 Valentina Di Liberto1†
Valentina Di Liberto1† Giuseppa Mudò1†
Giuseppa Mudò1† Roberta Garozzo2
Roberta Garozzo2 Monica Frinchi1
Monica Frinchi1 Víctor Fernandez-Dueñas3
Víctor Fernandez-Dueñas3 Patrizia Di Iorio4
Patrizia Di Iorio4 Renata Ciccarelli4
Renata Ciccarelli4 Francesco Caciagli4
Francesco Caciagli4 Daniele F. Condorelli2
Daniele F. Condorelli2 Francisco Ciruela3*
Francisco Ciruela3* Natale Belluardo1*
Natale Belluardo1*- 1Department of Experimental Biomedicine and Clinical Neurosciences, University of Palermo, Palermo, Italy
- 2Department of Biomedical and Biotechnological Sciences, Unit of Medical Biochemistry, University of Catania, Catania, Italy
- 3Department of Pathology and Experimental Therapeutics, Faculty of Medicine, Bellvitge Biomedical Research Institute, Institute of Neurosciences, University of Barcelona, Barcelona, Spain
- 4Department of Medical, Oral and Biotecnological Sciences, University of Chieti-Pescara, Chieti, Italy
Guanine-based purines (GBPs) have been recently proposed to be not only metabolic agents but also extracellular signaling molecules that regulate important functions in the central nervous system. In such way, GBPs-mediated neuroprotection, behavioral responses and neuronal plasticity have been broadly described in the literature. However, while a number of these functions (i.e., GBPs neurothophic effects) have been well-established, the molecular mechanisms behind these GBPs-dependent effects are still unknown. Furthermore, no plasma membrane receptors for GBPs have been described so far, thus GBPs are still considered orphan neuromodulators. Interestingly, an intricate and controversial functional interplay between GBPs effects and adenosine receptors activity has been recently described, thus triggering the hypothesis that GBPs mechanism of action might somehow involve adenosine receptors. Here, we review recent data describing the GBPs role in the brain. We focus on the involvement of GBPs regulating neuronal plasticity, and on the new hypothesis based on putative GBPs receptors. Overall, we expect to shed some light on the GBPs world since although these molecules might represent excellent candidates for certain neurological diseases management, the lack of putative GBPs receptors precludes any high throughput screening intent for the search of effective GBPs-based drugs.
Introduction
Guanine-based purines (GBPs), including the nucleotides guanosine 5′-triphosphate (GTP), guanosine 5′-diphosphate (GDP) and guanosine 5′-monophosphate (GMP), the nucleoside guanosine (GUO) and the nucleobase guanine (GUA) have been traditionally characterized as modulators of intracellular processes, especially considering their role in G protein dependent signal transduction. In the last two decades, GBPs have been also shown to exert extracellular effects and to act as neuromodulator pleiotropic molecules affecting several cellular processes, including growth, differentiation and survival, in both the central and the peripheral nervous system (CNS and PNS, respectively; Schmidt et al., 2007; Rathbone et al., 2008). However, despite accumulated experimental evidences supporting their extracellular role, putative receptor sites have not been characterized, thus GBPs are still orphan neuromodulators. In this article, apart from reviewing recent data illustrating the involvement of GBPs in the nervous system, we especially discuss the new hypothesis concerning the existence of putative GBPs receptors.
Purinergic System and “Purinome”
The term “purinergic system” usually refers to a complex system composed by purines bases, such as adenine (ADA) and GUA, their corresponding nucleotides adenosine 5′-triphosphate (ATP) and GTP, adenosine 5′-diphosphate (ADP) and GDP, adenosine 5′-monophosphate (AMP) and GMP, and nucleosides adenosine (ADO), inosine (INO) and GUO. In addition, the purinergic system includes the metabolites xanthine (XAN), hypoxanthine (HYPO) and uric acid (UA), together with the receptors, transporters and enzymes involved in purinergic transmission (Schmidt et al., 2007). Classically, purinergic signaling has been proposed to be regulated by the interaction of natural purines with a large number of specific own receptors, by a complex machinery assuring cell signaling transmission as well as by purine clarification from the extracellular medium by ecto-enzymes and/or bidirectional carriers (see below). Noteworthy, an emerging theory postulates that a broad-spectrum of direct/indirect interactions also exist within a cooperative synergic network, defined as “purinome” (Volonte and D’Ambrosi, 2009). The functions of the purinome involve the interaction and heteromerization between purinergic and other receptors. Interestingly, it is nowadays widely accepted that direct receptor-receptor interactions (i.e., oligomerization) permit to integrate information from different sources, thus providing a fine and complex control over various regulatory mechanisms (Ferre, 2007; Borroto-Escuela et al., 2012; Fuxe et al., 2012; Di Liberto et al., 2014). Regarding purinergic receptors, it has been shown, for instance, that adenosine 1 receptor (A1R) heterodimerizes with adenosine 2A receptor (A2AR), and also with dopamine D1, metabotropic glutamate mGlu1α, cannabinoid CB1 and adrenergic β1-and β2-adrenergic receptors, leading to diverse functional outcomes (Gines et al., 2000; Ciruela et al., 2001, 2006a,b; Chandrasekera et al., 2013). Recently, based on experimental evidence, Borroto-Escuela et al. (2014) proposed a network representation of G-protein coupled receptors (GPCR) heterodimers in the brain, in which A1R receptor emerges as a hub receptor forming 11 different heteroreceptor complexes.
Extracellular Purines: Their Release and Re-Uptake.
The extracellular concentration of purines is dependent on the amount of released purines, on the efficiency of re-uptake mechanisms and on the activity of extracellular metabolizing enzymes. Purines can be released to the extracellular space by both neurons and glial cells, under physiological and, even more interestingly, in pathological conditions. Adenine-based purines (ABPs), for example ATP, can be released from neuronal presynaptic terminals and glial cells (Burnstock et al., 1972; Wagner et al., 1978; Burnstock, 2006), where they can be used as substrate for several ectonucleotidases (Zimmermann, 1996; Bonan, 2012; Zimmermann et al., 2012), which generate multiple breakdown products (ADP, AMP, and ADO) able to activate different receptors and to trigger several effects (Zimmermann, 2011).
GTP is co-stored together with ATP in neuronal synaptic vesicles from which it is released in association with ATP by exocytosis (Wagner et al., 1978; Santos et al., 2006). On the other hand, ATP is not involved in the outflow of GTP from glial cells. Nevertheless, GTP and its di- and mono-phosphate derivatives, together with the corresponding adenine-based counterparts, are substrates and thus competitors of the same ecto-nucleotidases while neurons seem to be able to release very small amounts of ADO per se, the presence of ADO outside glial cells is mainly due to the extracellular metabolism of released ATP. There are some peculiar differences concerning the other ADA- and GUA-based nucleosides, INO and GUO. Although these nucleosides derive from the hydrolysis of extracellular Inosine monophosphate (IMP)/GMP or from ADO deamination (for INO), they would also be constitutively released from neurons, astrocytes, C6 or U373 glioma cells, microglial cells or SH-SY5Y neuroblastoma cells (Ballerini et al., 2005, 2006a,b; Giuliani et al., 2012c). However, non-physiological and relatively extreme stimulations (e.g., seizure activity, hypoxia/hypoglycemia, and superfusion with high K+) seem to be required to elicit ADO efflux from glial cells; accordingly, in such conditions extracellular ADO would be that escaping from uptake and metabolism, thus representing a tissue overflow rather than a release per se (Di Iorio et al., 1998).
Finally, regarding purine metabolites such as HYPO, GUA, and XAN, which can be found in the medium of cultured glial cells and in the fluid from super-fused brain slices (Rathbone et al., 1999; Zamzow et al., 2008), it was thought during several years that they were uniquely transported outside the cells by specific transporters (NBTs), different from those for nucleosides (ENTs) (Sinclair et al., 2000; Dos Santos-Rodrigues et al., 2014). Thus, their presence outside of cells was considered, like for ADO, the result of cell overflow of substances to be eliminated (Parkinson et al., 2011). However, these purine metabolites would still be present in the extracellular milieu of cells treated with inhibitors of transporters, suggesting that they would also derive from the extracellular metabolism of nucleosides (Rathbone et al., 1999; Jiang et al., 2008b; Giuliani et al., 2012a; Caciagli et al., 2014).
Taken together, the previous findings indicate that the systems of ABPs and GBPs are physically and functionally present outside cells, where they operate simultaneously. In addition some peculiarities deserve to be emphasized. For instance: (i) the extracellular levels of GBPs are about 2-3-fold higher than those of their ADA-based counterparts (Ciccarelli et al., 1999); (ii) the metabolism of ADA and GUA nucleotides maintains constant the proportions of the components of the two systems; however, there are some differences in the respective nucleosides, such as the presence of only one GUA (GUO) instead of the two ADA nucleosides (ADO and INO), or the different affinities of these compounds for the trans-membrane transport systems, as well as for the enzymes deputed to their metabolism; and (iii) compared to ADO, the extracellular levels of GUO remain particularly elevated after an ischemic insult, a fact that has been shown both in cultured astrocytes (Ciccarelli et al., 1999) and in a model in vivo of focal cerebral ischemia (Uemura et al., 1991). To end with, it is important to underline that it is not an easy task to determine the exact concentrations of extracellular GBPs. Thus, the most common analytical assays, such as HPLC or capillary electrophoresis, do not permit to differentiate some peaks of ADA and GUA compounds, which are often overlapped (e.g., HYPO and GUA), thus their quantification cannot be carried out properly. Hence, to ameliorate the knowledge of GBPs system it is still necessary to improve the analytical methods, in order to separate these compounds from the ABPs counterparts (Ito et al., 2000; Stentoft et al., 2014).
Release of Enzymes Metabolizing Purines
It is well known that purine metabolism is mainly oriented to preserve, by multiple pathways, the levels of triphosphate nucleotides. The enzyme system regulating the homeostasis of extracellular purines largely corresponds to that responsible for the metabolism and salvage of intracellular ABPs and GBPs. Indeed, it is well known that a broad spectrum of membrane-bound nucleotidases contribute to the breakdown of extracellular nucleotides (Zimmermann, 1996; Zimmermann et al., 2012). Moreover, it has also been recently reported that some soluble nucleotide kinases are released from cells and contribute to restore ATP levels when the extracellular amount of AMP becomes elevated (Yegutkin, 2014). In contrast, no ecto-enzymes seem to be involved in the catabolism of extracellular ADA- and GUA-based nucleosides, which would likely and only depend on some actively released soluble enzymes.
A few number of reports have indicated that some enzymes of the metabolism of purine nucleosides, such as ADO deaminase, purine nucleoside phosphorylase (PNP), Guanase and Xanthine Oxidase (XO), are detectable in plasma (Roberts and Newton, 2004; Roberts et al., 2004; Karabulut et al., 2005; Chittiprol et al., 2007; Lopez-Cruz et al., 2014; Giuliani et al., 2016). Furthermore, some of these enzymes have been proposed as potential diagnostic or prognostic markers for different pathological conditions. For instance, PNP plasma levels have been associated with the malignant degree and the diffusion capability of some tumors, such as the melanoma or pancreatic adenocarcinoma (Roberts et al., 2004; Vareed et al., 2011); whereas XO activity, which is physiologically low in plasma and increases after ischemic events, has been considered predictive for cardiovascular disease, independently of uric acid plasma levels (Gondouin et al., 2015; Lopez-Cruz et al., 2014). Nevertheless, it is still not sufficiently elucidated the origin of these plasmatic enzymes, the mechanisms by which they are released from cells and their activity in the metabolism of the extracellular purine nucleosides, importantly in the proximity of membrane purine receptors.
Recently, it has been observed, either in cell lines or in primary cultures of neural cells, that glial cells, especially microglia, but not neurons are able to constitutively release PNP in the culture medium. Extracellular K+ or exogenous ATP, but no other types of cell stimulation, such as LPS or interferon-γ, enhance, in a dose-dependent manner and via the lysosomal system, PNP release in the culture medium, where the enzyme is detectable both as soluble fraction and inside of membrane-derived vesicles (Polazzi et al., 2011). Interestingly, similar results have been shown investigating Guanase (Caciagli et al., 2014). These findings suggest that at least glial cells release the key enzymes of the metabolism of ABP and GBP nucleosides into the corresponding bases, which may act as potent cell damaging pro-inflammatory agents via reactive oxygen species (ROS) production. Therefore, the complex interplay between the activity of these enzymes and the transport systems of nucleosides across membranes may control the cell-specific homeostasis of extracellular purines as well as their signaling.
In physiological conditions, the activity of these kinds of enzymes controls the production of purine bases maintaining their levels in a range of “limited ability” to generate ROS. This delicate equilibrium seems to be lost during degenerative processes, which are often associated with chronic inflammatory, or in hypoxic, events. Hypoxia, for example, by inducing the over-expression/activity of 5′-nucleotidase and a concomitant blockade of the PNP activity, accelerates the hydrolysis of the nucleotides, thus causing a consequent accumulation of intra- and extra-cellular nucleosides, especially GUO (Ciccarelli et al., 1999). Similarly, during post-ischemic reperfusion, the functional block of the PNP seems to be not sufficient, resulting in the formation of large amounts of purine bases, whose catalytic oxidation, mediated by XO, produces ROS that through the activation of Nuclear factor kappa B (NF-kB) produce pro-inflammatory cytokines (Lorne et al., 2008).
Noteworthy, further emerging evidences support a relevant patho-physiological significance for these enzymes, which may have also some functions independent from their enzymatic activity. Among these enzymes, it is important to mention Nucleotidase II (Tozzi et al., 2013), which is currently being investigated (looking for its putative role outside cells) in our laboratory. This enzyme seems to interact with the ice protease-activating factor (IPAF) in order to regulate its folding and conformation, thus acting as a sensor of the global health state of the cell, and capable of regulating its death (Cividini et al., 2015). On its own, Guanase, named also cypin, plays a trophic role on the development of brain by regulating the dendritic arborization and neuronal morphology. In addition, Guanase seems to be involved in the mechanisms responsible of liver transplant rejection (Fernandez et al., 2010). Overall, the above-mentioned non-enzymatic activities of enzymes involved in purine nucleosides may be even more attractive as potential targets for developing new drugs.
Purinergic Receptors
The present review is mainly focused on GBPs functions. However, we first provide a short overview of ABPs receptors, in order to contextualize the current knowledge of GBPs receptors into the purinergic field.
ABPs Receptors: Long Story Made Short
ABPs play a pivotal role in the modulation of neurotransmission and neurotrophism acting on two different families of receptors: P1, activated by ADO, and P2, activated by ATP/ADP nucleotides. P1 receptor family includes four different subtypes (A1R, A2AR, A2BR, and A3R) of GPCRs. A1Rs and A3Rs are typically coupled to Gi proteins and thus inhibit adenylyl cyclases, whereas A2ARs and A2BRs are coupled to Gs proteins and increase the production of cyclic AMP (cAMP) (Zimmermann, 2011). P1 receptors are expressed in neurons, astrocytes, oligodendrocytes and microglia and their stimulation activates multiple functions, such as synaptic plasticity (Dare et al., 2007; Burnstock et al., 2011a,b; Zimmermann, 2011; Del Puerto et al., 2013). The P2 receptors are subdivided in two different subfamilies: ionotropic and metabotropic receptors (P2XRs and P2Yrs, respectively). The P2XRs subfamily includes seven different subtypes, which are mostly expressed by neurons and astrocytes, and in a lesser extent by oligodendrocytes, Schwann cells, and microglia. ATP mediated activation of P2XRs enhances rapid changes in membrane potential by increasing Na+, K+, and Ca2+permeability (North, 2002; Roberts et al., 2006; Del Puerto et al., 2013). Thus, they are involved in the regulation of fast synaptic transmission, synaptic plasticity, and fast neuron–glia signaling (Silinsky et al., 1992; Pankratov et al., 1998, 2002, 2009; Burnstock et al., 2011a; Lalo et al., 2011a,b). Conversely, the P2Y subfamily is composed by eight different P2YRs subtypes (Burnstock, 2007b). Interestingly, while P2Y1R, P2Y2R, P2Y4R, P2Y6R, and P2Y11R are coupled to Gq proteins and activate phospholipase C (PLC), the P2Y12R, P2Y13R, and P2Y14R are coupled to Gi proteins and inhibit adenylyl cyclases (Abbracchio et al., 2006). Overall, most of these receptors are expressed in neurons and glial cells, and are involved in bi-directional neuron–glia communication, thus exerting long-term effects on proliferation, neurogenesis, differentiation, migration and apoptosis (Burnstock, 2007a; Neary and Zimmermann, 2009; Verkhratsky et al., 2009; Del Puerto et al., 2013). For a more detailed introduction to ABPs receptors biology several well-documented reviews are available (Burnstock, 2007a,b; Burnstock et al., 2011a,b).
GBPs Receptors: Short Story Made Long
At present, despite the plethora of experimental evidences pointing to the existence of putative GBPs receptors in the brain, a specific receptor has not been still identified; accordingly, GBPs are orphan neuromodulators. However, a number of cellular effects of GBPs might only be explained by the activation of different extracellular binding sites, since most of them still persist in the presence of inhibitors of GBPs transporters (Gysbers and Rathbone, 1992; Di Iorio et al., 2004). Actually, specific binding sites for GTP in PC12 cells (Gysbers et al., 2000) and the presence of a single high affinity binding site for [3H]-GUO in rat brain membranes (Traversa et al., 2002, 2003) have been described, supporting the possible existence of GBPs transmembrane receptors. GUO binding on rat membranes is saturable, reversible and is not displaced by other naturally occurring purines, such as ADO, HYPO, XAN, caffeine, theophylline, GDP, GMP, and ATP, thus suggesting that this binding site does not involve receptors for ABPs (Traversa et al., 2002). Indeed, GUO or GTP do not bind with high-affinity to ABPs receptors (Muller and Scior, 1993). The available data is in agreement with the fact that GBPs, in particular GUO, bind to metabotropic receptors and that many effects of GBPs are mediated through G protein-dependent signaling pathways; it has been shown, for instance, that the pertussis toxin-mediated inhibition of Gi/Go-protein reverses some of the effects of GUO on cell viability and glutamate uptake in hippocampal slices (Dal-Cim et al., 2013). Furthermore, by monitoring the binding of a non-hydrolysable-labeled GTP, it has been demonstrated that GUO is able to activate a putative not yet identified GPCR, different from the well-characterized ADO receptors, in rat brain membranes, (Volpini et al., 2011). Interestingly, we were able to obtain similar results, since we determined the activation of a putative unknown GPCR for GUO by means of binding experiments and in situ autoradiography of [35S]GTPγS, respectively, in membranes and slices from rat brain (Grillo et al., 2012).
In addition to the above-mentioned data showing the existence of a putative unknown GPCR for GBPs, other findings also indicate that GUO may signal through A1R and/or A2AR (Figure 1). For example, it has been observed that GUO protects hippocampal slices against oxidative and inflammatory processes in an A1R dependent manner, while GUO-induced neuroprotection and stimulation of glutamate uptake involves A2AR activation (Dal-Cim et al., 2013). A1R blockade or A2AR activation can reverse GUO-evoked neuroprotective effect, suggesting that GUO effect may involve an interaction with A1R-A2AR oligomers (Lanznaster et al., 2016). Furthermore, GUA inhibition of renal Na+-ATPase activity, which is dependent on the activation of a Gi-coupled receptor, is blocked by antagonists of A1R (Wengert et al., 2011). In addition, while P2 receptor antagonists significantly reduce the stimulation of cell proliferation induced by GTP, a preferential A2BR antagonist only partially decreases the mitogenic activity of GUO in astrocytes (Ciccarelli et al., 2000). Overall, these data could suggest that GUO, probably with low affinity, may also bind to ADO receptors and act as an agonist, triggering alternative pathways to those promoted by ADO. However, it is needed to say that GUO binding to ADO receptors cannot account for all GUO-mediated effects in brain, since many of them still persist in the presence of both P1 and/or P2 receptor antagonists (Gysbers and Rathbone, 1992; Tasca and Souza, 2000; Frizzo et al., 2001). Indeed, the neuritogenic activity of GTP is not reproduced by ADA nucleotides, whereas the GUO effect is synergistic with that produced by A1R/A2R agonists and it is P1 receptors independent, since it is not inhibited by A1R/A2R antagonists (Gysbers and Rathbone, 1996a). Overall, these results support either the existence of specific GPCRs for GUO and/or of receptor heterocomplexes between GUO and ADO receptors (Figure 1). This functional interplay between GUO and ADO suggests that putative GUO receptors might share some particular features, yet elusive to current experimental approaches, with ARs, or alternatively that they might form heterocomplexes with ARs modulating the reciprocal activity (Figure 1). Finally, an additional theory, according to the hypothesis previously reported (Ciruela, 2013), consists of the no existence of putative GUO receptors, thus this molecule would signal through existing receptor complexes containing A1R and/or A2R (Figure 1). The elucidation of the mechanism of GUO and ADO receptors interaction and the characterization of membrane-binding sites are under current investigation in our laboratory. Overall, these possibilities may partially explain as to why GUO receptors have not still been identified, but they may also spur research to explore among both cloned known (i.e., ARs) and orphan GPCRs to find a potential specific GUO receptor. To this aim, good GPCR candidates to explore as potential GUO receptors could be GPR174/LPS3 (Civelli et al., 2013), showing high homology with P2Y10, and GPR23/LPA4, which shares high homology with human P2Y5 receptor and seems to be a potential putative GUO receptor, as suggested by preliminary data. (Di Liberto et al., 2012; Lanznaster et al., 2016). In addition, based on the evidence that ADO receptors, mainly A1R and A2R, form heteroreceptor complexes with several members P2Y receptor family (Borroto-Escuela et al., 2014) and on the high-sequence homology between GPR23 and P2Y5 receptors, it is reasonable to hypothesize an interaction between GPR23 and ADO receptors.
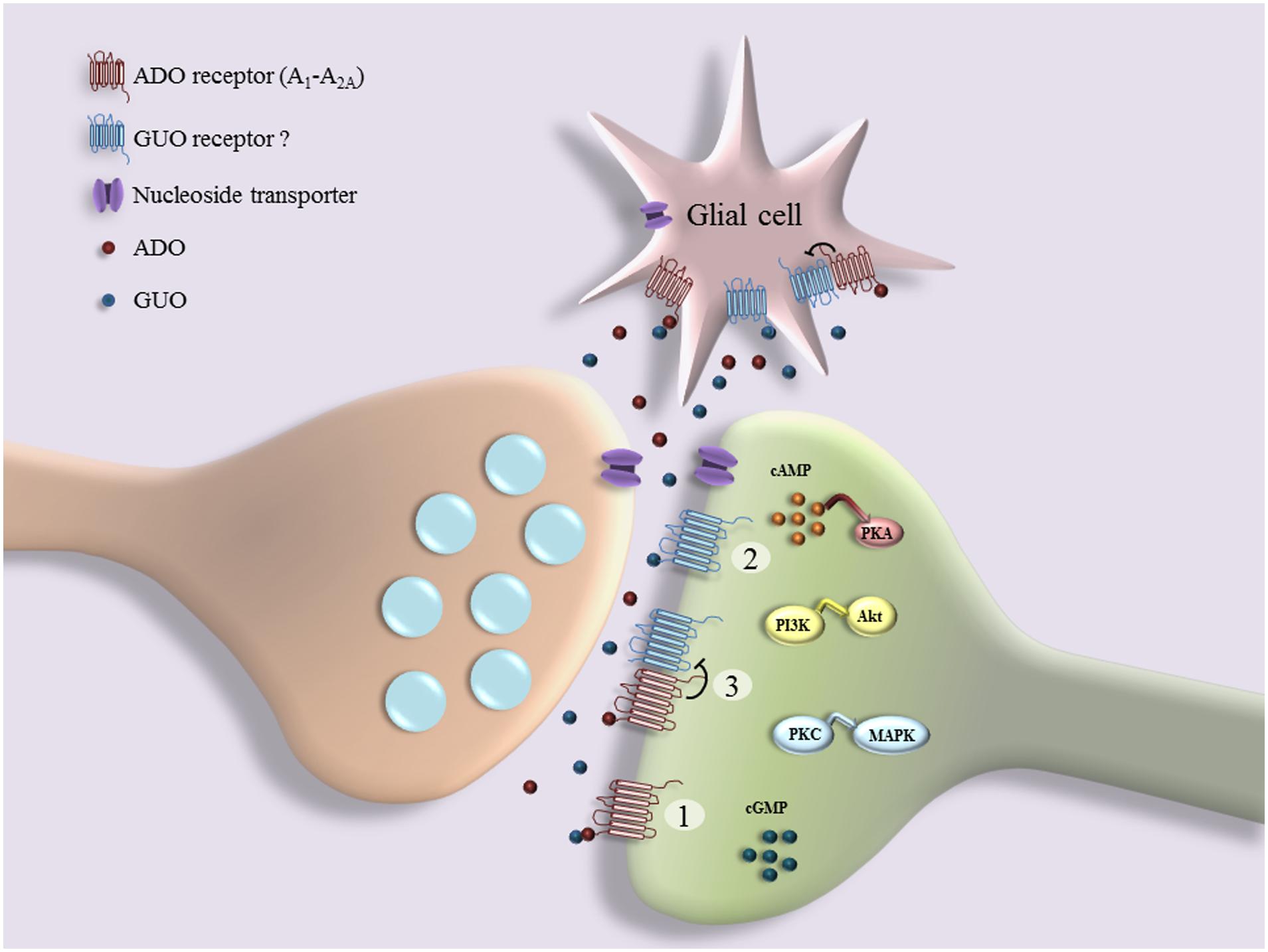
FIGURE 1. Schematic representation of interplay between guanosine (GUO) and adenosine (ADO) binding to receptors: GUO binds to ADO receptor (A1R and A2AR) in competitive manner with ADO (1); GUO binds to putative unknown GUO receptor (2), which may form heterocomplexes with ADO receptors (3). Several effects of GUO are dependent on cell signaling pathways downstream of receptors (e.g., cAMP/Protein Kinase A (PKA), PI3K/Akt, PKC/MAPK, and cGMP), culminating in functional cell responses such as cell proliferation, survival, neuritogenesis, neuroprotection, and cell migration.
GBPs and Cell Signaling
As stated above, several effects of GUO are mediated through the binding to unknown GPCRs, and therefore dependent on cell signaling pathways (Gysbers and Rathbone, 1996a,b; Traversa et al., 2003). Indeed, extracellular GUO enhances the neuritogenic effects of nerve growth factor (NGF) on PC12 cells through both cAMP-dependent (Gysbers and Rathbone, 1992; Gysbers and Rathbone, 1996a,b; Bau et al., 2005), and cAMP-independent mechanisms, such as the stimulation of soluble guanylate cyclase and the increase of intracellular cyclic GMP (cGMP; Bau et al., 2005). GUO is also able to stimulate, in vitro, neural stem cell proliferation by activating the cAMP response element binding (CREB) pathway (Su et al., 2013), whereas it protects hippocampal neurons against glutamate-induced cell death by a mechanism that involves the Phosphoinositide 3-kinase (PI3K)/Protein kinase B (PKB/Akt)/Glycogen Synthase Kinase3β (GSK3β) pathway (Molz et al., 2011). Similarly, in C6 glioma cells, GUO protects against 6-hydroxydopamine (6-OHDA)-induced neurotoxicity through the activation of the survival pathways regulated by extracellular signal-regulated kinases (ERK) and PI3K/Akt (Giuliani et al., 2015). As well, GUO is protective in different in vitro models against hypoglycemia by modulating oxidative and nitrosative stress and astroglial responses, such as glutamate uptake through different signaling involving protein kinase C (PKC), PI3K, p38, and ERK pathways (Quincozes-Santos et al., 2013). Additionally, GUO prevents in hippocampal slices mitochondrial membrane depolarization, inhibits (NF-kB) activation and reduces inducible nitric oxide synthase (iNOS) and ROS production via PI3K and mitogen-activated protein kinases (MAPK)/ERK (Dal-Cim et al., 2013). Also, it is neuroprotective against mitochondrial oxidative stress in neuroblastoma SH-SY5Y cells via PI3K/Akt/GSK-3β pathway (Dal-Cim et al., 2012). Again, anti-inflammatory effects of GUO on microglia are mediated by activation of the PI3K/Akt/MAPK (ERK1/2 and p38) pathways (D’Alimonte et al., 2007). Finally, GUO, even administered in vivo for a systemic treatment, is able to produce antidepressant-like effects, dependent on the modulation of NMDA receptors and mediated by cGMP and PI3K/mammalian target of rapamycin (mTOR) pathways (Bettio et al., 2012, 2014). Taken together, these data generated in different experimental models, mainly in in vitro studies, point out intracellular signaling events promoted by GUO and involving cAMP pathway as well as those linked to the PI3K and MAPK system. All these evidences may indirectly support the possibility that GBPs, particularly GUO, may activate cell surface receptors.
GBPs and Neuroprotection
Many studies have demonstrated that GBPs are able to exert neuroprotective effects on a wide variety of cell cultures and in vivo models (Schmidt et al., 2007; Ribeiro et al., 2015). These effects would be elicited through three main actions: by counteracting glutamate excitotoxicity; by preventing or attenuating neuroinflammation; by counteracting oxidative stress. In this part of the review we summarize recent research data investigating the neuroprotective effects of GBPs. Of note, we are not dealing here with the scarce clinical therapeutic available data.
Neuroprotection against Glutamate Toxicity
Different in vivo ischemic models have been used to show the neuroprotective role of GBPs, including oxygen and glucose deprivation, hypoxia models and in vivo transient and permanent ischemic stroke (Ribeiro et al., 2015). Generally, in ischemia conditions, glutamate released from neurons and astrocytes in the extracellular space, together with the ischemia-induced neuronal depolarization, leads to an abnormal neuronal firing and increased intracellular Ca2+ concentration with a subsequent loss of synaptic function and cell death (Brassai et al., 2015). In these models, the neuroprotective action of GBPs against glutamate toxicity and related intracellular signaling has been extensively characterized. In such way, it has been reported that GUO nucleotides, such as GTP, GDP and GMP, are able to inhibit NMDA-induced neurotoxicity in cultured hippocampal and neocortical neurons (Morciano et al., 2004), and that GMP is neuroprotective against glutamate or oxygen/glucose deprivation-induced neurotoxicity and against NMDA-induced apoptosis in hippocampal slices (Molz et al., 2005, 2008; Chang et al., 2008). Furthermore, in hippocampal slices and SH-SY5Y cells exposed to oxygen/glucose deprivation, GUO is neuroprotective against glutamate induced excitotoxicity via PI3K/Akt/GSK3β pathway and attenuates glutamate uptake impairment by promoting the activation of K+ channels and activating Gi/Go-proteins-coupled signaling (Dal-Cim et al., 2011, 2012, 2013). In addition, there is evidence that GUO is effective in counteracting the glutamate toxicity, thus preventing most of the effects promoted by hyperprolinemia, such as alteration of glutamatergic homeostasis due to a reduction of glutamate uptake and coupled to a decrease of membrane Na+, K+-ATPase activity and of intracellular ATP levels (Ferreira et al., 2012). As well, GUO increases glutamate uptake in hippocampal slices of neonatal rats exposed to a hypoxic-ischemic insult (Moretto et al., 2009). Similarly, it has been shown that GUO protects hippocampal neuron against ischemia induced by bilateral common carotid artery occlusion (Ganzella et al., 2012), decreases infarct volume and improves neurological function following ischemic stroke in rats (Rathbone et al., 2011). Also, GUO has been shown to protect neurons against cortical focal ischemia (Hansel et al., 2014, 2015) and prolong rat survival by decreasing neurological deficits following transient middle cerebral artery occlusion (Chang et al., 2008; Connell et al., 2013). Finally, GUO shows marked anticonvulsant/antiepileptic effects in several models of epilepsy (Kovacs et al., 2015a). For example, GUO and other GBPs attenuate the lipopolysaccharide-evoked increase in spike-wave discharges number in rats (Kovacs et al., 2015a,b). Also, GBPs are able to prevent seizures (de Oliveira et al., 2004; Schmidt et al., 2005) or to counteract electrophysiological spectral changes, including glutamate store into synaptic vesicles, in a quinolinic acid-induced seizure model (Tavares et al., 2005, 2008; Torres et al., 2010). Taken together, the reported data indicate that GBPs exert a neuroprotective action against glutamate toxicity and that many of the related effects on the glutamatergic system are produced by increasing astrocytic glutamate uptake (de Oliveira et al., 2004; Frizzo et al., 2005; Moretto et al., 2005; Schmidt et al., 2007), thus avoiding excitotoxic glutamate accumulation (Deutsch et al., 2008). Since this neuroprotective mechanism is largely accepted, it has been suggested the development of GBPs for the treatment of disorders associated with glutamate excitotoxicity (Deutsch et al., 2005).
Neuroprotection against Neuroinflammation-Induced Neuronal Damage
Neuroinflammation is known to contribute to neuronal damage in different noxious brain conditions (Allan and Rothwell, 2003). Thus, the GBPs role in preventing or attenuating neuroinflammation has been extensively studied. In this context, GUO is able to prevent lipopolysaccharide-induced pro-inflammatory response and oxidative stress in hippocampal primary astrocytes through activation of the Heme Oxygenase-1 (HO-1) pathway and independently of ADO system (Bellaver et al., 2015). Also, in focal cerebral ischemia model, GUO administration reduces pro-inflammatory and increases anti-inflammatory cytokine levels with reduction of microglial/macrophage cell number, leading to a decrease in neuronal damage and infarct volume with a positive effect in functional recovery (Hansel et al., 2015). Additionally, GUO has been shown to be neuroprotective in an ischemic stroke model, by inhibiting proinflammatory interleukin-8 (Connell et al., 2013), and to be able to protect hippocampal slices against inflammatory processes induced by combined glucose and oxygen deprivation, via a mechanism that involves MAPK/ERK and A1R activation (Dal-Cim et al., 2013). Finally, systemic administration of GUO following spinal cord injury in rats significantly improves motor and sensory functions, in association with attenuation of the inflammatory response and apoptotic cell death, and with an increase in sparing of axons and myelin preservation (Jiang et al., 2008a). Taken together, the neuroprotective effects exerted by GBPs against neuroinflammation support the existence of a combined activity of these compounds on both neurons and glial cells, particularly microglia cells, as already well shown for ABPs.
Neuroprotection against Reactive Oxygen Species
Oxidative stress can be defined as an increase of pro-oxidative mechanisms due to a disturbance of the equilibrium between pro-oxidant and anti-oxidant systems, which leads to the production of peroxides and free radicals that damage all the cell components, including proteins, lipids, and DNA (Kohen and Nyska, 2002). A large number of experimental data support a sustained role of GBPs in neuroprotection against oxidative stress in different in vivo and in vitro models. For instance, in cortical brain slices, GUO is neuroprotective against methylmercury-induced oxidative stress (Roos et al., 2009), and it is also able to prevent the increase in oxidative damage as well as in the levels of glutamate in a rat model of chronic hepatic encephalopathy (Paniz et al., 2014). In C6 glioma cells, GUO is neuroprotective against hypoglycemia by modulating oxidative and nitrosative stress and astroglial responses, such as glutamate uptake, glutamine synthetase activity and glutathione levels (Quincozes-Santos et al., 2013). Similarly, GUO protects SH-SY5Y neuroblastoma cells against β-amyloid toxicity, by decreasing the formation of early ROS (Pettifer et al., 2004; Tarozzi et al., 2010) and against the mitochondrial oxidative stress, induced by the blockage of mitochondrial complexes, by inducing HO-1 via PI3K/Akt/GSK-3β (Dal-Cim et al., 2012). Also, in hippocampal slices GUO reduces ROS production, prevents mitochondrial membrane depolarization, inhibits NF-kB activation and reduces iNOS following oxygen/glucose deprivation (Dal-Cim et al., 2013). GUO neuroprotection against glutamate-induced cell death mediated by the inhibition of iNOS has been further documented in hippocampal neurons (Molz et al., 2011). GUO may also be effective in attenuating the MPP+-induced collapse of mitochondrial transmembrane potential and in preventing the subsequent activation of caspase-3, thus protecting dopaminergic neurons against mitochondrial stress-induced damage (Li et al., 2014). In addition, in in vivo studies, GUO is able to reduce hippocampal oxidative damage in rats submitted to sepsis by cecal ligation and perforation (Petronilho et al., 2012), in which it also causes a recovery of memory impairment, and in mice submitted to acute restraint stress, in which GUO produces antidepressant-like effects (Bettio et al., 2012). Furthermore, in focal cerebral ischemia model, GUO administration reduces ROS levels and increases superoxide dismutase levels in the brain, leading to a decrease in neuronal damage and infarct volume with a positive effect in functional recovery (Hansel et al., 2015).
Taken together all the described findings strongly suggest a relevant neuroprotective role of GBPs in the mammalian brain, providing new targets and strategies for the treatment of brain diseases.
Behavioral Response to GBPs
Guanine-based purine effects on behavioral response have been investigated with the limit imposed by the lack of specific receptors and thereby available antagonist compounds. Noteworthy, systemic treatments show that GUO, even at high doses, does not induce mortality and any obvious behavioral disturbances, such as alterations of coordination, locomotion, body weight, and core temperature. Additionally, GUO does not produce depressant activity but rather excitant effects that resembles those caused by some ARs antagonists (El Yacoubi et al., 2003; Vinade et al., 2003; Schmidt et al., 2010). On the other hand, in memory tests in rats, GUO pre-training administration is amnesic on inhibitory avoidance task (Roesler et al., 2000; Vinade et al., 2003, 2004, 2005; Saute et al., 2006). This amnesic effect is compatible with inhibition of glutamatergic system and seems to be independent from A1R and A2R activation, since it is not inhibited by the ADO receptor antagonist caffeine (Vinade et al., 2004, 2005). This GUO effect on memory is also present after chronic administration with anticonvulsant doses of this nucleoside and, again, it is not blocked by ADO receptor antagonists (Vinade et al., 2003, 2004). Additionally, GMP is able to counteract the facilitatory effect of post-training intra-hippocampal glutamate administration on inhibitory avoidance task (Rubin et al., 1996). In contrast, in a different memory model, GUA seems to prevent the amnesic effect caused by L-NG-nitro-L-arginine methyl ester (L-NAME), an inhibitor of NOS (Giuliani et al., 2012b).
Regarding studies concerning motor behavior, it has been shown that GUO does not modulate spontaneous locomotion (Tort et al., 2004), but decreases locomotor activity in the open field test (Vinade et al., 2005). Also, it has been described that GUO improves motor behavior both in rats with parkinsonism, by decreasing bradykinesia (Su et al., 2009), and in rats with spinal cord injury (Jiang et al., 2007). Interestingly, GUO is able to produce antidepressant-like effects evaluated by means of the forced swimming test and the tail suspension test in mice. This behavioral effect would be dependent on the modulation of NMDA receptors, nitric oxide-cGMP and PI3K/mTOR pathways (Bettio et al., 2012, 2014). Additionally, systemic administration of GMP induces anxiolytic-like behavior in rats (Almeida et al., 2010). This anxiolytic-like effect of GBPs is further supported by data showing that chronic administration of GUO in mice exhibits an anxiolytic effect in the hole board task (Vinade et al., 2003), in the tail suspension test and in the open field test (Bettio et al., 2016). Similarly, acute GUO administration has also been shown to induce robust anxiolytic-like effects. Of note, pretreatment with caffeine completely abolishes GUO anxiolytic effects (Almeida et al., 2016). Noteworthy, preliminary experiments in our laboratory confirm the anxiolytic effect of GUO in three different behavioral tests in rats: light/dark, elevated plus-maze and open field (personal observations). All these behavioral data support an anxiolytic effect of GBPs, in particular of GUO, which is opposite to the depressant action of AR activation (Krugel, 2015). However, in several of the reported investigations, it is not clear the A1R and A2AR involvement in the behavioral outcomes following GUO treatment (Roesler et al., 2000; Vinade et al., 2004). Therefore, this issue needs further investigations, which should take into account other possibilities (see the GBPs receptor section, e.g., GUO may bind to heteroreceptor complexes).
Neuronal Plasticity Mediated by GBPs
To deal with the effects of GBPs on neural plasticity we have grouped the existing experimental data in the following four subjects: cell proliferation; neurite growth; synaptogenesis and synaptic activity; and synaptic plasticity.
Cell Proliferation
Cell proliferation mediated by GBPs has been largely investigated in glial cells and in a lesser extent in neuronal cells. Concerning this latter aspect, it has been shown that GBPs exert antiproliferative effects in human SH-SY5Y neuroblastoma cells by inducing a S-phase cell-cycle arrest, with up-regulation of genes for S-phase entry (Cyclin E2) and down-regulation of genes promoting cell-cycle progression, such as the cyclin B1/B2 that prevents S-phase exit (Guarnieri et al., 2009). By contrast, GUO is able to stimulate in vitro neural stem cell proliferation by activating the cAMP-CREB pathway (Su et al., 2013) and to potentiate neurogenesis in the subventricular zone in a rat model of parkinsonism (Su et al., 2009). Chronic treatment of mice for 21 days with GUO results in a significant increase in the number of immature neurons in the ventral hippocampal, which is known to regulate emotional and motivational behaviors (Bettio et al., 2016). Finally, GMP or GUO treatment of co-culture cerebellar neurons/astrocytes would not induce proliferation of neurons or astrocytes although it promotes MAPK activation (Decker et al., 2007).
Differently from data available on neurons, evidences pointing to mitogenic effects of GBPs on astrocytes are broad. The first result, already published in 1991, showed that GUO, GMP, GDP and GTP are able to increase cAMP and to stimulate astroblast proliferation through, at least in part, the enhancement of ADO extracellular levels (Kim et al., 1991; Rathbone et al., 1991; Ciccarelli et al., 2000). In this context, GUO has also been shown to stimulate astrocytes proliferation by producing large amounts of neuroprotective factors (Rathbone et al., 1999; Ciccarelli et al., 2001). Also, it has been observed that the mitogenic effects of GUO on astrocytes is significantly enhanced by the co-presence in the culture of microglial cells releasing soluble factors (Ciccarelli et al., 2000). Additionally, after spinal cord injury, chronic treatment with GUO increases remyelination by increasing proliferation and differentiation of endogenous adult oligodendroglial progenitor cells (Jiang et al., 2008a). In contrast with data suggesting a positive role of GBPs in the regulation of glial cell proliferation, Garozzo et al. (Garozzo et al., 2010) have demonstrated that GBPs (GUO, GMP, and especially GUA) exert a marked growth inhibition in standard cell culture conditions, on the U87 and U373 glioma cell lines and on other cancer cell lines, by decreasing the rate of progression through the S-phase. Interestingly, this antiproliferative effect is antagonized by ABPs and it is in part dependent on the intracellular hypoxanthine-guanine phosphoribosyltransferase, an enzyme responsible for the conversion of GUA into GMP, suggesting the central role of the intracellular metabolism of GUA for these inhibitory effects on cell proliferation (Garozzo et al., 2010). A similar antiproliferative effect has been observed in human SH-SY5Y neuroblastoma cells following GTP or GUO treatment (Guarnieri et al., 2009). Taken together, the opposite effects of GUO on cell proliferation may be dependent on the experimental model and cell lines used, which subsequently can lead to the activation of different GBPs binding sites or receptor–receptor interactions, and consequently to different functional outcomes. Nevertheless, taking into consideration that GBPs exert antiproliferative effects in brain cancer cell lines, it would be relevant to underline the possible anti-tumoral effect exerted by GBPs. In our opinion, this aspect deserves further investigation.
Neurite Outgrowth
GBPs are able to play significant neurotrophic roles by stimulating synthesis and secretion of trophic factors, in both neuronal and glial cells, and also by inducing cell differentiation, including neuritogenesis (Schmidt et al., 2007; Rathbone et al., 2008). Extracellular GUO not only stimulates neurite outgrowth in primary cultures of rat hippocampal neurons and in pheochromocytoma PC12 cells (Gysbers and Rathbone, 1996b; Rathbone et al., 1999), but also it enhances the neuritogenic effects of NGF on PC12 cells through both cAMP-dependent and -independent mechanisms (Gysbers and Rathbone, 1996a,b; Bau et al., 2005). In the same cell culture model, GTP may also enhance, by increasing intracellular Ca2+, NGF-dependent neurite outgrowth (Gysbers and Rathbone, 1996a; Gysbers et al., 2000). In human SH-SY5Y neuroblastoma cells, both GUO and GTP induce an increase in the number of neurites as well as in neurite length, and they also promote cell differentiation (Guarnieri et al., 2009). Finally GUO, although in a less extent than ADO, counteracts axonal degeneration following axotomy in cultured dorsal root ganglia neurons (Press and Milbrandt, 2009). All these effects on neurites growth are probably linked to a GBPs direct role in the regulation of neurotrophic factors expression and release (Schmidt et al., 2007; Rathbone et al., 2008). Indeed, GUO and GTP enhance in astrocytes cultures the synthesis and release of NGF, fibroblast growth factor-2 and transforming growth factor β (Middlemiss et al., 1995; Rathbone et al., 1999; Ciccarelli et al., 2001; Schmidt et al., 2007).
Synaptogenesis and Synaptic Activity
In addition to neuritogenesis, GUO may play a role in synaptogenesis. In fact, in vivo administration of GUO to the rat visual cortex promotes an increase in the number and size of synaptic buttons along the axonal branches projecting from the rat visual cortex to other cortical areas and to subcortical structures (Gerrikagoitia and Martinez-Millan, 2009). Interestingly, GUO may also increase cholesterol effiux and apolipoprotein E expression in astrocytes, through the PI3K/ERK1/2 pathways (Ballerini et al., 2006a), which have been demonstrated to promote the development of new synapses in retinal ganglion cell cultures (Goritz et al., 2005). We think that this interesting GBPs effect on synaptogenesis deserves more attention and more molecular approaches.
Concerning GBPs role in neuronal synaptic activity, it is known that GTP is stored in synaptic vesicles and co-released with ATP by exocytosis (Wagner et al., 1978; Santos et al., 2006). Also, indirect evidences indicate that GUO can be released from neurons following depolarization (Fredholm and Vernet, 1979). However, as already stated above, glial cells are the principal source of extracellular GUO, in both physiological and pathological conditions (Ciccarelli et al., 1999). GUO may also accumulate extracellularly as a result of both metabolism of extracellular nucleotides, by ectonucleotidases, and transport from the cytoplasm through the bi-directional nucleoside transporters (Schmidt et al., 2007). Once in the extracellular space, GBPs may interact with neurons at the synaptic level. However, the role of GBPs, both as neurotransmitters or neuromodulators, is poorly understood due to the above-mentioned fact that their putative own receptors are still unknown. Nevertheless, GBPs appear to interfere with prejunctional acethylcoline release in the circular muscle layer of mouse colon, and their effects are dependent on their cellular uptake and independent from ABP receptors (Zizzo et al., 2011). Furthermore, GUO is able to induce muscular relaxation; this effect is dependent on GUO cellular uptake, adenylyl cyclase activation and increase in cAMP intracellular levels, while it is independent of neural action potentials, ARs and K+ channel activation (Zizzo et al., 2013). In several experimental conditions GBPs seem to modulate glutamatergic neurotransmission. Indeed, GBPs prevent cell responses to excitatory amino-acids by blocking glutamate and AMPA binding in membrane preparations without interacting with G-proteins (Souza and Ramirez, 1991; Paz et al., 1994; Tasca et al., 1995, 1998; Dev et al., 1996; Migani et al., 1997; Porciuncula et al., 2002). Also, GBPs may inhibit membrane ion currents induced by NMDA (Paas et al., 1996) or kainate (Rubin et al., 1996; Poulopoulou and Nowak, 1998). These effects suggest that GBPs may exert a competitive inhibitory mechanism, acting as antagonists of ionotropic glutamate receptors, thus antagonizing glutamatergic neurotoxicicity, as already discussed in a previous section. Furthermore, GBPs show anti-epileptic activity not only by inhibiting lipopolysaccharide-evoked increase in spike-wave discharges (Kovacs et al., 2015a,b) but also by counteracting electrophysiological spectral changes, including glutamate storage into synaptic vesicles (Tavares et al., 2005, 2008; Torres et al., 2010), and by preventing seizures (de Oliveira et al., 2004; Schmidt et al., 2005).
On the other hand, there are evidences supporting that, at high doses, GMP may induce activation of ionotropic glutamate receptors and inhibition of glutamate transporters activity (Molz et al., 2009). Furthermore, antinociceptive effects have been observed after systemic administration of GUO, which would be linked to the modulation of non-NMDA glutamate receptors, either by means of a direct interaction with receptors or with their signal transduction mechanisms (Schmidt et al., 2008, 2010). Finally, GUO has also been shown to modulate glutamate transporters activity by decreasing glutamate uptake into synaptic vesicles. Therefore, it would modulate the amount of transmitter inside the vesicles, which might influence synaptic strength as well as vulnerability to neural damage processes, by decreasing the amount of glutamate released to the synaptic cleft (Tasca et al., 2004). Overall, the above-mentioned works largely support the modulatory role of GBPs at the glutamatergic synaptic level. However, further investigations are needed, in particular regarding the modulation of other neurotransmitters by GBPs.
Synaptic Plasticity
In contrast to the large body of data existent supporting the regulation exerted by ABPs on synaptic plasticity through their broad family of receptors, very little is known, at present, about the GBPs role in synaptic plasticity. The principal reason of this limitation is consists, as broadly repeated, of the lack of known receptor(s). Consequently, it has not been possible the discovery and use of selective agonists/antagonists. Importantly, these compounds would be crucial for determining the exact role of GBPs in various body compartments, including the CNS. Nevertheless, despite this limitation, it has been possible to assess, as described above, the modulation of glutamatergic neurotransmission by GBPs in several experimental conditions, which is known to be relevant in synaptic plasticity. Noteworthy, the effects of GBPs on glutamatergic system are mainly attributed to the increase in astrocytic glutamate uptake, or to interference with the function of ionotropic glutamate receptors. However, no data is available supporting direct neuronal presynaptic modulation of glutamate release. Indeed, GBPs block glutamate and AMPA binding in membrane preparations (Souza and Ramirez, 1991; Paz et al., 1994; Tasca et al., 1995, 1998; Dev et al., 1996; Migani et al., 1997; Porciuncula et al., 2002). Also, GBPs inhibit membrane currents induced by NMDA (Paas et al., 1996) or kainate (Rubin et al., 1996; Poulopoulou and Nowak, 1998), as well as prevent seizures (de Oliveira et al., 2004; Schmidt et al., 2005). Finally, it has also been shown that GMP, at high doses, induce activation of ionotropic glutamate receptors (Molz et al., 2009). Taken together, the actual experimental data suggest that GBPs may play a role on synaptic plasticity by modulating, in different ways, the glutamatergic system.
Conclusion and Outlook
In the present article, we have extensively summarized the present data concerning the role of the GBPs system in regulating neuronal function and plasticity. On the other hand, we have also briefly discussed the scarce knowledge regarding the mechanisms of action of these orphan neuromodulators. However, we would not like to simply conclude with the habitual sentence, classically reported in previous reviews on this issue, encouraging the scientific community to advance in GBPs research. Thus, it is clear that further work is still necessary to elucidate the mechanisms of action of GBPs, this is, the cloning of specific receptors and characterization of second messengers related to their extracellular effects. But this review has been conceived and carried out with the purpose of acquiring a new consciousness. We honestly think that research aiming the identification of GBPs receptors may be based on a recent suggestion (Ciruela, 2013) and our preliminary data, evidencing that GUO is somehow linked to ARs. Thus, cooperative efforts should be directed to explore both new GBPs putative receptors and their possible interplay, especially in terms of oligomerization and/or allosteric modulation, with ARs, and also the possible direct binding of GBPs to ARs. Overall, a very exciting work is expected to be completed, in a cooperative and collaborative way, in the next years in order to fill this “big gap” in the purinergic transmission field.
Author Contributions
BN, CF, CAF, CDF: contributed to plan the review and to revision of manuscript DLV, CR, MG: contributed to search for specific references and prepare the figure BN, CR, MG, DLV, CF: contributed to write the review BN, CDF, MG, CR, DLV, GR, FM, FDV, DIP: contributed to experimental data reported in the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported (BN and MG) by grants from PRIN-MIUR 20085HBSWS_002. Also, supported by Ministerio de Economía y Competitividad/Instituto de Salud Carlos III (SAF2014-55700-P, PCIN-2013-019-C03-03 and PIE14/00034) and Agentschap voor Innovatie door Wetenschap en Technologie (SBO-140028) to FC. F.C. and V.F.-D. belong to the “Neuropharmacology and Pain” accredited research group (Generalitat de Catalunya, 2014; SGR 1251).
References
Abbracchio, M. P., Burnstock, G., Boeynaems, J. M., Barnard, E. A., Boyer, J. L., Kennedy, C., et al. (2006). International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 58, 281–341. doi: 10.1124/pr.58.3.3
Allan, S. M., and Rothwell, N. J. (2003). Inflammation in central nervous system injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 1669–1677. doi: 10.1098/rstb.2003.1358
Almeida, R. F., Cereser, V. H. Jr., Faraco, R. B., Böhmer, A. E., Souza, D. O., and Ganzella, M. (2010). Systemic administration of GMP induces anxiolytic-like behavior in rats. Pharmacol. Biochem. Behav. 96, 306–311. doi: 10.1016/j.pbb.2010.05.022
Almeida, R. F., Comasseto, D. D., Ramos, D. B., Hansel, G., Zimmer, E. R., Loureiro, S. O., et al. (2016). Guanosine anxiolytic-like effect involves adenosinergic and glutamatergic neurotransmitter systems. Mol. Neurobiol. doi: 10.1007/s12035-015-9660-x [Epub ahead of print].
Ballerini, P., Ciccarelli, R., Di Iorio, P., Buccella, S., D’Alimonte, I., Giuliani, P., et al. (2006a). Guanosine effect on cholesterol efflux and apolipoprotein E expression in astrocytes. Purinergic Signal. 2, 637–649. doi: 10.1007/s11302-006-9011-5
Ballerini, P., Di Iorio, P., Caciagli, F., Rathbone, M. P., Jiang, S., Nargi, E., et al. (2006b). P2Y2 receptor up-regulation induced by guanosine or UTP in rat brain cultured astrocytes. Int. J. Immunopathol. Pharmacol. 19, 293–308.
Ballerini, P., Di Iorio, P., Ciccarelli, R., Caciagli, F., Poli, A., Beraudi, A., et al. (2005). P2Y1 and cysteinyl leukotriene receptors mediate purine and cysteinyl leukotriene co-release in primary cultures of rat microglia. Int. J. Immunopathol. Pharmacol. 18, 255–268.
Bau, C., Middlemiss, P. J., Hindley, S., Jiang, S., Ciccarelli, R., Caciagli, F., et al. (2005). Guanosine stimulates neurite outgrowth in PC12 cells via activation of heme oxygenase and cyclic GMP. Purinergic Signal. 1, 161–172. doi: 10.1007/s11302-005-6214-0
Bellaver, B., Souza, D. G., Bobermin, L. D., Goncalves, C. A., Souza, D. O., and Quincozes-Santos, A. (2015). Guanosine inhibits LPS-induced pro-inflammatory response and oxidative stress in hippocampal astrocytes through the heme oxygenase-1 pathway. Purinergic Signal. 11, 571–580. doi: 10.1007/s11302-015-9475-2
Bettio, L. E., Cunha, M. P., Budni, J., Pazini, F. L., Oliveira, A., Colla, A. R., et al. (2012). Guanosine produces an antidepressant-like effect through the modulation of NMDA receptors, nitric oxide-cGMP and PI3K/mTOR pathways. Behav. Brain Res. 234, 137–148. doi: 10.1016/j.bbr.2012.06.021
Bettio, L. E., Freitas, A. E., Neis, V. B., Santos, D. B., Ribeiro, C. M., Rosa, P. B., et al. (2014). Guanosine prevents behavioral alterations in the forced swimming test and hippocampal oxidative damage induced by acute restraint stress. Pharmacol. Biochem. Behav. 127, 7–14. doi: 10.1016/j.pbb.2014.10.002
Bettio, L. E., Neis, V. B., Pazini, F. L., Brocardo, P. S., Patten, A. R., Gil-Mohapel, J., et al. (2016). The antidepressant-like effect of chronic guanosine treatment is associated with increased hippocampal neuronal differentiation. Eur. J. Neurosci. 43, 1006–1015. doi: 10.1111/ejn.13172
Bonan, C. D. (2012). Ectonucleotidases and nucleotide/nucleoside transporters as pharmacological targets for neurological disorders. CNS Neurol. Disord. Drug Targets 11, 739–750. doi: 10.2174/187152712803581092
Borroto-Escuela, D. O., Brito, I., Romero-Fernandez, W., Di, P. M., Oflijan, J., Skieterska, K., et al. (2014). The G protein-coupled receptor heterodimer network (GPCR-HetNet) and its hub components. Int. J. Mol. Sci. 15, 8570–8590. doi: 10.3390/ijms15058570
Borroto-Escuela, D. O., Romero-Fernandez, W., Mudo, G., Perez-Alea, M., Ciruela, F., Tarakanov, A. O., et al. (2012). Fibroblast growth factor receptor 1- 5-hydroxytryptamine 1A heteroreceptor complexes and their enhancement of hippocampal plasticity. Biol. Psychiatry 71, 84–91. doi: 10.1016/j.biopsych.2011.09.012
Brassai, A., Suvanjeiev, R. G., Ban, E. G., and Lakatos, M. (2015). Role of synaptic and nonsynaptic glutamate receptors in ischaemia induced neurotoxicity. Brain Res. Bull. 112, 1–6. doi: 10.1016/j.brainresbull.2014.12.007
Burnstock, G. (2006). Historical review: ATP as a neurotransmitter. Trends Pharmacol. Sci. 27, 166–176. doi: 10.1016/j.tips.2006.01.005
Burnstock, G. (2007a). Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 87, 659–797. doi: 10.1152/physrev.00043.2006
Burnstock, G. (2007b). Purine and pyrimidine receptors. Cell Mol. Life Sci. 64, 1471–1483. doi: 10.1007/s00018-007-6497-0
Burnstock, G., Fredholm, B. B., and Verkhratsky, A. (2011a). Adenosine and ATP receptors in the brain. Curr. Top. Med. Chem. 11, 973–1011. doi: 10.2174/156802611795347627
Burnstock, G., Krugel, U., Abbracchio, M. P., and Illes, P. (2011b). Purinergic signalling: from normal behaviour to pathological brain function. Prog. Neurobiol. 95, 229–274. doi: 10.1016/j.pneurobio.2011.08.006
Burnstock, G., Satchell, D. G., and Smythe, A. (1972). A comparison of the excitatory and inhibitory effects of non-adrenergic, non-cholinergic nerve stimulation and exogenously applied ATP on a variety of smooth muscle preparations from different vertebrate species. Br. J. Pharmacol. 46, 234–242. doi: 10.1111/j.1476-5381.1972.tb06868.x
Caciagli, F., Di Iorio, P., Ciccarelli, R., Condorelli, D. F., Belluardo, N., Mudo, G., et al. (2014). Abstracts from purines 2014, an international conference on nucleotides, nucleosides and nucleobases, held in Bonn, Germany, from July 23-27, 2014: putative involvement of novel receptors in the effects produced by guanine and its derivatives at the Central Nervous System. Purinergic Signal. 10, 657–854. doi: 10.1007/s11302-014-9430-7
Chandrasekera, P. C., Wan, T. C., Gizewski, E. T., Auchampach, J. A., and Lasley, R. D. (2013). Adenosine A1 receptors heterodimerize with beta1- and beta2-adrenergic receptors creating novel receptor complexes with altered G protein coupling and signaling. Cell. Signal 25, 736–742. doi: 10.1016/j.cellsig.2012.12.022
Chang, R., Algird, A., Bau, C., Rathbone, M. P., and Jiang, S. (2008). Neuroprotective effects of guanosine on stroke models in vitro and in vivo. Neurosci. Lett. 431, 101–105. doi: 10.1016/j.neulet.2007.11.072
Chittiprol, S., Satishchandra, P., Bhimasenarao, R. S., Rangaswamy, G. R., Sureshbabu, S. V., Subbakrishna, D. K., et al. (2007). Plasma adenosine deaminase activity among HIV1 Clade C seropositives: relation to CD4 T cell population and antiretroviral therapy. Clin. Chim. Acta 377, 133–137. doi: 10.1016/j.cca.2006.09.006
Ciccarelli, R., Ballerini, P., Sabatino, G., Rathbone, M. P., D’Onofrio, M., Caciagli, F., et al. (2001). Involvement of astrocytes in purine-mediated reparative processes in the brain. Int. J. Dev. Neurosci. 19, 395–414. doi: 10.1016/S0736-5748(00)00084-8
Ciccarelli, R., Di Iorio, P., D’Alimonte, I., Giuliani, P., Florio, T., Caciagli, F., et al. (2000). Cultured astrocyte proliferation induced by extracellular guanosine involves endogenous adenosine and is raised by the co-presence of microglia. Glia 29, 202–211. doi: 10.1002/(SICI)1098-1136(20000201)29:3<202::AID-GLIA2>3.0.CO;2-C
Ciccarelli, R., Di Iorio, P., Giuliani, P., D’Alimonte, I., Ballerini, P., Caciagli, F., et al. (1999). Rat cultured astrocytes release guanine-based purines in basal conditions and after hypoxia/hypoglycemia. Glia 25, 93–98. doi: 10.1002/(SICI)1098-1136(19990101)25:1<93::AID-GLIA9>3.3.CO;2-E
Ciruela, F., Casado, V., Rodrigues, R. J., Lujan, R., Burgueno, J., Canals, M., et al. (2006a). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 26, 2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006
Ciruela, F., Escriche, M., Burgueno, J., Angulo, E., Casado, V., Soloviev, M. M., et al. (2001). Metabotropic glutamate 1alpha and adenosine A1 receptors assemble into functionally interacting complexes. J. Biol. Chem. 276, 18345–18351. doi: 10.1074/jbc.M006960200
Ciruela, F., Ferre, S., Casado, V., Cortes, A., Cunha, R. A., Lluis, C., et al. (2006b). Heterodimeric adenosine receptors: a device to regulate neurotransmitter release. Cell Mol. Life Sci. 63, 2427–2431. doi: 10.1007/s00018-006-6216-2
Civelli, O., Reinscheid, R. K., Zhang, Y., Wang, Z., Fredriksson, R., and Schioth, H. B. (2013). G protein-coupled receptor deorphanizations. Annu. Rev. Pharmacol. Toxicol. 53, 127–146. doi: 10.1146/annurev-pharmtox-010611-134548
Cividini, F., Tozzi, M. G., Galli, A., Pesi, R., Camici, M., Dumontet, C., et al. (2015). Cytosolic 5′-nucleotidase II interacts with the leucin rich repeat of NLR family member Ipaf. PLoS ONE 10:e0121525. doi: 10.1371/journal.pone.0121525
Connell, B. J., Di Iorio, P., Sayeed, I., Ballerini, P., Saleh, M. C., Giuliani, P., et al. (2013). Guanosine protects against reperfusion injury in rat brains after ischemic stroke. J. Neurosci. Res. 91, 262–272. doi: 10.1002/jnr.23156
Dal-Cim, T., Ludka, F. K., Martins, W. C., Reginato, C., Parada, E., Egea, J., et al. (2013). Guanosine controls inflammatory pathways to afford neuroprotection of hippocampal slices under oxygen and glucose deprivation conditions. J. Neurochem. 126, 437–450. doi: 10.1111/jnc.12324
Dal-Cim, T., Martins, W. C., Santos, A. R., and Tasca, C. I. (2011). Guanosine is neuroprotective against oxygen/glucose deprivation in hippocampal slices via large conductance Ca(2)+-activated K+ channels, phosphatidilinositol-3 kinase/protein kinase B pathway activation and glutamate uptake. Neuroscience 183, 212–220. doi: 10.1016/j.neuroscience.2011.03.022
Dal-Cim, T., Molz, S., Egea, J., Parada, E., Romero, A., Budni, J., et al. (2012). Guanosine protects human neuroblastoma SH-SY5Y cells against mitochondrial oxidative stress by inducing heme oxigenase-1 via PI3K/Akt/GSK-3beta pathway. Neurochem. Int. 61, 397–404. doi: 10.1016/j.neuint.2012.05.021
D’Alimonte, I., Flati, V., D’Auro, M., Toniato, E., Martinotti, S., Rathbone, M. P., et al. (2007). Guanosine inhibits CD40 receptor expression and function induced by cytokines and beta amyloid in mouse microglia cells. J. Immunol. 178, 720–731. doi: 10.4049/jimmunol.178.2.720
Dare, E., Schulte, G., Karovic, O., Hammarberg, C., and Fredholm, B. B. (2007). Modulation of glial cell functions by adenosine receptors. Physiol. Behav. 92, 15–20. doi: 10.1016/j.physbeh.2007.05.031
de Oliveira, D. L., Horn, J. F., Rodrigues, J. M., Frizzo, M. E., Moriguchi, E., Souza, D. O., et al. (2004). Quinolinic acid promotes seizures and decreases glutamate uptake in young rats: reversal by orally administered guanosine. Brain Res. 1018, 48–54. doi: 10.1016/j.brainres.2004.05.033
Decker, H., Francisco, S. S., Mendes-de-Aguiar, C. B., Romao, L. F., Boeck, C. R., Trentin, A. G., et al. (2007). Guanine derivatives modulate extracellular matrix proteins organization and improve neuron-astrocyte co-culture. J. Neurosci. Res. 85, 1943–1951. doi: 10.1002/jnr.21332
Del Puerto, A., Wandosell, F., and Garrido, J. J. (2013). Neuronal and glial purinergic receptors functions in neuron development and brain disease. Front. Cell Neurosci. 7:197. doi: 10.3389/fncel.2013.00197
Deutsch, S. I., Long, K. D., Rosse, R. B., Mastropaolo, J., and Eller, J. (2005). Hypothesized deficiency of guanine-based purines may contribute to abnormalities of neurodevelopment, neuromodulation, and neurotransmission in Lesch-Nyhan syndrome. Clin. Neuropharmacol. 28, 28–37. doi: 10.1097/01.wnf.0000152043.36198.25
Deutsch, S. I., Rosse, R. B., Long, K. D., Gaskins, B. L., and Mastropaolo, J. (2008). Guanosine possesses specific modulatory effects on NMDA receptor-mediated neurotransmission in intact mice. Eur. Neuropsychopharmacol. 18, 299–302. doi: 10.1016/j.euroneuro.2007.07.010
Dev, K. K., Roberts, P. J., and Henley, J. M. (1996). Characterisation of the interaction between guanyl nucleotides and AMPA receptors in rat brain. Neuropharmacology 35, 1583–1593. doi: 10.1016/S0028-3908(96)00123-2
Di Iorio, P., Ballerini, P., Caciagli, F., and Ciccarelli, R. (1998). Purinoceptor-mediated modulation of purine and neurotransmitter release from nervous tissue. Pharmacol. Res. 37, 169–178. doi: 10.1006/phrs.1998.0286
Di Iorio, P., Ballerini, P., Traversa, U., Nicoletti, F., D’Alimonte, I., Kleywegt, S., et al. (2004). The antiapoptotic effect of guanosine is mediated by the activation of the PI 3-kinase/AKT/PKB pathway in cultured rat astrocytes. Glia 46, 356–368. doi: 10.1002/glia.20002
Di Liberto, V., Garozzo, R., Grillo, M., Mudò, G., Caciagli, F., Condorelli, D. F., et al. (2012). Identification of GPR23/LPA4 as a candidate G protein-coupled receptor for guanosine. Acta Physiol. 206(Suppl. 692), O.16. doi: 10.1089/adt.2009.0261
Di Liberto, V., Mudo, G., Fuxe, K., and Belluardo, N. (2014). Interactions between cholinergic and fibroblast growth factor receptors in brain trophism and plasticity. Curr. Protein Pept. Sci. 15, 691–702. doi: 10.2174/1389203715666140901112245
Dos Santos-Rodrigues, A., Grane-Boladeras, N., Bicket, A., and Coe, I. R. (2014). Nucleoside transporters in the purinome. Neurochem. Int. 73, 229–237. doi: 10.1016/j.neuint.2014.03.014
El Yacoubi, M., Costentin, J., and Vaugeois, J. M. (2003). Adenosine A2A receptors and depression. Neurology 61, S82–S87. doi: 10.1212/01.WNL.0000095220.87550.F6
Fernandez, J. R., Sweet, E. S., Welsh, W. J., and Firestein, B. L. (2010). Identification of small molecule compounds with higher binding affinity to guanine deaminase (cypin) than guanine. Bioorg. Med. Chem. 18, 6748–6755. doi: 10.1016/j.bmc.2010.07.054
Ferre, S. (2007). Heteromerization of G-protein-coupled receptors. Implications for central nervous system function and dysfunction. Sci. World J. 7, 46–47. doi: 10.1100/tsw.2007.215
Ferreira, A. G., da Cunha, A. A., Scherer, E. B., Machado, F. R., da Cunha, M. J., Braga, A., et al. (2012). Evidence that hyperprolinemia alters glutamatergic homeostasis in rat brain: neuroprotector effect of guanosine. Neurochem. Res. 37, 205–213. doi: 10.1007/s11064-011-0604-1
Fredholm, B. B., and Vernet, L. (1979). Release of 3H-nucleosides from 3H-adenine labelled hypothalamic synaptosomes. Acta Physiol. Scand. 106, 97–107. doi: 10.1111/j.1748-1716.1979.tb06377.x
Frizzo, M. E., Lara, D. R., Dahm, K. C., Prokopiuk, A. S., Swanson, R. A., and Souza, D. O. (2001). Activation of glutamate uptake by guanosine in primary astrocyte cultures. Neuroreport 12, 879–881. doi: 10.1097/00001756-200103260-00051
Frizzo, M. E., Schwalm, F. D., Frizzo, J. K., Soares, F. A., and Souza, D. O. (2005). Guanosine enhances glutamate transport capacity in brain cortical slices. Cell Mol. Neurobiol. 25, 913–921. doi: 10.1007/s10571-005-4939-5
Fuxe, K., Borroto-Escuela, D. O., Marcellino, D., Romero-Fernandez, W., Frankowska, M., Guidolin, D., et al. (2012). GPCR heteromers and their allosteric receptor-receptor interactions. Curr. Med. Chem. 19, 356–363. doi: 10.2174/092986712803414259
Ganzella, M., de Oliveira, E. D., Comassetto, D. D., Cechetti, F., Cereser, V. H. Jr., Moreira, J. D., et al. (2012). Effects of chronic guanosine treatment on hippocampal damage and cognitive impairment of rats submitted to chronic cerebral hypoperfusion. Neurol. Sci. 33, 985–997. doi: 10.1007/s10072-011-0872-1
Garozzo, R., Sortino, M. A., Vancheri, C., and Condorelli, D. F. (2010). Antiproliferative effects induced by guanine-based purines require hypoxanthine-guanine phosphoribosyltransferase activity. Biol. Chem. 391, 1079–1089. doi: 10.1515/BC.2010.106
Gerrikagoitia, I., and Martinez-Millan, L. (2009). Guanosine-induced synaptogenesis in the adult brain in vivo. Anat. Rec. (Hoboken) 292, 1968–1975. doi: 10.1002/ar.20999
Gines, S., Hillion, J., Torvinen, M., Le, C. S., Casado, V., Canela, E. I., et al. (2000). Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc. Natl. Acad. Sci. U.S.A. 97, 8606–8611. doi: 10.1073/pnas.150241097
Giuliani, P., Ballerini, P., Buccella, S., Ciccarelli, R., Rathbone, M. P., Romano, S., et al. (2015). Guanosine protects glial cells against 6-hydroxydopamine toxicity. Adv. Exp. Med. Biol. 837, 23–33. doi: 10.1007/5584_2014_73
Giuliani, P., Ballerini, P., Ciccarelli, R., Buccella, S., Romano, S., D’Alimonte, I., et al. (2012a). Tissue distribution and metabolism of guanosine in rats following intraperitoneal injection. J. Biol. Regul. Homeost. Agents 26, 51–65.
Giuliani, P., Buccella, S., Ballerini, P., Ciccarelli, R., D’Alimonte, I., Cicchitti, S., et al. (2012b). Guanine-based purines modulate the effect of L-NAME on learning and memory in rats. Panminerva Med. 54, 53–58.
Giuliani, P., Romano, S., Ballerini, P., Ciccarelli, R., Petragnani, N., Cicchitti, S., et al. (2012c). Protective activity of guanosine in an in vitro model of Parkinson’s disease. Panminerva Med. 54, 43–51.
Giuliani, P., Zuccarini, M., Buccella, S., Rossini, M., D’Alimonte, I., Ciccarelli, R., et al. (2016). Development of a new HPLC method using fluorescence detection without derivatization for determining purine nucleoside phosphorylase activity in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1009-1010, 114–121. doi: 10.1016/j.jchromb.2015.12.012
Gondouin, B., Jourde-Chiche, N., Sallee, M., Dou, L., Cerini, C., Loundou, A., et al. (2015). Plasma xanthine oxidase activity is predictive of cardiovascular disease in patients with chronic kidney disease, independently of uric acid levels. Nephron 131, 167–174. doi: 10.1159/000441091
Goritz, C., Mauch, D. H., and Pfrieger, F. W. (2005). Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell Neurosci. 29, 190–201. doi: 10.1016/j.mcn.2005.02.006
Grillo, M., di Liberto, V., Garozzo, R., Mudo, G., Caciagli, F., Condorelli, D. F., et al. (2012). Brain expression and 3H-guanosine binding analysis of novel G protein-coupled receptor for guanosine (GPR23/LPA4). Acta Physiol. 206(Suppl. 692), P4.19.
Guarnieri, S., Pilla, R., Morabito, C., Sacchetti, S., Mancinelli, R., Fano, G., et al. (2009). Extracellular guanosine and GTP promote expression of differentiation markers and induce S-phase cell-cycle arrest in human SH-SY5Y neuroblastoma cells. Int. J. Dev. Neurosci. 27, 135–147. doi: 10.1016/j.ijdevneu.2008.11.007
Gysbers, J. W., Guarnieri, S., Mariggio, M. A., Pietrangelo, T., Fano, G., and Rathbone, M. P. (2000). Extracellular guanosine 5′ triphosphate enhances nerve growth factor-induced neurite outgrowth via increases in intracellular calcium. Neuroscience 96, 817–824. doi: 10.1016/S0306-4522(99)00588-6
Gysbers, J. W., and Rathbone, M. P. (1992). Guanosine enhances NGF-stimulated neurite outgrowth in PC12 cells. Neuroreport 3, 997–1000. doi: 10.1097/00001756-199211000-00013
Gysbers, J. W., and Rathbone, M. P. (1996a). GTP and guanosine synergistically enhance NGF-induced neurite outgrowth from PC12 cells. Int. J. Dev. Neurosci. 14, 19–34. doi: 10.1016/0736-5748(95)00083-6
Gysbers, J. W., and Rathbone, M. P. (1996b). Neurite outgrowth in PC12 cells is enhanced by guanosine through both cAMP-dependent and -independent mechanisms. Neurosci. Lett. 220, 175–178. doi: 10.1016/S0304-3940(96)13253-5
Hansel, G., Ramos, D. B., Delgado, C. A., Souza, D. G., Almeida, R. F., Portela, L. V., et al. (2014). The potential therapeutic effect of guanosine after cortical focal ischemia in rats. PLoS ONE 9:e90693. doi: 10.1371/journal.pone.0090693
Hansel, G., Tonon, A. C., Guella, F. L., Pettenuzzo, L. F., Duarte, T., Duarte, M. M., et al. (2015). Guanosine Protects Against Cortical Focal Ischemia. Involvement of Inflammatory Response. Mol. Neurobiol. 52, 1791–1803. doi: 10.1007/s12035-014-8978-0
Ito, T., van Kuilenburg, A. B., Bootsma, A. H., Haasnoot, A. J., van cruchton, A., Wada, Y., et al. (2000). Rapid screening of high-risk patients for disorders of purine and pyrimidine metabolism using HPLC-electrospray tandem mass spectrometry of liquid urine or urine-soaked filter paper strips. Clin. Chem. 46, 445–452.
Jiang, S., Ballerini, P., Buccella, S., Giuliani, P., Jiang, C., Huang, X., et al. (2008a). Remyelination after chronic spinal cord injury is associated with proliferation of endogenous adult progenitor cells after systemic administration of guanosine. Purinergic Signal. 4, 61–71. doi: 10.1007/s11302-007-9093-8
Jiang, S., Bendjelloul, F., Ballerini, P., D’Alimonte, I., Nargi, E., Jiang, C., et al. (2007). Guanosine reduces apoptosis and inflammation associated with restoration of function in rats with acute spinal cord injury. Purinergic Signal. 3, 411–421. doi: 10.1007/s11302-007-9079-6
Jiang, S., Fischione, G., Giuliani, P., Romano, S., Caciagli, F., and Di Iorio, P. (2008b). Metabolism and distribution of guanosine given intraperitoneally: implications for spinal cord injury. Nucleosides Nucleotides Nucleic Acids 27, 673–680. doi: 10.1080/15257770802143962
Karabulut, A. B., Kafkasli, A., Burak, F., and Gozukara, E. M. (2005). Maternal and fetal plasma adenosine deaminase, xanthine oxidase and malondialdehyde levels in pre-eclampsia. Cell Biochem. Funct. 23, 279–283. doi: 10.1002/cbf.1152
Kim, J. K., Rathbone, M. P., Middlemiss, P. J., Hughes, D. W., and Smith, R. W. (1991). Purinergic stimulation of astroblast proliferation: guanosine and its nucleotides stimulate cell division in chick astroblasts. J. Neurosci. Res. 28, 442–455. doi: 10.1002/jnr.490280318
Kohen, R., and Nyska, A. (2002). Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 30, 620–650. doi: 10.1080/01926230290166724
Kovacs, Z., Kekesi, K. A., Juhasz, G., Barna, J., Heja, L., Lakatos, R., et al. (2015a). Non-adenosine nucleoside inosine, guanosine and uridine as promising antiepileptic drugs: a summary of current literature. Mini. Rev. Med. Chem. 14, 1033–1042. doi: 10.2174/1389557514666141107120226
Kovacs, Z., Kekesi, K. A., Juhasz, G., and Dobolyi, A. (2015b). Modulatory effects of inosine, guanosine and uridine on lipopolysaccharide-evoked increase in spike-wave discharge activity in Wistar Albino Glaxo/Rijswijk rats. Brain Res. Bull. 118, 46–57. doi: 10.1016/j.brainresbull.2015.09.003
Krugel, U. (2015). Purinergic receptors in psychiatric disorders. Neuropharmacology 104, 212–225. doi: 10.1016/j.neuropharm.2015.10.032
Lalo, U., Pankratov, Y., Parpura, V., and Verkhratsky, A. (2011a). Ionotropic receptors in neuronal-astroglial signalling: what is the role of “excitable” molecules in non-excitable cells. Biochim. Biophys. Acta 1813, 992–1002. doi: 10.1016/j.bbamcr.2010.09.007
Lalo, U., Verkhratsky, A., and Pankratov, Y. (2011b). Ionotropic ATP receptors in neuronal-glial communication. Semin. Cell Dev. Biol. 22, 220–228. doi: 10.1016/j.semcdb.2011.02.012
Lanznaster, D., Dal-Cim, T., Piermartiri, T. C. B., and Tasca, C. I. (2016). Guanosine: a neuromodulator with therapeutic potential in brain disorders. Aging Dis. 7. doi: 10.14336/AD.2016.0208
Li, D. W., Yao, M., Dong, Y. H., Tang, M. N., Chen, W., Li, G. R., et al. (2014). Guanosine exerts neuroprotective effects by reversing mitochondrial dysfunction in a cellular model of Parkinson’s disease. Int. J. Mol. Med. 34, 1358–1364. doi: 10.3892/ijmm.2014.1904
Lopez-Cruz, R. I., Perez-Milicua, M. B., Crocker, D. E., Gaxiola-Robles, R., Bernal-Vertiz, J. A., and de la Rosa, A. (2014). Purine nucleoside phosphorylase and xanthine oxidase activities in erythrocytes and plasma from marine, semiaquatic and terrestrial mammals. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 171, 31–35. doi: 10.1016/j.cbpa.2014.02.007
Lorne, E., Zmijewski, J. W., Zhao, X., Liu, G., Tsuruta, Y., Park, Y. J., et al. (2008). Role of extracellular superoxide in neutrophil activation: interactions between xanthine oxidase and TLR4 induce proinflammatory cytokine production. Am. J. Physiol. Cell Physiol. 294, C985–C993. doi: 10.1152/ajpcell.00454.2007
Middlemiss, P. J., Gysbers, J. W., and Rathbone, M. P. (1995). Extracellular guanosine and guanosine-5′-triphosphate increase: NGF synthesis and release from cultured mouse neopallial astrocytes. Brain Res. 677, 152–156. doi: 10.1016/0006-8993(95)00156-K
Migani, P., Fiorini, R., Ferretti, E., Manini, E., Chimichi, S., and Moneti, G. (1997). Role of guanine nucleotides as endogenous ligands of a kainic acid binding site population in the mammalian cerebellum. J. Neurochem. 68, 1648–1654. doi: 10.1046/j.1471-4159.1997.68041648.x
Molz, S., Dal-Cim, T., Budni, J., Martin-de-Saavedra, M. D., Egea, J., Romero, A., et al. (2011). Neuroprotective effect of guanosine against glutamate-induced cell death in rat hippocampal slices is mediated by the phosphatidylinositol-3 kinase/Akt/glycogen synthase kinase 3beta pathway activation and inducible nitric oxide synthase inhibition. J. Neurosci. Res. 89, 1400–1408. doi: 10.1002/jnr.22681
Molz, S., Dal-Cim, T., and Tasca, C. I. (2009). Guanosine-5′-monophosphate induces cell death in rat hippocampal slices via ionotropic glutamate receptors activation and glutamate uptake inhibition. Neurochem. Int. 55, 703–709. doi: 10.1016/j.neuint.2009.06.015
Molz, S., Decker, H., Oliveira, I. J., Souza, D. O., and Tasca, C. I. (2005). Neurotoxicity induced by glutamate in glucose-deprived rat hippocampal slices is prevented by GMP. Neurochem. Res. 30, 83–89. doi: 10.1007/s11064-004-9689-0
Molz, S., Tharine, D. C., Decker, H., and Tasca, C. I. (2008). GMP prevents excitotoxicity mediated by NMDA receptor activation but not by reversal activity of glutamate transporters in rat hippocampal slices. Brain Res. 1231, 113–120. doi: 10.1016/j.brainres.2008.07.009
Morciano, M., Ortinau, S., and Zimmermann, H. (2004). Guanine nucleotides inhibit NMDA and kainate-induced neurotoxicity in cultured rat hippocampal and neocortical neurons. Neurochem. Int. 45, 95–101. doi: 10.1016/j.neuint.2003.11.017
Moretto, M. B., Arteni, N. S., Lavinsky, D., Netto, C. A., Rocha, J. B., Souza, D. O., et al. (2005). Hypoxic-ischemic insult decreases glutamate uptake by hippocampal slices from neonatal rats: prevention by guanosine. Exp. Neurol. 195, 400–406. doi: 10.1016/j.expneurol.2005.06.005
Moretto, M. B., Boff, B., Lavinsky, D., Netto, C. A., Rocha, J. B., Souza, D. O., et al. (2009). Importance of schedule of administration in the therapeutic efficacy of guanosine: early intervention after injury enhances glutamate uptake in model of hypoxia-ischemia. J. Mol. Neurosci. 38, 216–219. doi: 10.1007/s12031-008-9154-7
Muller, C. E., and Scior, T. (1993). Adenosine receptors and their modulators. Pharm. Acta Helv. 68, 77–111. doi: 10.1016/0031-6865(93)90012-U
Neary, J. T., and Zimmermann, H. (2009). Trophic functions of nucleotides in the central nervous system. Trends Neurosci. 32, 189–198. doi: 10.1016/j.tins.2009.01.002
North, R. A. (2002). Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1067. doi: 10.1152/physrev.00015.2002
Paas, Y., Devillers-Thiery, A., Changeux, J. P., Medevielle, F., and Teichberg, V. I. (1996). Identification of an extracellular motif involved in the binding of guanine nucleotides by a glutamate receptor. EMBO J. 15, 1548–1556.
Paniz, L. G., Calcagnotto, M. E., Pandolfo, P., Machado, D. G., Santos, G. F., Hansel, G., et al. (2014). Neuroprotective effects of guanosine administration on behavioral, brain activity, neurochemical and redox parameters in a rat model of chronic hepatic encephalopathy. Metab. Brain Dis. 29, 645–654. doi: 10.1007/s11011-014-9548-x
Pankratov, Y., Castro, E., Miras-Portugal, M. T., and Krishtal, O. (1998). A purinergic component of the excitatory postsynaptic current mediated by P2X receptors in the CA1 neurons of the rat hippocampus. Eur. J. Neurosci. 10, 3898–3902. doi: 10.1046/j.1460-9568.1998.00419.x
Pankratov, Y., Lalo, U., Krishtal, O. A., and Verkhratsky, A. (2009). P2X receptors and synaptic plasticity. Neuroscience 158, 137–148. doi: 10.1016/j.neuroscience.2008.03.076
Pankratov, Y. V., Lalo, U. V., and Krishtal, O. A. (2002). Role for P2X receptors in long-term potentiation. J. Neurosci. 22, 8363–8369.
Parkinson, F. E., Damaraju, V. L., Graham, K., Yao, S. Y., Baldwin, S. A., Cass, C. E., et al. (2011). Molecular biology of nucleoside transporters and their distributions and functions in the brain. Curr. Top. Med. Chem. 11, 948–972. doi: 10.2174/156802611795347582
Paz, M. M., Ramos, M., Ramirez, G., and Souza, D. (1994). Differential effects of guanine nucleotides on kainic acid binding and on adenylate cyclase activity in chick optic tectum. FEBS Lett. 355, 205–208. doi: 10.1016/0014-5793(94)01208-3
Petronilho, F., Perico, S. R., Vuolo, F., Mina, F., Constantino, L., Comim, C. M., et al. (2012). Protective effects of guanosine against sepsis-induced damage in rat brain and cognitive impairment. Brain Behav. Immun. 26, 904–910. doi: 10.1016/j.bbi.2012.03.007
Pettifer, K. M., Kleywegt, S., Bau, C. J., Ramsbottom, J. D., Vertes, E., Ciccarelli, R., et al. (2004). Guanosine protects SH-SY5Y cells against beta-amyloid-induced apoptosis. Neuroreport 15, 833–836. doi: 10.1097/00001756-200404090-00019
Polazzi, E., Monti, B., Mengoni, I., Giuliani, P., Buccella, S., Cicchitti, S., et al. (2011). “Purine nucleoside phosphorylase (PNP) is released from cultured at astrocytes and microglial cells by lysosomal exocytosis,” in Proceedings of the 35th National Congress of the Italian Society of Pharmacology Abstract online, Bologna.
Porciuncula, L. O., Vinade, L., Wofchuk, S., and Souza, D. O. (2002). Guanine based purines inhibit [(3)H]glutamate and [(3)H]AMPA binding at postsynaptic densities from cerebral cortex of rats. Brain Res. 928, 106–112. doi: 10.1016/S0006-8993(01)03368-6
Poulopoulou, C., and Nowak, L. M. (1998). Extracellular 3′,5′ cyclic guanosine monophosphate inhibits kainate-activated responses in cultured mouse cerebellar neurons. J. Pharmacol. Exp. Ther. 286, 99–109.
Press, C., and Milbrandt, J. (2009). The purine nucleosides adenosine and guanosine delay axonal degeneration in vitro. J. Neurochem. 109, 595–602. doi: 10.1111/j.1471-4159.2009.06002.x
Quincozes-Santos, A., Bobermin, L. D., de Souza, D. G., Bellaver, B., Goncalves, C. A., and Souza, D. O. (2013). Gliopreventive effects of guanosine against glucose deprivation in vitro. Purinergic Signal. 9, 643–654. doi: 10.1007/s11302-013-9377-0
Rathbone, M., Pilutti, L., Caciagli, F., and Jiang, S. (2008). Neurotrophic effects of extracellular guanosine. Nucleosides Nucleotides Nucleic Acids 27, 666–672. doi: 10.1080/15257770802143913
Rathbone, M. P., Middlemiss, P. J., DeLuca, B., and Jovetich, M. (1991). Extracellular guanosine increases astrocyte cAMP: inhibition by adenosine A2 antagonists. Neuroreport 2, 661–664. doi: 10.1097/00001756-199111000-00007
Rathbone, M. P., Middlemiss, P. J., Gysbers, J. W., Andrew, C., Herman, M. A., Reed, J. K., et al. (1999). Trophic effects of purines in neurons and glial cells. Prog. Neurobiol. 59, 663–690. doi: 10.1016/S0301-0082(99)00017-9
Rathbone, M. P., Saleh, T. M., Connell, B. J., Chang, R., Su, C., Worley, B., et al. (2011). Systemic administration of guanosine promotes functional and histological improvement following an ischemic stroke in rats. Brain Res. 1407, 79–89. doi: 10.1016/j.brainres.2011.06.027
Ribeiro, F. F., Xapelli, S., Miranda-Lourenco, C., Tanqueiro, S. R., Fonseca-Gomes, J., Diogenes, M. J., et al. (2015). Purine nucleosides in neuroregeneration and neuroprotection. Neuropharmacology 104, 226–242. doi: 10.1016/j.neuropharm.2015.11.006
Roberts, E. L., and Newton, R. P. (2004). Estimation of guanine deaminase using guanosine as a “prosubstrate”. Anal. Biochem. 324, 250–257. doi: 10.1016/j.ab.2003.09.041
Roberts, E. L., Newton, R. P., and Axford, A. T. (2004). Plasma purine nucleoside phosphorylase in cancer patients. Clin. Chim. Acta 344, 109–114. doi: 10.1016/j.cccn.2004.02.008
Roberts, J. A., Vial, C., Digby, H. R., Agboh, K. C., Wen, H., Atterbury-Thomas, A., et al. (2006). Molecular properties of P2X receptors. Pflugers Arch. 452, 486–500. doi: 10.1007/s00424-006-0073-6
Roesler, R., Vianna, M. R., Lara, D. R., Izquierdo, I., Schmidt, A. P., and Souza, D. O. (2000). Guanosine impairs inhibitory avoidance performance in rats. Neuroreport 11, 2537–2540. doi: 10.1097/00001756-200008030-00038
Roos, D. H., Puntel, R. L., Santos, M. M., Souza, D. O., Farina, M., Nogueira, C. W., et al. (2009). Guanosine and synthetic organoselenium compounds modulate methylmercury-induced oxidative stress in rat brain cortical slices: involvement of oxidative stress and glutamatergic system. Toxicol. In Vitro 23, 302–307. doi: 10.1016/j.tiv.2008.12.020
Rubin, M. A., Jurach, A., da Costa Junior, E. M., Lima, T. T., Jimenez-Bernal, R. E., Begnini, J., et al. (1996). GMP reverses the facilitatory effect of glutamate on inhibitory avoidance task in rats. Neuroreport 7, 2078–2080. doi: 10.1097/00001756-199609020-00004
Santos, T. G., Souza, D. O., and Tasca, C. I. (2006). GTP uptake into rat brain synaptic vesicles. Brain Res. 1070, 71–76. doi: 10.1016/j.brainres.2005.10.099
Saute, J. A., da Silveira, L. E., Soares, F. A., Martini, L. H., Souza, D. O., and Ganzella, M. (2006). Amnesic effect of GMP depends on its conversion to guanosine. Neurobiol. Learn. Mem. 85, 206–212. doi: 10.1016/j.nlm.2005.10.006
Schmidt, A. P., Avila, T. T., and Souza, D. O. (2005). Intracerebroventricular guanine-based purines protect against seizures induced by quinolinic acid in mice. Neurochem. Res. 30, 69–73. doi: 10.1007/s11064-004-9687-2
Schmidt, A. P., Bohmer, A. E., Leke, R., Schallenberger, C., Antunes, C., Pereira, M. S., et al. (2008). Antinociceptive effects of intracerebroventricular administration of guanine-based purines in mice: evidences for the mechanism of action. Brain Res. 1234, 50–58. doi: 10.1016/j.brainres.2008.07.091
Schmidt, A. P., Bohmer, A. E., Schallenberger, C., Antunes, C., Tavares, R. G., Wofchuk, S. T., et al. (2010). Mechanisms involved in the antinociception induced by systemic administration of guanosine in mice. Br. J. Pharmacol. 159, 1247–1263. doi: 10.1111/j.1476-5381.2009.00597.x
Schmidt, A. P., Lara, D. R., and Souza, D. O. (2007). Proposal of a guanine-based purinergic system in the mammalian central nervous system. Pharmacol. Ther. 116, 401–416. doi: 10.1016/j.pharmthera.2007.07.004
Silinsky, E. M., Gerzanich, V., and Vanner, S. M. (1992). ATP mediates excitatory synaptic transmission in mammalian neurones. Br. J. Pharmacol. 106, 762–763. doi: 10.1111/j.1476-5381.1992.tb14408.x
Sinclair, C. J., LaRiviere, C. G., Young, J. D., Cass, C. E., Baldwin, S. A., and Parkinson, F. E. (2000). Purine uptake and release in rat C6 glioma cells: nucleoside transport and purine metabolism under ATP-depleting conditions. J. Neurochem. 75, 1528–1538. doi: 10.1046/j.1471-4159.2000.0751528.x
Souza, D. O., and Ramirez, G. (1991). Effects of guanine nucleotides on kainic acid binding and on adenylate cyclase in chick optic tectum and cerebellum. J. Mol. Neurosci. 3, 39–45. doi: 10.1007/BF02896847
Stentoft, C., Vestergaard, M., Lovendahl, P., Kristensen, N. B., Moorby, J. M., and Jensen, S. K. (2014). Simultaneous quantification of purine and pyrimidine bases, nucleosides and their degradation products in bovine blood plasma by high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 1356, 197–210. doi: 10.1016/j.chroma.2014.06.065
Su, C., Elfeki, N., Ballerini, P., D’Alimonte, I., Bau, C., Ciccarelli, R., et al. (2009). Guanosine improves motor behavior, reduces apoptosis, and stimulates neurogenesis in rats with parkinsonism. J. Neurosci. Res. 87, 617–625. doi: 10.1002/jnr.21883
Su, C., Wang, P., Jiang, C., Ballerini, P., Caciagli, F., Rathbone, M. P., et al. (2013). Guanosine promotes proliferation of neural stem cells through cAMP-CREB pathway. J. Biol. Regul. Homeost. Agents 27, 673–680.
Tarozzi, A., Merlicco, A., Morroni, F., Bolondi, C., Di Iorio, P., Ciccarelli, R., et al. (2010). Guanosine protects human neuroblastoma cells from oxidative stress and toxicity induced by Amyloid-beta peptide oligomers. J. Biol. Regul. Homeost. Agents 24, 297–306.
Tasca, C. I., Cardoso, L. F., Martini, L. H., Ramirez, G., and Souza, D. O. (1998). Guanine nucleotides inhibit cAMP accumulation induced by metabotropic glutamate receptor activation. Neurochem. Res. 23, 183–188. doi: 10.1023/A:1022480825290
Tasca, C. I., Santos, T. G., Tavares, R. G., Battastini, A. M., Rocha, J. B., and Souza, D. O. (2004). Guanine derivatives modulate L-glutamate uptake into rat brain synaptic vesicles. Neurochem. Int. 44, 423–431. doi: 10.1016/j.neuint.2003.08.001
Tasca, C. I., and Souza, D. O. (2000). Interaction of adenosine and guanine derivatives in the rat hippocampus: effects on cyclic AMP levels and on the binding of adenosine analogues and GMP. Neurochem. Res. 25, 181–188. doi: 10.1023/A:1007557600687
Tasca, C. I., Wofchuk, S. T., Souza, D. O., Ramirez, G., and Rodnight, R. (1995). Guanine nucleotides inhibit the stimulation of GFAP phosphorylation by glutamate. Neuroreport 6, 249–252. doi: 10.1097/00001756-199501000-00006
Tavares, R. G., Schmidt, A. P., Abud, J., Tasca, C. I., and Souza, D. O. (2005). In vivo quinolinic acid increases synaptosomal glutamate release in rats: reversal by guanosine. Neurochem. Res. 30, 439–444. doi: 10.1007/s11064-005-2678-0
Tavares, R. G., Schmidt, A. P., Tasca, C. I., and Souza, D. O. (2008). Quinolinic acid-induced seizures stimulate glutamate uptake into synaptic vesicles from rat brain: effects prevented by guanine-based purines. Neurochem. Res. 33, 97–102. doi: 10.1007/s11064-007-9421-y
Torres, F. V., da Silva, F. M., Antunes, C., Kalinine, E., Antoniolli, E., Portela, L. V., et al. (2010). Electrophysiological effects of guanosine and MK-801 in a quinolinic acid-induced seizure model. Exp. Neurol. 221, 296–306. doi: 10.1016/j.expneurol.2009.11.013
Tort, A. B., Mantese, C. E., dos Anjos, G. M., Dietrich, M. O., Dall’Igna, O. P., Souza, D. O., et al. (2004). Guanosine selectively inhibits locomotor stimulation induced by the NMDA antagonist dizocilpine. Behav. Brain Res. 154, 417–422. doi: 10.1016/j.bbr.2004.03.008
Tozzi, M. G., Pesi, R., and Allegrini, S. (2013). On the physiological role of cytosolic 5′-nucleotidase II (cN-II): pathological and therapeutical implications. Curr. Med. Chem. 20, 4285–4291. doi: 10.2174/0929867311320340007
Traversa, U., Bombi, G., Camaioni, E., Macchiarulo, A., Costantino, G., Palmieri, C., et al. (2003). Rat brain guanosine binding site. Biological studies and pseudo-receptor construction. Bioorg. Med. Chem. 11, 5417–5425. doi: 10.1016/j.bmc.2003.09.043
Traversa, U., Bombi, G., Di, I. P., Ciccarelli, R., Werstiuk, E. S., and Rathbone, M. P. (2002). Specific [(3)H]-guanosine binding sites in rat brain membranes. Br. J. Pharmacol. 135, 969–976. doi: 10.1038/sj.bjp.0704542
Uemura, Y., Miller, J. M., Matson, W. R., and Beal, M. F. (1991). Neurochemical analysis of focal ischemia in rats. Stroke 22, 1548–1553. doi: 10.1161/01.STR.22.12.1548
Vareed, S. K., Bhat, V. B., Thompson, C., Vasu, V. T., Fermin, D., Choi, H., et al. (2011). Metabolites of purine nucleoside phosphorylase (NP) in serum have the potential to delineate pancreatic adenocarcinoma. PLoS. ONE 6:e17177. doi: 10.1371/journal.pone.0017177
Verkhratsky, A., Krishtal, O. A., and Burnstock, G. (2009). Purinoceptors on neuroglia. Mol. Neurobiol. 39, 190–208. doi: 10.1007/s12035-009-8063-2
Vinade, E. R., Izquierdo, I., Lara, D. R., Schmidt, A. P., and Souza, D. O. (2004). Oral administration of guanosine impairs inhibitory avoidance performance in rats and mice. Neurobiol. Learn. Mem. 81, 137–143. doi: 10.1016/j.nlm.2003.12.003
Vinade, E. R., Schmidt, A. P., Frizzo, M. E., Izquierdo, I., Elisabetsky, E., and Souza, D. O. (2003). Chronically administered guanosine is anticonvulsant, amnesic and anxiolytic in mice. Brain Res. 977, 97–102. doi: 10.1016/S0006-8993(03)02769-0
Vinade, E. R., Schmidt, A. P., Frizzo, M. E., Portela, L. V., Soares, F. A., Schwalm, F. D., et al. (2005). Effects of chronic administered guanosine on behavioral parameters and brain glutamate uptake in rats. J. Neurosci. Res. 79, 248–253. doi: 10.1002/jnr.20327
Volonte, C., and D’Ambrosi, N. (2009). Membrane compartments and purinergic signalling: the purinome, a complex interplay among ligands, degrading enzymes, receptors and transporters. FEBS J. 276, 318–329. doi: 10.1111/j.1742-4658.2008.06793.x
Volpini, R., Marucci, G., Buccioni, M., Dal, B. D., Lambertucci, C., Lammi, C., et al. (2011). Evidence for the existence of a specific g protein-coupled receptor activated by guanosine. Chem. Med. Chem. 6, 1074–1080. doi: 10.1002/cmdc.201100100
Wagner, J. A., Carlson, S. S., and Kelly, R. B. (1978). Chemical and physical characterization of cholinergic synaptic vesicles. Biochemistry 17, 1199–1206. doi: 10.1021/bi00600a010
Wengert, M., Adao-Novaes, J., Leao-Ferreira, L. R., and Caruso-Neves, C. (2011). Guanine-induced inhibition of renal Na(+)-ATPase activity: evidence for the involvement of the Gi protein-coupled receptor. Arch. Biochem. Biophys. 513, 126–130. doi: 10.1016/j.abb.2011.07.007
Yegutkin, G. G. (2014). Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol. 49, 473–497. doi: 10.3109/10409238.2014.953627
Zamzow, C. R., Xiong, W., and Parkinson, F. E. (2008). Adenosine produced by neurons is metabolized to hypoxanthine by astrocytes. J. Neurosci. Res. 86, 3447–3455. doi: 10.1002/jnr.21789
Zimmermann, H. (1996). Biochemistry, localization and functional roles of ecto-nucleotidases in the nervous system. Prog. Neurobiol. 49, 589–618. doi: 10.1016/0301-0082(96)00026-3
Zimmermann, H. (2011). Purinergic signaling in neural development. Semin. Cell Dev. Biol. 22, 194–204. doi: 10.1016/j.semcdb.2011.02.007
Zimmermann, H., Zebisch, M., and Strater, N. (2012). Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 8, 437–502. doi: 10.1007/s11302-012-9309-4
Zizzo, M. G., Mule, F., Amato, A., Maiorana, F., Mudo, G., Belluardo, N., et al. (2013). Guanosine negatively modulates the gastric motor function in mouse. Purinergic Signal. 9, 655–661. doi: 10.1007/s11302-013-9378-z
Keywords: guanine-based purines, guanosine, neuroprotection, synaptic plasticity, purinergic receptors, adenosine
Citation: Di Liberto V, Mudò G, Garozzo R, Frinchi M, Fernández-Dueñas V, Di Iorio P, Ciccarelli R, Caciagli F, Condorelli DF, Ciruela F and Belluardo N (2016) The Guanine-Based Purinergic System: The Tale of An Orphan Neuromodulation. Front. Pharmacol. 7:158. doi: 10.3389/fphar.2016.00158
Received: 12 April 2016; Accepted: 30 May 2016;
Published: 17 June 2016.
Edited by:
Luis F. Callado, University of the Basque Country, SpainReviewed by:
Dasiel Oscar Borroto-Escuela, Karolinska Institutet, SwedenCarina Rodrigues Boeck, Centro Universitário Franciscano, Brazil
Copyright © 2016 Di Liberto, Mudò, Garozzo, Frinchi, Fernández-Dueñas, Di Iorio, Ciccarelli, Caciagli, Condorelli, Ciruela and Belluardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natale Belluardo, bmF0YWxlLmJlbGx1YXJkb0B1bmlwYS5pdA==; Francisco Ciruela, ZmNpcnVlbGFAdWIuZWR1
†Co-first authors