 Li-Wei Zou1,2†Tong-Yi Dou3†Ping Wang1,2Wei Lei2,4
Li-Wei Zou1,2†Tong-Yi Dou3†Ping Wang1,2Wei Lei2,4 Zi-Miao Weng4Jie Hou4Dan-Dan Wang1Yi-Ming Fan3
Zi-Miao Weng4Jie Hou4Dan-Dan Wang1Yi-Ming Fan3 Wei-Dong Zhang1
Wei-Dong Zhang1 Guang-Bo Ge1,2*Ling Yang1*
Guang-Bo Ge1,2*Ling Yang1*- 1Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Laboratory of Pharmaceutical Resource Discovery, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China
- 3School of Life Science and Medicine, Dalian University of Technology, Panjin, China
- 4Biotechnology Department, College of Basic Medical Sciences, Dalian Medical University, Dalian, China
Human carboxylesterase 1 (hCE1), one of the most important serine hydrolases distributed in liver and adipocytes, plays key roles in endobiotic homeostasis and xenobiotic metabolism. This study aimed to find potent and selective inhibitors against hCE1 from phytochemicals and their derivatives. To this end, a series of natural triterpenoids were collected and their inhibitory effects against human carboxylesterases (hCEs) were assayed using D-Luciferin methyl ester (DME) and 6,8-dichloro-9,9-dimethyl-7-oxo-7,9-dihydroacridin-2-yl benzoate (DDAB) as specific optical substrate for hCE1, and hCE2, respectively. Following screening of a series of natural triterpenoids, oleanolic acid (OA), and ursolic acid (UA) were found with strong inhibitory effects on hCE1 and relative high selectivity over hCE2. In order to get the highly selective and potent inhibitors of hCE1, a series of OA and UA derivatives were synthesized from OA and UA by chemical modifications including oxidation, reduction, esterification, and amidation. The inhibitory effects of these derivatives on hCEs were assayed and the structure-activity relationships of tested triterpenoids as hCE1 inhibitors were carefully investigated. The results demonstrated that the carbonyl group at the C-28 site is essential for hCE1 inhibition, the modifications of OA or UA at this site including esters, amides and alcohols are unbeneficial for hCE1 inhibition. In contrast, the structural modifications on OA and UA at other sites, such as converting the C-3 hydroxy group to 3-O-β-carboxypropionyl (compounds 20 and 22), led to a dramatically increase of the inhibitory effects against hCE1 and very high selectivity over hCE2. 3D-QSAR analysis of all tested triterpenoids including OA and UA derivatives provide new insights into the fine relationships linking between the inhibitory effects on hCE1 and the steric-electrostatic properties of triterpenoids. Furthermore, both inhibition kinetic analyses and docking simulations demonstrated that compound 22 was a potent competitive inhibitor against hCE1-mediated DME hydrolysis. All these findings are very helpful for medicinal chemists to design and develop highly selective and more potent hCE1 inhibitors for biomedical applications.
Introduction
Mammalian carboxylesterases (CEs) are important members of the serine hydrolase superfamily (E.C. 3.1.1.1), which catalyze the hydrolysis of a wide variety of endogenous and xenobiotics ester compounds (Satoh and Hosokawa, 1998; Redinbo and Potter, 2005). In human, two primary carboxylesterases including human carboxylesterase 1 (hCE1) and human carboxylesterase 2 (hCE2), have been found and extensively studied in the past decade (Imai, 2006). These two isoforms share 47% amino acid sequence identity, but exhibit differential tissue distribution and distinct substrate and inhibitor specificities (Hosokawa, 2008). Generally, hCE1 is primarily expressed in the liver and adipocytes, and demonstrates substrate specificity for a large, bulky acyl group and a small alcohol group (Satoh et al., 2002; Imai et al., 2006). In contrast, hCE2 is highly expressed in the intestine and colon, and displays the opposite substrate preference for as mall acyl group and a large alcohol group (Xu et al., 2002; Sanghani et al., 2003; Kobayashi et al., 2012). The distribution and the catalytic property of hCE1 make this enzyme a key determinant for the bioactivation of numerous ester-containing drugs including oseltamivir (Shi et al., 2006), dabigatran etexilate (Hu et al., 2013), mycophenolate mofetil (Fujiyama et al., 2010), and trandolapril (Zhu et al., 2009), as well as for the metabolic inactivation and clearance of some esterified drugs, such as clopidogrel (Tang et al., 2006), methylphenidate (Sun et al., 2004), rufinamide (Williams et al., 2010), and oxybutynin (Sato et al., 2012).
As one of the most abundant esterases distributed in human liver and adipocytes, hCE1 participates in a wide range of physiological or pathological processes via hydrolysis of endogenous esters (such as cholesteryl esters and triacylglycerols) and thus plays key roles in cholesterol homeostasis and fatty acid metabolism (Crow et al., 2008; Li et al., 2016). Recent studies have revealed that the activities of hCE1 are markedly elevated in obese individuals and patients with type 2 diabetes, and the treatment of hCE1 inhibitors displayed multiple beneficial effects in both lipid and glucose homeostasis in genetic and diet-induced mouse models of obesity, insulin resistance and type 2 diabetes (Dominguez et al., 2014). Furthermore, hCE1 has been recognized as a therapeutic target for hypertriglyceridaemia, due to the key roles of this enzyme responsible for the enzymatic cleaving of triglyceride stores in hepatocytes (Gilham et al., 2003). The key roles of hCE1 in human diseases make the discovery of potent and selective inhibitors of hCE1 as drug candidates is of immense significance in both basic researches and clinical applications. However, the highly selective and potent inhibitors of hCE1 have been rarely reported. To data, only one hCE1 inhibitor termed GR148672X is in preclinical development for the treatment of hypertriglyceridaemia, but the selectivity and molecular interactions of this agent have not been disclosed (Gilham et al., 2003; Bachovchin and Cravatt, 2012). Thus, it is highly desirable to find more potent and selective hCE1 inhibitors for potential biomedical applications, including exploring the functions of hCE1 in biological systems and serving as therapeutic agents for the treatment of obese, type 2 diabetes and hypertriglyceridaemia.
In recent years, screening of the specific and potent inhibitors toward CEs from phytochemicals in medicinal plants or herbs has attracted increasing attentions (Liu et al., 2016; Wang et al., 2017), owing to most of phytochemicals display satisfying safety during long history of use for medical treatments (Li and Vederas, 2009; Ngo et al., 2013; Shen, 2015). To data, many phytochemicals including flavonoids (Li et al., 2015; Sun et al., 2016), tanshinones (Hatfield et al., 2013), and triterpenoids (Mai et al., 2015; Zou et al., 2016) have been reported with inhibitory effects against human carboxylesterases. However, most of these natural compounds demonstrated more potent inhibitory effects against hCE2 in contrast to hCE1 (Hatfield and Potter, 2011; Umehara et al., 2016; Xu et al., 2016; Wang et al., 2017). Thus, it is urgently necessary to find more potent and selective hCE1 inhibitors from phytochemicals. More recently, we have developed a highly specific bioluminescent probe substrate (termed DME) for hCE1 and a highly selective near-infrared fluorescent probe (termed DDAB) for hCE2, which have been successfully used for rapid screening and characterization of inhibitors against hCEs using cell or tissue preparations as enzyme sources (Jin et al., 2016; Wang et al., 2016). In the present study, DME and DDAB were used as the highly selective optical substrates for human CEs to rapidly screen hCE1 inhibitors from natural triterpenoid compounds. After preliminary screening, we found that two pentacyclic triterpenoids including oleanolic acid (OA) and ursolic acid (UA) displayed potent inhibitory effects against hCE1, with the IC50 values of 0.28 μM (for OA) and 0.24 μM (for UA), as well as relatively high selectivity over hCE2 (>19-folds). These findings promoted us to develop more potent and highly selective inhibitors against hCE1, using these two natural compounds as lead compounds. Hence, a series of OA and UA derivatives were semi-synthesized and assayed, while the structure-activity relationships (SAR) and 3D-QSAR analysis for all tested triterpenoids including OA and UA derivatives as hCE1 inhibitors were carefully studied. The obtained SAR was very helpful for the development of more potent and highly selective inhibitors against hCE1.
Materials and Methods
Chemicals and Reagents
Natural triterpenoids were purchased from Chengdu Pufei De Biotech Co., Ltd. (Chengdu, Sichuan, China). The purities of all tested compounds were determined by HPLC-UV, which were greater than 98%. Bis-p-nitrophenyl phosphate (BNPP) was purchased from TCI (Tokyo, Japan). Stock solutions of compound 1–27 were prepared in DMSO and stored at 4°C until use. Phosphate buffer (100 mM, pH 7.4 and pH 6.5) was prepared by using Millipore water and stored at 4°C until use. Human liver microsomes (HLMs) were obtained from Celsis (Shanghai, China). The specific probes DME (hCE1) and DDAB (hCE2) were synthesized in our lab as described previously (Jin et al., 2016; Wang et al., 2016). Millipore water (Millipore, Bedford, USA), HPLC grade acetonitrile, methanol, and formic acid (Tedia company, USA) were employed throughout the study. The Luciferin Detection Reagent (LDR) was obtained from Promega Corporation (USA). All 1H NMR (400 MHz) and 13C NMR (101 MHz) were recorded on a VARIAN INOVA-400 spectrometer with chemical shifts reported as ppm (in CDCl3, TMS as the internal standard). High resolution MS data were obtained with the LTQ Orbitrap mass spectrometer (Orbitrap Elite).
Instrument and Analytical Methods
All fluorescence-based assays were performed on Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, USA). The measurements for the hydrolytic metabolite of DME were set as follow, luminescence, gain = 135. The measurements for the hydrolytic metabolite of DDAB were set as follow, fluorescence detection with the excitation wavelength at 600 nm and the emission wavelength at 662 nm, gain = 100.
Enzyme Inhibition Assays
The inhibitory effects toward CEs were assayed using DME and DDAB as the specific probe substrates for hCE1 and hCE2, respectively. In brief, for the inhibition assays with DDAB, the incubation mixture (200 μL total volume) consisted of PBS, enzyme sources (0.5 and 5 μg/mL, HLM), inhibitors (compounds 1–27 or BNPP) and DDAB (both 10 μM, final concentration). Control incubations without BNPP were also performed. After incubation, ice-cold acetonitrile (equal volume of incubation mixture, 200 μL) was added to terminate the reaction. The mixture was then centrifuged at 20,000 g for 20 min at 4°C. All these compounds dissolved in DMSO, and the final concentration of DMSO was <2% (V/V). Aliquots of the supernatant were then taken for further microplate reader analyses. For inhibition assays with probe DME, the incubation mixture (50 μL total volume) consisted of PBS, enzyme sources (10 μg/mL, HLM), inhibitors (compounds 1–27 or BNPP) and DME (3 μM, final concentration). First, each of the potential esterase inhibitors was pre-incubated with HLM at 37°C for 10 min with shaking in microplate reader. Then DME was added to start the reaction. After incubation at 37°C for 10 min in a shaking bath, LDR (equal volume of incubation mixture, 50 μL) was added to terminate the reaction, and luminescence measurements were conducted as described above. The residual enzymatic activity (%) was determined by the percent of D-fluorescein production in the presence of known selective esterase inhibitors to the control (in the absence of inhibitors).
Inhibition Kinetic Analyses
The half maximal inhibitory concentration (IC50) of each compound was determined using various inhibitor concentrations under the same incubation conditions as mentioned above. The inhibition constant (Ki) values of compound 22 against hCE1 in HLM were determined using varied concentrations of DME in the presence or absence of each inhibitor. To determine the inhibition kinetic types (competitive inhibition, non-competitive inhibition, or uncompetitive type) of tested compounds, multiple concentrations of DME and varied concentrations of inhibitor were utilized to determine the corresponding reaction rates. Dixon plot and Lineweaver-Burk plots were used to fit the data. The inhibition kinetic type was evaluated by determining the intersection point in the Dixon and Lineweaver-Burk plots. The second plots based on the slopes from Lineweaver-Burk plot vs. inhibitor concentrations were utilized to calculate each the inhibition constant (Ki) value (Wang et al., 2015; Zhu et al., 2015, 2016).
Statistical Analysis
All values obtained from experiments were expressed as mean ± SD. The IC50 values which concentration of inhibitor that reduces enzyme activity by 50% and the Ki values were evaluated by nonlinear regression using GraphPad Prism 6.0 software (GraphPad Software, Inc., La Jolla, USA).
Molecular Docking
The protein structures of hCE1 were taken from the Protein Data Bank (PDB ID: 2DQY; Bencharit et al., 2003). The whole molecular docking process in this work was performed using Discovery Studio (BIOVIA Discovery Studio 2016, Dassault Systèmes, San Diego, USA). The “Prepare Protein” procedure was used to prepare the input protein structures for docking. Tasks including inserting missing atoms in incomplete residues, modeling missing loop regions, deleting alternate conformations (disorder), removing waters, standardizing atom names, and protonating titratable residues using predicted pKs were performed. Meanwhile, the “Prepare Ligand” procedure was used to prepare the input ligands for docking. Tasks including removing duplicates, enumerating isomers and tautomers, and generating 3D conformations were performed. The CHARMM 40.1 force field was used to represent the protein and ligand structures. Docking simulations were performed by a standard LibDock protocol, where protein features are referred to as hotspots. After a final energy-minimization step (allowing the ligand poses to be flexible), the top scoring ligand poses are saved. The rigid poses are placed into the active site of hCE1 and the hotspots are matched as triplets. For docking process, all ligands including DME and compound 22 are inputted, totally 431 conformers are generated, and finally 548 poses are successfully docked into the active site of hCE1. The protein-ligand complexes with the highest LibDock score were taken from the docking results and depicted in full text.
3D-QSAR Models
Three 3D-QSAR models were built using the corresponding package of Drug Discovery Studio. Primarily, compound 1–27 were aligned by consensus on both steric and electrostatic fields, with relative weight of 50–50%. The aligned molecules were placed in a 3D grid space, with grid spacing of 1.5 Å. The extent of the grid was set to the bounding box of all the ligands plus 6.0 Å of extension. The CHARMm force field was used. The electrostatic potential and the van der Waals potential were treated as separate terms. A +1e point charge was used as the electrostatic potential probe. Distance-dependent dielectric constant was used to mimic the solvation effect. For the van der Waals potential, a carbon atom with a 1.73 Å radius was used as a probe. The energy grid potentials were filtered to remove highly correlated descriptors (maximum descriptor correlation was set as 0.9). Rather than the full potential, a soft-core potential suggested in CDOCKER (Wu et al., 2003) was used. The energy grid potentials were filtered to remove highly correlated descriptors. Partial least squares (PLS) models were then built using these remaining descriptors, and Log(IC50 for hCE1) was used as activity properties. Cross validation were performed by splitting the training data into five groups.
General Procedures for the Synthesis of Compounds 15–27
The NMR data of synthetic compounds are provided in the Supplementary Material.
3-Oxo-olean-12-en-28-oic Acid (15)
To a solution of oleanoic acid 1 (456.7 mg, 1 mmol) in acetone (10 mL) was added Jones reagent (prepared from 107.9 mg of CrO3) at 0°C over a period of 30 min till the brown color persisted. The resulting solution was stirred for further 30 min. Progress of the reaction was monitored by TLC. After completion of the reaction, isopropanol (0.5 mL) was added. After evaporation of the solvent, the crude residue was diluted with dichloromethane (100 mL). The organic layer was washed with water (25 mL × 2), brine (25 mL) and dried over sodium sulfate. The solvent was concentrated under vacuum and the residual solid was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 20/1) to give the compound 15 (383.6 mg, 82%) as a white solid.
3β-Olean-12-ene-3,28-diol (16)
To a cooled (0°C) solution of LiAlH4 (75.9 mg, 2.0 mmol) in dry THF (8 mL) was added dropwise oleanoic acid 1 (235.4 mg, 0.5 mmol) in dry THF (5 mL) under Ar2 atmosphere. Suspension was stirred at 0°C for 1 h, allowed to rise to room temperature. Progress of the reaction was monitored by TLC. After completion of the reaction, the reaction was quenched with a solution of 1N NaOH (5 mL). Precipitate was filtered and washed with ethyl acetate. Organic phase was separated and aqueous layer was further extracted with ethyl acetate (25 mL × 2). The organic phase was washed with water (25 mL), brine (25 mL), and dried over sodium sulfate. After evaporation of the solvent, the crude residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 10/1) to give the compound 16 (123.9 mg, 56%) as a white solid.
3β-Hydroxy-olean-12-en-28-oic Acidmethyl Ester (17)
To a stirred solution of oleanoic acid 1 (228.4 mg, 0.5 mmol) in acetone (10 ml), anhydrous K2CO3 (69.1 mg, 0.5 mmol), and CH3I (46.7 μL, 0.75 mmol) were added at room temperature. The resulting solution was stirred for 12 h. Progress of the reaction was monitored by TLC. After completion of the reaction, the acetone was distilled off. The resulting mixture was diluted with water (15 mL) and extracted with dichloromethane (30 mL × 2). The combined organic layers were washed with brine (10 mL) and dried over sodium sulfate. After evaporation of the solvent, the crude residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 10/1) to give the compound 17 (203 mg, 86%) as a white solid.
3β-Hydroxy-olean-12-en-28-amide (18)
To a solution of compound 19 (260 mg, 0.52 mmol) in dichloromethane (10 mL), oxalylchloride (262 μL, 3.12 mmol) was added dropwise at room temperature. After stirring at room temperature for 2 h, the solvent was removed under reduced pressure and the residue was dissolved in toluene (10 mL). Then a conc. solution of ammonia (7 mL) was added at 4–8°C and the mixture was stirred for further 1 h. The mixture was extracted with dichloromethane (25 mL × 2). The combined organic layers were washed with water (10 mL), brine (10 mL), and dried over sodium sulfate. After evaporation of the solvent, the crude residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 5/1) to give a white solid. To a solution of the white solid (135 mg, 0.28 mmol) in methanol (6 mL) and tetrahydrofuran (2 mL), 1 M NaOH aq. (840 μL, 0.84 mmol) was added dropwise at room temperature. The resulting solution was stirred at 40°C for 5 h. Progress of the reaction was monitored by TLC. After completion of the reaction, the solvent was removed under reduced pressure. The resulting mixture was diluted with water (20 mL) and extracted with dichloromethane (30 mL × 2). The combined organic layers were washed with brine (15 mL) and dried over sodium sulfate. After evaporation of the solvent, the crude residue was purified by column chromatography on silica gel (dichloromethane/ methanol = 20/1) to give the compound 18 (100 mg, 42%) as a white solid.
3β-O-Acetyl-olean-12-en-28-oic Acid (19)
To a stirred solution of oleanoic acid 1 (500 mg, 1.09 mmol) in pyridine (7.5 ml), acetic anhydride (5.0 mL) was added dropwise at 0°C. The resulting solution was stirred at room temperature for 24 h and then poured into ice water (75 mL), resulting in the compound 19 (514 mg, 92%) as white solid precipitate.
3β-O-(β-Carboxypropionyl)-olean-12-en-28-oic Acid (20)
To a solution of oleanoic acid 1 (137 mg, 0.3 mmol) in dichloromethane (3 ml), succinic anhydride (150.1 mg, 1.5 mmol) and DMAP (73.3 mg, 0.6 mmol) were added at room temperature. The resulting solution was stirred at room temperature for 48 h. Progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was acidified with 1 N HCl to pH~3 and extracted with ethyl acetate (30 mL × 3). The combined organic layers were washed with water (10 mL), brine (10 mL), and dried over sodium sulfate. After evaporation of the solvent, the crude residue was purified by column chromatography on silica gel (dichloromethane/ methanol = 20/1) to give the compound 20 (155 mg, 93%) as a white solid.
3-Oxo-urs-12-en-28-oic Acid (21)
The preparation was performed as described above for compound 15 starting from ursoic acid 2 (182.8 mg, 0.4 mmol) to give compound 21 (134.2 mg, 74%) as a white solid.
3β-O-(β-Carboxypropionyl)-urs-12-en-28-oic Acid (22)
The preparation was performed as described above for compound 20 starting from ursoic acid 2 (137 mg, 0.3 mmol) to give compound 22 (145 mg, 87%) as a white solid.
3β-Hydroxy-urs-12-en-28-oic Acid Methyl Ester (23)
The preparation was performed as described above for compound 17 starting from ursoic acid 2 (456.7 mg, 1.0 mmol) to give compound 23 (450 mg, 95%) as a white solid.
3-Oxo-urs-12-en-28-oic Acid Methyl Ester (24)
The preparation was performed as described above for compound 15 starting from compound 23 (94.2 mg, 0.2 mmol) to give compound 24 (92 mg, 98%) as a white solid.
3,11-Dioxo-urs-12-en-28-oic Acid Methyl Ester (25)
To a solution of compound 24 (60 mg, 0.13 mmol) inacetic acid (2 ml) and acetic anhydride (2 ml), chromium trioxide (39.6 mg, 0.39 mmol) were added at room temperature. The resulting solution was stirred at rt for 3 h. Progress of the reaction was monitored by TLC. After completion of the reaction, the mixture was poured into a cooling water (20 mL). The mixture was adjusted to pH = 7~8 with 1 N NaOH, and then extracted with ethyl acetate (20 mL × 3). The combined organic layers were washed with water (10 mL), brine (10 mL), and dried over sodium sulfate. After evaporation of the solvent, the crude residue was purified by column chromatography on silica gel (dichloromethane/ methanol = 10/1) to give the compound 25 (31.3 mg, 50%) as a white solid.
3β-O-(β-Carboxypropionyl)-olean-12-en-30-oic Acid (27)
Compound 26 was prepared from glycyrrhetinic acid according to the published literature (Zou et al., 2016). The preparation of compound 27 was performed as described above for compound 20 starting from compound 26 (68.5 mg, 0.15 mmol) to give a white solid (81.4 mg, 97%).
Results and Discussion
Screening of hCE1 Inhibitors from Natural Triterpenoids
Triterpenes, a class of widespread natural compounds containing six isoprene units, are an excellent reservoir of biologically active compounds (Sheng and Sun, 2011; Hill and Connolly, 2017). In this study, a series of natural triterpenoids were collected and their inhibitory effects against human carboxylesterases were assayed by using DME and DDAB as specific optical substrate for hCE1 and hCE2, respectively (Figure 1). The bioassay results are summarized in Table 1. It is evident from Table 1 that two natural triterpenoids including oleanolic acid (OA, 1) and ursolic acid (UA, 2) displayed potent inhibitory effects against hCE1 (IC50, 0.28 and 0.24 μM, respectively) and relative high selectivity over hCE2 (19.6- and 25.2-fold against hCE2, respectively). In contrast, other natural triterpenoids (3–14) displayed both poor selectivity and specificity toward hCE1.
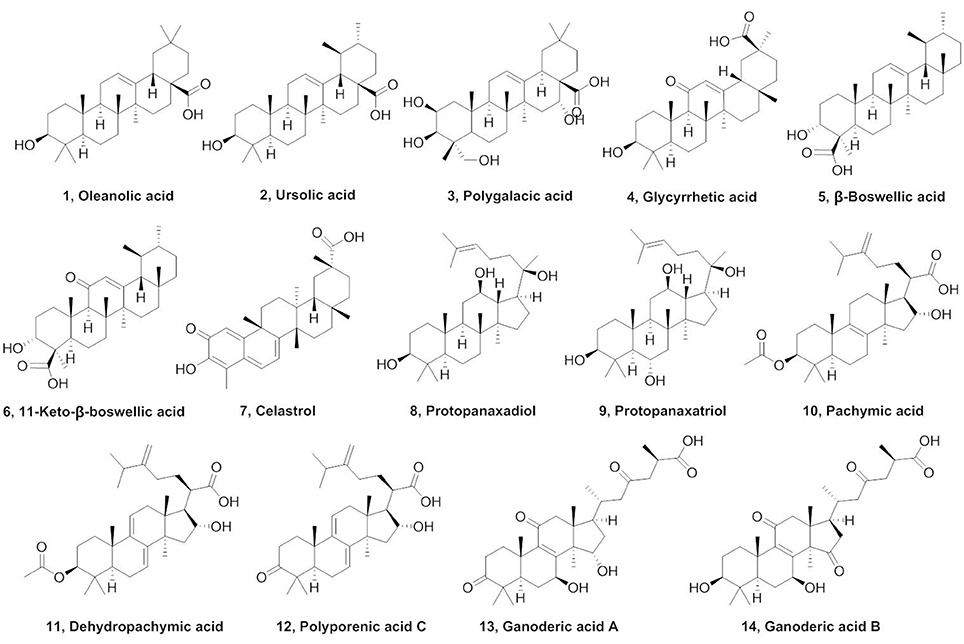
Figure 1. Chemical structures of tested natural triterpenoids.
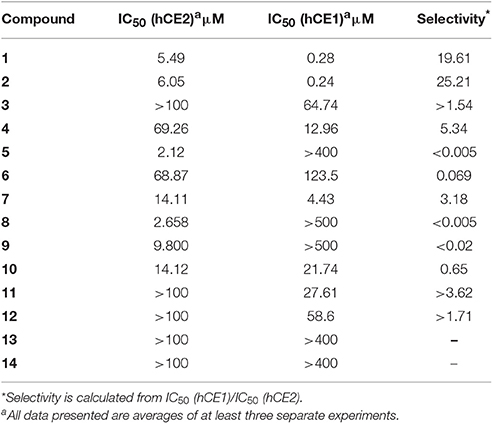
Table 1. The IC50 values of natural triterpenoids toward hCE1 and hCE2.
Chemistry
Compounds 15–27 were semi-synthesized according to Figure 2. 3-Keto compounds 15 and 21 were obtained with the Jones' reagent in high yield from 1 (OA), and 2 (UA), respectively. The OA was reduced with lithium aluminum hydride to afford the 28-hydroxymethyl compound 16. Reaction of the iodomethane with OA and UA furnished the target compounds 17 and 23. OA was acetylated in C-3 with acetic anhydride in pyridine to obtain ester 19 with high yield (92%). Compound 19, the acetate of OA, was then treated with oxalyl chloride without isolation, and further reacted with concentrated ammonia to afford amide compound which was hydrolyzed by NaOH to afford compound 18 in a yield of 42% over two steps. OA, UA and compound 26 were reacted with succinic anhydride in the presence of 4-dimethylaminopyridine to obtain the target product 20, 22, and 27, respectively. Compound 24 was synthesized from 23 by the same method as compound 15, with 98% yield. Compound 24 was oxidated with chromium trioxide to afford compound 25.
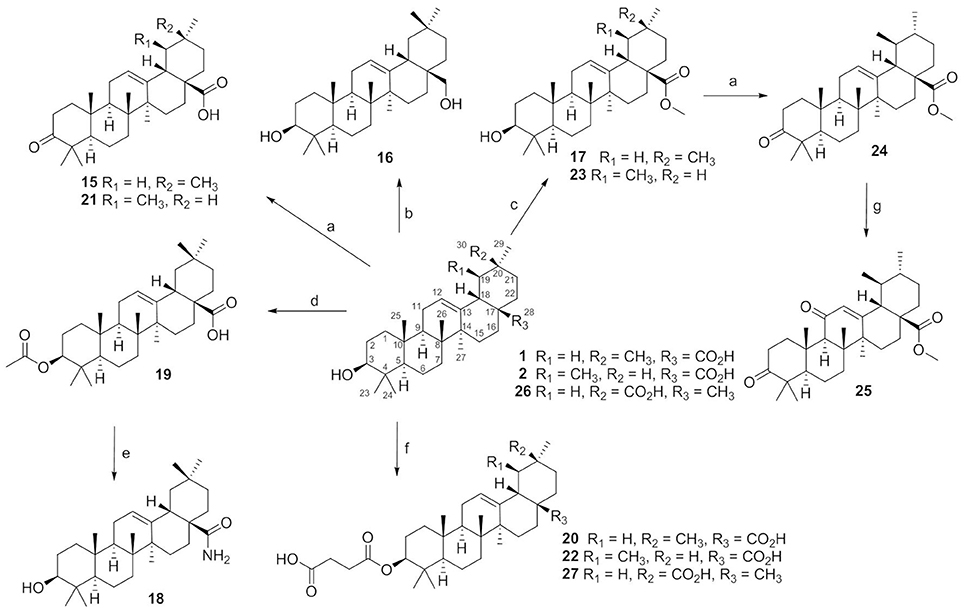
Figure 2. The synthesis routes for compounds 15–27. Reagents and conditions: (a) Jones reagent, acetone, 0°C, 1 h, 74–98%; (b) LiALH4, THF, rt, 24 h, 56%; (c) CH3I, K2CO3, acetone, rt, 12 h, 86–95%; (d) acetic anhydride, pyridine, rt, 12 h, 92%; (e) (COCl)2, CH2Cl2, rt, 2 h, then conc. ammonia, toluene, 4–8°C, NaOH, MeOH/THF, 40°C, 5 h, 42%; (f) succinic anhydride, DMAP, CH2Cl2, rt, 24 h, 87–97%; (g) CrO3, Ac2O, AcOH, rt, 3 h,50%.
SAR Study
Compounds 15–27 were assayed for their inhibitory effects against human CEs, including hCE1 and hCE2. The bioassay results were summarized in Table 2. Compound 15 exhibited relatively high selectivity toward hCE1 as compared with OA, suggesting that the introduction of carbonyl group at the C-3 site resulted in an increase of selectivity toward hCE1. Further modifications on the carbonyl group at the C-28 site of OA were conducted, and the alcohols (16), esters (17), and amides (18) derivatives were synthesized. As shown in Table 2, compounds 16–18 displayed both poor selectivity and specificity toward hCE1 compared with OA, suggesting that the carboxyl group at the C-28 site was very essential for hCE1 inhibition. Replacement of the C-3 hydroxyl group of OA with ethyl ester in compound 19 led to an increase of the inhibitory effects on hCE1 and a considerable selectivity toward hCE1 rather than hCE2, as compared with OA. Notably, replacement of the C-3 ethyl ester group with 3-O-β-carboxypropionyl in compound 20 led to a dramatically increase in the inhibitory effects against hCE1 (IC50, 17 nM) and the high selectivity over hCE2 (3296-fold against hCE2). These results suggested that the structural modifications on the C-3 hydroxyl group of OA were more feasible for the development of potent and highly selective inhibitors against hCE1.
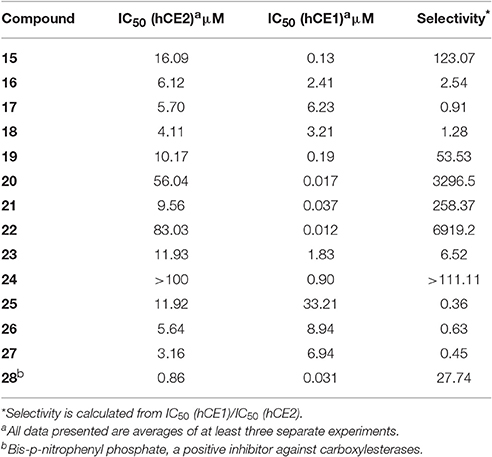
Table 2. The IC50 values of OA, UA, and their derivatives toward hCE1 and hCE2.
Consistently, 3-keto-UA derivative (21) exhibited similar trends in hCE1inhibition as 3-keto-OA derivative (15). Compound 22 (converting the C-3 hydroxyl group of UA to 3-O-β-carboxypropionyl group) showed excellent inhibitory effect against hCE1 with much lower IC50 value of 12 nM, which was 23-fold more potent than the parent compound UA and was 6,919-fold more selective over hCE2. Compound 23, a UA derivative bearing an ester group at the C-28 site, showed reduced inhibitory effect and poor selectivity toward hCE1 in contrast to compound 2. Compound 25, an 11-keto-UA derivative, displayed moderate inhibitory effect on hCE2 but its inhibition against hCE1 was significantly reduced compared to compound 24, suggesting that the carbonyl group introduced in such position was unbeneficial for hCE1 inhibition. Compounds 26 and 27, converting the C-28 carboxyl group of compound 1 and 20 to C-30 carboxyl moiety, respectively, displayed enhanced inhibitory effects against hCE2, while their inhibitory effects toward hCE1 were dramatically decreased. These results suggested that C-30 carboxyl group was beneficial for hCE2 inhibition but not good for hCE1 inhibition. In addition, a known inhibitor bis-p-nitrophenyl phosphate (BNPP) was tested under identical conditions as a positive control (Umehara et al., 2016). The result indicated that BNPP showed inferior inhibitory effect and poor selectivity in contrast to compounds 20 and 22 (Table 2). To the best of our knowledge, compound 22 is the most potent and highly selective inhibitor against hCE1 reported to date.
The structure-activity relationships (SAR) of these pentacyclic triterpenoids as hCE1 inhibitors have been summarized in Figure 3, which are very helpful for medical chemists to design and develop more potent and highly selective hCE1 inhibitors for biomedical applications.
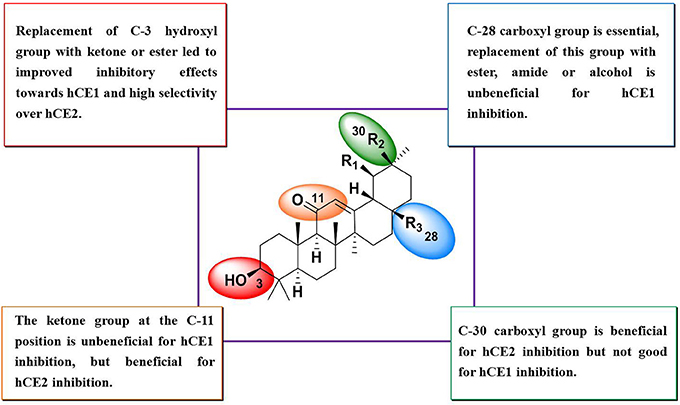
Figure 3. SAR summary of OA derivatives.
3D-QSAR Analysis
Quantitative Structure-Activity Relationship (QSAR) studies have been extensively applied to explore the correlations between biological activities and molecular descriptors for different classes of compounds (Soderholm et al., 2006; Vujasinovic et al., 2012). In this study, standard comparative molecular field analysis (CoMFA) was used to explore the relationships between structural features of all tested triterpenoids and their inhibitory effects on hCE1 (Table S1). The CoMFA steric and electrostatic fields based on PLS analysis were presented as 3-D contour plots in Figure 4. As shown in Figure 4A, the large red areas indicated that such regions with negative charges were favorable for hCE1 inhibition, while the blue are as implied that such regions with positive charges were favorable for hCE1 inhibition. The green-yellow steric contours depicted in Figure 4B illustrated that the steric bulks in green areas were beneficial for hCE1 inhibition, while the steric bulks in yellow areas were unbeneficial for hCE1 inhibition. The resulting CoMFA models suggested that both 3-D steric and electronic interactions could strongly affect the inhibitory effects of pentacyclic triterpenoids on hCE1. Notably, these findings agreed well with the experimental data, such as the electronegative group (red contour) at the C-28 position with the carboxyl group was beneficial for hCE1 inhibition, and the electropositive groups (blue contour) at the C-3 position could enhance the inhibitory effects against hCE1. The best CoMFA model was used to predict the inhibitory effects of all tested compounds in this study, which gave good statistical results (with the cross-validated q2 value of 0.554) and shown a strong correlation coefficient (R2 = 0.968) with experimental data (Figure 5). These findings provide new insights into the fine correlations between the inhibitory effects against hCE1 and the steric-electrostatic properties of triterpenoids, which are extremely helpful for rational design of novel pentacyclic triterpenoids as potent and selective hCE1 inhibitors.
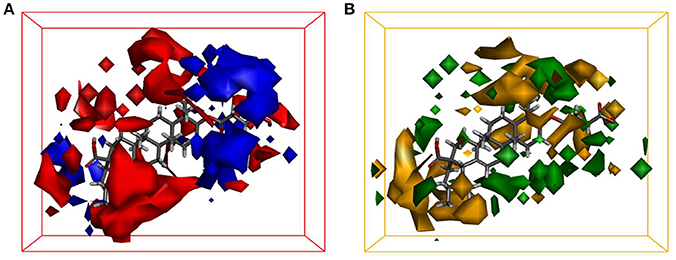
Figure 4. CoMFA steric and electrostatic contours displayed with most potent compound 22. (A) The red contours indicate the regions where substitution with more electronegative substituent are beneficial for hCE1 inhibition, whereas the blue contour shows the reverse; (B) The green areas indicate that the steric bulks are positively correlated with inhibitory activity, whereas the yellow are as indicate the steric bulks are negatively correlated with inhibitory activity.
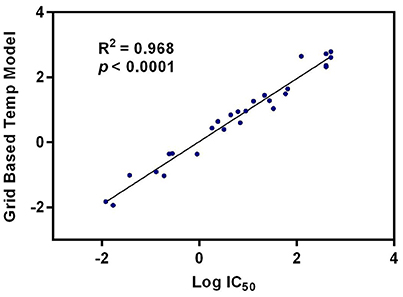
Figure 5. The correlation between the predictive and experimental inhibitory effects against hCE1 of compounds 1–27.
Inhibition Kinetic Analyses of Compound 22 toward hCE1
To further investigate the inhibitory behaviors of compound 22 on the catalytic activity of hCE1, the inhibition behaviors of this compound against hCE1 were performed. As shown in Figure 6A, Lineweaver-Burk plots demonstrated that compound 22 could inhibit hCE1 in HLMs via competitive inhibition, with the Ki values of 12.6 nM. In addition, the inhibitory tendency and potency of compound 22 against DME-hydrolysis in both HLMs and hCE1 were much closed (Figure S1). The IC50 value of compound 22 against recombinant hCE1 was evaluated as 9.2 nM, which has the similar value of inhibitory effect against hCE1 in HLMs (12.6 nM). These results demonstrated that compound 22 is highly selective and potent hCE1 inhibitors, which might be used as promising tools for exploring the biological functions of hCE1 in complex biological systems.
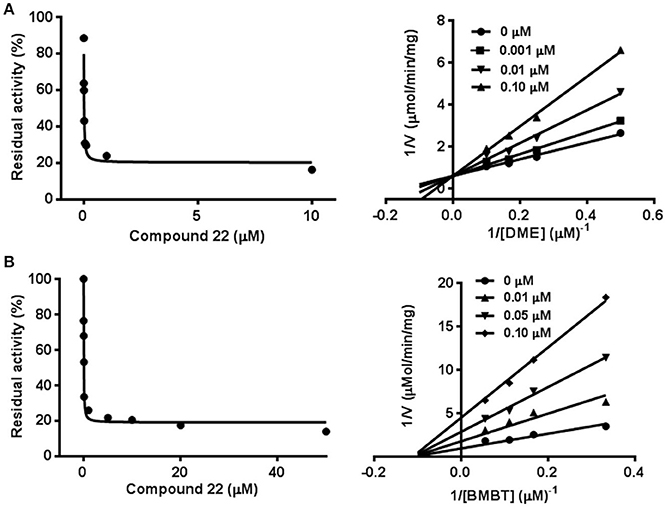
Figure 6. Inhibition behaviors of compound 22 against hCE1 mediated DME (A) and BMBT (B) hydrolysis. Left: the dose-dependent inhibition curves. Right: the Lineweaver-Burk plots. All data represent the mean of triplicate determinations.
Taking into account that hCE1 had at least two different ligand-binding sites, it is necessary to identify the ligand-binding sites of compound 22 and to investigate whether compound 22 displayed potent inhibitory effect against hCE1-meidated hydrolysis of other substrates (Lei et al., 2017). To this end, another optical probe substrate (BMBT) for hCE1 was used for further investigation on the inhibition behaviors of compound 22 (Liu et al., 2014). As shown in Figure 6B, Lineweaver-Burk plot demonstrated that compound 22 could inhibit hCE1-mediated BMBT hydrolysis in HLMs via non-competitive manner, with the IC50 and Ki values as 30.3 and 35.5 nM in HLMs, respectively. These findings clearly demonstrated that compound 22 could bind on the same ligand-binding site as DME rather than BMBT site, but this compound also displayed potent inhibitory effects against hCE1-meidated BMBT hydrolysis.
Molecular Docking Simulations
In order to gain a deep understanding of the inhibitory behavior of compound 22 against hCE1 from the view of ligand-enzyme interactions, molecular docking simulations were performed using a previous reported crystal structure of hCE1 (PDB ID: 2DQY) as the macromolecular model. As shown in Figure 7, Figures S2, S3 (Supplementary Data), DME and compound 22 could bind on hCE1 at the same site (also called ligand-bind site I) which was a hydrophobic pocket located at the surface of the catalytic domain of hCE1 (Figures 7A,C), while the BMBT (also called ligand-bind site II) bound on hCE1 at another site which was surrounded by Ser221, Ile359, and His468, etc. These findings agreed well with the experimental results from inhibition kinetic analyses, in which compound 22 functioned as a competitive inhibitor against hCE1-mediated DME hydrolysis but was a non-competitive inhibitor against hCE1-mediated BMBT hydrolysis.
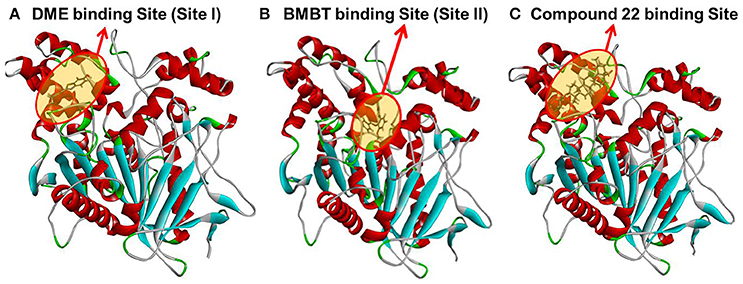
Figure 7. A stereo view of the crystal structure of hCE1 and the stereo diagram of each ligand aligned in its active site. (A) DME could bind on hCE1 at the ligand binding site I; (B) BMBT could bind on hCE1 at the ligand binding site II; (C) compound 22 could bind on hCE1 at the ligand binding site I (DME site).
Conclusions
In summary, a series of natural triterpenoids were collected and their inhibitory effects against human carboxylesterases were assayed. Two natural pentacyclic triterpenoids including OA and UA were found to display strong inhibitory effects on hCE1. Other natural pentacyclic triterpenoids, such as polygalacic acid (3) with more hydroxyl groups, glycyrrhetic acid (4), and celastrol (7) with carboxyl group at the C-30 site, displayed poor selectivity toward hCE1. Furthermore, in contrast to UA, β-boswellic acid (5) and 11-keto-β-boswellic acid (6) with carboxyl group at the C-23 site, demonstrated strong inhibitory effects on hCE2 and high selectivity over hCE1. Natural tetracyclic triterpenoids (8–14) exhibit less potentency and poor selectivity on hCE1, suggesting that the long alkyl chain at the C-20 site is unbeneficial for hCE1 inhibition. With the help of a series of semi-synthetic derivatives of OA and UA, the structure-activity relationships (SAR) analysis revealed that the carboxyl group at the C-28 site of OA and UA is very essential for hCE1 inhibition, and any modifications on this group with ester, amide or alcohol are unbeneficial for hCE1 inhibition. In contrast, the modifications of C-3 hydroxyl group are beneficial for hCE1 inhibition, and the replacement of C-3 hydroxyl group with a ketone or ester can lead to improvement of hCE1 inhibitory effects and high selectivity over hCE2. Guided by these SARs, the structural modifications of OA and UA, converting the C-3 hydroxyl group to 3-O-β-carboxypropionyl in compounds 20 and 22 lead to a dramatically increase of the inhibitory effects against hCE1 and high selectivity over hCE2. 3D-QSAR analysis demonstrated that electrostatic field and steric field are key factors affecting the inhibitory effects of OA and UA derivatives on hCE1. Further investigations demonstrated that compound 22 is a potent competitive inhibitor against hCE1-mediated DME hydrolysis but functioned as a noncompetitive inhibitor against BMBT hydrolysis. Molecular docking revealed that compound 22 could bind on the active site of hCE1 at the same ligand-binding site as DME, but this ligand-binding site is different from that of BMBT. All these findings will be very helpful for medicinal chemists to design and develop more potent and selective hCE1 inhibitors as drug candidates in future.
Author Contributions
LZ and GG were involved in the project design, carried out most of the experiments, and drafted the manuscript. TD contributed to the Molecular Docking simulations and 3D-QSAR analysis. PW, WL, ZW, JH, DW, and YF participated in the bioassays and inhibition kinetic analyses. WZ, GG, and LY designed and supervised this study. All authors read and approved the manuscript finally.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Basic Research Program of China (2013CB531805), NSF of China (21602219, 31600641, 81672961, 81573501, 81473181, and 81402822) and Innovative entrepreneurship program of high-level talents in Dalian (2016RQ025).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphar.2017.00435/full#supplementary-material
Abbreviations
BMBT, 2-(2-Hydroxy-3-methoxyphenyl) benzothiazole; BNPP, Bis-p-nitrophenyl phosphate; CEs, Mammalian carboxylesterases; CoMFA, Comparative molecular field analysis; DDAB, 6,8-dichloro-9,9-dimethyl-7-oxo-7,9-dihydroacridin-2-yl benzoate; DMAP, 4-Dimethylaminopyridine; DME, D-Luciferin methyl ester; hCE1, Human carboxylesterase 1; hCE2, Human carboxylesterase 2; HLM, Human liver microsomes; OA, Oleanolic acid; QSAR, Quantitative structure-activity relationship; SAR, Structure-activity relationships; TLC, Thin layer chromatography; UA, Ursolic acid.
References
Bachovchin, D. A., and Cravatt, B. F. (2012). The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov. 11, 52–68. doi: 10.1038/nrd3620
Bencharit, S., Morton, C. L., Xue, Y., Potter, P. M., and Redinbo, M. R. (2003). Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat. Struct. Biol. 10, 577–577. doi: 10.1038/nsb919
Crow, J. A., Middleton, B. L., Borazjani, A., Hatfield, M. J., Potter, P. M., and Ross, M. K. (2008). Inhibition of carboxylesterase 1 is associated with cholesteryl ester retention in human THP-1 monocyte/macrophages. Biochim. Biophys. Acta 1781, 643–654. doi: 10.1016/j.bbalip.2008.07.005
Dominguez, E., Galmozzi, A., Chang, J. W., Hsu, K. L., Pawlak, J., Li, W. W., et al. (2014). Integrated phenotypic and activity-based profiling links Ces3 to obesity and diabetes. Nat. Chem. Biol. 10, 113–121. doi: 10.1038/nchembio.1429
Fujiyama, N., Miura, M., Kato, S., Sone, T., Isobe, M., and Satoh, S. (2010). Involvement of carboxylesterase 1 and 2 in the hydrolysis of mycophenolate mofetil. Drug Metab. Dispos. 38, 2210–2217. doi: 10.1124/dmd.110.034249
Gilham, D., Ho, S., Rasouli, M., Martres, P., Vance, D. E., and Lehner, R. (2003). Inhibitors of hepatic microsomal triacylglycerol hydrolase decrease very low density lipoprotein secretion. Faseb J. 17, 1685–1687. doi: 10.1096/fj.02-0728fje
Hatfield, M. J., and Potter, P. M. (2011). Carboxylesterase inhibitors. Expert Opin. Ther. Pat. 21, 1159–1171. doi: 10.1517/13543776.2011.586339
Hatfield, M. J., Tsurkan, L. G., Hyatt, J. L., Edwards, C. C., Lemoff, A., Jeffries, C., et al. (2013). Modulation of esterified drug metabolism by tanshinones from Salvia miltiorrhiza (“Danshen”). J. Nat. Prod. 76, 36–44. doi: 10.1021/np300628a
Hill, R. A., and Connolly, J. D. (2017). Triterpenoids. Nat. Prod. Rep. 34, 90–122. doi: 10.1039/C6NP00094K
Hosokawa, M. (2008). Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules 13, 412–431. doi: 10.3390/molecules13020412
Hu, Z. Y., Parker, R. B., Herring, V. L., and Laizure, S. C. (2013). Conventional liquid chromatography/triple quadrupole mass spectrometry based metabolite identification and semi-quantitative estimation approach in the investigation of in vitro dabigatran etexilate metabolism. Anal. Bioanal. Chem. 405, 1695–1704. doi: 10.1007/s00216-012-6576-4
Imai, T. (2006). Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab. Pharmacokinet. 21, 173–185. doi: 10.2133/dmpk.21.173
Imai, T., Taketani, M., Shii, M., Hosokawa, M., and Chiba, K. (2006). Substrate specificity of carboxylesterase isozymes and their contribution to hydrolase activity in human liver and small intestine. Drug Metab. Dispos. 34, 1734–1741. doi: 10.1124/dmd.106.009381
Jin, Q., Feng, L., Wang, D. D., Wu, J. J., Hou, J., Dai, Z. R., et al. (2016). A highly selective near-infrared fluorescent probe for carboxylesterase 2 and its bioimaging applications in living cells and animals. Biosens. Bioelectron. 83, 193–199. doi: 10.1016/j.bios.2016.04.075
Kobayashi, Y., Fukami, T., Shimizu, M., Nakajima, M., and Yokoi, T. (2012). Contributions of arylacetamide deacetylase and carboxylesterase 2 to flutamide hydrolysis in human liver. Drug Metab. Dispos. 40, 1080–1084. doi: 10.1124/dmd.112.044537
Lei, W., Wang, D. D., Dou, T. Y., Hou, J., Feng, L., Yin, H., et al. (2017). Assessment of the inhibitory effects of pyrethroids againsthuman carboxylesterases. Toxicol. Appl. Pharmacol. 321, 48–56. doi: 10.1016/j.taap.2017.02.018
Li, J. B., Wang, Y. F., Matye, D., and Li, T. G. (2016). Sortilin 1 modulates hepatic cholesterol lipotoxicity in mice via functional interaction with liver carboxylesterase 1. Hepatology 63, 96a–97a.
Li, J. W. H., and Vederas, J. C. (2009). Drug discovery and natural products: end of an era or an endless frontier? Science 325, 161–165. doi: 10.1126/science.1168243
Li, Y. G., Hou, J., Li, S. Y., Lv, X., Ning, J., Wang, P., et al. (2015). Fructus Psoraleae contains natural compounds with potent inhibitory effects towards human carboxylesterase 2. Fitoterapia 101, 99–106. doi: 10.1016/j.fitote.2015.01.004
Liu, Y. J., Li, S. Y., Hou, J., Liu, Y. F., Wang, D. D., Jiang, Y. S., et al. (2016). Identification and characterization of naturally occurring inhibitors against human carboxylesterase 2 in White Mulberry Root-bark. Fitoterapia 115, 57–63. doi: 10.1016/j.fitote.2016.09.022
Liu, Z. M., Feng, L., Ge, G. B., Lv, X., Hou, J., Cao, Y. F., et al. (2014). A highly selective ratiometric fluorescent probe for in vitro monitoring and cellular imaging of human carboxylesterase 1. Biosens. Bioelectron. 57, 30–35. doi: 10.1016/j.bios.2014.01.049
Mai, Z. P., Zhou, K., Ge, G. B., Wang, C., Huo, X. K., Dong, P. P., et al. (2015). Protostane triterpenoids from the rhizome of alisma orientale exhibit inhibitory effects on human carboxylesterase 2. J. Nat. Prod. 78, 2372–2380. doi: 10.1021/acs.jnatprod.5b00321
Ngo, L. T., Okogun, J. I., and Folk, W. R. (2013). 21st Century natural product research and drug development and traditional medicines. Nat. Prod. Rep. 30, 584–592. doi: 10.1039/c3np20120a
Redinbo, M. R., and Potter, P. M. (2005). Mammalian carboxylesterases: from drug targets to protein therapeutics. Drug Discov. Today 10, 313–325. doi: 10.1016/S1359-6446(05)03383-0
Sanghani, S. P., Quinney, S. K., Fredenburg, T. B., Sun, Z. J., Davis, W. I., Murry, D. J., et al. (2003). Carboxylesterases expressed in human colon tumor tissue and their role in CPT-11 hydrolysis. Clin. Cancer Res. 9, 4983–4991.
Sato, Y., Miyashita, A., Iwatsubo, T., and Usui, T. (2012). Conclusive identification of the oxybutynin-hydrolyzing enzyme in human liver. Drug Metab. Dispos. 40, 902–906. doi: 10.1124/dmd.111.043208
Satoh, T., and Hosokawa, M. (1998). The mammalian carboxylesterases: from molecules to functions. Annu. Rev. Pharmacol. Toxicol. 38, 257–288. doi: 10.1146/annurev.pharmtox.38.1.257
Satoh, T., Taylor, P., Bosron, W. F., Sanghani, S. P., Hosokawa, M., and La Du, B. N. (2002). Current progress on esterases: from molecular structure to function. Drug Metab. Dispos. 30, 488–493. doi: 10.1124/dmd.30.5.488
Shen, B. (2015). A new golden age of natural products drug discovery. Cell 163, 1297–1300. doi: 10.1016/j.cell.2015.11.031
Sheng, H. M., and Sun, H. B. (2011). Synthesis, biology and clinical significance of pentacyclic triterpenes: a multi-target approach to prevention and treatment of metabolic and vascular diseases. Nat. Prod. Rep. 28, 543–593. doi: 10.1039/c0np00059k
Shi, D. S., Yang, J., Yang, D. F., Lecluyse, E. L., Black, C., You, L., et al. (2006). Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J. Pharmacol. Exp. Ther. 319, 1477–1484. doi: 10.1124/jpet.106.111807
Soderholm, A. A., Lehtovuori, P. T., and Nyronen, T. H. (2006). Docking and three-dimensional quantitative structure-activity relationship (3D QSAR) analyses of nonsteroidal progesterone receptor ligands. J. Med. Chem. 49, 4261–4268. doi: 10.1021/jm060234e
Sun, D. X., Ge, G. B., Dong, P. P., Cao, Y. F., Fu, Z. W., Ran, R. X., et al. (2016). Inhibition behavior of fructus psoraleae's ingredients towards human carboxylesterase 1 (hCES1). Xenobiotica 46, 503–510. doi: 10.3109/00498254.2015.1091521
Sun, Z. J., Murry, D. J., Sanghani, S. P., Davis, W. I., Kedishvili, N. Y., Zou, Q., et al. (2004). Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J. Pharmacol. Exp. Ther. 310, 469–476. doi: 10.1124/jpet.104.067116
Tang, M., Mukundan, M., Yang, J., Charpentier, N., Lecluyse, E. L., Black, C., et al. (2006). Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel is transesterificated in the presence of ethyl alcohol. J. Pharmacol. Exp. Ther. 319, 1467–1476. doi: 10.1124/jpet.106.110577
Umehara, K. I., Zollinger, M., Kigondu, E., Witschi, M., Juif, C., Huth, F., et al. (2016). Esterase phenotyping in human liver in vitro: specificity of carboxylesterase inhibitors. Xenobiotica 46, 862–867. doi: 10.3109/00498254.2015.1133867
Vujasinovic, I., Paravic-Radicevic, A., Mlinaric-Majerski, K., Brajsa, K., and Bertosa, B. (2012). Synthesis and biological validation of novel pyrazole derivatives with anticancer activity guided by 3D-QSAR analysis. Bioorg. Med. Chem. 20, 2101–2110. doi: 10.1016/j.bmc.2012.01.032
Wang, D. D., Jin, Q., Zou, L. W., Hou, J., Lv, X., Lei, W., et al. (2016). A bioluminescent sensor for highly selective and sensitive detection of human carboxylesterase 1 in complex biological samples. Chem. Commun. 52, 3183–3186. doi: 10.1039/C5CC09874B
Wang, D. D., Zou, L. W., Jin, Q., Hou, J., Ge, G. B., and Yang, L. (2017). Recent progress in the discovery of natural inhibitors against human carboxylesterases. Fitoterapia 117, 84–95. doi: 10.1016/j.fitote.2017.01.010
Wang, X. X., Lv, X., Li, S. Y., Hou, J., Ning, J., Wang, J. Y., et al. (2015). Identification and characterization of naturally occurring inhibitors against UDP-glucuronosyltransferase 1A1 in Fructus Psoraleae (Bu-gu-zhi). Toxicol. Appl. Pharmacol. 289, 70–78. doi: 10.1016/j.taap.2015.09.003
Williams, E. T., Carlson, J. E., Lai, W. G., Wong, Y. N., Yoshimura, T., Critchley, D. J., et al. (2010). Human in-vitro hydrolysis of rufinamide is primarily mediated by the liver-dominant carboxylesterase 1 and hydrolysis is inhibited by valproate and valproyl-CoA. Ann. Neurol. 68, S126–S126.
Wu, G. S., Robertson, D. H., Brooks, C. L., and Vieth, M. (2003). Detailed analysis of grid-based molecular docking: a case study of CDOCKER - A CHARMm-based MD docking algorithm. J. Comput. Chem. 24, 1549–1562. doi: 10.1002/jcc.10306
Xu, G., Zhang, W. H., Ma, M. K., and Mcleod, H. L. (2002). Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin. Cancer Res. 8, 2605–2611.
Xu, Y. J., Zhang, C. L., He, W. X., and Liu, D. (2016). Regulations of Xenobiotics and endobiotics on carboxylesterases: a comprehensive review. Eur. J. Drug Metab. Pharmacokinet. 41, 321–330. doi: 10.1007/s13318-016-0326-5
Zhu, H. J., Appel, D. I., Johnson, J. A., Chavin, K. D., and Markowitz, J. S. (2009). Role of carboxylesterase 1 and impact of natural genetic variants on the hydrolysis of trandolapril. Biochem. Pharmacol. 77, 1266–1272. doi: 10.1016/j.bcp.2008.12.017
Zhu, L. L., Xiao, L., Li, W., Zhang, Y., Han, W., Zhu, Y., et al. (2016). Human UDP-Glucuronosyltransferases 1A1, 1A3, 1A9, 2B4 and 2B7 are inhibited by Diethylstilbesterol. Basic Clin. Pharmacol. Toxicol. 119, 505–511. doi: 10.1111/bcpt.12618
Zhu, L. L., Xiao, L., Xia, Y., Zhou, K., Wang, H., Huang, M., Ge, G. B., et al. (2015). Diethylstilbestrol can effectively accelerate estradiol-17-O-glucuronidation, while potently inhibiting estradiol-3-O-glucuronidation. Toxicol. Appl. Pharmacol. 283, 109–116. doi: 10.1016/j.taap.2015.01.003
Zou, L. W., Li, Y. G., Wang, P., Zhou, K., Hou, J., Jin, Q., et al. (2016). Design, synthesis, and structure-activity relationship study of glycyrrhetinic acid derivatives as potent and selective inhibitors against human carboxylesterase 2. Eur. J. Med. Chem. 112, 280–288. doi: 10.1016/j.ejmech.2016.02.020
Keywords: human carboxylesterase 1 (hCE1), ursolic acid, oleanolic acid, structure-activity relationship (SAR), selective inhibitors
Citation: Zou L-W, Dou T-Y, Wang P, Lei W, Weng Z-M, Hou J, Wang D-D, Fan Y-M, Zhang W-D, Ge G-B and Yang L (2017) Structure-Activity Relationships of Pentacyclic Triterpenoids as Potent and Selective Inhibitors against Human Carboxylesterase 1. Front. Pharmacol. 8:435. doi: 10.3389/fphar.2017.00435
Received: 28 April 2017; Accepted: 16 June 2017;
Published: 30 June 2017.
Edited by:
Jianbo Xiao, University of Macau, ChinaReviewed by:
Kai Xiao, Second Military Medical University, ChinaChao Zhao, Fujian Agriculture and Forestry University, China
Copyright © 2017 Zou, Dou, Wang, Lei, Weng, Hou, Wang, Fan, Zhang, Ge and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang-Bo Ge, Z2VndWFuZ2JvQGRpY3AuYWMuY24=
Ling Yang, eWxpbmdkaWNwQHNvaHUuY29t
†These authors have contributed equally to this work.