 Nabil Eid
Nabil Eid Yuko Ito1
Yuko Ito1- 1Department of Anatomy and Cell Biology, Division of Life Sciences, Osaka Medical College, Osaka, Japan
- 2Osaka Medical College, Osaka, Japan
A growing body of evidence based on in vitro studies indicates that mitophagy (selective autophagic clearance of damaged mitochondria) is a prosurvival mechanism associated with cellular exposure to various mitochondrial stressors. Very recently, a limited number of publications on animal-based models of alcoholic fatty liver diseases have reported that Parkin-mediated mitophagy may mitigate hepatocyte apoptosis, improve mitochondrial quality and suppress steatosis (lipid accumulation). From this perspective, the authors focus on the mechanisms of Parkin mitochondrial translocation (a key consideration in mitophagy activation) and therapeutic implications of mitophagy in liver disease. DNA repair and other functions of Parkin beyond mitophagy are also briefly discussed. The paper additionally shows original data from the authors’ current research indicating enhanced hepatic mitophagy in ethanol-treated rats, which is associated with Parkin mitochondrial translocation triggered by oxidative mitochondrial DNA damage. Natural or pharmaceutical products that may trigger Parkin mitochondrial translocation in hepatocytes and/or suppress repressors of such translocation could be a potential therapeutic target in alcoholic and non-alcoholic fatty liver disease.
The Pink1–Parkin Pathway in Mitophagy
Autophagy (macroautophagy) is a prosurvival pathway for lysosomal degradation of most cellular components in response to diverse conditions of stress, such as oxidative stress, DNA damage, and lipid overload. Selective autophagic elimination of proapoptotic mitochondria is called mitophagy (Eid et al., 2013b; Lemasters, 2014). The PINK1/Parkin pathway involves the interplay of two recessive Parkinson’s-linked genes [PTEN-induced kinase 1 (PINK1) and Parkin (an E3 ubiquitin ligase)], which maintain mitochondrial homeostasis and clear dysfunctional mitochondria via mitophagy. Mutations affecting PINK1–Parkin genes cause Parkinson’s disease (PD; a neurodegenerative illness characterized by accumulation of dysfunctional mitochondria). In mammals, various effectors (including the mitophagy receptors NIX and BNIP3 and the PINK1–Parkin pathway) contribute to the elimination of damaged mitochondria under exposure to mitochondrial damaging agents (Youle and Narendra, 2011; Amadoro et al., 2014).
In healthy mammalian cells, the mitochondrial level of PINK1 is very low, while Parkin normally resides in the cytoplasm (Youle and Narendra, 2011; Eid et al., 2013a; Amadoro et al., 2014). Mitophagy is initiated by accumulation of PINK1 at the outer membrane of damaged mitochondria, resulting in the recruitment of cytoplasmic Parkin to those mitochondria. The PINK1–Parkin interaction in damaged mitochondria promotes mitophagy through protein ubiquitination and subsequent mitochondrial fragmentation and engulfment of mitochondria by LC3-mediated autophagosomes forming mitophagosomes. The latter fuse with lysosomes forming mitophagolysosomes with specific perinuclear localization (Eid et al., 2013a; Kim et al., 2013; Amadoro et al., 2014; Lemasters, 2014). Based mostly on in vitro studies, it is considered that various mechanisms (such as oxidative stress and mitochondrial depolarization/fission) and mitochondrial DNA (mtDNA) damage may activate mitophagy (Amadoro et al., 2014; Schneider and Cuervo, 2014; Zhang, 2015). Importantly, in consideration of similar mechanisms in various animal-based models, acute and chronic ethanol consumption has recently been reported to stimulate hepatic mitophagy resulting in reduction of steatosis and apoptosis (Ding et al., 2010; Eid et al., 2013a,b; Lin et al., 2013; Zhong et al., 2014; Williams et al., 2015).
Ethanol-Induced Hepatic Mitophagy is Associated with Parkin Mitochondrial Translocation Triggered by Oxidative DNA Damage
Immunoelectron microscopy (IEM) techniques play important roles in the exploration of components and trafficking in autophagic machinery, especially in relation to membranous structures that can only be clearly identified and localized using electron microscopes (Eskelinen et al., 2011; Lenzi et al., 2012; Eid et al., 2013a). As most research on Parkin-related mitophagy has involved the examination of cultured neuronal cells with focus on downstream signaling events in mitophagy, upstream signaling pathways remain comparatively poorly characterized (Grenier et al., 2013). Based on various light and electron microscopic techniques, the authors recently investigated the mechanism behind the triggering of Parkin mitochondrial translocation and mitophagy induction using a model rat liver with exposure to ethanol binge conditions (Nogales et al., 2014; Eid et al., 2016). As shown in Figure 1, compared to the control hepatocyte (Figure 1A), a marked increase in the number of mitophagic vacuoles (mitophagosomes and mitophagolysosomes; Figure 1B) was observed in the majority of normal hepatocytes in ethanol-treated rats (ETRs) within 6 h of a single intraperitoneal injection of ethanol (5 g/kg). However, the level of apoptotic hepatocytes was very low (data not shown). This enhanced mitophagy of ETRs hepatocytes was associated Parkin mitochondrial translocation as shown by IEM (Figures 1C,D) and immunofluorescence double labeling of Parkin and cytochrome c (Figures 1E,F). Also, Parkin also was detected clearly in mitophagosomes as shown in Supplementary Figure S1. Interestingly in ETRs, and as a novel unreported finding revealed by IEM, Parkin was observed to be selectively translocated to hepatocyte mitochondria and mitophagosomes enriched with 8-OHdG, which is a marker of oxidative mtDNA damage and mutagenicity (Gao et al., 2004; Guo et al., 2008) (Figure 2). We confirmed this colocalization of Parkin and 8-OHdG using immunofluorescence double labeling technique (Eid et al., 2016). Moreover, as shown in the control hepatocytes, Parkin expression was low and cytoplasmic, while the expression of 8-OHdG was very weak. Accordingly, consideration is required to determine the significance of Parkin co-localization with accumulated 8-OHdG in hepatocyte mitochondria of ETRs. In particular, there is a need for further research to establish whether there is simply a relation to the activation of cytoprotective mitophagy or whether Parkin serves other functions beyond mitophagy.
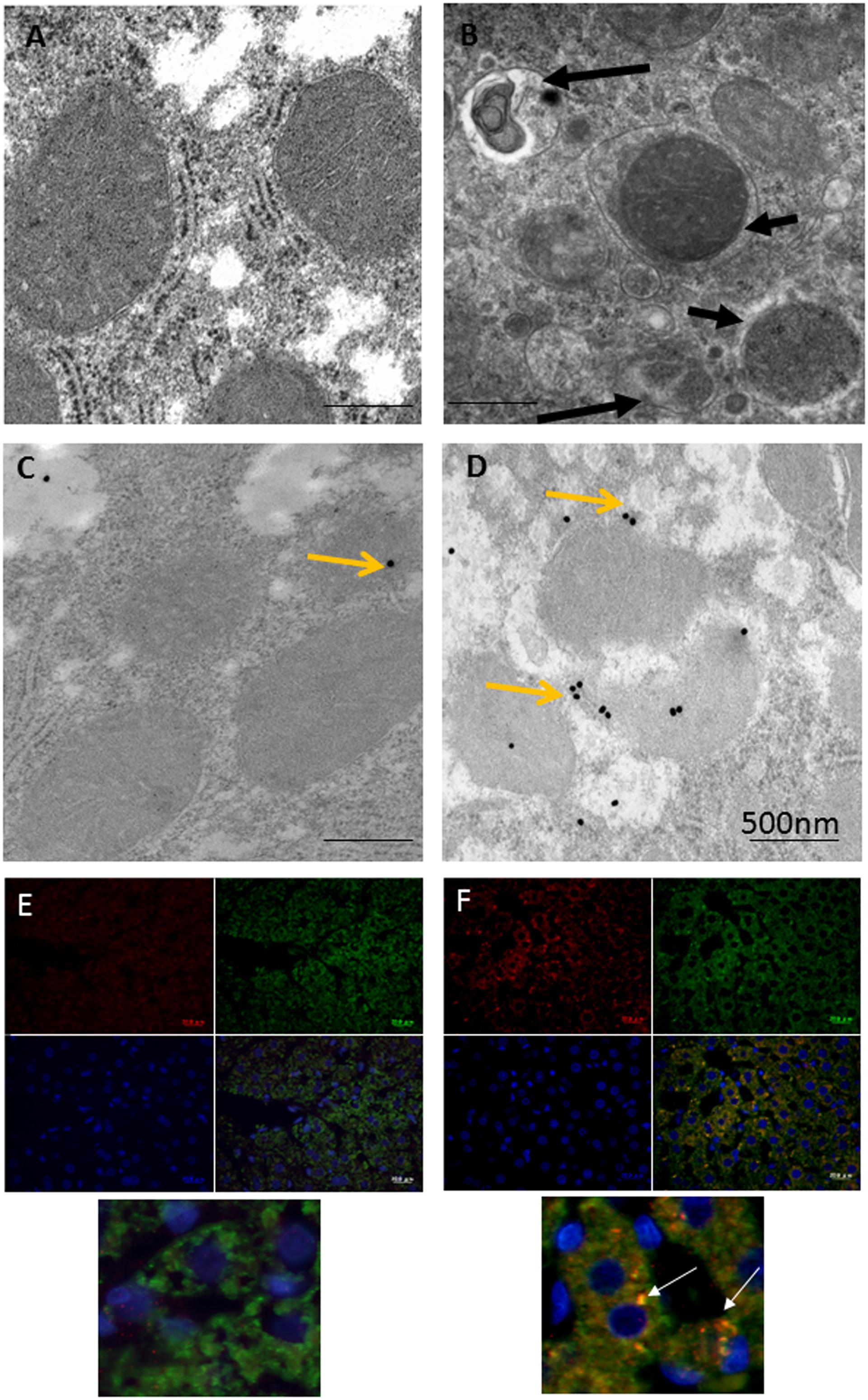
FIGURE 1. Enhanced mitophagy in hepatocytes of ETRs. (A,B) TEM of control (A) and ETRs (B). The short black arrows indicate mitophagosomes, while the long black arrows show mitophagolysosomes. (C,D) IEM of Parkin in control (C) and ETRs (D). Yellow arrows indicate 25 nm Parkin immunogold particles. (E,F) Immunofluorescence double labeling of Parkin (Red) and cytochrome c (green) in control (E) and ETRs (F). The white arrows show colocalization signals on merging (yellow) in magnified areas below. Note that DAPI (blue) is for nuclear counterstaining.
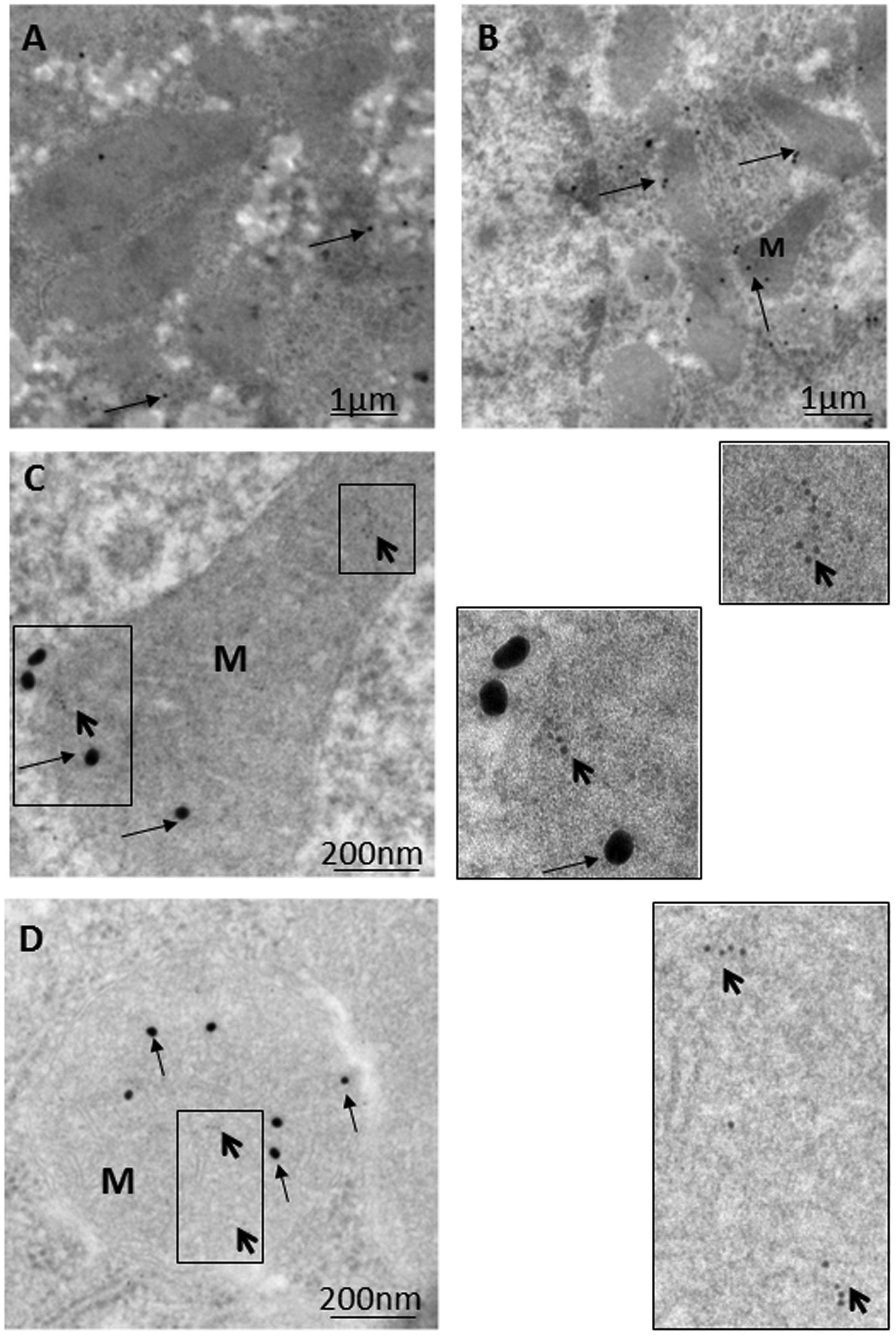
FIGURE 2. Parkin translocation to mitochondria and mitophagosomes with accumulated 8-OHdG in hepatocytes of ETRs. (A) Control while (B–D), ETRs. The boxed areas in C and D are magnified on the right. The long and short arrows indicate Parkin (25 nm) and 8-OHdG (6 nm) immunogold particles, respectively. M, mitochondrion. The method of post-embedding immunogold double labeling of Parkin (NB100-91921) and 8-OHdG (N45.1) was performed according to the manufacturer’s protocols (Aurion, Wageningen, Netherlands; http://www.aurion.nl/products/gold_sols.php) and recent publications (Eid et al., 2013a, 2016).
Parkin Co-Localization with 8-OHdG in ETR Hepatocytes: Relevance to Mitophagy
Parkin co-localization with accumulated 8-OHdG in hepatocyte mitochondria of ETRs (Figure 2) may be a signal for mitophagy induction and formation of mitophagosomes via the triggering of Parkin mitochondrial translocation (Youle and Narendra, 2011; Zhang, 2015; Eid et al., 2016). This may be supported by the study of Gmitterová et al. (2009), who reported an increase in 8-OHdG levels in the brain and peripheral tissues of PD patients, where Parkin mutations are common (Youle and Narendra, 2011; Amadoro et al., 2014). In addition, the co-localization of Parkin and 8-OHdG may represent an ideal method for monitoring mitophagy compared with other methods involving Parkin co-localization with outer mitochondrial proteins because the latter proteins could be degraded by proteasome rather than mitophagy (Yoshii et al., 2011).
Possible Functions of Parkin Beyond Mitophagy
Parkin co-localization with accumulated 8-OHdG in hepatocyte mitochondria of ETRs may be a stimulus for DNA repair and prevention of oncogenesis, as endogenous Parkin has a reported physical association with mtDNA (Rothfuss et al., 2009) and translocates to nuclei in cultured neuronal cells affected by oxidative DNA damage (Kao, 2009). Recent studies have also revealed further functions of mitochondrial Parkin in cell lines exposed to various stressors, including the suppression of mitochondrial spheroid formation (Yin and Ding, 2013; Eid et al., 2015; Khalil et al., 2015) and enhancement of mitochondrial-derived vesicle formation under oxidative stress (McLelland et al., 2014), stimulation of the selective escape of antiapoptotic proteins from mitochondria to the endoplasmic reticulum during mitophagy in stressed cells (Saita et al., 2013), and donation of mitochondrial-derived autophagosomal membranes in drug-treated breast cancer cells (Cook et al., 2014). Further studies in animal models of fatty liver disease are needed to investigate these functions of Parkin beyond mitophagy.
Pharmacological Manipulation of Parkin as a Potential Therapeutic Target in Fatty Liver Disease
A cumulative body of evidence indicates that the mitochondrion is the main target for alcohol toxicity (Hoek et al., 2002). Accordingly, the use of autophagy and/or mitophagy inducers may represent a suitable strategy for improving mitochondrial function in alcoholic fatty liver (AFL) and non-alcoholic fatty liver (NAFL) disease. The activation of autophagy through mTOR inhibitor rapamycin has been shown to enhance autophagic removal of damaged mitochondria in PD (Siddiqui et al., 2012). Lin et al. (2013) found that pharmacological promotion of autophagy by carbamazepine or rapamycin enhanced lipophagy and possibly mitophagy in animal models of AFL and NAFL disease, and that it subsequently alleviated steatosis and hepatocyte damage. Natural or pharmacological stimulation of mitophagy via the upregulation of Parkin expression and/or its mitochondrial translocation may represent a promising therapeutic target in relation to AFL and NAFL disease. The specific transcription factors that may upregulate Parkin expression in ETR hepatocytes may be linked to the FOXO3a signaling pathway, as FOXO3a has been reported to enhance the expression of LC3 in hepatocytes of ethanol-treated mice (Ni et al., 2013) and to stimulate PINK1–Parkin-mediated mitophagy with grape-derived antioxidant in stressed heart tissue (Das et al., 2014). The results of a recent study (Yu et al., 2016) indicated that quercetin suppressed chronic ethanol-induced hepatic mitochondrial damage in mice by activating mitophagy via Parkin overexpression, which was mediated by increased nuclear translocation of FOXO3a. As recently reported by the authors of this paper, PINK1 overexpression on hepatocyte mitochondrial outer membranes of acute and chronic ETRs may be a major sensor for Parkin mitochondrial translocation and recognition of damaged mitochondria by autophagic machinery (Eid et al., 2013a, 2015, 2016). Meanwhile, it has also been found that Parkin mitochondrial translocation may be repressed by cytoplasmic P53, thus preventing mitophagy induction in the myocardial muscle of stressed mice (Hoshino et al., 2013). Suppression of Parkin mitochondrial translocation repressors may therefore stimulate mitophagy and could be of therapeutic importance in hepatosteatosis. Treatment of ob/ob mice (a genetic model for NAFL) with metformin was found to enhance prosurvival mitophagy in hepatocytes by suppressing the inhibitory interaction of cytosolic p53 with Parkin, allowing Parkin mitochondrial translocation and increasing the degradation of mitofusins (Song et al., 2016). However, certain precautions should be considered in relation to the stimulation of mitophagy in AFL associated with viral infection because some studies have found that the hepatitis C virus may induce mitophagy in hepatocytes as a prosurvival mechanism, conferring protection and stimulating viral multiplication (Osna et al., 2011; Kim et al., 2013).
Conclusion
The proper understanding of the molecular mechanisms of mitophagy may be essential for the treatment of fatty liver disease induced by or associated with mitochondrial damage. IEM may be a powerful tool for detecting changes in subcellular localization of mitophagy proteins under various conditions, which may have diagnostic and therapeutic implications. Selective stimulation of Parkin-mediated mitophagy via the enhancement of its expression and/or mitochondrial translocation using natural or pharmaceutical products may have therapeutic potential for improving mitochondrial quality and survival, suppressing steatosis and preventing mutagenicity in fatty liver disease.
Ethics Statement
The animals were maintained and treated according to the guidelines set by the Experimental Animal Research Committee of Osaka Medical College.
Author Contributions
NE performed the experimental work, electron microscopic studies, and wrote the manuscript, YI participated in experimental work and design, YO participated in design of experiment and revised the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphar.2016.00100
FIGURE S1 | Immunoelectron microscopy (IEM) of Parkin in mitophagosome of ethanol-treated rat (ETR) hepatocytes. Black arrow indicate mitophagosome. White arrows in the inset indicate 6 nm Parkin immunogold particles. M, mitochondrion.
References
Amadoro, G., Corsetti, V., Florenzano, F., Atlante, A., Bobba, A., Nicolin, V., et al. (2014). Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: the pink-parkin pathway. Front. Aging Neurosci. 6:18. doi: 10.3389/fnagi.2014.00018
Cook, K. L., Soto-Pantoja, D. R., Abu-Asab, M., Clarke, P. A., Roberts, D. D., and Clarke, R. (2014). Mitochondria directly donate their membrane to form autophagosomes during a novel mechanism of parkin-associated mitophagy. Cell Biosci. 4, 16. doi: 10.1186/2045-3701-4-16
Das, S., Mitrovsky, G., Vasanthi, H. R., and Das, D. K. (2014). Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell. Longev. 2014, 345105. doi: 10.1155/2014/345105
Ding, W. X., Li, M., Chen, X., Ni, H. M., Lin, C. W., Gao, W., et al. (2010). Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 139, 1740–1752. doi: 10.1053/j.gastro.2010.07.041
Eid, N., Ito, Y., Akio Horibe, A., and Otsuki, Y. (2016). Ethanol-induced mitophagy in liver is associated with activation of the PINK1-Parkin pathway triggered by oxidative DNA damage. Histol. Histopathol. doi: 10.14670/HH-11-747 [Epub ahead of print].
Eid, N., Ito, Y., Maemura, K., and Otsuki, Y. (2013a). Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol treated rats: an immunohistochemical and electron microscopic study. J. Mol. Histol. 44, 311–326. doi: 10.1007/s10735-013-9483-x
Eid, N., Ito, Y., and Otsuki, Y. (2013b). The autophagic response to alcohol toxicity: the missing layer. J. Hepatol. 59, 398. doi: 10.1016/j.jhep.2013.03.038
Eid, N., Ito, Y., and Otsuki, Y. (2015). Mitophagy in steatotic hepatocytes of ethanol-treated wild-type and Parkin knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 309, 513–514. doi: 10.1152/ajpgi.00254.2015
Eskelinen, E. L., Reggiori, F., Baba, M., Kovács, A. L., and Seglen, P. O. (2011). Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy 7, 935–956. doi: 10.4161/auto.7.9.15760
Gao, D., Wei, C., Chen, L., Huang, J., Yang, S., and Diehl, A. M. (2004). Oxidative DNA damage and DNA repair enzyme expression are inversely related in murine models of fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 287, 1070–1077. doi: 10.1152/ajpgi.00228.2004
Gmitterová, K., Heinemann, U., Gawinecka, J., Varges, D., Ciesielczyk, B., Valkovic, P., et al. (2009). 8-OHdG in cerebrospinal fluid as a marker of oxidative stress in various neurodegenerative diseases. Neurodegener. Dis. 6, 263–269. doi: 10.1159/000237221
Grenier, K., McLelland, G. L., and Fon, E. A. (2013). Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front. Neurol. 4:100. doi: 10.3389/fneur.2013.00100
Guo, L., Yang, J. Y., and Wu, C. F. (2008). Oxidative DNA damage induced by ethanol in mouse peripheral leucocytes. Basic Clin. Pharmacol. Toxicol. 103, 222–227. doi: 10.1111/j.1742-7843.2008.00258.x
Hoek, J. B., Cahill, A., and Pastorino, J. G. (2002). Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 122, 2049–2063. doi: 10.1053/gast.2002.33613
Hoshino, A., Mita, Y., Okawa, Y., Ariyoshi, M., Iwai-Kanai, E., Ueyama, T., et al. (2013). Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 4, 2308. doi: 10.1038/ncomms3308
Kao, S. Y. (2009). DNA damage induces nuclear translocation of parkin. J. Biomed. Sci. 16, 67. doi: 10.1186/1423-0127-16-67
Khalil, B., El Fissi, N., Aouane, A., Cabirol-Pol, M. J., Rival, T., and Liévens, J. C. (2015). PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 6:e1617. doi: 10.1038/cddis.2014.581
Kim, S. J., Syed, G. H., and Siddiqui, A. (2013). Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 9:e1003285. doi: 10.1371/journal.ppat.1003285
Lemasters, J. J. (2014). Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (type 3). Redox Biol. 2, 749–754. doi: 10.1016/j.redox.2014.06.004
Lenzi, P., Marongiu, R., Falleni, A., Gelmetti, V., Busceti, C. L., Michiorri, S., et al. (2012). Subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 150, 194–217. doi: 10.4449/aib.v150i2/3.1417
Lin, C. W., Zhang, H., Li, M., Xiong, X., Chen, X., Chen, X., et al. (2013). Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 58, 993–999. doi: 10.1016/j.jhep.2013.01.011
McLelland, G. L., Soubannier, V., Chen, C. X., McBride, H. M., and Fon, E. A. (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. doi: 10.1002/embj.201385902
Ni, H. M., Du, K., You, M., and Ding, W. X. (2013). Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 183, 1815–1825. doi: 10.1016/j.ajpath.2013.08.011
Nogales, F., Rua, R. M., Ojeda, M. L., Murillo, M. L., and Carreras, O. (2014). Oral or intraperitoneal binge drinking and oxidative balance in adolescent rats. Chem. Res. Toxicol. 27, 1926–1933. doi: 10.1021/tx5002628
Osna, N. A., Thomes, P. G., and Jr, T. M. (2011). Involvement of autophagy in alcoholic liver injury and hepatitis C pathogenesis. World J. Gastroenterol. 17, 2507–2514. doi: 10.3748/wjg.v17.i20.2507
Rothfuss, O., Fischer, H., Hasegawa, T., Maisel, M., Leitner, P., Miesel, F., et al. (2009). Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 18, 3832–3850. doi: 10.1093/hmg/ddp327
Saita, S., Shirane, M., and Nakayama, K. I. (2013). Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 4, 1410. doi: 10.1038/ncomms2400
Schneider, J. L., and Cuervo, A. M. (2014). Liver autophagy: much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 11, 187–200. doi: 10.1038/nrgastro.2013.211
Siddiqui, A., Hanson, I., and Andersen, J. K. (2012). Mao-B elevation decreases parkin’s ability to efficiently clear damaged mitochondria: protective effects of rapamycin. Free Radic. Res. 46, 1011–1018. doi: 10.3109/10715762.2012.662277
Song, Y. M., Lee, W. K., Lee, Y. H., Kang, E. S., Cha, B. S., and Lee, B. W. (2016). Metformin restores parkin-mediated mitophagy, suppressed by cytosolic p53. Int. J. Mol. Sci. 17:E122. doi: 10.3390/ijms17010122
Williams, J. A., Ni, H. M., Ding, Y., and Ding, W. X. (2015). Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 309, 324–340. doi: 10.1152/ajpgi.00108.2015
Yin, X. M., and Ding, W. X. (2013). The reciprocal roles of PARK2 and mitofusins in mitophagy and mitochondrial spheroid formation. Autophagy 9, 1687–1692. doi: 10.4161/auto.24871
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Yu, X., Xu, Y., Zhang, S., Sun, J., Liu, P., Xiao, L., et al. (2016). Quercetin attenuates chronic ethanol-induced hepatic mitochondrial damage through enhanced mitophagy. Nutrients 8:E27. doi: 10.3390/nu8010027
Zhang, J. (2015). Teaching the basics of autophagy and mitophagy to redox biologists—mechanisms and experimental approaches. Redox Biol. 4, 242–259. doi: 10.1016/j.redox.2015.01.003
Keywords: ethanol, liver, lipophagy, mitophagy, Parkin, spheroids, 8-OHdG
Citation: Eid N, Ito Y and Otsuki Y (2016) Triggering of Parkin Mitochondrial Translocation in Mitophagy: Implications for Liver Diseases. Front. Pharmacol. 7:100. doi: 10.3389/fphar.2016.00100
Received: 15 February 2016; Accepted: 04 April 2016;
Published: 29 April 2016.
Edited by:
Angelo A. Izzo, University of Naples Federico II, ItalyReviewed by:
Alessandro Rufini, University of Leicester, UKRalf J. Braun, Universität Bayreuth, Germany
Xiaoxiao Sun, University of Georgia, USA
Copyright © 2016 Eid, Ito and Otsuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nabil Eid, bmFiaWxAYXJ0Lm9zYWthLW1lZC5hYy5qcA==; bmFiaWxAb3Nha2EtbWVkLmFjLmpw