 Andrew L. Siebel
Andrew L. Siebel- Metabolic and Vascular Physiology Laboratory, Baker IDI Heart and Diabetes Institute, Melbourne, VIC, Australia
High-density lipoprotein (HDL) and its principal apolipoprotein A-I (ApoA-I) have now been convincingly shown to influence glucose metabolism through multiple mechanisms. The key clinically relevant observations are that both acute HDL elevation via short-term reconstituted HDL (rHDL) infusion and chronically raising HDL via a cholesteryl ester transfer protein (CETP) inhibitor reduce blood glucose in individuals with type 2 diabetes mellitus (T2DM). HDL may mediate effects on glucose metabolism through actions in multiple organs (e.g., pancreas, skeletal muscle, heart, adipose, liver, brain) by three distinct mechanisms: (i) Insulin secretion from pancreatic beta cells, (ii) Insulin-independent glucose uptake, (iii) Insulin sensitivity. The molecular mechanisms appear to involve both direct HDL signaling actions as well as effects secondary to lipid removal from cells. The implications of glucoregulatory mechanisms linked to HDL extend from glycemic control to potential anti-ischemic actions via increased tissue glucose uptake and utilization. Such effects not only have implications for the prevention and management of diabetes, but also for ischemic vascular diseases including angina pectoris, intermittent claudication, cerebral ischemia and even some forms of dementia. This review will discuss the growing evidence for a role of HDL in glucose metabolism and outline related potential for HDL therapies.
Introduction
There is accumulating experimental and clinical evidence that HDL particles can control glucose metabolism via a variety of mechanisms (Drew et al., 2012; von Eckardstein and Widmann, 2014). At a mechanistic level HDL particles can alter multiple aspects of glucose metabolism. These include insulin secretion from pancreatic beta cells, and both insulin-dependent and insulin-independent glucose uptake and utilization in tissues. Some of these effects are mediated by direct signaling actions of HDL and its components, whilst others appear to be secondary to other known HDL functions, including tissue lipid efflux.
The action of HDL to move glucose from the circulation and into tissues has potential clinical relevance in terms of both reducing vascular complications by removing excess glucose from the circulation in the setting of T2DM, and also in providing adequate glucose to tissues for energy production, particularly in the context of ischemia. HDL therapeutics may therefore have application not only for the prevention and management of T2DM (Drew et al., 2012; von Eckardstein and Widmann, 2014), but also for ischemic vascular diseases including angina pectoris, intermittent claudication, cerebrovascular disease (Kingwell and Chapman, 2013) and even some forms of dementia (Stukas et al., 2014b).
This review will discuss the growing evidence for a role of HDL in glucose metabolism in the context of the evolving HDL hypothesis. It is now recognized that the main clinical variable used to assess HDL, HDL cholesterol, fails to provide an accurate representation of the multiple functions of heterogeneous HDL particles, which are composed of many hundreds of lipids and multiple proteins (Khera et al., 2011; Kingwell et al., 2014; Rohatgi et al., 2014; Saleheen et al., 2015). The role of HDL in glucose metabolism will be explored from the perspective of both disease pathophysiology and the potential for HDL therapies targeting this mechanism.
HDL, Glucose Metabolism, and T2DM
T2DM, insulin resistance and glucose intolerance are associated with low HDL cholesterol levels (Ford et al., 2002; Drexel et al., 2005; Gatti et al., 2009). While there are clear mechanisms by which insulin resistance contributes to low HDL cholesterol, there is also evidence that low HDL may actually promote development of diabetes (Drew et al., 2012). Low HDL cholesterol levels predict development of T2DM in prediabetics (Haffner et al., 1990) and progression of glycemia in those with established T2DM (Waldman et al., 2014). With the recent recognition of the importance of HDL function above simple plasma HDL cholesterol concentration (Rohatgi et al., 2014), it is interesting to note that HDL cholesterol efflux capacity is inversely related to T2DM in the EPIC Norfolk study (Saleheen et al., 2015).
On the other hand, a recent Mendelian randomization study showed that genetically reduced HDL cholesterol does not associate with increased risk of T2DM (Haase et al., 2015). While this may reflect that the observational association is due to confounding and/or reverse causation, consideration must also be given to the fact that HDL cholesterol, while clinically accessible, does not accurately reflect HDL function and may not therefore be the best instrumental variable to test. As HDL function assays such as cholesterol efflux capacity become feasible in larger data sets, the hypothesis that HDL function may contribute causally to the risk of T2DM can be further explored using Mendelian approaches. In the meantime, mechanistic and intervention studies are uncovering links between HDL and glucose metabolism, which may have physiological and therapeutic relevance.
Insulin Secretion
High-density lipoprotein has been implicated in the modulation of insulin secretion in cellular and animal studies with corroborating evidence from human intervention trials. The diverse functionality of HDL to combat cellular lipid accumulation, endoplasmic reticulum (ER) stress and apoptosis are three potential mechanisms that may preserve pancreatic beta cell function.
Pancreatic lipid accumulation and lipotoxicity have been well-documented to inhibit insulin production and secretion (Hao et al., 2007). We have shown that impairment of glucose-stimulated insulin secretion induced by oxidized low-density lipoprotein (LDL) can be countered by native HDL (50μg/mL) treatment (Drew et al., 2009). Further to this, a single dose of either wild-type ApoA-I (major protein component of HDL) or ApoA-I Milano was able to potentiate glucose-stimulated insulin secretion in high-fat fed mice (Stenkula et al., 2014). These effects on beta cell function may be mediated by the bioactive lipid sphingosine-1-phosphate (S1P), which is primarily carried within HDL particles and is known to independently promote glucose-stimulated insulin secretion (Cantrell Stanford et al., 2012).
At the signaling level the HDL transporters ATP-binding cassette, sub-family A, member 1 (ABCA1) and ATP-binding cassette, sub-family G, member 1 (ABCG1) have both been implicated in HDL-mediated effects on insulin secretion. Beta cell-specific deletion of the ABCA1 (Brunham et al., 2007) and ABCG1 (Sturek et al., 2010) transporters in mice impairs both cholesterol homeostasis and insulin secretion. Combined inactivation of ABCA1 and ABCG1 in beta cells increased intracellular cholesterol accumulation and induced beta cell inflammation, exacerbating the insulin secretory defect found in the single transporter knockout mice (Kruit et al., 2012). Furthermore, post-transcriptional suppression of ABCA1 via over expression of miR33a and miR145 in beta cells increased islet cholesterol levels and decreased glucose-stimulated insulin secretion (Wijesekara et al., 2012; Kang et al., 2013); effects that could be rescued by cholesterol depletion.
HDL may also influence insulin secretion via mechanisms other than cholesterol depletion, including via actions on insulin transcription (Fryirs et al., 2009; Cochran et al., 2014). At doses of up to 900μg/mL, lipid-free recombinant ApoA-I, ApoA-II and discoidal rHDL dose-dependently increased basal and glucose-stimulated insulin secretion in addition to insulin transcription (Fryirs et al., 2009). Whereas other experiments in MIN6 cells, as well as in islets isolated from mice and humans, found no evidence that HDL can directly enhance insulin production, basal or glucose-stimulated insulin secretion (Abderrahmani et al., 2007; Rutti et al., 2009).
The ability of HDL to inhibit ER stress-induced beta cell apoptosis could be another important mechanism by which HDL may ameliorate beta cell dysfunction (Petremand et al., 2009; Rutti et al., 2009). Bioactive lipids associated with HDL, such as S1P may also mediate anti-apoptotic effects on beta cells indirectly by interfering with the release of proapoptotic signals from non beta-cells within the pancreatic islet (Vollenweider et al., 2015). While it is more challenging to directly examine the effects of HDL on insulin secretion in humans, acute infusion of rHDL in patients with T2DM increased plasma insulin concentrations (Drew et al., 2009). Chronic HDL elevation induced by a CETP inhibitor in healthy individuals was also associated with an increase in postprandial plasma insulin and C-peptide concentrations (Siebel et al., 2013). Furthermore, plasma taken from these CETP inhibitor-treated individuals increased glucose-stimulated insulin secretion and cholesterol efflux from MIN6 beta cells relative to plasma from placebo-treated individuals. In this study, the actions of CETP inhibition on glucose-stimulated, but not basal insulin secretion suggests that HDL may be important in minimizing postprandial glucose excursions without raising resting insulin levels. Future clinical studies utilizing stable isotope tracers and minimal modeling techniques (Cobelli et al., 2014) are warranted to more directly characterize the effects of HDL on postprandial insulin secretion and glucose metabolism.
Insulin-independent Glucose Uptake in Skeletal Muscle
We have previously shown that acute rHDL infusion can reduce blood glucose in individuals with T2DM (Drew et al., 2009). This reduction in glucose preceded an increase in plasma insulin, supporting the hypothesis that HDL may also mediate insulin-independent glucose uptake within skeletal muscle (Drew et al., 2009). This has been further investigated in primary human skeletal muscle cells and adipocytes where both HDL and ApoA-I promoted glucose uptake independently of insulin stimulation (Han et al., 2007; Drew et al., 2009; Zhang et al., 2011; Dalla-Riva et al., 2013). Figure 1 depicts the intracellular signaling pathways activated by HDL in skeletal muscle. HDL-associated ApoA-I binds to ABCA1 inducing intracellular Ca2+ influx and activation of calcium/calmodulin activated protein kinase kinase (CaMKK) (Drew et al., 2009). Subsequent activation and phosphorylation of AMP-activated protein kinase (AMPK) increases translocation of glucose transporter 4 (GLUT4) to the plasma membrane (Dalla-Riva et al., 2013) and facilitates glucose uptake.
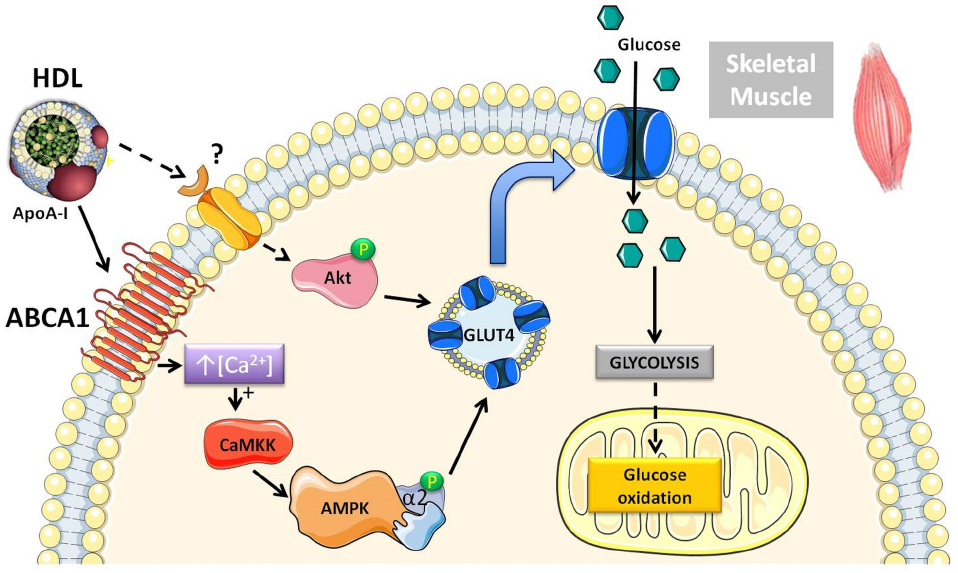
Figure 1. Schematic diagram of the pathways activated by HDL in skeletal muscle that modulate glucose metabolism. Both human and rodent studies have demonstrated that HDL can increase glucose uptake through an ABCA1/AMPK-dependent mechanism (Drew et al., 2009). Chronically elevating HDL levels pharmacologically or via genetic approaches in mice increases Akt phosphorylation, suggesting activation of an additional signaling pathway (Lehti et al., 2013). There is also more recent evidence in mouse muscle cells showing that HDL can directly stimulate anaerobic glycolysis and glucose oxidation.
Beyond glucose uptake, HDL has been shown to activate Akt in ApoA-I transgenic mice and directly enhance both glycolysis and glucose oxidation in mouse C2C12 muscle cells (Lehti et al., 2013). These insulin-independent effects of HDL on glucose metabolism, may actually account, at least partly, for in vivo effects of HDL on glucose disposal in the absence of a change in circulating insulin. Such effects may be mistakenly attributed to increased insulin sensitivity.
Insulin Sensitivity and Glucose Homeostasis
Tissue lipid deposition and chronic inflammation associated with obesity, metabolic syndrome and T2DM establishes a functional deficit in skeletal muscle and the heart, impairing how these metabolically active tissues respond to insulin. The insulin resistance associated with lipid accumulation and a pro-inflammatory milieu promotes lipolysis, increasing circulating free fatty acids, thus perpetuating dyslipidemia and insulin resistance (Hotamisligil, 2006; Klop et al., 2013). It is still unclear whether it is the persistent hyperglycemia associated with T2DM that leads to the chronic inflammation typical of metabolic syndrome or whether the inflammatory response triggers an imbalance in glucose control and leads to insulin resistance. Either way via its effects on reverse cholesterol transport, functional HDL can remove lipids from peripheral cells and potentially ameliorate inflammation (Reviewed, in Navab et al., 2011). There is some cellular evidence that HDL elevation can increase insulin sensitivity in peripheral tissues (Carey et al., 2013).
Examining insulin sensitivity in vivo is more complex due to the fact that increased glucose disposal relative to insulin may not necessarily reflect increased insulin sensitivity. As discussed in the previous section, such observations may actually be explained by direct actions of HDL on glucose disposal by mechanisms which do not involve insulin. Nevertheless, there are two recent in vivo studies consistent with the cellular studies, which suggest that HDL/ApoA-I can increase peripheral insulin sensitivity. The first of these employed intraperitoneal glucose and insulin tolerance tests in high fat diet fed mice treated for 2–4 weeks with lipid-free ApoA-I (8 mg/kg body weight) and showed improved glucose and insulin tolerance compared to untreated mice on the same high fat diet (McGrath et al., 2014). The second raised HDL cholesterol levels using the CETP inhibitor torcetrapib in insulin resistant hamsters and found that torcetrapib increased insulin-stimulated glucose uptake in skeletal muscle, liver and the heart, improving glucose homeostasis (Briand et al., 2014).
There are a number of animal studies that implicate HDL and/or ApoA-I in the maintenance of glucose homeostasis, with considerable conjecture over whether HDL has a direct effect on insulin sensitivity. The relevance of these findings is generally not clear due to a lack of insulin tolerance (or more accurately insulin responsiveness) testing or hyperinsulinemic-euglycemic clamp studies to examine direct effects on insulin sensitivity. Definitive evidence for an effect of HDL on insulin sensitivity in humans is also lacking. However, HDL cholesterol elevation by CETP inhibition has been shown to improve glycemic control in the face of reduced plasma insulin in people with T2DM in the ILLUMINATE trial (Barter et al., 2011). As evidence for a role of HDL in glucose metabolism accumulates, clinical trials of HDL therapies are now including glycemic endpoints to further test clinical relevance.
New Directions for HDL and Glucose Metabolism
The actions of HDL on glucose metabolism have potential benefit beyond vascular protection. The ability of HDL to stimulate tissue glucose uptake has implications for cellular metabolism in the brain, as well as for cell viability in other organs under ischemic/anoxic conditions where glucose becomes the predominant metabolic substrate. This latter mechanism may have particular relevance to ischemic vascular conditions, including coronary, cerebral and peripheral artery diseases.
Ischemic Heart Disease
We have recently established that HDL increases insulin-independent glucose uptake in rat cardiomyocytes via an Akt signaling pathway (Heywood et al., 2014). In this cardiomyocyte model, the AMPK pathway involved in human skeletal muscle cell glucose uptake is not activated by HDL. It is known that plasma levels of HDL cholesterol are strongly associated with increased glucose disposal during the acute phase of myocardial infarction (Carvalho et al., 2012, 2013). However, the relevance of HDL-mediated glucose uptake to myocardial protection in human ischemic heart disease has not been established.
Insulin resistance may directly contribute to the increased risk of ischemic heart disease and poorer outcomes after infarction in patients with T2DM (Icks et al., 2014). It is plausible that HDL may improve cardiac function by directly increasing glucose uptake in addition to promoting efficient coupling of anaerobic glycolysis to glucose oxidation in the heart. This is particularly relevant in the setting of acute coronary syndromes where the ischemic myocardium is exposed to an acute ATP deficit, which can be ameliorated by targeting glucose metabolism immediately post-event in mice (Ussher et al., 2012; Masoud et al., 2014). In addition, the well-established vasodilatory effects of HDL via nitric oxide production (Mineo et al., 2003; Drew et al., 2004) may also enhance glucose uptake via microcirculatory vasodilatation allowing insulin and glucose access to nutritive vascular beds.
The emerging actions of HDL and its bioactive components on glucose metabolism in the myocardium may contribute to the demonstrated protective effects of HDL in the context of ischemia-reperfusion injury (Calabresi et al., 2003; Theilmeier et al., 2006; Herzog et al., 2010; Tao et al., 2010; Brulhart-Meynet et al., 2015). Such actions may be particularly important in the setting of T2DM, where non-insulin mediated glucose uptake could support tissue viability in the face of insulin resistance. Further investigations are required to determine the relevance of HDL-mediated effects on glucose metabolism and their relevance under ischemic conditions both in the presence and absence of insulin resistance. In this regard, novel reconstituted and recombinant HDLs have particular potential as anti-ischemic therapeutics in the setting of both acute coronary syndromes and coronary surgeries involving bypass procedures including coronary artery grafts (Kingwell and Chapman, 2013). Current trials with HDL infusion therapies (NCT02108262 and NCT02484378) will provide further insights into the actions of HDL in the context of acute coronary syndromes, though the potential role of glucose metabolism will not be discernible.
Cerebrovascular Disease
For the same reasons that HDL-mediated effects on glucose metabolism may protect the ischemic myocardium, these actions may equally act in the brain to protect against transient ischemic attacks and stroke. In fact, the reliance of the brain on glucose for ATP production means that mechanisms controlling glucose uptake may be of even greater significance than in other organs. Even minor changes in cerebral blood flow have the potential to impact on neuronal viability via reduction in glucose supply. Indeed cerebral hypoperfusion and hypometabolism coincide in patients with dementia (Verfaillie et al., 2015).
The implications of reduced blood flow, and thus glucose supply to the brain, extend beyond acute stroke risk to Alzheimer’s disease, which has a significant vascular component (Di Marco et al., 2015). In this condition, cerebral blood flow is reduced by 10–30% in many brain regions, including those responsible for cognitive function and memory, with the extent of blood flow impairment correlating with disease severity (Jagust et al., 1997; Johnson et al., 1998; Hanyu et al., 2010; Kim et al., 2013). This reduction in cerebral blood flow is also detected prior to cognitive decline or amyloid accumulation. However, most relevant to this review, glucose uptake and utilization in the brain is also reduced in Alzheimer’s disease, mild cognitive impairment patients and transgenic animal models of Alzheimer’s disease (Herholz, 2010; Jagust et al., 2010; Shin et al., 2010; Nicolakakis and Hamel, 2011). Whether HDL can increase cerebral blood flow and glucose uptake in the central nervous system (CNS) is not currently known, but certainly worth exploring. Evidence to date has shown beneficial associations between brain health and HDL, but the contributing mechanisms have not been fully elucidated.
Given the well-established positive actions of HDL on vascular function, it is not surprising that several co-morbidities associated with increased Alzheimer’s disease risk are themselves associated with decreased HDL function (Riwanto and Landmesser, 2013). A role for HDL in glucose handling in the brain is an interesting candidate to partly explain the association of reduced circulating levels of HDL/ApoA-I with Alzheimer’s disease (Kuriyama et al., 1994; Kawano et al., 1995; Merched et al., 2000; Castano et al., 2006; Roher et al., 2009). Other mechanisms may relate to removal or breakdown of β-amyloid or reduction in its cytotoxicity (Paula-Lima et al., 2009; Lefterov et al., 2010). In this respect, ApoA-I may have actions analogous to apolipoprotein E (ApoE) where common genetic variants have directly opposing effects on β-amyloid deposition and clearance and therefore risk for Alzheimer’s disease (Corder et al., 1993). ApoE is the major apolipoprotein synthesized by astrocytes and microglia in the brain and forms lipoprotein particles that resemble HDLs found in plasma. Similarly to ApoA-I, ApoE is lipidated by ABCA1 (Pitas et al., 1987; LaDu et al., 1998; Fagan et al., 1999). Further work is required to understand the role of HDL and ApoA-I in brain lipid and glucose metabolism, both in the cerebral vasculature as well as neurons and glial cells and how this might relate to pathophysiology.
One important consideration for the potential application of HDL therapeutics to brain disorders is whether they can cross the blood-brain or blood-cerebral spinal fluid (CSF) barrier (Zlokovic, 2011). ApoA-I has been previously found in the CSF at relatively high levels (0.07–4 μg/mL or 0.1–0.5% plasma levels), (Koch et al., 2001) which is encouraging given the in vitro efficacy of ApoA-I at similar concentrations. More recently, Stukas et al. (2014a) have shown that intravenous injection of recombinant human ApoA-I specifically localizes to the choroid plexus. These data, along with in vitro primary cell culture experiments, suggest that plasma ApoA-I gains access to the CNS primarily by crossing the blood-CSF barrier via specific cellular mediated transport (Stukas et al., 2014a). The blood-CSF barrier is made up of highly fenestrated cuboidal epithelial cells within the choroid plexus (Lehtinen et al., 2013), which may explain how ApoA-I enters the CSF from the blood. This suggests that aside from the vasodilatory, anti-inflammatory and anti-oxidative actions of HDL/ApoA-I that could target cerebrovascular complications in Alzheimer’s disease, targeting brain-specific glucose metabolism may also be possible.
Conclusion
There is now convincing evidence that HDL modulates glucose metabolism in multiple tissues. These actions have deepened our understanding of the pathophysiology of a variety of disease states associated with low or dysfunctional HDL. While there are still many unanswered questions relating to the underlying mechanisms and key HDL component(s) responsible for the metabolic effects, this opens up the possibility of targeting glucose metabolism with HDL therapeutics currently in development. Future preclinical investigations and clinical trials will determine the relevance of HDL-mediated modulation of glucose metabolism to both glycemic control as well as tissue glucose supply to vital organs including the heart and the brain, especially under ischemic conditions.
Author Contributions
AS, SH, BK drafted, critically revised and approved the final version of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. BK has received product from CSL Behring and patient plasma from Hoffman La Roche both for investigator initiated clinical trials, with no associated research funding. She has also partnered with Resverlogix to fund an investigator-initiated clinical trial.
Funding
BK is a NHMRC of Australia Senior Principal Research Fellow (APP1059454).
Abbreviations
ABCA1 ATP-binding cassette, sub-family A (ABC1), member 1; ABCG1 ATP-binding cassette, sub-family G (ABC1), member 1; AMPK AMP-activated protein kinase; ApoA-I apolipoprotein A-I; ApoE apolipoprotein E; CaMKK calcium/calmodulin activated protein kinase; CETP cholesteryl ester transfer protein; CNS central nervous system; CSF cerebral spinal fluid; ER endoplasmic reticulum; GLUT4 glucose transporter 4; HDL high-density lipoprotein; LDL low-density lipoprotein; rHDL reconstituted HDL; S1P sphingosine-1-phosphate; T2DM type 2 diabetes mellitus.
References
Abderrahmani, A., Niederhauser, G., Favre, D., Abdelli, S., Ferdaoussi, M., Yang, J. Y., et al. (2007). Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 50, 1304–1314. doi: 10.1007/s00125-007-0642-z
Barter, P. J., Rye, K. A., Tardif, J. C., Waters, D. D., Boekholdt, S. M., Breazna, A., et al. (2011). Effect of torcetrapib on glucose, insulin, and hemoglobin a1c in subjects in the investigation of lipid level management to understand its impact in atherosclerotic events (ILLUMINATE) trial. Circulation 124, 555–562. doi: 10.1161/CIRCULATIONAHA.111.018259
Briand, F., Prunet-Marcassus, B., Thieblemont, Q., Costard, C., Muzotte, E., Sordello, S., et al. (2014). Raising HDL with CETP inhibitor torcetrapib improves glucose homeostasis in dyslipidemic and insulin resistant hamsters. Atherosclerosis 233, 359–362. doi: 10.1016/j.atherosclerosis.2014.01.028
Brulhart-Meynet, M. C., Braunersreuther, V., Brinck, J., Montecucco, F., Prost, J. C., Thomas, A., et al. (2015). Improving reconstituted HDL composition for efficient post-ischemic reduction of ischemia reperfusion injury. PLoS ONE 10:e0119664. doi: 10.1371/journal.pone.0119664
Brunham, L. R., Kruit, J. K., Pape, T. D., Timmins, J. M., Reuwer, A. Q., Vasanji, Z., et al. (2007). Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat. Med. 13, 340–347. doi: 10.1038/nm1546
Calabresi, L., Rossoni, G., Gomaraschi, M., Sisto, F., Berti, F., and Franceschini, G. (2003). High-density lipoproteins protect isolated rat hearts from ischemia-reperfusion injury by reducing cardiac tumor necrosis factor-alpha content and enhancing prostaglandin release. Circ. Res. 92, 330–337. doi: 10.1161/01.RES.0000054201.60308.1A
Cantrell Stanford, J., Morris, A. J., Sunkara, M., Popa, G. J., Larson, K. L., and Ozcan, S. (2012). Sphingosine 1-phosphate (S1P) regulates glucose-stimulated insulin secretion in pancreatic beta cells. J. Biol. Chem. 287, 13457–13464. doi: 10.1074/jbc.M111.268185
Carey, A. L., Siebel, A. L., Reddy-Luthmoodoo, M., Natoli, A. K., D’souza, W., Meikle, P. J., et al. (2013). Skeletal muscle insulin resistance associated with cholesterol-induced activation of macrophages is prevented by high density lipoprotein. PLoS ONE 8:e56601. doi: 10.1371/journal.pone.0056601
Carvalho, L. S., Cintra, R. M., Moura, F. A., Martins, N. V., Quinaglia, E. S. J. C., Coelho, O. R., et al. (2012). High plasma HDL-C attenuates stress hyperglycemia during acute phase of myocardial infarction. Atherosclerosis 220, 231–236. doi: 10.1016/j.atherosclerosis.2011.09.044
Carvalho, L. S., Martins, N. V., Moura, F. A., Cintra, R. M., Almeida, O. L., Quinaglia e Silva, J. C., et al. (2013). High-density lipoprotein levels are strongly associated with the recovery rate of insulin sensitivity during the acute phase of myocardial infarction: a study by euglycemic hyperinsulinemic clamp. J. Clin. Lipidol. 7, 24–28. doi: 10.1016/j.jacl.2012.10.003
Castano, E. M., Roher, A. E., Esh, C. L., Kokjohn, T. A., and Beach, T. (2006). Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer’s disease and non-demented elderly subjects. Neurol. Res. 28, 155–163. doi: 10.1179/016164106X98035
Cobelli, C., Dalla Man, C., Toffolo, G., Basu, R., Vella, A., and Rizza, R. (2014). The oral minimal model method. Diabetes Metab. Res. Rev. 63, 1203–1213. doi: 10.2337/db13-1198
Cochran, B. J., Bisoendial, R. J., Hou, L., Glaros, E. N., Rossy, J., Thomas, S. R., et al. (2014). Apolipoprotein A-I increases insulin secretion and production from pancreatic beta-cells via a G-protein-cAMP-PKA-FoxO1-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 34, 2261–2267. doi: 10.1161/ATVBAHA.114.304131
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Dalla-Riva, J., Stenkula, K. G., Petrlova, J., and Lagerstedt, J. O. (2013). Discoidal HDL and apoA-I-derived peptides improve glucose uptake in skeletal muscle. J. Lipid Res. 54, 1275–1282. doi: 10.1194/jlr.M032904
Di Marco, L. Y., Venneri, A., Farkas, E., Evans, P. C., Marzo, A., and Frangi, A. F. (2015). Vascular dysfunction in the pathogenesis of Alzheimer’s disease—a review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. doi: 10.1016/j.nbd.2015.08.014 [Epub ahead of print].
Drew, B. G., Duffy, S. J., Formosa, M. F., Natoli, A. K., Henstridge, D. C., Penfold, S. A., et al. (2009). High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 119, 2103–2111. doi: 10.1161/CIRCULATIONAHA.108.843219
Drew, B. G., Fidge, N. H., Gallon-Beaumier, G., Kemp, B. E., and Kingwell, B. A. (2004). High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 101, 6999–7004. doi: 10.1073/pnas.0306266101
Drew, B. G., Rye, K. A., Duffy, S. J., Barter, P., and Kingwell, B. A. (2012). The emerging role of HDL in glucose metabolism. Nat. Rev. Endocrinol. 8, 237–245. doi: 10.1038/nrendo.2011.235
Drexel, H., Aczel, S., Marte, T., Benzer, W., Langer, P., Moll, W., et al. (2005). Is atherosclerosis in diabetes and impaired fasting glucose driven by elevated LDL cholesterol or by decreased HDL cholesterol? Diabetes Care 28, 101–107. doi: 10.2337/diacare.28.1.101
Fagan, A. M., Holtzman, D. M., Munson, G., Mathur, T., Schneider, D., Chang, L. K., et al. (1999). Unique lipoproteins secreted by primary astrocytes from wild type, apoE (–/–), and human apoE transgenic mice. J. Biol. Chem. 274, 30001–30007. doi: 10.1074/jbc.274.42.30001
Ford, E. S., Giles, W. H., and Dietz, W. H. (2002). Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287, 356–359. doi: 10.1001/jama.287.3.356
Fryirs, M., Barter, P. J., and Rye, K. A. (2009). Cholesterol metabolism and pancreatic beta-cell function. Curr. Opin. Lipidol. 20, 159–164. doi: 10.1097/MOL.0b013e32832ac180
Gatti, A., Maranghi, M., Bacci, S., Carallo, C., Gnasso, A., Mandosi, E., et al. (2009). Poor glycemic control is an independent risk factor for low HDL cholesterol in patients with type 2 diabetes. Diabetes Care 32, 1550–1552. doi: 10.2337/dc09-0256
Haase, C. L., Tybjaerg-Hansen, A., Nordestgaard, B. G., and Frikke-Schmidt, R. (2015). High-density lipoprotein cholesterol and risk of type 2 diabetes: a Mendelian randomization study. Diabetes 64, 3328–3333. doi: 10.2337/db14-1603
Haffner, S. M., Stern, M. P., Hazuda, H. P., Mitchell, B. D., and Patterson, J. K. (1990). Cardiovascular risk factors in confirmed prediabetic individuals. Does the clock for coronary heart disease start ticking before the onset of clinical diabetes? JAMA 263, 2893–2898. doi: 10.1001/jama.1990.03440210043030
Han, R., Lai, R., Ding, Q., Wang, Z., Luo, X., Zhang, Y., et al. (2007). Apolipoprotein A-I stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia 50, 1960–1968. doi: 10.1007/s00125-007-0752-7
Hanyu, H., Sato, T., Hirao, K., Kanetaka, H., Iwamoto, T., and Koizumi, K. (2010). The progression of cognitive deterioration and regional cerebral blood flow patterns in Alzheimer’s disease: a longitudinal SPECT study. J. Neurol. Sci. 290, 96–101. doi: 10.1016/j.jns.2009.10.022
Hao, M., Head, W. S., Gunawardana, S. C., Hasty, A. H., and Piston, D. W. (2007). Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic beta-cell dysfunction. Diabetes Metab. Res. Rev. 56, 2328–2338. doi: 10.2337/db07-0056
Herholz, K. (2010). Cerebral glucose metabolism in preclinical and prodromal Alzheimer’s disease. Expert. Rev. Neurother. 10, 1667–1673. doi: 10.1586/ern.10.136
Herzog, C., Schmitz, M., Levkau, B., Herrgott, I., Mersmann, J., Larmann, J., et al. (2010). Intravenous sphingosylphosphorylcholine protects ischemic and postischemic myocardial tissue in a mouse model of myocardial ischemia/reperfusion injury. Mediators Inflamm. 2010, 425191. doi: 10.1155/2010/425191
Heywood, S. E., Henstridge, D. C., Carey, A. L., Delbridge, L. M., Kingwell, B. A., and Siebel, A. L. (2014). “High-density lipoprotein modulates cardiomyocyte glucose metabolism via an insulin-independent mechanism involving Akt,” in American Heart Association Conference, Chicago, IL.
Hotamisligil, G. S. (2006). Inflammation and metabolic disorders. Nature 444, 860–867. doi: 10.1038/nature05485
Icks, A., Claessen, H., Kirchberger, I., Heier, M., Peters, A., Trentinaglia, I., et al. (2014). Mortality after first myocardial infarction in diabetic and non-diabetic people between 1985 and 2009. The MONICA/KORA registry. Eur. J. Epidemiol. 29, 899–909. doi: 10.1007/s10654-014-9964-6
Jagust, W. J., Bandy, D., Chen, K., Foster, N. L., Landau, S. M., Mathis, C. A., et al. (2010). The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement 6, 221–229. doi: 10.1016/j.jalz.2010.03.003
Jagust, W. J., Eberling, J. L., Reed, B. R., Mathis, C. A., and Budinger, T. F. (1997). Clinical studies of cerebral blood flow in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 826, 254–262. doi: 10.1111/j.1749-6632.1997.tb48477.x
Johnson, K. A., Jones, K., Holman, B. L., Becker, J. A., Spiers, P. A., Satlin, A., et al. (1998). Preclinical prediction of Alzheimer’s disease using SPECT. Neurology 50, 1563–1571. doi: 10.1212/WNL.50.6.1563
Kang, M. H., Zhang, L. H., Wijesekara, N., De Haan, W., Butland, S., Bhattacharjee, A., et al. (2013). Regulation of ABCA1 protein expression and function in hepatic and pancreatic islet cells by miR-145. Arterioscler. Thromb. Vasc. Biol. 33, 2724–2732. doi: 10.1161/ATVBAHA.113.302004
Kawano, M., Kawakami, M., Otsuka, M., Yashima, H., Yaginuma, T., and Ueki, A. (1995). Marked decrease of plasma apolipoprotein AI and AII in Japanese patients with late-onset non-familial Alzheimer’s disease. Clin. Chim. Acta 239, 209–211. doi: 10.1016/0009-8981(95)06115-T
Khera, A. V., Cuchel, M., De La Llera-Moya, M., Rodrigues, A., Burke, M. F., Jafri, K., et al. (2011). Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 364, 127–135. doi: 10.1056/NEJMoa1001689
Kim, S. M., Kim, M. J., Rhee, H. Y., Ryu, C. W., Kim, E. J., Petersen, E. T., et al. (2013). Regional cerebral perfusion in patients with Alzheimer’s disease and mild cognitive impairment: effect of APOE epsilon4 allele. Neuroradiology 55, 25–34. doi: 10.1007/s00234-012-1077-x
Kingwell, B. A., and Chapman, M. J. (2013). Future of high-density lipoprotein infusion therapies: potential for clinical management of vascular disease. Circulation 128, 1112–1121. doi: 10.1161/CIRCULATIONAHA.113.002683
Kingwell, B. A., Chapman, M. J., Kontush, A., and Miller, N. E. (2014). HDL-targeted therapies: progress, failures and future. Nat. Rev. Drug Discov. 13, 445–464. doi: 10.1038/nrd4279
Klop, B., Elte, J. W., and Cabezas, M. C. (2013). Dyslipidemia in obesity: mechanisms and potential targets. Nutrients 5, 1218–1240. doi: 10.3390/nu5041218
Koch, S., Donarski, N., Goetze, K., Kreckel, M., Stuerenburg, H. J., Buhmann, C., et al. (2001). Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 42, 1143–1151.
Kruit, J. K., Wijesekara, N., Westwell-Roper, C., Vanmierlo, T., De Haan, W., Bhattacharjee, A., et al. (2012). Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired beta-cell function. Diabetes Metab. Res. Rev. 61, 659–664. doi: 10.2337/db11-1341
Kuriyama, M., Takahashi, K., Yamano, T., Hokezu, Y., Togo, S., Osame, M., et al. (1994). Low levels of serum apolipoprotein A I and A II in senile dementia. Jpn. J. Psychiatry Neurol. 48, 589–593.
LaDu, M. J., Gilligan, S. M., Lukens, J. R., Cabana, V. G., Reardon, C. A., Van Eldik, L. J., et al. (1998). Nascent astrocyte particles differ from lipoproteins in CSF. J. Neurochem. 70, 2070–2081. doi: 10.1046/j.1471-4159.1998.70052070.x
Lefterov, I., Fitz, N. F., Cronican, A. A., Fogg, A., Lefterov, P., Kodali, R., et al. (2010). Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J. Biol. Chem. 285, 36945–36957. doi: 10.1074/jbc.M110.127738
Lehti, M., Donelan, E., Abplanalp, W., Al-Massadi, O., Habegger, K. M., Weber, J., et al. (2013). High-density lipoprotein maintains skeletal muscle function by modulating cellular respiration in mice. Circulation 128, 2364–2371. doi: 10.1161/CIRCULATIONAHA.113.001551
Lehtinen, M. K., Bjornsson, C. S., Dymecki, S. M., Gilbertson, R. J., Holtzman, D. M., and Monuki, E. S. (2013). The choroid plexus and cerebrospinal fluid: emerging roles in development, disease, and therapy. J. Neurosci. 33, 17553–17559. doi: 10.1523/JNEUROSCI.3258-13.2013
Masoud, W. G., Ussher, J. R., Wang, W., Jaswal, J. S., Wagg, C. S., Dyck, J. R., et al. (2014). Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc. Res. 101, 30–38. doi: 10.1093/cvr/cvt216
McGrath, K. C., Li, X. H., Whitworth, P. T., Kasz, R., Tan, J. T., Mclennan, S. V., et al. (2014). High density lipoproteins improve insulin sensitivity in high-fat diet-fed mice by suppressing hepatic inflammation. J. Lipid Res. 55, 421–430. doi: 10.1194/jlr.M043281
Merched, A., Xia, Y., Visvikis, S., Serot, J. M., and Siest, G. (2000). Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol. Aging 21, 27–30. doi: 10.1016/S0197-4580(99)00103-7
Mineo, C., Yuhanna, I. S., Quon, M. J., and Shaul, P. W. (2003). High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J. Biol. Chem. 278, 9142–9149. doi: 10.1074/jbc.M211394200
Navab, M., Reddy, S. T., Van Lenten, B. J., and Fogelman, A. M. (2011). HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 8, 222–232. doi: 10.1038/nrcardio.2010.222
Nicolakakis, N., and Hamel, E. (2011). Neurovascular function in Alzheimer’s disease patients and experimental models. J. Cereb. Blood Flow Metab. 31, 1354–1370. doi: 10.1038/jcbfm.2011.43
Paula-Lima, A. C., Tricerri, M. A., Brito-Moreira, J., Bomfim, T. R., Oliveira, F. F., Magdesian, M. H., et al. (2009). Human apolipoprotein A-I binds amyloid-beta and prevents Abeta-induced neurotoxicity. Int. J. Biochem. Cell Biol. 41, 1361–1370. doi: 10.1016/j.biocel.2008.12.003
Petremand, J., Bulat, N., Butty, A. C., Poussin, C., Rutti, S., Au, K., et al. (2009). Involvement of 4E-BP1 in the protection induced by HDLs on pancreatic beta-cells. Mol. Endocrinol. 23, 1572–1586. doi: 10.1210/me.2008-0448
Pitas, R. E., Boyles, J. K., Lee, S. H., Hui, D., and Weisgraber, K. H. (1987). Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J. Biol. Chem. 262, 14352–14360.
Riwanto, M., and Landmesser, U. (2013). High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. J. Lipid Res. 54, 3227–3243. doi: 10.1194/jlr.R037762
Rohatgi, A., Khera, A., Berry, J. D., Givens, E. G., Ayers, C. R., Wedin, K. E., et al. (2014). HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 371, 2383–2393. doi: 10.1056/NEJMoa1409065
Roher, A. E., Maarouf, C. L., Sue, L. I., Hu, Y., Wilson, J., and Beach, T. G. (2009). Proteomics-derived cerebrospinal fluid markers of autopsy-confirmed Alzheimer’s disease. Biomarkers 14, 493–501. doi: 10.3109/13547500903108423
Rutti, S., Ehses, J. A., Sibler, R. A., Prazak, R., Rohrer, L., Georgopoulos, S., et al. (2009). Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 150, 4521–4530. doi: 10.1210/en.2009-0252
Saleheen, D., Scott, R., Javad, S., Zhao, W., Rodrigues, A., Picataggi, A., et al. (2015). Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol. 3, 507–513. doi: 10.1016/S2213-8587(15)00126-6
Shin, J., Lee, S. Y., Kim, S. J., Kim, S. H., Cho, S. J., and Kim, Y. B. (2010). Voxel-based analysis of Alzheimer’s disease PET imaging using a triplet of radiotracers: PIB, FDDNP, and FDG. Neuroimage 52, 488–496. doi: 10.1016/j.neuroimage.2010.04.013
Siebel, A. L., Natoli, A. K., Yap, F. Y., Carey, A. L., Reddy-Luthmoodoo, M., Sviridov, D., et al. (2013). Effects of high-density lipoprotein elevation with cholesteryl ester transfer protein inhibition on insulin secretion. Circ. Res. 113, 167–175. doi: 10.1161/CIRCRESAHA.113.300689
Stenkula, K. G., Lindahl, M., Petrlova, J., Dalla-Riva, J., Goransson, O., Cushman, S. W., et al. (2014). Single injections of apoA-I acutely improve in vivo glucose tolerance in insulin-resistant mice. Diabetologia 57, 797–800. doi: 10.1007/s00125-014-3162-7
Stukas, S., Robert, J., Lee, M., Kulic, I., Carr, M., Tourigny, K., et al. (2014a). Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J. Am. Heart Assoc. 3, e001156. doi: 10.1161/JAHA.114.001156
Stukas, S., Robert, J., and Wellington, C. L. (2014b). High-density lipoproteins and cerebrovascular integrity in Alzheimer’s disease. Cell Metab. 19, 574–591. doi: 10.1016/j.cmet.2014.01.003
Sturek, J. M., Castle, J. D., Trace, A. P., Page, L. C., Castle, A. M., Evans-Molina, C., et al. (2010). An intracellular role for ABCG1-mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J. Clin. Invest. 120, 2575–2589. doi: 10.1172/JCI41280
Tao, R., Hoover, H. E., Honbo, N., Kalinowski, M., Alano, C. C., Karliner, J. S., et al. (2010). High-density lipoprotein determines adult mouse cardiomyocyte fate after hypoxia-reoxygenation through lipoprotein-associated sphingosine 1-phosphate. Am. J. Physiol. Heart Circ. Physiol. 298, H1022–H1028. doi: 10.1152/ajpheart.00902.2009
Theilmeier, G., Schmidt, C., Herrmann, J., Keul, P., Schafers, M., Herrgott, I., et al. (2006). High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 114, 1403–1409. doi: 10.1161/CIRCULATIONAHA.105.607135
Ussher, J. R., Wang, W., Gandhi, M., Keung, W., Samokhvalov, V., Oka, T., et al. (2012). Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc. Res. 94, 359–369. doi: 10.1093/cvr/cvs129
Verfaillie, S. C., Adriaanse, S. M., Binnewijzend, M. A., Benedictus, M. R., Ossenkoppele, R., Wattjes, M. P., et al. (2015). Cerebral perfusion and glucose metabolism in Alzheimer’s disease and frontotemporal dementia: two sides of the same coin? Eur. Radiol. 25, 3050–3059. doi: 10.1007/s00330-015-3696-1
Vollenweider, P., Von Eckardstein, A., and Widmann, C. (2015). HDLs, diabetes, and metabolic syndrome. Handb. Exp. Pharmacol. 224, 405–421. doi: 10.1007/978-3-319-09665-0_12
von Eckardstein, A., and Widmann, C. (2014). High-density lipoprotein, beta cells, and diabetes. Cardiovasc. Res. 103, 384–394. doi: 10.1093/cvr/cvu143
Waldman, B., Jenkins, A. J., Davis, T. M., Taskinen, M. R., Scott, R., O’connell, R. L., et al. (2014). HDL-C and HDL-C/ApoA-I predict long-term progression of glycemia in established type 2 diabetes. Diabetes Care 37, 2351–2358. doi: 10.2337/dc13-2738
Wijesekara, N., Zhang, L. H., Kang, M. H., Abraham, T., Bhattacharjee, A., Warnock, G. L., et al. (2012). miR-33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes Metab. Res. Rev. 61, 653–658. doi: 10.2337/db11-0944
Zhang, Q., Zhang, Y., Feng, H., Guo, R., Jin, L., Wan, R., et al. (2011). High density lipoprotein (HDL) promotes glucose uptake in adipocytes and glycogen synthesis in muscle cells. PLoS ONE 6:e23556. doi: 10.1371/journal.pone.0023556
Keywords: HDL, glucose uptake, insulin sensitivity, insulin secretion, glucose metabolism, apolipoprotein A-I, cholesterol efflux
Citation: Siebel AL, Heywood SE and Kingwell BA (2015) HDL and glucose metabolism: current evidence and therapeutic potential. Front. Pharmacol. 6:258. doi: 10.3389/fphar.2015.00258
Received: 10 August 2015; Accepted: 19 October 2015;
Published: 31 October 2015.
Edited by:
Norman E. Miller, Magdalen College, Oxford, UKReviewed by:
Rink-Jan Lohman, The University of Queensland, AustraliaGhanshyam Upadhyay, City College of New York-CUNY, USA
Copyright © 2015 Siebel, Heywood and Kingwell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bronwyn A. Kingwell, YnJvbnd5bi5raW5nd2VsbEBiYWtlcmlkaS5lZHUuYXU=