 Fernando Concha-Benavente1,2
Fernando Concha-Benavente1,2 Robert L. Ferris1,2*
Robert L. Ferris1,2*- 1Department of Otolaryngology, University of Pittsburgh, Pittsburgh, PA, United States
- 2University of Pittsburgh Cancer Institute, Pittsburgh, PA, United States
Uncontrolled growth is a signature of carcinogenesis, in part mediated by overexpression or overstimulation of growth factor receptors. The epidermal growth factor receptor (EGFR) mediates activation of multiple oncogenic signaling pathways and escape from recognition by the host immune system. We discuss how EGFR signaling downregulates tumor antigen presentation, upregulates suppressive checkpoint receptor ligand programmed death ligand (PD-L1), induces secretion of inhibitory molecules such as transforming growth factor beta (TGFβ) and reprograms the metabolic pathways in cancer cells to upregulate aerobic glycolysis and lactate secretion that ultimately lead to impaired cellular immunity mediated by natural killer (NK) cell and cytotoxic T lymphocytes (CTL). Ultimately, our understanding of EGFR-mediated escape mechanisms has led us to design EGFR-specific monoclonal antibody therapies that not only inhibit tumor cell metabolic changes and intrinsic oncogenic signaling but also activates immune cells that mediate tumor clearance. Importantly, targeted immunotherapy may also benefit from combination with antibodies that target other immunosuppressive pathways such PD-L1 or TGFβ and ultimately enhance clinical efficacy.
Introduction
Cancer is characterized by uncontrolled cellular proliferation. The malignant oncogenic transformation of cells is caused by deleterious mutations of genes that control cell growth or activating mutations of those that favor cell division. However, unrestrained oncogenic proliferation is also caused by overexpression of molecules that are normally present in cells under homeostatic conditions, such as growth factor receptors. In this setting, one of the most studied oncogenic signaling pathways that has been characterized in many types of cancer is the ErbB/Her family of growth factor receptors that belong to the super family of receptor tyrosine kinases (RTK), given their capacity of phosphorylating tyrosine resides in their cytoplasmic tail and transduce extracellular signals through the activation of intracellular messengers (Linggi and Carpenter, 2006). The ErbB/Her family comprises four members: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4) (Linggi and Carpenter, 2006). The EGFR is considered a prototypical oncogenic growth factor receptor, since it activates multiple intracellular signaling transduction cascades including the mitogen activated protein kinase (MAPK), phosphatidylinositol-3 kinase (PI3K/AKT), Janus kinase/signal transduced, and activator of transcription (JAK/STAT) and protein kinase C (PKC) pathways (Marmor et al., 2004; Warren and Landgraf, 2006). Furthermore, several studies associate EGFR signaling with tumor progression of cancer, including breast, lung, head, and neck squamous cell carcinoma (HNSCC) and glioblastoma (Wada et al., 1990; Harari and Yarden, 2000; Blume-Jensen and Hunter, 2001). In addition to overexpression of wild type EGFR, some tumors also exhibit activating mutant forms such as glioblastoma, where a variant called EGFRvIII has been reported (Gan et al., 2013) or non-small cell lung cancer (NSCLC) where mutations in the gene encoding the EGFR kinase domain (T790M) have been associated with tumor resistance (Ohashi et al., 2013). Interestingly, other mutations such as the L858R mutation in exon 21 of the kinase domain of EGFR in NSCLC increased sensitivity to tyrosine kinase inhibition, however they did not improve clinical outcome (Peng et al., 2015).
The overactivation of EGFR downstream signaling pathways induces malignant transformation of tumor cells through increased cell proliferation and survival, resistance to growth inhibition or apoptosis and increased invasion and metastasis, capabilities that are a common denominator to the majority of tumors (Hanahan and Weinberg, 2000). Importantly, recent work has shown evidence for adding to this list of tumor transforming competences the downregulation of tumor cell immunogenicity which is key mediator for immune evasion. Indeed, the immune system plays a major role in tumor progression, immune mediated inflammation and the recruitment of immune infiltrating cells and their interaction with tumor cells and surrounding stromal cells forms an intricate cellular and molecular network called the tumor microenvironment (TME). Therefore, cancer progression not only depends on intrinsic growth factor signals that provide uncontrolled proliferation but also evasion of the host's antitumor immunity. In this context, the concept of cancer immunoediting originates, where tumor infiltrating immune cells specifically recognize highly immunogenic tumor cells at early stages of cancerous transformation and eliminate them, however a subset of these transformed cells survives elimination and enters the editing phase termed equilibrium. Subsequently, evolutionary pressure selects tumor cells that can progressively evade immune detection, leading to the escape and tumor growth (Schreiber et al., 2011). Recognizing that the immune system acknowledges the presence of cancer and sculpts its progress, underlines the importance of investigating and understanding the mechanisms by which these complex interactions occur and justifies the development of strategies to manipulate the host immune system in order to promote tumor control and elimination.
In this review we will discuss how tumor cell intrinsic oncogenic signals downstream the EGFR can lead to immunoescape by downregulating tumor cell immunogenicity such as diminishing HLA class I mediated antigen presentation or providing inhibitory signals, such as checkpoint inhibition mediated by programmed death ligand 1 (PD-L1), suppressive cytokines that induce an exhausted phenotype or modifying the extracellular milieu by upregulating concentrations of lactate. Additionally, we will also discuss how monoclonal antibody mediated EGFR inhibition can reverse immunoescape and induce activation of effector immune cell subsets such as CD8+ T cells and NK cells.
EGFR Mediated Immunoescape, Abnormal Signals 1, 2, and 3
EGFR overexpression and overactivation of downstream pathways induces oncogenic transformation. In addition to its intrinsic oncogenic potential the EGFR also plays an important role in evasion of tumor immunosurveillance. Herein we present evidence to support the view that tumor immune evasion occurs by deregulating the three fundamental signals for an efficient immune activation: Signal 1, mediated by HLA class I dependent antigen presentation; signal 2, mediated by co-stimulatory receptor-ligand interaction and signal 3, mediated by secretion of immunostimulatory soluble cytokines.
Aberrant Signal 1
Proper antigen presentation is a major pre-requisite for appropriate T cell responses, especially because of the key role of this process in the generation of tumor antigen (TA)-specific adaptive immune responses (Meissner et al., 2005; Lopez-Albaitero et al., 2006). In the cancer setting, it has been recently reported that EGFR downregulates the expression of APM components and HLA class I via activation of protein phosphatase type 11 (PTNP11) best known as SHP2 which dephosphorylates signal transducer and activator of transcription 1 (STAT1). Less activated STAT1 translates into reduced expression of antigen presenting machinery (APM) and HLA class I dependent antigen presentation (Concha-Benavente et al., 2013; Leibowitz et al., 2013). Interestingly, inhibition or depletion of SHP2 in tumor cell lines or treatment with IFNγ induced upregulation of phosphorylated STAT1 (pSTAT1) and restored expression of HLA class I and APM components. Importantly, expansion of EGFR-specific CTL was noted upon an enhanced HLA class I restricted antigen presentation in vitro (Leibowitz et al., 2013). Likewise, SHP2-mediated pSTAT1 downregulation diminished the production of Th1 cytokines by tumor cells, since its inhibition induced increased secretion of interleukin-12 (IL-12) p35/p40 and IFNγ-dependent CXCR3 and CCR5 binding chemokines (Leibowitz et al., 2013). Furthermore, a second mechanism dependent on the MAPK pathway has been reported for downregulation of HLA class I and APM components downstream the EGFR, where activated SHP2 dephosphorylates GDP, inducing GTP-mediated RAS activation (Agazie and Hayman, 2003).
Aberrant Signal 2
An aberrant co-inhibitory signal 2 is represented by PD-L1/PD-1 pathway activation since recent studies have shown that this axis constitutes a major suppressive mechanism to evade immune activation and tumor clearance by downregulating T cell activation, proliferation, survival, cytotoxicity and cytokine release (Tseng et al., 2001; Dong et al., 2002). Moreover, inhibition of this pathway has proved to be clinically relevant since blocking antibodies against PD-1 or PD-L1 have demonstrated encouraging clinical activity in patients with metastatic melanoma, renal cell carcinoma (RCC), non-small cell lung cancer (NSCLC), and head and neck cancer (HNSCC), where PD-L1 tumor expression enriched for clinical responders (Brahmer et al., 2012; Topalian et al., 2012; Ferris et al., 2016). Importantly, a recent study demonstrated that PD-L1 expression in NSCLC cell lines is mediated by constitutively active mutant EGFR/KRAS-MAPK pathway (Akbay et al., 2013), whereas in the setting of HNSCC, overexpressed wild-type EGFR induced the expression of PD-L1 in a JAK2/STAT1 dependent manner. Curiously, although both cancers upregulate PD-L1 in an EGFR-dependent fashion, two different signaling pathways were involved JAK/STAT and MAPK pathways. This interesting finding could be explained by the unique biology of each cancer type, where mutant EGFR/KRAS would strongly activate MAPK pathway in NSLSC (Akbay et al., 2013). In contrast, HNSCC with a much lower EGFR/KRAS mutation burden (2%) (Stransky et al., 2011; McBride et al., 2014; Cancer Genome Atlas Netwrork, 2015) relies more on an overstimulated wild type EGFR/JAK2 pathway for oncogenic signaling (Concha-Benavente et al., 2016). Supporting these results is the recent finding by Zaretsky et al. where JAK2 inactivating mutations correlated with resistance to anti-PD-1 therapy in melanoma patients, in this setting we could speculate that tumor cells are sculpted by the immune system developing new strategies to evade anti-PD-L1/PD-1 pathway blocking therapy by downregulating JAK2-mediated expression of PD-L1 (Zaretsky et al., 2016). Interestingly, IFNγ, which restored of HLA class I expression and antigen presentation in tumor cells as discussed above is also a major inducer of PD-L1 expression as shown in multiple cancer types including HNSCC (Concha-Benavente et al., 2015), fibrosarcoma (Lee et al., 2005), glioblastoma (Han et al., 2009), and multiple myeloma cells (Liu et al., 2007). Notably, there appears to be a crosstalk between EGFR and IFNγ pathways regarding PD-L1 regulation at least in HNSCC, since EGFR blockade downregulated IFNγ-dependent PD-L1 expression (Concha-Benavente et al., 2016). Therefore, EGFR blockade may not only diminish the tumor cell intrinsic EGFR-induced PD-L1 upregulation but also the cell extrinsic IFNγ-mediated signals that have been associated with CD8+ T cell infiltration in the TME (Lyford-Pike et al., 2013; Topalian et al., 2015). Interestingly, EGFR signaling not only induces de novo expression of PD-L1 as shown in lung and head and neck cancer but also promotes stabilization of PD-L1 surface expression through glycosylation of its extracellular domain mediated by glycogen synthase kinase 3β (GSK3β) activation (Li et al., 2016). In this report authors show that EGFR may induce PD-L1 surface stabilization by inhibiting GSK3β in basal-like breast cancer cells. Moreover, gefitinib-mediated inhibition of EGFR destabilized PD-L1 surface expression in a GSK3β dependent fashion and enhanced tumor specific T cell immunity measured by CD8+ T cell IFNγ and granzyme B production. Importantly, in this setting EGFR inhibition increased efficacy of anti-PD-1 therapy in a syngeneic mouse model (Li et al., 2016).
Aberrant Signal 3
Signal 3 is required for an optimal effector T cell activation mediated by soluble cytokines and chemokines secreted in the milieu (Mescher et al., 2006). In the tumor microenvironment, the presence of suppressive cytokines promotes unresponsiveness of CD8+ T effector immune cell infiltrates and proliferation of suppressive cell subsets such as regulatory T cells (Treg), myeloid derived suppressive cells (MDSC) or tumor-associated macrophages (TAM) (Rabinovich et al., 2007; Burkholder et al., 2014). EGFR activation induces the constitutive activation of signal transducer and activator of transcription 3 (STAT3), a known oncogenic transcription factor (Grandis et al., 1998; Schrump and Nguyen, 2001; Kijima et al., 2002). STAT3 plays a major role in promoting tumor immune evasion since it not only has opposite effects to immunostimulatory STAT1 but also induces the secretion of immunosuppressive soluble cytokines that induce a tolerant TME (Wang et al., 2004; Kortylewski et al., 2005). Previous studies showed that STAT3 mediates the expression of vascular endothelial growth factor (VEGF), IL-6, and IL-10 in many cancer types, which induce T cell tolerance through inhibition of DC differentiation and maturation (Gabrilovich et al., 1996; Yang and Lattime, 2003). In addition, IL-6, IL-10, and VEGF activate STAT3 in tumor infiltrating immune cells, providing a feed forward mechanism for STAT3 activation in the tumor microenvironment. An important immunosuppressive cytokine, transforming growth factor beta (TGFβ), is secreted by many cancers including melanoma, breast and colon cancer and is known to prevent cytotoxic T cell (CTL) expansion and activation (Mooradian et al., 1990), TGFβ and IL-10 are involved in the generation of regulatory T cells (Treg) which in turn inhibit CD8+ T cell activation and IFNγ secretion (Nishikawa et al., 2005). Additionally, Tregs are an important source of TGFβ and IL-10, which once secreted to the TME further propagate the immunosuppressive signals (Figure 1; Zorn et al., 2006; Larmonier et al., 2007).
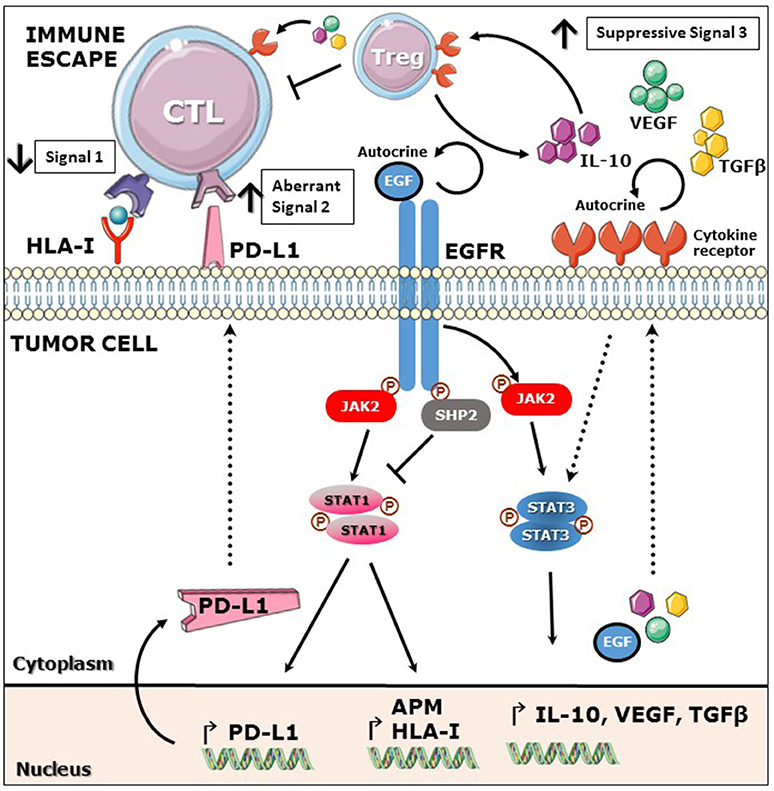
Figure 1. EGFR mediated immunoescape. EGFR stimulation induces activation of phosphatase SHP2 which decreases phosphorylation of STAT1 and subsequently expression of HLA class I and APM components. Additionally, EGFR mediated activation of JAK2-STAT1 induces expression of PD-L1. EGFR stimulation induces activation of STAT3 and production of immunosuppressive cytokines such as IL-10, VEGF and TGFβ which in turn induce Treg expansion and inhibition of CTL activation. Overall, EGFR signaling mediates downregulation of signal 1 and upregulation of suppressive signals 2 and 3, favoring escape from effector T cell recognition.
EGFR-Mediated Regulation of Tumor Cell Metabolism and Immunoescape
Tumor cells in contrast to most normal cells require large amounts of energy to support an increased rate of uncontrolled division. In this setting, one distinctive characteristic of cancer cells is to accelerate glucose degradation a metabolic process called glycolysis. Unlike normal tissues where glycolysis is limited by oxygen levels, tumor cells can reprogram their metabolic machinery to increase glycolysis and generate large amounts of lactate as a byproduct, a process that has been termed “aerobic glycolysis” and recognized as one of the new hallmarks of cancer (Hanahan and Weinberg, 2011; Ward and Thompson, 2012). Importantly, the lactate rich immunosuppressive TME impairs cytolytic functions of CD8+ T cells in vitro as well as their proliferation and cytokine production (Fischer et al., 2007). Interestingly, previous reports from more than a decade ago linked EGFR stimulation with enhanced aerobic glycolysis and lactate production in cancer cells including breast cancer (Kaplan et al., 1990; Baulida et al., 1992), however the mechanism by which the EGFR enhances glycolysis and lactate secretion was just recently reported. Lim et al. showed that EGFR stimulation in triple negative breast cancer (TNBC) cells induces activation of hexokinase 2 (HK2) and pyruvate kinase M2 (PKM2) an enzymatic isoform that is only expressed in embryonic cells and cancer cells. These two enzymes regulate the first and last steps of glycolysis respectively, and promote the accumulation of glycolytic intermediaries that ultimately enhance tumor cell proliferation. Interestingly, accumulation of the glycolytic intermediary fructose 1, 6 bisphosphate (F16BP) in TNBC cells via the activation of HK2 and PKM2 was found to directly engage the EGFR and enhance its phosphorylation and activation, thereby providing a positive feedback loop which ultimately increased lactate production and inhibition of CTL activity. Importantly, dual EGFR and glycolysis inhibition effectively suppressed TNBC cell proliferation and tumor growth (Lim et al., 2016). Similarly, work done by Makinoshima et al. showed that EGFR signaling maintained aerobic glycolysis in lung adenocarcinoma (LAD) cells and enhanced the extracellular acidification rate (ECAR) via lactate secretion. Moreover, specific EGFR inhibition with small molecule inhibitors gefitinib and erlotinib downregulated glucose transporter 3 (GLUT3) expression and that of genes involved in the pentose phosphate pathway (PPP) and pyrimidine synthesis. Overall, this study suggests that EGFR signaling regulates global metabolic pathways in EGFR-mutated LAD cells and may be an important target to reverse tumor cell–derived metabolic immunosuppression (Makinoshima et al., 2014).
Finally, EGFR activation has also been shown to upregulate hypoxia inducible factor 1 alpha (HIF-1α), a crucial molecule that reprograms cellular metabolism from oxidative phosphorylation to aerobic glycolysis. Interestingly, EGFR inhibition with monoclonal antibody cetuximab downregulated expression of HIF-1α and lactate dehydrogenase A (LDH-A), a crucial enzyme regulating the conversion of pyruvate to lactate. Interestingly, this inhibition only occurred in cetuximab-sensitive but not in cetuximab-resistant HNSCC cell lines, data that argues in favor of a metabolic immunoescape mechanism to EGFR inhibition of HNSCC cells mediated by lactate acidification of the tumor milieu (Lu et al., 2013). Overall these findings support the view that EGFR contributes to immune escape not only by downregulating tumor cell immunogenicity (signal 1) or providing aberrant signals 2 and 3 but also inducing an acidic, lactate rich milieu that impairs an effective adaptive antitumor immunity.
Activation of Anti-Tumor Immunity by Anti-EGFR Targeted Therapy
EGFR activation occurs upon ligand binding, therefore disruption of such interaction by targeted receptor-blocking specific agents is a logical strategy to shutdown not only its oncogenic metabolic shift and proliferative signaling but also the pathways involved in providing aberrant signals 1, 2, and 3. In this setting, small molecule inhibitors have been developed for inhibiting EGFR-induced phosphorylation of tyrosine residues of its cytoplasmic tail, two of such tyrosine kinase inhibitors (TKI) have been approved for clinical use, gefitinib and erlotinib (Stamos et al., 2002; Yun et al., 2007). In addition to small molecule inhibitors, two monoclonal antibodies (mAbs) targeting the EGFR have been approved for clinical use, cetuximab, an IgG1 chimeric mAb and panitumumab, a humanized IgG2 mAb, both inhibiting EGFR signaling to the same extent (Trivedi et al., 2016). Importantly, one added benefit of using EGFR blocking antibodies is that their interaction with EGFR extracellular domain may also induce endocytosis and degradation of the receptor (Wieduwilt and Moasser, 2008).
However, arguably the most important benefit of EGFR blocking mAbs is the activation of immune system effector cells via interaction of the antibody's Fc portion with the Fcγ receptors (FcγR) expressed on the surface of effector cells. In the case of cetuximab, this is supported by the observations that even though it interfered with growth signals, it did not induce cell death, probably because of activation of alternative survival pathways in cancer cells independent of the EGFR. Interestingly, cetuximab induced tumor cell death only when NK cells were added to in vitro co-cultures (Lopez-Albaitero and Ferris, 2007; Lopez-Albaitero et al., 2009; Taylor et al., 2009), therefore the major cetuximab antitumor effect seems to be immune mediated. Studies later revealed that cetuximab IgG1 framework allows its interaction with FcγRIIIA (CD16) on NK cells. Binding of cetuximab to CD16 on NK cells triggers a lytic process called antibody-dependent cellular cytotoxicity (ADCC) and IFNγ secretion. Moreover, NK cell-derived IFNγ mediates cross talk with DCs inducing their maturation and HLA class I antigen presentation which subsequently induce clonal expansion of EGFR-specific CD8+ T cells (Rafiq et al., 2002; Harbers et al., 2007; Banerjee et al., 2008; Marechal et al., 2010; Lee et al., 2011). Interestingly, the clinical activity of cetuximab is effective by increasing patient survival either as monotherapy or when added to radiation or platinum-based chemotherapy (Bonner et al., 2006; Vermorken et al., 2007, 2008). In addition to enhancing NK mediated cytotoxicity which is likely its most conspicuous effect, it has been recently shown that it could also activate neutrophils and mediate ADCC against EGFR expressing tumor cells via interaction with FcγRIIa, interestingly, this cytotoxic effect was FcγRIIIa genotype-dependent (Trivedi et al., 2016). On the other hand, panitumumab has shown less clinical efficacy than cetuximab (Mesia et al., 2015), this result may be explained by a less potent NK cell activation induced by panitumumab given its IgG2 framework, furthermore, its monocyte activation capability may be innocuous to EGFR expressing tumor targets given that monocytes are not capable of mediating ADCC. These preclinical findings are further supported by clinical data where patients treated with cetuximab had higher EGFR-specific cytotoxic CD8+ T cells when compared with those treated with panitumumab (Trivedi et al., 2016). Interestingly, it has also been reported that cetuximab-mediated EGFR blockade induced Treg expansion in head and neck cancer patients which correlated with resistance to cetuximab therapy (Jie et al., 2015), such Treg expansion was induced partially by DC maturation and T cell receptor stimulation in the presence of TGFβ. Interestingly, these in vitro expanded Tregs suppressed NK cell cytotoxicity against tumor cells providing evidence for Treg mediated immunosuppression in the tumor microenvironment.
Conclusions
Several studies support the view that tumors evolve intrinsic mechanisms to evade immune recognition. In this review we presented evidence for the ErbB/Her receptor family member the EGFR as an important driver of immunoescape by downregulating crucial immune activating signals 1, 2, and 3 and by inducing a metabolic shift of tumor cells to aerobic glycolysis and lactate secretion into the tumor microenvironment. Increasing our understanding of the mechanisms that tumor cells use to escape immunosurveillance will allow strategies to reverse EGFR mediated immune escape. Importantly, targeted mAb immunotherapy has the advantage of not only suppressing tumor intrinsic suppressive signals but also activating tumor infiltrating immune system cells which has shown clinical efficacy in many types of cancer. However, the majority of EGFR-overexpressing tumors have a complex genetic background with a significant level of compensatory oncogenic pathways regulating cell metabolism, proliferation, trafficking, and survival. Therefore, combination therapy not only targeting the EGFR but also other important molecules that regulate cellular immune responses in the TME such as the PD-L1/PD-1 axis or TGFβ and other metabolic immunosuppressive byproducts such as lactate should enhance immune responses and improve their clinical efficacy.
Author Contributions
FC wrote the manuscript and draw the figure, RF reviewed the manuscript and approved the final version.
Funding
This work was supported by National Institute of Health grants R01 DE19727, P50 CA097190, CA110249, and University of Pittsburgh Cancer Center Support Grant P30CA047904.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ZA and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
References
Agazie, Y. M., and Hayman, M. J. (2003). Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol. 23, 7875–7886. doi: 10.1128/MCB.23.21.7875-7886.2003
Akbay, E. A., Koyama, S., Carretero, J., Altabef, A., Tchaicha, J. H., Christensen, C. L., et al. (2013). Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 3, 1355–1363. doi: 10.1158/2159-8290.CD-13-0310
Banerjee, D., Matthews, P., Matayeva, E., Kaufman, J. L., Steinman, R. M., and Dhodapkar, K. M. (2008). Enhanced T-cell responses to glioma cells coated with the anti-EGF receptor antibody and targeted to activating FcgammaRs on human dendritic cells. J. Immunother. 31, 113–120. doi: 10.1097/CJI.0b013e31815a5892
Baulida, J., Onetti, R., and Bassols, A. (1992). Effects of epidermal growth factor on glycolysis in A431 cells. Biochem. Biophys. Res. Commun. 183, 1216–1223. doi: 10.1016/S0006-291X(05)80320-1
Blume-Jensen, P., and Hunter, T. (2001). Oncogenic kinase signalling. Nature 411, 355–365. doi: 10.1038/35077225
Bonner, J. A., Harari, P. M., Giralt, J., Azarnia, N., Shin, D. M., Cohen, R. B., et al. (2006). Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N.Engl. J. Med. 354, 567–578. doi: 10.1056/NEJMoa053422
Brahmer, J. R., Tykodi, S. S., Chow, L. Q., Hwu, W. J., Topalian, S. L., Hwu, P., et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N.Engl. J. Med. 366, 2455–2465. doi: 10.1056/NEJMoa1200694
Burkholder, B., Huang, R. Y., Burgess, R., Luo, S., Jones, V. S., Zhang, W., et al. (2014). Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 1845, 182–201. doi: 10.1016/j.bbcan.2014.01.004
Cancer Genome Atlas Network. (2015). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. doi: 10.1038/nature14129
Concha-Benavente, F., Srivastava, R. M., Ferrone, S., and Ferris, R. L. (2013). EGFR-mediated tumor immunoescape: the imbalance between phosphorylated STAT1 and phosphorylated STAT3. Oncoimmunology 2:e27215. doi: 10.4161/onci.27215
Concha-Benavente, F., Srivastava, R. M., Trivedi, S., Lei, Y., Chandran, U., Seethala, R. R., et al. (2015). Identification of the cell-intrinsic and extrinsic pathways downstream of EGFR and IFNγ that induce PD-L1 expression in head and neck cancer. Cancer Res. 76, 1031–1043. doi: 10.1158/0008-5472.CAN-15-2001
Concha-Benavente, F., Srivastava, R. M., Trivedi, S., Lei, Y., Chandran, U., Seethala, R. R., et al. (2016). Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFN γ that induce PD-L1 expression in head and neck cancer. Cancer Res. 76, 1031–1043. doi: 10.1158/0008-5472.CAN-15-2001
Dong, H., Strome, S. E., Salomao, D. R., Tamura, H., Hirano, F., Flies, D. B., et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800. doi: 10.1038/nm0902-1039c
Ferris, R. L., Blumenschein, G. Jr., Fayette, J., Guigay, J., Colevas, A. D., Licitra, L., et al. (2016). Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N.Engl. J. Med. 375, 1856–1867. doi: 10.1056/NEJMoa1602252
Fischer, K., Hoffmann, P., Voelkl, S., Meidenbauer, N., Ammer, J., Edinger, M., et al. (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819. doi: 10.1182/blood-2006-07-035972
Gabrilovich, D. I., Chen, H. L., Girgis, K. R., Cunningham, H. T., Meny, G. M., Nadaf, S., et al. (1996). Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 2, 1096–1103. doi: 10.1038/nm1096-1096
Gan, H. K., Cvrljevic, A. N., and Johns, T. G. (2013). The epidermal growth factor receptor variant III (EGFRvIII). where wild things are altered. FEBS J. 280, 5350–5370. doi: 10.1111/febs.12393
Grandis, J. R., Drenning, S. D., Chakraborty, A., Zhou, M. Y., Zeng, Q., Pitt, A. S., et al. (1998). Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth in vitro. J. Clin. Invest. 102, 1385–1392. doi: 10.1172/JCI3785
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57–70. doi: 10.1016/S0092-8674(00)81683-9
Han, S. J., Ahn, B. J., Waldron, J. S., Yang, I., Fang, S., Crane, C. A., et al. (2009). Gamma interferon-mediated superinduction of B7-H1 in PTEN-deficient glioblastoma. a paradoxical mechanism of immune evasion. Neuroreport 20, 1597–1602. doi: 10.1097/WNR.0b013e32833188f7
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Harari, D., and Yarden, Y. (2000). Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 19, 6102–6114. doi: 10.1038/sj.onc.1203973
Harbers, S. O., Crocker, A., Catalano, G., D'Agati, V., Jung, S., Desai, D. D., et al. (2007). Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. J. Clin. Invest. 117, 1361–1369. doi: 10.1172/JCI29470
Jie, H. B., Schuler, P. J., Lee, S. C., Srivastava, R. M., Argiris, A., Ferrone, S., et al. (2015). CTLA-4+ regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 75, 2200–2210. doi: 10.1158/0008-5472.CAN-14-2788
Kaplan, O., Jaroszewski, J. W., Faustino, P. J., Zugmaier, G., Ennis, B. W., Lippman, M., et al. (1990). Toxicity and effects of epidermal growth factor on glucose metabolism of MDA-468 human breast cancer cells. J. Biol. Chem. 265, 13641–13649.
Kijima, T., Niwa, H., Steinman, R. A., Drenning, S. D., Gooding, W. E., Wentzel, A. L., et al. (2002). STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 13, 355–362.
Kortylewski, M., Kujawski, M., Wang, T., Wei, S., Zhang, S., Pilon-Thomas, S., et al. (2005). Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321. doi: 10.1038/nm1325
Larmonier, N., Marron, M., Zeng, Y., Cantrell, J., Romanoski, A., Sepassi, M., et al. (2007). Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol. Immunother. 56, 48–59. doi: 10.1007/s00262-006-0160-8
Lee, S. C., Srivastava, R. M., Lopez-Albaitero, A., Ferrone, S., and Ferris, R. L. (2011). Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol. Res. 50, 248–254. doi: 10.1007/s12026-011-8231-0
Lee, S. K., Seo, S. H., Kim, B. S., Kim, C. D., Lee, J. H., Kang, J. S., et al. (2005). IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J. Dermatol. Sci. 40, 95–103. doi: 10.1016/j.jdermsci.2005.06.008
Leibowitz, M. S., Srivastava, R. M., Andrade Filho, P. A., Egloff, A. M., Wang, L., Seethala, R. R., et al. (2013). SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T-cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin. Cancer Res. 19, 798–808. doi: 10.1158/1078-0432.CCR-12-1517
Li, C. W., Lim, S. O., Xia, W., Lee, H. H., Chan, L. C., Kuo, C. W., et al. (2016). Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 7:12632. doi: 10.1038/ncomms12632
Lim, S. O., Li, C. W., Xia, W., Lee, H. H., Chang, S. S., Shen, J., et al. (2016). EGFR signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 76, 1284–1296. doi: 10.1158/0008-5472.CAN-15-2478
Linggi, B., and Carpenter, G. (2006). ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 16, 649–656. doi: 10.1016/j.tcb.2006.10.008
Liu, J., Hamrouni, A., Wolowiec, D., Coiteux, V., Kuliczkowski, K., Hetuin, D., et al. (2007). Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 110, 296–304. doi: 10.1182/blood-2006-10-051482
Lopez-Albaitero, A., and Ferris, R. L. (2007). Immune activation by epidermal growth factor receptor specific monoclonal antibody therapy for head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 133, 1277–1281. doi: 10.1001/archotol.133.12.1277
Lopez-Albaitero, A., Lee, S. C., Morgan, S., Grandis, J. R., Gooding, W. E., Ferrone, S., et al. (2009). Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol. Immunother. 58, 1853–1864. doi: 10.1007/s00262-009-0697-4
Lopez-Albaitero, A., Nayak, J. V., Ogino, T., Machandia, A., Gooding, W., DeLeo, A. B., et al. (2006). Role of antigen-processing machinery in the in vitro resistance of squamous cell carcinoma of the head and neck cells to recognition by CTL. J. Immunol. 176, 3402–3409. doi: 10.4049/jimmunol.176.6.3402
Lu, H., Li, X., Luo, Z., Liu, J., and Fan, Z. (2013). Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol. Cancer Ther. 12, 2187–2199. doi: 10.1158/1535-7163.MCT-12-1245
Lyford-Pike, S., Peng, S., Young, G. D., Taube, J. M., Westra, W. H., Akpeng, B., et al. (2013). Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 73, 1733–1741. doi: 10.1158/0008-5472.CAN-12-2384
Makinoshima, H., Takita, M., Matsumoto, S., Yagishita, A., Owada, S., Esumi, H., et al. (2014). Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J. Biol. Chem. 289, 20813–20823. doi: 10.1074/jbc.M114.575464
Marechal, R., De Schutter, J., Nagy, N., Demetter, P., Lemmers, A., Deviere, J., et al. (2010). Putative contribution of CD56 positive cells in cetuximab treatment efficacy in first-line metastatic colorectal cancer patients. BMC Cancer 10:340. doi: 10.1186/1471-2407-10-340
Marmor, M. D., Skaria, K. B., and Yarden, Y. (2004). Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Phys. 58, 903–913. doi: 10.1016/j.ijrobp.2003.06.002
McBride, S. M., Rothenberg, S. M., Faquin, W. C., Chan, A. W., Clark, J. R., Ellisen, L. W., et al. (2014). Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma. Head Neck 36, 1181–1188. doi: 10.1002/hed.23430
Meissner, M., Reichert, T. E., Kunkel, M., Gooding, W., Whiteside, T. L., Ferrone, S., et al. (2005). Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clin. Cancer Res. 11, 2552–2560. doi: 10.1158/1078-0432.CCR-04-2146
Mescher, M. F., Curtsinger, J. M., Agarwal, P., Casey, K. A., Gerner, M., Hammerbeck, C. D., et al. (2006). Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 211, 81–92. doi: 10.1111/j.0105-2896.2006.00382.x
Mesia, R., Henke, M., Fortin, A., Minn, H., Yunes Ancona, A. C., Cmelak, A., et al. (2015). Chemoradiotherapy with or without panitumumab in patients with unresected, locally advanced squamous-cell carcinoma of the head and neck (CONCERT-1): a randomised, controlled, open-label phase 2 trial. Lancet Oncol. 16, 208–220. doi: 10.1016/S1470-2045(14)71198-2
Mooradian, D. L., Purchio, A. F., and Furcht, L. T. (1990). Differential effects of transforming growth factor beta 1 on the growth of poorly and highly metastatic murine melanoma cells. Cancer Res. 50, 273–277.
Nishikawa, H., Kato, T., Tawara, I., Ikeda, H., Kuribayashi, K., Allen, P. M., et al. (2005). IFN-gamma controls the generation/activation of CD4+ CD25+ regulatory T cells in antitumor immune response. J. Immunol. 175, 4433–4440. doi: 10.4049/jimmunol.175.7.4433
Ohashi, K., Maruvka, Y. E., Michor, F., and Pao, W. (2013). Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J. Clin. Oncol. 31, 1070–1080. doi: 10.1200/JCO.2012.43.3912
Peng, L., Song, Z., and Jiao, S. (2015). Comparison of uncommon EGFR exon 21 L858R compound mutations with single mutation. Onco. Targets. Ther. 8, 905–910. doi: 10.2147/OTT.S78984
Rabinovich, G. A., Gabrilovich, D., and Sotomayor, E. M. (2007). Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 25, 267–296. doi: 10.1146/annurev.immunol.25.022106.141609
Rafiq, K., Bergtold, A., and Clynes, R. (2002). Immune complex-mediated antigen presentation induces tumor immunity. J. Clin. Invest. 110, 71–79. doi: 10.1172/JCI0215640
Schreiber, R. D., Old, L. J., and Smyth, M. J. (2011). Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331, 1565–1570. doi: 10.1126/science.1203486
Schrump, D. S., and Nguyen, D. M. (2001). The epidermal growth factor receptor-STAT pathway in esophageal cancer. Cancer J. 7, 108–111.
Stamos, J., Sliwkowski, M. X., and Eigenbrot, C. (2002). Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277, 46265–46272. doi: 10.1074/jbc.M207135200
Stransky, N., Egloff, A. M., Tward, A. D., Kostic, A. D., Cibulskis, K., Sivachenko, A., et al. (2011). The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160. doi: 10.1126/science.1208130
Taylor, R. J., Chan, S. L., Wood, A., Voskens, C. J., Wolf, J. S., Lin, W., et al. (2009). FcgammaRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol. Immunother. 58, 997–1006. doi: 10.1007/s00262-008-0613-3
Topalian, S. L., Drake, C. G., and Pardoll, D. M. (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. doi: 10.1016/j.ccell.2015.03.001
Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott, D. F., et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454. doi: 10.1056/NEJMoa1200690
Trivedi, S., Srivastava, R. M., Concha-Benavente, F., Ferrone, S., Garcia-Bates, T. M., Li, J., et al. (2016). Anti-EGFR targeted monoclonal antibody isotype influences antitumor cellular immunity in head and neck cancer patients. Clin. Cancer Res. 22, 5229–5237. doi: 10.1158/1078-0432.CCR-15-2971
Tseng, S. Y., Otsuji, M., Gorski, K., Huang, X., Slansky, J. E., Pai, S. I., et al. (2001). B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193, 839–846. doi: 10.1084/jem.193.7.839
Vermorken, J. B., Mesia, R., Rivera, F., Remenar, E., Kawecki, A., Rottey, S., et al. (2008). Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 359, 1116–1127. doi: 10.1056/NEJMoa0802656
Vermorken, J. B., Trigo, J., Hitt, R., Koralewski, P., Diaz-Rubio, E., Rolland, F., et al. (2007). Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J. Clin. Oncol. 25, 2171–2177. doi: 10.1200/JCO.2006.06.7447
Wada, T., Qian, X. L., and Greene, M. I. (1990). Intermolecular association of the p185neu protein and EGF receptor modulates EGF receptor function. Cell 61, 1339–1347. doi: 10.1016/0092-8674(90)90697-D
Wang, T., Niu, G., Kortylewski, M., Burdelya, L., Shain, K., Zhang, S., et al. (2004). Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10, 48–54. doi: 10.1038/nm976
Ward, P. S., and Thompson, C. B. (2012). Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308. doi: 10.1016/j.ccr.2012.02.014
Warren, C. M., and Landgraf, R. (2006). Signaling through ERBB receptors: multiple layers of diversity and control. Cell. Signal. 18, 923–933. doi: 10.1016/j.cellsig.2005.12.007
Wieduwilt, M. J., and Moasser, M. M. (2008). The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell. Mol. Life Sci. 65, 1566–1584. doi: 10.1007/s00018-008-7440-8
Yang, A. S., and Lattime, E. C. (2003). Tumor-induced interleukin 10 suppresses the ability of splenic dendritic cells to stimulate CD4 and CD8 T-cell responses. Cancer Res. 63, 2150–2157.
Yun, C. H., Boggon, T. J., Li, Y., Woo, M. S., Greulich, H., Meyerson, M., et al. (2007). Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11, 217–227. doi: 10.1016/j.ccr.2006.12.017
Zaretsky, J. M., Garcia-Diaz, A., Shin, D. S., Escuin-Ordinas, H., Hugo, W., Hu-Lieskovan, S., et al. (2016). Mutations associated with acquired resistance to pd-1 blockade in melanoma. N.Engl. J. Med. 375, 819–829. doi: 10.1056/NEJMoa1604958
Keywords: EGFR, PD-L1, PD-1, HLA class I, Immunoescape, aerobic glycolysis, T cells
Citation: Concha-Benavente F and Ferris RL (2017) Reversing EGFR Mediated Immunoescape by Targeted Monoclonal Antibody Therapy. Front. Pharmacol. 8:332. doi: 10.3389/fphar.2017.00332
Received: 19 January 2017; Accepted: 16 May 2017;
Published: 30 May 2017.
Edited by:
Francois X. Claret, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Elizabeth J. Ryan, St Vincent's University Hospital, IrelandClaudia Palena, National Cancer Institute, United States
Zhiqiang An, University of Texas Health Science Center at Houston, United States
Copyright © 2017 Concha-Benavente and Ferris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert L. Ferris, ZmVycmlzcmxAdXBtYy5lZHU=