Abstract
Neurodegenerative diseases are intricate in nature because of the involvement of the multiple pathophysiological events including mitochondrial dysfunction, neuroinflammation and oxidative stress. Alzheimer’s disease (AD) is a neurodegenerative disease explained by extracellular amyloid β deposits, intracellular neurofibrillary tangles and mitochondrial dysfunction. Increasing evidence has indicated that mitochondrial dysfunction displays significant role in the pathophysiological processes of AD. Mitochondrial dysfunction involves alterations in mitochondrial respiratory enzyme complex activities, oxidative stress, opening of permeability transition pore, and enhanced apoptosis. Various bioenergetics and antioxidants have been tried or under different investigational phase against AD and other neurodegenerative disorders (Parkinson’s disease, Huntington’s disease, and Amyotrophic lateral sclerosis) because of their complex and multiple site of action. These mitochondrial-targeting bioenergetics and antioxidant compounds such as coenzyme Q10, idebenone, creatine, mitoQ, mitovitE, MitoTEMPOL, latrepirdine, methylene blue, triterpenoids, SS peptides, curcumin, Ginkgo biloba, and omega-3 polyunsaturated fatty acids with potential efficacy in AD have been identified. Present review is intent to discuss mitochondrial restorative mechanisms of these bioenergetics and antioxidants as a potential alternative drug strategy for effective management of AD.
Introduction
Alzheimer’s disease (AD) common incapacitating neurodegenerative disease, identified by the occurrence of senile plaques extracellularly and neurofibrillary tangles intracellularly (Alzheimer’s, 2015; Kumar et al., 2015). Globally AD is becoming epidemic because of new cases in every 7 s and more than 36.5 million individuals are affected worldwide (Prince et al., 2014; Alzheimer’s, 2015). Senile plaque consists of amyloid β (Aβ) peptides and tangles are made from tau protein. It was found that Aβ peptides are produced by the proteolytic segmentation of the protein known as amyloid precursor protein (APP). Other characteristic features of AD include progressive and neuronal synaptic loss, mitochondrial dysfunction, oxidative stress and inflammatory responses (Iqbal and Grundke-Iqbal, 2010; Swerdlow et al., 2014; Alzheimer’s, 2015). An increasing body of evidences indicates that Aβ enhances neuronal vulnerability to mitochondrial dysfunction via an impairment of electron transport chain (ETC) and oxidative stress (Davis et al., 1995; Reddy and Beal, 2008; Reddy et al., 2010; Calkins et al., 2011; Swerdlow et al., 2014).
Till date, rigorous efforts have been made to understand the complex pathophysiological mechanisms’ underlying AD and other neurodegenerative disorders including Parkinson’s disease (PD), Huntington’s disease (HD), and Amyotrophic lateral sclerosis (ALS) but still it remains ill-defined and poorly understood disease. There have been many hypotheses put forward to explain their complex pathophysiology such as inflammatory, mitochondrial dysfunction and oxidative stress hypotheses (Kumar and Singh, 2015). Out of these hypotheses, mitochondrial dysfunction and oxidative stress hypotheses are the most argued one (Davis et al., 1995; Swerdlow et al., 2014). As these two events are occurring at very early stages of neurodegenerative diseases so these can be one of the important and potential therapeutic targets in the current scenario (Swerdlow et al., 2014).
Mitochondria, the major organelles in neurons (Chaturvedi and Beal, 2013) and via oxidative phosphorylation (OXPHOS) or mitochondrial respiratory chain produce energy as adenosine triphosphate (ATP; Schapira, 2012; Zeviani et al., 2012). Other functions of mitochondria are the regulation of calcium homeostasis, generation of free radicals and apoptosis (Swerdlow, 2011). Strong evidence indicated that mitochondrial dysfunction involves alterations of mitochondrial respiratory chain enzymes, generation of reactive oxygen species (ROS), opening of mitochondrial permeability transition pore (mPTP), structural abnormalities of mitochondria, oxidative stress and apoptosis (Hauptmann et al., 2006; Lin and Beal, 2006; Eckert and Müller, 2014). And these mitochondrial abnormalities are known to occur early in AD before Aβ deposition and are closely related to Aβ- or tau- pathology (Swerdlow et al., 2010; Maruszak and Żekanowski, 2011).
Various compounds have been demonstrated to possess mitochondrial restoring and anti-oxidant properties such as coenzyme Q10 (CoQ 10), vitamin E, curcumin, Gingko biloba, melatonin and lipoic acid (Du and Yan, 2010). Therapeutic potentials of these compounds have been suggested to reduce Aβ peptides accumulations, restoring mitochondrial functions, transport and synaptic plasticity, protect mitochondria from Aβ toxicity, attenuate cognitive impairment in AD, inhibit dopaminergic neuronal loss in PD and showed neuroprotective role in other neurodegenerative disorders like ALS, HD (Hauptmann et al., 2006; Lin and Beal, 2006; Pieczenik and Neustadt, 2007; Moreira et al., 2010; Maruszak and Żekanowski, 2011).
One of the important tasks of this review is to discuss the growing evidences demonstrating the importance of mitochondria and related features in the pathogenesis of AD. Finally, we will discuss mitochondrial dysfunction as a potential drug target for AD management. The authors also projected various drug strategies targeting mitochondrial dysfunction and oxidative stress which may help in attenuation of AD, PD, HD, and ALS pathologies. Also, an attempt has been made to discuss various compounds targeting mitochondria and oxidative stress as a future approach with major focus on AD pathology.
Mitochondrial Cascade Hypothesis
As discussed earlier, that the pathology of AD involves the extracellular aggregation of Aβ plaques and intracellular neurofibrillary tangles (Kumar and Singh, 2015). It was first suggested by Swerdlow in 2004 (Swerdlow and Khan, 2004) and according to this hypothesis; mitochondrial dysfunction is considered to be an early as well as a primary event in the pathophysiological cascade of AD (Swerdlow et al., 2010). Also it was proposed that genetic hereditary regulate mitochondrial functions and membrane strength, which changes with age and hence results in the development of AD related symptoms (Swerdlow and Khan, 2004; Swerdlow et al., 2010). This hypothesis assumed that autosomal dominant and sporadic AD are not etiologically same (Swerdlow et al., 2014). Mitochondrial dysfunction presents a connecting link between sporadic AD and autosomal dominant. In autosomal dominant AD, excessive Aβ accumulation slowly impairs mitochondrial functions which further initiate other AD related pathologies such as oxidative stress or neuroinflammation. In sporadic AD, age related occurrence of mitochondrial dysfunctions causes a variety of pathologies including oxidative stress and apoptosis (Hauptmann et al., 2006; Lin and Beal, 2006; Moreira et al., 2010; Witte et al., 2010).
Mechanism of Mitochondrial Dysfunction in AD
As reported earlier, in AD there is abnormal APP metabolism and an excessive Aβ accumulation (Kumar and Singh, 2015). It has been reported that Aβ peptides are present in the neuronal cells as well as in mitochondria (Swerdlow and Khan, 2004). When Aβ peptides accumulates in mitochondria it causes inhibition of mitochondrial respiratory enzyme complex-II and IV, causes decreased production of ATP and an increased production of ROS mitochondrial dysfunction in AD (Figure 1, Rhein et al., 2009; Swerdlow et al., 2010). Accumulation of Aβ peptides also known to reduce activity of enzyme of the tricarboxylic acid (TCA) cycle, α-ketoglutarate dehydrogenase (αKGD), pyruvate dehydrogenase and isocitrate dehydrogenase (Huang et al., 2003; Bubber et al., 2005). It is reported that, Aβ peptides interact with the Aβ binding site known as Aβ binding alcohol dehydrogenase (ABAD) which are present in the mitochondrial membranes and causes mitochondrial dysfunction (Lustbader et al., 2004), abnormal mitochondrial trafficking and decreased mitochondrial movement finally leads to synaptic degeneration (Calkins and Reddy, 2011). Further, Aβ peptide accumulation leads to dysfunctioning of mitochondrial Ca2+ channels, opening of mPTP and enhancement of cytochrome C (CytC) release (Calkins et al., 2011). Moreover, Aβ peptide accumulation inhibits protein import inside the mitochondria, which leads to mutation of mitochondrial DNA (mtDNA) and its damage (Lakatos et al., 2010). Mutations in APP also cause alterations of Ca2+ homeostasis leading to apoptosis (Khan et al., 2000). Accumulation of Aβ peptide and hyperphosphorylation of tau causes increased DRP-1nitrosylation which in turn causes abrupt mitochondrial fission and neurodegeneration (Manczak et al., 2011). Accumulation of soluble Aβ peptide and mutant APP impairs mitochondrial fusion and fission functions, abnormal mitochondrial movement, morphology and degradation of mitochondria (Manczak et al., 2011). Besides, it has also been reported that Aβ peptide accumulation causes abnormal expression of mitochondrial fission (Fis1) and fusion (mfn1/2 and OPA1) proteins which are involved in mitochondrial fission and fusion machinery (Manczak et al., 2011). These impaired dynamics causes decreased clearance of defective mitochondria which further enhanced neurodegeneration (Manczak et al., 2011). It has been studied that Aβ peptides induced hyperphosphorylation of tau causes inhibition of fission protein DRP1 which leads to abnormal mitochondrial elongation (Wang et al., 2008). In another study, Aβ peptides decrease proliferator-activated receptor-γ coactivator-1 α (PGC-1α) expressions, which leads to decreased mitochondrial biogenesis, mitochondrial DNA content (mtDNA) and increased neurodegeneration (McGill and Beal, 2006). Activation of PGC-1α causes increased non-amyloidogenic processing of APP, reduction in Aβ levels leading to increased survival of neuronal cells (McGill and Beal, 2006).
FIGURE 1
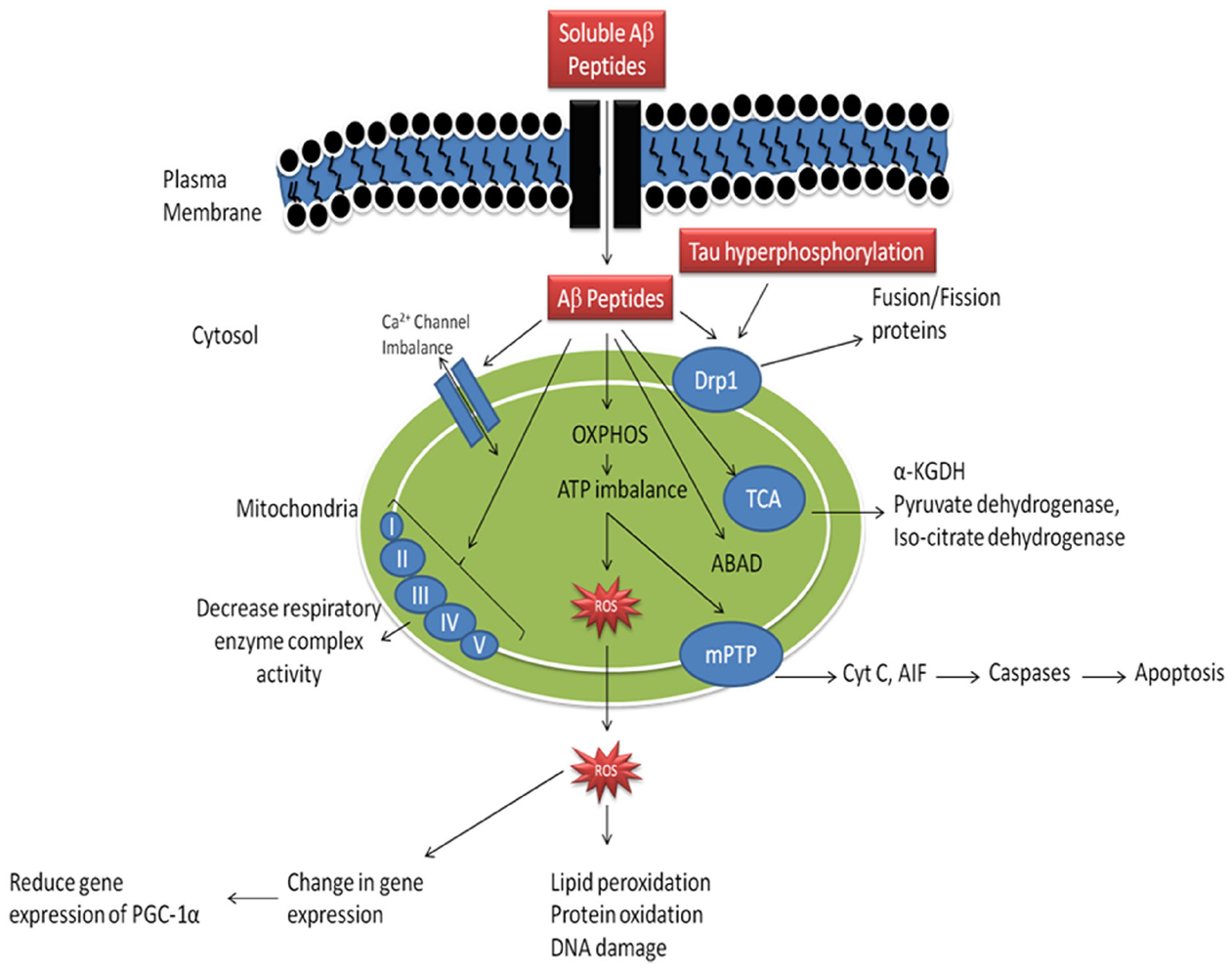
Mechanism of mitochondrial dysfunction in Alzheimer’s disease. (Aβ-amyloid β, OXPHOS- oxidative phosphorylation, ROS- reative oxygen species, mPTP- mitochondrial permeability transition pore, Cyt C- cytochrome C, Drp1- dynamin—related protein-1, ABAD- amyloid β binding alcohol dehydrogenase, α-KGDH- α-ketoglutarate dehydrogenase complex, PGC-1α- peroxisome proliferator activated receptor-γ-coactivator-1-α). Adopted and modified from Reddy and Beal (2008).
Mitochondria in AD
Mitochondria are the major energy producing cell organelles and also known as the power house of the cell (Du and Yan, 2010). Studies have expanded the role of the mitochondrial genome, consisting of maternally-inherited multiple copies of semi-autonomous genome mtDNA along with thousands of nuclear DNA (nDNA)-encoded genes (Castellani et al., 2002; Selfridge et al., 2013). The mtDNA encodes major components of mitochondrial OXPHOS which includes specifically ETC (Castellani et al., 2002). On the other side, nDNA encodes the genes which are required for the assembly of mitochondria and its related structural elements (Castellani et al., 2002; Selfridge et al., 2013). Therefore, mitochondrial respiratory rate, oxidative stress and apoptosis, which constitute important byproduct of OXPHOS (Selfridge et al., 2013).
In 1995, Jane C. Chisholm and his team proposed “mitochondrial bottle neck hypothesis.” According to this hypothesis, mitochondria represent a unique target for therapeutic interventions for all forms of AD (Davis et al., 1995). Besides, mitochondrial functions are essential as well as their dysfunction are sufficient enough to cause neurodegenerative disorder, including AD and thus providing mechanistic bottleneck in neurodegenerative diseases. In AD, multiple heterogeneous natures cause the same pathological phenotype in mitochondria. This hypothesis also proposed that mitochondrial dysfunction is a common pathway in different neurodegenerative disorders (Davis et al., 1995).
It has been reported that several mitochondrial respiratory enzymes (pyruvate dehydrogenase complex, ketoglutarate dehydrogenase complex, and cytochrome oxidase) are altered in AD (Moreira et al., 2010). According to the mitochondrial cascade hypothesis, defect in cytochrome oxidase is considered to be central. Cytochrome oxidase is the major enzymes of the terminal end of mitochondrial ETC. It accepts electron from cytochrome c, which in turn receive it from the upstream part of ETC. Cytochrome oxidase transfer electron to oxygen to form H2O rather than ROS. And this enzyme is the common site for all cellular oxygen consumption (Swerdlow et al., 2014).
It is well documented that reduced cytochrome oxidase activity is correlated with an increased defects in mtDNA in AD (Swerdlow et al., 2014). The brain, due to the presence of high lipid content, high oxygen consumption and low antioxidant defenses, remains the most vulnerable organ to oxidative stress. It is also well documented that free radical generation by mitochondrial dysfunction affect both in neurons and astrocytes during AD. There is also generation of free radicals () produced in mitochondrial ETC complexes I and III and enzymes of the TCA (α-ketoglutarate dehydrogenase) in AD. From inner mitochondrial membranes hydrogen peroxide (H2O2) and is released to the outer side (cytoplasm) which finally causes oxidation of cytoplasmic proteins (Figure 2; Ylikallio and Suomalainen, 2012). Figure 2 has been adopted and the modified from Reddy and Beal (2008).
FIGURE 2
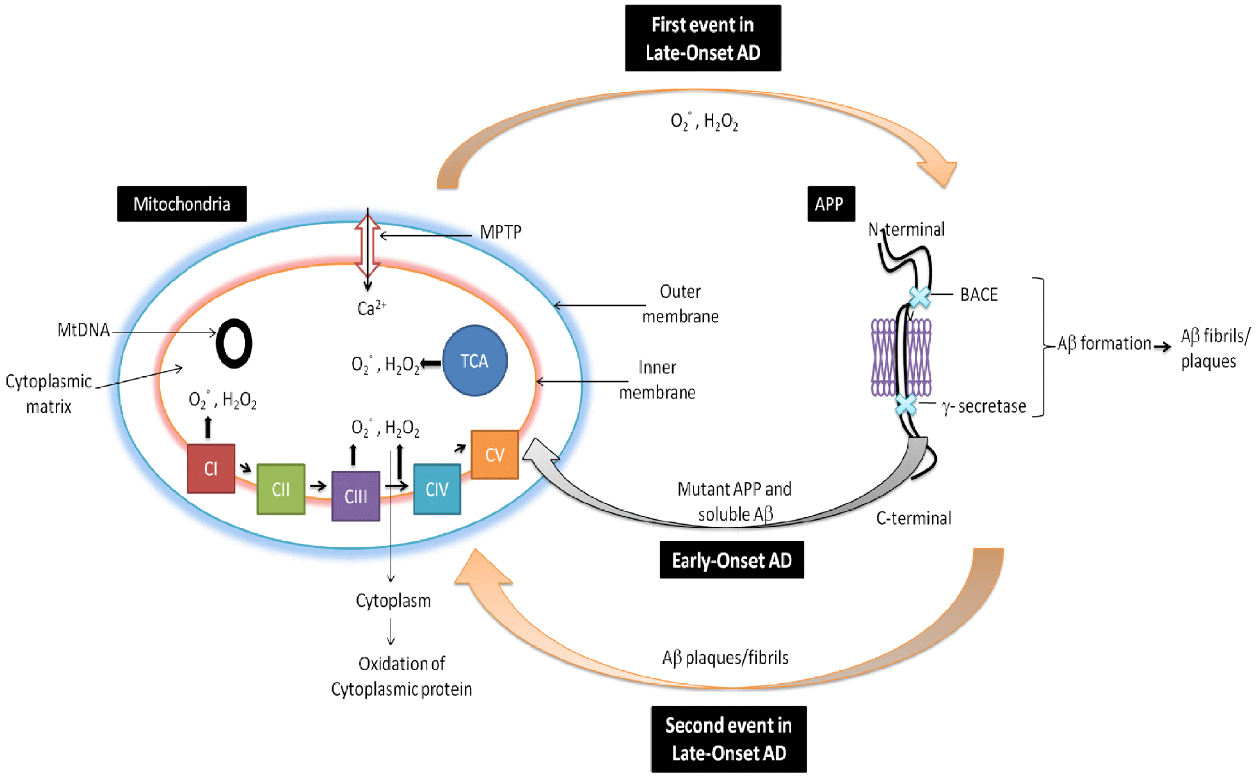
Role of mitochondria in AD. In early-onset AD, it is hypothesized that mitochondria leads to the generation of free radicals (, H2O2), which in turn decrease cytochrome oxidase activity and inhibit cellular ATP generation. Further in late-onset AD, free generation actives BACE mediated cleavage of Aβ. Again Aβ enter into the mitochondria and induces free radicals that leads to disruptions of ETC, decrease in cytochrome oxidase activity and the inhibition of ATP which finally leads to neuronal damage and cognitive decline in AD.
Various studies have been performed to understand the relationship between Aβ and mitochondria (Rhein et al., 2009; Ren et al., 2011). Study in transgenic mice (tgAPP/PS1) has shown that the soluble form of Aβ peptide causes a reduced mitochondrial membrane potential (MMP) and ATP levels (Ren et al., 2011). Another study on triple transgenic (APP/TAU/PS2) AD mice showed that decreased mitochondrial protein levels include reduction of the MMP and ATP synthesis (Rhein et al., 2009). In vivo study showed that intrahippocampal injection of Aβ peptides damaged mitochondria, causes a decreased Ca2+ dependent ATPase activity, MMP and an increased Ca2+ levels (Calkins et al., 2011). It has also been reported the correlation between Aβ and tau, it is described that both Aβ and tau work synergistically and leads to the impairment of oxidative phosphorylation (Rhein et al., 2009). Due to the accumulation of Aβ peptides in the mitochondrial import channels (TIM23 and TOM40) and mutant APP in AD brain causes mitochondrial dysfunction (Devi et al., 2006). A study on transgenic mice showed that APP (full length segment) binds to the mitochondria in neuronal cells, causes impaired energy metabolism and mitochondrial dysfunction (Anandatheerthavarada et al., 2003). It has been observed that during AD there is decreased genetic expression of oxidative phosphorylation and energy consumption (Chandrasekaran et al., 1996). A study on triple transgenic mouse (APP/PS1/Tau) showed dysregulated oxidative phosphorylation, glycolysis, TCA cycle, pyruvate metabolism, and mitochondrial protein synthetic pathways by the help of mitochondrial proteome analysis (Chou et al., 2011). Similarly, a study in Aβ transgenic mouse model of AD showed an impaired mitochondrial functions, ROS production, MMP and cytochrome c oxidase (COX) activity (Dragicevic et al., 2010).
Mitochondrial Therapeutics in Neurodegenerative Diseases
As previously discussed, alteration in mitochondrial bioenergetic defects, mitochondrial dynamics and mitochondrial trafficking and oxidative stress ROS mediated mitochondria damage plays a key role in AD, PD, HD, and ALS pathogenesis (Reddy and Beal, 2008; Chaturvedi and Beal, 2013). So, strategies that target mitochondrial dysfunction or the agents which enhance mitochondrial bioenergetics and reversed oxidative stress are potentially needed as therapeutics in AD, PD, HD, and ALS. Here we discussed the therapeutic potential of various bioenergetics and antioxidants (Figure 3) which are either have been used or being in different phases of clinical trials.
FIGURE 3
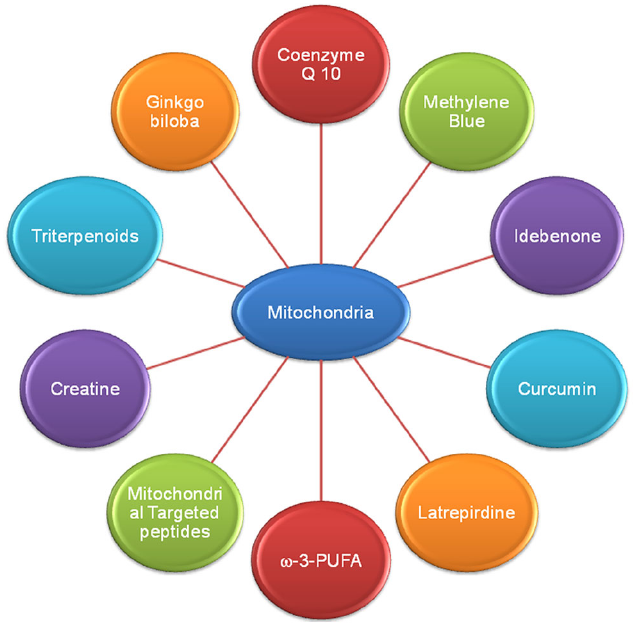
Various drugs and natural compounds with common potential therapeutic target mitochondria in Alzheimer’s disease.
Coenzyme Q 10
Mechanism of Action
It is an important cofactor of the ETC, known as ubiquinone. It functions as an electron acceptor for complex I and II and also act as an antioxidant in mitochondria and its membranes (Hargreaves, 2014). It is present in the inner mitochondrial membrane as a cofactor for three mitochondrial complexes (complexes I, II, and III) which play major role in OXPHOS (Hargreaves, 2014). According to Muller, one of the major functions of CoQ10, non-protein component of ETC, is to move and transfer electrons between flavoproteins and cytochromes (Müller et al., 2003). Further, it has been proposed that electrons of ETC must first be interact with CoQ10, a central rate-limiting step, hence considered as major component of ETC (Cleren et al., 2008). The major role of CoQ10 is ATP production. It also possesses antiapoptotic activity by inhibiting activation of the mitochondrial permeability independent of its free radical scavenging property (Matthews et al., 1998). It also acts as a cofactor as well as obligatory cofactor of mitochondrial uncoupling proteins whose activation reduces free radical generation from mitochondrial (Beal and Matthews, 1997; Kašparová et al., 2006).
Preclinical Studies
In vitro and in vivo analysis have suggested the neuroprotective potentials of CoQ10 in AD (Ishrat et al., 2006; Choi et al., 2012). It is a lipophilic anti-oxidant compound that improves cognitive functions, facilitates ATP synthesis and up-regulates mitochondrial function (Turunen et al., 2004). It is well documented that CoQ10 supplementation significantly increases endogenous brain CoQ10 content and provides protection from free radicals mediated oxidative damage biomolecules (Hargreaves, 2014). It also acts as cofactor of dehydrogenase in the ATP production and electrons and proton transport. Previous study on primary neuron culture suggests that CoQ10 significantly inhibit chemically (like paraquat and rotenone) induced mitochondrial dysfunction, maintained MMP, inhibit the mitochondrial ROS generation and neurodegeneration (Chaturvedi and Beal, 2008). CoQ10 also protects cultured cerebellar neurons against excitotoxin induced degeneration. A study on ICV-STZ infused rat showed that CoQ10 supplementation significantly restored choline acetyl transferase activity (Ishrat et al., 2006).
Clinical Studies
It is further evident from clinical studies that high doses of CoQ10 are beneficial for the treatment of neurodegenerative diseases (Crane, 2007; Galpern and Cudkowicz, 2007; Lenaz et al., 2007). Clinical study for the evaluation of safety and tolerability of CoQ10 and its combination treatment of mild to moderate AD patients (NCT00117403) is currently in ongoing phase (Müller et al., 2003). In this study, seventy five subjects received CoQ10 2400 mg daily; vitamin E 2400 IU, vitamin C 600 mg and α-lipoic acid 1800 mg; or placebo for a period of 4 months. Various parameters like safety, tolerability, CSF biomarkers of oxidative stress as well as CSF concentration of Aβ (1–40) and (1–42) were evaluated (Müller et al., 2003). Another study, phase-I pilot trial, an open label trial in 15 PD patients, was conducted for the safety and CoQ10 tolerability (Shults et al., 1998). In this study, CoQ10 at different doses 400, 600, and 800 mg/day for a period of 1 month showed significant safety and tolerability profiles along with dose-dependent increase in plasma CoQ10 levels (Shults et al., 1998). Another clinical trial (open label placebo controlled) in ALS patients showed that COQ10 (3000 mg/day) is safe and well tolerated (Ferrante et al., 2005). Recent clinical trial (multicenter, two-stage) in Phase II on CoQ10 showed no significant results between CoQ10 and placebo (Kaufmann et al., 2009).
Creatine
Mechanism of Action
Creatine, a nitrogenous guanidino compound, provides energy to the nerve and muscle cells due to its high energy requirement (Beal, 2011). It is a potent antioxidant and acts as an effective mPTP opening inhibitor and mitochondrial iron accumulation (Beal, 2011). It is present in the form of free creatine and phosphocreatine (PCr) in the human body. It is further gets transformed into PCr by the help of creatine kinase (CK) in skeletal muscle and brain. CK maintains cellular homeostasis by creating a pool of PCr for ATP generation (Adhihetty and Beal, 2008).
Preclinical Studies
It exert neuroprotective potential in different neurodegenerative disorders like AD, PD, HD, and ALS (Adhihetty and Beal, 2008; Beal, 2011). In vitro studies on neuronal cells have shown that it protects against neurotoxicity induced by 3-nitropropionic acid (3-NP), 1-methyl-4-phenylpyridinium (MPP+), and 6-hydroxydopamine (6-OHDA), glucose and serum deprivation (Chaturvedi and Beal, 2008). Creatine administration also known to inhibit the degeneration of dopaminergic neurons and reduced depletion of dopamine levels in PD (Matthews et al., 1999). Besides, creatine supplementation has been proved to be effective and protective against neuronal death caused by NMDA, malonate, Aβ and ibotenic acid induced neurotoxic injuries (Chaturvedi and Beal, 2008). Study on transgenic G93A ALS mice showed that creatine treatment protects motor neurons, brain atrophy and reduced mitochondrial dysfunction (Klivenyi et al., 1999). Besides, creatine in combination with the other bioenergetics compound CoQ10 produces a neuroprotective effects in neurodegenerative diseases (Yang et al., 2009b). Taken together, these studies may suggest creatine, a promising therapeutic agent for various neurodegenerative disorders including AD.
Clinical Studies
A clinical study (randomized, double-blind) of creatine in patients with mitochondrial cytopathies, showed constructive effects of creatine (Rodriguez et al., 2007). Another pilot study using creatine treatment showed advantageous effects on the mood swings of PD patients (Bender et al., 2006). In this study creatine (4 g/day) was safe and well tolerated in aged PD patients (Bender et al., 2008). A clinical study (randomized, double-blind) in phase II, on HD patients, showed that creatine (8 g/day) for 16 weeks was found to be safe and well tolerated, and decreased serum 8-hydroxy-2-deoxyguanosine which is the neuropathological marker for oxidative stress (Hersch et al., 2006). Taken together, these clinical studies may suggest a promising, beneficial and neuroprotective role of creatine in neurodegenerative diseases.
Idebenone
Mechanism of Action
It is an analog of CoQ10 consists of short chains of isoprene units, also known as ubiquinone. It has been reported that Idebenone crosses blood brain barrier easily and is well tolerated in humans (Senin et al., 1992). It is also known to possess good anti-oxidant properties (Senin et al., 1992). It belongs to the quinone family and is structurally similar to CoQ10 (Senin et al., 1992; Weyer et al., 1997; Gutzmann and Hadler, 1998).
Preclinical Studies
It has been reported that Idebenone produced neuroprotection against Aβ induced neurotoxicity both in vitro and in vivo (Weyer et al., 1997).
Clinical Studies
Clinical study of Idebenone showed its neuroprotective effects in AD patients on Alzheimer’s Disease Assessment Scale (ADAS) score (Gutzmann and Hadler, 1998). It has been proposed that Idebenone (360 mg/day) treatment was safe and well tolerated in AD patients (Gutzmann et al., 2002).
Latrepirdine
Mechanism of Action
Latrepirdine previously known as Dimebon (or Dimebolin), is a non-selective antihistamine (Sachdeva and Burns, 2011). Latrepirdine inhibits weakly acetylcholinesterase and butyrylcholinesterase (Bachurin et al., 2001). Latrepirdine also has known as inhibitor of NMDA receptor and voltage-gated calcium channels. It also possesses the neuroprotective effect mainly by maintaining mitochondrial structure and function (Sachdeva and Burns, 2011). Latrepirdine under both stress and non-stress conditions enhances mitochondrial function (Bachurin et al., 2001; Sachdeva and Burns, 2011). Latrepirdine has also been reported to inhibit Aβ-induced activation of the mPTP which can lead to apoptosis and hence protect neuronal mitochondria from Aβ-induced toxicity (Bachurin et al., 2001). Latrepirdine other functions include an increase in MMP and ATP production (Bachurin et al., 2001; Sachdeva and Burns, 2011).
Preclinical Studies
Latrepirdine has been shown to be effective in cognition in animal studies on young/adult mice or rats (Kieburtz et al., 2010; Vignisse et al., 2011). Latrepirdine also known to improve impaired mitochondrial function in AD (Reddy, 2009; Leuner et al., 2012). Latrepirdine (25 μmol/l) has been reported to be protective against the Aβ induced mitochondrial dysfunctions (Reddy, 2009; Leuner et al., 2012). A study on human SH-SY5Y neuroblastoma cells and primary rat cortical neurons showed that latrepirdine (0.1–10 nmol/l) improve mitochondrial function, such as MMP, ATP production and apoptosis (Zhang et al., 2009).
Clinical Studies
Phase 2 randomized controlled trial showed that, latrepirdine was well tolerated, safe and significantly enhanced the clinical outcomes in patients suffering from mild-to-moderate AD (Reddy and Reddy, 2011).
Triterpenoids
Mechanism of Action
Triterpenoids are derivatives of oleanolic acid and known to inhibit oxidative stress. They also possess anti-inflammatory properties via inhibition of inflammatory processes; by activation of antioxidant response element (ARE)-Nrf2-Keap1 signaling pathway (Liby et al., 2005). Activation of this pathway leads to the dissociation of Nrf2 from Keap1, translocation to the nucleus and then binding to the ARE promoter sequences which further causes induction of antioxidant and anti-inflammatory genes (Yates et al., 2007). It has recently reported that synthetic triterpenoids (CDDO) causes transcriptional activation of Nrf2, NQO-1, HO-1, glutathione transferase and other cytoprotective enzymes (Liby et al., 2005; Yates et al., 2007). CDDO-methyl amide (2-cyano-N-methyl-3,12-dioxooleana-1,9 (11)-dien-28 amide; CDDO-MA), synthetic triterpenoid, has been discovered as 200,000 times more potent inducer of NQO-1 or suppressor of iNOS than naturally occurring oleanolic acid (Yang et al., 2009a). Further study on same compound CDDO-MA showed that it is a potent and selective activator of the Nrf2/ARE pathway which is neuroprotective in nature (Kaidery et al., 2013).
Preclinical Studies
Experimental studies on 3-NP rat model and 1-methyl-4 phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model showed that synthetic triterpenoid CDDO-MA, exerts significant neuroprotective effects by potently activating Nrf2/ ARE signaling pathway (Yang et al., 2009c; Kaidery et al., 2013). CDDO-MA has also been reported to inhibit ROS generation, MPTP induced neurodegeneration and dopamine depletion and 3-NP induced striatal lesions (Yang et al., 2009c). It has also been observed that triterpenoids improve the behavioral parameters and survival in transgenic mouse models of variety of neurodegenerative diseases including AD, HD and ALS (Neymotin et al., 2011). These studies may suggest that targeting neuroprotective pathways (Nrf2/ARE) through synthetic triterpenoids (CDDO-MA) could be used as a better therapeutic approach in the treatment and management of neurodegenerative disorders (Dumont et al., 2009; Stack et al., 2010).
MitoQ
Mechanism of Action
MitoQ is formed by coenzyme Q or ubiquinone which is linked to triphosphonium ions via covalent bonding to form mitoquinone (Murphy and Smith, 2007). It is the most widely used antioxidant to target mitochondria. MitoQ demonstrated neuroprotection due to its direct effect on scavenging peroxynitrite, and superoxide, and thus protect mitochondria against lipid peroxidation (Manczak et al., 2010; Calkins et al., 2011; Jin et al., 2014).
Preclinical Studies
Studies on both in vitro and in vivo models showed that it exerts neuroprotective effects in various experimental models (Murphy and Smith, 2007; Jin et al., 2014). It also inhibits mitochondrial fission protein and translocation of pro-apoptotic protein (Bax) in the mitochondria in cellular models of PD (Jin et al., 2014). Although its efficacy in neurodegenerative diseases conditions needs to be further explored and understood.
Clinical Studies
A clinical study (double blind), in 128 newly diagnosed untreated patients with PD, MitoQ for 12 months with two doses did not produce any significant improvement according to United Parkinson Disease Rating Scale and PD progression when compared with the placebo control (Snow et al., 2010).
MitoVitE
Mechanism of Action
MitoVitE is also known as Mito tocopherol. Structurally, it is triphenylphosphonium (TPP) conjugated to α-tocopherol moiety of vitamin E via two-carbon chain. It also protects mitochondria from oxidative stress via inhibition of lipid peroxidation (Smith et al., 1999).
Preclinical Studies
In vivo study, showed that intravenous injection of MitoVitE accumulated rapidly in the heart, brain, muscle, liver, and kidney tissues which are mostly affected by mitochondrial dysfunction and oxidative stress (Smith et al., 2003). An in vitro cellular model also demonstrated its efficacy against mitochondrial oxidative stress, reduction of peroxide mediated oxidative stress, peroxide-induced caspase activation and oxidative stress-induced cell death (Jauslin et al., 2003; Dhanasekaran et al., 2004; Hughes et al., 2005). But till date, its efficacy and therapeutic potential in PD patients has not been investigated.
MitoTEMPOL
Mechanism of Action
It is another TPP+ derivative which consists of stable piperidine nitroxide radical TEMPOL (4-hydroxy-2,2,6,6,-tetramethylpiperidine- 1-oxyl). The most important property is to accept an electron from hydroxylamine (potent radical scavenger). It also acts as a SOD mimetic, whose function is to converts superoxide molecules into water to detoxify ferrous iron into ferric iron.
Preclinical Studies
In vitro study has shown its beneficial effects against mitochondrial dysfunction and mitochondria mediated oxidative stress (Trnka et al., 2008). However, its neuroprotective potential in different neurological problem is yet to be explored and understood.
SS (Szeto-Schiller) Peptides
Mechanism of Action
They are mitochondrial targeted peptides; act as novel anti-oxidants, helpful in restoring mitochondrial functions (Szeto, 2008). A study in isolated mitochondria showed that these peptides decreases mitochondrial ROS generation; inhibit mitochondrial swelling and cytochrome c release (Szeto, 2008; Manczak et al., 2010; Calkins et al., 2011). Further, the modification in their structure via addition of a tyrosine or modified tyrosine moiety causes improvement in their free radical scavenging properties, and; apoptosis (Manczak et al., 2010; Calkins et al., 2011; Smith and Murphy, 2011). Besides, their related peptides (SS-31 and SS-20) have also been shown to restore mitochondrial functional properties by reducing inhibition of the mitochondrial ETC, apoptosis and oxidative stress (Smith and Murphy, 2011).
Preclinical Studies
A study showed that SS peptides decrease mitochondrial ROS generation, inhibit mitochondrial swelling, and reduce cytochrome c release from mitochondria in different experimental models (Szeto, 2008). These findings have suggested their therapeutic potential in neurodegenerative disorders including AD.
Methylene Blue
Mechanism of Action
Methylene blue (MB) is an FDA-approved drug used for the treatment of various diseases like malaria and some psychiatric disorder for more than 100 years (Naylor et al., 1988; de-Oliveira and Guimarães, 1999; de Oliveira et al., 2000; Oz et al., 2012). MB possess better pharmacokinetic profile, readily absorbed in blood and quickly distributed to various organs (Peter et al., 2000; Rengelshausen et al., 2004). Other functions of MB includes cognition enhancing properties and increase oxygen consumption efficacy in isolated mitochondria (Zhang et al., 2006). MB (1 mg/kg) has also reported to increase COX activity and thereby improving energy functions in AD brains (Gonzalez-Lima et al., 1997; Valla et al., 2001; Callaway et al., 2004; Hauptmann et al., 2009).
Preclinical Studies
It is now well known that complex I of ETC is the most susceptible to oxidative stress in AD (Leuner et al., 2007). A study on mice showed that MB (0.07 mg/kg) when administered in the eyes of mice (intravitreally) by the help of microinjector significantly reverses rotenone induced mitochondrial dysfunction (Complex I inhibitors; Zhang et al., 2006). Neuroprotective effect of MB (0.15–4.0 mg/kg b.w, i.p.) has been well demonstrated in animal models of cognitive dysfunction (Deiana et al., 2009). It has also been reported that disturbances in the ETC generally elevate ROS level, which in turn increases Aβ production, Aβ impairs mitochondrial function, and hence finally a vicious cycle is initiated (Leuner et al., 2012). MB (as a redox compound), prevents the reduction of molecular oxygen to superoxide (Leuner et al., 2007). Thus MB acts as both mitochondrial restorer as well as antioxidant.
Ginkgo biloba
Mechanism of Action
EGb761® is a standardized extracts of Ginkgo biloba, herbal drug, used for the improvement of cognitive dysfunction. The extract consists of 24% flavonoids and 6% terpenes (Bedir et al., 2002; Sastre et al., 2002; Abdel-Kader et al., 2007).
Preclinical Studies
Study on PC12 cells showed that EGb761® significantly improved MMP and restored ATP levels after sodium nitroprusside (a nitric oxide donor) induced mitochondrial damage (Abdel-Kader et al., 2007). Similar results have been observed in isolated brain cells and brain mitochondria after treatment with EGb761® (Eckert et al., 2005). In vitro studies on PC12 cells having mutant APP, showed that EGb761® (0.01 mg/ml) treatment leads to enhanced Aβ production and increase mitochondrial functions (Eckert et al., 2005; Abdel-Kader et al., 2007). Animal study involving EGb761® (100 mg/kg) treatment for 14 days significantly improved complexes I, IV, and V activities of the mitochondria and alleviated nitrosative stress (Abdel-Kader et al., 2007). Further, protective mechanism is due to the presence of terpene lactones that restore mitochondrial functional properties and acts as a free radical scavenger (Abdel-Kader et al., 2007). Recently EGb761® showed an improvement in neuroplasticity, oxidative stress, long-term potentiation, spine density, neurogenesis (Müller et al., 2012). Besides, EGb761® alters APP processing, by upregulating the activity of α-secretase in hippocampal region (Colciaghi et al., 2004).
Clinical Studies
Clinically Ginkgo biloba has been well studied in dementia due to mitochondrial restoring properties (Stoll et al., 1996; Tang et al., 2002; Abdel-Kader et al., 2007; Wang et al., 2010; Weinmann et al., 2010). However, its neuroprotective mechanism is still not clear.
Curcumin
Mechanism of Action
Also known as diferuloylmethane, yellow pigment derived from the rhizome part of the turmeric plant (Curcuma longa, Sikora et al., 2010). It is lipophilic phenolic diferuloylmethane and possesses varieties of pharmacological functions, including anti-inflammatory, antioxidative, anti-proliferative, cholesterol- lowering, and neuroprotective (Bengmark, 2006; Lapchak, 2011; Vauzour, 2012). Curcumin affects various pathways involved in AD like neuroprotective processing of APP, tau phosphorylation, neuroinflammation, or oxidative stress (Zhu et al., 2004).
Preclinical Studies
A preclinical study on PC12 neuronal cells, showed that curcumin (25 μmol/l) given for 2 h maintained redox potential and respiratory functions of mitochondria after hydroxynonenal (4-HNE) treatment (Raza et al., 2008). Another study on neurons of rat cortices treated with tert-butyl hydroperoxide (t-BHP) to induce oxidative stress showed that curcumin (2.5–20 μmol/l) improves MMP and cytochrome c release, inhibits the activation of caspase-3, and altered the expression of Bcl-2 (Zhu et al., 2004). A study on homocystein-induced rat aging model showed that curcumin (5, 15, or 45 mg/kg) treatment significantly decreased MDA and superoxide anion levels and finally improves learning and memory (Ataie et al., 2010). Another study in aged mice showed curcumin’s antioxidative effects with significant decrease in ROS level and protein carbonylation (Dkhar and Sharma, 2010). In another in vivo study, chronic administration of d-galactose causes cognitive dysfunction, oxidative stress, and impaired mitochondrial enzyme complex I, II, and III levels and curcumin treatment (15 and 30 mg/kg) for 6 weeks significantly improved cognition, oxidative stress, and restored mitochondrial enzyme complex activity as compared to control (Kumar et al., 2011). Curcumin (120 mg/kg) in a diabetic rat model, up- regulate mitochondrial complex activities and increase ATP level in the brain (Rastogi et al., 2008).
Clinical Studies
Curcumin, due to its poor bioavailability and poor water solubility, may have limited clinical trials (Belkacemi et al., 2011). So, for effective therapy, new delivery strategies may need to be developed.
Omega-3 Polyunsaturated Fatty Acids
Mechanism of Action
Omega-3 polyunsaturated fatty acids (ω-3 PUFAs) are groups of essential fatty acids. It functions as energy substrates and is a part of integral membrane components. Therefore, plays major roles in the management of neurological function (Cole et al., 2009). A recent study showed that ω-3 PUFAs exerts neuroprotective role in the cognitive dysfunction (Eckert et al., 2011). Brain fulfills the need of ω-3 PUFAs by the delivery through blood because of their limited synthesis (Cole et al., 2009). It has been reported that decreased levels of ω-3 PUFAs or fish consumption increases risk for age-related cognitive deficits such as AD (Cole et al., 2009).
Preclinical Studies
In vitro studies in HEK-APP cells showed beneficial effects of ω-3 PUFAs (20 μmol/l) as it significantly increased mitochondrial membrane properties and processing of non-amyloidogenic APP, which causes enhanced secretion of sAPPα, that in turn protect against mitochondrial dysfunction and apoptosis (Eckert et al., 2011). It has also been reported that supplementations with ω-3 PUFAs increase membrane phospholipid docosahexaenoic acid (DHA). ω-3 PUFAs also known to improve the functions of complexes I and IV of mitochondrial respiratory chain, mitochondrial respiration and lipid metabolism (Barceló-Coblijn et al., 2003; Stanley et al., 2012).
Current Trends in Management of Mitochondrial Related Diseases
Mitochondrial Replacement Therapy
It has been reported that each cell contains almost 1,000 to 100, 000 copies of mtDNA which via maternal inheritance transfers to the offspring (Swerdlow et al., 2014). Various techniques have been introduced like genetic screening of embryos, and have been reported to partially decrease the risk of mitochondrial diseases transmitted from mother to offspring being (Tachibana et al., 2009; Reinhardt et al., 2013). Recently a new technique is introduced, mitochondrial replacement therapy, in which mitochondria healthy in nature are taken from a donor, is under investigation. A limitation of this approach is the combination of the genetic material from three different people, which causes array of ethical, safety and medical expostulations (McNamee, 2015).
Mitochondrial Editing Technique
It has been reported first time the gene-editing technology, the technique used to prevent mitochondrial diseases of humans from being transferred from female their offspring. Researchers from Salk Institute for Biological Studies in La Jolla, CA, USA discover mitochondrial editing technique for the treatment of various neurodegenerative disorders. It is safe, simple and more ethical as compared to mitochondrial replacement therapy as it does not involve donor DNA. It is an alternative approach which involves editing the mutated DNA of mouse using enzymes called as restriction endonucleases and transcription activator-like effector nucleases (TALENs). According to the author of the Salk Institute for Biological Studies, “this technique is based on a single injection of mRNA into a mother’s oocytes or early embryos and therefore could be easily implemented in IVF (in vitro fertilization) clinics throughout the world.” As mutation in mitochondrial DNA is involved in a variety of neurodegenerative disorders, cancer and aging, this technology may have broader potential therapeutic significance for the prevention of transmission of disease-causing mutations from mother to future generations, said by one of the author Belmonte (McNamee, 2015).
Conclusion
As in neurodegenerative diseases the two pathophysiological hallmarks mitochondrial dysfunction and oxidative stress were involved. In AD, apart from theses two other hallmarks are impaired energy metabolism, excessive ROS generation, impaired Ca2+ signaling, and increased mtDNA mutations and disturbance in OXPHOS. Mitochondrial dysfunction plays critical role in AD and can be considered as an important target for the treatment of clinical symptoms of AD. Previous evidences have shown the potential efficacy of various bioenergetics and antioxidants having in the treatment of AD, for example coenzyme Q10, carnitine, α-lipoic acid, Idebenone, mito-targeted compounds like Mito Q, Mito Vit E and Mito TEMPOL, Ginkgo biloba, curcumin and omega-3 polyunsaturated fatty acids. Preclinical data of the above mentioned drugs indicated beneficial effects, whereas most of the clinical trials did not. This might be because mitochondrial dysfunction represents an early event in AD progression and pharmacological interventions might came in the later event in AD. Targeting abnormal mitochondrial dynamics in AD may represent a novel therapeutic strategy which leads to the development of new mitochondrial targeted bioenergetics and antioxidants. Mitochondrial dysfunction and oxidative stress are also useful in defining the complex pathophysiology of this disabling disease. Still, more studies are urgently required to focus on the therapeutic interventions before the disease progresses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Abdel-KaderR.HauptmannS.KeilU.ScherpingI.LeunerK.EckertA.et al (2007). Stabilization of mitochondrial function by Ginkgo biloba extract (EGb 761). Pharmacol. Res.56, 493–502. 10.1016/j.phrs.2007.09.011
2
AdhihettyP. J.BealM. F. (2008). Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. Neuromolecular Med.10, 275–290. 10.1007/s12017-008-8053-y
3
Alzheimer’sA. (2015). 2015 Alzheimer’s disease facts and figures. Alzheimer’s Dement.11, 332–384. 10.1016/j.jalz.2015.02.003
4
AnandatheerthavaradaH. K.BiswasG.RobinM.-A.AvadhaniN. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol.161, 41–54. 10.1083/jcb.200207030
5
AtaieA.SabetkasaeiM.HaghparastA.MoghaddamA. H.KazeminejadB. (2010). Neuroprotective effects of the polyphenolic antioxidant agent, Curcumin, against homocysteine-induced cognitive impairment and oxidative stress in the rat. Pharmacol. Biochem. Behav.96, 378–385. 10.1016/j.pbb.2010.06.009
6
BachurinS.BukatinaE.LermontovaN.TkachenkoS.AfanasievA.GrigorievV.et al (2001). Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann. N. Y. Acad. Sci.939, 425–435. 10.1111/j.1749-6632.2001.tb03654.x
7
Barceló-CoblijnG.KitajkaK.PuskásL. G.HogyesE.ZvaraA.HacklerL.et al (2003). Gene expression and molecular composition of phospholipids in rat brain in relation to dietary n- 6 to n- 3 fatty acid ratio. Biochim. Biophys. Acta1632, 72–79. 10.1016/S1388-1981(03)00064-7
8
BealM. F. (2011). Neuroprotective effects of creatine. Amino Acids40, 1305–1313. 10.1007/s00726-011-0851-0
9
BealM. F.MatthewsR. T. (1997). Coenzyme Q 10 in the central nervous system and its potential usefulness in the treatment of neurodegenerative diseases. Mol. Aspects Med.18, 169–179. 10.1016/S0098-2997(97)00024-1
10
BedirE.TatliI. I.KhanR. A.ZhaoJ.TakamatsuS.WalkerL. A.et al (2002). Biologically active secondary metabolites from Ginkgo biloba. J. Agric. Food Chem.50, 3150–3155. 10.1021/jf011682s
11
BelkacemiA.DogguiS.DaoL.RamassamyC. (2011). Challenges associated with curcumin therapy in Alzheimer disease. Expert Rev. Mol. Med.13, e34. 10.1017/S1462399411002055
12
BenderA.BeckersJ.SchneiderI.HölterS.HaackT.RuthsatzT.et al (2008). Creatine improves health and survival of mice. Neurobiol. Aging29, 1404–1411. 10.1016/j.neurobiolaging.2007.03.001
13
BenderA.KochW.ElstnerM.SchombacherY.BenderJ.MoeschlM.et al (2006). Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial. Neurology67, 1262–1264. 10.1212/01.wnl.0000238518.34389.12
14
BengmarkS. (2006). Impact of nutrition on ageing and disease. Curr. Opin. Clin. Nutr. Metab. Care9, 2–7. 10.1097/01.mco.0000171129.29278.26
15
BubberP.HaroutunianV.FischG.BlassJ. P.GibsonG. E. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol.57, 695–703. 10.1002/ana.20474
16
CalkinsM. J.ManczakM.MaoP.ShirendebU.ReddyP. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet.20, 4515–4529. 10.1093/hmg/ddr381
17
CalkinsM. J.ReddyP. H. (2011). Amyloid β impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta1812, 507–513. 10.1016/j.bbadis.2011.01.007
18
CallawayN. L.RihaP. D.BrucheyA. K.MunshiZ.Gonzalez-LimaF. (2004). Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacol. Biochem. Behav.77, 175–181. 10.1016/j.pbb.2003.10.007
19
CastellaniR.HiraiK.AlievG.DrewK. L.NunomuraA.TakedaA.et al (2002). Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res.70, 357–360. 10.1002/jnr.10389
20
ChandrasekaranK.HatanpääK.BradyD. R.RapoportS. I. (1996). Evidence for physiological down-regulation of brain oxidative phosphorylation in Alzheimer’s disease. Exp. Neurol.142, 80–88. 10.1006/exnr.1996.0180
21
ChaturvediR. K.BealM. F. (2008). Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci.1147, 395–412. 10.1196/annals.1427.027
22
ChaturvediR. K.BealM. F. (2013). Mitochondrial diseases of the brain. Free Radic. Biol. Med.63, 1–29. 10.1016/j.freeradbiomed.2013.03.018
23
ChoiH.ParkH.-H.KohS.-H.ChoiN.-Y.YuH.-J.ParkJ.et al (2012). Coenzyme Q10 protects against amyloid β-induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology33, 85–90. 10.1016/j.neuro.2011.12.005
24
ChouJ. L.ShenoyD. V.ThomasN.ChoudharyP. K.LaferlaF. M.GoodmanS. R.et al (2011). Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J. Proteom.74, 466–479. 10.1016/j.jprot.2010.12.012
25
ClerenC.YangL.LorenzoB.CalingasanN. Y.SchomerA.SireciA.et al (2008). Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem.104, 1613–1621. 10.1111/j.1471-4159.2007.05097.x
26
ColciaghiF.BorroniB.ZimmermannM.BelloneC.LonghiA.PadovaniA.et al (2004). Amyloid precursor protein metabolism is regulated toward α-secretase pathway by Ginkgo biloba extracts. Neurobiol. Dis.16, 454–460. 10.1016/j.nbd.2004.03.011
27
ColeG. M.MaQ.-L.FrautschyS. A. (2009). Omega-3 fatty acids and dementia. Prostaglandins Leukot. Essent. Fatty Acids81, 213–221. 10.1016/j.plefa.2009.05.015
28
CraneF. L. (2007). Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion7, S2–S7. 10.1016/j.mito.2007.02.011
29
DavisJ. N.HunnicuttE. J.ChisholmJ. C. (1995). A mitochondrial bottleneck hypothesis of Alzheimer’s disease. Mol. Med. Today1, 240–247. 10.1016/S1357-4310(95)91532-X
30
de-OliveiraR.GuimarãesF. (1999). Anxiolytic effect of methylene blue microinjected into the dorsal periaqueductal gray matter. Braz. J. Med. Biol. Res.32, 1529–1532. 10.1590/S0100-879X1999001200012
31
de OliveiraR. W.Del BelE. A.GuimarãesF. S. (2000). Behavioral and c-fos expression changes induced by nitric oxide donors microinjected into the dorsal periaqueductal gray. Brain Res. Bull.51, 457–464. 10.1016/S0361-9230(99)00248-8
32
DeianaS.HarringtonC. R.WischikC. M.RiedelG. (2009). Methylthioninium chloride reverses cognitive deficits induced by scopolamine: comparison with rivastigmine. Psychopharmacology202, 53–65. 10.1007/s00213-008-1394-2
33
DeviL.PrabhuB. M.GalatiD. F.AvadhaniN. G.AnandatheerthavaradaH. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci.26, 9057–9068. 10.1523/JNEUROSCI.1469-06.2006
34
DhanasekaranA.KotamrajuS.KalivendiS. V.MatsunagaT.ShangT.KeszlerA.et al (2004). Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J. Biol. Chem.279, 37575–37587. 10.1074/jbc.M404003200
35
DkharP.SharmaR. (2010). Effect of dimethylsulphoxide and curcumin on protein carbonyls and reactive oxygen species of cerebral hemispheres of mice as a function of age. Int. J. Dev. Neurosci.28, 351–357. 10.1016/j.ijdevneu.2010.04.005
36
DragicevicN.MamcarzM.ZhuY.BuzzeoR.TanJ.ArendashG. W.et al (2010). Mitochondrial amyloid-β levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimer’s Dis.20, S535–S550. 10.3233/JAD-2010-100342
37
DuH.YanS. S. (2010). Mitochondrial medicine for neurodegenerative diseases. Int. J. Biochem. Cell Biol.42, 560–572. 10.1016/j.biocel.2010.01.004
38
DumontM.WilleE.CalingasanN. Y.TampelliniD.WilliamsC.GourasG. K.et al (2009). Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem.109, 502–512. 10.1111/j.1471-4159.2009.05970.x
39
EckertA.KeilU.ScherpingI.HauptmannS.MüllerW. E. (2005). Stabilization of mitochondrial membrane potential and improvement of neuronal energy metabolism by Ginkgo biloba extract EGb 761. Ann. N. Y. Acad. Sci.1056, 474–485. 10.1196/annals.1352.023
40
EckertG. P.ChangS.EckmannJ.CopanakiE.HaglS.HenerU.et al (2011). Liposome-incorporated DHA increases neuronal survival by enhancing non-amyloidogenic APP processing. Biochim. Biophys. Acta1808, 236–243. 10.1016/j.bbamem.2010.10.014
41
EckertG. P.MüllerW. E. (2014). Mitochondrial dysfunction: cause and consequence of Alzheimer’s. Mitochondrion Aging Dis.127, 183–210.
42
FerranteK.ShefnerJ.ZhangH.BetenskyR.O’brienM.YuH.et al (2005). Tolerance of high-dose (3,000 mg/day) coenzyme Q10 in ALS. Neurology65, 1834–1836. 10.1212/01.wnl.0000187070.35365.d7
43
GalpernW. R.CudkowiczM. E. (2007). Coenzyme Q treatment of neurodegenerative diseases of aging. Mitochondrion7, S146–S153. 10.1016/j.mito.2007.01.004
44
Gonzalez-LimaF.VallaJ.Matos-CollazoS. (1997). Quantitative cytochemistry of cytochrome oxidase and cellular morphometry of the human inferior colliculus in control and Alzheimer’s patients. Brain Res.752, 117–126. 10.1016/S0006-8993(96)01464-3
45
GutzmannH.HadlerD. (1998). Sustained Efficacy and Safety of Idebenone in the Treatment of Alzheimer’s Disease: Update on a 2-year Double-blind Multicentre Study. Springer. 10.1007/978-3-7091-7508-8_30
46
GutzmannH.KühlK.HadlerD.RappM. A. (2002). Safety and efficacy of idebenone versus tacrine in patients with Alzheimer’s disease: results of a randomized, double-blind, parallel-group multicenter study. Pharmacopsychiatry35, 12–18. 10.1055/s-2002-19833
47
HargreavesI. (2014). Coenzyme Q 10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Biol.49, 105–111. 10.1016/j.biocel.2014.01.020
48
HauptmannS.KeilU.ScherpingI.BonertA.EckertA.MüllerW. E. (2006). Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp. Gerontol.41, 668–673. 10.1016/j.exger.2006.03.012
49
HauptmannS.ScherpingI.DröseS.BrandtU.SchulzK.JendrachM.et al (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging30, 1574–1586. 10.1016/j.neurobiolaging.2007.12.005
50
HerschS.GevorkianS.MarderK.MoskowitzC.FeiginA.CoxM.et al (2006). Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2′ dG. Neurology66, 250–252. 10.1212/01.wnl.0000194318.74946.b6
51
HuangH.-M.ZhangH.XuH.GibsonG. E. (2003). Inhibition of the α-ketoglutarate dehydrogenase complex alters mitochondrial function and cellular calcium regulation. Biochim. Biophys. Acta1637, 119–126. 10.1016/S0925-4439(02)00222-3
52
HughesG.MurphyM. P.LedgerwoodE. C. (2005). Mitochondrial reactive oxygen species regulate the temporal activation of nuclear factor kappaB to modulate tumour necrosis factor-induced apoptosis: evidence from mitochondria-targeted antioxidants. Biochem. J.389, 83–89. 10.1042/BJ20050078
53
IqbalK.Grundke-IqbalI. (2010). Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimer’s Dementia6, 420–424. 10.1016/j.jalz.2010.04.006
54
IshratT.KhanM. B.HodaM. N.YousufS.AhmadM.AnsariM. A.et al (2006). Coenzyme Q10 modulates cognitive impairment against intracerebroventricular injection of streptozotocin in rats. Behav. Brain Res.171, 9–16. 10.1016/j.bbr.2006.03.009
55
JauslinM. L.MeierT.SmithR. A.MurphyM. P. (2003). Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J.17, 1972–1974. 10.1096/fj.03-0240fje
56
JinH.KanthasamyA.GhoshA.AnantharamV.KalyanaramanB.KanthasamyA. G. (2014). Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim. Biophys. Acta1842, 1282–1294. 10.1016/j.bbadis.2013.09.007
57
KaideryN. A.BanerjeeR.YangL.SmirnovaN. A.HushpulianD. M.LibyK. T.et al (2013). Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal.18, 139–157. 10.1089/ars.2011.4491
58
KašparováS.SumbalováZ.BystrickýP.KucharskáJ.LiptajT.MlynárikV.et al (2006). Effect of coenzyme Q 10 and vitamin E on brain energy metabolism in the animal model of Huntington’s disease. Neurochem. Int.48, 93–99. 10.1016/j.neuint.2005.09.002
59
KaufmannP.ThompsonJ. L.LevyG.BuchsbaumR.ShefnerJ.KrivickasL. S.et al (2009). Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol.66, 235–244. 10.1002/ana.21743
60
KhanS. M.CassarinoD. S.AbramovaN. N.KeeneyP. M.BorlandM. K.TrimmerP. A.et al (2000). Alzheimer’s disease cybrids replicate β-amyloid abnormalities through cell death pathways. Ann. Neurol.48, 148–155.
61
KieburtzK.McdermottM. P.VossT. S.Corey-BloomJ.DeuelL. M.DorseyE. R.et al (2010). A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch. Neurol.67, 154–160. 10.1001/archneurol.2009.334
62
KlivenyiP.FerranteR. J.MatthewsR. T.BogdanovM. B.KleinA. M.AndreassenO. A.et al (1999). Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat. Med.5, 347–350. 10.1038/6568
63
KumarA.DhullD. K.MishraP. S. (2015). Therapeutic potential of mGluR5 targeting in Alzheimer’s disease. Front. Neurosci.9:215. 10.3389/fnins.2015.00215
64
KumarA.PrakashA.DograS. (2011). Protective effect of curcumin (Curcuma longa) against D-galactose-induced senescence in mice. J. Asian. Nat. Prod. Res.13, 42–55. 10.1080/10286020.2010.544253
65
KumarA.SinghA. (2015). A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol. Rep.67, 195–203. 10.1016/j.pharep.2014.09.004
66
LakatosA.DerbenevaO.YounesD.KeatorD.BakkenT.LvovaM.et al (2010). Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol. Aging31, 1355–1363. 10.1016/j.neurobiolaging.2010.04.031
67
LapchakP. A. (2011). Neuroprotective and neurotrophic curcuminoids to treat stroke: a translational perspective. Expert Opin. Investig. Drugs20, 13–22. 10.1517/13543784.2011.542410
68
LenazG.FatoR.FormigginiG.GenovaM. L. (2007). The role of Coenzyme Q in mitochondrial electron transport. Mitochondrion7, S8–S33. 10.1016/j.mito.2007.03.009
69
LeunerK.HauptmannS.Abdel-KaderR.ScherpingI.KeilU.StrosznajderJ. B.et al (2007). Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease?Antioxid. Redox Signal.9, 1659–1676. 10.1089/ars.2007.1763
70
LeunerK.SchüttT.KurzC.EckertS. H.SchillerC.OcchipintiA.et al (2012). Mitochondrion-derived reactive oxygen species lead to enhanced amyloid β formation. Antioxid. Redox Signal.16, 1421–1433. 10.1089/ars.2011.4173
71
LibyK.HockT.YoreM. M.SuhN.PlaceA. E.RisingsongR.et al (2005). The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res.65, 4789–4798. 10.1158/0008-5472.CAN-04-4539
72
LinM. T.BealM. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature443, 787–795. 10.1038/nature05292
73
LustbaderJ. W.CirilliM.LinC.XuH. W.TakumaK.WangN.et al (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s Disease. Science304, 448–452. 10.1126/science.1091230
74
ManczakM.MaoP.CalkinsM. J.CorneaA.ReddyA. P.MurphyM. P.et al (2010). Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis.20, 609–631. 10.3233/JAD-2010-100564
75
ManczakM.CalkinsM. J.ReddyP. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid β with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet.20, 2495–2509. 10.1093/hmg/ddr139
76
MaruszakA.ŻekanowskiC. (2011). Mitochondrial dysfunction and Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry35, 320–330. 10.1016/j.pnpbp.2010.07.004
77
MatthewsR. T.FerranteR. J.KlivenyiP.YangL.KleinA. M.MuellerG.et al (1999). Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp. Neurol.157, 142–149. 10.1006/exnr.1999.7049
78
MatthewsR. T.YangL.BrowneS.BaikM.BealM. F. (1998). Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. U.S.A.95, 8892–8897. 10.1073/pnas.95.15.8892
79
McGillJ. K.BealM. F. (2006). PGC-1α, a new therapeutic target in Huntington’s disease?Cell127, 465–468. 10.1016/j.cell.2006.10.023
80
McNameeD. (2015). Breakthrough in ‘Editing’ Mitochondrial Disease DNA. Medical News Today. Available at: http://www.medicalnewstoday.com/articles/293033.php (accessed June 13, 2015).
81
MoreiraP. I.CarvalhoC.ZhuX.SmithM. A.PerryG. (2010). Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta1802, 2–10. 10.1016/j.bbadis.2009.10.006
82
MüllerT.BüttnerT.GholipourA.-F.KuhnW. (2003). Coenzyme Q 10 supplementation provides mild symptomatic benefit in patients with Parkinson’s disease. Neurosci. Lett.341, 201–204. 10.1016/S0304-3940(03)00185-X
83
MüllerW. E.HeiserJ.LeunerK. (2012). Effects of the standardized Ginkgo biloba extract EGb 761® on neuroplasticity. Int. Psychogeriatr.24, S21–S24. 10.1017/s1041610212000592
84
MurphyM. P.SmithR. A. (2007). Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol.47, 629–656. 10.1146/annurev.pharmtox.47.120505.105110
85
NaylorG.SmithA.ConnellyP. (1988). Methylene blue in mania. Biol. Psychiatry24, 941–942. 10.1016/0006-3223(88)90229-6
86
NeymotinA.CalingasanN. Y.WilleE.NaseriN.PetriS.DamianoM.et al (2011). Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med.51, 88–96. 10.1016/j.freeradbiomed.2011.03.027
87
OzM.IsaevD.LorkeD. E.HasanM.PetroianuG.ShippenbergT. S. (2012). Methylene blue inhibits function of the 5-HT transporter. Br. J. Pharmacol.166, 168–176. 10.1111/j.1476-5381.2011.01462.x
88
PeterC.HongwanD.KüpferA.LauterburgB. (2000). Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur. J. Clin. Pharmacol.56, 247–250. 10.1007/s002280000124
89
PieczenikS. R.NeustadtJ. (2007). Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol.83, 84–92. 10.1016/j.yexmp.2006.09.008
90
PrinceM.AlbaneseE.GuerchetM. (2014). World Alzheimer Report 2014. London: Alzheimer’s Disease International.
91
RastogiM.OjhaR. P.RajamanickamG.AgrawalA.AggarwalA.DubeyG. (2008). Curcuminoids modulates oxidative damage and mitochondrial dysfunction in diabetic rat brain. Free Radic. Res.42, 999–1005. 10.1080/10715760802571988
92
RazaH.JohnA.BrownE. M.BenedictS.KambalA. (2008). Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol. Appl. Pharmacol.226, 161–168. 10.1016/j.taap.2007.09.002
93
ReddyP. H. (2009). Amyloid β, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol.218, 286–292. 10.1016/j.expneurol.2009.03.042
94
ReddyP. H.BealM. F. (2008). Amyloid β, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med.14, 45–53. 10.1016/j.molmed.2007.12.002
95
ReddyP. H.ManczakM.MaoP.CalkinsM. J.ReddyA. P.ShirendebU. (2010). Amyloid-β and mitochondria in aging and Alzheimer’s disease: implications for synaptic damage and cognitive decline. J. Alzheimer’s Dis.20, S499–S512. 10.3233/JAD-2010-100504
96
ReddyP. H.ReddyT. P. (2011). Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res.8, 393–409. 10.2174/156720511795745401
97
ReinhardtK.DowlingD. K.MorrowE. H. (2013). Mitochondrial replacement, evolution, and the clinic. Science341, 1345–1346. 10.1126/science.1237146
98
RengelshausenJ.BurhenneJ.FröhlichM.TayrouzY.SinghS. K.RiedelK.-D.et al (2004). Pharmacokinetic interaction of chloroquine and methylene blue combination against malaria. Eur. J. Clin. Pharmacol.60, 709–715. 10.1007/s00228-004-0818-0
99
RenH.FuK.WangD.MuC.WangG. (2011). Oxidized DJ-1 interacts with the mitochondrial protein BCL-XL. J Biol Chem.286, 35308–35317. 10.1074/jbc.M110.207134
100
RheinV.SongX.WiesnerA.IttnerL. M.BaysangG.MeierF.et al (2009). Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. U.S.A.106, 20057–20062. 10.1073/pnas.0905529106
101
RodriguezM. C.MacdonaldJ. R.MahoneyD. J.PariseG.BealM. F.TarnopolskyM. A. (2007). Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve35, 235–242. 10.1002/mus.20688
102
SachdevaD.BurnsA. (2011). Dimebolin in dementia. CNS Neurosci. Ther.17, 199–205. 10.1111/j.1755-5949.2010.00156.x
103
SastreJ.LloretA.BorrásC.PeredaJ.García-SalaD.Droy-LefaixM.-T.et al (2002). Ginkgo biloba extract EGb 761 protects against mitochondrial aging in the brain and in the liver. Cell Mol. Biol.48, 685–692.
104
SchapiraA. H. (2012). Mitochondrial diseases. Lancet379, 1825–1834. 10.1016/S0140-6736(11)61305-6
105
SelfridgeJ. E.LeziE.LuJ.SwerdlowR. H. (2013). Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis.51, 3–12. 10.1016/j.nbd.2011.12.057
106
SeninU.ParnettiL.Barbagallo-SangiorgiG.BartorelliL.BocolaV.CapursoA.et al (1992). Idebenone in senile dementia of Alzheimer type: a multicentre study. Arch. Gerontol. Geriatr.15, 249–260. 10.1016/0167-4943(92)90060-H
107
ShultsC. W.BealM. F.FontaineD.NakanoK.HaasR. (1998). Absorption, tolerability, and effects on mitochondrial activity of oral coenzyme Q10 in parkinsonian patients. Neurology50, 793–795. 10.1212/WNL.50.3.793
108
SikoraE.Bielak-ZmijewskaA.MosieniakG.PiwockaK. (2010). The promise of slow down ageing may come from curcumin. Curr. Pharm. Des.16, 884–892. 10.2174/138161210790883507
109
SmithR. A.PorteousC. M.CoulterC. V.MurphyM. P. (1999). Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem.263, 709–716. 10.1046/j.1432-1327.1999.00543.x
110
SmithR. A.PorteousC. M.GaneA. M.MurphyM. P. (2003). Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U.S.A.100, 5407–5412. 10.1073/pnas.0931245100
111
SmithR.MurphyM. P. (2011). Mitochondria-targeted antioxidants as therapies. Dis. Med.11, 106–114.
112
SnowB. J.RolfeF. L.LockhartM. M.FramptonC. M.O’sullivanJ. D.FungV.et al (2010). A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord.25, 1670–1674. 10.1002/mds.23148
113
StackC.HoD.WilleE.CalingasanN. Y.WilliamsC.LibyK.et al (2010). Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med.49, 147–158. 10.1016/j.freeradbiomed.2010.03.017
114
StanleyW. C.KhairallahR. J.DabkowskiE. R. (2012). Update on lipids and mitochondrial function: impact of dietary n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. Metab. Care15, 122–126. 10.1097/MCO.0b013e32834fdaf7
115
StollS.ScheuerK.PohlO.MüllerW. (1996). Ginkgo biloba extract (EGb 761) independently improves changes in passive avoidance learning and brain membrane fluidity in the aging mouse. Pharmacopsychiatry29, 144–149. 10.1055/s-2007-979561
116
SwerdlowR. H. (2011). Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta1812, 1630–1639. 10.1016/j.bbadis.2011.08.012
117
SwerdlowR. H.BurnsJ. M.KhanS. M. (2010). The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis.20, 265–279. 10.3233/JAD-2010-100339
118
SwerdlowR. H.BurnsJ. M.KhanS. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta1842, 1219–1231. 10.1016/j.bbadis.2013.09.010
119
SwerdlowR. H.KhanS. M. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses63, 8–20. 10.1016/j.mehy.2003.12.045
120
SzetoH. H. (2008). Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid. Redox Signal.10, 601–620. 10.1089/ars.2007.1892
121
TachibanaM.SparmanM.SritanaudomchaiH.MaH.ClepperL.WoodwardJ.et al (2009). Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature461, 367–372. 10.1038/nature08368
122
TangF.NagS.ShiuS.PangS. (2002). The effects of melatonin and Ginkgo biloba extract on memory loss and choline acetyltransferase activities in the brain of rats infused intracerebroventricularly with β-amyloid 1–40. Life Sci.71, 2625–2631. 10.1016/S0024-3205(02)02105-7
123
TrnkaJ.BlaikieF. H.SmithR. A.MurphyM. P. (2008). A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med.44, 1406–1419. 10.1016/j.freeradbiomed.2007.12.036
124
TurunenM.OlssonJ.DallnerG. (2004). Metabolism and function of coenzyme Q. Biochim. Biophys. Acta1660, 171–199. 10.1016/j.bbamem.2003.11.012
125
VallaJ.BerndtJ. D.Gonzalez-LimaF. (2001). Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci.21, 4923–4930.
126
VauzourD. (2012). Dietary polyphenols as modulators of brain functions: biological actions and molecular mechanisms underpinning their beneficial effects. Oxid. Med. Cell. Longev.2012:914273. 10.1155/2012/914273
127
VignisseJ.SteinbuschH. W.BolkunovA.NunesJ.SantosA. I.GrandfilsC.et al (2011). Dimebon enhances hippocampus-dependent learning in both appetitive and inhibitory memory tasks in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry35, 510–522. 10.1016/j.pnpbp.2010.12.007
128
WangB.WangH.SongY.QiH.RongZ.ZhangL.et al (2010). Effectiveness of standardized Ginkgo biloba extract on cognitive symptoms of dementia with a six-month treatment: a bivariate random effect meta-analysis. Pharmacopsychiatry43, 86–91. 10.1055/s-0029-1242817
129
WangX.SuB.SiedlakS. L.MoreiraP. I.FujiokaH.WangY.et al (2008). Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U.S.A.105, 19318–19323. 10.1073/pnas.0804871105
130
WeinmannS.RollS.SchwarzbachC.VauthC.WillichS. N. (2010). Effects of Ginkgo biloba in dementia: systematic review and meta-analysis. BMC Geriat.10:14. 10.1186/1471-2318-10-14
131
WeyerG.Babej-DölleR.HadlerD.HofmannS.HerrmannW. (1997). A controlled study of 2 doses of idebenone in the treatment of Alzheimer’s disease. Neuropsychobiology36, 73–82. 10.1159/000119366
132
WitteM. E.GeurtsJ. J.De VriesH. E.Van Der ValkP.Van HorssenJ. (2010). Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration?Mitochondrion10, 411–418. 10.1016/j.mito.2010.05.014
133
YangL.CalingasanN. Y.ThomasB.ChaturvediR. K.KiaeiM.WilleE. J.et al (2009a). Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS ONE4:e5757. 10.1371/journal.pone.0005757
134
YangL.CalingasanN. Y.WilleE. J.CormierK.SmithK.FerranteR. J.et al (2009b). Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem.109, 1427–1439. 10.1111/j.1471-4159.2009.06074.x
135
YangL.ZhaoK.CalingasanN. Y.LuoG.SzetoH. H.BealM. F. (2009c). Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal.11, 2095–2104. 10.1089/ars.2009.2445
136
YatesM. S.TauchiM.KatsuokaF.FlandersK. C.LibyK. T.HondaT.et al (2007). Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer Ther.6, 154–162. 10.1158/1535-7163.MCT-06-0516
137
YlikallioE.SuomalainenA. (2012). Mechanisms of mitochondrial diseases. Ann. Med.44, 41–59. 10.3109/07853890.2011.598547
138
ZevianiM.TaroniF.GelleraC.DidonatoS. (2012). Molecular pathogenesis of mitochondrial diseases. Prog. Mitochondrial Bioenerg. Mol. Biol.5, 223–224. 10.1016/B978-0-444-82235-2.50041-2
139
ZhangS.HedskogL.PetersenC.WinbladB.AnkarcronaM. (2009). Dimebon (latrepirdine) enhances mitochondrial function and protects neuronal cells from death. J. Alzheimer’s Dis.21, 389–402. 10.3233/JAD-2010-100174
140
ZhangX.RojasJ. C.Gonzalez-LimaF. (2006). Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox. Res.9, 47–57. 10.1007/BF03033307
141
ZhuY.-G.ChenX.-C.ChenZ.-Z.ZengY.-Q.ShiG.-B.SuY.-H.et al (2004). Curcumin protects mitochondria from oxidative damage and attenuates apoptosis in cortical neurons. Acta Pharmacol. Sin.25, 1606–1612.
Summary
Keywords
Alzheimer’s disease, mitochondria, mitochondrial dysfunction, oxidative stress, coenzyme Q10
Citation
Kumar A and Singh A (2015) A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 6:206. doi: 10.3389/fphar.2015.00206
Received
28 July 2015
Accepted
07 September 2015
Published
24 September 2015
Volume
6 - 2015
Edited by
Alfredo Meneses, Center for Research and Advanced Studies of the National Polytechnic Institute, Mexico
Reviewed by
P. H. Reddy, Texas Tech University, USA; Gopalkumar Rakesh, Duke University, USA
Updates
Copyright
© 2015 Kumar and Singh.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anil Kumar, Pharmacology Division, University Institute of Pharmaceutical Sciences, UGC Centre of Advanced Study, Panjab University, 14th Sector, Chandigarh 160014, India, kumaruips@yahoo.com
This article was submitted to Neuropharmacology, a section of the journal Frontiers in Pharmacology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.