 Kirstin Hobiger
Kirstin Hobiger Thomas Friedrich
Thomas Friedrich- 1Department of Neurophysiology, Institute of Physiology and Pathophysiology, Philipps-Universität Marburg, Marburg, Germany
- 2Max-Volmer-Laboratory of Biophysical Chemistry, Institute of Chemistry, Technische Universität Berlin, Berlin, Germany
The transmembrane protein Ci-VSP from the ascidian Ciona intestinalis was described as first member of a fascinating family of enzymes, the voltage sensitive phosphatases (VSPs). Ci-VSP and its voltage-activated homologs from other species are stimulated by positive membrane potentials and dephosphorylate the head groups of negatively charged phosphoinositide phosphates (PIPs). In doing so, VSPs act as control centers at the cytosolic membrane surface, because they intervene in signaling cascades that are mediated by PIP lipids. The characteristic motif CX5RT/S in the active site classifies VSPs as members of the huge family of cysteine-based protein tyrosine phosphatases (PTPs). Although PTPs have already been well-characterized regarding both, structure and function, their relationship to VSPs has drawn only limited attention so far. Therefore, the intention of this review is to give a short overview about the extensive knowledge about PTPs in relation to the facts known about VSPs. Here, we concentrate on the structural features of the catalytic domain which are similar between both classes of phosphatases and their consequences for the enzymatic function. By discussing results obtained from crystal structures, molecular dynamics simulations, and mutagenesis studies, a possible mechanism for the catalytic cycle of VSPs is presented based on that one proposed for PTPs. In this way, we want to link the knowledge about the catalytic activity of VSPs and PTPs.
Introduction
Phosphorylation and dephosphorylation of cellular substrates, such as proteins, lipids, carbohydrates or nucleic acids, are crucial for the precise spatiotemporal transduction of signals across the cellular space. While phosphorylation of substrates is mediated by kinases, phosphatases attack their substrates by hydrolyzing covalently attached phosphate groups. In this context, protein tyrosine phosphatases (PTPs1) come into play with their dephosphorylation activity toward a diverse pool of cellular substrates. Members of this huge protein family are involved in multiple processes like endo- and exocytosis, cell differentiation, cell proliferation and migration – to list only a few examples. Because of this broad spectrum in activity, it is not surprising that malfunctions of PTPs are associated with various diseases, for example neurological disorders, diabetes, or cancer (Cohen, 2001; Tautz et al., 2013).
Most PTPs are very similar in the structure of their catalytic domain. Consequently, a medicinal treatment that should be specific just for one phosphatase bears always the risk of unwanted side effects since the drug may act on more than one of these enzymes. Therefore, the development of new medications requires a detailed knowledge about the individuality of PTPs, and here – in particular – about the molecular details of substrate recognition, activation and catalysis, about the mechanisms that regulate their activity, and about their functional role in specific signaling cascades (Zhang, 2001; Scott et al., 2010; He et al., 2014).
Voltage-sensitive phosphatases (VSPs) belong to the PTP family due to their homology in amino acid sequence of the active site and similarities in their structure of the catalytic domain. In VSPs, the phosphatase activity is directly coupled to the membrane potential. This voltage-switchable activity qualifies VSPs as ideal model systems to study enzymatic mechanisms of PTPs under defined experimental conditions.
Since the first identification of a PTP in Tonks et al. (1988a,b), a lot of studies have been performed leading to a huge amount of detailed information about these enzymes. However, although VSPs are members of the PTP family, their relationship has only been marginally discussed so far. In this review, we will address structural and mechanistic similarities of VSPs and PTPs. By linking the knowledge about these phosphatases, we want to answer the question if the results achieved for VSPs can be integrated into the set of principles which have already been described in more detail for PTPs.
General Classification of PTPs
The classification of PTPs is primarily based on their catalytic mechanism. Two different kinds of PTP subfamilies exist, the so called cysteine-based and the aspartate-based PTPs. Here, the name implies the key residue that mediates the dephosphorylation step in the reaction cycle of the respective class of enzymes. Although using a similar pool of substrates, cysteine- and aspartate-based PTPs are completely different in structure and the enzymatic reaction mechanism. Because this review will focus on cysteine-based PTPs, the reader interested in aspartate-based PTPs is referred to the excellent reviews by Moorhead et al. (2007, 2009).
The hallmark of all cysteine-based PTPs is the CX5RS/T sequence motif in their active site. Besides this so-called PTP recognition loop or P-loop, cysteine-based PTPs can be further categorized into three subclasses, I to III, based on their similarities in amino acid sequence, structure, and substrate specificity. The excellent overview of Alonso et al. (2004) is recommended here for a detailed classification of cysteine-based PTPs. However, for the sake of completeness, a short summary about the three PTP subclasses is also given here.
Class I PTPs – the classical and dual-specific PTPs
Class I PTPs form the largest subfamily of cysteine-based PTPs known so far. Their expression has been detected in all forms of life, from bacteria, yeast, plants to mammals (Pawson and Scott, 2005; Moorhead et al., 2009). This class of phosphatases comprises two subfamilies, the “classical” and the “dual-specific” PTPs (Figure 1).
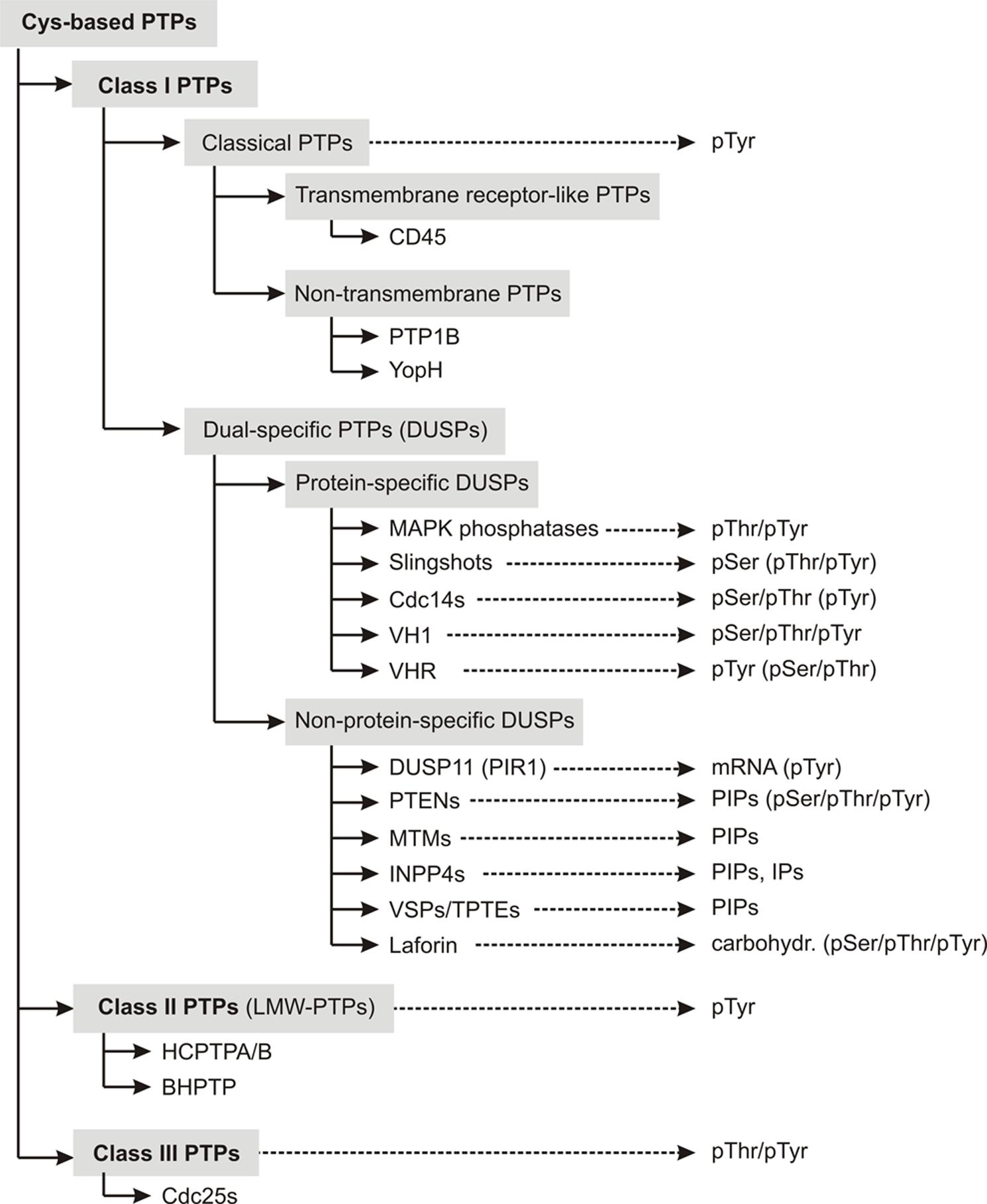
FIGURE 1. Overview about the classification scheme of cysteine-based PTPs. Members of individual subfamilies, which were exemplarily chosen and discussed in the main text, are listed in their respective PTP subclass. The preferred substrates of these phosphatases are given. Substrates that can be dephosphorylated, but with less preference are mentioned in parentheses. Abbreviations used here are explained in Table S1.
Classical PTPs show strict substrate specificity for phosphotyrosine (pTyr) residues. They are further categorized into receptor-like PTPs (rPTPs), which are anchored in the membrane, and non-transmembrane PTPs, which are located intracellularly.
A famous example for a receptor-like classical PTP is CD45, a transmembrane PTP that is expressed on the surface of leucocytes. Particularly in T lymphocytes, CD45 plays an essential role in the signal transduction that is initiated through the antigen-stimulation of the T cell receptor at the membrane surface (Altin and Sloan, 1997; Hermiston et al., 2003).
One prominent example for a non-transmembrane classical PTP is PTP1B, the founding member of the cysteine-based PTP superfamily (Tonks et al., 1988a,b). This soluble enzyme has attracted special attention in recent years, because it is well-known to act as negative regulator in the insulin signaling pathway. Therefore, it is a core subject of current pharmaceutical research as potential target for the treatment of type 2 diabetes and obesity (Goldstein, 2001; Zhang and Zhang, 2007; Combs, 2010). Besides that, PTP1B has also been discussed as putative drug target for breast cancer therapies (Yip et al., 2010; Feldhammer et al., 2013). However, interpretation of experimental results in this field needs careful consideration since there are hints that PTP1B might act in both directions, tumor suppression and promotion (Lessard et al., 2010).
In contrast to classical PTPs, phosphatases with the CX5RT/S motif in their active site and with activity toward more than pTyr substrates are categorized as dual-specific PTPs (DUSPs). DUSPs are the most diverse subclass among cysteine-based PTPs in terms of their substrate specificity. As described in more detail in the reviews of Alonso et al. (2004) and Tautz et al. (2013), they comprise phosphatases with activity toward protein- and/or non-protein substrates (Figure 1). Examples for protein-specific DUSPs are the phosphothreonine (pThr)-/pTyr-specific MAPK phosphatases (MKPs), the pSer-specific slingshots (SSHs), and the pSer-/pThr-specific Cdc14s.
Several DUSPs dephosphorylate more than protein-derived substrates. Examples for these phosphatases are the mRNA-specific phosphatase DUSP11 (also referred to as PIR1; Yuan et al., 1998), the phosphoinositol-specific PTENs (phosphatase and tensin homologs), MTMs (myotubularins) and INPP4s (inositol-4-phosphatases; Liu and Bankaitis, 2010; Kim et al., 2013), and the carbohydrate-specific phosphatase laforin (Ganesh et al., 2000).
For the following discussion, we selected only a few examples to illustrate general features of DUSPs regarding their structural, catalytic, and functional properties. We are aware that this can just give a rough insight into this field, because of the huge amount of information being available about this subclass of enzymes.
The first member of DUSPs described was VH1 from the vaccinia virus (Guan et al., 1991). This enzyme dephosphorylates pSer, pThr, and pTyr residues in vitro and in vivo (Guan and Dixon, 1991; Derrien et al., 1999). Included in the encapsidated fluid of vaccinia virions, VH1 is released into the intracellular space during the infection of a cell. Here, it dephosphorylates Stat1, an effector molecule of the host cell, which otherwise would initiate the antiviral immune response (Najarro and Traktman, 2001; Mann et al., 2008). In addition to that, VH1 activates own viral proteins by dephosphorylating them during the infection of a cell, i. a. those ones that mediate the viral morphogenesis (Liu et al., 1995; Derrien et al., 1999). In this way, VH1 is crucial for maintaining the viability of the virus.
The first homolog to VH1 found in humans was VHR (Ishibashi et al., 1992). This phosphatase hydrolyzes pTyr-/pThr-residues derived from MAP-kinases, such as Erk and Jnk and, thus, downregulates signaling cascades that are associated with cell proliferation and differentiation (Ishibashi et al., 1992; Denu et al., 1995b; Todd et al., 1999; Alonso et al., 2001).
Another human VH1-related phosphatase, DUSP11, specifically dephosphorylates mRNA and shows less activity toward phosphoproteins. Because DUSP11 interacts with the RNA/ribonucleoprotein-complex, it is proposed to be involved in the cellular mRNA metabolism (Yuan et al., 1998; Deshpande et al., 1999; Sankhala et al., 2014).
One of the most prominent members of non-protein-specific DUSPs is the tumor suppressor phosphatase PTEN. This enzyme was initially identified by screening diverse tumor-derived tissues as one of the most frequently mutated proteins (Li et al., 1997). Based on the P-loop motif in its active site, the cytosolic phosphatase was primarily believed to be a cysteine-based PTP specific for phosphorylated protein substrates. Shortly after its discovery, it has been demonstrated that PTEN is indeed a dual-specific class I PTP with dephosphorylation activity toward pTyr, pSer, and pThr residues in vitro (Furnari et al., 1997; Li and Sun, 1997; Myers et al., 1997). However, follow-up studies demonstrated that PTEN’s main substrate preference in vivo is toward the 3′-position of the phosphoinositide (PI) species PI(3,4,5)P3 at the inner cell membrane surface (Myers et al., 1997, 1998). Thus, PTEN counteracts the PI3K-signaling cascade, which otherwise leads to unrestricted cell growth with significantly increased susceptibility for tumor genesis (Knobbe et al., 2002; Leslie and Downes, 2002; Zhang and Yu, 2010; Miller et al., 2011).
One last example for DUSPs should be mentioned here: laforin. This human enzyme is the only phosphatase known so far that dephosphorylates complex carbohydrates (Worby et al., 2006; Tagliabracci et al., 2007). Laforin is further capable to dephosphorylate pTyr, pSer, and pThr residues in vitro (Ganesh et al., 2000) and pSer in vivo (Lohi et al., 2005). Missense mutations in the respective gene, EMP2A, which disrupt the activity of the phosphatase, are associated with a fatal progressive autosomal recessive form of myoclonic epilepsy, the Lafora disease (Tagliabracci et al., 2007). The mechanism by which laforin prevents the disease is only poorly understood. First results indicate that the phosphatase intervenes in the glycogen metabolism and prevents the formation of polyglucosan inclusion bodies in the cytosol of almost all cell types. These so-called “Lafora bodies” are a typical symptom of the disease (Wang et al., 2002; Fernández-Sánchez et al., 2003; Tagliabracci et al., 2007; Roach et al., 2012).
All these facts illustrate the broad substrate spectrum of DUSPs and their crucial relevance in human physiology. For further information about these phosphatases, the reader is referred to the detailed reviews listed here (Tronchère et al., 2003; Pulido and van Huijsduijnen, 2008; Liu and Bankaitis, 2010; Pulido et al., 2013; Tautz et al., 2013; He et al., 2014).
It should be noted, that the similarity of classical, dual-specific, and non-protein-/dual-specific phosphatases is largely restricted to the P-loop in the active site and the conservation of the folding pattern of the catalytic domain. The domains and regions that flank the phosphatase domain are diverse among class I PTPs, and specify their differences in cellular localization, activation, and regulation.
Class II PTPs – the low molecular weight PTPs
Class II PTPs are the so-called “low molecular weight” PTPs (LMW-PTPs). With ∼15-18 kDa, these phosphatases are smaller than class I PTPs that have a molecular weight of at least ∼30–50 kDa. Due to their catalytic activity at pH values ≤6, LMW-PTPs have also been classified as acid phosphatases (ACPs; Hopkinson et al., 1963; Lawrence and Van Etten, 1981; Lucentini et al., 2003). LMW-PTPs are soluble phosphatases with substrate specificity for pTyr substrates (Zhang and Van Etten, 1991; Stefani et al., 1993), but not for pSer or pThr residues (Ramponi et al., 1989; Wo et al., 1992).
Originally isolated from human red cells (Hopkinson et al., 1963), LMW-PTPs have been found in numerous prokaryotes and eukaryotes (Ramponi and Stefani, 1997a; Raugei et al., 2002). In mammals, they are ubiquitously expressed with no particular tissue-specific distribution (de Araujo et al., 1976; Zhang and Van Etten, 1990; Ramponi and Stefani, 1997b). Up to now, four human isoforms have been identified. All of them are splice variants of the transcription product of the ACP1 gene (Raugei et al., 2002). Two catalytically active isoforms, IF1 and IF2 (also named as HCPTPA and HCPTPB), differ only in a short sequence that spans one of the loop regions surrounding the active site. This variation in structure causes differences in both, substrate specificity and binding affinities against modulating ligands (Wo et al., 1992; Cirri et al., 1996; Ramponi and Stefani, 1997a). Apart from that, two other human splice variants of the ACP1 transcription product have been described, SV3 and LMW-PTP-C. Both isoforms seem to be catalytically inactive, but might regulate the active isoenzymes through the competition for substrate binding (Modesti et al., 1998; Tailor et al., 1999).
In mammals, catalytically active LMW-PTPs downregulate the activity of several tyrosine kinase receptors, for example the insulin receptor (Chiarugi et al., 1997), the vascular endothelial (Huang et al., 1999), the epidermal (Ramponi et al., 1989), and the fibroblast growth factor receptor (Park et al., 2002). Moreover, LMW-PTPs also regulate the cellular reaction to platelet-derived growth factor stimulation (Chiarugi et al., 2000). Because LMW-PTPs are involved in a variety of signaling cascades, the reduction or loss of their activity leads to different pathologic manifestations (Bottini et al., 2002), including allergy and asthma (Bottini et al., 2007), autoimmune diseases (Bottini et al., 2000), and neurodegenerative disorders like the Alzheimer’s disease (Shimohama et al., 1993, 1995). In particular, since LMW-PTPs play a crucial role in mitogenic signaling pathways, their malfunction entails high oncogenic potential (Malentacchi et al., 2005; Alho et al., 2008). Therefore, LMW-PTPs form another class of target molecules for therapeutics against serious diseases, including cancer (Homan et al., 2010; Mascarello et al., 2010; Seiler et al., 2013).
Class III PTPs – the Cdc25 phosphatases
Class III PTPs belong to the family of cell division control proteins (Cdc). According to this nomenclature, class III PTPs are named as Cdc25. Initially characterized in yeast (Russell and Nurse, 1984), these phosphatases are expressed in all eukaryotes (Boutros et al., 2007; Rudolph, 2007) except plants (Boudolf et al., 2006). Up to now, three isoforms have been identified in humans: Cdc25A, B and C (Galaktionov and Beach, 1991).
Cdc25 isoforms specifically dephosphorylate cyclin-dependent kinases (Cdks) at their inhibitory, dually phosphorylated N-terminal Thr-Tyr motif (Gautier et al., 1991; Krek and Nigg, 1991; Honda et al., 1993). In turn, these phosphatases show dual-specific substrate specificity. Cdks require for their activation the dephosphorylation by Cdc25 and, additionally, a phosphorylation mediated by Cdk-activating kinases (Kaldis, 2014). Through this enzymatic interplay, Cdks initiate the progression through the cell cycle (Gautier et al., 1991; Honda et al., 1993; Lammer et al., 1998). Apart from that, Cdc25s are also involved in preventing Cdk activation upon cellular DNA damage or incomplete replication (Kristjánsdóttir and Rudolph, 2004; Karlsson-Rosenthal and Millar, 2006). Acting as regulator of the eukaryotic cell cycle qualifies Cdc25 phosphatases as promising targets for anti-cancer therapeutics (Kristjánsdóttir and Rudolph, 2004; Rudolph, 2007).
Because Cdc25s dephosphorylate more than pTyr substrates, they are often called dual-specific phosphatases. This nomenclature might cause confusion by mixing up Cdc25s with dual-specific phosphatases from class I PTPs. Here, it should be noted, that despite the activity toward more than pTyr substrates, dual-specific PTPs of class I and III are individual subfamilies with significant differences in protein structure.
Interestingly, Cdc25s have evolved as a unique subclass of PTPs, but the reason for this uniqueness has been controversially discussed. As mentioned by Alonso et al. (2004), one possible explanation could be that ancestral Cdc25s had already acted as rhodanese-type enzymes – a class of sulfur-transfer proteins (Cipollone et al., 2008) – on Cdks, but were finally transformed into cysteine-based PTPs when the Tyr/Thr phosphorylation of Cdks appeared. This idea is supported by the fact that the overall structure of Cdc25s is rather similar to rhodanese enzymes than to class I and II PTPs (Fauman et al., 1998; Reynolds et al., 1999; Bordo and Bork, 2002).
Although all three subclasses I to III of the cysteine-based PTP family share similarities regarding their catalytic mechanism and active site structure, all of them have evolved independently from each other (Alonso et al., 2004; Moorhead et al., 2009). From that, the question arises: to which subclass of PTPs does the family of VSPs belong?
Integration of Voltage Sensitive Phosphatases Into the Classification System of PTPs
The first mammalian variants of VSPs were described in the early 2000s with publications about transmembrane phosphatases with tensin homology (TPTEs2; Chen et al., 1999; Guipponi et al., 2000, 2001; Walker et al., 2001; Wu et al., 2001; Tapparel et al., 2003). In these early studies, the research had been focused only on the biochemical characterization of the enzymatic activity, because the proteins share a high sequence homology with the tumor suppressor PTEN. Finally, bioinformatic analyses suggested that the transmembrane part of these phosphatases might act as voltage sensor domain (Kumanovics et al., 2002). However, no functional characterization of a voltage-dependent activity was carried out at this point. With the description of Ci-VSP (VSP from Ciona intestinalis) in Murata et al. (2005), the first experimental evidence for the existence of a transmembrane phosphatase with voltage-driven activity was published.
Ci-VSP, the most prominent example of VSPs to date, can be easily expressed in heterologous expression systems; a fact that facilitates the electrophysiological characterization of this protein. As other VSPs, Ci-VSP consists of a membrane-spanning voltage sensor domain and a cytosolic catalytic domain. The voltage sensor domain consists of four putatively α-helical transmembrane segments (Li et al., 2014). The fourth segment contains positively charged residues arranged in a periodical order as it can be found in voltage-gated ion channels (Villalba-Galea, 2012b). The cytosolic domain consists of two subdomains, the phosphatase and an adjacent C2 domain (Matsuda et al., 2011; Liu et al., 2012). The phosphatase-/C2-complex of Ci-VSP shows ∼40% amino acid homology to the tumor suppressor PTEN. Due to this fact, the phosphatase domain of Ci-VSP is structurally related to that one of class I PTPs (Okamura and Dixon, 2011; Villalba-Galea, 2012a).
Generally, VSPs are capable to “translate” voltage stimuli at the plasma membrane directly into enzymatic activity in the cytosol. Positive membrane potentials lead to conformational changes in the voltage sensor domain that activate the intracellular catalytic domain. In the activated state, the enzymatic domain cleaves phosphate groups from the inositol ring of phosphoinositide phosphates (PIPs). In particular, Ci-VSP specifically hydrolyzes the 5′-phosphate of the PIP-species PI(3,4,5)P3 and PI(4,5)P2 (Iwasaki et al., 2008; Halaszovich et al., 2009). Furthermore, several results indicated a 3′-site activity toward PI(3,4)P2 for Ci-VSP when the phosphatase was expressed in Xenopus oocytes, but with a decrease in efficiency compared to the 5′-activitiy (Sakata et al., 2011; Sakata and Okamura, 2013). Controversially, such a 3′-specificity could not be detected when Ci-VSP was expressed in mammalian cells (Lacroix et al., 2011; Halaszovich et al., 2012). This discrepancy raised the question whether the 3′-site activity of Ci-VSP is specific for the oocyte expression system (Halaszovich et al., 2012).
After the discovery of Ci-VSP, several homologs have been described from other species, such as zebrafish (Danio rerio, Dr-VSP; Hossain et al., 2008), chicken (Gallus gallus, Gg-VSP; Neuhaus and Hollemann, 2009; Kurokawa et al., 2012), the African clawed frog (Xenopus laevis, Xl-VSP; Ratzan et al., 2011), and urodele amphibians (Mutua et al., 2014). In addition, transmembrane phosphatases found in mammals have been characterized in more detail in the recent years (Halaszovich et al., 2012; Kurokawa et al., 2012; Sutton et al., 2012). Although several studies demonstrated the expression of VSPs in testis, intestine, stomach, kidney, and the nervous system (Chen et al., 1999; Walker et al., 2001; Tapparel et al., 2003; Murata et al., 2005; Neuhaus and Hollemann, 2009; Ogasawara et al., 2011), the physiological role of VSPs in their native organisms is still elusive.
There are several isoforms of VSPs that are catalytically inactive toward their presumed PIP-substrates, for example TPTE/hVSP2 (Walker et al., 2001; Leslie et al., 2007). However, all active isoenzymes share the common property of dephosphorylation activity at positive membrane potentials. Moreover, all homologs characterized so far show substrate specificity against PIP-lipids, whereas the preference either for the 3′- or the 5′-phosphate at the inositol ring seems to differ among VSPs (Walker et al., 2001; Murata et al., 2005; Iwasaki et al., 2008; Halaszovich et al., 2009, 2012; Lacroix et al., 2011; Kurokawa et al., 2012).
In conclusion, because of the CX5RS/T motif in the active site, VSPs belong to the family of cysteine-based PTPs. Furthermore, the catalytic domain of VSPs shows a three-dimensional folding that is characteristic for class I PTPs. Together with the fact that VSPs specifically dephosphorylate PIP-lipids, the criteria are met to classify VSPs as dual-specific class I PTPs (Figure 1).
General Folding of the Catalytic Domain of PTPs
The catalytic domains of cysteine-based PTPs have various folding properties in common. For example, all of them contain a central core consisting of highly twisted β-sheets, which are flanked by several α-helices (Figure 2A). One of the β-sheet-α-helix-loops contains the active site motif CX5RS/T. This P-loop is highly conserved and shares a homolog folding pattern for all cysteine-based PTPs (Figure 2B). Although the residues in the X5-segment vary largely between the different PTP subclasses (Figure 3A), the superposition of the P-loop structures evinces only minor deviations in the Cα-trace of the protein backbones of less than 1 Å.
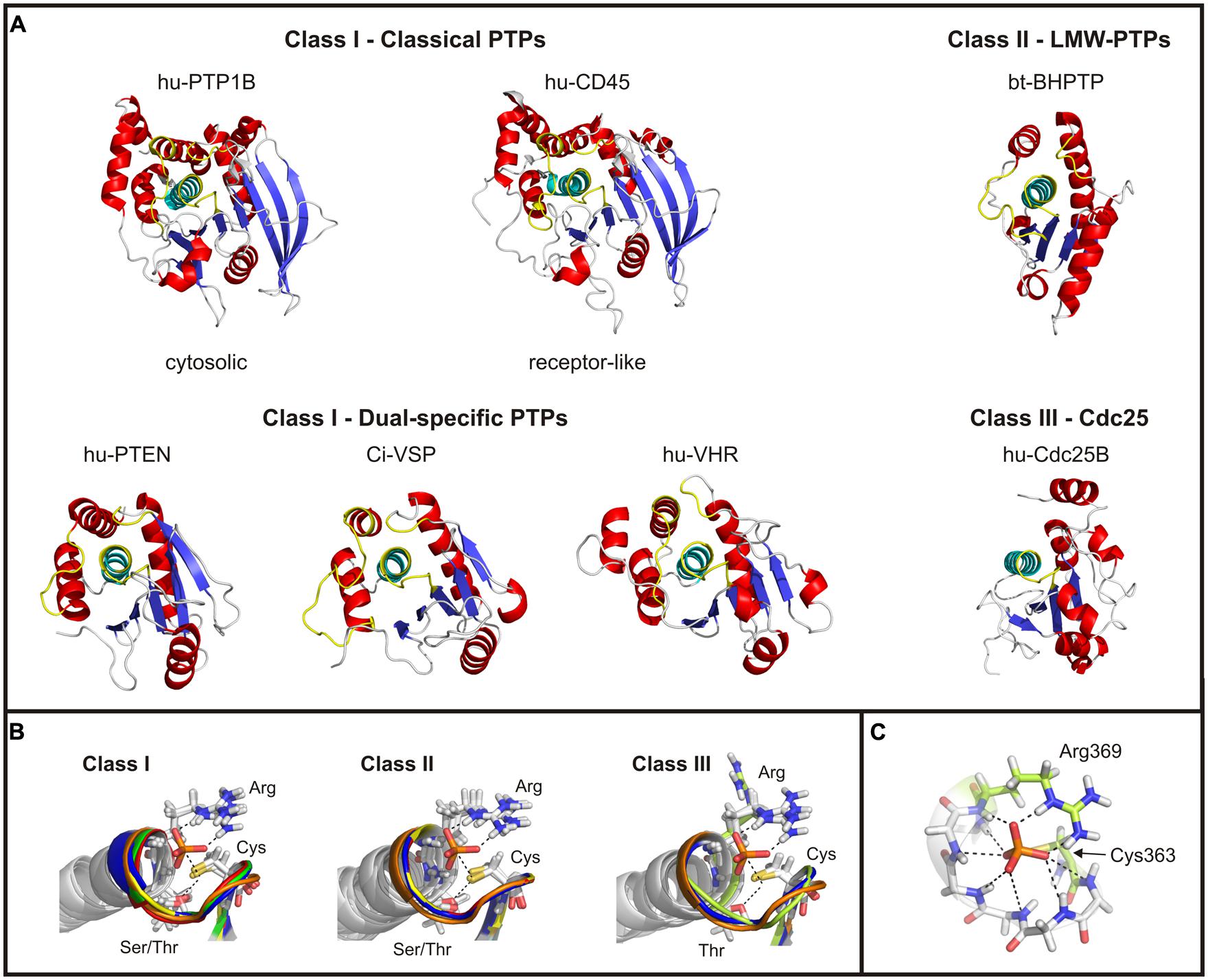
FIGURE 2. Structural comparison of the catalytic domains of cysteine-based PTPs. (A) Catalytic domains of exemplary chosen phosphatases are shown. The twisted β-sheet core is colored in blue; the α-helices surrounding the core are shown in red. The α-helix C-terminal to the active site loop is depicted in cyan, and the regions corresponding to the P-, TI- and WPD-loop of class I PTPs in yellow (see text for details). In case of Cdc25s, no TI- and WPD-loop exist. The structure of Ci-VSP was taken from the MD simulation (Hobiger et al., 2013) which was performed on the basis of the crystal structure by Matsuda et al. (2011; PDB accession number 3awe). All other structures were created based on the crystal structures available in the RCSB protein data bank. The accession numbers are: 1nwe (hu-PTP1B), 1ygu (hu-CD45. Here, the catalytically active domain D1 is shown.), 1d5r (hu-PTEN), 1vhr (hu-VHR), 1dg9 (bt-BHPTP), and 1qb0 (hu-Cdc25B). Abbreviations are: hu, human; bt, bos taurus; and BHPTP, bovine heart PTP. (B) The P-loop motif and the following α-helix are structurally aligned for all classes of cysteine-based PTPs. For comparable reasons, the P-loop structure of Ci-VSP has been integrated in all panels (colored in orange). Class I PTPs used for the alignment were the human isoforms of PTP1B (1nwe: green), of the D1 domain of CD45 (1ygu, red), of PTEN (1d5r, blue), and of VHR (1vhr, yellow). Class II PTPs are the bovine BHPTP (1dg9, yellow), and the human isoforms HCPTPA (5pnt, blue) and HCPTPB (1xww, red). Class III PTPs are the human isoforms Cdc25A (1c25, yellowish) and Cdc25B (1qb0, blue). In all structures a phosphate anion is indicated in the active site. (C) The phosphate group of the substrate is coordinated by hydrogen bonds with backbone atoms of the P-loop residues, as demonstrated here for Ci-VSP. Residues crucial in catalysis, Cys363 and Arg369, are particularly denoted.
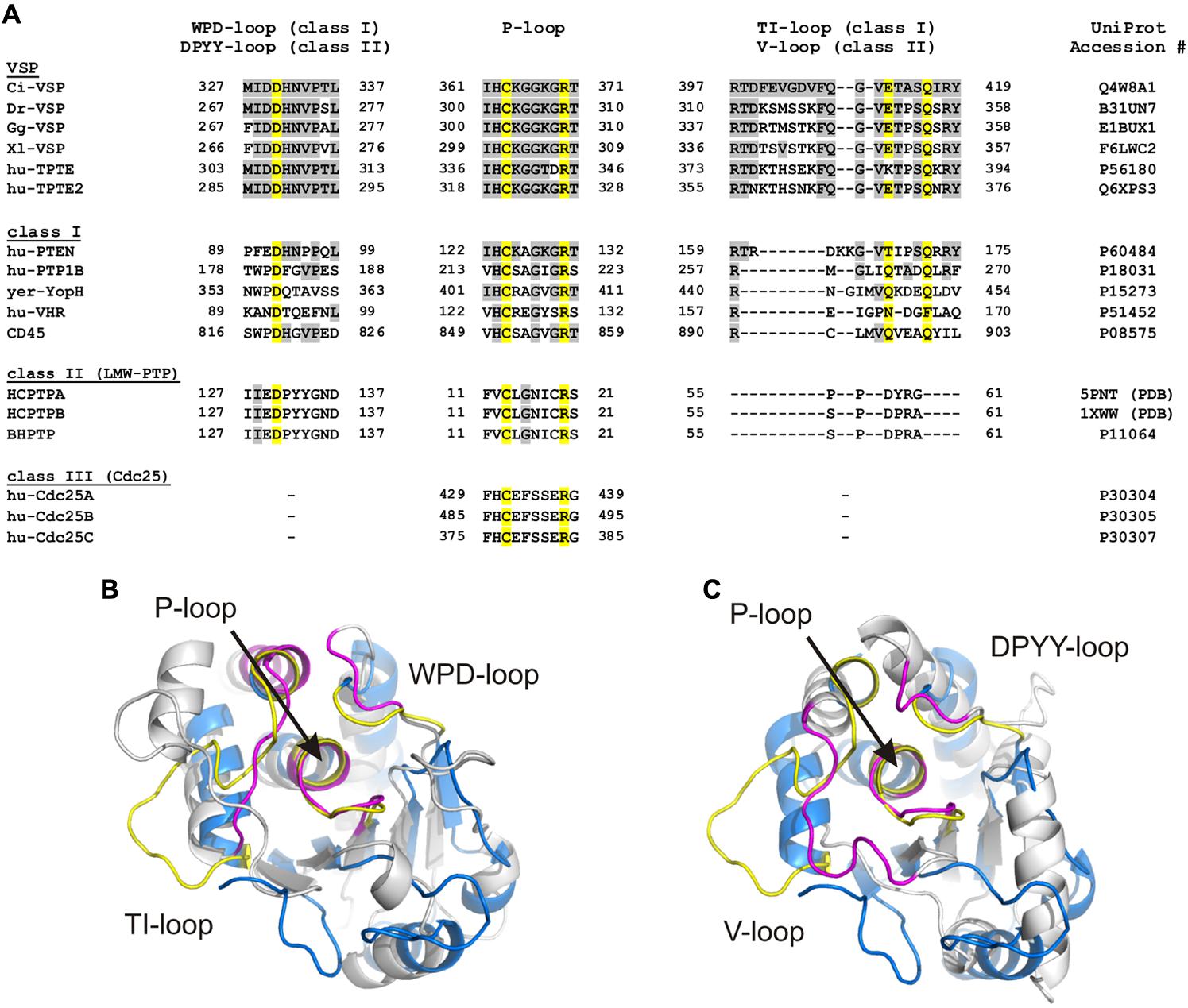
FIGURE 3. Loop regions determine the conformation of the substrate binding pocket in cysteine-based PTPs. (A) Sequence alignment of the three loops which specify the conformation of the substrate binding pocket. The P-loop contains the highly conserved CX5R motif that serves as active site. The WPD-/DPYY- and the TI-/V-loops form the side walls of the substrate binding pocket in class I and II PTPs. Because class III PTPs lack the two latter loops, no sequence alignment could be performed for these regions. Amino acids identified as identical to Ci-VSP are marked in gray. Yellow colored residues are crucial for catalysis and widely conserved among PTPs. UniProt accession numbers are indicated for all sequences used for the alignment; in case of HCPTPA and HCPTPB, accession numbers in the PDB database are given, since UniProt contains the same reference sequence for both PTPs. (B,C) Structural alignment of the phosphatase domain of Ci-VSP (colored in blue) with (B) the catalytic domain of the human class I PTP, VHR (PDB accession number 1vhr) and (C) the class II PTP, BHPTP (bovine heart PTP, PDB 1dg9). VHR and BHPTP are colored in gray. Loops are highlighted in yellow for Ci-VSP and in magenta for the respective PTP. The catalytic domain of Ci-VSP was obtained by MD simulations (Hobiger et al., 2013) based on the crystal structure of Matsuda et al. (2011; PDB 3awe).
For all cysteine-based PTPs, the active site is located within a crevice with the conserved cysteine at its bottom. Together with the P-loop arginine, the catalytically active residue forms a cradle-like structure (Figure 2B). This conformation is important for the correct binding of the substrate and for stabilizing the active site structure during the whole catalytic cycle. A network of hydrogen bridges between the phosphate group of the substrate and the backbone atoms of the P-loop further facilitates this stabilization (Figure 2C).
The active site pocket of class I and II PTPs is flanked by two loop regions, which are named TI- and WPD-loop for class I PTPs, and variable (or V-) and DPYY-loop for class II PTPs (Figure 3). In both classes, these regions contain amino acids that are crucial for the enzymatic reaction. Although the loop regions in class I and II PTPs differ in length and sequence (Figure 3A), their existence indicates a common principle for cysteine-based PTPs regarding the regulation of their activity.
In class III PTPs, none of these two loops exists (Figure 2A). However, these phosphatases use mechanisms that compensate for the lack of the loop structures flanking the active site. This will be discussed later in this review.
Despite several similarities, class I to III PTPs show significant structural differences which will be discussed in the following.
The Catalytic Domain of Class I PTPs
Catalytic domains of classical PTPs contain ∼240–280 amino acids, and are the largest among the cysteine-based PTPs (Figure 2A). The catalytic domains of dual-specific PTPs are smaller; for example, the phosphatase of PTEN contains ∼180 amino acids. In particular, class I PTPs differ in the amount of β-sheets in the protein core and of the α-helices flanking the core. These structural variations are one of the reasons for differences in regulation of the catalytic activity and the cellular targeting of class I PTPs (Tabernero et al., 2008).
Each class I PTP contains at least one catalytic domain. Examples for single phosphatase PTPs are PTP1B, VHR, laforin and PTEN (Tonks, 2006; Pulido et al., 2013). Most of the receptor-like PTPs contain two tandemly repeated catalytic domains, called D1 and D2 (Alonso et al., 2004), where the latter is located more distally to the membrane and has no catalytic activity (Streuli et al., 1990). As shown for CD45, the D2 domain is proposed to play a regulatory role for the phosphatase activity of D1 (Nam et al., 2005).
Another characteristic feature that differs among class I PTPs is the depth of the substrate binding pocket, with ∼9 Å for classical and ∼6 Å for dual-specific PTPs (Barford et al., 1994; Stuckey et al., 1994; Yuvaniyama et al., 1996). This difference is proposed to determine the substrate specificity of the phosphatases (Barford et al., 1994; Jia et al., 1995; Yuvaniyama et al., 1996). The deeper pocket in classical PTPs permits the access to the active site only for longer pTyr residues, whereas dual-specific phosphatases are capable to bind pSer, pThr, and/or pTyr residues.
In case of the PIP-specific phosphatase PTEN, a crevice of 8 Å in depth was observed in the crystal structure with a wider access than in classical PTPs. This structural feature was suggested to be the reason that PTEN preferentially accommodates the sterically demanding PIP-substrates in its active site instead of pTyr, pSer, or pThr substrates (Lee et al., 1999).
The Catalytic Domain of Class II PTPs
LMW-PTPs contain one catalytic domain that consists of four β-sheets flanked by at least five α-helices on both sides (Figure 2A; Su et al., 1994; Zhang et al., 1994a). In contrast to class I PTPs, the P-loop is located at the N-terminal part of the phosphatase connecting sheet β1 with helix α1. For example, the catalytically active cysteine of the bovine liver LMW-PTP is located at position 12 in contrast to the soluble PTP1B, a class I PTP, which has the cysteine at position 215 (Figure 3A). As mentioned above, the P-loop in LMW-PTPs is surrounded by two loops: (i) the V-loop, which differs in length and sequence among the phosphatases, and (ii) the DPYY-loop. Both loops are as closely positioned to the active site as the TI- and WPD-loops in class I PTPs (Figures 3B,C). This configuration leads to a compact substrate binding pocket in LMW-PTPs, which is smaller, but similar to that of class I PTPs. This is an interesting fact, since the overall amino acid sequences of both PTP subclasses show no significant homology except for the P-loop motif (Figure 3A; Su et al., 1994).
The Catalytic Domain of Class III PTPs
The full length of Cdc25 phosphatases ranges from 300 to 600 amino acids (Kristjánsdóttir and Rudolph, 2004). Structurally, the enzymes are divided into an N- and C-terminal region. The N-terminal part is highly variant among Cdc25 phosphatases and contains several regulatory sites, which are involved in interactions with other proteins (Conklin et al., 1995; Chen et al., 2000a; Gnesutta et al., 2001; Kawabe et al., 2002), in phosphorylation or ubiquitination (Hoffmann et al., 1993, 1994; Gabrielli et al., 1997; Bernardi et al., 2000; Mailand et al., 2000; Donzelli et al., 2002).
The C-terminal catalytic domain consists of about 200 residues. It contains the active site motif CX5R, but lacks the serine or threonine seven positions more C-terminal to the catalytically active cysteine as found in other PTPs (Figure 3A). However, the motif adopts the cradle-shaped conformation that is typical for cysteine-based PTPs, with the cysteine and the arginine at positions that allow for binding of the substrate in the active site and for passing through the catalytic cycle (Figure 2B; Fauman et al., 1998; Reynolds et al., 1999). Here, it should be noted that in the crystal structure of Cdc25A (Fauman et al., 1998), the arginine points away from the substrate binding pocket (Figure 2B); this configuration would be unfavorable for substrate binding. However, in a structure subsequently described for the Cdc25A-homolog Cdc25B, the arginine was positioned in a way that resembles that of other PTPs (Reynolds et al., 1999). Additionally, molecular dynamics (MDs) simulations carried out on Cdc25A in presence of an oxyanion revealed a spontaneous flip of the P-loop segment during docking of the ligand into the substrate binding pocket. In this way, the active site of Cdc25A adopts a conformation similar to other PTPs (Kolmodin and Åqvist, 2000).
Apart from the active site motif, the α-helix following C-terminally after the P-loop in Cdc25 matches well with the configuration found in class I and II PTPs (Figure 2B). However, the catalytic domain of Cdc25 contains a smaller number of β-strands and α-helices compared to class I PTPs (Figure 2A). Moreover, the loop regions surrounding the substrate binding pocket in class I and II PTPs are absent in Cdc25 phosphatases. Interestingly, the overall structure of class III PTPs is less homologous to other cysteine-based PTP subfamilies, but shows more similarities to their ancestral relatives, the rhodanese enzymes (Fauman et al., 1998; Reynolds et al., 1999).
General Catalytic Mechanism of PTPs
Since the three-dimensional structure of the active site is similar for all three PTP subclasses, it is reasonable to assume that these phosphatases share a similar enzymatic reaction mechanism. Indeed, results from numerous studies about different cysteine-based PTPs converge on the same general two-step catalytic mechanism that applies for all class I to III phosphatases (Figure 4; Zhang et al., 1994c; Denu and Dixon, 1995, 1998; Tonks and Neel, 2001; Rudolph, 2002; Wang et al., 2003).
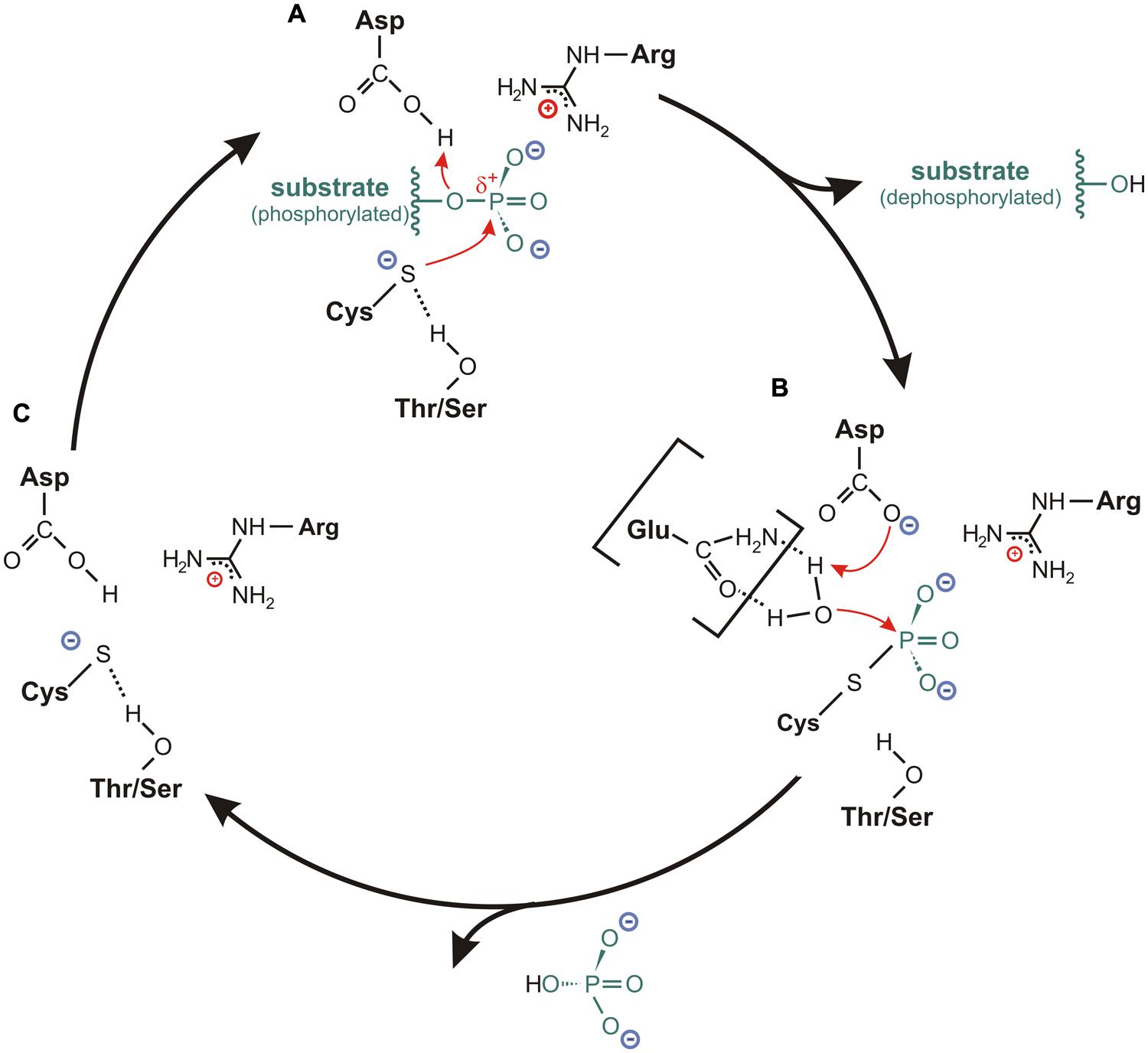
FIGURE 4. Catalytic cycle for cysteine-based PTPs. (A) In the resting state, the sulfur of the catalytic cysteine is stabilized in an anionic thiolate form. This is mediated by a hydrogen bond between the sulfur and the hydroxyl oxygen of the conserved Thr-/Ser-side chain located in the P-loop. Upon binding of the substrate, where the P-loop arginine is particularly involved in the positioning of the substrate, the thiolate acts as catalytic nucleophile by attacking the phosphorus atom of the substrate. The formation of a cysteinyl-phosphate intermediate is facilitated by a general acid (usually an aspartate from the WPD- or DPYY-loop flanking the active site), which donates a proton to the substrate leaving group. Subsequently, the dephosphorylated substrate dissociates from the substrate binding pocket. (B) The hydrolysis of the cysteinyl-phosphate intermediate is mediated by an activated water molecule. Here, the aspartate or a corresponding residue serves as general base by accepting one proton of the water molecule. In several class I PTPs, the water molecule is activated through hydrogen bonds with one or two polar amino acids in the TI-loop, as it is indicated here for an interaction with a glutamine. After accepting the proton from the general base, the phosphate dissociates from the catalytic cysteine, so that (C) the resting state conformation of the active site is reconfigured.
The reaction cycle starts with a nucleophilic attack on the phosphorus atom of the substrate; a step that is mediated by the cysteine in the PTP signature motif (Figure 4A). Here, a general acid located in close vicinity to the catalytic cysteine donates a proton to the substrate leaving group. In this way, a cysteinyl-phosphate intermediate is formed and the dephosphorylated substrate dissociates from the active site cavity (Figure 4A; Guan and Dixon, 1991; Jia et al., 1995; Denu et al., 1996; Pannifer et al., 1998).
The second reaction step requires the presence of an activated water molecule. This is achieved by a general base (Figure 4B). The residue that serves as general acid during the first reaction step usually acts as general base in this second step by accepting a proton from the activated water molecule (Taddei et al., 1994; Denu et al., 1995a, 1996; Flint et al., 1997; Pannifer et al., 1998). The resulting configuration in the active site promotes the hydrolysis of the cysteinyl-phosphate intermediate, whereby the phosphate group is released. This step finalizes the reaction and restores the conformation of the active site for a new catalytic cycle (Figure 4C).
Differences in the Catalytic Cycle Between Class I, II, and III PTPs
A close look into the reaction scheme of cysteine-based PTPs implies that not only the active site motif participates in catalysis, but that all conserved loop regions are involved in the reaction. In particular, for class I and II PTPs, the WPD- or DPYY-loop contains the general acid for the first reaction step (Figure 3A). Due to the lack of this loop in class III PTPs, the existence of a general acid has been controversial. An early study indicated that an aspartate localized in the protein apart from the P-loop serves as general acid in Cdc25 (Eckstein et al., 1996). However, the crystal structure of the human isoform Cdc25A had been resolved shortly afterwards, and it demonstrated a structural rather than a catalytic role for this aspartate (Fauman et al., 1998). Based on this and the Cdc25B structure, one of the two glutamates found in the X5-segment of the active site motif (Figure 3A) were proposed to act as general acid (Fauman et al., 1998; Reynolds et al., 1999). These results were further supported by MD simulations carried out on Cdc25B in presence of its natural substrate (Arantes, 2008). In contrast to that, it has been further suggested that Cdc25 phosphatases might not require a general acid, because changes in pH in the active site during the catalytic cycle could compensate for the lack of a proton donor (Rudolph, 2002, 2007). Controversially, several studies imply that the general acid might be provided by the substrate itself instead of being contributed by the phosphatase (Chen et al., 2000b). However, up to now this question has not been completely resolved.
For the second catalytic step in class I and II PTPs, the amino acid that served as general acid in the previous reaction step is now involved in the activation of a water molecule by acting as general base. A different mechanism can be assumed for class III PTPs again, because of the lack of the WPD- or a WPD-equivalent loop. Studies addressing the influence of the pH on the active site suggest that, also here, the two glutamates in the P-loop might be involved in the activation of a critical water molecule (Rudolph, 2002, 2007). However, also this issue is still under controversial discussion.
In the following, we want to elucidate in more detail the role of the loop regions surrounding the active site. Because Cdc25 phosphatases are such a distinct subclass of cysteine-based PTPs with large differences in the amino acids that might participate in the catalytic reaction, we will exclude them from further discussion. Readers interested in details of the catalytic mechanism of class III PTPs are referred to the reviews of Rudolph (2002, 2007).
Structural Elements Defining the Phosphatase Activity of PTPs
The P-Loop
As mentioned above, the active site motif CX5RS/T is highly conserved among all cysteine-based PTPs (Figure 3A). This loop region is located at the bottom of the substrate binding pocket and is surrounded by other loop regions forming the side walls of the pocket. Polar or charged residues in the P-loop are involved in substrate docking and in the stabilization of the cysteinyl-phosphate intermediate during the catalytic cycle. In the following, we will discuss the functional relevance of critical amino acids in this loop region.
The catalytic cysteine
The cysteine within the CX5RS/T-motif of the P-loop is the most crucial residue for the catalytic reaction. The exchange of this residue by serine results in the inactivation of all cysteine-based PTPs. The cysteine-to-serine substitution in the active site is one of the best characterized mutations of PTPs, since it is generally used as negative control to analyze the catalytic activity of these enzymes.
Apart from that, it has to be considered that the nucleophilic attack mediated by the P-loop cysteine requires a correct positioning of the sulfhydryl-side chain with respect to the size of the substrate binding pocket and the adjacent residues being involved in the reaction steps. In this context, it has been frequently described that the nucleophilic attack requires the sulfur of the cysteine to be in an anionic thiolate configuration (Zhang, 1993; Denu and Dixon, 1995; Evans et al., 1996; Lohse et al., 1997). Crystal structures or results obtained by MD simulations showed that this configuration is enabled by a hydrogen bond between the sulfur of the cysteine and the hydroxyl oxygen of the conserved P-loop threonine or serine seven positions C-terminally to the cysteine (Figures 2B, 3A, and 4A; Barford et al., 1994; Stuckey et al., 1994; Yuvaniyama et al., 1996; Barford et al., 1998). As proposed by Evans et al. (1996) for BHPTP (a class II PTP), the hydrogen bond between the cysteine and the serine causes an unusually low pKa of the sulfhydryl-side chain, with less than 4. Similarly, reduced pKa values were also described for other cysteine-based phosphatases, such as PTP1B (Lohse et al., 1997) or YopH from Yersinia pestis (Zhang, 1993). This condition is proposed to stabilize the cysteine in the thiolate configuration, so that the nucleophilic attack on the substrate can proceed (Dillet et al., 2000). In addition to that, Denu and Dixon (1995) proposed that the hydrogen bond between the cysteine and the Thr/Ser might not only stabilize the conformation of the enzyme’s resting state, but also that one of the cysteinyl-phosphate intermediate. In this way, the hydrolysis of the phosphate during the second reaction step might be facilitated (Denu and Dixon, 1995).
Analogously in Ci-VSP, the catalytic cysteine and the P-loop threonine are closely located to each other (Figure 2A); a configuration that was found in crystal structures of the cytosolic domain of Ci-VSP (Matsuda et al., 2011; Liu et al., 2012) and in a structural model of the protein obtained by MD simulations (Hobiger et al., 2013). These results suggest the existence of a hydrogen bond between the two residues also for Ci-VSP. However, it is still elusive if this configuration has any influence on the pKa value of the cysteine side chain. Therefore, the question remains if the sulfur is in an anionic thiolate form in VSPs, and which role this configuration plays in catalysis.
Another interesting feature about the cysteine in the P-loop is its sensitivity to redox modification. Several studies demonstrated that the presence of redox-reactive agents, such as H2O2 or NO, reversibly eliminates the enzymatic activity of cysteine-based PTPs (Denu and Tanner, 1998; Xu et al., 2002; Salmeen and Barford, 2005; Tonks, 2005; Ross et al., 2007; Conway et al., 2010; Ostman et al., 2011; Miki and Funato, 2012). For most PTPs, this inhibition is caused by the formation of a disulfide bond between the P-loop cysteine and another cysteine in its immediate environment, as e.g., observed in PTEN (class I; Lee et al., 2002), HCPTP (class II; Chiarugi et al., 2001), and Cdc25B (class III PTP; Buhrmann et al., 2005). In case of PTP1B, the cysteine forms a sulphenyl-amide bond under oxidizing conditions with the nitrogen atom in the backbone of a serine that is located next to the catalytically active side chain (Figure 5C; Salmeen et al., 2003; van Montfort et al., 2003). When bound either in the sulphenyl-amide or in the disulfide bond, the sulfhydryl group of the P-loop cysteine is incapable of performing the nucleophilic attack on the phosphate substrate, thus preventing catalysis.
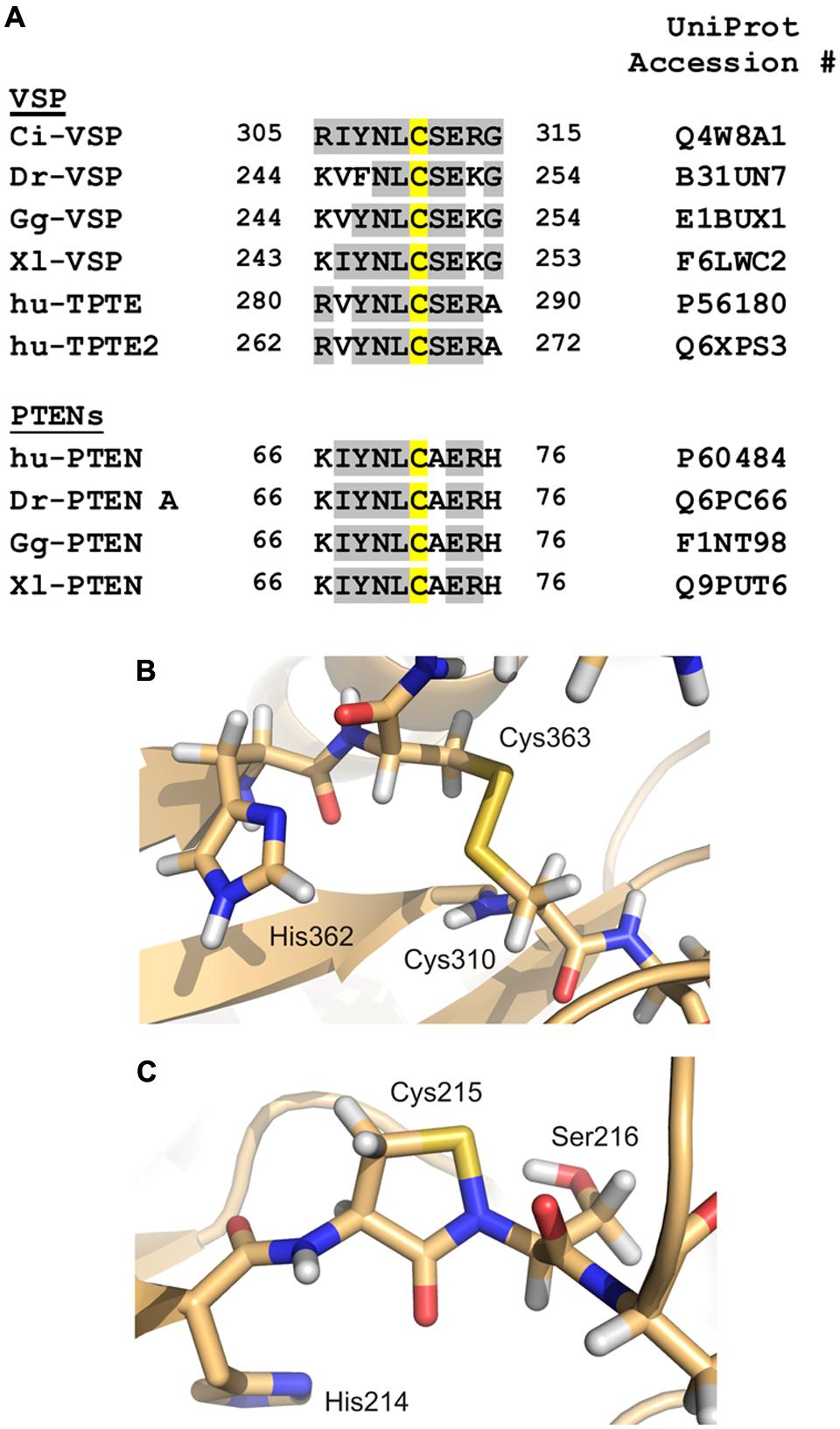
FIGURE 5. Redox modification of the catalytically active side chain in cysteine-based PTPs. (A) The amino acid motif of Ci-VSP, which contains Cys310 as putative interaction partner for the disulfide-bridge with the catalytic Cys363, is aligned with sequences of VSP homologs from various species (Dr, Danio rerio; Gg, Gallus gallus; Xl, Xenopus laevis; hu, human). Additionally, PTEN isoforms from different species are included in this alignment. Amino acid identities to Ci-VSP are colored in gray. Cys310 and the corresponding position in the respective proteins are marked in yellow. (B) For Ci-VSP, the disulfide bridge between Cys310 and the catalytic Cys363 is shown as it has been identified in a crystal structure of the cytosolic domain (PDB accession number 3awg; Matsuda et al., 2011). (C) In crystal structures of the human PTP1B, the formation of a sulphenyl-amide bond has been observed between the P-loop Ser216 and the catalytically active Cys215 (PDB 1oes; van Montfort et al., 2003).
For Ci-VSP, a disulfide bond has been found in one of the crystal structures of the cytosolic domain (Figure 5B; Matsuda et al., 2011). This bond was present between the catalytically active residue Cys363 and the neighboring cysteine, Cys310. Accordingly to other PTPs, Ci-VSP does not show phosphatase activity toward PIP substrates in presence of H2O2 (Matsuda et al., 2011), which demonstrates the inhibiting effect of oxidizing conditions on catalysis.
The alignment of amino acid sequences shows that the cysteine corresponding to position 310 in Ci-VSP is highly conserved in VSPs and also in PTENs (Figure 5A). For the latter enzymes, the redox regulation has been well-characterized (Lee et al., 2002; Leslie et al., 2003; Cho et al., 2004; Yu et al., 2005; Kim et al., 2010; Kitagishi and Matsuda, 2013). These facts indicate that the redox regulation of VSPs might follow a similar pattern as described for other PTPs.
The invariant arginine
Apart from the P-loop cysteine, the invariant arginine in the active site motif has frequently been found to be critical for the correct binding of the substrate in the active site (Figure 2B; Stuckey et al., 1994; Jia et al., 1995; Barford et al., 1998). Furthermore, this residue has been proposed to be responsible for the stabilization of the cysteinyl-phosphate intermediate during the reaction cycle (Zhang et al., 1994c; Pannifer et al., 1998). Based on crystal structure analyses of the soluble phosphatase domain of Ci-VSP, Liu et al. (2012) suggested that this arginine probably marks the position of the phosphate at the substrate that has to be cleaved.
Glycine versus alanine and the influence on substrate specificity
A further interesting residue in the P-loop motif is the amino acid two positions more C-terminally from the catalytic cysteine. While PTEN carries an alanine at this position, a glycine is found there in VSPs (Figure 3A). PTEN specifically dephosphorylates the 3′-phosphate from PI(3,4,5)P3 (Maehama and Dixon, 1998; Maehama et al., 2001; Iwasaki et al., 2008), whereas Ci-VSP shows a broader substrate spectrum by cleaving the 5′-phosphate from PI(3,4,5)P3 and PI(4,5)P2 (Iwasaki et al., 2008; Halaszovich et al., 2009) and – presumably with less efficiency – the 3′-phosphate from PI(3,4)P2 (Sakata et al., 2011; Liu et al., 2012; Sakata and Okamura, 2013). This difference raises the question whether the substrate specificity of VSPs will be changed if the glycine in the active site is substituted against alanine to mimic the substrate binding pocket of PTEN.
In Ci-VSP, the mutation G365A significantly decreases the phosphatase activity toward PI(3,4,5)P3 and PI(4,5)P2 in vitro and in vivo (Iwasaki et al., 2008; Liu et al., 2012). However, results about an apparent 3′-phosphatase activity of the G365A mutant are ambivalent. The purified catalytic domain of Ci-VSP which lacks the transmembrane part of the protein shows increased activity toward PI(3,4)P2 and PI(3,5)P2 in vitro in comparison to the wild type (Iwasaki et al., 2008). In contrast to that, the phosphatase activity of the G365A full-length protein was significantly reduced in vivo toward PI(3,4)P2 compared to the wild type (Liu et al., 2012). These results clearly demonstrate that data obtained by in vitro and in vivo assays can diverge for VSPs. The reason for this could be that the active site might adopt a different conformation under in vitro conditions than in vivo, since the structural stabilization of the substrate binding pocket through interactions with the membrane surface or the voltage sensor domain cannot be achieved in these experiments.
Interestingly, the G365A mutant was still capable to deplete PI(4,5)P2 in vivo (Liu et al., 2012). Thus, the mutation in the P-loop alone is not efficient to convert Ci-VSP into an exclusive 3′-phosphatase like PTEN. Further studies are required to identify the structural determinants for the substrate specificity in VSPs.
Specificity regarding protein- or lipid-derived substrates
Last but not least, the amino acid five residues C-terminally from the active site cysteine (Figure 3A) also exhibits interesting properties. In PTEN, the G129E mutation eliminates PIP-specific phosphatase activity without affecting the activity toward peptide-derived substrates (Furnari et al., 1997; Myers et al., 1998; Ramaswamy et al., 1999). Gly129 is located at the bottom of the substrate binding pocket. On the basis of the crystal structure of PTEN, it was proposed that the G129E mutation might result in the reduction of the wide access to the active site. In turn, this might prevent the accommodation of PIP substrates, but does not eliminate the capacity to bind phosphoprotein-substrates (Lee et al., 1999).
In case of VSPs, no activity toward peptide-derived substrates has been demonstrated so far. Because of the structural similarity to PTEN, it would be interesting to see if VSPs are also capable to dephosphorylate this kind of substrates in addition to PIP molecules.
The WPD- or DPYY-Loop
The so-called WPD-loop of class I PTPs is located several amino acid positions N-terminal to the P-loop (Figure 3). The name of this region is derived from three amino acids in this motif, which are conserved in several phosphatases, as for example in PTP1B, YopH, and CD45 (Figure 3A). The nomenclature of this loop persists for historical reasons, although some members of the PTP family have been discovered that show considerable variations in the WPD-loop sequence.
In case of class II PTPs, the motif which corresponds to the WPD-loop is also highly conserved. Here, this region is termed DPYY-loop, again because of its conserved sequence pattern (Figure 3A; Tabernero et al., 1999).
The invariant aspartate
The most important residue in the WPD- or the DPYY-loop is the invariant aspartate. Besides the cysteine and arginine conserved in the P-loop, this residue is the third one within the group of most crucial actors in the enzymatic reaction. It mediates the formation of the cysteinyl-phosphate intermediate by acting as general acid during the first step of the catalytic cycle. In the second step, it acts as general base by accepting the proton from an activated water molecule, which enables the reconfiguration of the active site for a new reaction cycle (Figure 4; Taddei et al., 1994; Zhang et al., 1994b; Denu et al., 1995a, 1996).
For class I PTPs, mutations of the invariant aspartate reduce or abolish the phosphatase activity (Zhang et al., 1994c; Lohse et al., 1997; Denu and Dixon, 1998). In case of rPTPα, a receptor-like classical PTP, the substitution by glutamate preserves the charge at this position, but changes the steric conditions in such a way that the catalytic activity is decreased at least by two orders of magnitude (Flint et al., 1997; Wu et al., 1997). However, substitution of the aspartate in rPTPα by alanine completely eliminates the catalytic activity toward pTyr (Wu et al., 1997).
Interestingly, it is known from studies on different PTPs that an aspartate-to-alanine mutation in the WPD- or DYPP-loop generates highly efficient substrate-trapping mutants (Garton et al., 1996; Flint et al., 1997; Tonks and Neel, 2001; Blanchetot et al., 2005; Monteleone et al., 2012). Garton et al. (1996) suggested that the mutation enhances the hydrophobic properties of the active site cleft and removes the repulsive electrostatic potential between the aspartate and the phosphate moiety of the substrate. In this way, the substrate-bound conformation could be stabilized (Garton et al., 1996).
In agreement with these results, the corresponding D331A mutation in Ci-VSP drastically slows down the off-motion of the voltage sensor domain after a voltage stimulation (Kohout et al., 2010). This indicates a high energy barrier for the conformational transition of the sensor domain into the resting state, which could be due to the fact that the WPD mutation leads to a trapping of the catalytic domain at its substrate at the membrane surface. Interestingly, the D331A mutation shows a residual phosphatase activity toward PI(3,4,5)P3 vesicles in vitro, but was completely inactive toward PI(4,5)P2 in vivo (Kohout et al., 2010). These results imply a drastic slow-down of the catalytic turnover rate, which might arise from substrate trapping also in Ci-VSP.
Since the results obtained from Ci-VSP agree with those from other PTPs, it can be assumed that the aspartate in the WPD-loop of VSPs might play a similar role in catalysis as it does for class I and II PTPs.
A glutamate instead of an aspartate with consequence for substrate specificity
Quite recently, an “unusual” PTP has been identified that does not fit into this strict pattern of PTPs with the invariant aspartate in the WPD-loop: the receptor-like class I phosphatase PTPRQ. This enzyme contains a glutamate at the position corresponding to the aspartate in other PTPs. Remarkably, receptor-like PTPs had only been classified as classical PTPs so far. However, PTPRQ has only little activity toward pTyr substrates, but displays specificity for phosphoinositides (Oganesian et al., 2003; Seifert et al., 2003; Park et al., 2013; Yu et al., 2013). Furthermore, in contrast to PIP-specific PTPs like VSPs and PTENs, which show a substrate preference for one position at the inositol ring, PTPRQ dephosphorylates the 3′- and 5′- phosphates of the PIP head group (Oganesian et al., 2003). Notably, if the glutamate in the WPE-loop of PTPRQ is substituted by an aspartate mimicking the sequence pattern of other PTPs, the phosphatase is converted into a pTyr-specific phosphatase with no activity toward PIP substrates (Oganesian et al., 2003).
The TI-loop in Class I PTPs Versus the V-Loop in Class II PTPs
The third important loop in the catalytic domain of class I PTPs is the TI-loop. Also here, the name results from a Thr-Ile pair in this region that is conserved in many (including PTEN), but not all PTPs (Figure 3A). Remarkably, the overall amino acid sequence of the TI-loop is not as highly conserved as the one of the P- and WPD-loop, which makes it difficult to create a reliable sequence alignment of this region. A structural alignment of different crystallized phosphatase domains on the basis of the P-loop conformation finally enables the identification of the region that corresponds to the TI-loop (Figures 3B,C). The motives found by this structural comparison are aligned in Figure 3A. This alignment illustrates the high sequence variation in this loop. However, it further shows a notable similarity for class I PTPs: despite the sequence variation, most of these PTPs contain at least one (i.e., PTEN) or even two glutamines (i.e., PTP1B, YopH, CD45) in the TI-loop. Therefore, this region is often also referred to as Q-loop for class I PTPs (Tabernero et al., 2008).
For class II PTPs, little homology of the V-loop to the corresponding TI-loop of class I PTPs can be observed (Figure 3A). An additional structural alignment of both regions revealed also no significant similarity, which makes the informational content of the amino acid alignment in Figure 3A vague. However, as the TI-loop in class I PTPs, the V-loop in class II PTPs contributes to one of the side walls of the substrate binding pocket (Figure 3C). In addition to that, class II PTPs differ in their capability to respond to activators and inhibitors, which is caused by variations in the amino acid sequence of the V-loop regions (Zhang et al., 1998; Madhurantakam et al., 2005). Taken together, the V-loop plays an important role for catalysis in class II PTPs, although it seems to be completely different from the role of the TI-loop in class I PTPs.
Glutamine versus threonine versus glutamate in the TI-loop in class I PTPs
There are many crystal structures of class I PTPs available that contain a compound in the active site that mimics the bound substrate. In these structures, the conserved glutamines of the TI-loop stabilize a water molecule, which is required for the hydrolysis of the cysteinyl-phosphate intermediate during the second step of the enzymatic reaction. For example, in structures of PTP1B with a ligand bound to the active site, the two glutamines Gln262 and Gln266 coordinate the water molecule (Figure 6C; Brandao et al., 2010). This configuration has been further proposed to be crucial in class I PTPs for positioning the WPD-loop in an optimal conformation for catalysis (Pannifer et al., 1998; Brandao et al., 2010).
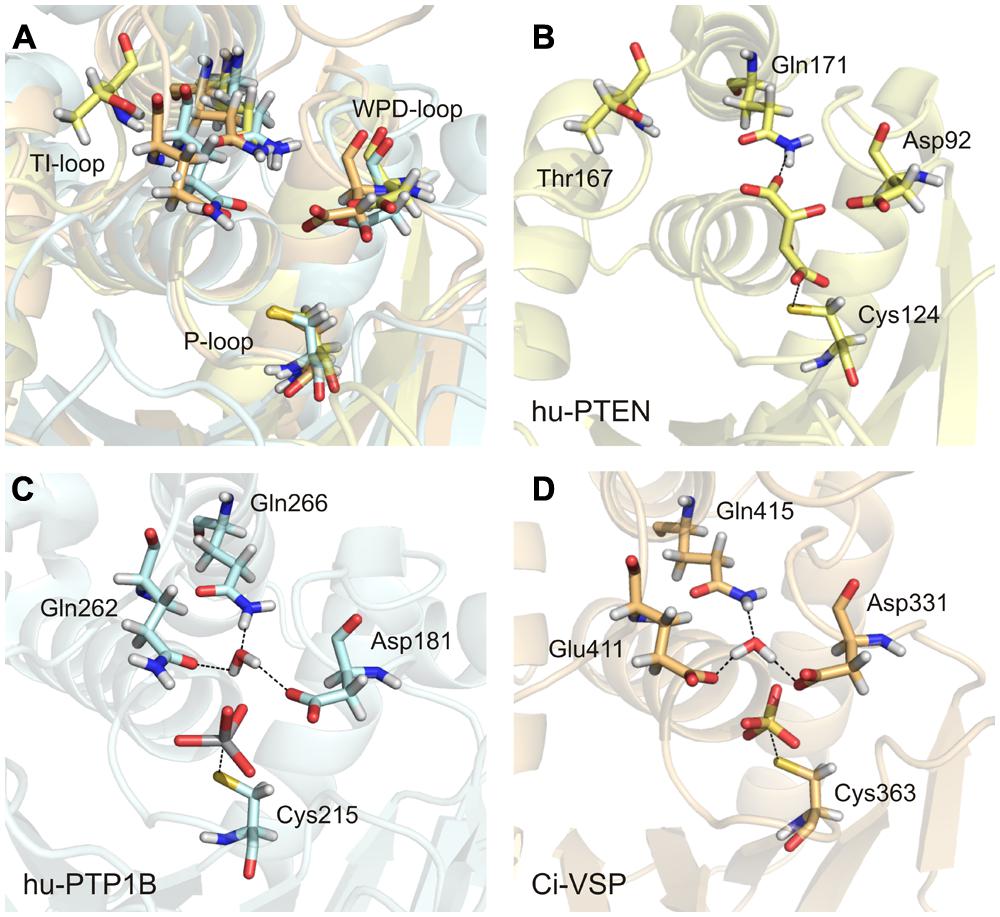
FIGURE 6. An activated water molecule is required for the second step in catalysis of class I PTPs. (A) The substrate binding pocket is structurally aligned for the human PTP1B (in light blue, PDB accession number 3ib0), human PTEN (in yellow, PDB 1d5r), and Ci-VSP [in orange, modeled by MD simulation based on the crystal structure of Matsuda et al. (2011) and Hobiger et al. (2013) PDB 3awe]. The glutamines from the TI-loop of hu-PTP1B and the corresponding amino acids in the other PTPs, the invariant aspartate from the WPD-loop, and the catalytic cysteine from the P-loop are represented as sticks. (B–D) The substrate binding pockets are shown individually for the three PTPs with the following ligands bound to the active site, as found in the respective crystal structures: L(+)-tartrate in hu-PTEN, vanadate in hu-PTP1B, and sulfate in Ci-VSP. One water molecule is coordinated adjacently to the aspartate and the ligand by the TI-loop glutamines in hu-PTP1B (C) and by corresponding residues in Ci-VSP (D). (B) This water molecule is absent in hu-PTEN. Thr167 that corresponds to Gln262 in hu-PTP1B and Glu411 in Ci-VSP is oriented away from the active site. One of the oxygen atoms of the bound tartrate forms a hydrogen bond with the nitrogen of Gln171.
With this knowledge at hand, the question remains: what role does the TI-loop play in VSPs? In these phosphatases, two conserved glutamines exist in the TI-loop, e.g., Gln408 and Gln415 in Ci-VSP (Figure 3A). During the catalytic reaction, both residues could putatively act as interaction partners for the coordination of the water molecule. Therefore, we performed a structural alignment of the phosphatase domains of Ci-VSP and PTP1B (Figure 6). This comparison reveals that in Ci-VSP, Glu411 instead of Gln408 overlaps with Gln262 in PTP1B (Figure 6A). In this configuration, Gln415 of Ci-VSP is positioned in such a way that it corresponds to Gln266 in PTP1B (Figures 6C,D). Furthermore, the model of Ci-VSP, which was obtained by MD simulations (Hobiger et al., 2013) predicts the coordination of a water molecule by Glu411 and Gln415 in the immediate environment to the phosphate group of the substrate (Figure 6D). The invariant aspartate in the WPD-loop of Ci-VSP, Asp331, additionally participates in this configuration; analogously to the aspartate in the WPD-loop of PTP1B (Figures 6C,D).
It should be noted here, that no direct experimental evidence exists so far, whether this configuration is indeed adopted during the catalytic cycle of Ci-VSP. Thus, our proposal just illustrates a hypothetical scenario based on modeling data. However, studies on Glu411 mutants of Ci-VSP support our hypothesis. Recently, Glu411 has been shown to be critical for catalysis in Ci-VSP (Matsuda et al., 2011; Liu et al., 2012). Substitution of Glu411 against hydrophobic residues (Ile, Leu, or Phe) reduced the activity toward PI(3,4,5)P3 in vitro and almost abolished activity toward PI(4,5)P2 in vivo (Liu et al., 2012). Crystal structures of the phosphatase domain of Ci-VSP suggested that Glu411 could compete with the substrate for binding to the active site (Matsuda et al., 2011; Liu et al., 2012). Even more so, Liu et al. (2012) proposed that Glu411 may act as molecular switch by changing its position in such a way that the active site opens or closes; a mechanism that was similarly described for Gln262 in PTP1B (Brandao et al., 2010).
Interestingly, in the phosphatase domain of PTEN, which is structurally highly homologous to that one of Ci-VSP, a threonine is located at the position corresponding to Glu411 (Figure 3A). Substitution of Glu411 against threonine in Ci-VSP causes a moderate decrease in the phosphatase activity toward PI(3,4,5)P3 in vitro and PI(4,5)P2 in vivo, and a strong reduction of its activity toward PI(3,4)P2 in vivo (Liu et al., 2012). Taken together, these findings suggest that a hydrophobic residue is less well-tolerated at this position for the catalytic activity of Ci-VSP compared to the polar side chain of threonine. This supports our hypothesis that Glu411 might participate in coordinating the water molecule by electrostatic interactions during the second step of catalysis.
But can the threonine in PTEN participate in the coordination of a water molecule? Such a configuration can be studied in the crystal structure of PTEN (Lee et al., 1999). A view into the active site structure of PTEN with a bound tartrate molecule shows that Thr167, which corresponds to Glu411 in Ci-VSP and Gln262 in PTP1B, is oriented away from the substrate binding pocket (Figure 6B). Such a configuration would contradict the assumption that Thr167 is involved in the water molecule coordination. However, it should be noted that the tartrate bound in the active site inhibits the catalytic activity of PTEN (Lee et al., 1999). This might be caused by the fact that upon binding of tartrate, Thr167 could be pushed into a position that prevents its participation in catalysis and, consequently, eliminates phosphatase activity. Therefore, it is possible that the crystal structure of PTEN does not represent the catalytically active conformation of the active site. Moreover, mutations T167A and Q171A in PTEN, which would correspond to Q262A and Q266A in PTP1B, and E411A and Q415A in Ci-VSP, reduced phosphatase activity in vitro by 60 to 75%, respectively (Lee et al., 1999). These results suggest a functional relevance for both amino acids in the enzymatic reaction of PTEN.
In conclusion, we suggest that the TI-loop in Ci-VSP is directly involved in catalysis since it contains residues that might coordinate the water molecule for the second step in the reaction cycle, namely Glu411 and Gln415. Changing the electrostatic properties by introducing a hydrophobic residue instead of Glu411 eliminates or significantly reduces catalytic activity, whereas the substitution by a polar side chain such as threonine does not abolish enzymatic function, but causes changes in substrate specificity. However, further studies are required to elucidate exactly the interactions in the active site of VSPs.
Other charged positions in the TI-loop of class I PTPs
Apart from Glu411 in Ci-VSP, at least one additional charged side chain is located at the N-terminal part of the TI-loop in VSPs. Again, a sequence comparison with other PTPs reveals large variations in this region (Figure 3A), which makes it difficult to speculate about the role of these charged residues for VSPs in comparison to the results obtained for PTPs.
In case of Ci-VSP, recent studies suggested that Asp400 in the N-terminal part of the TI-loop interacts with the region that links the phosphatase to the adjacent voltage sensor domain (Liu et al., 2012; Hobiger et al., 2013). Notably, the linker motif of Ci-VSP is assumed to be critical for the coupling between the catalytic and the voltage sensor domain (Murata et al., 2005; Hossain et al., 2008; Villalba-Galea et al., 2009; Kohout et al., 2010; Lacroix et al., 2011; Hobiger et al., 2012). In particular, the interaction between the linker and Asp400 was suggested to be crucial for stabilizing the substrate binding pocket in a voltage-dependent manner (Liu et al., 2012; Hobiger et al., 2013). This process might be one critical step in transferring the conformational change from the voltage sensor to the catalytic domain upon voltage stimulation.
In PTEN, the region that is homologous to the linker of VSPs has been proposed to act as phospholipid binding motif (PBM), because it mediates the targeting of the enzyme to PIP-containing membranes (Campbell et al., 2003; Das et al., 2003; Iijima et al., 2004; Walker et al., 2004; Vazquez and Devreotes, 2006; Redfern et al., 2008). Analogously, this function has also been suggested for the linker in Ci-VSP (Villalba-Galea et al., 2009; Kohout et al., 2010; Hobiger et al., 2012). Deletion of the PBM retains phosphatase activity of PTEN toward water-soluble substrates, but eliminates the activity toward membrane-bound substrates (Iijima et al., 2004; Vazquez et al., 2006). In contrast to that, the purified catalytic domain of Ci-VSP does not show phosphatase activity in vitro if the linker is deleted, neither toward membrane-bound substrates nor the soluble PIP head group Ins(1,3,4,5)P4 (Kohout et al., 2010).
Although the PBM in PTEN shares structural and functional similarities with the linker in Ci-VSP, no direct interactions between the PBM and regions in the catalytic domain have been described for PTEN up to now. Upon membrane binding of the PBM, conformational changes occur in the catalytic domain leading to the assumption of an allosteric activation mechanism in PTEN (Campbell et al., 2003; McConnachie et al., 2003; Iijima et al., 2004; Vazquez and Devreotes, 2006; Redfern et al., 2008). However, the details of this process are still elusive and the question remains, whether the interaction between the linker and the TI-loop observed for Ci-VSP is an exclusive property of this protein.
A Model for the Catalytic Mechanism in VSPs
Including all results discussed above, we suggest the following two-step catalytic reaction mechanism for Ci-VSP as representative for all VSPs.
During the first step in catalysis, the binding of the phosphorylated substrate is enabled by defined structural properties of the active site. Here, the phosphate group that will be cleaved from the substrate is positioned by the P-loop Arg369, and a network of hydrogen bonds formed by the backbone atoms of the active site loop. The catalytic Cys363 is oriented in its position by a hydrogen bond with the adjacent Thr370. This configuration stabilizes the sulfur of the cysteine in an anionic thiolate form. All these factors ensure optimal reaction conditions, so that Cys363 acts as nucleophile attacking the phosphate group of the substrate. With the assistance of Asp331 from the WPD-loop, which donates a proton to the substrate leaving group, a cysteinyl-phosphate intermediate is formed, and the dephosphorylated substrate dissociates from the active site pocket.
To return into the resting state, the phosphorus in the reaction intermediate is attacked by an activated water molecule. The activation of the water molecule is achieved by the coordination through Glu411 and Gln415 from the TI-loop. Furthermore, one of the protons of the water molecule is accepted by Asp331. In this way, the phosphate group gets hydrolyzed, so that the initial conformation of the active site is restored to start a new catalytic cycle.
Summary
Research on VSPs is still an emerging field in membrane biophysics and cell biology. Since the activity of VSPs is switchable by voltage stimulation, these enzymes have been mainly characterized by electrophysiological methods. Taking their structural identity and sequence pattern into account, VSPs can be classified as members of the huge protein family of cysteine-based PTPs. They dephosphorylate specifically PIP-lipids instead of phosphotyrosine substrates. Therefore, and because of the structural properties of their catalytic domain, VSPs belong to the subclass of dual-specific class I PTPs.
The aim of this review is to point out that the information presently available about VSPs and cysteine-based PTPs can be well-integrated into a set of common principles. In particular, we showed that all cysteine-based PTPs including VSPs share a common folding pattern of their catalytic domain. In addition, the two-step reaction mechanisms proposed for class I to III PTPs can also be assumed for VSPs, since all results obtained so far from this latter phosphatase family match well with the results about well-characterized cysteine-based PTPs, such as PTP1B, CD45, VHR, or VH1. However, many of the suggestions made here still need experimental verification. By linking the knowledge about VSPs and PTPs, we want to inspire further research on VSPs to contribute to and to benefit from efforts in the field of the large phosphatase family to which they belong.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Tillmann Utesch for support with his expertise in protein structures and dynamics. We further thank Dominik Oliver and Aileen Franke for critical reading of the manuscript and fruitful discussions. This work was supported by grants from the German Research Foundation (Cluster of Excellence “Unifying Concepts in Catalysis,” the Berlin International Graduate School of Natural Sciences and Engineering BIG-NSE) and by a postdoctoral funding from the TU Berlin.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fphar.2015.00020/abstract
Footnotes
- ^ A list of all abbreviations used in this text is given in Table S1.
- ^ A variant of the TPTE family has been initially named as TPIP (TPTE and PTEN homologous inositol lipid phosphatase; Walker et al., 2001), nowadays also called TPTE2. To introduce a more consistent nomenclature, the human isoforms of TPTEs, TPTE2 and TPTE, have also been referred to as hVSP1 and hVSP2, respectively (Halaszovich et al., 2012).
References
Alho, I., Clara Bicho, M., Carvalho, R., da Silva, A. P., Costa, L., and Bicho, M. (2008). Low molecular weight protein tyrosine phosphatase genetic polymorphism and susceptibility to cancer development. Cancer Genet. Cytogenet. 181, 20–24. doi: 10.1016/j.cancergencyto.2007.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alonso, A., Sasin, J., Bottini, N., Friedberg, I., Friedberg, I., Osterman, A.,et al. (2004). Protein tyrosine phosphatases in the human genome. Cell 117, 699–711. doi: 10.1016/j.cell.2004.05.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alonso, A., Saxena, M., Williams, S., and Mustelin, T. (2001). Inhibitory role for dual specificity phosphatase VHR in T cell antigen receptor and CD28-induced Erk and Jnk activation. J. Biol. Chem. 276, 4766–4771. doi: 10.1074/jbc.M006497200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Altin, J. G., and Sloan, E. K. (1997). The role of CD45 and CD45-associated molecules in T cell activation. Immunol. Cell Biol. 75, 430–445. doi: 10.1038/icb.1997.68
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arantes, G. M. (2008). The catalytic acid in the dephosphorylation of the Cdk2-pTpY/CycA protein complex by Cdc25B phosphatase. J. Phys. Chem. B 112, 15244–15247. doi: 10.1021/jp8070019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barford, D., Das, A. K., and Egloff, M. P. (1998). The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 27, 133–164. doi: 10.1146/annurev.biophys.27.1.133
Barford, D., Flint, A. J., and Tonks, N. K. (1994). Crystal structure of human protein tyrosine phosphatase 1B. Science 263, 1397–1404. doi: 10.1126/science.8128219
Bernardi, R., Liebermann, D. A., and Hoffmann, B. (2000). Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progression and terminal differentiation. Oncogene 19, 2447–2454. doi: 10.1038/sj.onc.1203564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blanchetot, C., Chagnon, N., Dubé, N., Hallé, M., and Tremblay, M. L. (2005). Substrate-trapping techniques in the identification of cellular PTP targets. Methods 35, 44–53. doi: 10.1016/j.ymeth.2004.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bordo, D., and Bork, P. (2002). The rhodanese/Cdc25 phosphatase superfamily. EMBO Rep. 3, 741–746. doi: 10.1093/embo-reports/kvf150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bottini, E., Bergamaschi, A., Magrini, A., Claudio, S., Ammendola, L., Grassi, S.,et al. (2007). Allergy and ACP1 genetic polymorphism. Allergy Asthma Proc. 28, 87–92. doi: 10.2500/aap.2007.28.2916
Bottini, N., Bottini, E., Gloria-Bottini, F., and Mustelin, T. (2002). Low-molecular-weight protein tyrosine phosphatase and human disease: in search of biochemical mechanisms. Arch. Immunol. Ther. Exp. 50, 95–104.
Bottini, N., Gloria-Bottini, F., Lucarini, N., Ronchetti, P. G., and Fontana, L. (2000). Inflammatory bowel disease: are there gender differences in the genetics of signal transduction? A preliminary study of cytosolic low molecular weight protein tyrosine phosphatase. Dis. Markers 16, 163–166. doi: 10.1155/2000/101739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boudolf, V., Inzé, D., and De Veylder, L. (2006). What if higher plants lack a CDC25 phosphatase? Trends Plant Sci. 11, 474–479. doi: 10.1016/j.tplants.2006.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boutros, R., Lobjois, V., and Ducommun, B. (2007). CDC25 phosphatases in cancer cells: key players? Good targets? Nat. Rev. Cancer 7, 495–507. doi: 10.1038/nrc2169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brandao, T. A. S., Hengge, A. C., and Johnson, S. J. (2010). Insights into the reaction of protein-tyrosine phosphatase 1B: crystal structures for transition state analogs of both catalytic steps. J. Biol. Chem. 285, 15874–15883. doi: 10.1074/jbc.M109.066951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buhrmann, G., Parker, B., Sohn, J., Rudolph, J., and Mattos, C. (2005). Structural mechanism of oxidative regulation of the phosphatase cdc25b via an intramolecular disulfide bond. Biochemistry 44, 5307–5316. doi: 10.1021/bi047449f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Campbell, R. B., Liu, F., and Ross, A. H. (2003). Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 278, 33617–33620. doi: 10.1074/jbc.C300296200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, H., Rossier, C., Morris, M. A., Scott, H. S., Gos, A., Bairoch, A.,et al. (1999). A testis-specific gene, TPTE, encodes a putative transmembrane tyrosine phosphatase and maps to the pericentromeric region of human chromosomes 21 and 13, and to chromosomes 15, 22, and Y. Hum. Genet. 105, 399–409. doi: 10.1007/s004390051122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, R. A., Michaeli, T., van Aelst, L., and Ballester, R. (2000a). A role for the noncatalytic n terminus in the function of cdc25, a saccharomyces cerevisiae ras-guanine nucleotide exchange factor. Genetics 154, 1473–1484.
Chen, W., Wilborn, M., and Rudolph, J. (2000b). Dual-Specific Cdc25B phosphatase: in search of the catalytic acid. Biochemistry 39, 10781–10789. doi: 10.1021/bi000909u
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiarugi, P., Cirri, P., Giannoni, E., Camici, G., Manao, G., Raugei, G.,et al. (2000). The low M(r) protein-tyrosine phosphatase is involved in Rho-mediated cytoskeleton rearrangement after integrin and platelet-derived growth factor stimulation. J. Biol. Chem. 275, 4640–4646. doi: 10.1074/jbc.275.7.4640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiarugi, P., Cirri, P., Marra, F., Raugei, G., Camici, G., Manao, G.,et al. (1997). LMW-PTP is a negative regulator of insulin-mediated mitotic and metabolic signalling. Biochem. Biophys. Res. Commun. 238, 676–682. doi: 10.1006/bbrc.1997.7355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiarugi, P., Fiaschi, T., Taddei, M. L., Talini, D., Giannoni, E., Raugei, G.,et al. (2001). Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet-derived growth factor receptor stimulation. J. Biol. Chem. 276, 33478–33487. doi: 10.1074/jbc.M102302200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, S.-H., Lee, C.-H., Ahn, Y., Kim, H., Kim, H., Ahn, C.-Y.,et al. (2004). Redox regulation of PTEN and protein tyrosine phosphatases in H2O2-mediated cell signaling. FEBS Lett. 560, 7–13. doi: 10.1016/S0014-5793(04)00112-7
Cipollone, R., Ascenzi, P., Tomao, P., Imperi, F., and Visca, P. (2008). Enzymatic detoxification of cyanide: clues from Pseudomonas aeruginosa Rhodanese. J. Mol. Microbiol. Biotechnol. 15, 199–211. doi: 10.1159/000121331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cirri, P., Fiaschi, T., Chiarugi, P., Camici, G., Manao, G., Raugei, G.,et al. (1996). The molecular basis of the differing kinetic behavior of the two low molecular mass phosphotyrosine protein phosphatase isoforms. J. Biol. Chem. 271, 2604–2607. doi: 10.1074/jbc.271.5.2604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, P. (2001). The role of protein phosphorylation in human health and disease. The sir hans krebs medal lecture. Eur. J. Biochem. 268, 5001–5010. doi: 10.1046/j.0014-2956.2001.02473.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Combs, A. P. (2010). Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem. 53, 2333–2344. doi: 10.1021/jm901090b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conklin, D. S., Galaktionov, K., and Beach, D. (1995). 14-3-3 proteins associate with Cdc25 phosphatases. Proc. Natl. Acad. Sci. U.S.A. 92, 7892–7896. doi: 10.1073/pnas.92.17.7892
Conway, S. J., Gardiner, J., Grove, S. J. A., Johns, M. K., Lim, Z.-Y., Painter, G. F.,et al. (2010). Synthesis and biological evaluation of phosphatidylinositol phosphate affinity probes. Org. Biomol. Chem. 8, 66–76. doi: 10.1039/b913399b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Das, S., Dixon, J. E., and Cho, W. (2003). Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. U.S.A. 100, 7491–7496. doi: 10.1073/pnas.0932835100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Araujo, P. S., Mies, V., and Miranda, O. (1976). Subcellular distribution of low- and high-molecular weight acid phosphatases. Biochim. Biophys. Acta 452, 121–130. doi: 10.1016/0005-2744(76)90063–90062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denu, J. M., and Dixon, J. E. (1995). A catalytic mechanism for the dual-specific phosphatases. Proc. Natl. Acad. Sci. U.S.A. 92, 5910–5914. doi: 10.1073/pnas.92.13.5910
Denu, J. M., and Dixon, J. E. (1998). Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol. 2, 633–641. doi: 10.1016/S1367-5931(98)80095-1
Denu, J. M., Lohse, D. L., Vijayalakshmit, J., Saper, M. A., and Dixon, J. E. (1996). Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis. Proc. Natl. Acad. Sci. U.S.A. 93, 2493–2498. doi: 10.1073/pnas.93.6.2493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denu, J. M., and Tanner, K. G. (1998). Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642. doi: 10.1021/bi973035t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denu, J. M., Zhou, G., Guo, Y., and Dixon, J. E. (1995a). The catalytic role of aspartic acid-92 in the human dual-specific protein-tyrosine-phosphatase vaccinia h1-related. Biochemistry 34, 3396–3403. doi: 10.1021/bi00010a031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denu, J. M., Zhou, G., Wu, L., Zhao, R., Yuvaniyama, J., Saper, M. A.,et al. (1995b). The purification and characterization of a human dual-specific protein tyrosine phosphatase. J. Biol. Chem. 27, 3796–3803.
Derrien, M., Punjabi, A., Khanna, M., Grubisha, O., and Traktman, P. (1999). Tyrosine Phosphorylation of A17 during Vaccinia virus infection: involvement of the H1 phosphatase and the F10 kinase. J. Virol. 73, 7287–7296.
Deshpande, T., Takagi, T., Hao, L., Buratowski, S., and Charbonneau, H. (1999). Human PIR1 of the protein-tyrosine phosphatase superfamily has RNA 5’-triphosphatase and diphosphatase activities. J. Biol. Chem. 274, 16590–16594. doi: 10.1074/jbc.274.23.16590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dillet, V., Van Etten, R. L., and Bashford, D. (2000). Stabilization of charges and protonation states in the active site of the protein tyrosine phosphatases: a computational study. J. Phys. Chem. B 104, 11321–11333. doi: 10.1021/jp001575l
Donzelli, M., Squatrito, M., Ganoth, D., Hershko, A., Pagano, M., and Draetta, G. F. (2002). Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 21, 4875–4884. doi: 10.1093/emboj/cdf491
Eckstein, J. W., Beer-Romero, P., and Berdo, I. (1996). Identification of an essential acidic residue in Cdc25 protein phosphatase and a general three-dimensional model for a core region in protein phosphatases. Protein Sci. 51, 5–12. doi: 10.1002/pro.5560050102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evans, B., Tishmack, P. A., Pokalsky, C., Zhang, M., and Van Etten, R. L. (1996). Site-directed mutagenesis, kinetic, and spectroscopic studies of the P-loop residues in a low molecular weight protein tyrosine phosphatase. Biochemistry 2960, 13609–13617. doi: 10.1021/bi9605651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fauman, E. B., Cogswell, J. P., Lovejoy, B., Rocque, W. J., Holmes, W., Montana, V. G.,et al. (1998). Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 93, 617–625. doi: 10.1016/S0092-8674(00)81190-93
Feldhammer, M., Uetani, N., Miranda-Saavedra, D., and Tremblay, M. L. (2013). PTP1B: a simple enzyme for a complex world. Crit. Rev. Biochem. Mol. Biol. 48, 430–445. doi: 10.3109/10409238.2013.819830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernández-Sánchez, M. E., Criado-García, O., Heath, K. E., García-Fojeda, B., Medraño-Fernández, I., Gomez-Garre, P.,et al. (2003). Laforin, the dual-phosphatase responsible for Lafora disease, interacts with R5 (PTG), a regulatory subunit of protein phosphatase-1 that enhances glycogen accumulation. Hum. Mol. Genet. 12, 3161–3171. doi: 10.1093/hmg/ddg340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flint, A. J., Tiganis, T., Barford, D., and Tonks, N. K. (1997). Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 94, 1680–1685. doi: 10.1073/pnas.94.5.1680
Furnari, F. B., Lin, H., Huang, H.-J. S., and Cavenee, W. K. (1997). Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc. Natl. Acad. Sci. U.S.A. 94, 12479–12484. doi: 10.1073/pnas.94.23.12479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gabrielli, B. G., Clark, J. M., McCormack, A. K., and Ellem, K. A. O. (1997). Hyperphosphorylation of the N-terminal domain of Cdc25 regulates activity toward cyclin B1/Cdc2 but not cyclin A/Cdk2. J. Biol. Chem. 272, 28607–28614. doi: 10.1074/jbc.272.45.28607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galaktionov, K., and Beach, D. (1991). Specific activation of Cdc25 tyrosine phosphatases by B-type cyclins: evidence for multiple role of mitotic cyclins. Cell 67, 1181–1194. doi: 10.1016/0092-8674(91)90294-9
Ganesh, S., Agarwala, K. L., Ueda, K., Akagi, T., Shoda, K., Usui, T.,et al. (2000). Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 9, 2251–2261. doi: 10.1093/oxfordjournals.hmg.a018916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garton, A. J., Flint, A. J., and Tonks, N. K. (1996). Identification of p130cas as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol. Cell. Biol. 16, 6408–6418.
Gautier, J., Solomon, M. J., Booher, R. N., Bazan, J. F., and Kirschner, M. W. (1991). Cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell 67, 197–211. doi: 10.1016/0092-8674(91)90583-K
Gnesutta, N., Ceriani, M., Innocenti, M., Mauri, I., Zippel, R., Sturani, E.,et al. (2001). Cloning and characterization of mouse UBPy, a deubiquitinating enzyme that interacts with the Ras guanine nucleotide exchange factor CDC25(Mm)/Ras-GRF1. J. Biol. Chem. 276, 39448–39454. doi: 10.1074/jbc.M103454200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goldstein, B. J. (2001). Protein-tyrosine phosphatase 1B (PTP1B): a novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr. Drug Targets Immune Endocr. Metab. Disord. 1, 265–275. doi: 10.2174/1568008013341163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guan, K., Broyles, S. S., and Dixon, J. E. (1991). A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature 350, 359–362. doi: 10.1038/350359a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guan, K. L., and Dixon, J. E. (1991). Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem. 266, 17026–17030.
Guipponi, M., Tapparel, C., Jousson, O., Scamuffa, N., Mas, C., Rossier, C.,et al. (2001). The murine orthologue of the Golgi-localized TPTE protein provides clues to the evolutionary history of the human TPTE gene family. Hum. Genet. 109, 569–575. doi: 10.1007/s004390100607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guipponi, M., Yaspo, M. L., Riesselman, L., Chen, H., De Sario, A., Roizes, G.,et al. (2000). Genomic structure of a copy of the human TPTE gene which encompasses 87 kb on the short arm of chromosome 21. Hum. Genet. 107, 127–131. doi: 10.1007/s004390000343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Halaszovich, C. R., Leitner, M. G., Mavrantoni, A., Le, A., Frezza, L., Feuer, A.,et al. (2012). A human phospholipid phosphatase activated by a transmembrane control module. J. Lipid Res. 53, 2266–2274. doi: 10.1194/jlr.M026021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Halaszovich, C. R., Schreiber, D. N., and Oliver, D. (2009). Ci-VSP is a depolarization-activated phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-trisphosphate 5’-phosphatase. J. Biol. Chem. 284, 2106–2113. doi: 10.1074/jbc.M803543200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, R.-J., Yu, Z.-H., Zhang, R.-Y., and Zhang, Z.-Y. (2014). Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 35, 1227–1246. doi: 10.1038/aps.2014.80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hermiston, M. L., Xu, Z., and Weiss, A. (2003). CD45: a critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 21, 107–137. doi: 10.1146/annurev.immunol.21.120601.140946
Hobiger, K., Utesch, T., Mroginski, M. A., and Friedrich, T. (2012). Coupling of Ci-VSP modules requires a combination of structure and electrostatics within the linker. Biophys. J. 102, 1313–1322. doi: 10.1016/j.bpj.2012.02.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hobiger, K., Utesch, T., Mroginski, M. A., Seebohm, G., and Friedrich, T. (2013). The linker pivot in Ci-VSP: the key to unlock catalysis. PLoS ONE 8:e70272. doi: 10.1371/journal.pone.0070272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoffmann, I., Clarke, P. R., Marcote, M. J., Karsenti, E., and Draetta, G. (1993). Phosphorylation and activation of human cdc25-C by cdc2-cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J. 12, 53–63.
Hoffmann, I., Draetta, G., and Karsenti, E. (1994). Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 13, 4302–4310.
Homan, K. T., Balasubramaniam, D., Zabell, A. P. R., Wiest, O., Helquist, P., and Stauffacher, C. V. (2010). Identification of novel inhibitors for a low molecular weight protein tyrosine phosphatase via virtual screening. Bioorg. Med. Chem. Lett. 18, 5449–5456. doi: 10.1016/j.bmc.2010.04.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Honda, R., Ohba, Y., Nagata, A., Okayama, H., and Yasuda, H. (1993). Dephosphorylation of human p34cdc2 kinase on both Thr-14 and Tyr-15 by human cdc25B phosphatase. FEBS Lett. 318, 331–334. doi: 10.1016/0014-5793(93)80540-B
Hopkinson, D. A., Spencer, N., and Harris, H. (1963). Red cell acid phosphatase variants: a new human polymorphism. Nature 199, 969–971. doi: 10.1038/199969a0
Hossain, M. I., Iwasaki, H., Okochi, Y., Chahine, M., Higashijima, S., Nagayama, K.,et al. (2008). Enzyme domain affects the movement of the voltage sensor in ascidian and zebrafish voltage-sensing phosphatases. J. Biol. Chem. 283, 18248–18259. doi: 10.1074/jbc.M706184200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, L., Sankar, S., Lin, C., Kontos, C. D., Schroff, A. D., Cha, E. H.,et al. (1999). HCPTPA, a protein tyrosine phosphatase that regulates vascular endothelial growth factor receptor-mediated signal transduction and biological activity. J. Biol. Chem. 274, 38183–38188. doi: 10.1074/jbc.274.53.38183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iijima, M., Huang, Y. E., Luo, H. R., Vazquez, F., and Devreotes, P. N. (2004). Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J. Biol. Chem. 279, 16606–16613. doi: 10.1074/jbc.M312098200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ishibashi, T., Bottaro, D. P., Chan, A., Miki, T., and Aaronson, S. A. (1992). Expression cloning of a human dual-specificity phosphatase. Proc. Natl. Acad. Sci. U.S.A. 89, 12170–12174. doi: 10.1073/pnas.89.24.12170
Iwasaki, H., Murata, Y., Kim, Y., Hossain, M. I., Worby, C. A., Dixon, J. E.,et al. (2008). A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. U.S.A. 105, 7970–7975. doi: 10.1073/pnas.0803936105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jia, Z., Barford, D., Flint, A., and Tonks, N. (1995). Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 268, 1754–1758. doi: 10.1126/science.7540771
Kaldis, P. (2014). The cdk-activating kinase (CAK): from yeast to mammals. Cell. Mol. Life Sci. C. 55, 284–296. doi: 10.1007/s000180050290
Karlsson-Rosenthal, C., and Millar, J. B. A. (2006). Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 16, 285–292. doi: 10.1016/j.tcb.2006.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawabe, T., Masashi, S., Ando, T., Kimura, M., Hori, H., and Okamoto, T. (2002). Cdc25C interacts with PCNA at G2/M transition. Oncogene 21, 1717–1726. doi: 10.1038/sj/onc/1205229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, Y., Song, Y. B., Kim, T.-Y., Kim, I., Han, S.-J., Ahn, Y.,et al. (2010). Redox regulation of the tumor suppressor PTEN by glutathione. FEBS Lett. 584, 3550–3556. doi: 10.1016/j.febslet.2010.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, Y. J., Jahan, N., and Bahk, Y. Y. (2013). Biochemistry and structure of phosphoinositide phosphatases. BMB Rep. 46, 1–8. doi: 10.5483/BMBRep.2013.46.1.261
Kitagishi, Y., and Matsuda, S. (2013). Redox regulation of tumor suppressor PTEN in cancer and aging (Review). Int. J. Mol. Med. 31, 511–515. doi: 10.3892/ijmm.2013.1235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knobbe, C. B., Merlo, A., and Reifenberger, G. (2002). Pten signaling in gliomas. Neuro Oncol. 4, 196–211. doi: 10.1215/15228517-4-3-196
Kohout, S. C., Bell, S. C., Liu, L., Xu, Q., Minor, D. L., and Isacoff, E. Y. (2010). Electrochemical coupling in the voltage-dependent phosphatase Ci-VSP. Nat. Chem. Biol. 6, 369–375. doi: 10.1038/nchembio.349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kolmodin, K., and Åqvist, J. (2000). Prediction of a ligand-induced conformational change in the catalytic core of Cdc25A. FEBS Lett. 465, 8–11. doi: 10.1016/S0014-5793(99)01718-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krek, W., and Nigg, E. A. (1991). Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 10, 3331–3341.
Kristjánsdóttir, K., and Rudolph, J. (2004). Cdc25 Phosphatases and Cancer. Chem. Biol. 11, 1043–1051. doi: 10.1016/j.chembiol.2004.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumanovics, A., Levin, G., and Blount, P. (2002). Family ties of gated pores: evolution of the sensor module. FASEB J. 16, 1623–1629. doi: 10.1096/fj.02-0238hyp
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurokawa, T., Takasuga, S., Sakata, S., Yamaguchi, S., Horie, S., Homma, K. J.,et al. (2012). 3’ Phosphatase activity toward phosphatidylinositol 3,4-bisphosphate [PI(3,4)P2] by voltage-sensing phosphatase (VSP). Proc. Natl. Acad. Sci. U.S.A. 109, 10089–10094. doi: 10.1073/pnas.1203799109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lacroix, J. J., Halaszovich, C. R., Schreiber, D. N., Leitner, M. G., Bezanilla, F., Oliver, D.,et al. (2011). Controlling the activity of a phosphatase and tensin homolog (PTEN) by membrane potential. J. Biol. Chem. 286, 17945–17953. doi: 10.1074/jbc.M110.201749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lammer, C., Wagerer, S., Saffrich, R., Mertens, D., Ansorge, W., and Hoffmann, I. (1998). The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J. Cell Sci. 111(Pt. 16), 2445–2453.
Lawrence, G. L., and Van Etten, R. L. (1981). The low-molecular weight acid phosphatase from bovine liver: isolations, amino acid composition, and chemical modification studies. Arch. Biochem. Biophys. 206, 122–131. doi: 10.1016/0003-9861(81)90073-4
Lee, J. O., Yang, H., Georgescu, M. M., Cristofano, A., Di, Maehama, T., Shi, Y.,et al. (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334. doi: 10.1016/S0092-8674(00)81663-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S.-R., Yang, K.-S., Kwon, J., Lee, C., Jeong, W., and Rhee, S. G. (2002). Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342. doi: 10.1074/jbc.M111899200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leslie, N. R., Bennett, D., Lindsay, Y. E., Stewart, H., Gray, A., and Downes, C. P. (2003). Redox regulation of PI3-kinase signalling via inactivation of PTEN. EMBO J. 22, 5501–5510. doi: 10.1093/emboj/cdg513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leslie, N. R., and Downes, C. P. (2002). PTEN: the down side of PI3-kinase signalling. Cell. Signal. 14, 285–295. doi: 10.1016/S0898-6568(01)00234-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leslie, N. R., Yang, X., Downes, C. P., and Weijer, C. J. (2007). PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr. Biol. 17, 115–125. doi: 10.1016/j.cub.2006.12.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lessard, L., Stuible, M., and Tremblay, M. L. (2010). The two faces of PTP1B in cancer. Biochim. Biophys. Acta 1804, 613–619. doi: 10.1016/j.bbapap.2009.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, D.-M., and Sun, H. (1997). TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 57, 2124–2129.
Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S. I.,et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. doi: 10.1126/science.275.5308.1943
Li, Q., Wanderling, S., Paduch, M., Medovoy, D., Singharoy, A., McGreevy, R.,et al. (2014). Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat. Struct. Mol. Biol. 21, 244–252. doi: 10.1038/nsmb.2768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, K., Lemon, B., and Traktman, P. (1995). The dual-specificity phosphatase encoded by vaccinia virus, vh1, is essential for viral transcription in vivo and in vitro. J. Virol. 69, 7823–7834.
Liu, L., Kohout, S. C., Xu, Q., Müller, S., Kimberlin, C. R., Isacoff, E. Y.,et al. (2012). A glutamate switch controls voltage-sensitive phosphatase function. Nat. Struct. Mol. Biol. 19, 633–641. doi: 10.1038/nsmb.2289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y., and Bankaitis, V. A. (2010). Phosphoinositide phosphatases in cell biology and disease. Prog. Lipid Res. 49, 201–217. doi: 10.1016/j.plipres.2009.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lohi, H., Ianzano, L., Zhao, X.-C., Chan, E. M., Turnbull, J., Scherer, S. W.,et al. (2005). Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 14, 2727–2736. doi: 10.1093/hmg/ddi306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lohse, D. L., Denu, J. M., Santoro, N., and Dixon, J. E. (1997). Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1. Biochemistry 36, 4568–4575. doi: 10.1021/bi963094r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucentini, L., Fulle, S., Ricciolini, C., Lancioni, H., and Panara, F. (2003). Low molecular weight phosphotyrosine protein phosphatase from PC12 cells: purification, some properties and expression during neurogenesis in vitro and in vivo. Int. J. Biochem. Cell Biol. 35, 1378–1387. doi: 10.1016/S1357-2725(03)00099-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madhurantakam, C., Rajakumara, E., Mazumdar, A., Saha, B., Mitra, D., Wiker, H. G.,et al. (2005). Crystal structure of low-molecular-weight protein tyrosine phosphatase from Mycobacterium tuberculosis at 1.9-A resolution. J. Bacteriol. 187, 2175–2181. doi: 10.1128/JB.187.6.2175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maehama, T., and Dixon, J. E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378. doi: 10.1074/jbc.273.22.13375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maehama, T., Taylor, G. S., and Dixon, J. E. (2001). PTEN and myotubularin: novel phosphoinositide phosphatases. Annu. Rev. Biochem. 70, 247–279. doi: 10.1146/annurev.biochem.70.1.247
Mailand, N., Falck, J., Lukas, C., Syljuåsen, R. G., Welcker, M., Bartek, J.,et al. (2000). Rapid destruction of human Cdc25A in response to DNA damage. Science 288, 1425–1429. doi: 10.1126/science.288.5470.1425
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malentacchi, F., Marzocchini, R., Gelmini, S., Orlando, C., Serio, M., Ramponi, G.,et al. (2005). Up-regulated expression of low molecular weight protein tyrosine phosphatases in different human cancers. Biochem. Biophys. Res. Commun. 334, 875–883. doi: 10.1016/j.bbrc.2005.06.176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mann, B. A., Huang, J. H., Li, P., Chang, H.-C., Slee, R. B., O’Sullivan, A.,et al. (2008). Vaccinia virus blocks stat1-dependent and stat1-independent gene expression induced by type i and type ii interferons. J. Interf. Cytokine Res. 28, 367–380. doi: 10.1089/jir.2007.0113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mascarello, A., Chiaradia, L. D., Vernal, J., Villarino, A., Guido, R. V. C., Perizzolo, P.,et al. (2010). Inhibition of Mycobacterium tuberculosis tyrosine phosphatase PtpA by synthetic chalcones: kinetics, molecular modeling, toxicity, and effect on growth. Bioorg. Med. Chem. 18, 3783–3789. doi: 10.1016/j.bmc.2010.04.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuda, M., Takeshita, K., Kurokawa, T., Sakata, S., Suzuki, M., Yamashita, E.,et al. (2011). Crystal structure of the cytoplasmic phosphatase and tensin homolog (PTEN)-like region of Ciona intestinalis voltage-sensing phosphatase provides insight into substrate specificity and redox regulation of the phosphoinositide phosphatase activity. J. Biol. Chem. 286, 23368–23377. doi: 10.1074/jbc.M110.214361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McConnachie, G., Pass, I., Walker, S. M., and Downes, C. P. (2003). Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN: evidence for activation by anionic phospholipids. Biochem. J. 371, 947–955. doi: 10.1042/BJ20021848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miki, H., and Funato, Y. (2012). Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J. Biol. Chem. 151, 255–261. doi: 10.1093/jb/mvs006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, T. W., Rexer, B. N., Garrett, J. T., and Arteaga, C. L. (2011). Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 13, 224. doi: 10.1186/bcr3039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Modesti, A., Marzocchini, R., Raugei, G., Chiti, F., Sereni, A., Magherini, F.,et al. (1998). Cloning, expression, and characterisation of a new human low Mr phosphotyrosine protein phosphatase originating by alternative splicing. FEBS Lett. 431, 111–115. doi: 10.1016/S0014-5793(98)00732-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monteleone, M. C., González Wusener, A. E., Burdisso, J. E., Conde, C., Cáceres, A., and Arregui, C. O. (2012). ER-bound protein tyrosine phosphatase PTP1B interacts with Src at the plasma membrane/substrate interface. PLoS ONE 7:e38948. doi: 10.1371/journal.pone.0038948
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moorhead, G. B., De Wever, V., Templeton, G., and Kerk, D. (2009). Evolution of protein phosphatases in plants and animals. Biochem. J. 417, 401–409. doi: 10.1042/BJ20081986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moorhead, G. B. G., Trinkle-Mulcahy, L., and Ulke-Lemée, A. (2007). Emerging roles of nuclear protein phosphatases. Nat. Rev. Mol. Cell Biol. 8, 234–244. doi: 10.1038/nrm2126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murata, Y., Iwasaki, H., Sasaki, M., Inaba, K., and Okamura, Y. (2005). Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243. doi: 10.1038/nature03650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mutua, J., Jinno, Y., Sakata, S., Okochi, Y., Ueno, S., Tsutsui, H.,et al. (2014). Functional diversity of voltage-sensing phosphatases in two urodele amphibians. Physiol. Rep. 2, e12061. doi: 10.14814/phy2.12061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Myers, M. P., Pass, I., Batty, I. H., van der Kaay, J., Stolarov, J. P., Hemmings, B. A.,et al. (1998). The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. U.S.A. 95, 13513–13518. doi: 10.1073/pnas.95.23.13513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Myers, M. P., Stolarov, J. P., Eng, C., Li, J., Wang, S. I., Wigler, M. H.,et al. (1997). P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. U.S.A. 94, 9052–9057. doi: 10.1073/pnas.94.17.9052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Najarro, P., and Traktman, P. (2001). Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J. Virol. 75, 3185–3196. doi: 10.1128/JVI.75.7.3185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nam, H.-J., Poy, F., Saito, H., and Frederick, C. A. (2005). Structural basis for the function and regulation of the receptor protein tyrosine phosphatase CD45. J. Exp. Med. 201, 441–452. doi: 10.1084/jem.20041890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neuhaus, H., and Hollemann, T. (2009). Kidney specific expression of cTPTE during development of the chick embryo. Gene Expr. Patterns 9, 568–571. doi: 10.1016/j.gep.2009.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oganesian, A., Poot, M., Daum, G., Coats, S. A., Wright, M. B., Seifert, R. A.,et al. (2003). Protein tyrosine phosphatase RQ is a phosphatidylinositol phosphatase that can regulate cell survival and proliferation. Proc. Natl. Acad. Sci. U.S.A. 100, 7563–7568. doi: 10.1073/pnas.1336511100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogasawara, M., Sasaki, M., Nakazawa, N., Nishino, A., and Okamura, Y. (2011). Gene expression profile of Ci-VSP in juveniles and adult blood cells of ascidian. Gene Expr. Patterns 11, 233–238. doi: 10.1016/j.gep.2010.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Okamura, Y., and Dixon, J. E. (2011). Voltage-sensing phosphatase: its molecular relationship with PTEN. Physiology 26, 6–13. doi: 10.1152/physiol.00035.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ostman, A., Frijhoff, J., Sandin, A., and Böhmer, F.-D. (2011). Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 150, 345–356. doi: 10.1093/jb/mvr104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pannifer, A. D., Flint, A. J., Tonks, N. K., and Barford, D. (1998). Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by X-ray crystallography. J. Biol. Chem. 273, 10454–10462. doi: 10.1074/jbc.273.17.10454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, E. K., Warner, N., Mood, K., Pawson, T., and Daar, I. O. (2002). Low-molecular-weight protein tyrosine phosphatase is a positive component of the fibroblast growth factor receptor signaling pathway. Mol. Cell. Biol. 22, 3404–3414. doi: 10.1128/MCB.22.10.3404-3414.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, H., Yu, K. R., Ku, B., Kim, B. Y., and Kim, S. J. (2013). Identification of novel PTPRQ phosphatase inhibitors based on the virtual screening with docking simulations. Theor. Biol. Med. Model. 10:49. doi: 10.1186/1742-4682-10-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pawson, T., and Scott, J. D. (2005). Protein phosphorylation in signaling – 50 years and counting. Trends Biochem. Sci. 30, 286–290. doi: 10.1016/j.tibs.2005.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pulido, R., Stoker, A. W., and Hendriks, W. J. A. J. (2013). PTPs emerge as PIPs: protein tyrosine phosphatases with lipid-phosphatase activities in human disease. Hum. Mol. Genet. 22, R66–R76. doi: 10.1093/hmg/ddt347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pulido, R., and van Huijsduijnen, R. H. (2008). Protein tyrosine phosphatases: dual-specificity phosphatases in health and disease. FEBS J. 275, 848–866. doi: 10.1111/j.1742-4658.2008.06250.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramaswamy, S., Nakamura, N., Vazquez, F., Batt, D. B., Perera, S., Roberts, T. M.,et al. (1999). Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 2110–2115. doi: 10.1073/pnas.96.5.2110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramponi, G., Manao, G., Camici, G., Cappugi, G., Ruggiero, M., and Bottaro, D. P. (1989). The 18 kDa cytosolic acid phosphatase from bovine liver has phosphotyrosine phosphatase activity on the autophosphorylated epidermal growth factor receptor. FEBS Lett. 250, 469–473. doi: 10.1016/0014-5793(89)80778-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramponi, G., and Stefani, M. (1997a). Structural, catalytic, and functional properties of low Mr phosphotyrosine protein phosphatases. Evidence of a long evolutionary history. Int. J. Biochem. Cell Biol. 29, 279–292. doi: 10.1016/S1357-2725(96)00109-4
Ramponi, G., and Stefani, M. (1997b). Structure and function of the low Mr phosphotyrosine protein phosphatases. Biochim. Biophys. Acta 1341, 137–156. doi: 10.1016/S0167-4838(97)00087-83
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ratzan, W. J., Evsikov, A. V., Okamura, Y., and Jaffe, L. A. (2011). Voltage sensitive phosphoinositide phosphatases of Xenopus: their tissue distribution and voltage dependence. J. Cell. Physiol. 226, 2740–2746. doi: 10.1002/jcp.22854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raugei, G., Ramponi, G., and Chiarugi, P. (2002). Low molecular weight protein tyrosine phosphatases: small, but smart. Cell Mol. Life Sci. 59, 941–949. doi: 10.1007/s00018-002-8481-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Redfern, R. E., Redfern, D., Furgason, M. L. M., Munson, M., Ross, A. H., and Gericke, A. (2008). PTEN phosphatase selectively binds phosphoinositides and undergoes structural changes. Biochemistry 47, 2162–2171. doi: 10.1021/bi702114w
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reynolds, R. A., Yem, A. W., Wolfe, C. L., Deibel, M. R., Chidester, C. G., and Watenpaugh, K. D. (1999). Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J. Mol. Biol. 293, 549–566. doi: 10.1006/jmbi.1999.3168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roach, P. J., Depaoli-Roach, A. A., Hurley, T. D., and Tagliabracci, V. S. (2012). Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441, 763–787. doi: 10.1042/BJ20111416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ross, S. H., Lindsay, Y., Safrany, S. T., Lorenzo, O., Villa, F., Toth, R.,et al. (2007). Differential redox regulation within the PTP superfamily. Cell. Signal. 19, 1521–1530. doi: 10.1016/j.cellsig.2007.01.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rudolph, J. (2002). Catalytic Mechanism of Cdc25. Biochemistry 41, 14613–14623. doi: 10.1021/bi0263513
Rudolph, J. (2007). Cdc25 Phosphatases: structure, Specificity, and Mechanism. Biochemistry 46, 3595–3604. doi: 10.1021/bi700026j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Russell, P., and Nurse, P. (1984). cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell 45, 145–153. doi: 10.1016/0092-8674(86)90546-90545
Sakata, S., Hossain, M. I., and Okamura, Y. (2011). Coupling of the phosphatase activity of Ci-VSP to its voltage sensor activity over the entire range of voltage sensitivity. J. Physiol. 589, 2687–2705. doi: 10.1113/jphysiol.2011.208165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakata, S., and Okamura, Y. (2013). Phosphatase activity of the voltage-sensing phosphatase, VSP, shows graded dependence on the extent of activation of the voltage sensor. J. Physiol. 1–54. doi: 10.1113/jphysiol.2013.263640.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Salmeen, A., Andersen, J. N., Myers, M. P., Meng, T.-C., Hinks, J. A., Tonks, N. K.,et al. (2003). Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773. doi: 10.1038/nature01680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Salmeen, A., and Barford, D. (2005). Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox. Signal. 7, 560–577. doi: 10.1089/ars.2005.7.560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sankhala, R. S., Lokareddy, R. K., and Cingolani, G. (2014). Structure of human PIR1, an atypical dual-specificity phosphatase. Biochemistry 53, 862–871. doi: 10.1021/bi401240x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scott, L. M., Lawrence, H. R., Sebti, S. M., Lawrence, N. J., and Wu, J. (2010). Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr. Pharm. Des. 16, 1843–1862. doi: 10.2174/138161210791209027
Seifert, R. A., Coats, S. A., Oganesian, A., Wright, M. B., Dishmon, M., Booth, C. J.,et al. (2003). PTPRQ is a novel phosphatidylinositol phosphatase that can be expressed as a cytoplasmic protein or as a subcellularly localized receptor-like protein. Exp. Cell Res. 287, 374–386. doi: 10.1016/S0014-4827(03)00121-126
Seiler, C. L., Richards, K. A., Jakubowski, H. V., and McIntee, E. J. (2013). Identification of new inhibitors for low molecular weight protein tyrosine phosphatase isoform B. Bioorg. Med. Chem. Lett. 23, 5912–5914. doi: 10.1016/j.bmcl.2013.08.079 doi: 10.1016/j.bmcl.2013.08.079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shimohama, S., Fujimoto, S., Chachin, M., Taniguchi, T., Perry, G., Whitehouse, P. J.,et al. (1995). Alterations of low molecular weight acid phosphatase protein level in Alzheimer’s disease. Brain Res. 699, 125–129. doi: 10.1016/0006-8993(95)00995-993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shimohama, S., Fujimoto, S., Taniguchi, T., Karneyama, M., and Kimura, J. (1993). Reduction of low-molecular-weight acid phosphatase activity in Alzheimer brains. Ann. Neurol. 33, 616–621. doi: 10.1002/ana.410330610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefani, M., Caselli, A., Bucciantini, M., Pazzagli, L., Dolfi, F., Camici, G.,et al. (1993). Dephosphorylation of tyrosine phosphorylated synthetic peptides by rat liver phosphotyrosine protein phosphatase isoenzymes. FEBS Lett. 326, 131–134. doi: 10.1016/0014-5793(93)81776-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Streuli, M., Krueger, N. X., Thai, T., Tang, M., and Saito, H. (1990). Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J. 9, 2399–2407.
Stuckey, J. A., Schubert, H. L., Fauman, E. B., Zhang, Z. Y., Dixon, J. E., and Saper, M. A. (1994). Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature 370, 571–575. doi: 10.1038/370571a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Su, X.-D., Taddei, N., Stefani, M., Ramponi, G., and Nordlund, P. (1994). The crystal structure of a low-molecular-weight phosphotyrosine protein phosphatase. Nature 370, 575–578. doi: 10.1038/370575a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sutton, K. A., Jungnickel, M. K., Jovine, L., and Florman, H. M. (2012). Evolution of the voltage sensor domain of the voltage-sensitive phosphoinositide phosphatase VSP/TPTE suggests a role as a proton channel in eutherian mammals. Mol. Biol. Evol. 29, 2147–2155. doi: 10.1093/molbev/mss083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tabernero, L., Aricescu, A. R., Jones, E. Y., and Szedlacsek, S. E. (2008). Protein tyrosine phosphatases: structure-function relationships. FEBS J. 275, 867–882. doi: 10.1111/j.1742-4658.2008.06251.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tabernero, L., Evans, B. N., Tishmack, P. A., Van Etten, R. L., and Stauffacher, C. V. (1999). The structure of the bovine protein tyrosine phosphatase dimer reveals a potential self-regulation mechanism. Biochemistry 38, 11651–11658. doi: 10.1021/bi990381x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taddei, N., Chiarugi, P., Cirri, P., Fiaschi, T., Stefani, M., Camici, G.,et al. (1994). Aspartic-129 is an essential residue in the catalytic mechanism of the low Mr phosphotyrosine protein phosphatase. FEBS Lett. 350, 328–332. doi: 10.1016/0014-5793(94)00805-801
Tagliabracci, V. S., Turnbull, J., Wang, W., Girard, J.-M., Zhao, X., Skurat, A. V.,et al. (2007). Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 19262–19266. doi: 10.1073/pnas.0707952104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tailor, P., Gilman, J., Williams, S., and Mustelin, T. (1999). A novel isoform of the low molecular weight phosphotyrosine phosphatase, LMPTP-C, arising from alternative mRNA splicing. Eur. J. Biochem. 262, 277–282. doi: 10.1046/j.1432-1327.1999.00353.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tapparel, C., Reymond, A., Girardet, C., Guillou, L., Lyle, R., Lamon, C.,et al. (2003). The TPTE gene family: cellular expression, subcellular localization, and alternative splicing. Gene 323, 189–199. doi: 10.1016/j.gene.2003.09.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tautz, L., Critton, D. A., and Grotegut, S. (2013). Protein tyrosine phosphatases: structure, function, and implication in human disease. Methods Mol. Biol. 1053, 179–221. doi: 10.1007/978-1-62703-562-0-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Todd, J. L., Tanner, K. G., and Denu, J. M. (1999). Extracellular regulated kinases (Erk) 1 and Erk2 Are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR: a novel role in down-regulating the Erk pathway. J. Biol. Chem. 274, 13271–13280. doi: 10.1074/jbc.274.19.13271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tonks, N. K. (2005). Redox redux: revisiting PTPs and the control of cell signaling. Cell 121, 667–670. doi: 10.1016/j.cell.2005.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tonks, N. K. (2006). Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846. doi: 10.1038/nrm2039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tonks, N. K., Diltz, C. D., and Fischer, E. H. (1988a). Characterization of the Major Protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 263, 6731–6737.
Tonks, N. K., Diltz, C. D., and Fischer, E. H. (1988b). Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 263, 6722–6730.
Tonks, N. K., and Neel, B. G. (2001). Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 13, 182–195. doi: 10.1016/S0955-0674(00)00196-194
Tronchère, H., Buj-Bello, A., Mandel, J.-L., and Payrastre, B. (2003). Implication of phosphoinositide phosphatases in genetic diseases: the case of myotubularin. Cell Mol. Life Sci. 60, 2084–2099. doi: 10.1007/s00018-003-3062-3063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Montfort, R. L. M., Congreve, M., Tisi, D., Carr, R., and Jhoti, H. (2003). Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423, 773–777. doi: 10.1038/nature01681.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vazquez, F., and Devreotes, P. (2006). Regulation of PTEN function as a PIP3 gatekeeper through membrane interaction. Cell Cycle 5, 1523–1527. doi: 10.4161/cc.5.14.3005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vazquez, F., Matsuoka, S., Sellers, W. R., Yanagida, T., Ueda, M., and Devreotes, P. N. (2006). Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 103, 3633–3638. doi: 10.1073/pnas.0510570103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villalba-Galea, C. A. (2012a). New insights in the activity of voltage sensitive phosphatases. Cell. Signal. 24, 1541–1547. doi: 10.1016/j.cellsig.2012.03.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villalba-Galea, C. A. (2012b). Voltage-controlled enzymes: the new JanusBifrons. Front. Pharmacol. 3:161. doi: 10.3389/fphar.2012.00161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villalba-Galea, C. A., Miceli, F., Taglialatela, M., and Bezanilla, F. (2009). Coupling between the voltage-sensing and phosphatase domains of Ci-VSP. J. Gen. Physiol. 134, 5–14. doi: 10.1085/jgp.200910215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walker, S. M., Downes, C. P., and Leslie, N. R. (2001). TPIP: a novel phosphoinositide 3-phosphatase. Biochem. J. 360, 277–283. doi: 10.1042/0264-6021:3600277
Walker, S. M., Leslie, N. R., Perera, N. M., Batty, I. H., and Downes, C. P. (2004). The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem. J. 379, 301–307. doi: 10.1042/BJ20031839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, J., Stuckey, J. A., Wishart, M. J., and Dixon, J. E. (2002). A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J. Biol. Chem. 277, 2377–2380. doi: 10.1074/jbc.C100686200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, W.-Q., Sun, J.-P., and Zhang, Z.-Y. (2003). An overview of the protein tyrosine phosphatase superfamily. Curr. Top. Med. Chem. 3, 739–748. doi: 10.2174/1568026033452302
Wo, Y.-Y. P., McCormack, A. L., Shabanowitz, J., Hunt, D. F., Davis, J. P., Mitchell, G. L.,et al. (1992). Sequencing, cloning, and expression of human red cell-type acid phosphatase, a cytoplasmic phosphotyrosyl protein phosphatase. J. Biol. Chem. 267, 10856–10865.
Worby, C. A., Gentry, M. S., and Dixon, J. E. (2006). Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 281, 30412–30418. doi: 10.1074/jbc.M606117200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, L., Buist, A., den Hertog, J., and Zhang, Z.-Y. (1997). Comparative kinetic analysis and substrate specificity of the tandem catalytic domains of the receptor-like protein-tyrosine phosphatase. J. Biol. Chem. 272, 6994–7002. doi: 10.1074/jbc.272.11.6994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, Y., Dowbenko, D., Pisabarro, M. T., Dillard-Telm, L., Koeppen, H., and Lasky, L. A. (2001). PTEN 2, a Golgi-associated testis-specific homologue of the PTEN tumor suppressor lipid phosphatase. J. Biol. Chem. 276, 21745–21753. doi: 10.1074/jbc.M101480200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, D., Rovira, I. I., and Finkel, T. (2002). Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev. Cell 2, 251–252. doi: 10.1016/S1534-5807(02)00132-136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yip, S.-C., Saha, S., and Chernoff, J. (2010). PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem. Sci. 35, 442–449. doi: 10.1016/j.tibs.2010.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, C.-X., Li, S., and Whorton, A. R. (2005). Redox regulation of PTEN by S-nitrosothiols. Mol. Pharmacol. 68, 847–854. doi: 10.1124/mol.104.010504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, K. R., Kim, Y. J., Jung, S.-K., Ku, B., Park, H., Cho, S. Y.,et al. (2013). Structural basis for the dephosphorylating activity of PTPRQ towards phosphatidylinositide substrates. Acta Crystallogr. D. Biol. Crystallogr. 69, 1522–1529. doi: 10.1107/S0907444913010457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuan, Y., Li, D.-M., and Sun, H. (1998). PIR1, a novel phosphatase that exhibits high affinity to RNA-ribonucleoprotein complexes. J. Biol. Chem. 273, 20347–20353. doi: 10.1074/jbc.273.32.20347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuvaniyama, J., Denu, J. M., Dixon, J. E., and Saper, M. A. (1996). Crystal structure of the dual specificity protein phosphatase VHR. Science 272, 1328–1331. doi: 10.1126/science.272.5266.1328
Zhang, M., Stauffacher, C. V., Lin, D., and Van Etten, R. L. (1998). Crystal structure of a human low molecular weight phosphotyrosyl phosphatase: implications for substrate specificity. J. Biol. Chem. 273, 21714–21720. doi: 10.1074/jbc.273.34.21714
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, M., Van Etten, R. L., and Stauffacher, C. V. (1994a). Crystal structure of bovine heart phosphotyrosyl phosphatase at 2.2-A resolution. Biochemistry 33, 11097–11105. doi: 10.1021/bi00203a006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Z., Harms, E., and Van Etten, R. L. (1994b). Asp129 of low molecular weight protein tyrosine phosphatase is involved in leaving group protonation. J. Biol. Chem. 269, 25947–25950.
Zhang, Z. Y., Wang, Y., and Dixon, J. E. (1994c). Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 91, 1624–1627. doi: 10.1073/pnas.91.5.1624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, S., and Yu, D. (2010). PI(3)King Apart PTEN’s role in cancer. Clin. Cancer Res. 16, 4325–4330. doi: 10.1158/1078-0432.CCR-09-2990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, S., and Zhang, Z.-Y. (2007). PTP1B as a drug target: recent developments in PTP1B inhibitor discovery. Drug Discov. Today 12, 373–381. doi: 10.1016/j.drudis.2007.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Z.-Y. (1993). Active Site Labeling of the Yersinia Protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 32, 9340–9345. doi: 10.1021/bi00087a012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Z.-Y. (2001). Protein tyrosine phosphatases: prospects for therapeutics. Curr. Opin. Cell Biol. 5, 416–423. doi: 10.1016/S1367-5931(00)00223-4
Zhang, Z.-Y., and Van Etten, R. L. (1990). Purification and characterization of a low-molecular-weight acid phosphatase—A phosphotyrosyl-protein phosphatase from bovine heart. Arch. Biochem. Biophys. 282, 39–49. doi: 10.1016/0003-9861(90)90084-C
Zhang, Z. Y., and Van Etten, R. L. (1991). Leaving group dependence and proton inventory studies of the phosphorylation of a cytoplasmic phosphotyrosyl protein phosphatase from bovine heart. Biochemistry 30, 8954–8959. doi: 10.1021/bi00101a006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cysteine-based phosphatase, dual-specific phosphatase, acid phosphatase, low molecular weight phosphatase, Cdc25, phosphoinositide, phosphotyrosine, phosphoproteins
Citation: Hobiger K and Friedrich T (2015) Voltage sensitive phosphatases: emerging kinship to protein tyrosine phosphatases from structure-function research. Front. Pharmacol. 6:20. doi: 10.3389/fphar.2015.00020
Received: 04 December 2014; Accepted: 21 January 2015;
Published online: 10 February 2015.
Edited by:
Carlos Alberto Villalba-Galea, Virginia Commonwealth University School of Medicine, USAReviewed by:
Michel Vivaudou, Institut de Biologie Structurale, FranceAlonzo Harvey Ross, University of Massachusetts Medical School, USA
Copyright © 2015 Hobiger and Friedrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirstin Hobiger, Department of Neurophysiology, Institute of Physiology and Pathophysiology, Philipps-Universität Marburg, Deutschhausstraße 1-2, 35037 Marburg, Germany e-mail:a2lyc3Rpbi5ob2JpZ2VyQHN0YWZmLnVuaS1tYXJidXJnLmRl