 Eng Wee Chua
Eng Wee Chua Pei Yuen Ng
Pei Yuen Ng- Faculty of Pharmacy, National University of Malaysia, Kuala Lumpur, Malaysia
The launch of the MinION Access Program has caused much activity within the scientific community. MinION represents a keenly anticipated, novel addition to the current melange of commercial sequencers. Driven by the nanopore sequencing mechanism that requires minimal sample manipulation, the device is capable of generating long sequence reads in sizes (up to or exceeding 50 kb) that surpass those of all other platforms. One notable advantage of this feature is that long-range haplotypes can be more accurately resolved; such advantage is particularly pertinent to the genotyping of complex loci such as genes encoding the human leukocyte antigens, which are pivotal determinants of drug hypersensitivity. With this timely, albeit brief, review, we set out to examine the applications on which MinION has been tested thus far, the bioinformatics workflow tailored to the unique characteristics of its extended sequence reads, the device’s potential utility in the detection of genetic markers for drug hypersensitivity, and how it may eventually evolve to become fit for diagnostic purposes in the clinical setting.
Introduction
The launch of the MinION Access Program by Oxford Nanopore Technologies (ONT), a UK-based company specializing in nanopore sequencing, has caused much activity within the scientific community. The device is a miniature, third-generation sequencer in which 512 nanopores are housed and responsible for sensing single-stranded DNAs. With steadily improving sequencing accuracy, MinION has been a much welcome addition to the melange of tools deployed for diagnosing inherited drug hypersensitivity. The compact measurements of the device confer it such a degree of portability that is unsurpassable by other platforms. There has even been speculation that MinION could be transported to Mars and used to probe the existence of alien life forms (Check Hayden, 2015). Thus far, the use of MinION has been directed largely toward DNA sequencing; however, a broader range of MinION applications for RNA, microRNA, and protein analysis are being explored. A scaled-up version of MinION, which is composed of 48 flow cells and designated PromethION, has also been made available via another program, granting the participants early access to the platform (Karow, 2015).
MinION: History and Applications
The characteristics of ion channels as nanopores for DNA molecule detection have been extensively investigated since two decades ago (Kasianowicz et al., 1996; Howorka et al., 2001). When purified, single-stranded DNAs are passed through an array of pores embedded onto a membrane, characteristic current patterns that reflect the identities of DNA bases are produced (Clarke et al., 2009; Ip et al., 2015). Various improvements in the experimental setup have increased the accuracy of base-reading; these have included modification of the structure of α-hemolysin channels, addition of bulky cyclodextrin molecules to reduce the speed of DNAs translocating through the pores, introduction of hairpin polynucleotides to connect unzipped, double-stranded DNAs, and adjustment of salt concentrations in the buffer to alter the voltage across the membrane (Clarke et al., 2009; Brown et al., 2012). These changes have ultimately led to the release of MinION. Compared with existing second-generation sequencers such as MiSeq and Ion Torrent that sequence up to 400 bp, MinION is deemed ideal for DNA sequencing as it can generate long reads up to or exceeding 50 kb (Jain et al., 2015). Moreover, it does not require pre-amplification of the sample, which removes potential bias in the data that could be introduced by polymerase chain reactions (PCRs).
MinION has been tested in diverse applications, ranging from early work on bacterial sequencing and identification, to recent discovery of its ability to detect pathogens in human plasma and that to distinguish methylated DNA bases from their unmethylated counterparts (Figure 1). Extended sequences generated by MinION have been used to supplement better quality MiSeq data in constructing the genomes of Bacteroides fragilis and Saccharomyces cerevisiae (Goodwin et al., 2015; Risse et al., 2015). Further enhancement of bioinformatics workflow has allowed de novo genome assembly for the Escherichia coli K-12 MG1655 strain, whereby 98.4% nucleotide accuracy was noted across the 4.6-mb reconstructed genome (Loman et al., 2015). Such high-quality data output from the device has later been translated into differentiating three closely related poxviruses, namely cowpox, vaccinia-MVA, and vaccinia-Lister (Kilianski et al., 2015). The portability of MinION and relatively rapid sample preparation and data generation associated with the sequencer have positioned it to be a useful tool in the recent Ebola epidemic, wherein it was used on-site for monitoring the evolution of Ebola virus in a series of clinical samples. Similar principles of viral identification from human blood have also been applied to sequencing chikungunya and hepatitis C viruses (Greninger et al., 2015; Quick et al., 2016).
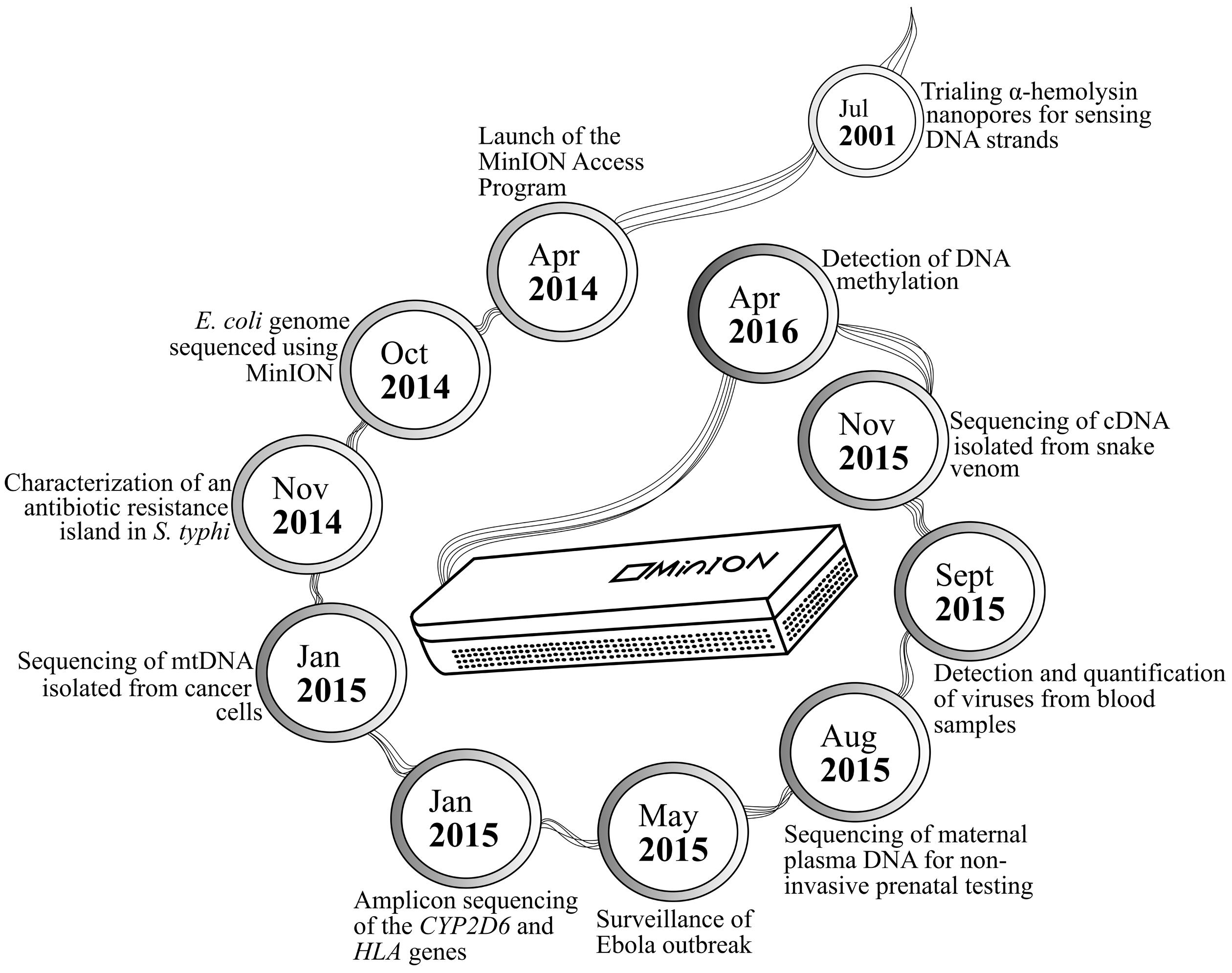
FIGURE 1. MinION applications presented in chronological order.
In the detection of DNA methylation patterns in human samples, the subtle differences in the electrical signals produced by methylated cytosine bases could also be picked up by MinION (Simpson et al., 2016). This is an exciting breakthrough as the conventional identification of these molecular modifications requires a more complex and specialized sample preparation approach, such as methylated DNA immunoprecipitation followed by sequencing, or bisulfite genomic sequencing (Frommer et al., 1992; Corley et al., 2015). Bisulfite sequencing, the gold standard for detecting DNA methylation, indirectly allows differentiation of methylated cytosines via selective conversion of unmethylated cytosines to uracils, which are then detected as thymine post-PCR (Li and Tollefsbol, 2011). In contrast, the MinION-based workflow is much simpler: a statistical model has been developed and optimized for direct identification of 5-methylcytosine from samples prepared by ONT’s standard protocol (Simpson et al., 2016). Though a direct causal relationship between drug hypersensitivity and DNA methylation has not been observed, the epigenetic phenomenon has been proven to mediate other processes such as disease pathogenesis and the effectiveness of a number of drugs (Beyrouthy et al., 2009; Anier et al., 2010).
Processing of Nanopore Sequencing Data
Central to the processing of nanopore sequencing data is the hidden Markov model (Eddy, 2004), which has been applied to various stages of sequence analysis from base-calling, fine-tuning alignment, to variant discovery (Jain et al., 2015; Szalay and Golovchenko, 2015). For base-calling, this model yields statistical deductions about the underlying DNA sequences (hidden state) based on a series of emitted observations. The successive ionic perturbations (events) caused by 6-mer DNAs (or 5-mer DNAs for now-obsolete workflows) that are drawn through the nanopores are rendered into DNA sequences based on known pairs of DNA sextets and corresponding current values (Timp et al., 2012; Szalay and Golovchenko, 2015). A major obstacle to this approach is that the current levels for all possible 6-mer combinations comprise a continuum of electrical signals rather than segregate into discrete patterns that could be unambiguously interpreted. To tease out these ionic signatures, additional clues are gained from neighboring sequences (Timp et al., 2012). For instance, suppose we were to infer a 6-mer DNA from an electrical signal, which indicated there were two possibilities: TACGTA and TACGTT. We knew that, on most occasions, the preceding sequence, ATACGT, was likely to transition to TACGTA; thus TACGTA should have been the sequence motif from which the signal had originated (Timp et al., 2012). Though this example is rather simplistic, it serves to illustrate the effectiveness of the Markov model. The emission and transition probabilities can be derived from datasets that are used to train the model.
Similarly, for sequence alignment, maximum likelihood estimates can be computed for all nanopore sequencing error types within a Markov network, i.e., insertions, deletions, and substitutions. These estimates are then used to ascertain whether a reference-discordant read is indeed misaligned. For instance, as A–T or T–A miscalls are unlikely, sequence alignments containing these mismatches may well have been incorrectly placed. Alternately, the aligner may not be at fault and the discrepancies may have arisen from inherent DNA variation (Jain et al., 2015). This strategy has been extrapolated to enhance alignments generated by existing aligners and subsequently detect variants deliberately introduced into a phage genome reference (Jain et al., 2015).
An example of a bioinformatics pipeline for MinION-generated data is shown in Figure 2. Raw electrical signals can be base-called by a tool provided by ONT, Metrichor, or other open-source software (Boža et al., 2016; David et al., 2016). DNA sequences produced from the nanopores are stored in the FAST5 format, alongside other types of data such as the run statistics. All data are stratified and placed within predefined categories. Pre-alignment processing typically requires extraction of FASTA or FASTQ sequences from the FAST5 files, using Poretools (Python-based; Loman and Quinlan, 2014) or poRe (written in R; Watson et al., 2015). A number of long-read aligners have been tested thus far on nanopore sequences, including BLASR, BWA-MEM, LASTZ, and LAST. One of the challenges unique to long-read alignment lies in quickly finding short matches, termed seeds or anchors, between two sequences during a preliminary round of alignment (Li and Durbin, 2009; Chaisson and Tesler, 2012; Li, 2013). With longer sequences, the numbers of possible matches and mismatches are larger; hence, the operation of the aligners would be affected, to a greater extent, by the efficiency of their seed-and-extend algorithm.
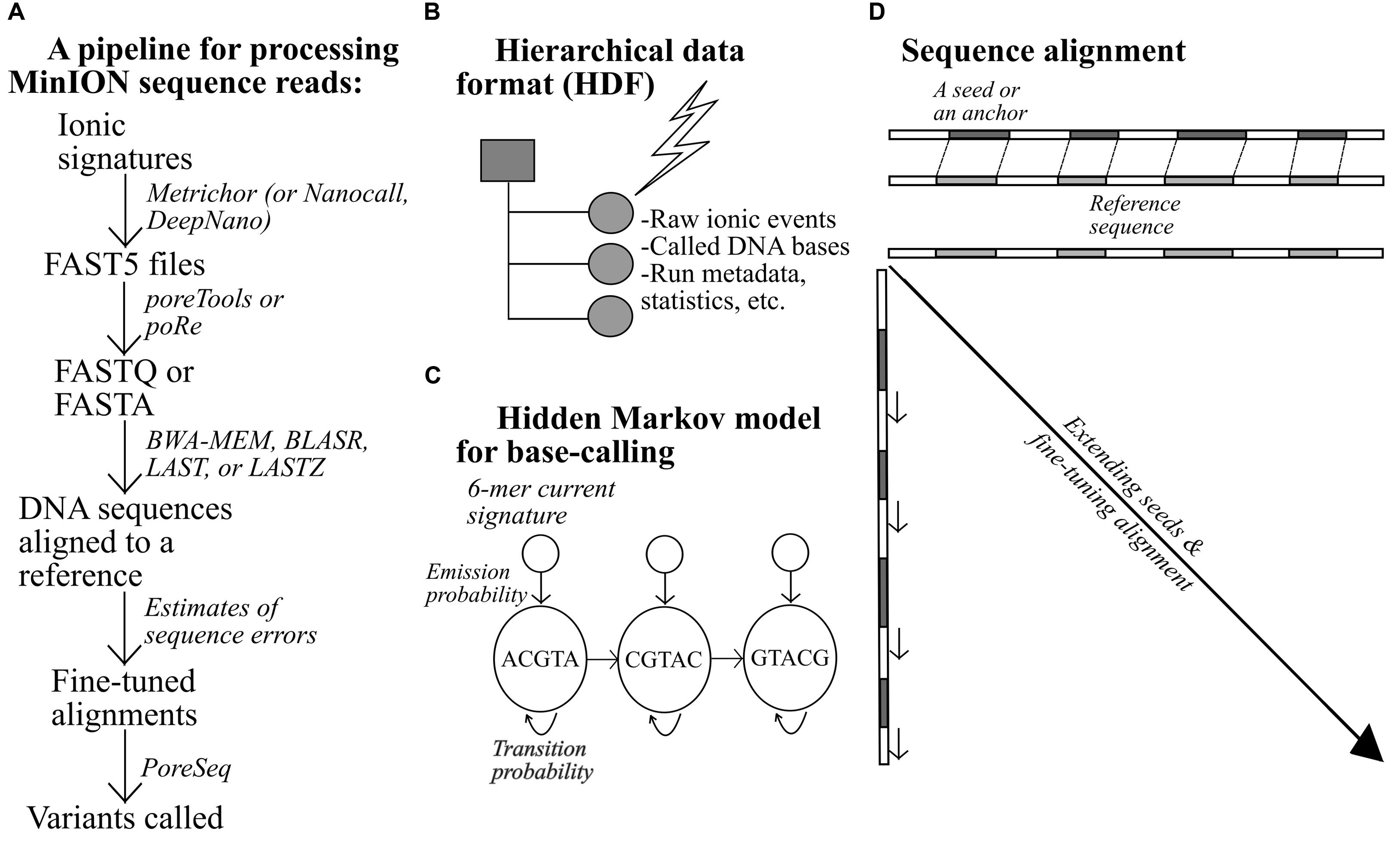
FIGURE 2. An example of a pipeline for the processing of MinION data (A), (B–D) alongside additional notes for relevant bioinformatics-related concepts.
BLASR and BWA-MEM first invoke Burrow-Wheelers transform to create easily searchable, sorted strings (index) of a reference genome to facilitate initial side-by-side check that pinpoints short exact matches, which are subsequently extended or refined to form longer DNA stretches (Chaisson and Tesler, 2012; Li, 2013). LASTZ resembles BLASR and BWA-MEM but with two principal variations. First, the aligner employs a different indexing mechanism and dissects the reference sequence into equally sized, overlapping segments to ease sequence comparison. Second, it does not require perfect similarity for qualifying a short match as an acceptable seed; some degree of discrepancy is permitted (Harris, 2007). LAST differs from LASTZ in that it can resolve repeat-rich sequences more successfully (Kiełbasa et al., 2011). When aligned, the sequences can be further scrutinized using NanoOK, which works out the read-length distribution, the occurrence of k-mers, the depths of coverage across targets, and other relevant statistics (Leggett et al., 2015). With accurate alignments, high-confidence DNA variant calls can then be generated.
Pharmacogenetics: A Classic Case of Abacavir Hypersensitivity
Abacavir, a nucleoside reverse-transcriptase inhibitor used to treat HIV-1 infection, causes a potentially fatal hypersensitivity reaction, to which Caucasians are notably susceptible, in 4–9% of individuals exposed to the drug (Hetherington et al., 2002; Mallal et al., 2002; Symonds et al., 2002; Martin et al., 2004). Abacavir-induced hypersensitivity remains, to date, one of the few rewarding examples of pharmacogenetics-guided therapy. Genetic predisposition, specifically the presence of the HLA-B∗57:01 allele (human leukocyte antigen), is a strong predictor of abacavir hypersensitivity. The association, first reported in a Western Australian population (Mallal et al., 2002), has since been replicated in many other studies (Hetherington et al., 2002; Hughes et al., 2004a; Rauch et al., 2006; Rodriguez-Novoa et al., 2007; Waters et al., 2007; Zucman et al., 2007; Mallal et al., 2008). The HLA-B∗57:01 allele has a high degree of penetrance; the pooled odds ratio from three studies was computed to be 29. Nevertheless, not all HLA-B∗57:01 carriers would be hypersensitive toward abacavir; of every ten individuals harboring the allele, only five would indeed develop a reaction (Hetherington et al., 2002; Mallal et al., 2002; Hughes et al., 2004a). It is likely that other gene loci or pathways, particularly those implicated in the conversion of abacavir into allergenic metabolites, have also contributed to the pathogenesis of abacavir hypersensitivity (Martin et al., 2004, 2012).
Screening for HLA-B∗57:01 carriage, and using this information to preclude susceptible individuals from receiving abacavir, substantially reduced the incidence of abacavir hypersensitivity, hence relieving the costs that would have been incurred by the management of these reactions (Hughes et al., 2004a; Schackman et al., 2008). The decrease in the incidence of hypersensitivity reactions ranged from two-fold (Waters et al., 2007; Mallal et al., 2008) or four-fold (Rauch et al., 2006) to complete eradication (Zucman et al., 2007). The cost-efficiency of HLA-B∗57:01 testing is ethnicity-dependent, being largest in an all-Caucasian or a predominantly Caucasian population. Individuals of other ethnic origins, such as Africans (Hetherington et al., 2002; Hughes et al., 2004b) and Taiwanese (Sun et al., 2007), derive little benefit from such a discriminative strategy. The risk of developing abacavir hypersensitivity is considerably lower in both populations (Symonds et al., 2002; Sun et al., 2007). For instance, the incidence of abacavir-induced hypersensitivity is only 0.9% among Taiwanese individuals, coinciding with their equally rare carriage of the HLA-B∗57:01 allele (0.3% versus about 8% in Whites; Rauch et al., 2006; Sun et al., 2007). Despite the obvious influence of individual ethnic backgrounds, the Clinical Pharmacogenetics Implementation Consortium has recommended HLA-B∗57:01 screening for all patients before they are given abacavir (Martin et al., 2012, 2014). Distinguishing HLA alleles could be challenging, as these alleles may vary at only a few positions within their second and third exons (Robinson et al., 2015).
Use of the MinION Device to Genotype HLA Alleles
Several test options exist for HLA-B∗57:01 screening, with sequence-based methods being most technically complex and deemed unsuited for routine use (Martin et al., 2012). These methods often rely on PCRs to enrich the desired HLA regions (Carapito et al., 2016). In a bespoke, PCR-based pipeline (Ammar et al., 2015), the HLA-A and HLA-B regions were amplified in two long-range PCRs and the resultant products (~4 kb each) were sequenced on MinION. The accuracy of the nanopore reads was expectedly low, with the proportion of reference-discordant bases nearing 30%. The authors surmised that this could have adversely affected the genotyping results: the two samples trialed using this method were both erroneously typed. The possibility of the HLA haplotypes being obscured by PCR recombination or chimerism was alluded to but not thoroughly discussed (Ammar et al., 2015; Laver et al., 2016).
Cross-over extension or template switching is a well-known PCR artifact whereby incompletely synthesized PCR products (mega-primers) anneal to new templates giving rise to chimeric alleles (Odelberg et al., 1995). Recombined alleles would confound haplotype interpretation and it is noteworthy that such errors are clinically significant and could diminish the efficacy of MinION-centric approaches for HLA genotyping. For instance, in the case of characterizing the BCR-ABL1 gene in chronic myeloid leukemia, the occurrence of PCR recombination was found to create artificial compound BCR-ABL1 mutations, potentially misguiding anti-cancer therapy. After two rounds of PCR, totalling 80 cycles, nearly 50% of the amplicons were noted to be chimeric (Parker et al., 2014).
Several alterations could be made to the PCR protocol to attenuate template switching. For instance, the extension time could be prolonged to ensure complete synthesis of PCR products in each cycle; or the number of PCR cycles curtailed to reduce the probability of PCR recombination (McDonald et al., 2002; Laver et al., 2016). Cross-over events are more likely to occur near the end of PCR cycling or following numerous rounds of extension, during which the synthesized products are most concentrated. Alternatively, a different mechanism of enrichment could be adopted to obviate the need for these changes. Short, complementary oligonucleotides, acting as probes, can efficiently capture target HLA regions from a pool of genomic DNAs that have been trimmed to a pre-defined size range (Wittig et al., 2015). For MinION, it may be necessary to opt for longer probes (Karamitros and Magiorkinis, 2015) and a much less severe DNA fragmentation protocol, in order to preserve the capacity of the device to produce very long reads for the assembly of large-scale haplotypes.
Presently, a more pressing concern over MinION is perhaps its low base-calling accuracy; the error rate has frequently been estimated to exceed 10% (Check Hayden, 2015). Previous attempts at overcoming this problem have tackled chiefly two aspects of the sequencing workflow: conversion of input DNAs into sequenceable templates; and translation of ionic events into DNA bases. The first tactic derives benefit from redundant sequencing in that the input DNA is circularized and amplified in a segmental manner (rolling circle amplification) to generate tandem copies (≥6) of a segment for consensus sequence determination (Li et al., 2016). This has resulted in greatly increased read accuracy (>97%) that approach those attainable by the second-generation platforms (Quail et al., 2012). However, the cost of such improved accuracy is a reduced net output from MinION; in other words, fewer bases are ultimately emitted per run. The second tactic is based on statistical learning enhanced by the rapidly expanding MinION datasets that are openly accessible. This has enabled mature hidden Markov models to be built for base-calling, establishing more precise event-to-sequence patterns. The construction of a sophisticated artificial neural network for DNA sequence deduction has also been suggested as a solution to the conundrum (Jain et al., 2015; Boža et al., 2016; David et al., 2016). Currently, it is uncertain which of the two strategies is superior: modified template preparation which adds some degree of complexity, or better trained in silico algorithms?
The relatively time-consuming nature of sequence-based HLA typing constitutes another limitation that must be overcome to enable routine use of the technique. For instance, it may not be uncommon for a MinION sequencing run to take one day, though it is possible to analyze the data prior to conclusion of the run. The requirement for long-range PCR amplification to isolate HLA genes could easily lengthen the procedure to span two days. For the synthesis of long amplicons, protracted elongation time is an inevitable bottleneck; 30–60 s are typically needed for the polymerization of ~1000 nucleotides. A probe-based protocol may entail an even longer time of 3–4 days; but it offers the advantage of eradicating PCR chimerism by needing only 18 cycles of amplification for the enrichment of captured fragments (Karamitros and Magiorkinis, 2015; Laver et al., 2016). On the other hand, an expedited long PCR protocol may be formulated from the following ingredients: whole-blood PCR which eliminates the need for DNA extraction (Mercier et al., 1990), ultra-fast PCR empowered by prompt temperature switches (Wheeler et al., 2011), and highly processive Taq polymerases capable of incorporating nucleotides at a faster rate (Böhlke et al., 2000).
Conclusion
As we have pointed out, several issues warrant deliberation before MinION-based HLA typing could be considered for clinical use. The unsatisfactory data accuracy is still an unresolved issue. A modified preparatory protocol that unifies sequence information from tandem copies of a DNA segment has been shown to augment base-calling accuracy (Li et al., 2016). It may be worthwhile to compare the performance of this technique with other in silico approaches. The turnaround time for sequence-based tests needs to be drastically shortened; also, the testing process should be complemented by a streamlined mechanism for data analysis, interpretation, and reporting. Spartan RX, a panel indicated for the identification of CYP2C19 poor metabolizers, can generate the required result within 1 h of sample acquisition (Spartan Bioscience Inc., 2016). Above all, the actual utility of sequence-based tests begs the question as to whether the level of HLA genotype resolution achieved by other quicker and less laborious methods, such as allele-specific PCR, is already sufficient in the clinical setting (Martin et al., 2012). Despite these uncertainties, the diverse utility of MinION is evident in the assortment of applications on which it has been trialed. We are optimistic that MinION will be eventually morphed into a potent tool for the diagnosis of drug hypersensitivity.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ammar, R., Paton, T. A., Torti, D., Shlien, A., and Bader, G. D. (2015). Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Res. 4:17. doi: 10.12688/f1000research.6037.1
Anier, K., Malinovskaja, K., Aonurm-Helm, A., Zharkovsky, A., and Kalda, A. (2010). DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology 35, 2450–2461. doi: 10.1038/npp.2010.128
Beyrouthy, M. J., Garner, K. M., Hever, M. P., Freemantle, S. J., Eastman, A., Dmitrovsky, E., et al. (2009). High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 69, 9360–9366. doi: 10.1158/0008-5472.CAN-09-1490
Böhlke, K., Pisani, F. M., Vorgias, C. E., Frey, B., Sobek, H., Rossi, M., et al. (2000). PCR performance of the B-type DNA polymerase from the thermophilic euryarchaeon Thermococcus aggregans improved by mutations in the Y-GG/A motif. Nucleic Acids Res. 28, 3910–3917. doi: 10.1093/nar/28.20.3910
Boža, V., Brejová, B., and Vinař, T. (2016). DeepNano: deep recurrent neural networks for base calling in MinION nanopore reads. arXiv:1603.09195.
Brown, C. G., Clarke, J. A., and Heron, A. J. (2012). Hairpin Loop Method for Double Strand Polynucleotide Sequencing using Transmembrane Pores. U.S. Patent no 14/234,698. Washington, DC: U.S. Patent and Trademark Office.
Carapito, R., Radosavljevic, M., and Bahram, S. (2016). Next-generationsequencing of the HLA locus: methods and impacts on HLA typing, population genetics and disease association studies. Hum. Immunol. doi: 10.1016/j.humimm.2016.04.002.
Chaisson, M. J., and Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13:238. doi: 10.1186/1471-2105-13-238
Check Hayden, E. (2015). Pint-sized DNA sequencer impresses first users. Nature 521, 15–16. doi: 10.1038/521015a
Clarke, J., Wu, H. C., Jayasinghe, L., Patel, A., Reid, S., and Bayley, H. (2009). Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 4, 265–270. doi: 10.1038/nnano.2009.12
Corley, M. J., Zhang, W., Zheng, X., Lum-Jones, A., and Maunakea, A. K. (2015). Semiconductor-based sequencing of genome-wide DNA methylation states. Epigenetics 10, 153–166. doi: 10.1080/15592294.2014.1003747
David, M., Dursi, L. J., Yao, D., Boutros, P. C., and Simpson, J. T. (2016). Nanocall: an open source basecaller for Oxford nanopore sequencing data. bioRxiv 1:46086.
Eddy, S. R. (2004). What is a hidden Markov model? Nat. Biotechnol. 22, 1315–1316. doi: 10.1038/nbt1004-1315
Frommer, M., McDonald, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., et al. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U.S.A. 89, 1827–1831. doi: 10.1073/pnas.89.5.1827
Goodwin, S., Gurtowski, J., Ethe-Sayers, S., Deshpande, P., Schatz, M. C., and McCombie, W. R. (2015). Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 25, 1750–1756. doi: 10.1101/gr.191395.115
Greninger, A. L., Naccache, S. N., Federman, S., Yu, G., Mbala, P., Bres, V., et al. (2015). Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 7:99. doi: 10.1186/s13073-015-0220-9
Harris, R. S. (2007). Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis. Pennsylvania, PA: The Pennsylvania State University.
Hetherington, S., McGuirk, S., Powell, G., Cutrell, A., Naderer, O., Spreen, B., et al. (2002). Hypersensitivity reactions during therapy with the nucleoside reverse transcriptase inhibitor abacavir. Clin. Ther. 23, 1603–1614. doi: 10.1016/S0149-2918(01)80132-6
Howorka, S., Cheley, S., and Bayley, H. (2001). Sequence-specific detection of individual DNA strands using engineered nanopores. Nat. Biotechnol. 19, 636–639. doi: 10.1038/90236
Hughes, A. R., Mosteller, M., Bansal, A. T., Davies, K., Haneline, S. A., Lai, E. H., et al. (2004b). Association of genetic variations in HLA-B region with hypersensitivity to abacavir in some, but not all, populations. Pharmacogenomics 5, 203–211. doi: 10.1517/phgs.5.2.203.27481
Hughes, D. A., Vilar, F. J., Ward, C. C., Alfirevic, A., Park, B. K., and Pirmohamed, M. (2004a). Cost-effectiveness analysis of HLA B∗5701 genotyping in preventing abacavir hypersensitivity. Pharmacogenet. Genomics 14, 335–342. doi: 10.1097/00008571-200406000-00002
Ip, C. L., Loose, M., Tyson, J. R., de Cesare, M., Brown, B. L., Jain, M., et al. (2015). MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Res. 4:1075. doi: 10.12688/f1000research.7201.1
Jain, M., Fiddes, I. T., Miga, K. H., Olsen, H. E., Paten, B., and Akeson, M. (2015). Improved data analysis for the MinION nanopore sequencer. Nat. Methods 12, 351–356. doi: 10.1038/nmeth.3290
Karamitros, T., and Magiorkinis, G. (2015). A novel method for the multiplexed target enrichment of MinION next generation sequencing libraries using PCR-generated baits. Nucleic Acids Res. 43, e152. doi: 10.1093/nar/gkv773
Karow, J. (2015). Oxford Nanopore Outlines Specs for New Sequencers, Automated Sample Prep System, Pay-As-Go Pricing. Genomeweb, May 14, 2015. Available at: https://www.genomeweb.com/sequencing-technology/oxford-nanopore-outlines-specs-new-sequencers-automated-sample-prep-system-pay [accessed May 24, 2016].
Kasianowicz, J. J., Brandin, E., Branton, D., and Deamer, D. W. (1996). Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. U.S.A. 93, 13770–13773. doi: 10.1073/pnas.93.24.13770
Kiełbasa, S. M., Wan, R., Sato, K., Horton, P., and Frith, M. C. (2011). Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493. doi: 10.1101/gr.113985.110
Kilianski, A., Haas, J. L., Corriveau, E. J., Liem, A. T., Willis, K. L., Kadavy, D. R., et al. (2015). Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. Gigascience 4:12. doi: 10.1186/s13742-015-0051-z
Laver, T. W., Caswell, R. C., Moore, K. A., Poschmann, J., Johnson, M. B., Owens, M. M., et al. (2016). Pitfalls of haplotype phasing from amplicon-based long-read sequencing. Sci. Rep. 6:21746. doi: 10.1038/srep21746
Leggett, R. M., Heavens, D., Caccamo, M., Clark, M. D., and Davey, R. P. (2015). NanoOK: multi-reference alignment analysis of nanopore sequencing data, quality and error profiles. Bioinformatics 32, 142–144. doi: 10.1093/bioinformatics/btv540
Li, C., Chng, K. R., Boey, J. H., Ng, H. Q., Wilm, A., and Nagarajan, N. (2016). INC-Seq: accurate single molecule reads using nanopore sequencing. bioRxiv 1:038042.
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Y., and Tollefsbol, T. O. (2011). DNA methylation detection: bisulfite genomic sequencing analysis. Methods Mol. Biol. 791, 11–21. doi: 10.1007/978-1-61779-316-5_2
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Loman, N. J., and Quinlan, A. R. (2014). Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30, 3399–3401. doi: 10.1093/bioinformatics/btu555
Mallal, S., Nolan, D., Witt, C., Masel, G., Martin, A. M., Moore, C., et al. (2002). Association between presence of HLA-B∗5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reversetranscriptase inhibitor abacavir. Lancet 359, 727–732. doi: 10.1016/S0140-6736(02)07873-X
Mallal, S., Phillips, E., Carosi, G., Molina, J.-M., Workman, C., Tomažič, J., et al. (2008). HLA-B∗5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 358, 568–579. doi: 10.1056/NEJMoa0706135
Martin, A. M., Nolan, D., Gaudieri, S., Almeida, C. A., Nolan, R., James, I., et al. (2004). Predisposition to abacavir hypersensitivity conferred by HLA-B∗5701 and a haplotypic Hsp70-Hom variant. Proc. Natl. Acad. Sci. U.S.A. 101, 4180–4185. doi: 10.1073/pnas.0307067101
Martin, M. A., Hoffman, J. M., Freimuth, R. R., Klein, T. E., Dong, B. J., Pirmohamed, M., et al. (2014). Clinical Pharmacogenetics Implementation Consortium guidelines for HLA-B genotype and abacavir dosing: 2014 update. Clin. Pharmacol. Ther. 95, 499–500. doi: 10.1038/clpt.2014.38
Martin, M. A., Klein, T. E., Dong, B. J., Pirmohamed, M., Haas, D. W., and Kroetz, D. L. (2012). Clinical Pharmacogenetics Implementation Consortium guidelines for HLA-B genotype and abacavir dosing. Clin. Pharmacol. Ther. 91, 734–738. doi: 10.1038/clpt.2011.355
McDonald, O. G., Krynetski, E. Y., and Evans, W. E. (2002). Molecular haplotyping of genomic DNA for multiple single-nucleotide polymorphisms located kilobases apart using long-range polymerase chain reaction and intramolecular ligation. Pharmacogenetics 12, 93–99. doi: 10.1097/00008571-200203000-00003
Mercier, B., Gaucher, C., Feugeas, O., and Mazurier, C. (1990). Direct PCR from whole blood, without DNA extraction. Nucleic Acids Res. 18, 5908. doi: 10.1093/nar/18.19.5908
Odelberg, S. J., Weiss, R. B., Hata, A., and White, R. (1995). Template-switching during DNA synthesis by Thermus aquaticus DNA polymerase I. Nucleic Acids Res. 23, 2049–2057. doi: 10.1093/nar/23.11.2049
Parker, W. T., Phillis, S. R., Yeung, D. T., Hughes, T. P., Scott, H. S., and Branford, S. (2014). Many BCR-ABL1 compound mutations reported in chronic myeloid leukemia patients may actually be artifacts due to PCR-mediated recombination. Blood 124, 153–155. doi: 10.1182/blood-2014-05-573485
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13-341
Quick, J., Loman, N. J., Duraffour, S., Simpson, J. T., Severi, E., Cowley, L., et al. (2016). Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232. doi: 10.1038/nature16996
Rauch, A., Nolan, D., Martin, A., McKinnon, E., Almeida, C., and Mallal, S. (2006). Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study. Clin. Infect. Dis. 43, 99–102. doi: 10.1086/504874
Risse, J., Thomson, M., Patrick, S., Blakely, G., Koutsovoulos, G., Blaxter, M., et al. (2015). A single chromosome assembly of Bacteroides fragilis strain BE1 from Illumina and MinION nanopore sequencing data. Gigascience 4:60. doi: 10.1186/s13742-015-0101-6
Robinson, J., Halliwell, J. A., Hayhurst, J. H., Flicek, P., Parham, P., and Marsh, S. G. E. (2015). The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43, D423–D431. doi: 10.1093/nar/gku1161
Rodriguez-Novoa, S., Garcia-Gasco, P., Blanco, F., Gonzalez-Pardo, G., Castellares, C., Moreno, V., et al. (2007). Value of the HLA-B∗5701 allele to predict abacavir hypersensitivity in Spaniards. AIDS Res. Hum. Retroviruses 23, 1374–1376. doi: 10.1089/aid.2006.0244
Schackman, B. R., Scott, C. A., Walensky, R. P., Losina, E., Freedberg, K. A., and Sax, P. E. (2008). The cost effectiveness of HLA-B∗5701 genetic screening to guide initial antiretroviral therapy for HIV. AIDS 15, 2025–2033. doi: 10.1097/QAD.0b013e3283103ce6
Simpson, J. T., Workman, R., Zuzarte, P. C., David, M., Dursi, L. J., and Timp, W. (2016). Detecting DNA methylation using the Oxford Nanopore Technologies MinION sequencer. bioRxiv 1:047142.
Spartan Bioscience Inc. (2016). Spartan RX [Internet]. Available at: http://www.spartanbio.com/products/spartan-rx/demo-video/
Sun, H. Y., Hung, C. C., Lin, P. H., Chang, S. F., Yang, C. Y., Chang, S. Y., et al. (2007). Incidence of abacavir hypersensitivity and its relationship with HLA-B∗5701 in HIV-infected patients in Taiwan. J. Antimicrob. Chemother. 60, 599–604. doi: 10.1093/jac/dkm243
Symonds, W., Cutrell, A., Edwards, M., Steel, H., Spreen, B., Powell, G., et al. (2002). Risk factor analysis of hypersensitivity reactions to abacavir. Clin. Ther. 24, 565–573. doi: 10.1016/S0149-2918(02)85132-3
Szalay, T., and Golovchenko, J. A. (2015). De novo sequencing and variant calling with nanopores using PoreSeq. Nat. Biotechnol. 33, 1087–1091. doi: 10.1038/nbt.3360
Timp, W., Comer, J., and Aksimentiev, A. (2012). DNA base-calling from a nanopore using a Viterbi algorithm. Biophys. J. 102, L37–L39. doi: 10.1016/j.bpj.2012.04.009
Waters, L. J., Mandalia, S., Gazzard, B., and Nelson, M. (2007). Prospective HLA-B∗5701 screening and abacavir hypersensitivity: a single centre experience. AIDS 21, 2533–2534. doi: 10.1097/QAD.0b013e328273bc07
Watson, M., Thomson, M., Risse, J., Talbot, R., Santoyo-Lopez, J., Gharbi, K., et al. (2015). poRe: an R package for the visualization and analysis of nanopore sequencing data. Bioinformatics 31, 114–115. doi: 10.1093/bioinformatics/btu590
Wheeler, E. K., Hara, C. A., Frank, J., Deotte, J., Hall, S. B., Benett, W., et al. (2011). Under-three minute PCR: probing the limits of fast amplification. Analyst 136, 3707–3712. doi: 10.1039/c1an15365j
Wittig, M., Anmarkrud, J. A., Kässens, J. C., Koch, S., Forster, M., Ellinghaus, E., et al. (2015). Development of a high-resolution NGS-based HLA-typing and analysis pipeline. Nucleic Acids Res. 43, e70. doi: 10.1093/nar/gkv184
Zucman, D., Truchis, P. D., Majerholc, C., Stegman, S., and Caillat-Zucman, S. (2007). Prospective screening for human leukocyte antigen-B*5701 avoids abacavir hypersensitivity reaction in the ethnically mixed French HIV population. J. Acquir. Immune Defic. Syndr. 45, 1–3. doi: 10.1097/QAI.0b013e318046ea31
Keywords: MinION, nanopore sequencing, drug hypersensitivity, sequence-based HLA typing, bioinformatics analysis
Citation: Chua EW and Ng PY (2016) MinION: A Novel Tool for Predicting Drug Hypersensitivity? Front. Pharmacol. 7:156. doi: 10.3389/fphar.2016.00156
Received: 28 April 2016; Accepted: 30 May 2016;
Published: 14 June 2016.
Edited by:
Emanuela Corsini, Università degli Studi di Milano, ItalyReviewed by:
Melih O. Babaoglu, Hacettepe University Faculty of Medicine, TurkeyMartin Lennard, University of Sheffield, UK
Copyright © 2016 Chua and Ng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eng Wee Chua, Y2V3ODU5MTFAdWttLmVkdS5teQ==