 Amal Kamal Abdel-Aziz
Amal Kamal Abdel-Aziz Ashraf B. Abdel-Naim2
Ashraf B. Abdel-Naim2 Saverio Minucci
Saverio Minucci- 1Department of Experimental Oncology, European Institute of Oncology, Milan, Italy
- 2Department of Pharmacology and Toxicology, Faculty of Pharmacy, Ain Shams University, Cairo, Egypt
- 3Cancer Biology Department, National Cancer Institute, Cairo University, Cairo, Egypt
- 4Department of Biosciences, University of Milan, Milan, Italy
- 5Max F. Perutz Laboratories, Department of Microbiology and Immunobiology, University of Vienna, Vienna, Austria
Sunitinib, a multityrosine kinase inhibitor, is currently the standard first-line therapy in metastatic renal cell carcinoma (mRCC) and is also used in treating patients with pancreatic neuroendocrine and imatinib-resistant gastrointestinal stromal tumors (GIST). Nevertheless, most patients eventually relapse secondary to intrinsic or acquired sunitinib resistance. Autophagy has been reported to contribute to both chemo-sensitivity and -resistance. However, over the last few years, controversial regulatory effects of sunitinib on autophagy have been reported. Since gaining insights into the underlying molecular insights and clinical implications is indispensible for achieving optimum therapeutic response, this minireview article sheds light on the role of a network of prosurvival signaling pathways recently identified as key mediators of sunitinib resistance with established and emerging functions as autophagy regulators. Furthermore, we underscore putative prognostic biomarkers of sunitinib responsiveness that could guide clinicians toward patient stratification and more individualized therapy. Importantly, innovative therapeutic strategies/approaches to overcome sunitinib resistance both evaluated in preclinical studies and perspective clinical trials are discussed which could ultimately be translated to better clinical outcome.
Introduction
Following the initial breakthrough success of imatinib, the first FDA-approved tyrosine kinase inhibitor(TKI), TKIs were deemed to revolutionize cancer therapy. Nevertheless, emergence of imatinib-resistance prompted development of novel structurally distinct TKIs (Abdel-Aziz et al., 2015; Aziz et al., 2016). Among these second-generation TKIs, sunitinib, was designed as an oral small molecule ATP mimetic which competes with endogenous ATP for binding at the catalytic site of several tyrosine kinase receptors including vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), fms-like tyrosine kinase-3 receptor (FLT3) and stem cell factor receptor (c-kit) which are preferentially overexpressed in diverse types of cancer (Cella et al., 2015). In 2006, sunitinib became the first drug jointly approved by the FDA for treating both metastatic renal cell carcinoma (mRCC) and imatinib-resistant gastrointestinal stromal tumor(GIST) patients. Five years later, treatment of progressive pancreatic neuroendocrine tumors(pNET) was added to sunitinib indications. Since then, several clinical trials were initiated to evaluate its efficacy against diverse cancer types including those with limited therapeutic options as differentiated thyroid and invasive lower urothelial cancer. Nonetheless, intrinsic as well as acquired resistance to sunitinib rapidly emerged as a challenge restraining its clinical benefit (Table 1) (Adelaiye et al., 2014). In fact, almost one-third of sunitinib-treated patients are intrinsically resistant whereas the initially responders eventually relapse and develop progressive disease resulting in modest overall survival benefit (Stacchiotti et al., 2012; Adelaiye et al., 2014). Given the complexity of the target spectrum modulated by sunitinib, deeper understanding of the contribution of those targets to the sensitivity or resistance to sunitinib is cardinal for its optimal clinical use. Among chemoresistance mechanisms, other than mutation or amplification of drug targets, activation of prosurvival signaling pathways is a frequently exploited strategy by cancer cells to evade cell death and sustain their proliferation (Hammers et al., 2010; Shojaei et al., 2010).
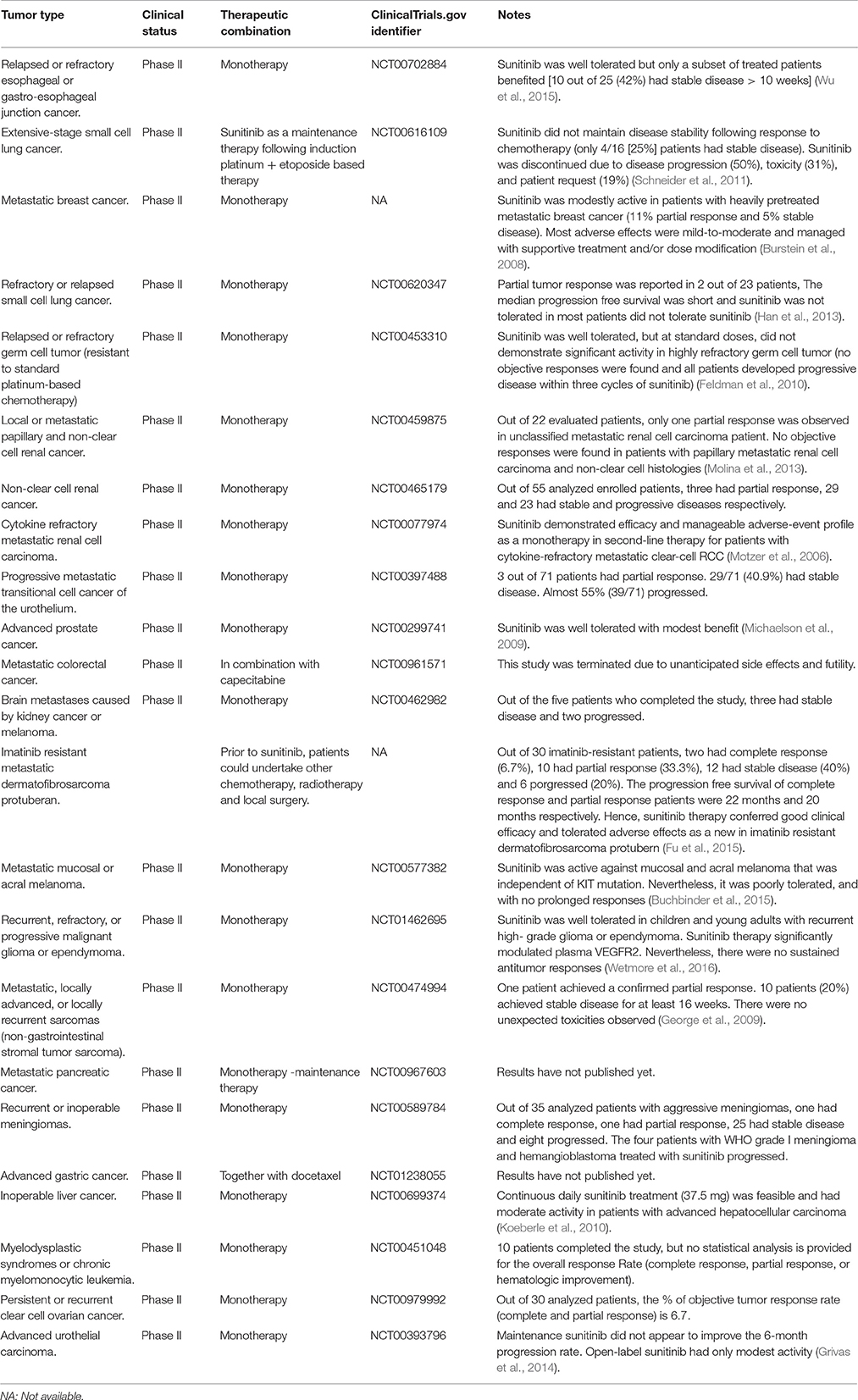
Table 1. Clinical trials investigating the efficacy and safety/tolerability of sunitinib against different cancer types.
Biphasic Modulation of Autophagy by Sunitinib
The role of autophagy in tumorigenesis has been somewhat controversial (Yang et al., 2011; Abdel-Aziz et al., 2015). In addition to the well-established pro-survival functions during nutrient-deprivation, evidence suggests cytotoxic effect of excessive autophagy triggered by conditions other than starvation. Overwhelming autophagy induction may contribute to cell death through digesting essential cellular macromolecules and organelles and hence, is classified as programmed cell death type II (Maiuri et al., 2007). In line with this controversy, autophagy modulation has been suggested to contribute to both resistance and cytotoxicity of several chemotherapeutics (Yang et al., 2011; Abdel-Aziz et al., 2015). Throughout the last few years, controversial regulatory effects of sunitinib on autophagy have been reported. For instance, sunitinib activated autophagy in RCC, phaeochromocytoma, thyroid and breast cancer (Lin et al., 2012; Ikeda et al., 2013; Abdel-Aziz et al., 2014a,b). In contrast, others have reported negative regulatory effects on autophagic flux in human urinary bladder and medullary thyroid cancer (Santoni et al., 2013; Lopergolo et al., 2014).
In our recently published study, we aimed to systematically address this discrepancy (Elgendy et al., 2016). We primarily screened the anticancer efficacy of a broad range of sunitinib concentrations against a panel of human cancer cell lines representative of diverse types; renal cell carcinoma, pancreatic neuroendocrine tumor, colorectal cancer and osteosarcoma (Elgendy et al., 2016). Each cancer cell type had its unique “sunitinib-tolerance threshold” beyond which its viability was dramatically compromised. Strikingly, most cancer cell types tolerated sunitinib levels analogous to that of treated cancer patients. In accordance with our preclinical findings where higher sunitinib doses were cytotoxic, cancer patients who primarily progressed in response to standard sunitinib doses and during “sunitinib-off” period were resensitized by escalating sunitinib dose (Stacchiotti et al., 2012; Mitchell et al., 2015). Intriguingly, sunitinib tuned the activity of the autophagic machinery in a biphasic pattern. In response to tolerated doses—regardless of cancer type—sunitinib inhibited autophagy as evidenced by decreased GFP-LC3 puncta, LC3II/I ratio and increased p62/SQTSM1 levels. In contrast, autophagy was triggered in cancer cells challenged with cytotoxic sunitinib doses (Elgendy et al., 2016) (Figure 1).
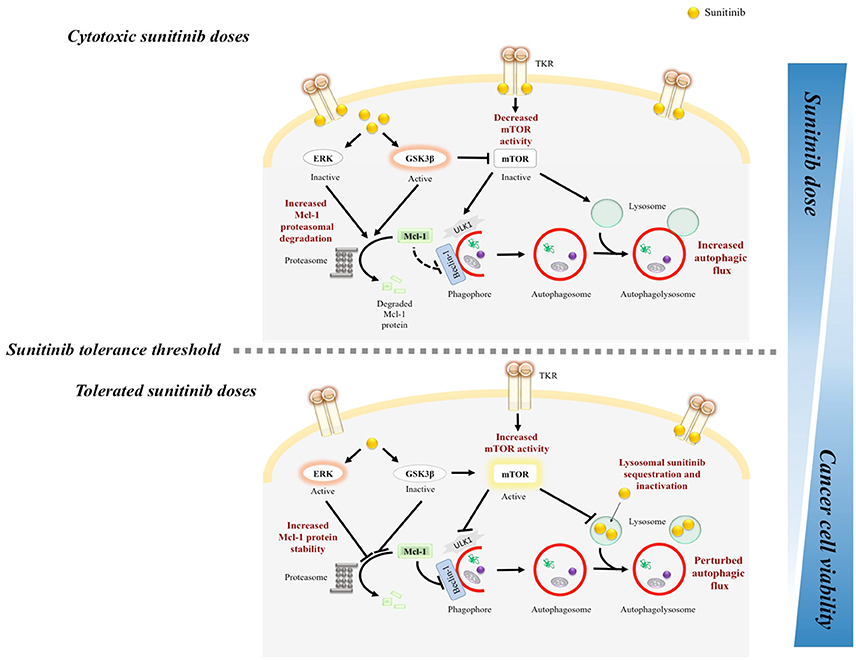
Figure 1. Mechanistic illustration of sunitinib dose-range dependent biphasic regulation of autophagy in cancer cells. TKR, tyrosine kinase receptor. Autophagy was inhibited in cancer cells that resist the anticancer activity of clinically relevant sunitinib levels. In response to tolerated doses, sunitinib increased ERK, and GSK3β phosphorylation which increased Mcl-1 stability, and mTOR activity. In addition, given its lysomotropic property, sunitinib resistance has been linked to its lysosomal sequestration, and hence, inactivation. In contrast, escalating sunitinib dose or pharmacologically targeting Mcl-1/mTOR restored cancer cell responsiveness/sensitivity to sunitinib.
Below, we review the differential modulatory effects of sunitinib on autophagy and their link to sunitinib resistance.
Biphasic mTORC1 and Mcl-1 Modulation Mediates Dual Regulation of Autophagy by Sunitinib
Autophagy is regulated by a complex network of molecular switches, among which mammalian target of rapamycin (mTOR) is a central player (Yang et al., 2011). mTOR exists in two distinct complexes, mTORC1 and mTORC2. Unlike the relatively unexplored role of mTORC2 in cancer biology, mTORC1 is a nutrient sensitive sensor that orchestrates cell metabolism, cell cycle progression and autophagy (Yang et al., 2011). mTORC1 represses the latter at both initiation and degradation stages via inhibiting ULK1 complex and lysosomal function respectively (Elgendy et al., 2014; Puertollano, 2014). Furthermore, antiapoptotic B-cell lymphoma 2 (Bcl-2) family members such as Bcl-2, myeloid cell leukemia 1 (Mcl-1), and B-cell lymphoma-X large (Bcl-xL) - through their direct and indirect interactions with Beclin-1/Atg6 - have emerged as autophagy regulators which mediate the regulatory crosstalks between apoptosis and autophagy (Elgendy et al., 2014).
Intriguingly, in our study, cancer cells responded to subcytotoxic sunitinib doses by activating mTORC1 and increasing Mcl-1 protein levels (Elgendy et al., 2016) (Figure 1). Importantly, increased mTORC1 activity and Mcl-1 level represented a pro-survival cellular response to the mild to moderate stress triggered by these sunitinib doses since inhibiting mTORC1 or depleting Mcl-1 sensitized cancer cells to tolerated doses (Elgendy et al., 2016). Notably, mTORC1 activity and Mcl-1 levels were found to be higher in sunitinib-resistant compared to parental melanoma cells further confirming their role in mediating sunitinib resistance (Elgendy et al., 2016). Finally, analysis of pNET and RCC samples obtained from sunitinib-resistant patients showed a significant correlation between post-sunitinib increase in Mcl-1 levels and mTORC1 activity and resistance to treatment with sunitinib (Elgendy et al., 2016). In line with our observations, Makhov et al. has shown that deletion of PTEN (phosphatase and a tensin homolog deleted from chromosome 10), a negative regulator of PI3K/AKT/mTOR signaling, correlated with sunitinib resistance in renal and prostate cancer (Makhov et al., 2012). Conversely, cytotoxic doses dramatically diminished mTORC1 activity and Mcl-1 levels. In alignment with our findings, sunitinib was active against acute myeloid leukemias (AML) possessing activating PDGFR, FLT-3, and c-kit mutations through inhibiting mTOR and reducing Mcl-1 levels (Ikezoe et al., 2006; Teng et al., 2013).
Interestingly, in addition to genetic tools, our results showed that pharmacological mTOR and/or Mcl-1 inhibition using rapamycin and sorafenib respectively sensitized cancer cells to primarily tolerated doses of sunitinib (Elgendy et al., 2016). In accordance with our findings, Lin and colleagues demonstrated that while silencing Atg5 abrogated sunitinib cytotoxicity, everolimus and trehalose, mTOR-dependent and independent autophagy activators respectively, boosted the antiproliferative activity of sunitinib in medullary thyroid cancer cells (Lin et al., 2012). Furthermore, Atg5 silencing antagonized everolimus- and trehalose-triggered increase in sunitinib efficacy (Lin et al., 2012). Additionally, our data provide mechanistic rationale for the previously reported synergy between sunitinib and mTOR inhibitors identified through unbiased binary screening (Li et al., 2014).
Sunitinib-Induced ERK and GSK3β Modulation Lead to mTORC1 and Mcl-1 Regulation
Deeper mechanistic analysis showed that the increase in Mcl-1 protein levels upon treatment with tolerated doses of sunitinib was associated with decreased Mcl-1 proteasomal degradation and enhanced stability, rather than modulation of gene expression (Elgendy et al., 2016). One distinct feature of Mcl-1 among other antiapoptotic Bcl-2 family members is its short half-life. This has been linked to its tight regulation by complex mechanisms on several levels including phosphorylation in its unique PEST region at Thr163 and Ser159 residues by extracellular signal regulated kinase (ERK) and glycogen synthase kinase 3β (GSK3β) respectively which in turn, diminishes and enhances the rate of Mcl-1 proteasomal degradation (Domina et al., 2004; Yan et al., 2017). Enhanced Mcl-1 stability in response to lower doses of sunitinib was the net result of both ERK activation and GSK3β inhibition (Elgendy et al., 2016). Conversely, higher “cytotoxic” doses led to opposite effects where ERK was inhibited and GSK3β was activated, resulting in Mcl-1 destabilization (Elgendy et al., 2016). Furthermore, dual modulation of GSK3β activity by lower and higher doses of sunitinib also mediated the differential effect on mTORC1 activity (Figure 1).
Regulatory Effects of Sunitinib on AXL and MET Signaling Modulate ERK and GSK3β Activity
Induced AXL and MET signaling which in turn increased ERK and GSK3β phosphorylation has been linked to sunitinib resistance in RCC (Zhou et al., 2016). Intriguingly, Qu et al. have lately identified a novel long non-coding RNA called lncARSR (lncRNA Activated in RCC with Sunitinib Resistance) which promoted sunitinib resistance via acting as a competing endogenous RNA. Indeed, IncARSR through sequestering miR-34 and miR-449 upregulated AXL/c-MET expression and activated ERK, STAT3 and AKT signaling. In a positive regulatory feedback loop, activated AKT boosted IncARSR expression. Strikingly, sunitinib-resistant RCC cells secreted IncARSR via exosomes which reached out to sunitinib sensitive cells and transformed them into resistant ones (Rna et al., 2016). Sunitinib-induced ERK activation secondary to increased EGFR phosphorylation was also linked to sunitinib resistance mediated by epithelial-to-mesenchymal transition (EMT) in mRCC cells (Mizumoto et al., 2015). In addition, sphingosine kinase-1 (SK1)-mediated ERK activation was triggered in sunitinib-resistant RCC cell lines which were resensitized to sunitinib using SK1 and ERK pharmacological inhibitors (Gao and Deng, 2014). Ras/Raf activating mutations and subsequent constitutive MEK/ERK activation has also been speculated to contribute to sunitinib resistance in thyroid carcinoma cells (Piscazzi et al., 2012). Moreover, sunitinib-induced apoptosis in medulloblastoma was associated with GSK3β and mTOR dephosphorylation (Yang et al., 2010).
Lysosomal Sequestration of Sunitinib Impedes Autophagic Flux
mRCC resistance to sunitinib was reported to be secondary to impedance of autophagy at its termination stage. Sunitinib neutralized the acidic lysosomal pH and hence, inactivated one of the major lysosome-associated proteases, cathepsin B (Giuliano et al., 2015). In light of the observations illustrating the tightly regulated crosstalk between lysosomes and mTORC1 where active mTORC1 negatively impacted lysosomal biogenesis, function and autophagosomes-lysosomes fusion (Puertollano, 2014), we postulate that subcytotoxic sunitinib doses-induced mTOR activation could also contribute to lysosomal dysfunction associated with the “chemoresistance phenotype.” Accordingly, it has been proposed that increasing sunitinib availability at its target TKRs via preventing its lysosomal trapping using agents which increase lysosomal membrane permeability could resensitize mRCC to sunitinib.
Modulatory Effects of Sunitinib on ATP-Binding Cassette (ABC) Transporters and Autophagy
The role of multidrug resistance proteins has been linked to chemoresistance (Giuliano et al., 2015). Indeed, sunitinib increased ABCB1 expression favoring further lysosomal accumulation of sunitinib and its cellular efflux. Inhibiting this transporter by elacridar restored sunitinib senstivity in resistant mRCC (Giuliano et al., 2015; Sato et al., 2015). Sunitinib also increased ABCG2 levels in RCC cells (Sato et al., 2015) and glioma xenografts (Shojaei et al., 2010). Such increment is thought to compensate for the inhibitory effects of sunitinib on ABCG2 function (Dai et al., 2009; Shukla et al., 2009). While targeting ABC transporters might be useful in reversing sunitinib resistance, attention should be directed toward the bioavailability of co-administered drugs (Shukla et al., 2009; Sato et al., 2015). Interestingly, ABCG2 has also been shown to modulate autophagy (Ding et al., 2016). Compared to parental cells, ABCG2 overexpression—which was associated with boosted autophagy—rendered cancer cells more rendered cancer cells more resistant to stress inducers. Consistently, knocking down ABCG2 inhibited autophagy (Ding et al., 2016). Yet, it is still unknown whether modulating ABC transporters function/expression by sunitinib mediates its regulatory effects on autophagy and cancer cell sensitivity/resistance to sunitinib.
Antiangiogenic Effects of Sunitinib, Tumor Microenvironment, and Autophagy
As tumors progress, their oxygen and nutrients demands substantially outweigh their supply. To address these needs, “angiogenesis” is switched on generating tumor-associated neovasculature. Within this context, sunitinib emerged as an effective antiangiogenic agent via inhibiting VEGFR, PDGFR, and c-KIT, which was then indicated for treating tumors with high angiogenic burden as mRCC. Although an interrupted sunitinib treatment schedule of 4-weeks ON/2-weeks OFF was adopted to reduce its side effects, rapid tumor regrowth and metastasis considerably ensued during the 2-week break period (Ebos et al., 2009; Griffioen et al., 2012). While the underlying mechanisms remain elusive, microenvironmental changes are speculated to condition/acclimatize body organs to be more permissive to tumor extravasation by acting as “premetastatic niche” (Loges et al., 2009). Anti-angiogenic therapy—via triggering hypoxia—might induce autophagy in both tumor cells and microenvironment. While activating autophagy in cancer cells might retard their proliferation, stromal cancer-associated fibroblasts autodigest themselves into basic nutrients for epithelial cancer cells. This host-parasitic like paradigm is known as “Battery-Operated Tumor Growth” or “The Autophagic Tumor Stroma Model of Cancer Cell Metabolism” (Martinez-outschoorn et al., 2010). Cancer-associated cachexia is also hypothesized to start as local stromal autophagy, and then disseminates systemically. Consistently, increased metabolic rate of cachexic cancer patients is restored to their normal levels following surgical tumor excision (Martinez-outschoorn et al., 2010). Furthermore, sunitinib resistance is linked to persistence of intratumor myeloid derived suppressor cells (MDSCs) with dominating subset of neutrophilic population that produces proangiogenic MMP9, MMP8, and IL-8 (Finke et al., 2011). Activation of proangiogenic hepatocyte growth factor/c-Met signaling pathway also contributed to sunitinib resistance which was reversed using c-Met inhibitor (Shojaei et al., 2010). Notably, inhibiting c-Met signaling induced autophagic cell death in glioblastoma (Liu et al., 2013). Collectively, these divergent effects of antiangiogenics on primary tumor and its microenvironment necessitates elemental consideration of prominent issues while administering anti-VEGF therapy including; relative benefit-to-risk of “continuous vs. intermittent” treatment schedules, optimal dose, duration of treatment and tumor stage. This also emphasizes the importance of combination therapy as a possible approach to abrogate resistance to anti-VEGF therapy.
Mitophagy, Effects of Sunitinib on Mitochondrial Structure/Function and Apoptosis
Mitophagy (or selective autophagic degradation of mitochondria) has been linked to tumorigenesis (Hjelmeland and Zhang, 2016). One of the key mitophagy mediators, Parkin, putative tumor suppressor gene, translocates to the mitochondria secondary to loss of mitochondrial membrane potential, ubiquitinating mitochondrial proteins and recruiting p62-LC3 and autophagosomes to the mitochondria (Hjelmeland and Zhang, 2016). Melatonin synergistically sensitized human heptocellular carcinoma cells to sorafenib through its pro-oxidant capacity and activating mitophagy (Prieto-domínguez et al., 2016). In fact, oxidative stress and hypoxia are fundamental autophagy inducers (Kulikov et al., 2017). Hypoxia - via hypoxia inducible factor 1α (HIF1α)- induced the expression of mitophagy receptors; BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) and its homolog NIX (Kulikov et al., 2017). Though sunitinib blocked HIF1α translation and hence, repressed the expression of its downstream target genes in colorectal and renal carcinoma cells (Shin et al., 2010). Severe mitochondrial structural abnormalities was also reported in the heart of a patient with sunitinib-induced heart failure (Kerkela et al., 2008). This has been linked to its off-target inhibitory effect on AMPK (Kerkela et al., 2008). Sunitinib also triggered mitochondrial damage, cytochrome C release, caspase 9 activation and apoptotic cell death in cardiomyocytes in vitro and in vivo (Chu et al., 2007). Sunitinib increased both death receptor and mitochondrial-dependent apoptosis in AML cells (Teng et al., 2013). Nonetheless, it still remains to be elucidated whether sunitinib modulates mitophagy and therapeutic intervention with mitophagy could sensitize cancer cells to sunitinib.
Linking the Modulatory Effects of Sunitinib on Autophagy to Genomic Instability
Dysfunctional autophagy has been connected to increased genomic instability. Intriguingly, sunitinib-induced increased autophagic flux concurred with increased micronuclei, diagnostic marker of nuclear instability, in human RCC (Yan et al., 2017). Nuclear LC3, phosphorylated Ulk1 and p62 interacted with Rad51 or PARP-1, which are both engaged in maintaining genomic stability (Yan et al., 2017). Sunitinib was incapable of accumulating micronuclei in p62/LC3-depleted cells. Depleting Rad51/PARP-1 abolished sunitinib-induced autophagy (Yan et al., 2017). Since p62 acts as the connecting bridge between ubiquitin and autophagic machineries, both systems are speculated to coordinate genomic stability. Despite being a marker of DNA damage, γ-H2AX actively participates in DNA repair. γ-H2AX and PARP-1/Rad51 interaction was disrupted following p62 depletion (Yan et al., 2017). Although sunitinib elevated γ-H2AX level, it decreased Rad51 expression which is essential for homologous recombination repair, Accordingly, while sunitinib induced acute DNA damage may lead to cancer cell death, it might also trigger non-homologous end joint DNA repair mechanisms. Collectively, these findings proposed a mechanistic link between the modulatory effects of sunitinib on autophagy and nuclear instability.
Adverse Effects of Sunitinib and Autophagy
Clinical trials and post-marketing surveillance have reported that sunitinib use is associated with several adverse effects including cardiac failure and cognitive impairment. In this regards, it has been shown that sunitinib-induced autophagic cell death contributed to its cardiotoxicity (Zhao et al., 2010). Impeded autophagic flux has been associated with sunitinib-induced chemobrain (chemotherapy-related cognitive impairment) (Abdel-Aziz et al., 2016). As our data strongly suggest a potential therapeutic synergy of a combination of sunitinib with Mcl-1/mTORC1 inhibitors such as sorafenib and rapalogues which are known to induce autophagy, this could be of crucial clinical relevance especially concerning the toxicity of such combination. Attempts to combine other drugs with sunitinib have thus been so far unsuccessful, largely due to toxicity. However, our in vivo results demonstrated a strong synergy on tumor xenograft growth of such combinations at doses lower that those used clinically with favorable tolerability/no sign of toxicity.
Translating Preclinical Findings to Bedside
Novel Predictive Markers of Sunitinib Responsiveness
Canonical clinicopathological evaluation is unable to predict the therapeutic response to sunitinib-treated cancer patients. Identification of novel molecular prognostic markers is therefore urgently needed. Based on the present findings, immunohistochemical assessment of ribosomal S6 phosphorylation (as readout of mTORC1 activity) and Mcl-1 protein levels could serve as markers that predict sunitinib response. Additionally, elevated IncARSR levels in pre-treatment RCC patients significantly correlated with poor sunitinib response. In contrast, low IncARSR levels conferred improved progression-free survival and favorable prognosis following sunitinib therapy (Rna et al., 2016). Notably, IncARSR levels were remarkably increased in patients who regressed and relapsed post-sunitinib therapy compared with pre-therapy levels. Hence, IncARSR is proposed as an independent predictor for sunitinib response in RCC patients (Rna et al., 2016).
Emerging Therapeutic Modalities to Overcome Sunitinib Resistance
Additionally, the present findings provide a rationale for the lack of cytotoxic effects of clinically relevant doses of sunitinib, and suggest novel strategies -in addition to its anti-angiogenic effects- to directly induce tumor cell death, and overcome sunitinib resistance;
(i) Ideally, though not easily achievable in clinical practice, tailoring sunitinib dose per each patient based on their response should select patients that need escalation of sunitinib dose to reach cytotoxic effects at tolerable doses. Rovithi et al. showed that an alternating schedule of high sunitinib efficiently impaired tumor growth in vivo and maintained significantly higher plasma and intratumoral sunitinib levels compared to the standard, daily regimen (Rovithi et al., 2016). Accordingly a phase I clinical trial (NCT02058901) was initiated to investigate the safety, tolerability and efficacy of intermittent, high dose sunitinib schedules (300 mg sunitinib, administered once weekly) in patients with advanced solid tumors, with preliminary indications of prolonged disease stabilization and tolerability in a heavily pretreated group of patients (Rovithi et al., 2016).
(ii) Alternatively, pharmacological targeting of Mcl-1 and mTOR presents a promising strategy in combating/reversing sunitinib resistance. It is worth mentioning that FDA has already approved sequential treatment of sunitinib-resistant mRCC patients with everolimus, mTOR inhibitor. In addition, a phase I clinical trial aiming at determining the highest tolerable dose of “sunitinib and temsirolimus (mTOR inhibitor)” combination that could be administered to mRCC patients has lately been completed. There are also parallel clinical trials testing the efficacy of sorafenib in treating sunitinib-resistant mRCC and GIST patients. Yet, the clinical outcome of combining sunitinib and Mcl-1 and/or mTOR inhibitor in sunitinib-resistant patients remains to be investigated. Consistent with the emerging role of MET and AXL signaling in mediating sunitinib resistance, combining a small molecule inhibitior of both MET and AXL, BMS777607, with sunitinib both suppressed the growth of sunitinib-resistant RCC xenografts and prevented the emergence of sunitinib resistance (Rna et al., 2016). In support with this notion, a randomized Phase III clinical trial has further confirmed the clinical value of cabozantinib, a multityrosine kinase inhibitor targeting MET, VEGFR, and AXL in RCC patients who had progressed following VEGFR inhibitor therapy including sunitinib (Géczi et al., 2015).
Open Questions
In a broader scope beyond sunitinib, accumulating evidence reported compensatory activation of PI3K/AKT/mTOR signaling pathway and/or heightened Mcl-1 expression in diverse cancer types resistant to other TKIs including imatinib, dasatinib, erlotinib, and gefitinib (Burchert et al., 2005; Wu et al., 2016). This raises questions:
• Whether activation of mTOR/Mcl-1 signaling is a common adaptive resistance mechanism exploited by cancer cells—regardless of their origin and type—to evade the anticancer activity of TKIs.
• And accordingly, whether pharmacological targeting of mTOR/Mcl-1 could be adopted in TKIs-resistant patients. In line with this, two Phase I/II clinical trials assessing the safety and efficacy of combining imatinib and everolimus in treating imatinib-resistant CML and GIST patients have been launched.
Concluding Remarks
Sunitinib resistance in patients correlated with reinforced negative regulation of autophagy (increased Mcl-1 stability and mTORC1 activity) and lysosomal dysfunction (owing to sunitinib sequestration and hence inactivation). Both events acted as pro-survival adaptive responses that compromised the anticancer activity of sunitinib. Conversely, cytotoxic sunitinib levels destabilized Mcl-1, inhibited mTORC1 and activated autophagy. Hence, this may mechanistically resolve the previously described discrepancy in terms of “ON/OFF” autophagy regulation by sunitinib. Gaining deeper mechanistic insights into sunitinib resistance would provide better prognostic biomarkers that could guide clinicians toward patient stratification and more individualized therapy. Importantly, this would offer more innovative therapeutic strategies/approaches to overcome sunitinib resistance which could ultimately be translated to a better clinical outcome.
Author Contributions
AKA conceived and wrote the manuscript. ABA, SS, SM, and ME revised the manuscript. SM and ME reviewed the outline and content of the manuscript. All authors have read and approved the submitted manuscript for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in SM's lab is supported by AIRC, FIRC, CNR (Epigen), and Horizon 2020 grants. AKA has been awarded a fellowship by AIRC. AKA has been awarded a fellowship by AIRC. ME received funding from the Mahlke-Obermann Stiftung and the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 609431/INDICAR (Interdisciplinary Cancer Research) Postdoctoral Fellowship Programme of the University of Vienna.
Abbreviations
ERK, extracellular regulated kinase; EMT, epithelial to mesenchymal transition; FLT3, fms-like tyrosine kinase-3 receptor; GSK3β, glycogen synthase kinase 3β; GIST, gastrointestinal stromal tumor; HIF1α, hypoxia inducible factor 1α; lncARSR, lncRNA Activated in RCC with Sunitinib Resistance; MDSCs, myeloid derived suppressor cells; mRCC, metastatic renal cell carcinoma; mTOR, mammalian target of rapamycin; PDGFR, platelet derived growth factor receptor; pNET, progressive pancreatic neuroendocrine tumor; TKI, tyrosine kinase inhibitor; VEGFR, vascular endothelial growth factor receptor.
References
Abdel-Aziz, A. K., Mantawy, E. M., Said, R. S., and Helwa, R. (2016). The tyrosine kinase inhibitor, sunitinib malate, induces cognitive impairment in vivo via dysregulating VEGFR signaling, apoptotic and autophagic machineries. Exp. Neurol. 283, 129–141. doi: 10.1016/j.expneurol.2016.06.004
Abdel-Aziz, A. K., Saad, S., Azab, E., Youssef, S. S., El-sayed, A. M., and El-demerdash, E. (2015). Modulation of imatinib cytotoxicity by selenite in HCT116 colorectal cancer cells. Basic Clin. Pharmacol. Toxicol. 116, 37–46. doi: 10.1111/bcpt.12281
Abdel-Aziz, A. K., Shouman, S., El-demerdash, E., Elgendy, M., and Abdel-naim, A. B. (2014a). Chloroquine as a promising adjuvant chemotherapy together with sunitinib. Sci. Proc. 1, 1–3. doi: 10.14800/sp.384
Abdel-Aziz, A. K., Shouman, S., El-Demerdash, E., Elgendy, M., and Abdel-Naim, A. B. (2014b). Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem. Biol. Interact. 217, 28–40. doi: 10.1016/j.cbi.2014.04.007
Adelaiye, R., Ciamporcero, E., Miles, K. M., Sotomayor, P., Bard, J., Tsompana, M., et al. (2014). Sunitinib dose escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications. Mol. Cancer Ther. 14, 513–522. doi: 10.1158/1535-7163.MCT-14-0208
Aziz, M. A., Serya, R. A. T., Lasheen, D. S., Abdel-Aziz, A. K., Esmat, A., Mansour, A. M., et al. (2016). Discovery of Potent VEGFR-2 inhibitors based on furopyrimidine and thienopyrimidne scaffolds as cancer targeting agents. Sci. Rep. 6:24460. doi: 10.1038/srep24460
Buchbinder, E. I., Sosman, J. A., Lawrence, D. P., and Mcdermott, D. F. (2015). Phase 2 study of sunitinib in patients with metastatic mucosal or acral melanoma. Cancer 121, 4007–4015. doi: 10.1002/cncr.29622
Burchert, A., Wang, Y., Cai, D., von Bubnoff, N., Paschka, P., Müller-Brüsselbach, S., et al. (2005). Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia 19, 1774–1782. doi: 10.1038/sj.leu.2403898
Burstein, H. J., Elias, A. D., Rugo, H. S., Cobleigh, M. A., Wolff, A. C., Eisenberg, P. D., et al. (2008). Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J. Clin. Oncol. 26, 1810–1816. doi: 10.1200/JCO.2007.14.5375
Cella, C. A., Minucci, S., Spada, F., Galdy, S., Elgendy, M., Ravenda, P. S., et al. (2015). Dual inhibition of mTOR pathway and VEGF signalling in neuroendocrine neoplasms: from bench to bedside. Cancer Treat. Rev. 15, 30–39. doi: 10.1016/j.ctrv.2015.06.008
Chu, T. F., Rupnick, M. A., Kerkela, R., Dallabrida, S. M., Zurakowski, D., Nguyen, L., et al. (2007). Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370, 2011–2019. doi: 10.1016/S0140-6736(07)61865-0
Dai, C., Liang, Y., Wang, Y., Tiwari, A. K., Yan, Y., Wang, F., et al. (2009). Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 279, 74–83. doi: 10.1016/j.canlet.2009.01.027
Ding, R., Jin, S., Pabon, K., and Scotto, K. W. (2016). A role for ABCG2 beyond drug transport : regulation of autophagy. Autophagy 12, 737–751. doi: 10.1080/15548627.2016.1155009
Domina, A. M., Vrana, J. A., Gregory, M. A., Hann, S. R., and Craig, R. W. (2004). MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 23, 5301–5315. doi: 10.1038/sj.onc.1207692
Ebos, J. M. L., Lee, C. R., Cruz-munoz, W., Bjarnason, G. A., Christensen, J. G., and Kerbel, R. S. (2009). Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239. doi: 10.1016/j.ccr.2009.01.021
Elgendy, M., Abdel-Aziz, A. K., Renne, S. L., Bornaghi, V., Procopio, G., Colecchia, M., et al. (2016). Dual modulation of MCL-1 and mTOR determines the response to sunitinib. J. Clin. Invest. 127, 23–25. doi: 10.1172/JCI84386
Elgendy, M., Ciro, M., Abdel-Aziz, A. K., Belmonte, G., Dal Zuffo, R., Mercurio, C., et al. (2014). Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat. Commun. 5, 5637. doi: 10.1038/ncomms6637
Feldman, D. R., Turkula, S., Ginsberg, M. S., Ishill, N., Patil, S., Carousso, M., et al. (2010). Phase II trial of sunitinib in patients with relapsed or refractory germ cell tumors. Invest. New Drugs. 28, 523–528. doi: 10.1007/s10637-009-9280-2
Finke, J., Ko, J., Rini, B., Rayman, P., Ireland, J., and Cohen, P. (2011). International immunopharmacology MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy Int. Immunopharmacol. 11, 856–861. doi: 10.1016/j.intimp.2011.01.030
Fu, Y., Kang, H., Zhao, H., Hu, J., Zhang, H., Li, X., et al. (2015). Sunitinib for patients with locally advanced or distantly metastatic dermatofibrosarcoma protuberans but resistant to imatinib. Int. J. Clin. Exp. Med. 8, 8288–8294.
Gao, H., and Deng, L. (2014). Sphingosine kinase-1 activation causes acquired resistance against sunitinib in renal cell carcinoma cells. Cell Biochem. Biophys. 68, 419–425. doi: 10.1007/s12013-013-9723-4
Géczi, L., Keam, B., Maroto, P., Heng, D. Y. C., Schmidinger, M., and Kantoff, P. W. (2015). Cabozantinib vs. everolimus in advanced renal-cell carcinoma. New Engl. J. Med. Engl. J. Med. 373, 1814–1823. doi: 10.1056/NEJMoa1510016
George, S., Merriam, P., Maki, R. G., Abbeele, A. D., Van den Abbeele, A. D., Yap, J. T., et al. (2009). Multicenter phase ii trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J. Clin. Oncol. 27, 3154–3160. doi: 10.1200/JCO.2008.20.9890
Giuliano, S., Cormerais, Y., Dufies, M., Grépin, R., Colosetti, P., Belaid, A., et al. (2015). Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 11, 1891–1904. doi: 10.1080/15548627.2015.1085742
Griffioen, A. W., Mans, L. A., Graaf, A. M. A., and De Nowak-sliwinska, P. (2012). Rapid angiogenesis onset after discontinuation of sunitinib treatment of renal cell carcinoma patients. Clin. Cancer Res. 18, 3961–3971. doi: 10.1158/1078-0432.CCR-12-0002
Grivas, P. D., Daignault, S., Tagawa, S. T., and Nanus, D. M. (2014). Double-blind, randomized, phase 2 trial of maintenance sunitinib vs. placebo after response to chemotherapy in patients with advanced urothelial carcinoma. Cancer 120, 692–701. doi: 10.1002/cncr.28477
Hammers, H. J., Verheul, H. M., Salumbides, B., Sharma, R., and Rudek, M. (2010). Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma : evidence from a xenograft study. Mol. Cancer Ther. 9, 1525–1535. doi: 10.1158/1535-7163.MCT-09-1106
Han, J. Y., Kim, H. Y., Lim, K. Y., Han, J. H., Lee, Y. J., Kwak, M. H., et al. (2013). A phase II study of sunitinib in patients with relapsed or refractory small cell lung cancer. Lung Cancer. 79, 137–142. doi: 10.1016/j.lungcan.2012.09.019
Hjelmeland, A., and Zhang, J. (2016). Metabolic, autophagic, and mitophagic activities in cancer initiation and progression. Biomed. J. 39, 98–106. doi: 10.1016/j.bj.2015.10.002
Ikeda, T., Ishii, K., Saito, Y., Miura, M., Otagiri, A., Kawakami, Y., et al. (2013). Inhibition of autophagy enhances sunitinib-induced cytotoxicity in rat pheochromocytoma PC12 cells. J. Pharmacol. Sci. 121, 67–73. doi: 10.1254/jphs.12158FP
Ikezoe, T., Nishioka, C., and Tasaka, T. (2006). The antitumor effects of sunitinib (formerly SU11248) against a variety of human hematologic malignancies : enhancement of growth inhibition via inhibition of mammalian target of rapamycin signaling. Mol. Cancer Ther. 5, 2522–2530. doi: 10.1158/1535-7163.MCT-06-0071
Kerkela, R., Woulfe, K. C., Durand, J., Vagnozzi, R., Kramer, D., Chu, T. F., et al. (2008). Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin. Transl. Sci. 2, 15–25. doi: 10.1111/j.1752-8062.2008.00090.x
Koeberle, D., Montemurro, M., Samaras, P., Majno, P., Simcock, M., Limacher, A., et al. (2010). Continuous sunitinib treatment in patients with advanced hepatocellular carcinoma: a Swiss Group for Clinical Cancer Research (SAKK) and Swiss Association for the Study of the Liver (SASL) multicenter phase II trial (SAKK 77/06). Oncologist 15, 285–292. doi: 10.1634/theoncologist.2009-0316
Kulikov, A. V., Luchkina, E. A., Gogvadze, V., and Zhivotovsky, B. (2017). Mitophagy : link to cancer development and therapy. Biochem. Biophys. Res. Commun. 482, 432–439. doi: 10.1016/j.bbrc.2016.10.088
Li, X., Tong, L., Ding, J., and Meng, L. (2014). Systematic combination screening reveals synergism between rapamycin and sunitinib against human lung cancer. Cancer Lett. 342, 159–166. doi: 10.1016/j.canlet.2013.08.046
Lin, C. I., Whang, E. E., Lorch, J. H., and Ruan, D. T. (2012). Autophagic activation potentiates the antiproliferative effects of tyrosine kinase inhibitors in medullary thyroid cancer. Surgery 152, 1142–1149. doi: 10.1016/j.surg.2012.08.016
Liu, Y., Liu, J., Chai, K., Tashiro, S., Onodera, S., and Ikejima, T. (2013). Inhibition of c-met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. J. Pharm. Pharmacol. 65, 1622–1642. doi: 10.1111/jphp.12140
Loges, S., Mazzone, M., Hohensinner, P., and Carmeliet, P. (2009). Silencing or fueling metastasis with VEGF inhibitors : antiangiogenesis revisited. Cancer Cell 15, 167–170. doi: 10.1016/j.ccr.2009.02.007
Lopergolo, A., Nicolini, V., Favini, E., Bo, L. D., Tortoreto, M., Cominetti, D., et al. (2014). Synergistic cooperation between sunitinib and cisplatin promotes apoptotic cell death in human medullary thyroid cancer. J. Clin. Endocrin. Metab. 99, 498–509. doi: 10.1210/jc.2013-2574
Maiuri, M. C., Zalckvar, E., Kimchi, A., and Kroemer, G. (2007). Self-eating and self-killing: crosstalk between autophagy and apoptosis. Mol. Cell. Biol. 8, 741–752. doi: 10.1038/nrm2239
Makhov, P. B., Golovine, K., Kutikov, A., Teper, E., Canter, D. J., Simhan, J., et al. (2012). Modulation of Akt/mTOR signaling overcomes sunitinib resistance in renal and prostate cancer cells. Mol. Cancer Ther. 11, 1510–1517. doi: 10.1158/1535-7163.MCT-11-0907
Martinez-outschoorn, U. E., Whitaker-menezes, D., Pavlides, S., Chiavarina, B., Bonuccelli, G., Trimmer, C., et al. (2010). The autophagic tumor stroma model of cancer or “battery-operated tumor growth” A simple solution to the autophagy paradox. Cell Cycle 9, 4297–4306. doi: 10.4161/cc.9.21.13817
Michaelson, M. D., Regan, M. M., Oh, W. K., Kaufman, D. S., Olivier, K., Michaelson, S. Z., et al. (2009). Phase II study of sunitinib in men with advanced prostate cancer. Ann. Oncol. 20, 913–920. doi: 10.1093/annonc/mdp111
Mitchell, N., Fong, P. C., and Broom, R. J. (2015). Clinical experience with sunitinib dose escalation in metastatic renal cell carcinoma. Asia Pac. J. Clin. Oncol. 11, e1–5. doi: 10.1111/ajco.12296
Mizumoto, A., Yamamoto, K., Nakayama, Y., Takara, K., Nakagawa, T., Hirano, T., et al. (2015). Induction of epithelial-mesenchymal transition via activation of epidermal growth factor receptor contributes to sunitinib resistance in human renal cell carcinoma cell lines. J. Pharmacol. Exp. Ther. 355, 152–158. doi: 10.1124/jpet.115.226639
Molina, A. M., Feldman, D. R., Ginsberg, M. S., Kroog, G., Tickoo, S. K., Jia, X., et al. (2013). Phase II trial of sunitinib in patients with metastatic non-clear cell renal cell carcinoma. Invest. New Drugs. 30, 335–340. doi: 10.1007/s10637-010-9491-6
Motzer, R. J., Rini, B. I., Bukowski, R. M., Curti, B. D., George, D. J., Hudes, G. R., et al. (2006). Sunitinib in patients with metastatic renal cell carcinoma. JAMA 295, 2516–2524. doi: 10.1001/jama.295.21.2516
Piscazzi, A., Costantino, E., Maddalena, F., Natalicchio, M. I., Gerardi, A. M. T., Antonetti, R., et al. (2012). Activation of the RAS/RAF/ERK signaling pathway contributes to resistance to sunitinib in thyroid carcinoma cell lines. J. Clin. Endocrinol. Metab. 97, 898–906. doi: 10.1210/jc.2011-3269
Prieto-domínguez, N., Ordóñez, R., Fernández, A., Méndez-blanco, C., Baulies, A., Garcia-ruiz, C., et al. (2016). Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 61, 396–407. doi: 10.1111/jpi.12358
Rna, C. E., Qu, L., Ding, J., Chen, C., Sun, Y., Wang, H., et al. (2016). Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a article exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous, RNA. Cancer Cell 29, 653–668. doi: 10.1016/j.ccell.2016.03.004
Rovithi, M., Haas, R. R., De Honeywell, R. J., Poel, D., Peters, G. J., and Griffioen, A. W. (2016). Alternative scheduling of pulsatile, high dose sunitinib efficiently suppresses tumor growth. J. Exp. Clin. Cancer Res. 35, 1–11. doi: 10.1186/s13046-016-0411-2
Santoni, M., Amantini, C., Morelli, M. B., Liberati, S., Farfariello, V., Nabissi, M., et al. (2013). Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br. J. Cancer 109, 1040–1050. doi: 10.1038/bjc.2013.420
Sato, H., Siddig, S., Uzu, M., Suzuki, S., and Nomura, Y. (2015). Elacridar enhances the cytotoxic effects of sunitinib and prevents multidrug resistance in renal carcinoma cells. Eur. J. Pharmacol. 746, 258–266. doi: 10.1016/j.ejphar.2014.11.021
Schneider, B. J., Gadgeel, S. M., Ramnath, N., Wozniak, A. J., Dy, G. K., and Daignault, S. K. G. (2011). Phase II trial of sunitinib maintenance therapy after platinum-based chemotherapy in patients with extensive-stage small cell lung cancer. J. Thorac. Oncol. 6, 1117–1120. doi: 10.1097/JTO.0b013e31821529c3
Shin, H., Cho, C., Kim, T., and Park, J. (2010). Sunitinib deregulates tumor adaptation to hypoxia by inhibiting HIF-1 a synthesis in HT-29 colon cancer cells. Biochem. Biophys. Res. Commun. 398, 205–211. doi: 10.1016/j.bbrc.2010.06.060
Shojaei, F., Lee, J. H., Simmons, B. H., Wong, A., Esparza, C. O., Plumlee, P. A., et al. (2010). HGF / c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 70, 10090–10100. doi: 10.1158/0008-5472.CAN-10-0489
Shukla, S., Robey, R. W., Bates, S. E., and Ambudkar, S. V. (2009). Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-Binding Cassette (ABC) Transporters P-Glycoprotein (ABCB1) and ABCG2 ABSTRACT. Drug Metab. Dispos. 37, 359–365. doi: 10.1124/dmd.108.024612
Stacchiotti, S., Negri, T., Libertini, M., Palassini, E., Marrari, A., De Troia, B., et al. (2012). Sunitinib malate in solitary fibrous tumor (SFT). Ann. Oncol. 23, 3171–3179. doi: 10.1093/annonc/mds143
Teng, C. L. J., Yu, C. T. R., Hwang, W. L., Tsai, J. R., Liu, H. C., Hwang, G. Y., et al. (2013). Effector mechanisms of sunitinib-induced G1 cell cycle arrest, differentiation, and apoptosis in human acute myeloid leukaemia HL60 and KG-1 cells. Ann. Hematol. 92, 301–313. doi: 10.1007/s00277-012-1627-7
Wetmore, C., Daryani, V. M., Billups, C. A., Boyett, J. M., Leary, S., Tanos, R., et al. (2016). Phase II evaluation of sunitinib in the treatment of recurrent or refractory high- grade glioma or ependymoma in children: a children's Oncology Group Study ACNS1021. Cancer Med. 5, 1416–1424. doi: 10.1002/cam4.713
Wu, C., Mikhail, S., Wei, L., Timmers, C., Tahiri, S., Neal, A., et al. (2015). A phase II and pharmacodynamic study of sunitinib in relapsed/refractory oesophageal and gastro-oesophageal cancers. Br. J. Cancer. 113, 220–225. doi: 10.1038/bjc.2015.197
Wu, D.-W., Chen, C.-Y., Chu, C.-L., and Lee, H. (2016). Paxillin confers resistance to tyrosine kinase inhibitors in EGFR-mutant lung cancers via modulating BIM and Mcl-1 protein stability. Oncogene 35, 621–630. doi: 10.1038/onc.2015.120
Yan, S., Liu, L., Ren, F., Gao, Q., Xu, S., Hou, B., et al. (2017). Sunitinib induces genomic instability of renal carcinoma cells through affecting the interaction of LC3-II and PARP-1. Cell Death Dis. 8:e2988. doi: 10.1038/cddis.2017.387
Yang, F., Jove, V., Xin, H., Hedvat, M., Van Meter, T. E., and Yu, H. (2010). Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways. Mol. Cancer Res. 8, 35–45. doi: 10.1158/1541-7786.MCR-09-0220
Yang, Z. J., Chee, C. E., Huang, S., and Sinicrope, F. A. (2011). Autophagy modulation for cancer therapy. Cancer Biol. Ther. 11, 169–176. doi: 10.4161/cbt.11.2.14663
Zhao, Y., Xue, T., Yang, X., Zhu, H., Ding, X., Lou, L., et al. (2010). Autophagy plays an important role in Sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol. Appl. Pharmacol. 248, 20–27. doi: 10.1016/j.taap.2010.07.007
Keywords: autophagy, cancer, Mcl-1, mTOR, resistance, Sunitinib
Citation: Abdel-Aziz AK, Abdel-Naim AB, Shouman S, Minucci S and Elgendy M (2017) From Resistance to Sensitivity: Insights and Implications of Biphasic Modulation of Autophagy by Sunitinib. Front. Pharmacol. 8:718. doi: 10.3389/fphar.2017.00718
Received: 20 June 2017; Accepted: 25 September 2017;
Published: 10 October 2017.
Edited by:
Amit K. Tiwari, University of Toledo, United StatesReviewed by:
Nor Eddine Sounni, University of Liège, BelgiumKetan Shirish Patil, St. John's University, Taiwan
Copyright © 2017 Abdel-Aziz, Abdel-Naim, Shouman, Minucci and Elgendy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saverio Minucci, c2F2ZXJpby5taW51Y2NpQGllby5pdA==
Mohamed Elgendy, bW9oYW1lZC5lbGdlbmR5QHVuaXZpZS5hYy5hdA==