Abstract
Incorporating phenotypic screening as a key strategy enhances predictivity and translatability of drug discovery efforts. Cellular imaging serves as a “phenotypic anchor” to identify important toxicologic pathology that encompasses an array of underlying mechanisms, thus provides an effective means to reduce drug development failures due to insufficient safety. This mini-review highlights the latest advances in hepatotoxicity, cardiotoxicity, and genetic toxicity tests that utilized cellular imaging as a screening strategy, and recommends path forward for further improvement.
Introduction
Modern medicines have saved countless lives. One needs only to trace the discovery of antiretroviral drugs to marvel at its contribution to human health (Broder, 2010). However, new therapies are necessary to address still unmet medical needs while adhering to the timeless adage of “first doing no harm” to patients. Predictive toxicology meets the latter challenge by offering accurate and timely predictions to minimize drug toxicity (Xu and Urban, 2010). It is recognized as a frontier for identifying better medicines in a more cost-effective manner by biopharmaceutical research and regulatory communities (FDA, 2010).
To successfully practice predictive toxicology, one must adopt an integrated approach utilizing innovations from multiple fields of science and engineering. In recent years there is a resurgence of phenotypic screens in drug efficacy discovery (Moffat et al., 2014). Likewise to improve confidence in the translatability of toxicity predictions, one should also incorporate phenotypic screens as a key strategy. This is because the “phenotype” of a specific safety signal can be caused by many underlying mechanisms and pathways, thus a reductionist “one gene/protein/mechanism to one toxicity” approach most likely will not be sufficient. This does not mean that one should revert to simple cell death measurements. Instead, more specific cellular functions that are vital to sustain physiological homeostasis can serve as translational links between non-clinical tests and clinical observations. Among many practical choices of phenotypic screens, cellular imaging technologies can be applied as “phenotypic anchors” to identify the same histopathology that occurs in vivo. Cellular imaging encompasses the techniques that allow quantitative detection and measurement of cellular structures and components including organelles and biomolecules ranging from macromolecules to small ions, often aided by automated microscopes, digitized cameras, and image analysis algorithms. Since “seeing is believing,” this “phenotype first” approach assures an unbiased study for compounds that affect important cellular homeostasis without exhausting all possible mechanistic tests. Of course, “phenotype first” does not preclude mechanistic investigations to better understand the underlying reason(s) of drug safety findings. Often it is a holistic approach relying on mechanism-informed phenotypic test, coupled with pharmacokinetic and pharmacodynamics (PKPD) modeling that produces the best overall prediction toward clinical outcome.
Hepatic toxicity imaging
Hepatic toxicity ranked highest in research priorities among adverse drug reactions in the pharmaceutical industry, and current detection systems remain imperfect especially with regard to idiosyncratic hepatotoxicity (Opar, 2012). Key mechanisms of drug-induced liver injury (DILI) include oxidative stress and/or mitochondrial damage leading to apoptosis or steatosis, and hepatobiliary transporter inhibition leading to cholestasis (Kaplowitz, 2005; Tujios and Fontana, 2011).
Mechanism-informed phenotypic tests using cellular imaging have been developed to encompass these key DILI mechanisms. For example, a panel of fluorescent imaging probes has been applied to measure oxidative stress, mitochondrial function, glutathione content, and hepatocellular lipidosis simultaneously in primary human hepatocytes (Xu et al., 2008). It has demonstrated ~60% sensitivity and ~95% specificity for drugs that have caused idiosyncratic DILI. This hepatocyte imaging assay technology has been successfully applied to both differentiate compounds for the same pharmacologic target (e.g., p38 MAP kinase inhibitors), and study the mechanism of DILI signals observed in the clinic (e.g., Her2 receptor antagonist) (Xu et al., 2011). A similar imaging approach measuring oxidative stress in primary human hepatocytes resulted in 41% sensitivity and 86% specificity, and was shown to have better predictivity than HepG2 cell lines or HepG2 plus hepatic metabolism from the liver S9 fractions (Garside et al., 2013). These studies demonstrated the application of cellular imaging technology to predict DILI, and highlighted the need to enhance test sensitivity by recapitulating additional DILI mechanisms and pathways.
Since inhibition of a panel of hepatobiliary transporters is another known pathway of drug-induced cholestasis (Morgan et al., 2013), cellular imaging was applied to assess transporter functions. The assay utilized quantitative imaging of fluorescent bile acid and its disposition in the bile canaliculi compartment of the hepatocyte cultures (Xu et al., 2011). As protein trafficking and sorting disturbances also play an integral part of intrahepatic cholestasis (Hayashi and Sugiyama, 2013), whole cell systems as opposed to isolated membrane vesicles over-expressing one transporter at a time should continue to play a key role in phenotypic screening of cholestasis-inducing drugs.
Several improvements on these cellular imaging tests should be explored to further enhance accurate prediction of DILI:
Long-term: Adopt a longer-term test system that maintains differentiated metabolic functions of human liver. Recently with advancement in iPSC-derived hepatocyte cultures, multi-day testing appeared promising. However, these longer-term models still need to be assessed with more DILI negative drugs to fully assess specificity (Ware et al., 2015).
Diversity: Apply more than one human liver origin with defined genetic background to increase test sensitivity toward idiosyncratic DILI. Patient-derived hepatic stem cells that can be differentiated into adult hepatocyte cultures can be interesting models to explore idiosyncratic causes of DILI.
Probes: Expand fluorescent imaging probes to include additional bile acid analogs that are substrates of both NTCP and BSEP transporters, in addition to cholyl lysyl fluoresceine which is a more specific substrate for OATP/MRP transporters (de Waart et al., 2010).
Inflammatory and multi-cell systems: Explore the role of pro-inflammatory cytokines and/or Kupffer cells for possible synergistic effects in test sensitivity without sacrificing specificity (Cosgrove et al., 2009), and assess the potential benefit of 3D liver chip with controlled microfluidics.
Modeling: Mechanism-based PKPD modeling approaches should be applied to integrate any in vitro measurements into a holistic in vivo prediction (Woodhead et al., 2014). Predictions made by the more complex model should also be compared to simple in vitro-in vivo scaling or classification approaches based on exposure multiples or pre-defined safety margins.
Cardiac toxicity imaging
Adverse cardiovascular effects ranked highest among safety reasons for delayed approvals or non-approvals by the US Food and Drug Administration (FDA) between 2000 and 2012 (Sacks et al., 2014). Many drugs that prolong the QT interval in electrocardiograms block the delayed rectifier K+ current (Ikr) by blocking one key ion channel, the hERG channel. However, not all hERG channel blockers cause QT interval prolongation nor translate to the more severe clinical concern, torsade de pointes [TdP, or twisting of the points in electrocardiograms (Vargas, 2010)]. So QT prolongation is a surrogate biomarker that is not very specific to TdP. In addition to TdP there are other mechanisms of cardiotoxicity. Cardiovascular functional abnormalities can also arise through impaired left ventricular function characterized by changes in cardiomyocyte contractility (Doherty et al., 2013). Therefore, a more holistic screening approach is needed. Since 2013, the FDA working with other international research communities have an on-going initiative to develop better preclinical tools to evaluate cardiovascular safety with the ultimate goal of preventing early stage compounds from being unnecessarily discontinued, while still screening out molecules that are either pro-arrhythmic or lead to changes in cardiomyocyte contractility (Sager et al., 2014).
Synchronously beating human cardiomyocytes should be explored as a key phenotypic screening model. Despite some limitations of iPSC-derived cardiomyocytes, they are the future for large-scale functional cardiotoxicity screening models in preclinical drug evaluation (Puppala et al., 2013). This is because there is a virtually unlimited supply of functionally competent cells, and there is the possibility to create cells from defined genetic background thus lending the model amenable to further mechanistic studies. Researchers have studied 131 drugs using iPSC-derived cardiomyocytes, using fast kinetic imaging-based Ca2+ flux as continuously cell-beating measurements (Sirenko et al., 2013). Each output (beat rate, peak characteristics, and cell viability) generated as a result of such cellular imaging assay has yielded a panel of imaging “signatures” for understanding the type of pathophysiological effect that a chemical may have on the heart. Not surprisingly, beat rate and several peak shape parameters were found to be better predictors than simple cell viability. In another study, video-based microscopic imaging recapitulated expected drug effects in stem-cell-derived cardiomyocytes without the use of exogenous labels and produced the same beat traces as patch clamp (Maddah et al., 2015). Recently, the throughput and accuracy of iPSC-based model to detect changes in cardiomyocyte contraction was found applicable in drug discovery screening, with a sensitivity and specificity of 87 and 70%, respectively (Pointon et al., 2015).
Tyrosine kinase inhibitors (TKIs) are efficacious against tumors harboring mutations and/or over-expressing such kinases. However, TKIs have been hampered by cardiotoxicity issues detected most frequently as changes in left-ventricular ejection fraction (LVEF), which were not predicted by hERG inhibition per se. Using multi-parameter cellular imaging it was demonstrated while crizotinib, sunitinib, and nilotinib disrupted the normal beat pattern of human iPSC-derived cardiomyocytes, erlotinib did not. These alterations in iPSC-cardiomyocyte beat pattern correlated well with known clinical cardiac outcomes of these drugs, demonstrating the potential to use this phenotypic screen as a routine test to identify safer compounds in a class of drug candidates (Doherty et al., 2013).
Several improvements on these cellular imaging screens should be explored to further enhance the overall prediction of cardiotoxicity:
beating disturbances by a variety of drugs with different cardiotoxic mechanisms
longer-term cellular models to study cardiac hypertrophy
drug metabolites by introducing hepatic metabolism (e.g., either hepatic S9 fraction or multi-cell systems)
more test qualification with large compound sets consisting of an equal number of positive and negatives that have been rigorously adjudicated. A balanced test set of compounds with more negative or clean compounds is critical to assess test specificity.
Genetic toxicity imaging
Carcinogenicity remained a major nonclinical safety finding among FDA non-approvals and delayed approvals between 2000 and 2012 (Sacks et al., 2014). In one of the largest retrospective analyses of pharmaceutical compounds to date, it was demonstrated that significant R&D time, resources, and animal usage can be reduced by relying on the near 100% negative predictive values (NPV) of combined tests evaluating genetic toxicity, hormonal perturbation, and evidence of neoplasia in chronic rat toxicology studies (Sistare et al., 2011). To minimize the impact of carcinogenicity findings in late-stage drug development, both mutational and chromosomal changes need to be evaluated as they can be caused by different underlying mechanisms (FDA, 2012).
Cellular imaging has greatly enhanced the efficiency and accuracy of measuring both mutational and chromosomal changes. The Ames test remains a cost-effective stethoscope to study genetic toxicology (Claxton et al., 2010). In order to enable rapid scoring of mutant bacterial colonies, the Salmonella strains used in the Ames test were engineered to express bioluminescent proteins (Aubrecht et al., 2007). The high-throughput imaging based on automated counting of the number of surviving bioluminescent colonies offered a far more accurate assessment of genotoxicity than bulk biomass measurements (Xu and Aubrecht, 2010).
With regard to chromosomal changes, the in vitro micronucleus test is an accepted test by regulatory authorities (EMA and FDA) (FDA, 2012). While traditional micronucleus test relied on manual counting of the number of micronuclei in thousands of cells per treatment sample and hundreds of samples per test compound, cellular imaging has made this task more efficient. The increased throughput and accuracy of computerized analysis of micronuclei frequency enabled rapid structure-activity profiling of drug candidates during lead optimization (Xu and Aubrecht, 2010).
Conclusion
From a systems perspective, since in vitro genotoxicity tests have near 100% negative predictive value toward in vivo genotoxicity outcome, are among the most cost-effective to operate (i.e., using bacterial strains and mammalian cell lines), and include both general cytotoxicity and cell growth inhibition as integral part of assay readouts, in vitro Ames and micronucleus should be employed as the foundational tests to select those “clean” compounds for further R&D investments. In the lead optimization stage of small-molecule drug discovery, these genotoxicity tests should be followed by cardiac and hepatic toxicity screening using primary cells or iPSCs next, then other organ-specific tests on an ad hoc basis last. The ad hoc tests are reserved for program-specific purposes, e.g., triggered by hypotheses about the mechanism-based toxicity or prior in vivo findings for previous compounds in the program. Examples of such ad hoc assays include: muscle, kidney, neural, teratonenic, developmental, myelotoxicity, and lymphotoxicity. The potential for multi-organ chip to improve toxicity prediction should also fall into this last category until its predictivity and throughput make it possible for front-loading (Bhatia and Ingber, 2014). The ad hoc assays can also include further investigation of previous positive findings, e.g., micronucleus positives can be imaged for other markers to differentiate chromosome breaks (clastogens) vs. chromosome loss (aneuploidy), where an acceptable safety margin can allow for further drug development (Cheung et al., 2014). In the complex business of defining drug safety, “clean” compound in all test systems are not always realistic. One should therefore use a balanced and holistic approach starting with defining an acceptable safety profile for a drug candidate, understanding the predictive value and limitation of each test system, and a “weight-of-evidence” approach integrating findings from human cells, in vivo animal testing, and human clinical trials. Table 1 summarizes the imaging tests and their predictive values described in this mini-review.
Table 1
| Cell model (reference) | Imaging technique | Threshold for positives | Predictive value | Example image |
|---|---|---|---|---|
| Human hepatocyte (Xu et al., 2008) | Fluorescence imaging of oxidative stress, mitochondrial function, glutathione content and hepatocellular lipidosis | >2.5 fold above or < 2.5 fold below the vehicle control mean values (depending on which fluorescence channel). Appropriate cut-off levels were selected using ROC curves | ~60% sensitivity and ~95% specificity | 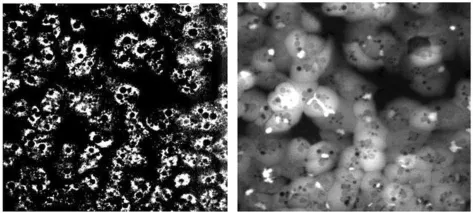 |
| Human hepatocyte (Garside et al., 2013) | Fluorescence imaging of oxidative stress | >6 SD of the vehicle control mean values | 41% sensitivity and 86% specificity | |
| Human hepatocyte (Xu et al., 2011) | Fluorescence imaging of bile acid and its disposition in bile canaliculi | < 2.5 fold below the vehicle control mean values | TBD | 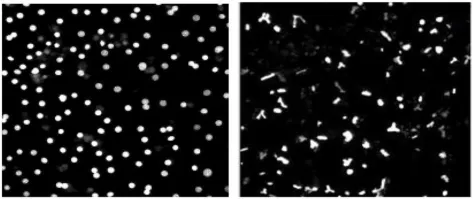 |
| iPSC-derived human cardiomyocytes (Sirenko et al., 2013) | Fast kinetic imaging-based Ca2+ flux as continuously cell-beating measurements (beat rate, amplitude, and other beat parameters) | >1 SD of the vehicle control mean values | TBD | 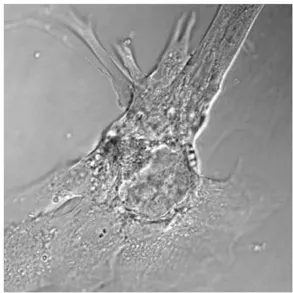 |
| iPSC-derived human cardiomyocytes (Pointon et al., 2015) | Fast kinetic imaging-based Ca2+ flux as continuously cell-beating measurements (peak count, average peak amplitude, average peak width, average peak rise time, average peak decay time, and average peak spacing) | The median of the positive and negative control wells were set at 0 and −100, respectively, and the signals from all wells scaled to this range. Appropriate cut-off levels were selected using ROC curves | 87% sensitivity and 70% specificity, using peak count | |
| Ames (Xu and Aubrecht, 2010) | Automated counting of the number of surviving bioluminescent colonies | Same as manual counting | 100% combined negative predictive value to carcinogenicity test |  |
| In vitro micronucleus (Xu and Aubrecht, 2010) | Automated scoring of the micronucleus frequency | Same as manual scoring | 100% combined negative predictive value to carcinogenicity test | 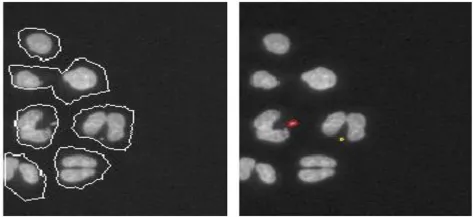 |
A summary of imaging tests and their predictive values reviewed by this article.
SD, standard deviation; ROC, receiver operator characteristic; TBD, to be determined.
Safety deficiencies account for more than half of delayed FDA approval or non-approval of new drug applications from 2000 to 2012 (Sacks et al., 2014). Predictive toxicology holds the key to reduce this attrition and make R&D investment more sustainable. To realize its promise practitioners of predictive toxicology must continue to integrate diverse innovations from multiple fields of biomedical engineering, including:
Predictive and reliable cellular models: esp., evaluation, characterization, production, and standardization of predictive human cells
Mechanism-informed phenotypic screening strategy, including cellular imaging as a key component as it provides a phenotypic anchor and direct linkage to in vivo histopathology
Quantitative therapeutic window predictions: e.g., using mechanism-based scaling or predictive PKPD modeling
One may envision a future when the banking of pluripotent stem cells from individual human donor with defined genetic makeup and subsequent expansion into fully-functional organ parenchymal cells as a desirable model toward predicting personalized drug responses, for both drug efficacy and safety. But before we achieve that dream, the holistic applications of an integrated approach as highlighted above have already marched us a long way toward transforming toxicology to improve product safety.
Conflict of interest statement
The author is an employee of Merck and Co. However, views expressed in this mini-review are the author's own and do not reflect positions of any company or organization.
Statements
Acknowledgments
The author acknowledges Drs. Timothy Johnson, Frank Sistare, Nicolau Beckmann, and three peer reviewers for critical review of the manuscript. The author sincerely apologizes that many scholars' publications cannot be cited in this mini-review due to space limitation.
Conflict of interest
The author is an employee of Merck and Co. However, views expressed in this mini-review are the author's own and do not reflect positions of any company or organization.
References
1
AubrechtJ.OsowskiJ. J.PersaudP.CheungJ. R.AckermanJ.LopesS. H.et al. (2007). Bioluminescent Salmonella reverse mutation assay: a screen for detecting mutagenicity with high throughput attributes. Mutagenesis22, 335–342. 10.1093/mutage/gem022
2
BhatiaS. N.IngberD. E. (2014). Microfluidic organs-on-chips. Nat. Biotechnol.32, 760–772. 10.1038/nbt.2989
3
BroderS. (2010). The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res.85, 1–18. 10.1016/j.antiviral.2009.10.002
4
CheungJ. R.DickinsonD. A.MossJ.SchulerM. J.SpellmanR. A.HeardP. L. (2014). Histone markers identify the mode of action for compounds positive in the TK6 micronucleus assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 777, 7–16. 10.1016/j.mrgentox.2014.11.002
5
ClaxtonL. D.Umbuzeiro GdeA.DeMariniD. M. (2010). The Salmonella mutagenicity assay: the stethoscope of genetic toxicology for the 21st century. Environ. Health Perspect.118, 1515–1522. 10.1289/ehp.1002336
6
CosgroveB. D.KingB. M.HasanM. A.AlexopoulosL. G.FaraziP. A.HendriksB. S.et al. (2009). Synergistic drug-cytokine induction of hepatocellular death as an in vitro approach for the study of inflammation-associated idiosyncratic drug hepatotoxicity. Toxicol. Appl. Pharmacol.237, 317–330. 10.1016/j.taap.2009.04.002
7
de WaartD. R.HäuslerS.VlamingM. L.KunneC.HanggiE.GrussH. J.et al. (2010). Hepatic transport mechanisms of cholyl-L-lysyl-fluorescein. J. Pharmacol. Exp. Ther.334, 78–86. 10.1124/jpet.110.166991
8
DohertyK. R.WappelR. L.TalbertD. R.TruskP. B.MoranD. M.KramerJ. W.et al. (2013). Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol.272, 245–255. 10.1016/j.taap.2013.04.027
9
FDA. (2010). Predictive Safety Testing Consortium. Available online at: http://c-path.org/programs/pstc/
10
FDA. (2012). International Conference on Harmonisation; guidance on S2(R1) Genotoxicity Testing and Data Interpretation for Pharmaceuticals intended for Human Use; availability. Notice. Fed. Regist.77, 33748–33749. Available online at: http://www.gpo.gov/fdsys/pkg/FR-2012-06-07/html/2012-13774.htm
11
GarsideH.MarcoeK. F.Chesnut-SpeelmanJ.FosterA. J.MuthasD.Gerry KennaJ. G.et al. (2013). Evaluation of the use of imaging parameters for the detection of compound-induced hepatotoxicity in 384-well cultures of HepG2 cells and cryopreserved primary human hepatocytes. Toxicol. In Vitro28, 171–181. 10.1016/j.tiv.2013.10.015
12
HayashiH.SugiyamaY. (2013). Bile salt export pump (BSEP/ABCB11): trafficking and sorting disturbances. Curr. Mol. Pharmacol.6, 95–103. 10.2174/18744672113069990036
13
KaplowitzN. (2005). Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov.4, 489–499. 10.1038/nrd1750
14
MaddahM.HeidmannJ. D.MandegarM. A.WalkerC. D.BoloukiS.ConklinB. R.et al. (2015). A non-invasive platform for functional characterization of stem-cell-derived cardiomyocytes with applications in cardiotoxicity testing. Stem Cell Reports4, 621–631. 10.1016/j.stemcr.2015.02.007
15
MoffatJ. G.RudolphJ.BaileyD. (2014). Phenotypic screening in cancer drug discovery—past, present and future. Nat. Rev. Drug Discov.13, 588–602. 10.1038/nrd4366
16
MorganR. E.van StadenC. J.ChenY.KalyanaramanN.KalanziJ.DunnR. T.et al. (2013). A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol. Sci.136, 216–241. 10.1093/toxsci/kft176
17
OparA. (2012). Overtaking the DILI Model-T. Nat. Rev. Drug Discov.11, 585–586. 10.1038/nrd3818
18
PointonA.HarmerA. R.DaleI. L.Abi-GergesN.BowesJ.PollardC.et al. (2015). Assessment of cardiomyocyte contraction in human-induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci.144, 227–237. 10.1093/toxsci/kfu312
19
PuppalaD.CollisL. P.SunS. Z.BonatoV.ChenX.AnsonB.et al. (2013). Comparative gene expression profiling in human-induced pluripotent stem cell-derived cardiocytes and human and cynomolgus heart tissue. Toxicol. Sci.131, 292–301. 10.1093/toxsci/kfs282
20
SacksL. V.ShamsuddinH. H.YasinskayaY. I.BouriK.LanthierM. L.ShermanR. E. (2014). Scientific and regulatory reasons for delay and denial of FDA approval of initial applications for new drugs, 2000-2012. JAMA311, 378–384. 10.1001/jama.2013.282542
21
SagerP. T.GintantG.TurnerJ. R.PettitS.StockbridgeN. (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the cardiac safety research consortium. Am. Heart J.167, 292–300. 10.1016/j.ahj.2013.11.004
22
SirenkoO.CromwellE. F.CrittendenC.WignallJ. A.WrightF. A.RusynI. (2013). Assessment of beating parameters in human induced pluripotent stem cells enables quantitative in vitro screening for cardiotoxicity. Toxicol. Appl. Pharmacol.273, 500–507. 10.1016/j.taap.2013.09.017
23
SistareF. D.MortonD.AldenC.ChristensenJ.KellerD.JongheS. D.et al. (2011). An analysis of pharmaceutical experience with decades of rat carcinogenicity testing: support for a proposal to modify current regulatory guidelines. Toxicol. Pathol.39, 716–744. 10.1177/0192623311406935
24
TujiosS.FontanaR. J. (2011). Mechanisms of drug-induced liver injury: from bedside to bench. Nat. Rev. Gastroenterol. Hepatol.8, 202–211. 10.1038/nrgastro.2011.22
25
VargasH. M. (2010). A new preclinical biomarker for risk of Torsades de Pointes: drug-induced reduction of the cardiac electromechanical window. Br. J. Pharmacol.161, 1441–1443. 10.1111/j.1476-5381.2010.00980.x
26
WareB. R.BergerD. R.KhetaniS. R. (2015). Prediction of drug-induced liver injury in micropatterned co-cultures containing iPSC-derived human hepatocytes. Toxicol. Sci.145, 252–262. 10.1093/toxsci/kfv048
27
WoodheadJ. L.YangK.SilerS. Q.WatkinsP. B.BrouwerK. L.BartonH. A.et al. (2014). Exploring BSEP inhibition-mediated toxicity with a mechanistic model of drug-induced liver injury. Front. Pharmacol.5:240. 10.3389/fphar.2014.00240
28
XuJ. J.AubrechtJ. (2010). Screening approaches for genetic toxicity, in Predictive Toxicology in Drug Safety, eds XuJ. J.UrbanL. (Cambridge, UK: Cambridge University Press), 18–33.
29
XuJ. J.UrbanL. (2010). Predictive Toxicology in Drug Safety.Cambridge, UK: Cambridge University Press.
30
XuJ. J.HenstockP. V.DunnM. C.SmithA. R.ChabotJ. R.de GraafD. (2008). Cellular imaging predictions of clinical drug-induced liver injury. Toxicol. Sci.105, 97–105. 10.1093/toxsci/kfn109
31
XuJ.DunnM. C.SmithA.TienE. (2011). Assessment of hepatotoxicity potential of drug candidate molecules including kinase inhibitors by hepatocyte imaging assay technology and bile flux imaging assay technology, in Kinase Inhibitors: Methods in Molecular Biology, Vol. 795, ed KusterB. (Freising: Humana Press), 83–107.
Summary
Keywords
hepatotoxicity, cardiotoxicity, genetic toxicology, cellular imaging, predictive toxicology, discovery toxicology, phenotypic screening
Citation
Xu JJ (2015) Cellular imaging: a key phenotypic screening strategy for predictive toxicology. Front. Pharmacol. 6:191. doi: 10.3389/fphar.2015.00191
Received
29 May 2015
Accepted
24 August 2015
Published
08 September 2015
Volume
6 - 2015
Edited by
Nicolau Beckmann, Novartis Institutes for BioMedical Research, Switzerland
Reviewed by
Peter James King, Queen Mary University of London, UK; Mikael Varakun Persson, H. Lundbeck A/S, Denmark; Paul Hockings, Antaros Medical, Sweden
Updates
Copyright
© 2015 Xu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinghai J. Xu, Merck and Co., 2000 Galloping Hill Road, Kenilworth, NJ 07033, USA jinghai_xu@merck.com
This article was submitted to Experimental Pharmacology and Drug Discovery, a section of the journal Frontiers in Pharmacology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.