 Shota Sasagawa1†
Shota Sasagawa1† Yuhei Nishimura1,2,3,4,5†Shiko Okabe2†Soichiro Murakami2Yoshifumi Ashikawa2Mizuki Yuge2Koki Kawaguchi1Reiko Kawase1Ryuji Okamoto6Masaaki Ito6
Yuhei Nishimura1,2,3,4,5†Shiko Okabe2†Soichiro Murakami2Yoshifumi Ashikawa2Mizuki Yuge2Koki Kawaguchi1Reiko Kawase1Ryuji Okamoto6Masaaki Ito6 Toshio Tanaka1,3,4,5*
Toshio Tanaka1,3,4,5*- 1Department of Systems Pharmacology, Mie University Graduate School of Medicine, Tsu, Japan
- 2Department of Molecular and Cellular Pharmacology, Pharmacogenomics and Pharmacoinformatics, Mie University Graduate School of Medicine, Tsu, Japan
- 3Mie University Medical Zebrafish Research Center, Tsu, Japan
- 4Department of Omics Medicine, Mie University Industrial Technology Innovation Institute, Tsu, Japan
- 5Department of Bioinformatics, Mie University Life Science Research Center, Tsu, Japan
- 6Department of Cardiology and Nephrology, Mie University Graduate School of Medicine, Tsu, Japan
Hypertrophic cardiomyopathy (HCM) is characterized by left ventricular hypertrophy and is associated with a number of potential outcomes, including impaired diastolic function, heart failure, and sudden cardiac death. Various etiologies have been described for HCM, including pressure overload and mutations in sarcomeric and non-sarcomeric genes. However, the molecular pathogenesis of HCM remains incompletely understood. In this study, we performed comparative transcriptome analysis to identify dysregulated genes common to five mouse HCM models of differing etiology: (i) mutation of myosin heavy chain 6, (ii) mutation of tropomyosin 1, (iii) expressing human phospholamban on a null background, (iv) knockout of frataxin, and (v) transverse aortic constriction. Gene-by-gene comparison identified five genes dysregulated in all five HCM models. Glutathione S-transferase kappa 1 (Gstk1) was significantly downregulated in the five models, whereas myosin heavy chain 7 (Myh7), connective tissue growth factor (Ctgf), periostin (Postn), and reticulon 4 (Rtn4) were significantly upregulated. Gene ontology comparison revealed that 51 cellular processes were significantly enriched in genes dysregulated in each transcriptome dataset. Among them, six processes (oxidative stress, aging, contraction, developmental process, cell differentiation, and cell proliferation) were related to four of the five genes dysregulated in all HCM models. GSTK1 was related to oxidative stress only, whereas the other four genes were related to all six cell processes except MYH7 for oxidative stress. Gene–gene functional interaction network analysis suggested correlative expression of GSTK1, MYH7, and actin alpha 2 (ACTA2). To investigate the implications of Gstk1 downregulation for cardiac function, we knocked out gstk1 in zebrafish using the clustered regularly interspaced short palindromic repeats/Cas9 system. We found that expression of the zebrafish homologs of MYH7, ACTA2, and actin alpha 1 were increased in the gstk1-knockout zebrafish. In vivo imaging of zebrafish expressing a fluorescent protein in cardiomyocytes showed that gstk1 deletion significantly decreased the end diastolic volume and, to a lesser extent, end systolic volume. These results suggest that downregulation of GSTK1 may be a common mechanism underlying HCM of various etiologies, possibly through increasing oxidative stress and the expression of sarcomere genes.
Introduction
Hypertrophic cardiomyopathy is characterized by thickening of the left ventricle and is associated with a range of potential outcomes, such as impaired diastolic function, heart failure, and sudden cardiac death (Semsarian et al., 2015). The prevalence of HCM is estimated to be ~1 in 500 people (Semsarian et al., 2015). HCM has multiple etiologies, including mutation in sarcomeric genes such as myosin heavy chain 7 (MYH7) and tropomyosin 1 (TPM1) and in non-sarcomeric genes such as PLN and FXN (Ho et al., 2015). HCM is also caused by pressure overload (Lai et al., 2014; Aubert et al., 2016). However, the molecular mechanisms underlying HCM remain incompletely understood (Force et al., 2010). In general, mutations in MYH7 and other myosin genes associated with HCM increase the force-generating capacity of the sarcomere rather than diminish its function (Poggesi and Ho, 2014). In addition, most HCM-associated mutations in thin filament regulatory proteins such as TPM1 increase the Ca2++ sensitivity of force production (Ashrafian et al., 2011). These findings suggest that compensatory hypertrophy is unlikely to be the cause of HCM induced by mutation of sarcomeric genes (Ashrafian et al., 2011). PLN regulates sarcoplasmic reticulum Ca2+ cycling in the heart through inhibition of ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (ATP2A2) (Wang et al., 2011). Mutation of PLN causing superinhibition of ATP2A2 can cause HCM (Wang et al., 2011). Haploinsufficiency of ATP2A2 can also cause HCM, possibly through mitochondrial dysfunction (Prasad et al., 2015), suggesting that mutation of PLN causing HCM may impair mitochondrial function. Haploinsufficiency of FXN is a major cause of FA (Payne and Wagner, 2012). FA is associated with progressive HCM, and this is a common cause of death in FA patients (Payne and Wagner, 2012). FXN is an iron-binding protein targeted to the mitochondrial matrix, and consistent with this, mitochondrial function is impaired in FA (Payne and Wagner, 2012). Mitochondrial dysfunction has also been detected in HCM caused by mutation in sarcomeric genes (Lucas et al., 2003) and pressure overload (Doenst et al., 2013). These findings suggest the existence of convergent pathways that cause HCM by impairment of mitochondrial function.
Comparative transcriptomics could represent a new frontier in the search for novel biomarkers and/or therapeutic targets in diseases with multiple etiologies because it facilitates the identification of dysregulated genes common to all disease etiologies (Sasagawa et al., 2016b). In this study, we sought to identify DEGs common to five different mouse models of HCM. The transcriptome datasets were downloaded from a public database (Barrett et al., 2009) and were derived from mouse models of HCM caused by: (i) mutation of myosin heavy chain 6 (Myh6) (Luczak et al., 2011), (ii) mutation of Tpm1 (Rajan et al., 2013), (iii) expressing human PLN on a null background (Wang et al., 2011), (iv) KO of Fxn (Huang et al., 2013), and (v) TAC, a model of pressure overload-induced HCM (Lai et al., 2014). We identified five genes dysregulated in all five HCM transcriptome datasets, among which glutathione S-transferase kappa 1 (Gstk1) was the only gene downregulated. We were particularly interested in this gene because Gstk1 is localized in mitochondria and peroxisomes (Petit et al., 2009). We examined the function of gstk1 in zebrafish, which has emerged as a useful in vivo model to study human genetic disorders including HCM (Becker et al., 2012). We demonstrate here that knockout of gstk1 in zebrafish increased the expression of HCM marker genes and decreased the cardiac EDV and, to a lesser extent, the ESV, suggesting that downregulation of GSTK1 may be a common mechanism underlying HCM of various etiologies.
Materials and Methods
Ethics Statement
This study was carried out in strict accordance with Japanese law [The Humane Treatment and Management of Animals (2014), Standards Relating to the Care and Management of Laboratory Animals and Relief of Pain (2013), and the Guidelines for Proper Conduct of Animal Experiments (Science Council of Japan, 2006)] (Science Council of Japan, 2006; Ministry of the Environment Japan, 2013, 2014). All efforts were made to minimize animal suffering. Mie University Institutional Animal Care and Use Committee guidelines state that no approval is required for experiments using zebrafish.
Comparative Transcriptome Analysis
Among the transcriptome datasets analyzing HCM caused by mutation or KO of 23 genes listed in Table 1 of Ho et al. (2015), including cardiac troponin T (TNNT2), cardiac troponin I (TNNI3), and cardiac myosin-binding protein C (MYBPC3), in the GEO (Barrett et al., 2009), we selected datasets satisfying all of the following criteria: (i) using mice 2–4 months old when RNA was extracted, (ii) using mice fed with a standard diet without any supplementation, (iii) analyzing the expression profile of mRNAs but not microRNAs or non-coding RNAs, (iv) with downloadable raw data or normalized data, with the quality of each probe signal available, and (v) having control and HCM groups with at least two samples in each group. Four transcriptome datasets passed these criteria. In the Myh6 model (GSE25700) (Luczak et al., 2011), C57BL/6 mice overexpressed a rat Myh6 transgene containing a point mutation (R403Q) and a deletion of amino acids 468–527 was replaced with nine non-myosin amino acids (Myh6-R403Q-d50) (Vikstrom et al., 1996). Hearts from wild-type or HCM C57BL/6 mice were excised at 2 months of age and subjected to transcriptome analysis. In the Tpm1 mutation model (GSE42892; Rajan et al., 2013), FVB/N mice overexpressed mouse Tpm1 containing a point mutation resulting in E180G (Tpm1-E180G). Hearts from wild-type or HCM FVB/N mice were excised at 4 months of age and subjected to transcriptome analysis. In the human PLN model (GSE20172) (Wang et al., 2011), hearts from wild-type or transgenic mice expressing human PLN on a null background were excised at 11 weeks of age and used for transcriptome analysis. In the Fxn-KO model (GSE31208; Huang et al., 2013), hearts from wild-type or muscle creatine kinase conditional Fxn-KO mice were excised at 10 weeks of age and subjected to transcriptome analysis. We also included a transcriptome dataset analyzing HCM caused by TAC, because it is a representative murine model of cardiac hypertrophy (Rockman et al., 1991). In the TAC model (GSE56348; Lai et al., 2014), hearts were excised from C57BL/6J mice 1 month after TAC or sham surgery performed at 8-week-old and subjected to transcriptome analysis. The raw data were normalized using “affy” (Gautier et al., 2004) for GSE25700 and GSE31208, “oligo” (Carvalho et al., 2007) for GSE42892 and GSE56348, and “limma” (Ritchie et al., 2015) for GSE20172 in Bioconductor (Gentleman et al., 2004). Probes with reliable signals were selected and subjected to “RankProd” (Hong et al., 2006) to identify DEGs in the HCM mice compared with the relevant control mice using a false discovery rate (FDR) of 20% as the threshold. The gene symbols of the DEGs in each model were converted to those of the human orthologous genes using Life Science Knowledge Bank (World Fusion, Tokyo, Japan). The list of DEGs in each HCM model is shown in Supplementary Tables S1-1–S1-5.
Bioinformatic Analysis of the DEGs in the Five HCM Models
To identify cellular processes significantly enriched for each DEG identified in each HCM model, we used Pathway Studio (Nikitin et al., 2003) that uses gene sets derived from natural language processing-based text mining of published literature in relation to biological functions such as cellular processes, expression targets, and binding partners. The lists of DEGs shown in Supplementary Tables S1-1–S1-5 were subjected to Pathway Studio and used to predict the cellular processes significantly enriched, using subnetwork enrichment analysis. The predicted cellular processes with p < 1.0 × 10-5 are shown in Supplementary Tables S2-1–S2-5. Common cellular processes among Supplementary Tables S2-1–S2-5 are shown in Supplementary Table S2-6.
To identify networks related to the five genes dysregulated in all five mouse HCM models, we used GeneMANIA in Cytoscape (Shannon et al., 2003) with the default settings. GeneMANIA uses a database of organism-specific weighted networks to construct a weighted composite functional interaction network between a pair of genes, including physical interaction, co-expression, pathway, co-localization, and shared protein domains, from a list of genes (Montojo et al., 2014). The networks related to the five common DEGs are shown in Supplementary Tables S3-1–S3-6. The gene score calculated based on these functional interactions is shown in Supplementary Table S3-7.
We used iRegulon to identify TFs potentially regulating the five DEGs common to the HCM models (Janky et al., 2014). iRegulon exploits the fact that genes co-regulated by the same TF often contain common TF-binding sites. iRegulon has been successfully used to identify TFs in gene lists (Sasagawa et al., 2016a) using ENCODE ChIP-seq data as a reference database (Gerstein et al., 2012). The five common DEGs were subjected to iRegulon using a normalized enrichment score of 5 as the threshold. The predicted TFs are listed in Supplementary Table S4.
Zebrafish Strains
We obtained Tg (myl7:mRFP) zebrafish, which express mRFP under the control of the myosin light chain 7 (myl7) promoter, a gene selectively expressed in cardiomyocytes (Kawahara et al., 2009), from the National BioResource Project Zebrafish (Saitama, Japan). Zebrafish were bred and maintained according to previously described methods (Westerfield, 2007; Nishimura et al., 2016). Briefly, zebrafish were raised at 28.5°C ± 0.5°C with a 14-h/10-h light/dark cycle. Embryos were obtained by natural mating and cultured in 0.3× Danieau’s solution (19.3 mM NaCl, 0.23 mM KCl, 0.13 mM MgSO4, 0.2 mM Ca(NO3)2, 1.7 mM HEPES, pH 7.2) until 5 dpf, at which time they were used for the in vivo imaging analyses or were processed for qPCR.
Knockout of gstk1 in Zebrafish
KO of gstk1 in zebrafish was performed by the ready-to-use CRISPR/Cas9 method (Aida et al., 2015). CRISPR RNA (crRNA) targeting a 5′-ATTGTCTCAAAAACGTTGGA-3′ sequence in the gstk1 genome and trans-activating crRNA (tracrRNA) were obtained from FASMAC (Kanagawa, Japan). Recombinant Cas9 protein was obtained from Toolgen (Seoul, South Korea). In brief, crRNA, tracrRNA, and Cas9 protein were dissolved in sterilized water at concentrations of 250, 1000, and 1000 ng/μL, respectively, and stored at -80°C until required. For microinjection, the crRNA, tracrRNA, Cas9 protein, and a lissamine-labeled control morpholino with no known target gene (Gene Tools, Philomath, OR, USA) were mixed in Yamamoto’s Ringer’s solution (0.75% NaCl, 0.02% KCl, 0.02% CaCl2, 0.002% NaHCO3) to final concentrations of 100, 100, 400 ng/μL, and 50 nM, respectively. The solution was injected into one- to four-cell-stage zebrafish embryos derived from the Tg (myl7:mRFP) line.
At 1 dpf, the embryos exhibiting bright lissamine fluorescence were selected and maintained until 5 dpf. At 5 dpf, the selected zebrafish were used for in vivo imaging of the cardiac ventricles or were processed for qPCR. After completion of the in vivo imaging experiments, genomic DNA was extracted from the zebrafish by incubation in 50 μL of lysis buffer (10 mM Tris–HCl, pH 8.0, 0.1 mM EDTA, 0.2% Triton X-100, 200 μg/mL proteinase K) at 55°C overnight, followed by incubation at 99°C for 10 min. The solution was then placed at 4°C and used as the template for PCR. To detect the crRNA-induced mutations, we performed a heteroduplex mobility assay (Kotani et al., 2015). Briefly, a short fragment of the gstk1 gene encompassing the crRNA target sites was amplified from the genomic DNA using gstk1_gF1 and gstk1_gR1 primers and QuickTaq (Toyobo, Osaka, Japan). PCR cycling conditions were: 94°C for 2 min followed by 40 cycles of 94°C for 30 s, 60°C for 30 s, and 68°C for 30 s. The PCR products were electrophoresed on 10% polyacrylamide gels (Wako Chemicals) and visualized by ethidium bromide staining. The result is shown in Supplementary Figure S2. The crRNA, tracrRNA, and PCR primer sequences are shown in Supplementary Table S5.
In Vivo Imaging of the Zebrafish Heart
Tg (myl7:mRFP) zebrafish at 5 dpf were transferred onto glass slides. A few drops of 3% low-melting point agarose were laid over the living larvae, which were immediately placed on their backs. The ventricles of the embedded larvae were observed using an epifluorescence microscope (SMZ25; Nikon, Tokyo, Japan) with RFP filters, and images were recorded at 100 frames/s for 10 s. Quantitative assessment of cardiac function was performed using ImageJ (Schneider et al., 2012) and Volocity software (Perkin Elmer, Cambridge, MA, USA). Briefly, the time-lapse images were processed using the Fast Fourier Transform package in ImageJ to reduce the background noise, and the long and short diastolic and systolic diameters of the ventricle were measured using Volocity. The EDV and ESV were calculated using the diameters. The EF was calculated from the EDV and ESV.
Quantitative PCR Analysis
Total RNA was extracted from control or gstk1-KO zebrafish at 5 dpf using an RNAqueous Micro Kit (Takara, Kyoto, Japan) according to the manufacturer’s protocol. RNA concentrations were determined using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA), and cDNAs were generated using a ReverTra Ace qPCR RT Kit (Toyobo). Quantitative PCR (qPCR) was performed using an ABI Prism 7300 (Life Technologies Carlsbad, CA, USA) with THUNDERBIRD SYBR qPCR Mix (Toyobo). The thermal cycling conditions were: 95°C for 1 min followed by 40 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 45 s. We measured the expression of actin alpha 1a (acta1a), actin alpha 1b (acta1b), ventricular myosin heavy chain (vmhc), actin-related protein 1 (arp1), connective tissue growth factor a (ctgfa), periostin, osteoblast specific factor b (postnb) and glyceraldehyde-3-phosphate dehydrogenase (gapdh) mRNA. The acta1a, acta1b, vmhc, arp1, ctgfa, and postnb mRNA levels were normalized to gapdh mRNA levels to correct for variability in the initial template concentration and the conversion efficiency of the reverse transcription reaction. The primer sequences are shown in Supplementary Table S5.
Statistical Analysis
Statistical analysis was performed using Prism 6 (GraphPad, La Jolla, CA, USA). Group means were compared by the Mann–Whitney U test with alpha set at 0.05. Data are shown as the mean ± standard error (SEM).
Results
Identification of Dysregulated Genes Common to the Five Mouse HCM Models
To identify genes dysregulated in HCM of differing etiologies, we downloaded transcriptome datasets from studies of five mouse models of HCM (Luczak et al., 2011; Wang et al., 2011; Huang et al., 2013; Rajan et al., 2013; Lai et al., 2014) from GEO (Barrett et al., 2009). We identified 966, 118, 866, 247, and 1349 DEGs in HCM caused by mutation of Myh6, mutation of Tpm1, a human PLN transgene, KO of Fxn1, and TAC, respectively, compared with the relevant controls (Supplementary Tables S1-1–S1-5). A Venn diagram showing unique and shared DEGs is shown in Figure 1. Five DEGs were either upregulated or downregulated in all five datasets (Table 1). Expression of glutathione S-transferase kappa 1 (Gstk1) was significantly decreased in the five HCM transcriptome datasets, whereas connective tissue growth factor (Ctgf), myosin heavy chain 7 (Myh7), periostin (Postn), and reticulon 4 (Rtn4) were significantly increased in all datasets. These results suggest that these five DEGs may be robust biomarkers of HCM caused by multiple mechanisms.
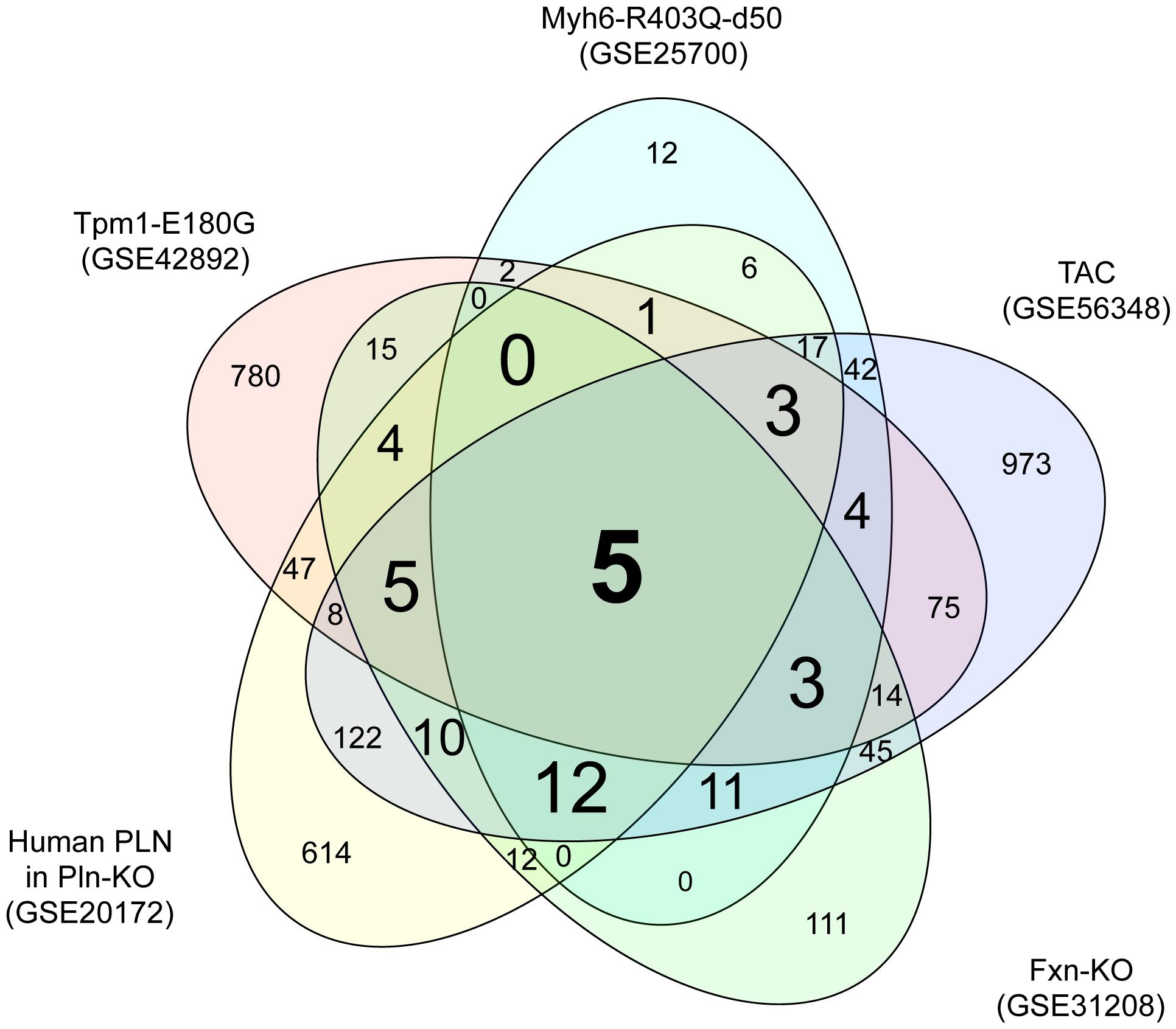
FIGURE 1. Venn diagram of differentially expressed genes in the five hypertrophic cardiomyopathy (HCM) transcriptome datasets. Transcriptome data from the mouse HCM models caused by (i) mutation of Myh6 (GSE25700), (ii) mutation of Tpm1 (GSE42892), (iii) human PLN transgene (GSE20172), (iv) KO of Fxn (GSE31208), and (v) TAC (GSE56348) were downloaded from GEO. Genes that were differentially expressed in the HCM and control groups of each dataset were identified using a FDR of 20% as the threshold. The numbers of differentially expressed genes unique to each transcriptome dataset and shared between datasets are shown.
Identification of Functional Interaction Networks Related to the Common DEGs
We then analyzed the cellular processes significantly enriched for the DEGs identified in each HCM transcriptome dataset. The identified processes are shown in Supplementary Tables S2-1–S2-5. Fifty-one cellular processes were common to the five transcriptome datasets (Figure 2A; Supplementary Table S2-6). Among these, six (oxidative stress, aging, contraction, developmental process, cell differentiation, and cell proliferation) were related to four of the five genes dysregulated in all HCM models (Figure 2B). GSTK1 was related to oxidative stress only, whereas the other four genes were related to all six cell processes, except MYH7 for oxidative stress. These results suggest that increased oxidative stress may be a common pathophysiological mechanism in HCM.
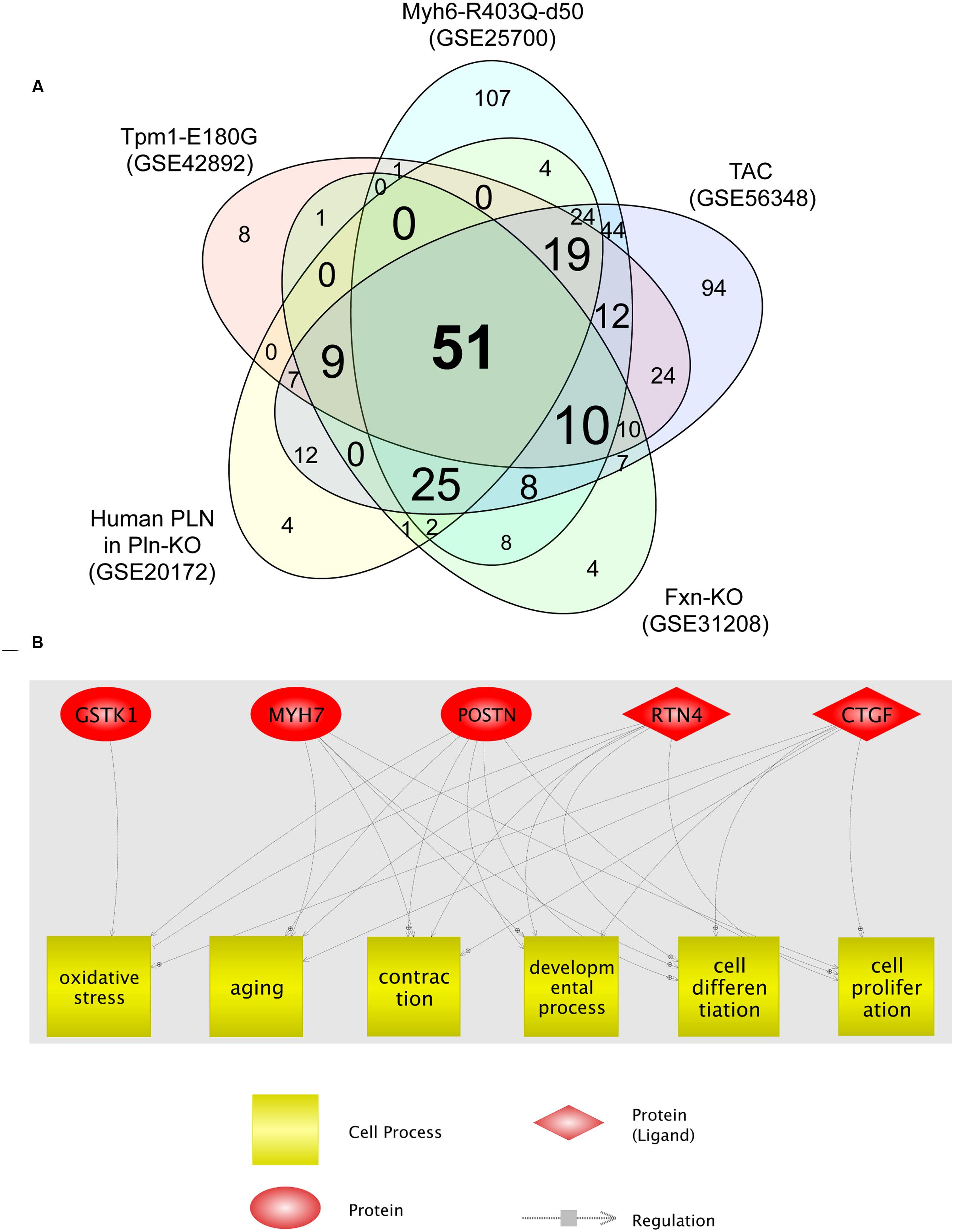
FIGURE 2. Cell processes related to the five genes dysregulated in all five HCM transcriptome datasets. (A) Venn diagram of cellular processes significantly enriched for differentially expressed genes identified in each HCM transcriptome dataset. The numbers of cellular processes unique to each transcriptome dataset and shared between datasets are shown. (B) Network among the five genes dysregulated in all five HCM transcriptome datasets, and cellular processes that connect four of the five dysregulated genes.
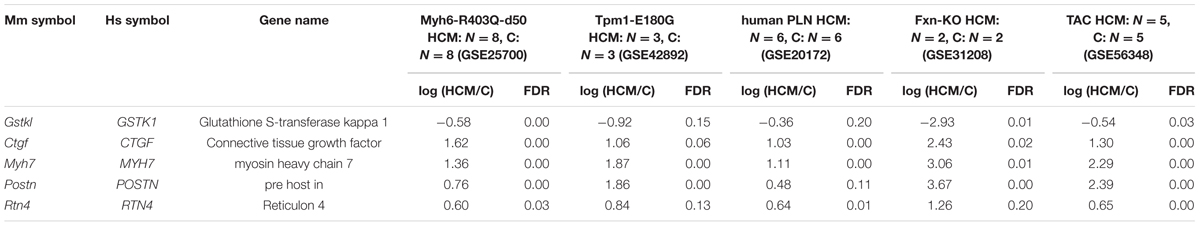
TABLE 1. Differentially expressed genes common to the five hypertrophic cardiomyopathy (HCM) transcriptome datasets.
We next analyzed the functional interaction networks related to the five common DEGs using GeneMANIA (Montojo et al., 2014). The networks identified by GeneMANIA are shown in Figure 3 and Supplementary Tables S3-1–S3-7. GSTK1 is connected to MYH7 and RTN4 through actin-related protein 1 homolog B (ACTR1B) and to CTGF and POSTN through actin α2 (ACTA2) as co-expressed genes (Supplementary Table S3-1). GSTK1 is also connected to adiponectin (ADIPOQ) by sharing a physical interaction (Supplementary Table S3-2). These results suggest that down-regulation of GSTK1 may affect the expression of co-expressed genes in the network and impair pathways related to ADIPOQ.
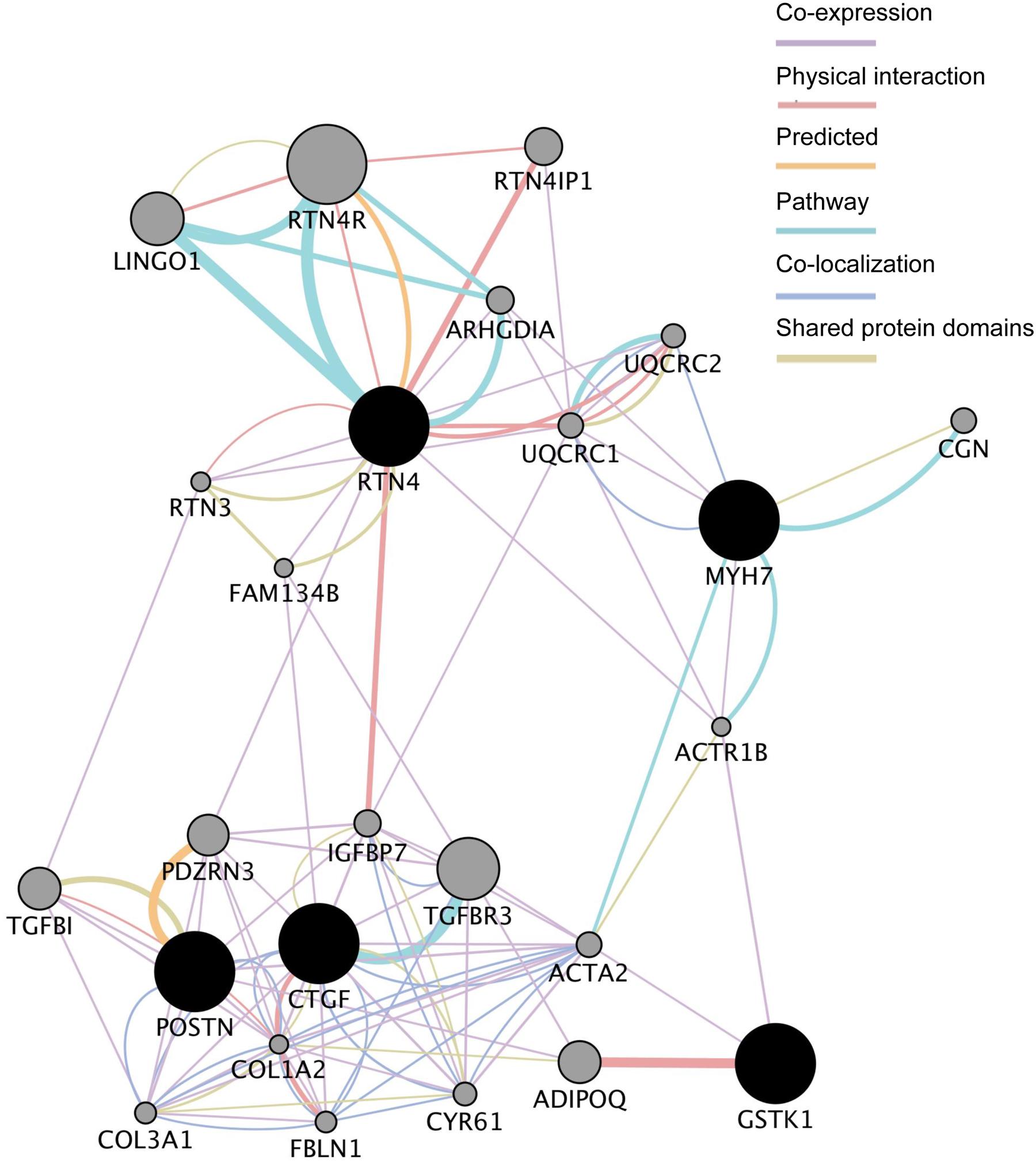
FIGURE 3. Functional interaction networks related to the five genes dysregulated in all five HCM transcriptome datasets. The five genes (shown in black circles) were subjected to GeneMANIA searches. The functional interaction networks identified by GeneMANIA are shown. Co-expression (Supplementary Table S3-1), physical interaction (Supplementary Table S3-2), predicted (Supplementary Table S3-3), pathway (Supplementary Table S3-4), co-localization (Supplementary Table S3-5), and shared protein domain (Supplementary Table S3-6) interactions are shown in purple, russet, orange, sky, aqua, and green lines, respectively. The size of the gray circles denotes the score in the functional network (Supplementary Table S3-7).
We also analyzed the TFs potentially regulating the common DEGs using iRegulon (Janky et al., 2014). The identified TFs are shown in Supplementary Figure S1 and Table S4. Eight TFs were identified, including TATA-binding protein (TBP), SRF, TEA domain family member 1 (TEAD1), and nuclear transcription factor Y beta (NFYB) as TFs possibly regulating the transcription of MYH7, CTGF, RTN4, and/or POSTN. SRF is known to be activated in HCM (Chai and Tarnawski, 2002). These results suggest that SRF may be activated in the five HCM models. However, no TFs were identified in this analysis as regulating GSTK1 (Supplementary Table S4).
gstk1 Knockout Increases Expression of the Zebrafish Homologs of ACTA2
To determine whether reduction of GSTK1 affects expression of genes co-expressed in the functional interaction networks identified by GeneMANIA, we knocked out gstk1 in zebrafish using the CRISPR/Cas9 system (Aida et al., 2015). Recent technological advances in genome editing have enabled us to make KO zebrafish for any gene of interest and validated the characterization of KO zebrafish at F0 (Kotani et al., 2015; Sasagawa et al., 2016b). As shown in Supplementary Figure S2, multiple heteroduplexes (red line) above the homoduplex (black arrowhead) were observed when PCR was performed using genomic DNA from zebrafish injected with crRNA for gstk1. These results suggest that the gstk1 crRNA efficiently edited the gstk1 gene, resulting in gstk1-KO. We then performed qPCR to examine the effect of gstk1-KO on the expression of acta1a, acta1b, vmhc, actr1, ctgfa, and postnb, the zebrafish homologs of ACTA2, ACTA1, MYH7, ACTR1B, CTGF, and POSTN, respectively. As shown in Figure 4, acta1a expression was significantly increased in gstk1-KO zebrafish compared with the control zebrafish line. The expression of acta1b and vmhc was slightly increased in the gstk1-KO zebrafish (p = 0.05 and 0.06, respectively). By contrast, there were no significant differences in actr1, ctgfa, and postnb expression between the gstk1-KO and control zebrafish. These results suggest that GSTK1 downregulation may increase the expression of sarcomeric genes, including ACTA2, ACTA1, and MYH7.
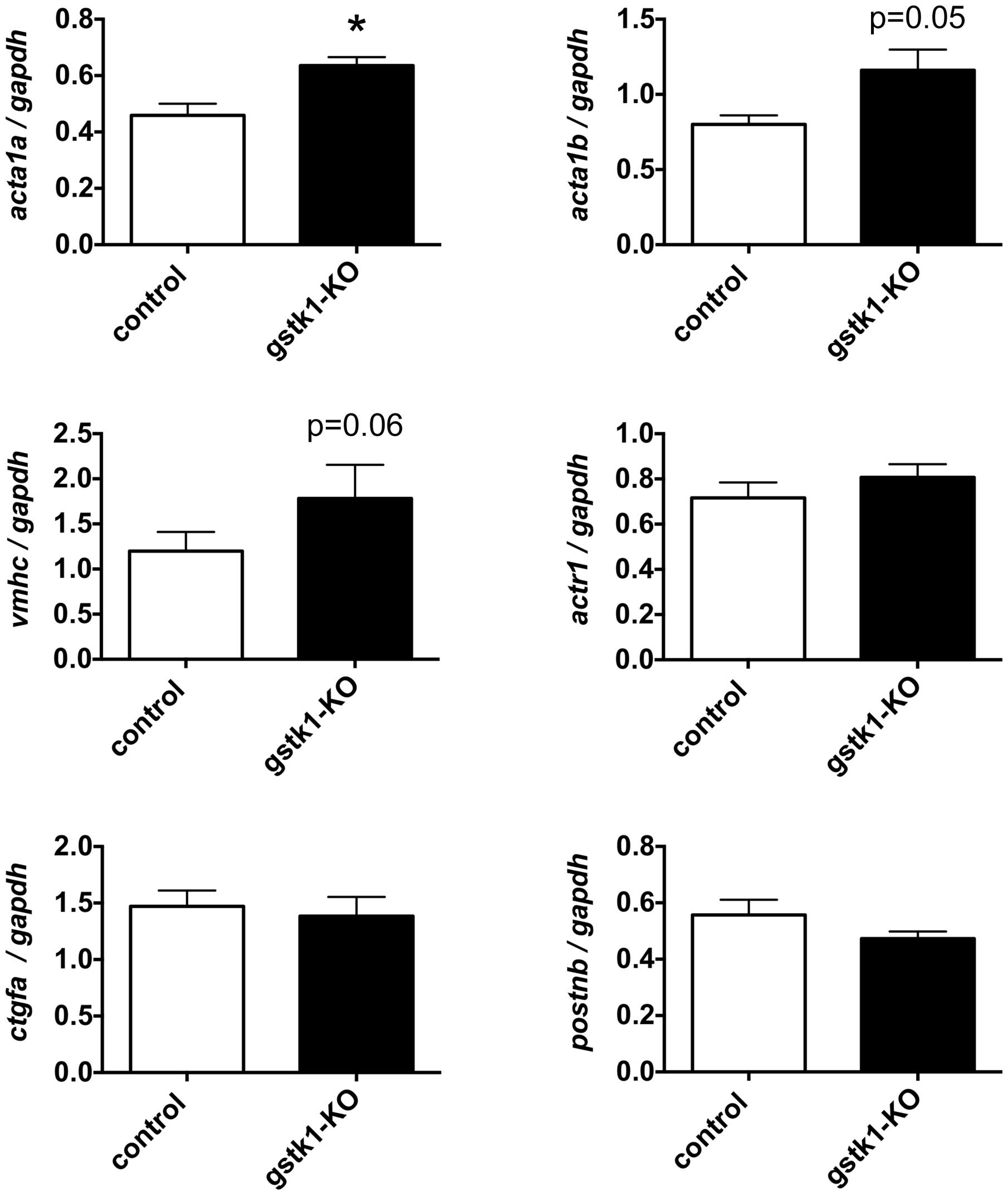
FIGURE 4. gstk1 knockout in zebrafish increases the expression of acta1a qPCR analysis of acta1a, acta1b, vmhc, actr1, ctgfa, and postnb mRNA levels in control and gstk1-knockout (KO) zebrafish at 5 dpf. Expression was normalized to gapdh mRNA levels. N = 8 per group. *p < 0.05 vs. control group.
gstk1 Knockout Decreases the Cardiac End Diastolic and Systolic Volume in Zebrafish
We next investigated the cardiac function of gstk1-KO zebrafish expressing mRFP in cardiomyocytes. The animals were examined by fluorescence microscopy and images were captured for measurement of diastolic and systolic diameters and calculation of EDV, ESV, and EF. We observed that the EDV in gstk1-KO zebrafish was significantly smaller than that of control zebrafish (Figure 5). The ESV was also significantly reduced in gstk1-KO zebrafish compared with that in control zebrafish, albeit to a lesser extent than the EDV. These results suggest that downregulation of GSTK1 and subsequent upregulation of sarcomeric genes may be a common pathophysiological mechanism in HCM.
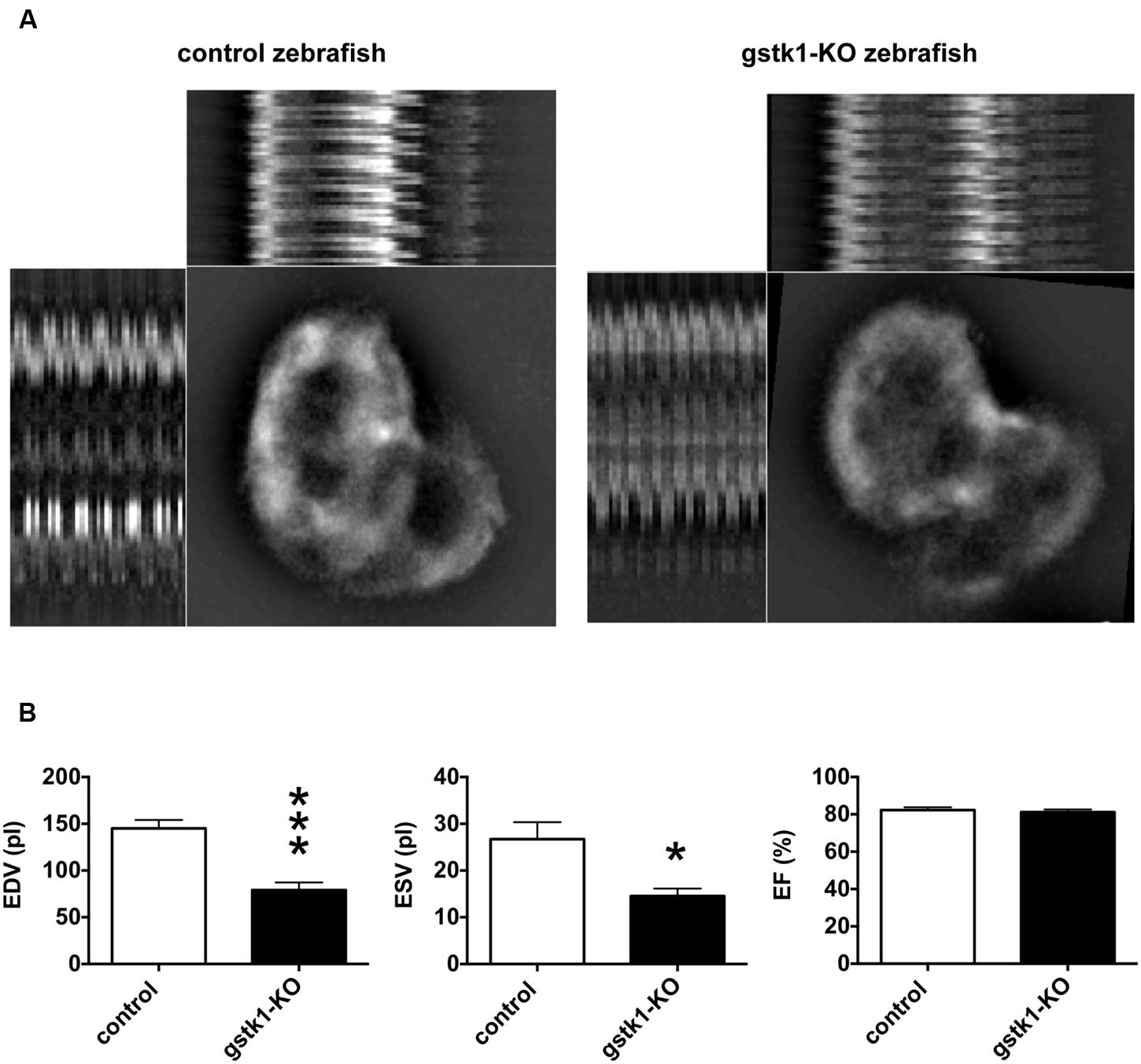
FIGURE 5. gstk1 knockout decreases the cardiac end diastolic and systolic volumes in zebrafish. (A) In vivo imaging of the hearts of control and gstk1-knockout (KO) Tg (myl7:mRFP) zebrafish at 5 dpf. Zebrafish were placed on slides on their backs, and the heart was imaged under a fluorescence microscope at 100 frames/s for 10 s. Image stack projections and the M-mode imaging of ventricles are shown. Bar, 100 μm. (B) Quantitative analysis of the in vivo imaging of zebrafish heart. The end diastolic volume (EDV) of gstk1-KO zebrafish was significantly smaller than that of control zebrafish, whereas the end systolic volume (ESV) and ejection fraction (EF) were not significantly different. N = 13 and 7 for the control and gstk1-KO groups, respectively. *p < 0.05, ***p < 0.001 vs. control group.
Discussion
MYH7, CTGF, POSTN, RTN4, and GSTK1 are Dysregulated in HCM
We demonstrated that expression of Myh7, Ctgf, Postn, and Rtn4 is significantly increased and expression of Gstk1 is significantly decreased in five mouse HCM models. These results suggest that the five genes may be robust biomarkers of disease and/or involved in the pathogenesis of HCM.
MYH7, which encodes cardiac muscle β myosin heavy chain, is the major sarcomeric protein. Expression of MYH7 is increased in the hearts of HCM patients compared with healthy subjects (Kontaraki et al., 2007). Our bioinformatic analysis revealed that SRF may increase the expression of MYH7, consistent with previous reports (Nelson et al., 2005). These findings suggest that MYH7 upregulation, possibly through SRF activation, may be a common pathophysiological pathway in HCM.
CTGF and POSTN are extracellular matrix proteins regulated by TGFβ signaling (Grotendorst, 1997; Horiuchi et al., 1999). Previous work has shown that Ctgf and Postn expression is increased in mouse models of HCM caused by Myh6-R403Q (Teekakirikul et al., 2010; Tsoutsman et al., 2013) and by KO of Mybpc3 (Judge et al., 2015). It was suggested that the source of Ctgf and Postn may be cardiac fibroblasts (Teekakirikul et al., 2010; Seidman and Seidman, 2011). TGFβ signaling is activated in various HCM models, and inhibition of TGFβ reduces fibrosis and limits hypertrophy remodeling (Teekakirikul et al., 2010; Seidman and Seidman, 2011). Thus, increased expression of CTGF and POSTN, potentially via TGFβ signaling, may underlie the fibrosis and collagen deposition associated with HCM of various etiologies.
RTN4, also called Nogo-A, is enriched in endoplasmic reticulum and known to be increased in genetic models of dilated cardiomyopathy and in end-stage heart failure in humans (Bullard et al., 2008). Knockdown of Nogo-A inhibited hypoxia/reoxygenation-induced activation of mitochondrial-dependent apoptosis in cardiomyocytes (Sarkey et al., 2011). These findings suggest that increased expression of RTN4 may also be associated with HCM through impairment of mitochondria.
GSTK1 is a member of the κ class of glutathione S-transferases and is localized in mitochondria and peroxisomes (Morel et al., 2004; Raza, 2011). Proteomic analysis revealed that Gstk1 expression is decreased in HCM caused by pressure overload (Aubert et al., 2016) and diabetic cardiomyopathy (Das et al., 2015). Gstk1 expression is increased by peroxisome proliferator-activated receptor α (PPARα) agonists (Knight et al., 2008). Given that PPARα signaling is impaired in HCM (Taegtmeyer et al., 2004), these observations suggest that GSTK1 expression may be reduced, possibly via inhibition of PPARα, in HCM caused by various mechanisms.
Reduction of GSTK1 Expression is Associated with HCM
In this study, we demonstrated that KO of gstk1 in zebrafish increased the expression of ACTA2, ACTA1, and MYH7 homologs and decreased the EDV and, to a lesser extent, the ESV, consistent with a possible causative role for GSTK1 downregulation in HCM.
GSTK1 is localized in mitochondria and peroxisomes (Raza, 2011), which are the important sites for lipid metabolism and oxygen consumption in cardiomyocytes (Colasante et al., 2015). Knockdown of gstk1 and gstk2 in Caenorhabditis elegans impairs oxygen consumption and lipid metabolism (Petit et al., 2009). In contrast, overexpression of GSTK1 reduces lipid peroxidation in peroxisomes (Wang et al., 2013). Peroxisome-derived oxidative stress can cause mitochondrial damage (Wang et al., 2013). Oxidative stress, lipid peroxidation, and mitochondrial dysfunction have been associated with HCM (Vakrou and Abraham, 2014); the suppression of each function mitigated HCM (Liu et al., 2012). These findings suggest that down-regulation of GSTK1 may cause HCM through increased oxidative stress, lipid peroxidation, and mitochondrial dysfunction. Increased oxidative stress activates SRF (Jin et al., 2011), suggesting that down-regulation of GSTK1 may activate SRF through increasing oxidative stress, resulting in increased SRF-target genes such as ACTA2, ACTA1, and MYH7 (Miano et al., 2007). However, the magnitude of the increase in the homologs of ACTA1 and MYH7 in gstk1-KO zebrafish was not strong. Moreover, the expression of the homologs of CTGF and POSTN was significantly increased in the five mouse HCM models but not in gstk1-KO zebrafish. These results suggest that down-regulation of GSTK1 and other signaling pathways may synergistically cause HCM in mammals. Further studies using various models such as Gstk1-KO mice and induced pluripotent stem cells derived from HCM patients will be necessary to elucidate the detailed mechanisms by which downregulation of GSTK1 may cause HCM.
Author Contributions
YN conceived the study, performed the bioinformatics analyses, and wrote the manuscript. SS performed the experiments using the gstk1-KO zebrafish. SO performed the quantitative analysis of in vivo imaging of zebrafish hearts. SM, YA, MY, KK, and RK provided assistance with experiments. RO and MI wrote the manuscript. TT conceived the study and wrote the manuscript.
Funding
This work was supported in part by the Japan Society for the Promotion of Science KAKENHI (25670127, 15K15051, 24510069, 16K08547) and the Long-range Research Initiative of the Japan Chemical Industrial Association (13_PT01-01).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Junko Koiwa, Hiroko Nakayama, Yuka Hayakawa, Yuka Takahashi, Chizuru Hirota, and Michiko Ariyoshi for assistance with the experiments, and Rie Ikeyama and Yuka Mizutani for administrative support.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphar.2016.00162
Abbreviations
ACTA1, actin alpha 1; ACTA2, actin alpha 2; CRISPR, clustered regularly interspaced short palindromic repeats; crRNA, CRISPR RNA; CTGF, connective tissue growth factor; dpf, days-post-fertilization; EDV, end diastolic volume; ESV, end systolic volume; EF, ejection fraction; DEG, differentially expressed gene; FA, Friedreich’s ataxia; FXN, frataxin; GEO, Gene Expression Omnibus; GSTK1, glutathione S-transferase kappa 1; HCM, hypertrophic cardiomyopathy; KO, knockout; mRFP, monomeric red fluorescent protein; MYH6, myosin heavy chain 6; MYH7, myosin heavy chain 7; MYL7, myosin light chain 7; PLN, phospholamban; POSTN, periostin; qPCR, quantitative polymerase chain reaction; RTN4, reticulon 4; SRF, serum response factor; TAC, transverse aortic constriction; TF, transcription factor; TPM1, tropomyosin 1.
References
Aida, T., Chiyo, K., Usami, T., Ishikubo, H., Imahashi, R., Wada, Y., et al. (2015). Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 16:87. doi: 10.1186/s13059-015-0653-x
Ashrafian, H., Mckenna, W. J., and Watkins, H. (2011). Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 109, 86–96. doi: 10.1161/CIRCRESAHA.111.242974
Aubert, G., Martin, O. J., Horton, J. L., Lai, L., Vega, R. B., Leone, T. C., et al. (2016). The failing heart relies on ketone bodies as a fuel. Circulation 133, 698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
Barrett, T., Troup, D. B., Wilhite, S. E., Ledoux, P., Rudnev, D., Evangelista, C., et al. (2009). NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 37, D885–D890. doi: 10.1093/nar/gkn764
Becker, J. R., Robinson, T. Y., Sachidanandan, C., Kelly, A. E., Coy, S., Peterson, R. T., et al. (2012). In vivo natriuretic peptide reporter assay identifies chemical modifiers of hypertrophic cardiomyopathy signalling. Cardiovasc. Res. 93, 463–470. doi: 10.1093/cvr/cvr350
Bullard, T. A., Protack, T. L., Aguilar, F., Bagwe, S., Massey, H. T., and Blaxall, B. C. (2008). Identification of Nogo as a novel indicator of heart failure. Physiol. Genomics 32, 182–189. doi: 10.1152/physiolgenomics.00200.2007
Carvalho, B., Bengtsson, H., Speed, T. P., and Irizarry, R. A. (2007). Exploration, normalization, and genotype calls of high-density oligonucleotide SNP array data. Biostatistics 8, 485–499. doi: 10.1093/biostatistics/kxl042
Chai, J., and Tarnawski, A. S. (2002). Serum response factor: discovery, biochemistry, biological roles and implications for tissue injury healing. J. Physiol. Pharmacol. 53, 147–157.
Colasante, C., Chen, J., Ahlemeyer, B., and Baumgart-Vogt, E. (2015). Peroxisomes in cardiomyocytes and the peroxisome / peroxisome proliferator-activated receptor-loop. Thromb. Haemost. 113, 452–463. doi: 10.1160/TH14-06-0497
Das, A., Durrant, D., Salloum, F. N., Xi, L., and Kukreja, R. C. (2015). PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Ther. 147, 12–21. doi: 10.1016/j.pharmthera.2014.10.003
Doenst, T., Nguyen, T. D., and Abel, E. D. (2013). Cardiac metabolism in heart failure: implications beyond ATP production. Circ. Res. 113, 709–724. doi: 10.1161/CIRCRESAHA.113.300376
Force, T., Bonow, R. O., Houser, S. R., Solaro, R. J., Hershberger, R. E., Adhikari, B., et al. (2010). Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation 122, 1130–1133.
Gautier, L., Cope, L., Bolstad, B. M., and Irizarry, R. A. (2004). affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315. doi: 10.1093/bioinformatics/btg405
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. doi: 10.1186/gb-2004-5-10-r80
Gerstein, M. B., Kundaje, A., Hariharan, M., Landt, S. G., Yan, K. K., Cheng, C., et al. (2012). Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100. doi: 10.1038/nature11245
Grotendorst, G. R. (1997). Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 8, 171–179. doi: 10.1016/S1359-6101(97)00010-5
Ho, C. Y., Charron, P., Richard, P., Girolami, F., Van Spaendonck-Zwarts, K. Y., and Pinto, Y. (2015). Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc. Res. 105, 397–408. doi: 10.1093/cvr/cvv025
Hong, F., Breitling, R., Mcentee, C. W., Wittner, B. S., Nemhauser, J. L., and Chory, J. (2006). RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 22, 2825–2827. doi: 10.1093/bioinformatics/btl476
Horiuchi, K., Amizuka, N., Takeshita, S., Takamatsu, H., Katsuura, M., Ozawa, H., et al. (1999). Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J. Bone. Miner. Res. 14, 1239–1249. doi: 10.1359/jbmr.1999.14.7.1239
Huang, M. L., Sivagurunathan, S., Ting, S., Jansson, P. J., Austin, C. J., Kelly, M., et al. (2013). Molecular and functional alterations in a mouse cardiac model of Friedreich ataxia: activation of the integrated stress response, eIF2alpha phosphorylation, and the induction of downstream targets. Am. J. Pathol. 183, 745–757. doi: 10.1016/j.ajpath.2013.05.032
Janky, R., Verfaillie, A., Imrichova, H., Van De Sande, B., Standaert, L., Christiaens, V., et al. (2014). iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 10:e1003731. doi: 10.1371/journal.pcbi.1003731
Jin, W., Goldfine, A. B., Boes, T., Henry, R. R., Ciaraldi, T. P., Kim, E. Y., et al. (2011). Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J. Clin. Invest. 121, 918–929. doi: 10.1172/JCI41940
Judge, D. P., Neamatalla, H., Norris, R. A., Levine, R. A., Butcher, J. T., Vignier, N., et al. (2015). Targeted Mybpc3 knock-out mice with cardiac hypertrophy exhibit structural mitral valve abnormalities. J. Cardiovasc. Dev. Dis. 2, 48–65. doi: 10.3390/jcdd2020048
Kawahara, A., Nishi, T., Hisano, Y., Fukui, H., Yamaguchi, A., and Mochizuki, N. (2009). The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 323, 524–527. doi: 10.1126/science.1167449
Knight, T. R., Choudhuri, S., and Klaassen, C. D. (2008). Induction of hepatic glutathione S-transferases in male mice by prototypes of various classes of microsomal enzyme inducers. Toxicol. Sci. 106, 329–338. doi: 10.1093/toxsci/kfn179
Kontaraki, J. E., Parthenakis, F. I., Patrianakos, A. P., Karalis, I. K., and Vardas, P. E. (2007). Altered expression of early cardiac marker genes in circulating cells of patients with hypertrophic cardiomyopathy. Cardiovasc. Pathol. 16, 329–335. doi: 10.1016/j.carpath.2007.04.004
Kotani, H., Taimatsu, K., Ohga, R., Ota, S., and Kawahara, A. (2015). Efficient multiple genome modifications induced by the crRNAs, tracrRNA and Cas9 protein complex in zebrafish. PLoS ONE 10:e0128319. doi: 10.1371/journal.pone.0128319
Lai, L., Leone, T. C., Keller, M. P., Martin, O. J., Broman, A. T., Nigro, J., et al. (2014). Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ. Heart Fail. 7, 1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469
Liu, X., Ye, B., Miller, S., Yuan, H., Zhang, H., Tian, L., et al. (2012). Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell. Biol. 32, 4493–4504. doi: 10.1128/MCB.01092-12
Lucas, D. T., Aryal, P., Szweda, L. I., Koch, W. J., and Leinwand, L. A. (2003). Alterations in mitochondrial function in a mouse model of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 284, H575–H583. doi: 10.1152/ajpheart.00619.2002
Luczak, E. D., Barthel, K. K., Stauffer, B. L., Konhilas, J. P., Cheung, T. H., and Leinwand, L. A. (2011). Remodeling the cardiac transcriptional landscape with diet. Physiol. Genom. 43, 772–780. doi: 10.1152/physiolgenomics.00237.2010
Miano, J. M., Long, X., and Fujiwara, K. (2007). Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am. J. Physiol. Cell Physiol. 292, C70–C81. doi: 10.1152/ajpcell.00386.2006
Ministry of the Environment Japan (2013). Standards Relating to the Care and Management of Laboratory Animals and Relief of Pain, Notice No.88. Chiyoda: Ministry of the Environment Japan.
Ministry of the Environment Japan (2014). The Law for the Humane Treatment and Management of Animals, Law No. 105. Chiyoda: Ministry of the Environment Japan.
Montojo, J., Zuberi, K., Rodriguez, H., Bader, G. D., and Morris, Q. (2014). GeneMANIA: fast gene network construction and function prediction for Cytoscape. F1000Res. 3:153. doi: 10.12688/f1000research.4572.1
Morel, F., Rauch, C., Petit, E., Piton, A., Theret, N., Coles, B., et al. (2004). Gene and protein characterization of the human glutathione S-transferase kappa and evidence for a peroxisomal localization. J. Biol. Chem. 279, 16246–16253. doi: 10.1074/jbc.M313357200
Nelson, T. J., Balza, R. Jr., Xiao, Q., and Misra, R. P. (2005). SRF-dependent gene expression in isolated cardiomyocytes: regulation of genes involved in cardiac hypertrophy. J. Mol. Cell Cardiol. 39, 479–489. doi: 10.1016/j.yjmcc.2005.05.004
Nikitin, A., Egorov, S., Daraselia, N., and Mazo, I. (2003). Pathway studio–the analysis and navigation of molecular networks. Bioinformatics 19, 2155–2157. doi: 10.1093/bioinformatics/btg290
Nishimura, Y., Inoue, A., Sasagawa, S., Koiwa, J., Kawaguchi, K., Kawase, R., et al. (2016). Using zebrafish in systems toxicology for developmental toxicity testing. Congenit. Anom. (Kyoto) 56, 18–27. doi: 10.1111/cga.12142
Payne, R. M., and Wagner, G. R. (2012). Cardiomyopathy in Friedreich ataxia: clinical findings and research. J. Child Neurol. 27, 1179–1186. doi: 10.1177/0883073812448535
Petit, E., Michelet, X., Rauch, C., Bertrand-Michel, J., Terce, F., Legouis, R., et al. (2009). Glutathione transferases kappa 1 and kappa 2 localize in peroxisomes and mitochondria, respectively, and are involved in lipid metabolism and respiration in Caenorhabditis elegans. FEBS J. 276, 5030–5040. doi: 10.1111/j.1742-4658.2009.07200.x
Poggesi, C., and Ho, C. Y. (2014). Muscle dysfunction in hypertrophic cardiomyopathy: what is needed to move to translation? J. Muscle Res. Cell Motil. 35, 37–45. doi: 10.1007/s10974-014-9374-0
Prasad, V., Lorenz, J. N., Lasko, V. M., Nieman, M. L., Huang, W., Wang, Y., et al. (2015). SERCA2 haploinsufficiency in a mouse model of darier disease causes a selective predisposition to heart failure. BioMed Res. Int. 2015:251598. doi: 10.1155/2015/251598
Rajan, S., Pena, J. R., Jegga, A. G., Aronow, B. J., Wolska, B. M., and Wieczorek, D. F. (2013). Microarray analysis of active cardiac remodeling genes in a familial hypertrophic cardiomyopathy mouse model rescued by a phospholamban knockout. Physiol. Genom. 45, 764–773. doi: 10.1152/physiolgenomics.00023.2013
Raza, H. (2011). Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J. 278, 4243–4251. doi: 10.1111/j.1742-4658.2011.08358.x
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Rockman, H. A., Ross, R. S., Harris, A. N., Knowlton, K. U., Steinhelper, M. E., Field, L. J., et al. (1991). Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 88, 8277–8281. doi: 10.1073/pnas.88.18.8277
Sarkey, J. P., Chu, M., Mcshane, M., Bovo, E., Ait Mou, Y., Zima, A. V., et al. (2011). Nogo-A knockdown inhibits hypoxia/reoxygenation-induced activation of mitochondrial-dependent apoptosis in cardiomyocytes. J. Mol. Cell Cardiol. 50, 1044–1055. doi: 10.1016/j.yjmcc.2011.03.004
Sasagawa, S., Nishimura, Y., Hayakawa, Y., Murakami, S., Ashikawa, Y., Yuge, M., et al. (2016a). E2F4 promotes neuronal regeneration and functional recovery after spinal cord injury in zebrafish. Front. Pharmacol. 7:119. doi: 10.3389/fphar.2016.00119
Sasagawa, S., Nishimura, Y., Sawada, H., Zhang, E., Murakami, S., Ashikawa, Y., et al. (2016b). Comparative transcriptome analysis identifies CCDC80 as a novel gene associated with pulmonary arterial hypertension. Front. Pharmacol. 7:142. doi: 10.3389/fphar.2016.00142
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Science Council of Japan (2006). Guidelines for Proper Conduct of Animal Experiments. Available at: www.scj.go.jp/ja/info/kohyo/pdf/kohyo-20-k16-2e.pdf
Seidman, C. E., and Seidman, J. G. (2011). Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ. Res. 108, 743–750. doi: 10.1161/CIRCRESAHA.110.223834
Semsarian, C., Ingles, J., Maron, M. S., and Maron, B. J. (2015). New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254. doi: 10.1016/j.jacc.2015.01.019
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Taegtmeyer, H., Golfman, L., Sharma, S., Razeghi, P., and Van Arsdall, M. (2004). Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann. N. Y. Acad. Sci. 1015, 202–213. doi: 10.1196/annals.1302.017
Teekakirikul, P., Eminaga, S., Toka, O., Alcalai, R., Wang, L., Wakimoto, H., et al. (2010). Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J. Clin. Invest. 120, 3520–3529. doi: 10.1172/JCI42028
Tsoutsman, T., Wang, X., Garchow, K., Riser, B., Twigg, S., and Semsarian, C. (2013). CCN2 plays a key role in extracellular matrix gene expression in severe hypertrophic cardiomyopathy and heart failure. J. Mol. Cell Cardiol. 62, 164–178. doi: 10.1016/j.yjmcc.2013.05.019
Vakrou, S., and Abraham, M. R. (2014). Hypertrophic cardiomyopathy: a heart in need of an energy bar? Front. Physiol. 5:309. doi: 10.3389/fphys.2014.00309
Vikstrom, K. L., Factor, S. M., and Leinwand, L. A. (1996). Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol. Med. 2, 556–567.
Wang, B., Van Veldhoven, P. P., Brees, C., Rubio, N., Nordgren, M., Apanasets, O., et al. (2013). Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic. Biol. Med. 65, 882–894. doi: 10.1016/j.freeradbiomed.2013.08.173
Wang, H. S., Arvanitis, D. A., Dong, M., Niklewski, P. J., Zhao, W., Lam, C. K., et al. (2011). SERCA2a superinhibition by human phospholamban triggers electrical and structural remodeling in mouse hearts. Physiol. Genom. 43, 357–364. doi: 10.1152/physiolgenomics.00032.2010
Keywords: hypertrophic cardiomyopathy, comparative transcriptomics, GSTK1, mitochondria, oxidative stress, zebrafish, CRISPR/Cas9, systems pharmacology
Citation: Sasagawa S, Nishimura Y, Okabe S, Murakami S, Ashikawa Y, Yuge M, Kawaguchi K, Kawase R, Okamoto R, Ito M and Tanaka T (2016) Downregulation of GSTK1 Is a Common Mechanism Underlying Hypertrophic Cardiomyopathy. Front. Pharmacol. 7:162. doi: 10.3389/fphar.2016.00162
Received: 22 April 2016; Accepted: 01 June 2016;
Published: 14 June 2016.
Edited by:
Aida Salameh, HELIOS Heart Center Leipzig, GermanyReviewed by:
Martina Krüger, Heinrich Heine University Düsseldorf, GermanySampath K. Gollapudi, Washington State University, USA
Copyright © 2016 Sasagawa, Nishimura, Okabe, Murakami, Ashikawa, Yuge, Kawaguchi, Kawase, Okamoto, Ito and Tanaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshio Tanaka, dGFuYWthQGRvYy5tZWRpYy5taWUtdS5hYy5qcA==
†These authors have contributed equally to this work.