, S. (2012a). Selective RNA vs. DNA G-quadruplex targeting by in situ click chemistry. Angew. Chem. Int. Ed. Engl. 51, 11073–11078. doi: 10.1002/anie.201206281
Di Antonio, M., Rodriguez, R., and Balasubramanian, S. (2012b). Experimental approaches to identify cellular G-quadruplex structures and functions. Methods 57, 84–92. doi: 10.1016/j.ymeth.2012.01.008
Didiot, M. C., Tian, Z., Schaeffer, C., Subramanian, M., Mandel, J. L., and Moine, H. (2008). The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 36, 4902–4912. doi: 10.1093/nar/gkn472
Do, N. Q., Lim, K. W., Teo, M. H., Heddi, B., and Phan, A. T. (2011). Stacking of G-quadruplexes: NMR structure of a G-rich oligonucleotide with potential anti-HIV and anticancer activity. Nucleic Acids Res. 39, 9448–9457. doi: 10.1093/nar/gkr539
Drygin, D., Siddiqui-Jain, A., O'brien, S., Schwaebe, M., Lin, A., Bliesath, J., et al. (2009). Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 69, 7653–7661. doi: 10.1158/0008-5472.CAN-09-1304
Durbin, R. M., Abecasis, G. R., Altshuler, D., Auton, A., Brooks, L. D., Gibbs, R. A., et al. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. doi: 10.1038/nature09534
Eddy, J., and Maizels, N. (2008). Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 36, 1321–1333. doi: 10.1093/nar/gkm1138
Endoh, T., Kawasaki, Y., and Sugimoto, N. (2013). Suppression of gene expression by G-quadruplexes in open reading frames depends on G-quadruplex stability. Angew. Chem. Int. Ed. Engl. 52, 5522–5526. doi: 10.1002/anie.201300058
Fekete, A., Kenesi, E., Hunyadi-Gulyas, E., Durgo, H., Berko, B., Dunai, Z. A., et al. (2012). The guanine-quadruplex structure in the human c-myc gene's promoter is converted into B-DNA form by the human poly(ADP-ribose)polymerase-1. PLoS ONE 7:e42690. doi: 10.1371/journal.pone.0042690
Fernando, H., Sewitz, S., Darot, J., Tavare, S., Huppert, J. L., and Balasubramanian, S. (2009). Genome-wide analysis of a G-quadruplex-specific single-chain antibody that regulates gene expression. Nucleic Acids Res. 37, 6716–6722. doi: 10.1093/nar/gkp740
Gonzalez, D. C., Machado, G. P., Guedin, A., Mergny, J. L., and And Cabrera-Perez, M. A. (2013). FDA-approved drugs selected using virtual screening bind specifically to G-Quadruplex DNA. Curr. Pharm. Des. 19, 2164. doi: 10.2174/1381612811319120004
Gonzalez, V., Guo, K., Hurley, L., and Sun, D. (2009). Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J. Biol. Chem. 284, 23622–23635. doi: 10.1074/jbc.M109.018028
Guedin, A., Gros, J., Alberti, P., and Mergny, J. L. (2010). How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 38, 7858–7868. doi: 10.1093/nar/gkq639
Gunaratnam, M., Swank, S., Haider, S. M., Galesa, K., Reszka, A. P., Beltran, M., et al. (2009). Targeting human gastrointestinal stromal tumor cells with a quadruplex-binding small molecule. J. Med. Chem. 52, 3774–3783. doi: 10.1021/jm900424a
Hai, Y., Cao, W., Liu, G., Hong, S. P., Elela, S. A., Klinck, R., et al. (2008). A G-tract element in apoptotic agents-induced alternative splicing. Nucleic Acids Res. 36, 3320–3331. doi: 10.1093/nar/gkn207
Heinrich, M. C., Corless, C. L., Demetri, G. D., Blanke, C. D., Von Mehren, M., Joensuu, H., et al. (2003). Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 21, 4342–4349. doi: 10.1200/JCO.2003.04.190
Huppert, J. L. (2007). Four-stranded DNA: cancer, gene regulation and drug development. Philos. Trans. A Math. Phys.Eng. Sci. 365, 2969–2984. doi: 10.1098/rsta.2007.0011
Huppert, J. L., and Balasubramanian, S. (2005). Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 33, 2908–2916. doi: 10.1093/nar/gki609
Huppert, J. L., and Balasubramanian, S. (2007). G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 35, 406–413. doi: 10.1093/nar/gkl1057
Jongbloet, P. H., Verbeek, A. L., Den Heijer, M., and Roeleveld, N. (2008). Methylenetetrahydrofolate reductase (MTHFR) gene polymorphisms resulting in suboptimal oocyte maturation: a discussion of folate status, neural tube defects, schizophrenia, and vasculopathy. J. Exp. Clin. Assis. Reprod. 5:5. doi: 10.1186/1743-1050-5-5
Kikin, O., D'antonio, L., and Bagga, P. S. (2006). QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 34, W676–W682. doi: 10.1093/nar/gkl253
Kim, M. Y., Vankayalapati, H., Shin-Ya, K., Wierzba, K., and Hurley, L. H. (2002). Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular g-quadruplex. J. Am. Chem. Soc. 124, 2098–2099. doi: 10.1021/ja017308q
Lennard, L. (2002). TPMT in the treatment of Crohn's disease with azathioprine. Gut 51, 143–146. doi: 10.1136/gut.51.2.143
Lymperopoulos, A., and Bathgate, A. (2012). Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: the known and the unknown. Pharmacogenomics 13, 323–341. doi: 10.2217/pgs.11.178
Ma, D. L., Chan, D. S., Lee, P., Kwan, M. H., and Leung, C. H. (2011). Molecular modeling of drug-DNA interactions: virtual screening to structure-based design. Biochimie 93, 1252–1266. doi: 10.1016/j.biochi.2011.04.002
Madian, A. G., Wheeler, H. E., Jones, R. B., and Dolan, M. E. (2012). Relating human genetic variation to variation in drug responses. Trends Genet. 28, 487–495. doi: 10.1016/j.tig.2012.06.008
Maizels, N., and Gray, L. T. (2013). The G4 Genome. PLoS Genet. 9:e1003468. doi: 10.1371/journal.pgen.1003468
Marcel, V., Tran, P. L., Sagne, C., Martel-Planche, G., Vaslin, L., Teulade-Fichou, M. P., et al. (2011). G-quadruplex structures in TP53 intron 3: role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis 32, 271–278. doi: 10.1093/carcin/bgq253
Marini, N. J., Gin, J., Ziegle, J., Keho, K. H., Ginzinger, D., Gilbert, D. A., et al. (2008). The prevalence of folate-remedial MTHFR enzyme variants in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 8055–8060. doi: 10.1073/pnas.0802813105
Membrino, A., Cogoi, S., Pedersen, E. B., and Xodo, L. E. (2011). G4-DNA formation in the HRAS promoter and rational design of decoy oligonucleotides for cancer therapy. PLoS ONE 6:e24421. doi: 10.1371/journal.pone.0024421
Mergny, J. L., Riou, J. F., Mailliet, P., Teulade-Fichou, M. P., and Gilson, E. (2002). Natural and pharmacological regulation of telomerase. Nucleic Acids Res. 30, 839–865. doi: 10.1093/nar/30.4.839
Morris, M. J., and Basu, S. (2009). An unusually stable G-quadruplex within the 5-UTR of the MT3 matrix metalloproteinase mRNA represses translation in eukaryotic cells. Biochemistry 48, 5313–5319. doi: 10.1021/bi900498z
Morris, M. J., Negishi, Y., Pazsint, C., Schonhoft, J. D., and Basu, S. (2010). An RNA G-quadruplex is essential for cap-independent translation initiation in human VEGF IRES. J. Am. Chem. Soc. 132, 17831–17839. doi: 10.1021/ja106287x
Murat, P., Zhong, J., Lekieffre, L., Cowieson, N. P., Clancy, J. L., Preiss, T., et al. (2014). G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 10, 358–364. doi: 10.1038/nchembio.1479
Nagatoishi, S., Isono, N., Tsumoto, K., and Sugimoto, N. (2011). Loop residues of thrombin-binding DNA aptamer impact G-quadruplex stability and thrombin binding. Biochimie 93, 1231–1238. doi: 10.1016/j.biochi.2011.03.013
Neidle, S. (2010). Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 277, 1118–1125. doi: 10.1111/j.1742-4658.2009.07463.x
Nelson, M. R., Wegmann, D., Ehm, M. G., Kessner, D., St Jean, P., Verzilli, C., et al. (2012). An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337, 100–104. doi: 10.1126/science.1217876
Ou, T. M., Lu, Y. J., Zhang, C., Huang, Z. S., Wang, X. D., Tan, J. H., et al. (2007). Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 50, 1465–1474. doi: 10.1021/jm0610088
Palumbo, S. L., Memmott, R. M., Uribe, D. J., Krotova-Khan, Y., Hurley, L. H., and Ebbinghaus, S. W. (2008). A novel G-quadruplex-forming GGA repeat region in the c-myb promoter is a critical regulator of promoter activity. Nucleic Acids Res. 36, 1755–1769. doi: 10.1093/nar/gkm1069
Postel, E. H., Berberich, S. J., Flint, S. J., and Ferrone, C. A. (1993). Human c-myc transcription factor PuF identified as nm23-H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science 261, 478–480. doi: 10.1126/science.8392752
Province, M. A., Goetz, M. P., Brauch, H., Flockhart, D. A., Hebert, J. M., Whaley, R., et al. (2014). CYP2D6 genotype and adjuvant tamoxifen: meta-analysis of heterogeneous study populations. Clin. Pharmacol. Ther. 95, 216–227. doi: 10.1038/clpt.2013.186
Qin, Y., Fortin, J. S., Tye, D., Gleason-Guzman, M., Brooks, T. A., and Hurley, L. H. (2010). Molecular cloning of the human platelet-derived growth factor receptor beta (PDGFR-beta) promoter and drug targeting of the G-quadruplex-forming region to repress PDGFR-beta expression. Biochemistry 49, 4208–4219. doi: 10.1021/bi100330w
Raiber, E. A., Kranaster, R., Lam, E., Nikan, M., and Balasubramanian, S. (2012). A non-canonical DNA structure is a binding motif for the transcription factor SP1 in vitro. Nucleic Acids Res. 40, 1499–1508. doi: 10.1093/nar/gkr882
Roberts, R. L., Gearry, R. B., Bland, M. V., Sies, C. W., George, P. M., Burt, M., et al. (2008). Trinucleotide repeat variants in the promoter of the thiopurine S-methyltransferase gene of patients exhibiting ultra-high enzyme activity. Pharmacogenet. Genomics 18, 434–438. doi: 10.1097/FPC.0b013e3282f85e47
Rodriguez, R. (2012). Small-molecule induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 8, 301–310. doi: 10.1038/nchembio.780
Sadee, W., Wang, D., Papp, A. C., Pinsonneault, J. K., Smith, R. M., Moyer, R. A., et al. (2011). Pharmacogenomics of the RNA world: structural RNA polymorphisms in drug therapy. Clin. Pharmacol. Ther. 89, 355–365. doi: 10.1038/clpt.2010.314
Salvati, E., Zizza, P., Rizzo, A., Iachettini, S., Cingolani, C., D'angelo, C., et al. (2014). Evidence for G-quadruplex in the promoter of vegfr-2 and its targeting to inhibit tumor angiogenesis. Nucleic Acids Res. 42, 2945–2957. doi: 10.1093/nar/gkt1289
Samanta, S., Anderson, K., Moran, S., Hawke, D., Gorenstein, D., and Fornage, M. (2012). Characterization of a human 12/15-lipoxygenase promoter variant associated with atherosclerosis identifies vimentin as a promoter binding protein. PLoS ONE 7:e42417. doi: 10.1371/journal.pone.0042417
Sandilands, A. J., and O'shaughnessy, K. M. (2005). The functional significance of genetic variation within the beta-adrenoceptor. Br. J. Clin. Pharmacol. 60, 235–243. doi: 10.1111/j.1365-2125.2005.02438.x
Sangkuhl, K., Berlin, D. S., Altman, R. B., and Klein, T. E. (2008). PharmGKB: understanding the effects of individual genetic variants. Drug Metab. Rev. 40, 539–551. doi: 10.1080/03602530802413338
Sen, D., and Gilbert, W. (1988). Formation of parallel four stranded complexes by guanine-rich motifs and its implications for meiosis. Nature 334, 364–366. doi: 10.1038/334364a0
Shin, Y. J., Kumarasamy, V., Camacho, D., and Sun, D. (2014). Involvement of G-quadruplex structures in regulation of human RET gene expression by small molecules in human medullary thyroid carcinoma TT cells. Oncogene. doi: 10.1038/onc.2014.65. [Epub ahead of print].
Siddiqui-Jain, A., Grand, C. L., Bearss, D. J., and Hurley, L. H. (2002). Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 99, 11593–11598. doi: 10.1073/pnas.182256799
Sim, S. C., Kacevska, M., and Ingelman-Sundberg, M. (2013). Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenomics J. 13, 1–11. doi: 10.1038/tpj.2012.45
Sundquist, W. I., and Klug, A. (1989). Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 342, 825–829. doi: 10.1038/342825a0
Tukey, R. H., and Strassburg, C. P. (2000). Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 40, 581–616. doi: 10.1146/annurev.pharmtox.40.1.581
Valton, A. L., Hassan-Zadeh, V., Lema, I., Boggetto, N., Alberti, P., Saintome, C., et al. (2014). G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 33, 732–746. doi: 10.1002/embj.201387506
Verma, A., Yadav, V. K., Basundra, R., Kumar, A., and Chowdhury, S. (2009). Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 37, 4194–4204. doi: 10.1093/nar/gkn1076
Wang, J., Lu, K., Xuan, S., Toh, Z., Zhang, D., and Shao, F. (2013). A Pt(II)-Dip complex stabilizes parallel c-myc G-quadruplex. Chem. Commun. 49, 4758–4760. doi: 10.1039/c3cc40868j
Wang, L., McLeod, H. L., and Weinshilboum, R. M. (2011). Genomics and drug response. New Engl. J. Med. 364, 1144–1153. doi: 10.1056/NEJMra1010600
Weinshilboum, R. (2001). Thiopurine pharmacogenetics: clinical and molecular studies of thiopurine methyltransferase. Drug Metab. Dispos 29, 601–605.
Wheeler, H. E., and Dolan, M. E. (2012). Lymphoblastoid cell lines in pharmacogenomic discovery and clinical translation. Pharmacogenomics 13, 55–70. doi: 10.2217/pgs.11.121
Whirl-Carrillo, M., McDonagh, E. M., Hebert, J. M., Gong, L., Sangkuhl, K., Thorn, C. F., et al. (2012). Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92, 414–417. doi: 10.1038/clpt.2012.96
Wong, H. M., Stegle, O., Rodgers, S., and Huppert, J. L. (2010). A toolbox for predicting g-quadruplex formation and stability. J. Nucleic Acids 2010:564946. doi: 10.4061/2010/564946
Wu, Y., and Brosh, R. M. Jr. (2010). G-quadruplex nucleic acids and human disease. FEBS J. 277, 3470–3488. doi: 10.1111/j.1742-4658.2010.07760.x
Xodo, L., Paramasivam, M., Membrino, A., and Cogoi, S. (2008). Protein hnRNPA1 binds to a critical G-rich element of KRAS and unwinds G-quadruplex structures: implications in transcription. Nucleic Acids Symp. Ser. 159–160. doi: 10.1093/nass/nrn081
Keywords: drug targets, G-quadruplex (G4), gene expression, gene regulation, secondary structure
Citation: Cree SL and Kennedy MA (2014) Relevance of G-quadruplex structures to pharmacogenetics. Front. Pharmacol. 5:160. doi: 10.3389/fphar.2014.00160
Received: 11 April 2014; Accepted: 19 June 2014;
Published online: 08 July 2014.
Edited by:
Marcelo Rizzatti Luizon, University of California, San Francisco, USAReviewed by:
Colin Ross, UBC, CanadaMarco Folini, Fondazione IRCCS Istituto Nazionale dei Tumori, Italy
Copyright © 2014 Cree and Kennedy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin A. Kennedy, Department of Pathology, Carney Centre for Pharmacogenomics, University of Otago, PO Box 4345, Christchurch, New Zealand e-mail:bWFydGluLmtlbm5lZHlAb3RhZ28uYWMubno=