 Chao Wang1†
Chao Wang1† Barbara K. Kemp-Harper1†
Barbara K. Kemp-Harper1† Martina Kocan2Sheng Yu Ang2
Martina Kocan2Sheng Yu Ang2 Tim D. Hewitson3,4
Tim D. Hewitson3,4 Chrishan S. Samuel1*†
Chrishan S. Samuel1*†
- 1Cardiovascular Disease Program, Biomedicine Discovery Institute, Department of Pharmacology, Monash University, Clayton, VIC, Australia
- 2Drug Discovery Biology, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC, Australia
- 3Department of Nephrology, Royal Melbourne Hospital, Parkville, VIC, Australia
- 4Department of Medicine, Royal Melbourne Hospital, University of Melbourne, Parkville, VIC, Australia
Introduction: The anti-fibrotic hormone, relaxin, has been inferred to disrupt transforming growth factor (TGF)-β1/Smad2 phosphorylation (pSmad2) signal transduction and promote collagen-degrading gelatinase activity via a nitric oxide (NO)-dependent pathway. Here, we determined the extent to which NO, soluble guanylate cyclase (sGC) and cyclic guanosine monophosphate (cGMP) were directly involved in the anti-fibrotic actions of relaxin using a selective NO scavenger and sGC inhibitor, and comparing and combining relaxin’s effects with that of an NO donor.
Methods and Results: Primary renal cortical myofibroblasts isolated from injured rat kidneys were treated with human recombinant relaxin (RLX; 16.8 nM), the NO donor, diethylamine NONOate (DEA/NO; 0.5–5 μM) or the combined effects of RLX (16.8 nM) and DEA/NO (5 μM) over 72 h. The effects of RLX (16.8 nM) and DEA/NO (5 μM) were also evaluated in the presence of the NO scavenger, hydroxocobalamin (HXC; 100 μM) or sGC inhibitor, ODQ (5 μM) over 72 h. Furthermore, the effects of RLX (30 nM), DEA/NO (5 μM) and RLX (30 nM) + DEA/NO (5 μM) on cGMP levels were directly measured, in the presence or absence of ODQ (5 μM). Changes in matrix metalloproteinase (MMP)-2, MMP-9 (cell media), pSmad2 and α-smooth muscle actin (α-SMA; a measure myofibroblast differentiation) (cell layer) were assessed by gelatin zymography and Western blotting, respectively. At the highest concentration tested, both RLX and DEA/NO promoted MMP-2 and MMP-9 levels by 25–33%, while inhibiting pSmad2 and α-SMA expression by up to 50% (all p < 0.05 vs. untreated and vehicle-treated cells). However, 5μM of DEA/NO was required to produce the effects seen with 16.8 nM of RLX over 72 h. The anti-fibrotic effects of RLX or DEA/NO alone were completely abrogated by HXC and ODQ (both p < 0.01 vs. RLX alone or DEA/NO alone), but were significantly enhanced when added in combination (all p < 0.05 vs. RLX alone). Additionally, the direct cGMP-promoting effects of RLX, DEA/NO and RLX+DEA/NO (which all increased cGMP levels by 12-16-fold over basal levels; all p < 0.01 vs. vehicle-treated cells) were significantly inhibited by pre-treatment of ODQ (all p < 0.05 vs. the respective treatments alone).
Conclusion: These findings confirmed that RLX mediates its TGF-β1-inhibitory and gelatinase-promoting effects via a NO-sGC-cGMP-dependent pathway, which was additively augmented by co-administration of DEA/NO.
Introduction
Fibrosis (organ scarring) is a universal manifestation of chronic or severe tissue injury and represents an aberrant wound healing response, which in effect results from the failure of the affected organ to repair and regenerate (Wynn and Ramalingam, 2012). The pathogenesis of fibrosis in several organs including the kidney involves consecutive but overlapping events in response to injury or stress, including inflammation, extracellular matrix (ECM) synthesis (fibrogenesis) and remodeling (Eddy, 2000; Hewitson, 2009; Wynn and Ramalingam, 2012). The rate of ECM synthesis which contributes to fibrosis is dependent on the activity of various pro-fibrotic cytokines and growth factors, primarily transforming growth factor (TGF)-β1, and on myofibroblast burden (Eddy, 2000; Hewitson, 2009; Wynn and Ramalingam, 2012). Recognized from their de novo expression of α-smooth muscle actin (α-SMA), myofibroblasts are mainly derived from the proliferation and differentiation of local fibroblasts following injury, and synthesize excessive amounts of collagen and other ECM proteins that form the basis of renal fibrosis.
In addition to synthesizing ECM components, myofibroblasts also secrete various ECM degrading proteinases, with the extent of ECM accumulation depending upon the balance between ECM synthesis and degradation. The latter can be regulated by several proteases, of which the matrix metalloproteinase (MMP) family, that are in turn regulated by tissue inhibitors of metalloproteinases (TIMPs), are the most studied (Catania et al., 2007; Hewitson, 2009). The gelatinases, MMP-2 (gelatinase A) and MMP-9 (gelatinase B) preferentially cleave basement membrane collagen IV within the kidney, while MMP-2 can also digest interstitial collagens (Aimes and Quigley, 1995). The effect of TGF-β1 on gelatinase activity has been demonstrated to be inhibitory (Howard et al., 2012), but can vary in a time- (Ogawa et al., 2011) and species/model (Saed et al., 2000; Ogawa et al., 2011; Ye et al., 2011; Bates et al., 2015)-dependent manner.
The naturally occurring hormone, relaxin, has emerged as an effective anti-fibrotic in several organs (reviewed in Samuel and Hewitson, 2006; Bennett, 2009; Du et al., 2014) including the aged (Samuel et al., 2004b; Danielson et al., 2006) and injured (Garber et al., 2001, 2003; McDonald et al., 2003; Lekgabe et al., 2005; Hewitson et al., 2010; Sasser et al., 2011; Yoshida et al., 2012, 2013) kidney. Relaxin has been well-demonstrated to inhibit TGF-β1-induced myofibroblast differentiation and myofibroblast-induced aberrant ECM/collagen production (Unemori and Amento, 1990; Unemori et al., 1996; Samuel et al., 2004a; Heeg et al., 2005; Mookerjee et al., 2009), while also being able to augment ECM/collagen degradation through its ability to up-regulate various collagenases (Unemori et al., 1996; Bennett et al., 2003; Kapila et al., 2009; Samuel et al., 2011; Ahmad et al., 2012; Chow et al., 2012; Bennett et al., 2014) and gelatinases (Lekgabe et al., 2005; Kapila et al., 2009; Conrad and Shroff, 2011; Ahmad et al., 2012; Chow et al., 2012; Sassoli et al., 2013; Frati et al., 2015; Sarwar et al., 2015) and/or inhibit TIMP activity (Unemori and Amento, 1990; Williams et al., 2001; Samuel et al., 2008; Sassoli et al., 2013; Bennett et al., 2014).
More recent studies aimed at elucidating the signal transduction mechanisms by which its TGF-β1-inhibitory effects are mediated in (myo)fibroblasts have shown that relaxin signals through its cognate G protein-coupled receptor, relaxin family peptide receptor 1 (RXFP1), to activate an extracellular signal-regulated kinase 1/2 phosphorylation (pERK1/2) and neuronal nitric oxide (NO) synthase (nNOS)-sGC/cyclic guanosine monophosphate (cGMP)-dependent pathway (Mookerjee et al., 2009; Chow et al., 2012; Sarwar et al., 2015). This primarily results in the relaxin-mediated down-regulation of Smad2 phosphorylation (pSmad2) in several organs (Heeg et al., 2005; Mookerjee et al., 2009; Bennett et al., 2014; Royce et al., 2014; Samuel et al., 2014), but also notch-1-induced down-regulation of pSmad3 in cardiac fibroblasts (Sassoli et al., 2013). The inhibition of these intracellular proteins, that promote TGF-β1 signal transduction, leads to suppression of TGF-β1-induced myofibroblast differentiation and aberrant ECM/collagen deposition. Furthermore, by down-regulating TGF-β1 activity at the pSmad2 level, relaxin has been shown to up-regulate iNOS contributing to its MMP-promoting effects (Chow et al., 2012).
As many of relaxin’s actions are mediated via NO (Baccari and Bani, 2008), studies investigating its anti-fibrotic actions have assumed a role for NO given relaxin’s ability to stimulate nNOS in cardiac and renal fibroblasts (Mookerjee et al., 2009; Chow et al., 2012; Sarwar et al., 2015) and/or up-regulate iNOS in renal and dermal fibroblasts (Chow et al., 2012). This is supported by reports of an inverse relationship between NO/cGMP and TGF-β1 (Chu and Prasad, 1999) as well as pSmad2/pSmad3 (Chen et al., 2011) activity. Moreover, both NO (Saura et al., 2005) and direct sGC stimulators (Beyer et al., 2015) have been demonstrated to inhibit TGF-β1 signal transduction. However, the actual involvement of NO in the anti-fibrotic actions of relaxin was not directly demonstrated in those studies. To address this limitation, we investigated the extent to which NO, sGC and cGMP were directly involved in the anti-fibrotic actions of relaxin, by evaluating the effects of a selective NO scavenger (hydroxocobalamin; HXC) and sGC inhibitor (ODQ) on four well-known end-points associated with the anti-fibrotic effects of relaxin: pSmad2, α-SMA (as a measure of myofibroblast differentiation), MMP-2 and MMP-9. Furthermore, we compared and combined the effects of relaxin with a NO donor (diethylamine NONOate; DEA/NO) on these four end-points, and additionally on cGMP levels, in the absence or presence of ODQ.
Materials and Methods
Cell Culture
Primary renal cortical myofibroblasts were isolated from fibrotic kidneys of Sprague-Dawley rats 3-days post-ureteric obstruction, characterized and identified as detailed before (Grimwood and Masterson, 2009; Mookerjee et al., 2009; Chow et al., 2012), and used between passages 30–35 (P30–35) for the outlined studies. Cells were re-characterized at the completion of the experiments. Based on cytochemical staining for alpha-smooth muscle actin, 100% of cells were myofibroblasts. Pericytes and smooth muscle cells were excluded by the absence of positive staining for the cytoskeletal protein desmin. Isolating cells from UUO-injured rats was approved by the appropriate Animal Ethics Committee within the University of Melbourne; which adheres to the Australian Code of Practice for the care and use of animals for scientific purposes. It has previously been shown that cells isolated from fibrotic kidneys are more active and a better reflection of their in vivo counterparts than fibroblasts that are cultured from normal kidneys (Rodemann and Muller, 1990). These cells are responsive to recombinant human relaxin (RLX) treatment as they specifically express RXFP1 (Masterson et al., 2004; Mookerjee et al., 2009; Chow et al., 2012). Rat renal myofibroblasts were cultured in Dulbecco’s Modified Eagles Medium (DMEM; GIBCO-Life Technologies, Grand Island, NY, USA) containing 10% (v/v) fetal calf serum (FCS; GIBCO-Life Technologies), penicillin (100 units/mL), streptomycin (100 μg/mL), 2% (v/v) 1M HEPES (GIBCO-Life Technologies) and 1% (v/v) 200 μM L-glutamine (Sigma–Aldrich, St Louis, MO, USA) (DMEM-FCS) and were maintained in a 37°C incubator with 95% O2/5% CO2. The FCS acted as a protein carrier to ensure the stability of rhRLX in the culture media, while the renal fibroblasts were found to grow/proliferate better in the presence of HEPES and L-glutamine. Cells were cultured to approximately 80–90% confluence before being split and passaged or seeded for experiments. All experiments described below were performed at least 3–4 separate times in duplicate.
Determining the Effects of an NO Donor, NO Scavenger and sGC Inhibitor on the Anti-fibrotic Effects of RLX
To evaluate the direct contributions of NO and sGC to the anti-fibrotic effects of RLX, renal myofibroblasts were seeded in 12-well plates at a density of 1–1.25 × 105 cells/well and treated with either RLX alone (16.8 nM or 100 ng/ml; which is an optimal concentration that had previously been shown to inhibit TGF-β1-induced pSmad2 and α-SMA expression (Masterson et al., 2004; Mookerjee et al., 2009), while promoting MMP-2 and MMP-9 levels (Chow et al., 2012) in these cells, increasing concentrations of the NO donor, DEA/NO (0.1, 1, 5 μM; Sigma–Aldrich) or the combined effects of RLX (16.8 nM) and DEA/NO (5 μM) for 72 h. DEA/NO was initially made up as a 10 μM stock solution in 0.01M NaOH (to prevent DEA/NO from decomposing) and diluted to the appropriate concentrations in DMEM-FCS immediately prior to addition. DEA/NO was added three times daily (at ~5–6 h intervals) over the 72 h treatment-period, while RLX was added once at the beginning of each experiment. Untreated cells and cells treated with 10 μM NaOH (vehicle) alone were used as controls.
In separate experiments, renal myofibroblasts were seeded as above and treated with DEA/NO (5 μM) or RLX (16.8 nM), in the absence or presence of the NO scavenger, HXC (100 μM; Sigma–Aldrich; Favaloro and Kemp-Harper, 2007; Andrews et al., 2009) or sGC inhibitor, ODQ (5 μM; Cayman Chemicals, Ann Arbor, MI, USA; Mookerjee et al., 2009; Chow et al., 2012) for 72 h. HXC or ODQ were added once daily over the 72 h treatment-period, and their effects alone were also evaluated to ensure that they did not affect basal expression of the various end-points measured. After 72 h in each case, the media and cell layers were collected and assayed as described below.
In another set of experiments, renal myofibroblasts were seeded in 48-well plates at a density of 1 × 105 cells/well and grown overnight in DMEM-FCS. Cells were serum starved for 6 h before addition of ligands, then pre-treated with or without ODQ (5 μM) for 15 min prior to stimulation with either RLX [30 nM; a concentration that had previously been used to stimulate cGMP levels in fibroblasts (Sarwar et al., 2015)] alone, DEA/NO (5 μM) alone or the combined effects of RLX (30 nM) and DEA/NO (5 μM) for 30 min. Myofibroblasts treated with vehicle (10 μM NaOH) were used to measure basal cGMP levels. cGMP accumulation was detected using the AlphaLISA cGMP Detection kit (Perkin Elmer, Hopkington, MA, USA). Cells were lysed in ice–cold 100% ethanol following addition of 80 μL of lysis buffer. Twenty microliter lysate was transferred to the 384-well optiplate (PerkinElmer) and analyzed according to the manufacturer’s instructions.
Protein Isolation and Western Blotting
After the media was removed, the cells within each well were washed with Dulbecco’s Phosphate-Buffered Saline (DPBS) (GIBCO-Life Technologies) to remove any residual media. After discarding the DPBS, cells were then treated with Accutase solution (Sigma–Aldrich, St Louis, MO, USA) and incubated at 37°C for 5 min to facilitate the harvest of cells from the 12-well plates. Harvested cells were then transferred to Eppendorf tubes and centrifuged at 7000 rpm for 10 min. Cell pellets from each well were then re-suspended in 20 μL of RIPA lysis buffer and incubated on ice for at least 30 min for cell lysis. After incubation, the cell lysates were centrifuged at 13200 rpm for 10 min at 4°C, thereafter the supernatants were collected and transferred to new Eppendorf tubes and stored at -80°C until required.
Equivalent one-third portions of each protein sample (for the analysis of pSmad2, total Smad2/3 and α-SMA) were separated on pre-cast 10% SDS-polyacrylamide separating gels (Bio-Rad, Philadelphia, PA, USA) as described before (Chow et al., 2012), and transferred to PVDF membranes using the Bio-Rad Trans-Blot Turbo transfer system (Bio-Rad, Philadelphia, PA, USA), before membranes were blocked for 1 h with 5% (w/v) skim milk powder. Membranes were then probed with primary rabbit monoclonal antibodies to either total Smad2/3 (#3102; 1:1000 dilution; Cell signaling, Danvers, MA, USA) or pSmad2 (#3108; 1:1000 dilution; Cell signaling), or with a primary mouse monoclonal antibody to α-SMA (M0851; 1:1000 dilution; DAKO, Glostrup, Denmark) for at least 16 h at 4°C. Membranes were then probed with a goat anti-rabbit antibody (1:2500 dilution; DAKO) to detect total Smad2/3 and pSmad2; while a goat anti-mouse antibody (1:2000 dilution; DAKO) was used to detect α-SMA. Membranes were then developed with the Bio-Rad Clarity Western ECL substrate kit (Bio-Rad) for 5 min according to the manufacturer’s protocol followed by visualization and imaging using the Bio-rad ChemicDoc MP. The optical density (OD) of the appropriate bands were then quantified and analyzed using Image Lab software (Bio-Rad). The measured density of pSmad2 and α-SMA were then corrected to the density of total Smad2/3, expressed as the relative ratio to that measured from the untreated or vehicle-treated control group (which was expressed as 1 in each case), and subsequently presented as the mean OD for pSmad2 or α-SMA from the n = 3–4 separate experiments performed per treatment group.
Gelatin Zymography
Changes in latent (L) and active (A) MMP-2 and MMP-9 levels that were secreted from rat renal myofibroblasts into the cell media over the 72 h experimental period, were assessed by gelatin zymography as described previously (Chow et al., 2012). Equal volumes of each sample were electrophoresed on 7.5% acrylamide gels containing 1mg/ml gelatin. Gelatinolytic activity was indicated by clear bands and densitometry of these MMP bands was performed using the Bio-rad ChemicDoc MP and Image Lab software (Bio-Rad). The mean ± SEM of each (combined latent and active) MMP was then determined and expressed as the relative ratio to that measured from the untreated or vehicle-treated control group (which was expressed as 1 in each case); from the n = 3–4 separate experiments performed per treatment group.
Statistical Analysis
All statistical analyses was performed using GraphPad Prism v6.0 (GraphPad Software Inc., La Jolla, CA, USA). All data were expressed as the mean ± SEM and analyzed by one-way ANOVA, using a Neuman–Keuls post hoc test to allow for multiple comparisons between groups; while differences in cGMP levels were analyzed by Student’s t-tests. P < 0.05 was considered statistically significant.
Results
The Individual vs. Combined Effects of RLX and DEA/NO on Measures of Renal Fibrosis
The effects of RLX (16.8 nM) on renal pSmad2, α-SMA expression, MMP-2 and MMP-9 levels were firstly compared to increasing concentrations of DEA/NO (0.1–5 μM) (Figure 1); and then combined with the highest concentration of DEA/NO evaluated (5 μM) (Figure 2). As previously demonstrated (Masterson et al., 2004; Mookerjee et al., 2009; Chow et al., 2012), RLX (16.8 nM) significantly inhibited pSmad2 and α-SMA expression by 35–50% (Figures 1A and 2A), but increased both MMP-2 and MMP-9 levels by 25–30% (Figures 1B and 2B) over 72 h in culture (all P < 0.05 vs. untreated control group). Although the magnitude of some of these RLX-induced effects were not as great as that reported previously (Masterson et al., 2004; Mookerjee et al., 2009; Chow et al., 2012), this may be due to the heterogeneity of primary cell cultures. In comparison, DEA/NO was found to concentration-dependently inhibit renal pSmad2 levels by ~10%, ~30% and ~33% when administered at 0.1, 1, and 5 μM, respectively, over 72 h in culture (all P < 0.05 vs. vehicle-treated group; Figure 1A). Likewise, DEA/NO significantly inhibited α-SMA expression by ~40 and ~60% at 1 and 5 μM, respectively, over the same time period (both P < 0.01 vs. untreated control and vehicle-treated groups; Figure 1A). On the other hand, the NO donor significantly increased MMP-2 and MMP-9 levels by ~33 and ~25%, respectively (both P < 0.05 vs. untreated control and vehicle-treated group), but only when administered at a concentration of 5 μM (Figure 1B).
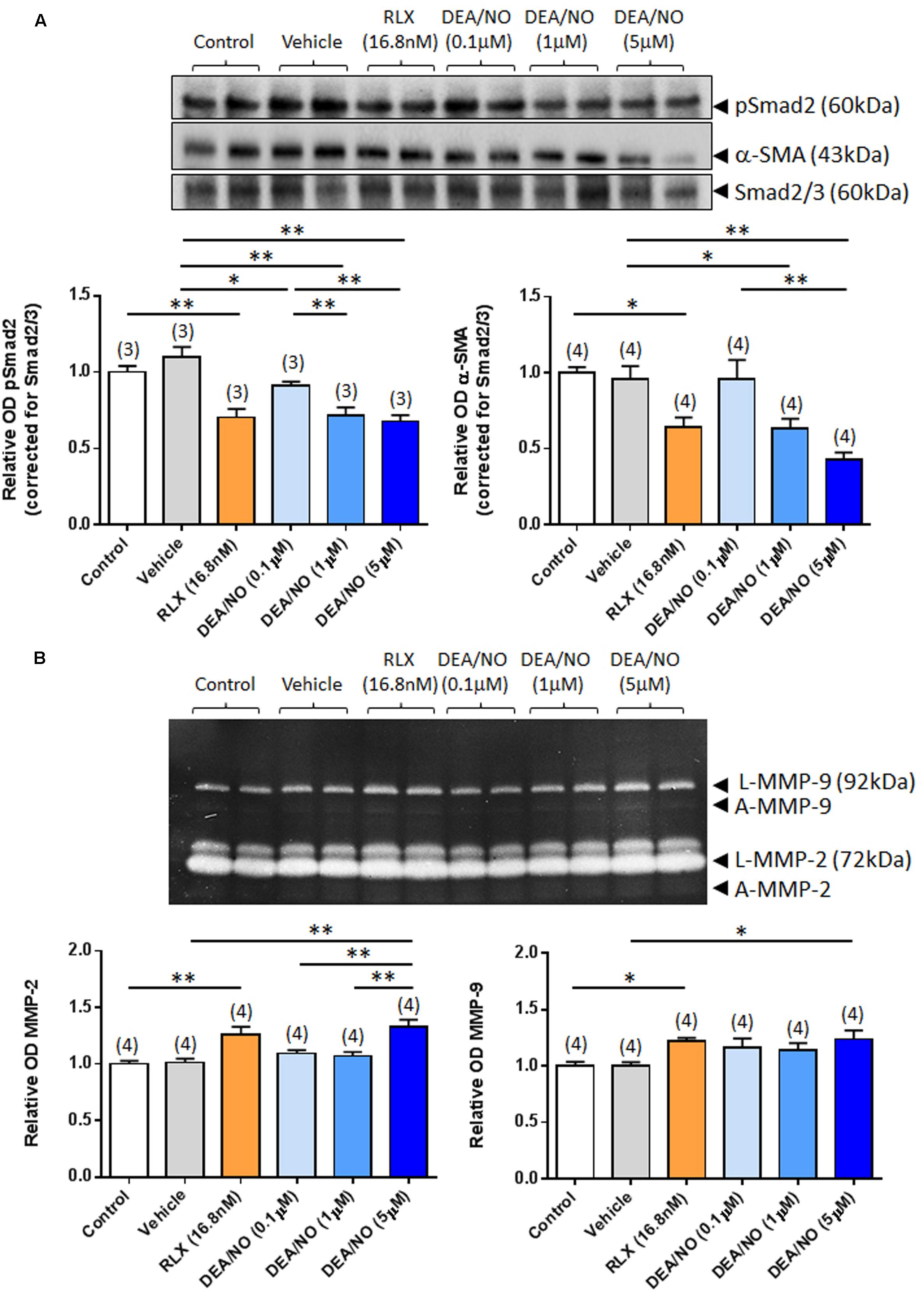
FIGURE 1. The effects of RLX vs. DEA/NO on pSmad2, α-SMA, MMP-2 and MMP-9. Shown are (A) representative (duplicate samples from one experiment) Western blots of pSmad2 and α-SMA; and (B) gelatin zymographs of latent (L) and active (A) MMP-2 and MMP-9 from untreated control rat renal myofibroblasts and cells treated with vehicle (10 μM NaOH) alone, RLX (16.8 nM) alone or increasing concentrations of DEA/NO (0.1–5 μM) alone, after 72 h in culture. Also shown are the mean ± SEM optical density (OD) of (A) pSmad2 and α-SMA (corrected for Smad2/3 loading); and (B) (latent and active) MMP-2 and MMP-9, as determined from densitometry measurements of the Western blots or zymographs. Numbers in parenthesis represent the number of independent experiments carried out in duplicate. *P < 0.05, **P < 0.01 vs. respective groups highlighted.
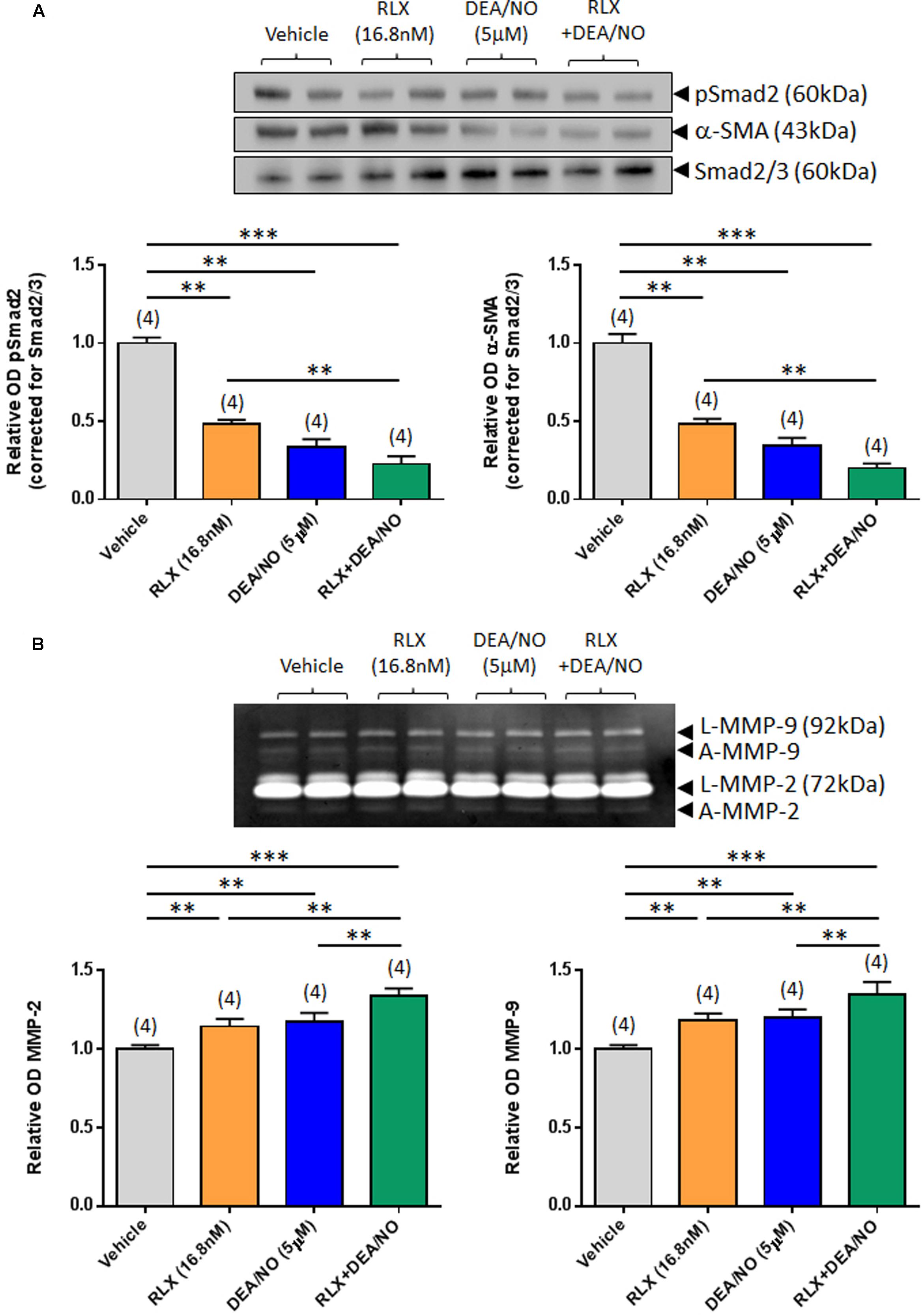
FIGURE 2. The combined effects of RLX and DEA/NO on pSmad2, α-SMA, MMP-2 and MMP-9. Shown are (A) representative (duplicate samples from one experiment) Western blots of pSmad2 and α-SMA; and (B) gelatin zymographs of latent (L) and active (A) MMP-2 and MMP-9 from vehicle (10 μM NaOH)-treated rat renal myofibroblasts and cells treated with RLX (16.8 nM) alone, DEA/NO (5 μM) alone or the combined effects of RLX (16.8 nM) alone and DEA/NO (5 μM) after 72 h in culture. Also shown are the mean ± SEM OD of (A) pSmad2 and α-SMA (corrected for Smad2/3 loading); and (B) (latent and active) MMP-2 and MMP-9, as determined from densitometry measurements of the Western blots or zymographs. Numbers in parenthesis represent the number of independent experiments carried out in duplicate. **P < 0.01, ***P < 0.001 vs. respective groups highlighted.
Combining RLX (16.8 nM) with DEA/NO (5 μM) resulted in a further additive reduction of pSmad2 and α-SMA expression over 72 h in culture, by 50–55% over the effects of RLX alone (both P < 0.001 vs. vehicle-treated group; P < 0.01 vs. RLX alone-treated group; Figure 2A). Similarly, combining RLX and DEA/NO additively promoted both MMP-2 and MMP-9 levels over 72 h, by ~15% over the effects of RLX alone or DEA alone (both P < 0.001 vs. vehicle-treated group; P < 0.05 vs. RLX alone-treated group; P < 0.05 vs. DEA/NO alone-treated group; Figure 2B).
The Effects of HXC and ODQ on the Anti-fibrotic Effects of DEA/NO and RLX
To further verify whether the effects of RLX were mediated via NO and sGC, its anti-fibrotic effects were next evaluated in the absence or presence of the NO scavenger, HXC (100 μM) and sGC inhibitor, ODQ (5 μM). The specificity of the effects of HXC and ODQ was first established in studies demonstrating that they were also able to completely abrogate the DEA/NO (5 μM)-induced down-regulation of pSmad2 and α-SMA expression (Figure 3A) in addition to the DEA/NO-mediated promotion of gelatinase expression (Figure 3B) (all P < 0.01 vs. DEA/NO alone-treated group), without affecting basal expression of these end-points over 72 h in culture.
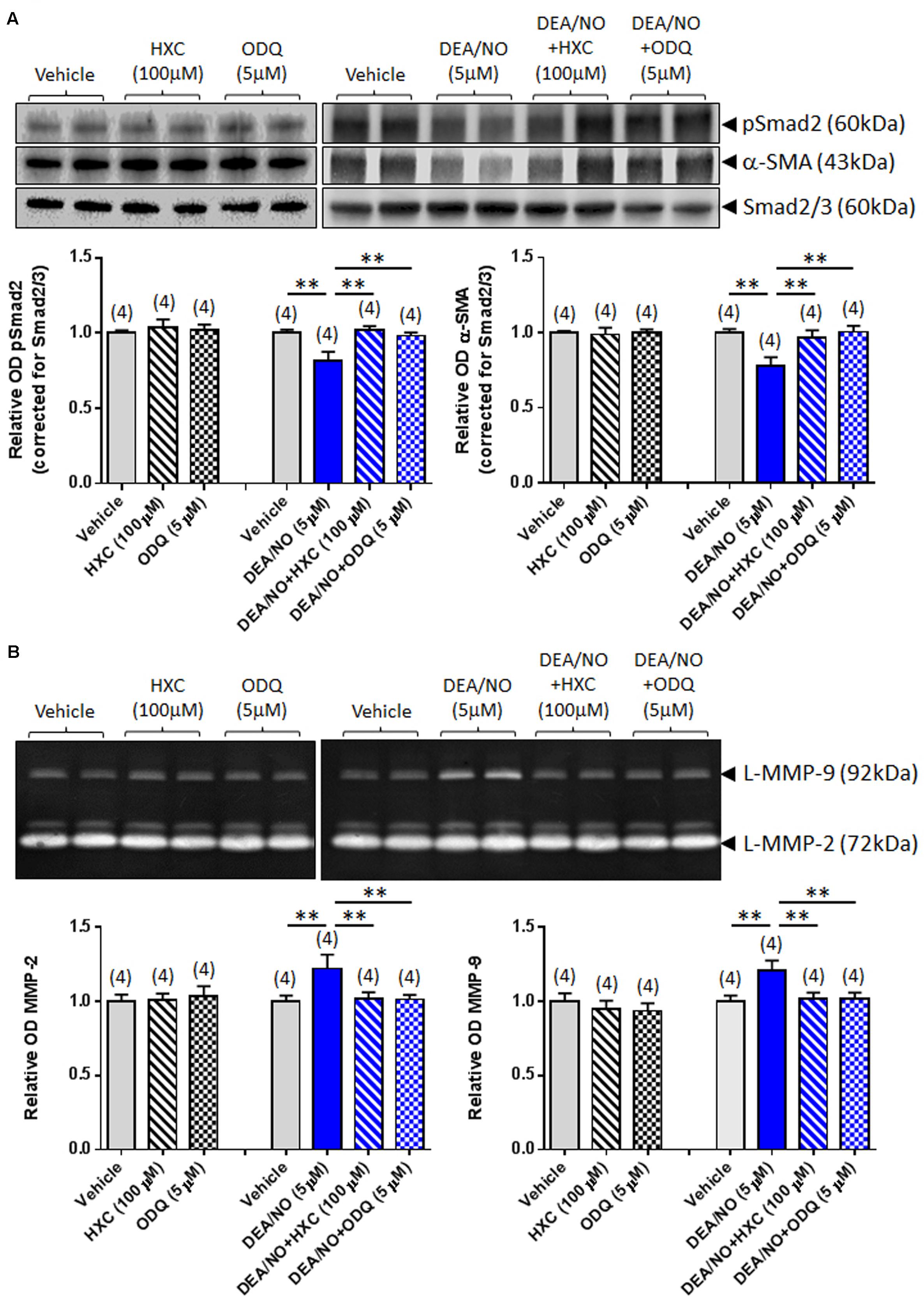
FIGURE 3. The effects of HXC and ODQ on basal and DEA/NO-mediated changes of pSmad2, α-SMA, MMP-2 and MMP-9. Shown are (A) representative (duplicate samples from one experiment) Western blots of pSmad2 and α-SMA; and (B) gelatin zymographs of latent (L) MMP-2 and MMP-9 from vehicle (10 μM NaOH)-treated rat renal myofibroblasts and cells treated with HXC (100 μM), ODQ (5 μM) alone, DEA/NO (5 μM) alone or the combined effects of DEA/NO (5 μM) and HXC (100 μM) or ODQ (5 μM), after 72 h in culture. Also shown are the mean ± SEM optical density (OD) of (A) pSmad2 and α-SMA (corrected for Smad2/3 loading); and (B) (latent) MMP-2 and MMP-9, as determined from densitometry measurements of the Western blots or zymographs. Numbers in parenthesis represent the number of independent experiments carried out in duplicate. **P < 0.01 vs. respective groups highlighted.
Similarly, the ability of RLX (16.8 nM) to down-regulate pSmad2 and α-SMA expression was completely abrogated by either co-administration of the NO scavenger, HXC (100 μM) or the sGC inhibitor, ODQ (5 μM) (both P < 0.05 vs. RLX alone-treated group; Figure 4A) over 72 h. Likewise, the ability of RLX to promote MMP-2 and MMP-9 levels was completely abrogated by co-administration of HXC or ODQ over the same time period (both P < 0.01 vs. RLX alone-treated group; Figure 4B).
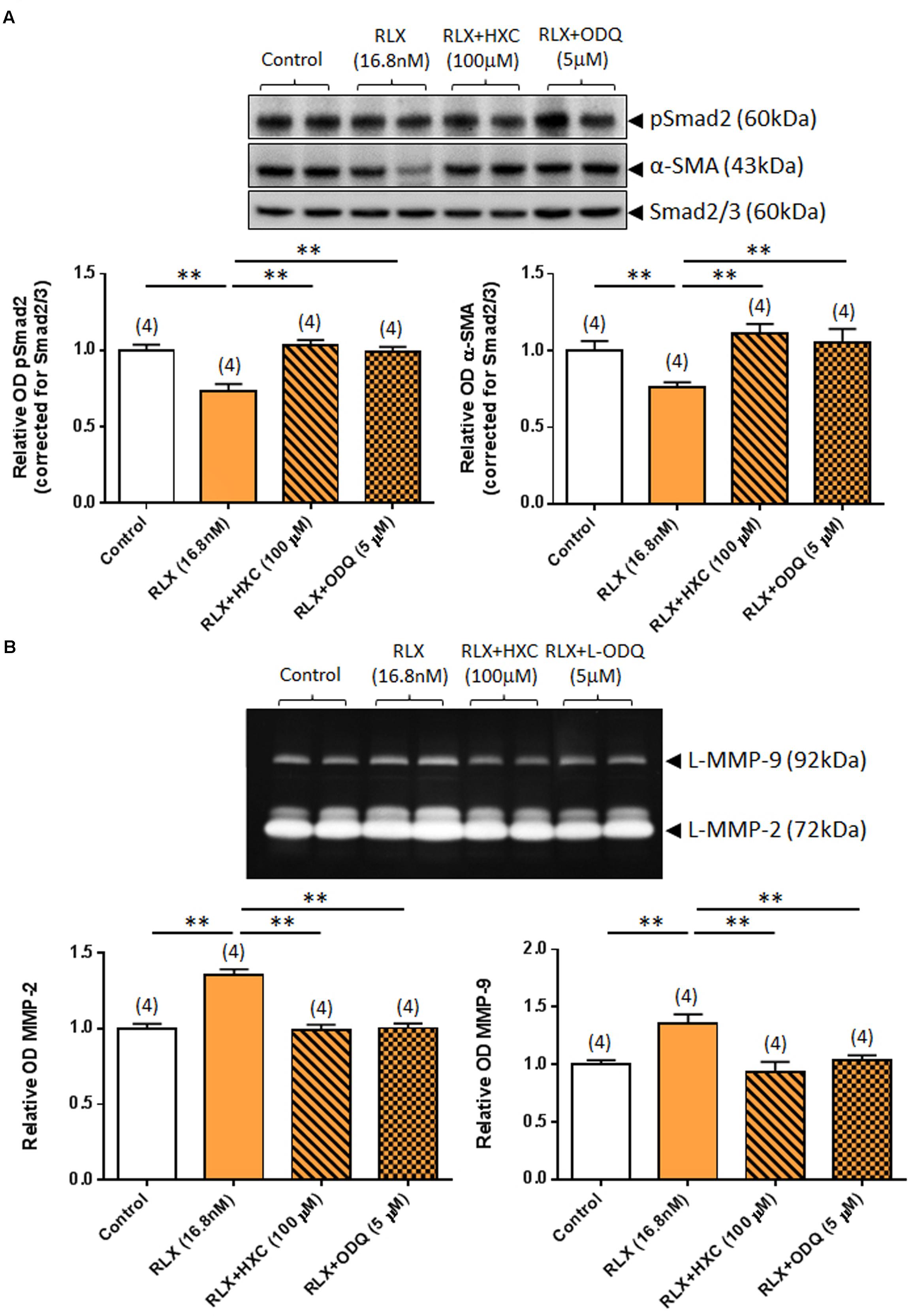
FIGURE 4. The effects of HXC and ODQ on RLX-mediated changes of pSmad2, α-SMA, MMP-2 and MMP-9. Shown are (A) representative (duplicate samples from one experiment) Western blots of pSmad2 and α-SMA; and (B) gelatin zymographs of latent (L) MMP-2 and MMP-9 from untreated (control) rat renal myofibroblasts and cells treated with RLX (16.8 nM) alone or the combined effects of RLX (16.8 nM) and HXC (100 μM) or ODQ (5 μM), after 72 h in culture. Also shown are the mean ± SEM optical density (OD) of (A) pSmad2 and α-SMA (corrected for Smad2/3 loading); and (B) (latent) MMP-2 and MMP-9, as determined from densitometry measurements of the Western blots or zymographs. Numbers in parenthesis represent the number of independent experiments carried out in duplicate. **P < 0.01 vs. respective groups highlighted.
The Effects of ODQ on the cGMP-Promoting Effects of RLX and DEA/NO
To further confirm that RLX was mediating its effects via cGMP, which is typically stimulated by sGC upon NO activation, direct measurement of cGMP accumulation from renal myofibroblasts was found to be strikingly increased by RLX (30 nM) or DEA/NO (5 μM) alone (by 12–13-fold; both P < 0.01 vs. basal (vehicle-treated) cGMP levels; Figure 5). Combining RLX and DEA/NO tended to increase cGMP levels further (by 16-fold), although this change was not statistically significant as compared to either treatment alone (Figure 5). These cGMP-promoting effects of RLX, DEA/NO or the combined effects of both were significantly inhibited by pre-administration of ODQ (5 μM) (all P < 0.05 vs. respective treatments alone; Figure 5).
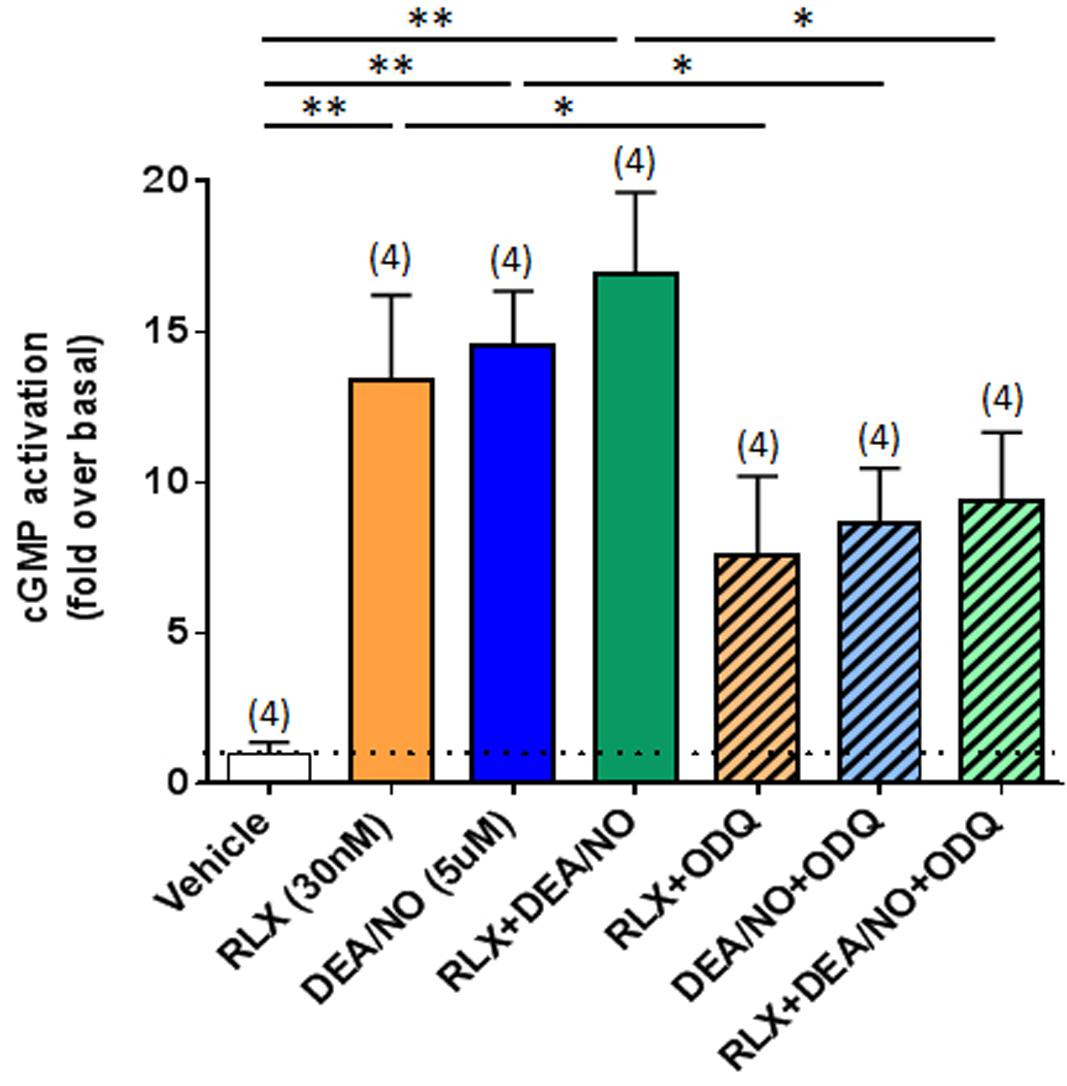
FIGURE 5. The effects of ODQ on RLX ± DEA/NO-mediated changes in cGMP accumulation. Shown are the mean ± SEM cGMP levels in rat renal myofibroblasts treated for 30 min with RLX (30 nM), DEA/NO (5 μM), or the combined effects of both; and sub-groups of correspondingly treated cells that were pre-exposed to ODQ (5 μM) for 15 min prior to administration of RLX ± DEA/NO. Numbers in parenthesis represent the number of independent experiments carried out. *P < 0.05, **P < 0.01 vs. respective groups highlighted.
Discussion
This study aimed to further elucidate and confirm the molecular mechanisms by which the anti-fibrotic hormone, relaxin, inhibited the TGF-β1/pSmad2 axis (Heeg et al., 2005; Mookerjee et al., 2009). As the effects of RLX were enhanced in the presence of an NO donor, but abrogated in the presence of an NO scavenger, to the best of our knowledge, this study has demonstrated for the first time a direct contribution of NO to the TGF-β1-inhibitory effects of RLX. Furthermore, for the first time, we have shown that RLX directly stimulates cGMP upon its administration to rat renal myofibroblasts. Previous studies have shown that RLX up-regulates nNOS expression (of the NOS isoforms) in RXFP1-expressing rat renal myofibroblasts (Masterson et al., 2004; Mookerjee et al., 2009; Chow et al., 2012) and human cardiac fibroblasts (Sarwar et al., 2015). In this context, our findings have confirmed that relaxin signals through a RXFP1-pERK1/2-nNOS-NO-sGC-cGMP-dependent pathway to mediate its anti-fibrotic actions. Additionally, this study provided the first evidence that the NO donor, DEA/NO, could also induce anti-fibrotic actions via a sGC-cGMP-dependent pathway, leading to inhibition of TGF-β1 signal transduction (at the level of pSmad2) and subsequently, the pro-fibrotic influence of TGF-β1 on renal myofibroblast differentiation, while being able to promote collagen-degrading gelatinases (MMP-2 and MMP-9). Importantly, from a therapeutic perspective, our findings strongly suggest that combining RLX with NO donors would enhance its anti-fibrotic efficacy.
The TGF-β1-inhibitory effects of RLX have been well-documented in various (myo)fibroblast culture models in vitro (Unemori and Amento, 1990; Unemori et al., 1996; Samuel et al., 2004a; Heeg et al., 2005) and animal models of disease in vivo (Garber et al., 2001; Mookerjee et al., 2009; Samuel et al., 2011, 2014; Bennett et al., 2014). As detailed in Section “Introduction,” RLX binding and activation of its cognate receptor, RXFP1, on renal myofibroblasts results in the phosphorylation of ERK1/2 and activation of nNOS to inhibit pSmad2 (Mookerjee et al., 2009; Chow et al., 2012), in the absence of any direct effects on Smad3, Smad4, or Smad7 (Heeg et al., 2005), to disrupt TGF-β1 signal transduction. The findings from this study also support previous reports (Mookerjee et al., 2009; Chow et al., 2012) in demonstrating that the selective sGC inhibitor, ODQ (at 5 μM) was able to abolish the RLX-mediated inhibition of pSmad2 and α-SMA expression, in addition to its ability to promote gelatinase levels in the rat renal myofibroblasts studied. For the first time, they now demonstrate that the cGMP-promoting effects of RLX in these cells are also inhibited by ODQ. Based on previous studies in endothelial cells, it is likely that in renal myofibroblasts cGMP generated by stimulation of sGC suppresses Smad2/3 phosphorylation via a cGMP-dependent protein kinase 1 (PKG-1)-dependent manner (Saura et al., 2005). However, this is yet to be confirmed. Subsequently, inhibition of TGF-β1 releases various MMPs including MMP-2 and MMP-9 via an iNOS-dependent mechanism (Chow et al., 2012), which collectively contributes to the anti-fibrotic actions of RLX.
The therapeutic potential of targeting the NO-sGC-cGMP signaling pathway in kidney fibrosis was further highlighted by our findings with the NO donor, DEA/NO. DEA/NO, which spontaneously decomposes to generate NO with a half-life of ~2.5 min, exhibited similar anti-fibrotic actions to that of RLX (but at relatively higher concentration), which were sensitive to HXC and ODQ and thus, mediated via the NO-sGC-cGMP signaling pathway. These findings are supported by several in vivo and in vitro studies which have indicated that other NO donors can reduce fibrosis in multiple organs. For example, the NO donors molsidomine and sodium nitroprusside (SNP) were found to protect against liver fibrosis in rodents (Ozturk et al., 2002; Ali et al., 2012), while SNP and diethylenetetra-amine NONOate (DETA/NO) were reported to modulate human lung fibroblast proliferation (Liu et al., 2001). In myofibroblast culture models, it has also been reported that exogenous NO, donated by S-nitroso-N-acetyl penicillamine (SNAP), could inhibit myofibroblast proliferation, differentiation and collagen production (Vernet et al., 2002). In accordance with the current findings, these studies attributed the anti-fibrotic actions of NO donors to inhibition of the TGF-β1/Smad2 axis (Saura et al., 2005), suggesting a potential common mode of action of NO in myofibroblasts.
In contrast, other studies have demonstrated that the exogenous NO donor, DETA/NO (125–500 μM; t1/2 ~20 h), promoted TIMP-1, TGF-β1 and collagen synthesis in fibrosis-related disorders such as keloid (hypertrophic scar) and pulmonary fibrosis (Hsu et al., 2007). Moreover, while others have reported MMP-inhibitory effects of DETA/NO (500 μM) in a vascular model (Gurjar et al., 1999), our study suggested that DEA/NO (5 μM; t1/2 ~2.5 min) was able to promote MMP-2 and MMP-9 levels in myofibroblasts (which lack eNOS Mookerjee et al., 2009). These different findings with respect to the impact of NO donors on measures of fibrosis may reflect the use of NO donors with varying half-lives and at different concentrations, modulation of distinct downstream targets of cGMP and the study of diverse disease etiologies. Although further work is required to investigate which NO donors mediate the most efficacious anti-fibrotic effects in various pathological scenarios, our study demonstrates that DEA/NO at least exerts its anti-fibrotic actions through a similar mode of action to that of RLX, and in fact additively potentiates the anti-fibrotic effects of RLX. Collectively our data highlights the therapeutic potential of targeting the NO-sGC-cGMP signalling pathway in the treatment of fibrosis. In view of this, it would of great interest to evaluate the combined anti-fibrotic effects of RLX with the new generation NO-independent sGC stimulators (BAY 41-2272; Beyer et al., 2015) and/or cGMP analogs (8-Br-cGMP; Rivero-Vilches et al., 2003), the former demonstrating excellent safety and tolerability in phase III trials in patients with pulmonary hypertension. In particular, greater synergy may be achieved between RLX and BAY 41-2272, which target canonical and non-cononical (Beyer et al., 2015) TGF-β1 signal transduction pathways, respectively.
Despite the positive implications of these findings, there were a number of limitations with this study that need to be addressed in future investigations to further validate and translate the therapeutic potential of the treatments that were investigated. Firstly, the end-points measured were all completed from a cell culture model; albeit from cells isolated from injured rat kidneys (which are more reflective of their in vivo counterparts). Having said that, the findings obtained were consistent with data obtained from TGF-β1-stimulated human renal fibroblast cell lines (Heeg et al., 2005) and RLX-treated mice in vivo (Chow et al., 2014). Secondly, only one NO donor and one NO scavenger were used. However, our findings that HXC and ODQ were able to negate the effects of both the NO donor, DEA/NO and RLX strongly suggests that the effects of RLX were mediated via a NO-sGC-dependent mechanism. Given that NO scavengers and sGC inhibitors cannot be administered to the intact animal, they cannot be used to delineate the contribution of the NO-sGC-cGMP signaling pathway to the anti-fibrotic actions of RLX in vivo. Finally, while NO and sGC levels were not directly determined, we were able to demonstrate that the RLX- and DEA/NO-induced increase in cGMP accumulation was blocked by ODQ, confirming that RLX was able to signal through a NO-sGC-cGMP-dependent mechanism to mediate its anti-fibrotic actions in vitro.
Despite these limitations, this study confirmed at least at the in vitro level, that RLX mediates its overall anti-fibrotic actions in primary renal myofibroblasts via a NO-sGC-cGMP-dependent mechanism. As RLX is currently being assessed in phase III clinical trials for its cardio-protective and vasodilatory properties (Teerlink et al., 2013) and NO donors have been evaluated for their ability to treat fibrosis (Ozturk et al., 2002; Vernet et al., 2002; Ali et al., 2012), there is tremendous potential for combining these therapies to achieve added organ protection and reduced mortality. Extending this combination approach to the in vivo level will be key to translating the findings demonstrated in the current study.
Author Contributions
Participated in research design: BK-H, CS. Conducted experiments: CW, BK-H, MK, SYA. Contributed reagents or tools: BK-H, TH, CS. Performed data analysis: CW, BK-H, MK, SA, TH, CS. Wrote or contributed to writing of manuscript: CW, BK-H, MK, TH, CS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank Corthera Inc (San Carlos, CA, USA; a subsidiary of Novartis AG, Basel, Switzerland) for providing the human recombinant relaxin used in this study. This study was supported by a National Health and Medical Research Council (NHMRC) of Australia Project Grant (GNT628634) to CS and TH, and by a NHMRC Senior Research Fellowship (GNT1041766) to CS.
References
Ahmad, N., Wang, W., Nair, R., and Kapila, S. (2012). Relaxin induces matrix-metalloproteinases-9 and -13 via RXFP1: induction of MMP-9 involves the PI3K, ERK, Akt and PKC-zeta pathways. Mol. Cell. Endocrinol. 363, 46–61. doi: 10.1016/j.mce.2012.07.006
Aimes, R. T., and Quigley, J. P. (1995). Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J. Biol. Chem. 270, 5872–5876.
Ali, G., Mohsin, S., Khan, M., Nasir, G. A., Shams, S., Khan, S. N., et al. (2012). Nitric oxide augments mesenchymal stem cell ability to repair liver fibrosis. J. Transl. Med. 10:75. doi: 10.1186/1479-5876-10-75
Andrews, K. L., Irvine, J. C., Tare, M., Apostolopoulos, J., Favaloro, J. L., Triggle, C. R., et al. (2009). A role for nitroxyl (HNO) as an endothelium-derived relaxing and hyperpolarizing factor in resistance arteries. Br. J. Pharmacol. 157, 540–550. doi: 10.1111/j.1476-5381.2009.00150.x
Baccari, M. C., and Bani, D. (2008). Relaxin and nitric oxide signalling. Curr. Prot. Pept. Sci. 9, 638–645. doi: 10.2174/138920308786733921
Bates, A. L., Pickup, M. W., Hallett, M. A., Dozier, E. A., Thomas, S., and Fingleton, B. (2015). Stromal matrix metalloproteinase 2 regulates collagen expression and promotes the outgrowth of experimental metastases. J. Path. 235, 773–783. doi: 10.1002/path.4493
Bennett, R. G. (2009). Relaxin and its role in the development and treatment of fibrosis. Transl. Res. 154, 1–6. doi: 10.1016/j.trsl.2009.03.007
Bennett, R. G., Heimann, D. G., Singh, S., Simpson, R. L., and Tuma, D. J. (2014). Relaxin decreases the severity of established hepatic fibrosis in mice. Liver Int. 34, 416–426. doi: 10.1111/liv.12247
Bennett, R. G., Kharbanda, K. K., and Tuma, D. J. (2003). Inhibition of markers of hepatic stellate cell activation by the hormone relaxin. Biochem. Pharmacol. 66, 867–874. doi: 10.1016/S0006-2952(03)00403-9
Beyer, C., Zenzmaier, C., Palumbo-Zerr, K., Mancuso, R., Distler, A., Dees, C., et al. (2015). Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFbeta signalling. Ann. Rheum. Dis. 74, 1408–1416. doi: 10.1136/annrheumdis-2013-204508
Catania, J. M., Chen, G., and Parrish, A. R. (2007). Role of matrix metalloproteinases in renal pathophysiologies. Am. J. Physiol. Renal Physiol. 292, F905–F911. doi: 10.1152/ajprenal.00421.2006
Chen, J., Shearer, G. C., Chen, Q., Healy, C. L., Beyer, A. J., Nareddy, V. B., et al. (2011). Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation 123, 584–593. doi: 10.1161/CIRCULATIONAHA.110.971853
Chow, B. S., Chew, E. G., Zhao, C., Bathgate, R. A., Hewitson, T. D., and Samuel, C. S. (2012). Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS ONE 7:e42714. doi: 10.1371/journal.pone.0042714
Chow, B. S., Kocan, M., Bosnyak, S., Sarwar, M., Wigg, B., Jones, E. S., et al. (2014). Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int. 86, 75–85. doi: 10.1038/ki.2013.518
Chu, A. J., and Prasad, J. K. (1999). Up-regulation by human recombinant transforming growth factor beta-1 of collagen production in cultured dermal fibroblasts is mediated by the inhibition of nitric oxide signaling. J. Am. Coll. Surg. 188, 271–280. doi: 10.1016/S1072-7515(98)00303-2
Conrad, K. P., and Shroff, S. G. (2011). Effects of relaxin on arterial dilation, remodeling, and mechanical properties. Curr. Hypertens. Reps. 13, 409–420. doi: 10.1007/s11906-011-0231-x
Danielson, L. A., Welford, A., and Harris, A. (2006). Relaxin improves renal function and histology in aging Munich Wistar rats. J. Am. Soc. Nephrol. 17, 1325–1333. doi: 10.1681/ASN.2005121307
Du, X. J., Hewitson, T. D., Nguyen, M. N., and Samuel, C. S. (2014). Therapeutic effects of serelaxin in acute heart failure. Circ. J. 78, 542–552. doi: 10.1253/circj.CJ-14-0014
Eddy, A. A. (2000). Molecular basis of renal fibrosis. Pediatr. Nephrol. 15, 290–301. doi: 10.1007/s004670000461
Favaloro, J. L., and Kemp-Harper, B. K. (2007). The nitroxyl anion (HNO) is a potent dilator of rat coronary vasculature. Cardiovasc. Res. 73, 587–596. doi: 10.1016/j.cardiores.2006.11.018
Frati, A., Ricci, B., Pierucci, F., Nistri, S., Bani, D., and Meacci, E. (2015). Role of sphingosine kinase/S1P axis in ECM remodeling of cardiac cells elicited by relaxin. Mol. Endocrinol. 29, 53–67. doi: 10.1210/me.2014-1201
Garber, S. L., Mirochnik, Y., Brecklin, C., Slobodskoy, L., Arruda, J. A., and Dunea, G. (2003). Effect of relaxin in two models of renal mass reduction. Am. J. Nephrol. 23, 8–12. doi: 10.1159/000066302
Garber, S. L., Mirochnik, Y., Brecklin, C. S., Unemori, E. N., Singh, A. K., Slobodskoy, L., et al. (2001). Relaxin decreases renal interstitial fibrosis and slows progression of renal disease. Kidney Int. 59, 876–882. doi: 10.1046/j.1523-1755.2001.059003876.x
Grimwood, L., and Masterson, R. (2009). Propogation and culture of renal fibroblasts. Methods Mol. Biol. 466, 25–37. doi: 10.1007/978-1-59745-352-3_3
Gurjar, M. V., Sharma, R. V., and Bhalla, R. C. (1999). eNOS gene transfer inhibits smooth muscle cell migration and MMP-2 and MMP-9 activity. Arterioscler. Thromb. Vasc. Biol. 19, 2871–2877. doi: 10.1161/01.ATV.19.12.2871
Heeg, M. H., Koziolek, M. J., Vasko, R., Schaefer, L., Sharma, K., Muller, G. A., et al. (2005). The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 68, 96–109. doi: 10.1111/j.1523-1755.2005.00384.x
Hewitson, T. D. (2009). Renal tubulointerstitial fibrosis: common but never simple. Am. J. Physiol. Renal Physiol. 296, F1239–F1244. doi: 10.1152/ajprenal.90521.2008
Hewitson, T. D., Ho, W. Y., and Samuel, C. S. (2010). Antifibrotic properties of relaxin: in vivo mechanism of action in experimental renal tubulointerstitial fibrosis. Endocrinology 151, 4938–4948. doi: 10.1210/en.2010-0286
Howard, E. W., Crider, B. J., Updike, D. L., Bullen, E. C., Parks, E. E., Haaksma, C. J., et al. (2012). MMP-2 expression by fibroblasts is suppressed by the myofibroblast phenotype. Exp. Cell Res. 318, 1542–1553. doi: 10.1016/j.yexcr.2012.03.007
Hsu, Y. C., Wang, L. F., Chien, Y. W., and Lee, W. R. (2007). Induction of TIMP-1 and HSP47 synthesis in primary keloid fibroblasts by exogenous nitric oxide. J. Dermatol. Sci. 45, 37–44. doi: 10.1016/j.jdermsci.2006.10.002
Kapila, S., Wang, W., and Uston, K. (2009). Matrix metalloproteinase induction by relaxin causes cartilage matrix degradation in target synovial joints. Ann. N. Y. Acad. Sci. 1160, 322–328. doi: 10.1111/j.1749-6632.2009.03830.x
Lekgabe, E. D., Kiriazis, H., Zhao, C., Xu, Q., Moore, X. L., Su, Y., et al. (2005). Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension 46, 412–418. doi: 10.1161/01.HYP.0000171930.00697.2f
Liu, X., Zhu, Y. K., Wang, H., Kohyama, T., Wen, F., and Rennard, S. I. (2001). EFfect of nitric oxide donors on human lung fibroblast proliferation in vitro. Chest 120, S13–S14. doi: 10.1378/chest.120.1_suppl.S13
Masterson, R., Hewitson, T. D., Kelynack, K., Martic, M., Parry, L., Bathgate, R., et al. (2004). Relaxin down-regulates renal fibroblast function and promotes matrix remodelling in vitro. Nephrol. Dial. Transpl. 19, 544–552. doi: 10.1093/ndt/gfg598
McDonald, G. A., Sarkar, P., Rennke, H., Unemori, E., Kalluri, R., and Sukhatme, V. P. (2003). Relaxin increases ubiquitin-dependent degradation of fibronectin in vitro and ameliorates renal fibrosis in vivo. Am. J. Physiol. Renal Physiol. 285, F59–F67. doi: 10.1152/ajprenal.00157.2002
Mookerjee, I., Hewitson, T. D., Halls, M. L., Summers, R. J., Mathai, M. L., Bathgate, R. A., et al. (2009). Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 23, 1219–1229. doi: 10.1096/fj.08-120857
Ogawa, K., Funaba, M., and Tsujimoto, M. (2011). The effects of TGF-beta1 on the expression of type IV collagenases in mouse peritoneal macrophages. Mol. Biol. Rep. 38, 1451–1456. doi: 10.1007/s11033-010-0250-8
Ozturk, H., Yagmur, Y., Buyukbayram, H., Dokucu, A. I., and Gurel, A. (2002). Effects of the nitric oxide donor molsidomine on the early stages of liver damage in rats with bile duct ligation: a biochemical and immunohistochemical approach. Eur. Surg. Res. 34, 285–290. doi: 10.1159/000063069
Rivero-Vilches, F. J., de Frutos, S., Saura, M., Rodriguez-Puyol, D., and Rodriguez-Puyol, M. (2003). Differential relaxing responses to particulate or soluble guanylyl cyclase activation on endothelial cells: a mechanism dependent on PKG-I alpha activation by NO/cGMP. Am. J. Physiol. Cell Physiol. 285, C891–C898. doi: 10.1152/ajpcell.00590.2002
Rodemann, H. P., and Muller, G. A. (1990). Abnormal growth and clonal proliferation of fibroblasts derived from kidneys with interstitial fibrosis. Proc. Soc. Exp. Biol. Med. 195, 57–63. doi: 10.3181/00379727-195-43118
Royce, S. G., Lim, C. X., Patel, K. P., Wang, B., Samuel, C. S., and Tang, M. L. (2014). Intranasally administered serelaxin abrogates airway remodelling and attenuates airway hyperresponsiveness in allergic airways disease. Clin. Exp. Allergy 44, 1399–1408. doi: 10.1111/cea.12391
Saed, G. M., Zhang, W., and Diamond, M. P. (2000). Effect of hypoxia on stimulatory effect of TGF-beta 1 on MMP-2 and MMP-9 activities in mouse fibroblasts. J. Soc. Gynecol. Invest. 7, 348–354. doi: 10.1016/S1071-5576(00)00079-4
Samuel, C. S., Bodaragama, H., Chew, J. Y., Widdop, R. E., Royce, S. G., and Hewitson, T. D. (2014). Serelaxin is a more efficacious antifibrotic than enalapril in an experimental model of heart disease. Hypertension 64, 315–322. doi: 10.1161/HYPERTENSIONAHA.114.03594
Samuel, C. S., Cendrawan, S., Gao, X. M., Ming, Z., Zhao, C., Kiriazis, H., et al. (2011). Relaxin remodels fibrotic healing following myocardial infarction. Lab. Invest. 91, 675–690. doi: 10.1038/labinvest.2010.198
Samuel, C. S., and Hewitson, T. D. (2006). Relaxin in cardiovascular and renal disease. Kidney Int. 69, 1498–1502. doi: 10.1038/sj.ki.5000264
Samuel, C. S., Hewitson, T. D., Zhang, Y., and Kelly, D. J. (2008). Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. Endocrinology 149, 3286–3293. doi: 10.1210/en.2008-0250
Samuel, C. S., Unemori, E. N., Mookerjee, I., Bathgate, R. A., Layfield, S. L., Mak, J., et al. (2004a). Relaxin modulates cardiac fibroblast proliferation, differentiation and collagen production and reverses cardiac fibrosis in vivo. Endocrinology 145, 4125–4133. doi: 10.1210/en.2004-0209
Samuel, C. S., Zhao, C., Bond, C. P., Hewitson, T. D., Amento, E. P., and Summers, R. J. (2004b). Relaxin-1-deficient mice develop an age-related progression of renal fibrosis. Kidney Int. 65, 2054–2064. doi: 10.1111/j.1523-1755.2004.00628.x
Sarwar, M., Samuel, C. S., Bathgate, R. A., Stewart, D. R., and Summers, R. J. (2015). Serelaxin-mediated signal transduction in human vascular cells: bell-shaped concentration-response curves reflect differential coupling to G proteins. Br. J. Pharmacol. 172, 1005–1019. doi: 10.1111/bph.12964
Sasser, J. M., Molnar, M., and Baylis, C. (2011). Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N(omega)-nitro-L-arginine methyl ester hypertensive rats. Hypertension 58, 197–204. doi: 10.1161/HYPERTENSIONAHA.110.164392
Sassoli, C., Chellini, F., Pini, A., Tani, A., Nistri, S., Nosi, D., et al. (2013). Relaxin prevents cardiac fibroblast-myofibroblast transition via notch-1-mediated inhibition of TGF-beta/Smad3 signaling. PLoS ONE 8:e63896. doi: 10.1371/journal.pone.0063896
Saura, M., Zaragoza, C., Herranz, B., Griera, M., Diez-Marques, L., Rodriguez-Puyol, D., et al. (2005). Nitric oxide regulates transforming growth factor-beta signaling in endothelial cells. Circ. Res. 97, 1115–1123. doi: 10.1161/01.RES.0000191538.76771.66
Teerlink, J. R., Cotter, G., Davison, B. A., Felker, G. M., Filippatos, G., Greenberg, B. H., et al. (2013). Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet 381, 29–39. doi: 10.1016/S0140-6736(12)61855-8
Unemori, E. N., and Amento, E. P. (1990). Relaxin modulates synthesis and secretion of procollagenase and collagen by human dermal fibroblasts. J. Biol. Chem. 265, 10681–10685.
Unemori, E. N., Pickford, L. B., Salles, A. L., Piercy, C. E., Grove, B. H., Erikson, M. E., et al. (1996). Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J. Clin. Invest. 98, 2739–2745. doi: 10.1172/JCI119099
Vernet, D., Ferrini, M. G., Valente, E. G., Magee, T. R., Bou-Gharios, G., Rajfer, J., et al. (2002). Effect of nitric oxide on the differentiation of fibroblasts into myofibroblasts in the Peyronie’s fibrotic plaque and in its rat model. Nitric Oxide 7, 262–276. doi: 10.1016/S1089-8603(02)00124-6
Williams, E. J., Benyon, R. C., Trim, N., Hadwin, R., Grove, B. H., Arthur, M. J., et al. (2001). Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut 49, 577–583. doi: 10.1136/gut.49.4.577
Wynn, T. A., and Ramalingam, T. R. (2012). Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040. doi: 10.1038/nm.2807
Ye, H., Cai, P. C., Zhou, Q., and Ma, W. L. (2011). Transforming growth factor-beta1 suppresses the up-regulation of matrix metalloproteinase-2 by lung fibroblasts in response to tumor necrosis factor-alpha. Wound Rep. Regen. 19, 392–399. doi: 10.1111/j.1524-475X.2011.00680.x
Yoshida, T., Kumagai, H., Kohsaka, T., and Ikegaya, N. (2013). Relaxin protects against renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 305, F1169–F1176. doi: 10.1152/ajprenal.00654.2012
Keywords: fibrosis, myofibroblasts, cell signaling, relaxin, transforming growth factor-β1, nitric oxide, cGMP
Citation: Wang C, Kemp-Harper BK, Kocan M, Ang SY, Hewitson TD and Samuel CS (2016) The Anti-fibrotic Actions of Relaxin Are Mediated Through a NO-sGC-cGMP-Dependent Pathway in Renal Myofibroblasts In Vitro and Enhanced by the NO Donor, Diethylamine NONOate. Front. Pharmacol. 7:91. doi: 10.3389/fphar.2016.00091
Received: 08 February 2016; Accepted: 21 March 2016;
Published: 31 March 2016.
Edited by:
Layton Harris Smith, Sanford Burnham Prebys Medical Discovery Institute, USAReviewed by:
Bin-Nan Wu, Kaohsiung Medical University, TaiwanGaetano Santulli, Columbia University, USA
Copyright © 2016 Wang, Kemp-Harper, Kocan, Ang, Hewitson and Samuel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chrishan S. Samuel, Y2hyaXNoYW4uc2FtdWVsQG1vbmFzaC5lZHU=
†These authors have contributed equally to this work.