 Xiaorei Sai
Xiaorei Sai Raj K. Ladher
Raj K. Ladher- Laboratory for Sensory Development, RIKEN Center for Developmental Biology, Kobe, Japan
Various cellular replacement therapies using in vitro generated cells to replace damaged tissue have been proposed as strategies to alleviate hearing loss. All such therapies must involve a complete understanding of the earliest steps in inner ear development; its induction as a thickened plate of cells in the non-neural, surface ectoderm of the embryo, to its internalization as an otocyst embedded in the head mesenchyme of the embryo. Such knowledge informs researchers addressing the feasibility of the proposed strategy and present alternatives if needed. In this review we describe the mechanisms of inner ear induction, concentrating on the factors that steer the fate of ectoderm into precursors of the inner ear. Induction then leads to inner ear morphogenesis and we describe the cellular changes that occur as the inner ear is converted from a superficial placode to an internalized otocyst, and how they are coordinated with a particular emphasis on how the signaling environment surrounding the inner ear influences these processes.
Introduction
The inner ear is a complex structure. It is composed of numerous cell types that include ciliated mechanoreceptors to detect mechanical stimuli associated with balance or sound detection, the neurons of the eight cranial nerve, the cochleovestibular nerve, that transmits the stimuli to the central nervous system, as well as other cell types to maintain the unique ionic composition of the inner ear (Slepecky, 1996; Highstein et al., 2004). All of these cell types are derived from the otic placode, a seemingly simple epithelial structure that is part of the surface non-neural ectoderm adjacent to the caudal part of the hindbrain (Barald and Kelley, 2004; Groves and Fekete, 2012). As is typical for most tissue induction during early development, secreted factors from surrounding tissue act on competent ectoderm to convert its fate (Ladher et al., 2010). This dynamic epigenetic control ensures that the otic placode forms at the right time and the right place, starting the train of events that will convert the placode into a fully functioning inner ear. The otic placode is induced superficial, yet the inner ear itself is embedded within the cephalic mesenchyme (Meier, 1978a,b). Thus, morphogenetic processes must take place that internalize this tissue. The process, called invagination occurs concomitantly with induction. Thus, changes in the shape of the cells are intimately linked to the inductive process. Indeed, changes in the cytoskeletal architecture of the otic placode are directly controlled by secreted factors, and it is clear that for complete and accurate morphogenesis the topographical relationship of the otic placode to the tissue emitting the inducing signal is critically important (Sai and Ladher, 2008).
Culture studies of isolated otic placodes at different stages have shown that once induction and invagination is complete, sensory cell and neuronal differentiation is autonomous in this tissue (Freter et al., 2008). This suggests that these early events by themselves are sufficient for at least some later differentiation. Thus, a deep understanding of the earliest events in inner ear development is necessary to inform cell differentiation protocols and attempts in vitro organ engineering that will form the next-generation of therapies for hearing loss.
Induction
The otic placode is part of a series of cranial placodes located in the head of all vertebrate animals. Placodes are thickened regions of ectoderm adjacent to the neural plate boundary that will give rise to sensory structures in the head as well as many of the cranial nerves (Baker and Bronner-Fraser, 2001; Schlosser, 2006, 2014; Graham and Shimeld, 2013). Anterior is the olfactory placode, which will generate the nasal epithelium as well as the first cranial nerve. Caudal to the olfactory placode is the lens placode. Uniquely, amongst the cranial placode, it produces neither neurons nor sensory cells. The two lobes of the trigeminal placode, the ophthalmic and the maxilla-mandibular are found around the eye. These placode are wholly neurogenic and form the sensory neurons of the fifth cranial nerve. As already stated, the otic placode will give rise to the inner ear as well as the eight cranial nerve. Lateral to the otic placode are the epibranchial series of placodes, the most anterior, the geniculate, forms the sensory components of the seventh cranial nerve, innervating the taste buds and conveying touch information from the ear lobes. The second epibranchial placode (EPD), the petrosal, will give rise to the ninth cranial nerve, innervating the tongue, as well as the carotid body. The most caudal is the nodose EPD, and will contribute to the tenth cranial nerve, the vagus (Baker and Bronner-Fraser, 2001; Ladher et al., 2010).
The otic placode, like all the cranial placodes, arises from the pre-placodal region (PPR); this can be thought of as ectoderm that is competent to develop into any placode given the correct signals, but has not yet committed to one (Bailey and Streit, 2006; Groves and LaBonne, 2014; Saint-Jeannet and Moody, 2014). Otic placode induction occurs by signals from adjacent tissues acting on the PPR. Otic induction itself can be though of as a progressive process, with a gradual restriction of possible fates as a function of time, under the control of signaling interactions (Figure 1). This section describes these processes, and the signals and transcription factors that control them, in more detail.
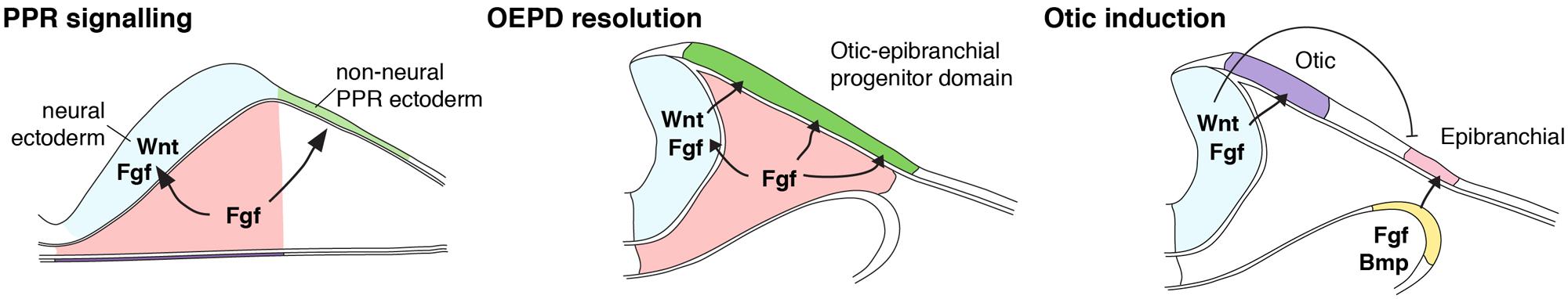
FIGURE 1. Model of inner ear induction. Shown is a scheme summarizing the induction of the inner ear, synthesizing data from zebrafish, chick, and mouse. At early neurulation stages, mesodermal source of FGF signals overlying pre-placodal ectoderm to adopt an otic-epibranchial placode (OEPD) fate. Mesodermal FGF also signals neural ectoderm to express FGF and Wnt signals. At later neurulation stages, soon after the neural tube has closed, Wnt then acts on OEPD to specify the inner ear from within the OEPD. (Modified from Ladher et al., 2010).
Establishing the Pre-Placodal Region
One of the first events in the patterning of the embryonic ectoderm is its separation into neural and non-neural domains in the head region of the embryo. This occurs at around the time of gastrulation and is directed by signals coming from the mesoderm and endoderm as well as signals acting within the ectoderm itself (Ahrens and Schlosser, 2005; Litsiou et al., 2005; Bailey and Streit, 2006). Subsequent interactions cause the border region between these two territories to become a region of competence for two different types of tissue; the region of the border within the neural plate is, given the right set of signals, able to generate neural crest. In contrast, border cells that are found in the non-neural ectoderm are competent to generate sensory placodes, and this is called the pre-PPR (Pieper et al., 2012).
Molecularly the PPR is defined by the expression of Six and Eya family transcription factors (Schlosser, 2007; Sato et al., 2012; Groves and LaBonne, 2014; Saint-Jeannet and Moody, 2014; Yajima et al., 2014). These describe a strip of expression wrapped around the neural plate, from roughly the first somite to the rostral tip of the neural plate. Mutations in these genes do lead to deficits in some placodal derivatives, but do not result in a complete absence of the placodes (Oliver et al., 1995; Ozaki et al., 2002, 2004; Laclef et al., 2003; Zheng et al., 2003; Konishi et al., 2006; Zou et al., 2006; Ikeda et al., 2007, 2010; Li et al., 2010; Suzuki et al., 2010a,b; Yajima et al., 2010). This could be due to redundancy amongst the Eya and Six family transcription factors, or it could suggest that the exact nature of the PPR is a more complicated than simply a region of competence for all placodes. The importance of the PPR in otic induction was demonstrated in the experiments of Martin and Groves (2006). In these experiments, competence of ectoderm to express the otic induction marker Pax2 in response to FGF2 was tested. The finding that ventral ectoderm could only respond if transplanted to the PPR/neural border region for 6 h strongly supports the idea that the PPR should be considered a region of competence to respond to placode inducing signals. Furthermore this competence is actively conferred.
Embryological experiments have suggested that the PPR is induced from signals that come from surrounding tissues (Ahrens and Schlosser, 2005; Litsiou et al., 2005; Bailey and Streit, 2006). In chick, the ablation of the head mesoderm abolishes the expression of PPR markers (Litsiou et al., 2005). Conversely, head, but not trunk mesoderm induces their expression in non-neural ectoderm. Fgf4 and Fgf8 are both expressed in the mesoderm that underlies the PPR, making them likely candidates to act as PPR inducing signals (Ahrens and Schlosser, 2005; Litsiou et al., 2005). Indeed, inhibition of FGF signaling can down-regulate some markers of the PPR in chick (Litsiou et al., 2005) and in embryos of the amphibian Xenopus (Ahrens and Schlosser, 2005). Furthermore, FGF8 protein is able to induce some PPR markers in competent non-neural ectoderm (Litsiou et al., 2005), however, it remains to be seen if this tissue is then competent to respond to FGF2 to induce otic markers. The cranial mesoderm also expresses inhibitors of Wnt and BMP (bone morphogenetic protein) signaling, suggesting that the activity of these proteins is inhibitory to PPR formation. Indeed, activation of Wnt or BMP signaling does inhibit the expression of PPR markers, whereas the inhibition of these signals can expand PPR marker gene expression. Thus it is likely that FGF signaling in combination with Wnt and BMP inhibition is necessary for complete PPR induction (Litsiou et al., 2005). What is not clear is the hierarchical organization of these signals; it is possible that some of these signals act upstream to promote the differentiation of non-neural ectoderm and that other signals confer PPR fate secondarily. One possible way of resolving this is to use in vitro cell systems, such as ES cells, to monitor the exact tissue induced by each kind of treatment over fine time points. If combined with a PPR specific reporter line (Sato et al., 2010, 2012), this experiment would also serve to greatly optimize the induction of inner ear tissues in ES cells.
As we will describe in the next section, FGF signaling is reused in the formation of the inner ear precursor domain. It is clear that FGF signaling has different consequences over a very narrow time window, and this raises the conundrum of how these responses are coordinated and spirited in time. One possible mechanism is by invoking the idea of feed-forward loops. Here, one can suggest that the action of FGF signaling in PPR induction also induces a transcription factor that is able to alter the response to FGF signaling. One example may be the Foxi3 transcription factor. Foxi3 is necessary for the response of PPR ectoderm to FGF signals to express Pax2 (Khatri et al., 2014). Interestingly, the expression of Foxi3 is controlled by the same tissues that control the expression of other PPR markers, suggesting not only a mechanism that ensures the correct temporal response to FGF signaling but also adds a molecular correlate to the idea that the PPR is a region of ectoderm competent to respond to placode inducing cues.
Induction of the Otic-Epibranchial Progenitor Domain
The mechanisms underlying the formation of the inner ear have been actively investigated for over 100 years. In this time researchers have used a variety of model systems, namely amphibians, fish, birds and mammals, and a variety of approaches, from experimental embryology to genetic studies, to ask how otic induction is controlled (reviewed in Ladher et al., 2010; Groves and Fekete, 2012). This work has provided a consensus view on how the posterior part of the PPR is specified to the inner ear fate.
One of the earliest markers that are induced during otic induction is the transcription factor Pax2 or, depending on the species, its close homolog Pax8 (Christophorou et al., 2010; Freter et al., 2012). Pax2/8 expression commences at mid-neurula stages (when the neural tube is closing) next to the caudal part of the future hindbrain in all vertebrate species that have been investigated (Plachov et al., 1990; Stoykova and Gruss, 1994; Lun and Brand, 1998; Pfeffer et al., 1998; Funahashi et al., 1999; Kozmik et al., 1999; Bouchard et al., 2002; Krelova et al., 2002; Hans et al., 2004; Mackereth et al., 2005). This location is at the junction of the head and the trunk of the animal, and extends anterior to the first somite. Lineage labeling of the Pax2 domain revealed that expression encompasses progenitors of not only the inner ear but also the EDPs (Bouchard et al., 2004; Ohyama and Groves, 2004; Streit, 2004; McCarroll et al., 2012). Thus, it is clear that the initial step of PPR restriction is the induction of a common progenitor domain for the otic and EDPs. This domain has been termed the otic-epibranchial progenitor domain (OEPD), the pre-otic field (POF), or the posterior placodal area (PPA). In this review, we will use the OEPD terminology.
Embryological experiments, defined the region of the embryo that was able to induce the OEPD. In particular, embryos from chick and the amphibian Xenopus showed that paraxial mesoderm plays a key role (reviewed in Ladher et al., 2010; Groves and Fekete, 2012). Chick experiments were able to resolve this domain to the mesoderm between the first somite and the level of the third rhombomere (Groves and Bronner-Fraser, 2000; Ladher et al., 2000; Kil et al., 2005). This underlies with the ectodermal domain of Pax2/8. However, it is also clear that the mesoderm is not the only source for signals. Pax2 can only be induced in non-neural ectodermal tissue by mesoderm when some neural ectoderm is also included; without this neural contribution, the OEPD is not induced (Ladher et al., 2000). Thus, it is likely that both mesodermal and neural ectodermal signals contribute to the induction of the OEPD.
Several lines of evidence suggest that members of the fibroblast growth factor (FGF) family mediate the induction of the OEPD (reviewed in Schimmang, 2007). In chick, knockdown studies (using antisense oligonucleotide or neutralizing antibodies) pointed to a role for FGF3 in OEPD induction (Represa et al., 1991; Vendrell et al., 2000). Indeed the expression of Fgf3 in the mesoderm and hindbrain of the chick embryo is consistent with it playing a major role in OEPD induction (Mahmood et al., 1995; Freter et al., 2008). However, knockouts in the mouse and further experiments in the chick seem to rule out an exclusive role for FGF3. In mouse Fgf3 mutants the inner ear is induced although later development is impaired (Mansour et al., 1993). In the mouse, Fgf3 is not expressed in the mesoderm and this has led to the idea that perhaps one or more FGF ligands are also required to mediate OEPD induction. In the chick, Fgf19 is expressed in a similar pattern to Fgf3, appearing first in the mesoderm and then in the hindbrain (Ladher et al., 2000). Knockdown of just Fgf19 has no real effect, however, when both Fgf19 and Fgf3 are knocked-down, OEPD induction does not take place (Freter et al., 2008). In the mouse, Fgf10 is expressed in the mesoderm, apparently taking the placode of chick Fgf19, and mutants of both Fgf3 and Fgf10 fail to induce the OEPD (Wright and Mansour, 2003).
Control of mesodermal Fgf10 (in the mouse) or Fgf19 (in the chick) seems to be, itself, under the control of signaling from the underlying endoderm, in this case by yet another member of the FGF family, Fgf8. The OEPD fails to be induced in mutants for Fgf3 and Fgf8. The situation is slightly different in the chick, where Fgf8 knockdown alone appears necessary for OEPD induction (Ladher et al., 2005).
One open question is what does the OEPD represent? Are OEPD cells equipotent, with individual cells able to give rise to both otic and epibranchial progenitors? Is it a mixed population of interspersed epibranchial and otic progenitors? Or are otic and epibranchial progenitors confined to distinct domains within the OEPD? Individual cell labeling and tracing will resolve this question, however, some clues exist when expression patterns are inspected. In particular the expression of Foxi1 is noteworthy. In chick Foxi1 is expressed around the OEPD at the time of OEPD induction, however, it’s expression begins to encroach into the OEPD such that it overlaps with the periphery of the OEPD at around the time the inner ear begins to segregate. Foxi1 remains absent from the putative otic placode, but is expressed in the EDP (Freter et al., 2008). One interpretation is that Foxi1 prevents commitment to the inner ear fate and that at early stages, both epibranchial and otic precursors do not express Foxi1 and have the potential to form either cell type.
Induction of the Inner Ear
As stated above the development of the inner ear is progressive, involving the gradual restriction of cell fate, from non-neural ectoderm, to PPR, to OEPD and then to inner ear fate. This has been clearly demonstrated in the chick, using cultures of the putative inner ear ectoderm isolated and cultured ex vivo: ectoderm taken at mid-neurula stages (when the embryo has between 1 and 3 somites) can only express Pax2, an OEPD marker. In contrast ectoderm taken at late neurula stages (when the embryo has between 4 and 6 somites) can express Soho1, a transcription factor that is associated with commitment to the otic fate. Furthermore, ectoderm taken at this stage can form hair cells (Freter et al., 2008). This suggests that signals from surrounding tissue are still required to enable the transition from the OEPD to committed inner ear precursors.
Wnt8a (which is known as Wnt8c in the chick) is expressed in the caudal hindbrain is necessary for the partitioning of the otic placode from the OEPD (Freter et al., 2008). The application of FGF19 (the mesodermal signal in chick) and WNT8a to competent ectoderm was sufficient to complete inner ear development in non-neural ectoderm (Ladher et al., 2000). Inhibition of the Wnt signal using the antagonist Dkk1 blocked the expression of the inner ear marker Soho1 but did not inhibit the expression of Pax2 (Freter et al., 2008). This experiment provides further support of the progressive nature of inner ear induction. In mice, an inner ear specific mutation of β-catenin, a down-stream transducer of the wnt pathway, results a block in the formation of the inner ear. Wnt8a is induced in both chick and mouse as a result of FGF signaling. In chick, FGF19 can induce Wnt8a and in mice mutant for Fgf3 fail to express Wnt8a (Urness et al., 2010). While these data suggest that Wnt8a in response to Fgf signaling is responsible for partitioning the inner ear from the OEPD, mouse mutants data presents a more confusing scenario. Mutant for Wnt8a do affect the otic placode (Vendrell et al., 2013). It is possible that other Wnts proteins expressed in the hindbrain, such as Wnt1, Wnt3a, and/or Wnt6 are all able to compensate for the loss of Wnt8. Thus although Wnt signaling, as determined by the over-expression of the Wnt antagonist Dkk1 and the loss of the Wnt signaling transducer β-catenin, does result in a failure of the inner ear to partition from the OEPD, the significance of the most likely ligand, Wnt8a, is less clear. It is likely that it acts to ensure the inner ear forms in the correct axial position, however, other Wnt genes can compensate for its absence.
Morphogenesis
The inner ear is a closed structure located inside the head, embedded within the mesenchyme of the head. However, as described above, the placode is a superficial structure. From its position on the surface of the embryo, the otic placode must undergo a series of morphogenetic changes so that it ends up as an isolated closed vesicle in the cephalic mesoderm (Meier, 1978a,b). In this section, we describe the morphogenetic changes that take place as the otic placode changes shape to become the inner ear precursor (Figure 2). As the later morphogenetic events that shape the otocyst to the final inner ear are not well known, these will not be described.
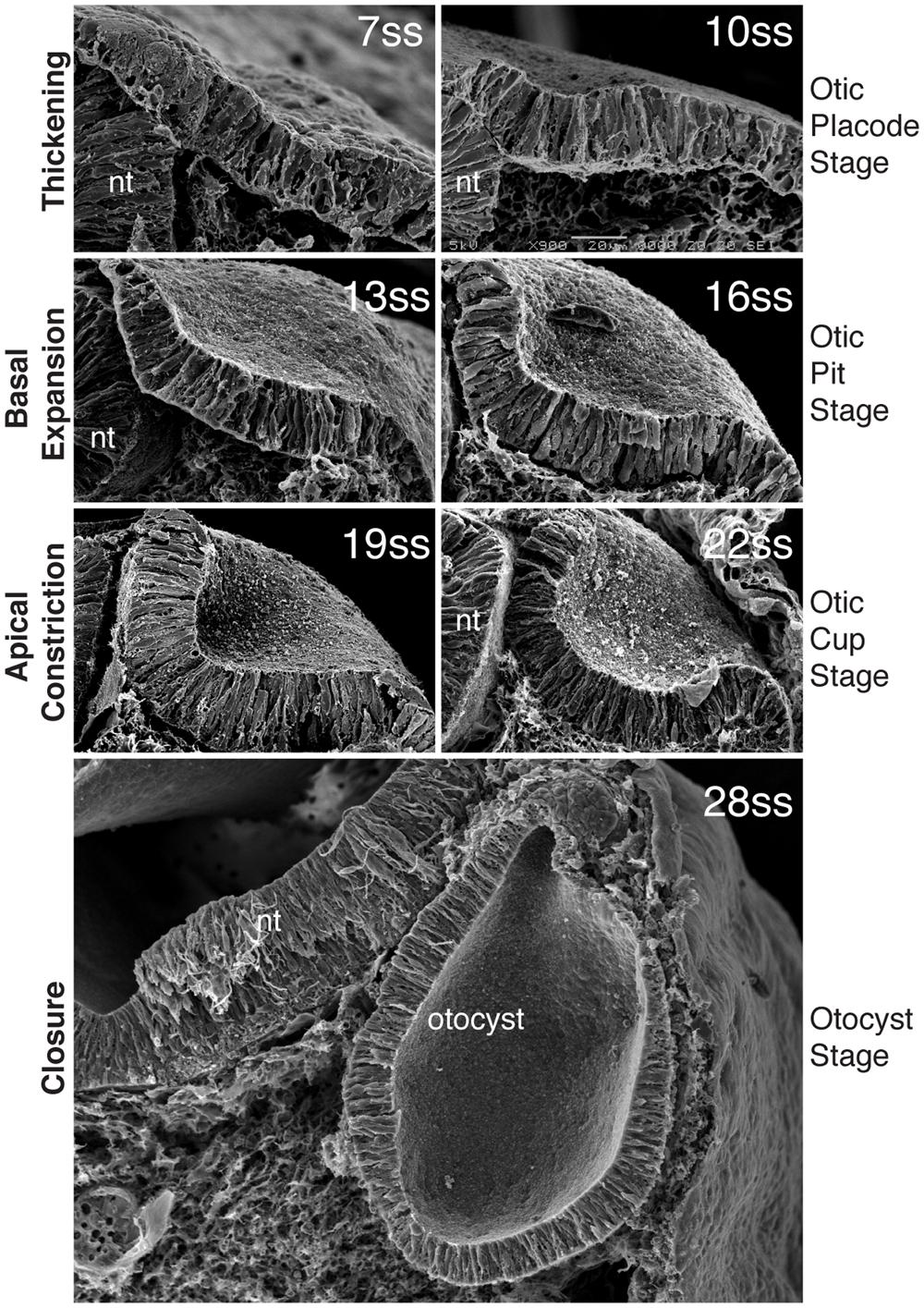
FIGURE 2. Morphogenesis of the inner ear. Scanning electron micrographs through the chick otic placode at different stages of development, show the stages of morphogenesis. By 7ss, the otic placode has segregated from the OEPD. At around 10ss of development, the placode begins to invaginates such that at 13ss and 16ss the placode forms a configuration known as the otic pit. Basal expansion predominates at this stage. After 16ss and until closure, apical constriction is the main processs driving invagination and the otic placode at these stages progressively deepens form the otic cup. With closure, the final form of the otocyst is apparent. (Modified from Sai and Ladher, 2008).
Epithelial Thickening
The otic placode itself already has a morphology that sets it apart from the rest of the ectoderm; it consists of a thickened columnar pseudo-stratified epithelium as opposed to the squamous-like epithelium surrounding it. In amphibians, chick and mouse, where this has been investigated, this thickening occurs around the time of otic induction, when the embryo has 4–6 somites. The mechanisms controlling the thickening of the placodal epithelium are not well known, although Pax2 has been proposed as a regulator of placodal thickening (Christophorou et al., 2010). The actual mechanics of placodal thickening are also unclear, however, studies investigating the function of members of the small GTPase family members, RhoA and Rac1 in the development of the lens placode suggest that Rac1 is a major effector of thickening (Chauhan et al., 2011) and is likely to be necessary for placodal thickening in the otic placode. Placodal thickening is likely to be necessary for the mode of proliferation in the inner ear. Cells within the otic placode divide by interkinetic nuclear migration (INM); Sauer, 1936). INM is thought to permit tighter packing of epithelial progenitors, which allows a higher concentration of cells (Fish et al., 2008). Furthermore, the packing density of pseudostratified epithelia is necessary for division. During INM, nuclei actively migrate apically to divide. Daughter nuclei are then passively pushed back to the basal side by the action of surrounding nuclei and by virtue of their packing density, and it is likely that if this packing density were not maintained division would be aberrant (Kosodo et al., 2011). It is also likely that thickened epithelia allow precise and separable control of both apical and basal domains of the placodal cells. As we discuss, this is particularly important in the control of invagination, in which signals act on the basal side and eventually result in apical changes.
Invagination
Soon after the partition of the otic placode from the OEPD, invagination begins to take place. By the time the embryo has between 8 and 10 somites, a slight depression in the epithelial plain is apparent. This depression continues to deform, deepening before pinching off and forming an enclosed vesicle within the cephalic mesoderm (Figure 2). The mechanisms behind otic invagination are only now starting to become clear. However, insights have been obtained using quite a simple analysis. Here, measurements of the apical and basal faces of the otic placode during its morphogenesis suggest two types of cell shape change (Alvarez and Navascués, 1990; Sai et al., 2014). Between 10 and 16 somites (in the chick), the apical face of the otic placode does not change, however, the basal side expands, the placode at this stage is also known as the otic pit. From 16 to 22 somites stages the apical length decreased dramatically, and the otic placode is driven deeper into the mesenchyme. The placode at this point adopts a configuration that has been termed the otic cup. Otic invagination itself can be considered as a biphasic process; phase one being basal expansion and phase two being apical constriction.
One of the driving forces behind changes in cell shape are alterations in the cytoskeleton, that is the network of actin fibers, microtubules, and intermediate filaments that provide structure and support to a cell (Lecuit and Lenne, 2007). Therefore, it is no surprise that during basal expansion clear difference in the distribution of actin are observed. At 10 somite stages (ss), actin fibers (or F-actin) can be detected in apically and basally in the otic placode cell. At 13ss, F-actin is depleted basally and enriched apically (Sai and Ladher, 2008). This is also when basal expansion occurs. Thus, the key to understanding basal expansion is to understand how basal F-actin is cleared. Furthermore, as basal expansion needs to occur in the whole of the otic placode, the changes in cell behavior must be coordinated across the tissue.
Clues about the mechanism came from experiments in which the otic placode was isolated and placed into culture. Explants of the otic placode, freed of underlying mesoderm and endoderm as well as adjacent neural tissue, are able to clear actin from the basal side and to round up and form otic vesicles if taken at 16 somites stage of development. However, otic explants taken at 10 somite stages of development neither clear actin from the basal side of the otic placode nor show any signs of epithelial deformation (Sai and Ladher, 2008). This suggests that extrinsic signals are required for invagination. One obvious candidate for the extrinsic signal that controls and coordinates invagination was the same signal controlling its induction, FGF. Indeed when 10ss explants were cultured with beads that locally delivered FGF the ability to clear basal actin was restored. Further experiments involving pharmacological inhibitors confirmed that the FGF pathway, through the activation of phospholipase C (PLC) gamma activated the motor protein myosin-II, by phosphorylating its regulatory subunit, myosin light chain. Active myosin-II exerted a non-canonical activity on the basal side that resulted in the depolymerization of F-actin. Thus, during basal expansion, active myosin light chain and F-actin are reciprocally localized; F-actin apically and phosphorylated myosin light chain basally. As we discuss later this is in contrast to apical constriction where both F-actin and active myosin-II co-localize to the apical face of the otic placode (Sai and Ladher, 2008).
While this mechanism for basal expansion has only been described during inner ear invagination, it is likely that it does occur in other epithelial tissues undergoing deformation. Prior to the elevation of the neural plate, the reciprocal localisation of phosphorylated myosin light chain and F-actin is also observed, suggestive of basal expansion (Sai and Ladher, 2008). Similarly, a transient reciprocal localisation is observed in the mesodermal progenitors about to invagination during Drosophila gastrulation (Dawes-Hoang et al., 2005; Fox and Peifer, 2007). In all cases it is likely that a signal acting on the basal side of the cell triggers this activity, ensuring not only a coordination amongst all of the cells within the tissue, but also direction to the deformation. With respect to in vitro tissue engineering approaches, this does suggest that signals need to be spatially confined and care must be given to which side of the cell is stimulated by ectopic signals.
From 16 somites stage of chick development, the apices of the otic placode cell begin to constriction, deepening the invagination and driving the internalization of the otocyst. The mechanisms of apical constriction are similar to those that have been determined for other epithelial remodeling events such as wound healing, neural tube formation, or dorsal closure in Drosophila (Sawyer et al., 2010; Martin and Goldstein, 2014). During otic morphogenesis, this phase is characterized by the co-expression of active myosin II and F-actin (Sai et al., 2014). Thus the key to understanding apical constriction in the otic placode is to understand how myosin II is activated apically.
Recent findings suggest that the mechanisms of apical constriction during otic invagination are analogous to those driving neural tube closure (Nishimura et al., 2012). Apical constriction is powered by the canonical activity of the myosin-II motor generating contractile tension in actin filaments. This activity is localized circumferentially in the apex of the cell, and is coincident with the apical junctional complex (AJC). The AJC includes both tight junctions and adherens junctions, which act to maintain the integrity of the epithelia. These junctions act as anchor points from actomyosin fibers, and it is from here that force can be more effectively transmitted across cells. Thus the activation of the myosin-II motor occurs here (Nishimura et al., 2012; Sai et al., 2014). The RhoGTPase family has been frequently implicated in the regulation of junctional actin by activating Myosin-II, and has been shown to be important in the regulation of apical constriction in many other systems (Martin and Goldstein, 2014). RhoGTPases act as switches, cycling between a GTP-bound “ON” state and a GDP-bound “OFF” state. This switch is control by the action of effectors of RhoGTPases; Rho Guanine exchange factor (RhoGEF) switches on Rho, and a Rho Guanine Activating Protein (RhoGAP) switches it off (Hall, 2012) and thus it is likely that upstream control of Rho activity is exerted by specific GEFs and GAPs. For example, studies in Drosophila gastrulation have shown that RhoGEF1 stimulates Rho1, which in turn activates the Drosophila homolog of the Rho-associated, coiled-coil protein kinase (ROCK). Drok is responsible for the activation of myosin-II (Mason et al., 2013). Similarly, in vertebrate neurulation RhoA activation by the GEF ArhGEF11 is necessary for neural tube closure through the activation of myosin light chain (Nishimura et al., 2012). In the otic placode, RhoA is localized apically and is similar to neural tube closure, is also activated by ArhGEF11 (Sai et al., 2014). Once active, RhoA is able to activate ROCK, which directly phosphorylates myosin light chain, thus activating myosin-II. This network, is likely to be conserved across a range of epithelial morphogenetic systems, and ensures apically restricted myosin-II.
One further consideration is the separation of basal expansion and apical constriction. ArhGEF11 is expressed at around 13ss, coincident with apical constriction. Furthermore it is localized to the otic placode, and is not found in surrounding epithelia, suggesting that its expression is likely to be regulated by otic induction cues (Sai et al., 2014). Thus, it is probable that signaling is not only responsible for phase 1 otic morphogenesis but also, indirectly, for phase 2. In phase 1 morphogenesis, actin is cleared from the basal side of the otic placode, through the direct action of FGF signaling, ensuring that the contractive activity of myosin-II and RhoA activity cannot act ectopically (Sai and Ladher, 2008). In phase 2, the induction and expression of ArhGEF11 activates RhoA and the thus myosin-II. The combination of RhoA and Myosin-II activity permits the contractive activity of this motor protein, and mediates apical constriction.
Once internalized the otic vesicle must then be closed off, with the two edges fusing, isolating the inner ear vesicle from the rest of the ectoderm. The mechanism of fusion is still not clear, but parallels can be drawn with wound healing and with the closure event that occurs once the neural tube has formed a tube (Colas and Schoenwolf, 2001; Martin and Parkhurst, 2004). Preliminary data does suggest that the edges of the otic epithelium send out protrusive processes, and these filopodia knit the knit the epithelium together such that it is now contiguous. Once closed, the isolated otocyst now is primed to undergo further shape changes to form the final structure of the membranous labyrinth. These morphogenetic mechanisms are only now beginning to be understood and are described elsewhere (Chang et al., 2004; Mansour and Schoenwolf, 2005; Abraira et al., 2008; Ohta et al., 2010). These processes may employ similar mechanisms or utilize extrinsic signals, also acting in a localized and directional fashion, to complete the final maturation of the inner ear.
Conclusion
Both induction and the early morphogenesis of the inner ear require signaling factors deployed in both a spatially and temporally restricted pattern. Mimicking these signals in vitro to generate in vitro inner ears may at first pass seem daunting. However, the therapeutic value in these studies will come from understanding the spatial and temporal effect of signaling and then to employ them when needed. It should be noted that for the differentiation of hair cells and of otic placode derived neurons, only the initial signals need to be recapitulated (Freter et al., 2008). Once the otic placode has been induced hair cell and neurons will differentiate without the need for further tissue interactions and additional signals. These kinds of ideas have formed the basis of differentiation protocols that steer ES cell fate to otic placodal fates, however, their efficacy as potential therapies remains to be validated rigorously (Li et al., 2003; Oshima et al., 2010; Chen et al., 2012; Koehler et al., 2013). In some of these differentiation protocols, in vitro generated otic progenitors have been used to repair lesions in the auditory nerve (Chen et al., 2012). These studies although promising, highlight the necessity to further improve and build on our knowledge of inner ear induction and early morphogenesis so that these finding can be used to deliver more efficient and effective therapeutic in vitro generated inner ear tissues, that many next-generation therapies will utilize.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraira, V. E., Del Rio, T., Tucker, A. F., Slonimsky, J., Keirnes, H. L., and Goodrich, L. V. (2008). Cross-repressive interactions between Lrig3 and netrin 1 shape the architecture of the inner ear. Development 135, 4091–4099. doi: 10.1242/dev.029330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ahrens, K., and Schlosser, G. (2005). Tissues and signals involved in the induction of placodal Six1 expression in Xenopus laevis. Dev. Biol. 288, 40–59. doi: 10.1016/j.ydbio.2005.07.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alvarez, I. S., and Navascués, J. (1990). Shaping, invagination, and closure of the chick embryo otic vesicle: scanning electron microscopic and quantitative study. Anat. Rec. 228, 315–326. doi: 10.1002/ar.1092280311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bailey, A. P., and Streit, A. (2006). Sensory organs: making and breaking the pre-placodal region. Curr. Top. Dev. Biol. 72, 167–204. doi: 10.1016/S0070-2153(05)72003-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baker, C. V., and Bronner-Fraser, M. (2001). Vertebrate cranial placodes I. Embryonic induction. Dev. Biol. 232, 1–61. doi: 10.1006/dbio.2001.0156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barald, K. F., and Kelley, M. W. (2004). From placode to polarization: new tunes in inner ear development. Development 131, 4119–4130. doi: 10.1242/dev.01339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouchard, M., Souabni, A., and Busslinger, M. (2004). Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis 38, 105–109. doi: 10.1002/gene.20008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouchard, M., Souabni, A., Mandler, M., Neubüser, A., and Busslinger, M. (2002). Nephric lineage specification by Pax2 and Pax8. Genes Dev. 16, 2958–2970. doi: 10.1101/gad.240102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, W., Brigande, J. V., Fekete, D. M., and Wu, D. K. (2004). The development of semicircular canals in the inner ear: role of FGFs in sensory cristae. Development 131, 4201–4211. doi: 10.1242/dev.01292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chauhan, B. K., Lou, M., Zheng, Y., and Lang, R. A. (2011). Balanced Rac1 and RhoA activities regulate cell shape and drive invagination morphogenesis in epithelia. Proc. Natl. Acad. Sci. U.S.A. 108, 18289–18294. doi: 10.1073/pnas.1108993108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, W., Jongkamonwiwat, N., Abbas, L., Eshtan, S. J., Johnson, S. L., Kuhn, S.,et al. (2012). Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature 490, 278–282. doi: 10.1038/nature11415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christophorou, N. A., Mende, M., Lleras-Forero, L., Grocott, T., and Streit, A. (2010). Pax2 coordinates epithelial morphogenesis and cell fate in the inner ear. Dev. Biol. 345, 180–190. doi: 10.1016/j.ydbio.2010.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colas, J. F., and Schoenwolf, G. C. (2001). Towards a cellular and molecular understanding of neurulation. Dev. Dyn. 221, 117–145. doi: 10.1002/dvdy.1144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dawes-Hoang, R. E., Parmar, K. M., Christiansen, A. E., Phelps, C. B., Brand, A. H., and Wieschaus, E. F. (2005). Folded gastrulation, cell shape change and the control of myosin localization. Development 132, 4165–4178. doi: 10.1242/dev.01938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fish, J. L., Dehay, C., Kennedy, H., and Huttner, W. B. (2008). Making bigger brains-the evolution of neural-progenitor-cell division. J. Cell Sci. 121, 2783–2793. doi: 10.1242/jcs.023465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fox, D. T., and Peifer, M. (2007). Abelson kinase (Abl) and RhoGEF2 regulate actin organization during cell constriction in Drosophila. Development 134, 567–578. doi: 10.1242/dev.02748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freter, S., Muta, Y., Mak, S. S., Rinkwitz, S., and Ladher, R. K. (2008). Progressive restriction of otic fate: the role of FGF and Wnt in resolving inner ear potential. Development 135, 3415–3424. doi: 10.1242/dev.026674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freter, S., Muta, Y., O’neill, P., Vassilev, V. S., Kuraku, S., and Ladher, R. K. (2012). Pax2 modulates proliferation during specification of the otic and epibranchial placodes. Dev. Dyn. 241, 1716–1728. doi: 10.1002/dvdy.23856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Funahashi, J., Okafuji, T., Ohuchi, H., Noji, S., Tanaka, H., and Nakamura, H. (1999). Role of Pax-5 in the regulation of a mid-hindbrain organizer’s activity. Dev. Growth Differ. 41, 59–72. doi: 10.1046/j.1440-169x.1999.00401.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graham, A., and Shimeld, S. M. (2013). The origin and evolution of the ectodermal placodes. J. Anat. 222, 32–40. doi: 10.1111/j.1469-7580.2012.01506.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groves, A. K., and Bronner-Fraser, M. (2000). Competence, specification and commitment in otic placode induction. Development 127, 3489–3499.
Groves, A. K., and Fekete, D. M. (2012). Shaping sound in space: the regulation of inner ear patterning. Development 139, 245–257. doi: 10.1242/dev.067074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groves, A. K., and LaBonne, C. (2014). Setting appropriate boundaries: fate, patterning and competence at the neural plate border. Dev. Biol. 389, 2–12. doi: 10.1016/j.ydbio.2013.11.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, A. (2012). Rho family GTPases. Biochem. Soc. Trans. 40, 1378–1382. doi: 10.1042/BST20120103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hans, S., Liu, D., and Westerfield, M. (2004). Pax8 and Pax2a function synergistically in otic specification, downstream of the Foxi1 and Dlx3b transcription factors. Development 131, 5091–5102. doi: 10.1242/dev.01346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Highstein, S. M., (2004). “Anatomy and physiology of the central and peripheral vestibular system: overview,” in The vestibular system, eds S. M. Highstein, R. R. Fay, and A. N. Poppe (Berlin: Springer), 24.
Ikeda, K., Kageyama, R., Suzuki, Y., and Kawakami, K. (2010). Six1 is indispensable for production of functional progenitor cells during olfactory epithelial development. Int. J. Dev. Biol. 54, 1453–1464. doi: 10.1387/ijdb.093041ki
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ikeda, K., Ookawara, S., Sato, S., Ando, Z., Kageyama, R., and Kawakami, K. (2007). Six1 is essential for early neurogenesis in the development of olfactory epithelium. Dev. Biol. 311, 53–68. doi: 10.1016/j.ydbio.2007.08.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khatri, S. B., Edlund, R. K., and Groves, A. K. (2014). Foxi3 is necessary for the induction of the chick otic placode in response to FGF signaling. Dev. Biol. 391, 158–169. doi: 10.1016/j.ydbio.2014.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kil, S. H., Streit, A., Brown, S. T., Agrawal, N., Collazo, A., Zile, M. H.,et al. (2005). Distinct roles for hindbrain and paraxial mesoderm in the induction and patterning of the inner ear revealed by a study of vitamin-A-deficient quail. Dev. Biol. 285, 252–271. doi: 10.1016/j.ydbio.2005.05.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koehler, K. R., Mikosz, A. M., Molosh, A. I., Patel, D., and Hashino, E. (2013). Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500, 217–221. doi: 10.1038/nature12298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konishi, Y., Ikeda, K., Iwakura, Y., and Kawakami, K. (2006). Six1 and Six4 promote survival of sensory neurons during early trigeminal gangliogenesis. Brain Res. 1116, 93–102. doi: 10.1016/j.brainres.2006.07.103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kosodo, Y., Suetsugu, T., Suda, M., Mimori-Kiyosue, Y., Toida, K., Baba, S. A.,et al. (2011). Regulation of interkinetic nuclear migration by cell cycle-coupled active and passive mechanisms in the developing brain. EMBO J. 30, 1690–1704. doi: 10.1038/emboj.2011.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kozmik, Z., Holland, N. D., Kalousova, A., Paces, J., Schubert, M., and Holland, L. Z. (1999). Characterization of an amphioxus paired box gene, AmphiPax2/5/8: developmental expression patterns in optic support cells, nephridium, thyroid-like structures and pharyngeal gill slits, but not in the midbrain-hindbrain boundary region. Development 126, 1295–1304.
Krelova, J., Holland, L. Z., Schubert, M., Burgtorf, C., Benes, V., and Kozmik, Z. (2002). Functional equivalency of amphioxus and vertebrate Pax258 transcription factors suggests that the activation of mid-hindbrain specific genes in vertebrates occurs via the recruitment of Pax regulatory elements. Gene 282, 143–150. doi: 10.1016/S0378-1119(01)00840-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laclef, C., Souil, E., Demignon, J., and Maire, P. (2003). Thymus, kidney and craniofacial abnormalities in Six 1 deficient mice. Mech. Dev. 120, 669–679. doi: 10.1016/S0925-4773(03)00065-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ladher, R. K., Anakwe, K. U., Gurney, A. L., Schoenwolf, G. C., and Francis-West, P. H. (2000). Identification of synergistic signals initiating inner ear development. Science 290, 1965–1967. doi: 10.1126/science.290.5498.1965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ladher, R. K., O’neill, P., and Begbie, J. (2010). From shared lineage to distinct functions: the development of the inner ear and epibranchial placodes. Development 137, 1777–1785. doi: 10.1242/dev.040055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ladher, R. K., Wright, T. J., Moon, A. M., Mansour, S. L., and Schoenwolf, G. C. (2005). FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 19, 603–613. doi: 10.1101/gad.1273605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lecuit, T., and Lenne, P. F. (2007). Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Nat. Rev. Mol. Cell Biol. 8, 633–644. doi: 10.1038/nrm2222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, H., Roblin, G., Liu, H., and Heller, S. (2003). Generation of hair cells by stepwise differentiation of embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 100, 13495–13500. doi: 10.1073/pnas.2334503100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y., Manaligod, J. M., and Weeks, D. L. (2010). EYA1 mutations associated with the branchio-oto-renal syndrome result in defective otic development in Xenopus laevis. Biol. Cell 102, 277–292. doi: 10.1042/BC20090098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Litsiou, A., Hanson, S., and Streit, A. (2005). A balance of FGF, BMP and WNT signalling positions the future placode territory in the head. Development 132, 4051–4062. doi: 10.1242/dev.01964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lun, K., and Brand, M. (1998). A series of no isthmus (noi) alleles of the zebrafish pax2.1 gene reveals multiple signaling events in development of the midbrain-hindbrain boundary. Development 125, 3049–3062.
Mackereth, M. D., Kwak, S. J., Fritz, A., and Riley, B. B. (2005). Zebrafish pax8 is required for otic placode induction and plays a redundant role with Pax2 genes in the maintenance of the otic placode. Development 132, 371–382. doi: 10.1242/dev.01587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mahmood, R., Kiefer, P., Guthrie, S., Dickson, C., and Mason, I. (1995). Multiple roles for FGF-3 during cranial neural development in the chicken. Development 121, 1399–1410.
Mansour, S. L., Goddard, J. M., and Capecchi, M. R. (1993). Mice homozygous for a targeted disruption of the proto-oncogene int-2 have developmental defects in the tail and inner ear. Development 117, 13–28.
Mansour, S. L., and Schoenwolf, G. C. (2005). “Morphogenesis of the inner ear,” in Development of the Inner Ear, eds M. W. Kelley, D. K. Wu, A. N. Popper, and R. R. Fay (Berlin: Springer), 26, 43–84. doi: 10.1007/0-387-30678-1_3
Martin, A. C., and Goldstein, B. (2014). Apical constriction: themes and variations on a cellular mechanism driving morphogenesis. Development 141, 1987–1998. doi: 10.1242/dev.102228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, K., and Groves, A. K. (2006). Competence of cranial ectoderm to respond to Fgf signaling suggests a two-step model of otic placode induction. Development 133, 877–887. doi: 10.1242/dev.02267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, P., and Parkhurst, S. M. (2004). Parallels between tissue repair and embryo morphogenesis. Development 131, 3021–3034. doi: 10.1242/dev.01253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mason, F. M., Tworoger, M., and Martin, A. C. (2013). Apical domain polarization localizes actin-myosin activity to drive ratchet-like apical constriction. Nat. Cell Biol. 15, 926–936. doi: 10.1038/ncb2796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCarroll, M. N., Lewis, Z. R., Culbertson, M. D., Martin, B. L., Kimelman, D., and Nechiporuk, A. V. (2012). Graded levels of Pax2a and Pax8 regulate cell differentiation during sensory placode formation. Development 139, 2740–2750. doi: 10.1242/dev.076075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meier, S. (1978a). Development of the embryonic chick otic placode. I. Light microscopic analysis. Anat. Rec. 191, 447–458. doi: 10.1002/ar.1091910405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meier, S. (1978b). Development of the embryonic chick otic placode. II. Electron microscopic analysis. Anat. Rec. 191, 459–477. doi: 10.1002/ar.1091910406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishimura, T., Honda, H., and Takeichi, M. (2012). Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell 149, 1084–1097. doi: 10.1016/j.cell.2012.04.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohta, S., Mansour, S. L., and Schoenwolf, G. C. (2010). BMP/SMAD signaling regulates the cell behaviors that drive the initial dorsal-specific regional morphogenesis of the otocyst. Dev. Biol. 347, 369–381. doi: 10.1016/j.ydbio.2010.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohyama, T., and Groves, A. K. (2004). Expression of mouse Foxi class genes in early craniofacial development. Dev. Dyn. 231, 640–646. doi: 10.1002/dvdy.20160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oliver, G., Mailhos, A., Wehr, R., Copeland, N. G., Jenkins, N. A., and Gruss, P. (1995). Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development 121, 4045–4055.
Oshima, K., Shin, K., Diensthuber, M., Peng, A. W., Ricci, A. J., and Heller, S. (2010). Mechanosensitive hair cell-like cells from embryonic and induced pluripotent stem cells. Cell 141, 704–716. doi: 10.1016/j.cell.2010.03.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ozaki, H., Nakamura, K., Funahashi, J., Ikeda, K., Yamada, G., Tokano, H.,et al. (2004). Six1 controls patterning of the mouse otic vesicle. Development 131, 551–562. doi: 10.1242/dev.00943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ozaki, H., Watanabe, Y., Ikeda, K., and Kawakami, K. (2002). Impaired interactions between mouse Eyal harboring mutations found in patients with branchio-oto-renal syndrome and Six, Dach, and G proteins. J. Hum. Genet. 47, 107–116. doi: 10.1007/s100380200011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeffer, P. L., Gerster, T., Lun, K., Brand, M., and Busslinger, M. (1998). Characterization of three novel members of the zebrafish Pax2/5/8 family: dependency of Pax5 and Pax8 expression on the Pax2.1 (noi) function. Development 125, 3063–3074.
Pieper, M., Ahrens, K., Rink, E., Peter, A., and Schlosser, G. (2012). Differential distribution of competence for panplacodal and neural crest induction to non-neural and neural ectoderm. Development 139, 1175–1187. doi: 10.1242/dev.074468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Plachov, D., Chowdhury, K., Walther, C., Simon, D., Guenet, J. L., and Gruss, P. (1990). Pax8, a murine paired box gene expressed in the developing excretory system and thyroid gland. Development 110, 643–651.
Represa, J., Leon, Y., Miner, C., and Giraldez, F. (1991). The int-2 proto-oncogene is responsible for induction of the inner ear. Nature 353, 561–563. doi: 10.1038/353561a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sai, X., and Ladher, R. K. (2008). FGF signaling regulates cytoskeletal remodeling during epithelial morphogenesis. Curr. Biol. 18, 976–981. doi: 10.1016/j.cub.2008.05.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sai, X., Yonemura, S., and Ladher, R. K. (2014). Junctionally restricted RhoA activity is necessary for apical constriction during phase 2 inner ear placode invagination. Dev. Biol. 394, 206–216. doi: 10.1016/j.ydbio.2014.08.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saint-Jeannet, J. P., and Moody, S. A. (2014). Establishing the pre-placodal region and breaking it into placodes with distinct identities. Dev. Biol. 389, 13–27. doi: 10.1016/j.ydbio.2014.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sato, S., Ikeda, K., Shioi, G., Nakao, K., Yajima, H., and Kawakami, K. (2012). Regulation of Six1 expression by evolutionarily conserved enhancers in tetrapods. Dev. Biol. 368, 95–108. doi: 10.1016/j.ydbio.2012.05.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sato, S., Ikeda, K., Shioi, G., Ochi, H., Ogino, H., Yajima, H.,et al. (2010). Conserved expression of mouse Six1 in the pre-placodal region (PPR) and identification of an enhancer for the rostral PPR. Dev. Biol. 344, 158–171. doi: 10.1016/j.ydbio.2010.04.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sauer, F. C. (1936). The interkinetic migration of embryonic epithelial nuclei. J. Morphol. 60, 1–11. doi: 10.1002/jmor.1050600102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sawyer, J. M., Harrell, J. R., Shemer, G., Sullivan-Brown, J., Roh-Johnson, M., and Goldstein, B. (2010). Apical constriction: a cell shape change that can drive morphogenesis. Dev. Biol. 341, 5–19. doi: 10.1016/j.ydbio.2009.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schimmang, T. (2007). Expression and functions of FGF ligands during early otic development. Int. J. Dev. Biol. 51, 473–481. doi: 10.1387/ijdb.072334ts
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlosser, G. (2006). Induction and specification of cranial placodes. Dev. Biol. 294, 303–351. doi: 10.1016/j.ydbio.2006.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlosser, G. (2007). How old genes make a new head: redeployment of Six and Eya genes during the evolution of vertebrate cranial placodes. Integr. Comp. Biol. 47, 343–359. doi: 10.1093/icb/icm031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlosser, G. (2014). Early embryonic specification of vertebrate cranial placodes. Wiley Interdiscip. Rev. Dev. Biol. 3, 349–363. doi: 10.1002/wdev.142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Slepecky, N. B. (1996). “Structure of the mammalian cochlea,” in The Cochlea, eds P. Dallos, A. N. Popper, and R. R. Fay (New York: Springer), 44–129.
Stoykova, A., and Gruss, P. (1994). Roles of Pax-genes in developing and adult brain as suggested by expression patterns. J. Neurosci. 14, 1395–1412.
Streit, A. (2004). Early development of the cranial sensory nervous system: from a common field to individual placodes. Dev. Biol. 276, 1–15. doi: 10.1016/j.ydbio.2004.08.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, Y., Ikeda, K., and Kawakami, K. (2010a). Expression of Six1 and Six4 in mouse taste buds. J. Mol. Histol. 41, 205–214. doi: 10.1007/s10735-010-9280-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, Y., Ikeda, K., and Kawakami, K. (2010b). Regulatory role of Six1 in the development of taste papillae. Cell Tissue Res. 339, 513–525. doi: 10.1007/s00441-009-0917-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Urness, L. D., Paxton, C. N., Wang, X., Schoenwolf, G. C., and Mansour, S. L. (2010). FGF signaling regulates otic placode induction and refinement by controlling both ectodermal target genes and hindbrain Wnt8a. Dev. Biol. 340, 595–604. doi: 10.1016/j.ydbio.2010.02.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vendrell, V., Carnicero, E., Giraldez, F., Alonso, M. T., and Schimmang, T. (2000). Induction of inner ear fate by FGF3. Development 127, 2011–2019.
Vendrell, V., Vazquez-Echeverria, C., Lopez-Hernandez, I., Alonso, B. D., Martinez, S., Pujades, C.,et al. (2013). Roles of Wnt8a during formation and patterning of the mouse inner ear. Mech. Dev. 130, 160–168. doi: 10.1016/j.mod.2012.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wright, T. J., and Mansour, S. L. (2003). Fgf3 and Fgf10 are required for mouse otic placode induction. Development 130, 3379–3390. doi: 10.1242/dev.00555
Yajima, H., Motohashi, N., Ono, Y., Sato, S., Ikeda, K., Masuda, S.,et al. (2010). Six family genes control the proliferation and differentiation of muscle satellite cells. Exp. Cell Res. 316, 2932–2944. doi: 10.1016/j.yexcr.2010.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yajima, H., Suzuki, M., Ochi, H., Ikeda, K., Sato, S., Yamamura, K.,et al. (2014). Six1 is a key regulator of the developmental and evolutionary architecture of sensory neurons in craniates. BMC Biol. 12:40. doi: 10.1186/1741-7007-12-40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, W., Huang, L., Wei, Z. B., Silvius, D., Tang, B., and Xu, P. X. (2003). The role of Six1 in mammalian auditory system development. Development 130, 3989–4000. doi: 10.1242/dev.00628
Zou, D., Silvius, D., Rodrigo-Blomqvist, S., Enerback, S., and Xu, P. X. (2006). Eya1 regulates the growth of otic epithelium and interacts with Pax2 during the development of all sensory areas in the inner ear. Dev. Biol. 298, 430–441. doi: 10.1016/j.ydbio.2006.06.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: inner ear, induction, FGF signaling, myosin-II, morphogenesis
Citation: Sai X and Ladher RK (2015) Early steps in inner ear development: induction and morphogenesis of the otic placode. Front. Pharmacol. 6:19. doi: 10.3389/fphar.2015.00019
Received: 20 December 2014; Accepted: 21 January 2015;
Published online: 10 February 2015.
Edited by:
Hiroshi Hibino, Niigata University, JapanReviewed by:
Takao Imai, Osaka University Graduate School of Medicine, JapanFumiaki Nin, Niigata University, Japan
Copyright © 2015 Sai and Ladher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raj K. Ladher, Laboratory for Sensory Development, RIKEN Center for Developmental Biology, 2-2-3 Minatojima-Minamimachi, Chuo-ku, Kobe 650-0047, Japan e-mail:cmFqLWxhZGhlckBjZGIucmlrZW4uanA=