Abstract
Cannabinoids include the active constituents of Cannabis or are molecules that mimic the structure and/or function of these Cannabis-derived molecules. Cannabinoids produce many of their cellular and organ system effects by interacting with the well-characterized CB1 and CB2 receptors. However, it has become clear that not all effects of cannabinoid drugs are attributable to their interaction with CB1 and CB2 receptors. Evidence now demonstrates that cannabinoid agents produce effects by modulating activity of the entire array of cellular macromolecules targeted by other drug classes, including: other receptor types; ion channels; transporters; enzymes, and protein- and non-protein cellular structures. This review summarizes evidence for these interactions in the CNS and in cancer, and is organized according to the cellular targets involved. The CNS represents a well-studied area and cancer is emerging in terms of understanding mechanisms by which cannabinoids modulate their activity. Considering the CNS and cancer together allow identification of non-cannabinoid receptor targets that are shared and divergent in both systems. This comparative approach allows the identified targets to be compared and contrasted, suggesting potential new areas of investigation. It also provides insight into the diverse sources of efficacy employed by this interesting class of drugs. Obtaining a comprehensive understanding of the diverse mechanisms of cannabinoid action may lead to the design and development of therapeutic agents with greater efficacy and specificity for their cellular targets.
Introduction
Cannabinoids are a broad and diverse class of drugs that are structurally- or functionally-related to those isolated from Cannabis (i.e., are “Cannabis-like”). Several structural classes of cannabinoid drugs have been identified, including: phytocannabinoids (related to those derived from plant material); endogenously-produced cannabinoids; and related eicosanoids that regulate vertebrate endocannabinoid signaling systems; synthetic and other types of cannabinoids (many developed seeking modulators of other signaling systems, but found to interact with the classic CB1 and/or CB2 receptors). Classical cannabinoid agonists (structures shown in Table 1) bind to and activate cannabinoid receptors 1 and 2 (CB1, CB2) that modulate signal transduction cascades to produce various physiological and pathological outcomes. The actions of cannabinoids are also regulated by the endocannabinoid system (ECS) which includes enzymes involved in synthesis, uptake and degradation of endogenous cannabinoid ligands, and the CB1 and CB2 receptors. Cannabinoid pharmacology is an active field, and new examples of cannabinoid drugs are identified regularly (Shevyrin et al., 2016) with the primary goal of discovering novel therapeutics.
Table 1
| Endocannabinoids (ECs) | |||
|---|---|---|---|
| Arachidonic acid-derived ECs | |||
AEA | Noladin Ether (2-AG ether) | 2-AG | Virodhamine |
NAGly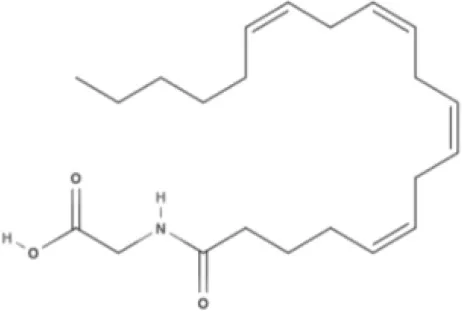 | 2A-LPA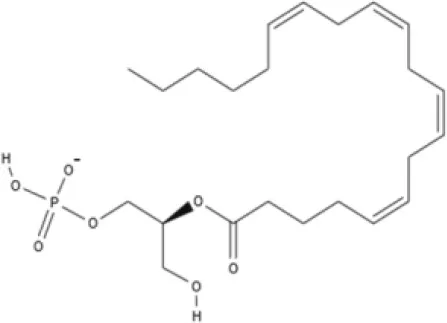 | Oleic acid-derived EC Oleoylethanolamide (OEA) | Palmitic acid-derived EC Palmitoylethanolamide (PEA) |
Other EC Lysophosphatidylinositol (LPI)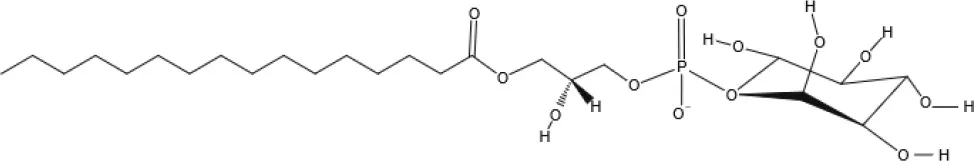 | |||
| Phytocannabinoids | |||
| Tricyclic phytocannabinoids | |||
THC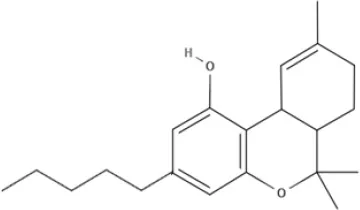 | THCA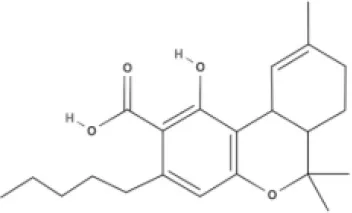 | THCV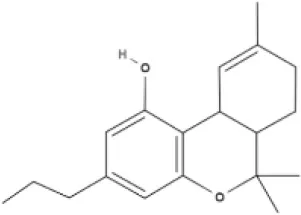 | THCVA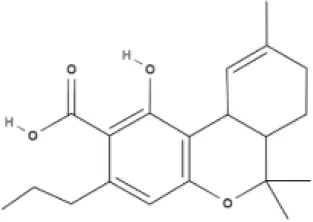 |
CBN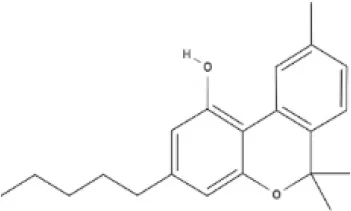 | |||
| Bicyclic phytocannabinoids | |||
CBD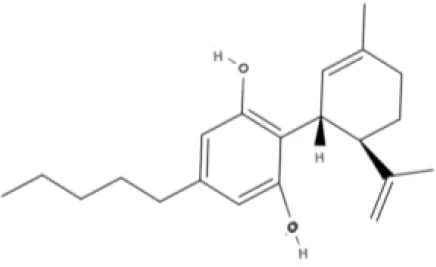 | CBDA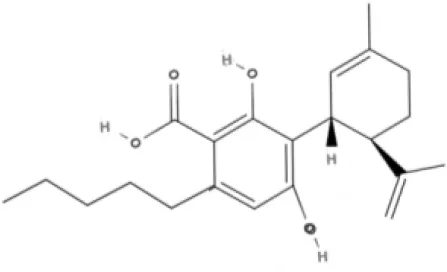 | CBDV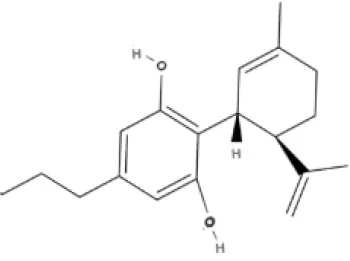 | CBDVA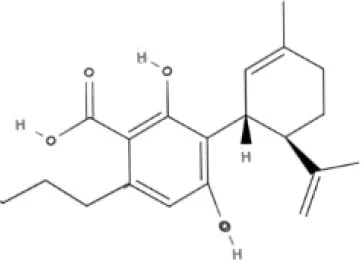 |
AbCBD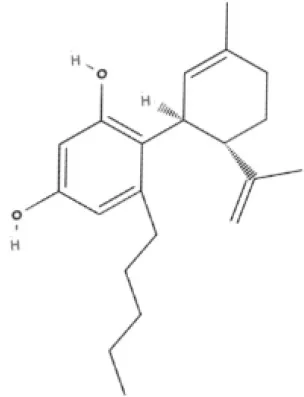 | |||
| Other phytocannabinoids | |||
CBG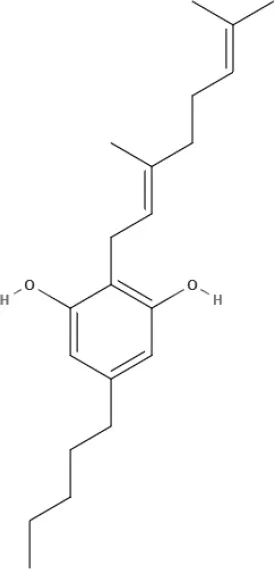 | CBGV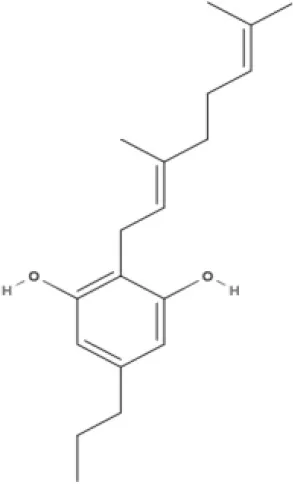 | CBGA | CBC |
| Synthetic cannabinoids | |||
| Dibenzopyran derivatives (THC related compounds) | Cyclohexylphenol derivatives | ||
HU-210 | Ajulemic acid | CP55940 | |
| Aminoalkylindoles | |||
JWH-015 | WIN | AM630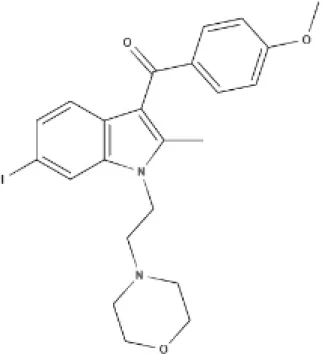 | |
| Synthetic analogs of ECs | |||
MA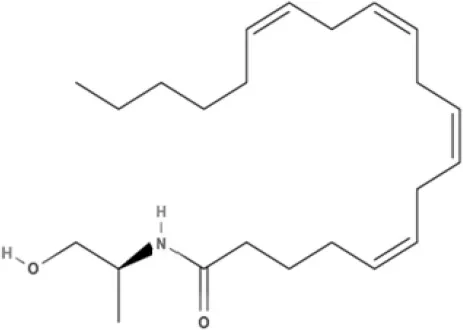 | ACPA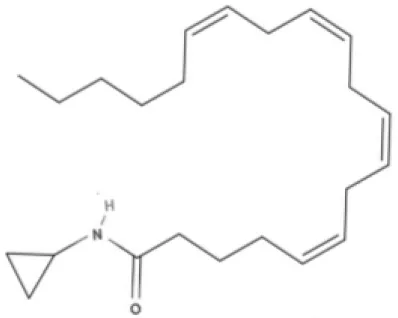 | ||
| Other synthetic cannabinoids | |||
O-1918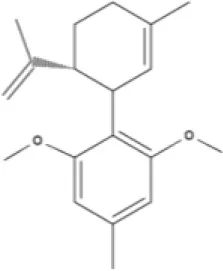 | O-1602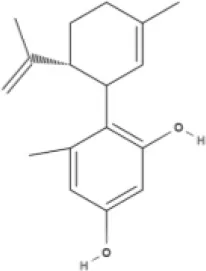 | HU-308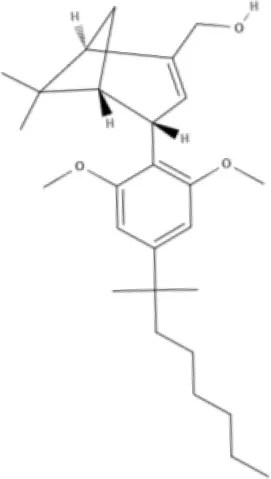 | LY320135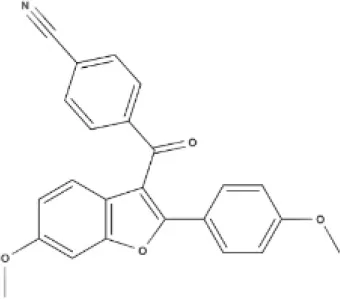 |
SR141716A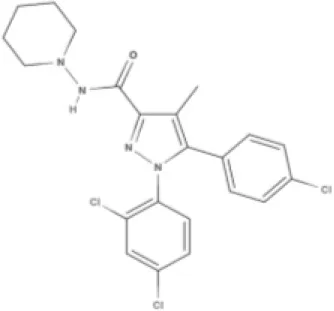 | AM251 | ||
Structures of cannabinoids reviewed, summarized by class.
AEA, arachidonoyl ethanolamide or anandamide; 2-AG, 2-arachidonoyl glycerol; OEA, oleoylethanolamine; PEA, palmitoylethanolamide; NAGly, N-arachidonoyl glycine; LPI, lysophosphatidylinositol; 2A-LPA, 2-arachidonoyl lysophosphatidic acid; THC, Δ9-tetrahydrocannabinol; THCA, THC acid; THCVA, tetrahydrocannabivarin acid; CBD, cannabidiol; CBDA, cannabidiol acid; CBDV, cannabidivarin; CBDVA, cannabidivarin acid; CBN, cannabinol; CBC, cannabichromene; CBG, cannabigerol; CBGV, cannabigerovarin; CBGA, cannabigerol acid; AbCBD, abnormal cannabidiol; WIN, WIN55212-2; MA, methanandamide; ACPA, arachidonylcyclopropylamide.
It is now clear that, in addition to the classic CB1 and CB2 receptors, cannabinoid-related agents interact with a spectrum of macromolecular targets, including: other receptors; ion channels; transporters; enzymes and; protein- and non-protein cellular structures. Evidence for cannabinoid modulation of these targets has already been the subject of excellent reviews (Kreitzer and Stella, 2009; De Petrocellis and Di Marzo, 2010; Pertwee, 2010). The purpose of this review is to examine current studies that focus on how off-receptor targets mediate the effects of classical cannabinoids in the CNS and in cancer. Cannabinoids that interact with cannabinoid receptors in an allosteric manner (allosteric modulators) are outside of the scope of this review but have been recently reviewed elsewhere (Busquets Garcia et al., 2016; Janero and Thakur, 2016; Khurana et al., 2017; Nguyen et al., 2017).
Active constituents of Cannabis produce CNS effects, and the endocannabinoid signaling system was discovered in CNS, therefore cannabinoid effects on neuronal activity have been more thoroughly and efficiently studied than in more peripheral processes like cancer. Although CB1 and/or CB2 receptors are known to be expressed in some cancers (Sarfaraz et al., 2008), their levels vary and may be either up- or down regulated (Bifulco et al., 2001; Begum et al., 2005). Variable receptor expression suggests that cannabinoid effects in cancer are more likely to involve non-receptor mechanisms than in CNS, making the system a promising area to examine for novel targets. Thus, focusing on both CNS and cancer will allow established central and emerging peripheral effects that are not mediated by CB1 or CB2 to be compared and contrasted, in order to appreciate common and divergent mechanisms that are involved. This comparison may also reveal important cannabinoid targets in the CNS and cancer that can form a basis for inquiry in other organ systems. In addition, awareness of non-cannabinoid receptor targets of cannabinoids may lead to the development of drugs with greater efficacy and specificity for their targets.
Roles of cannabinoids and the cannabinoid receptors in the CNS
CB1 receptors are abundant in CNS and expressed in brain at consistently high densities across vertebrate species within which levels have been measured (~2,000 fmol/mg protein, e.g., Pertwee, 1997; Soderstrom et al., 2000; Soderstrom and Johnson, 2001). Although expressed at much lower levels than CB1, CB2 receptors also play clear roles in reward-related CNS activity (Zhang et al., 2014) and immune responses (Cabral et al., 2008). High-level CB1 expression (approaching that of amino acid transmitter receptors) may have contributed to delayed appreciation of central cannabinoid effects produced following interaction with other cellular macromolecules. This is because the magnitude of non-receptor-mediated effects may be small relative to those that follow activation of the more abundant CB1 receptors. This is of therapeutic importance as when using drugs to treat disease often “less is more” and more modest indirect effects may be adequate to mitigate disease processes while avoiding toxicity. Thus, cannabinoid-related agents with targets independent of CB1 and CB2 receptors are currently being effectively employed or evaluated for management of a variety of CNS disorders (McPartland et al., 2014). The non-CB1/CB2 CNS-relevant targets reviewed here, along with the cannabinoid ligands that interact with them, are summarized in Table 2.
Table 2
| Target | Target type | Ligand | Ligand type | Activity | Potency (EC50, nM) | System | Assay | References |
|---|---|---|---|---|---|---|---|---|
| 5-HT1A | Receptor | CBD | Phyto | Agon | NA | Rat BNST | Anxiolysis | Gomes et al., 2011 |
| 5-HT1A | Receptor | CBD | Phyto | Agon | ~3,000 | LN 231 | Cell viab. | Ward et al., 2014 |
| 5-HT1A | Receptor | CBD | Phyto | Agon | NA | CHO | Cyclase inhib. | Russo et al., 2005 |
| 5-HT1A | Receptor | CBD | Phyto | Agon | ~1 μg/kg | Rat | Gaping | Rock et al., 2014 |
| 5-HT1A | Receptor | CBD | Phyto | Agon | NA | CHO | GTPγS | Russo et al., 2005 |
| 5-HT1A | Receptor | CBD | Phyto | Agon | NA | Mouse | Panic | Twardowschy et al., 2013 |
| 5-HT1A | Receptor | CBD | Phyto | Agon | NA | Rat MFB | Plus maze | Fogaça et al., 2014 |
| 5-HT1A | Receptor | CBD | Phyto | Antag | NA | Rat | ICSS | Katsidoni et al., 2013 |
| 5-HT1A | Receptor | CBD | Phyto | Ligand | NA | CHO | Lig. displ. | Russo et al., 2005 |
| 5-HT3 | Receptor | AEA | Endo | Antag | 190 | Rat NDG | Cat. current | Fan, 1995 |
| 5-HT3 | Receptor | AEA | Endo | NCAnt | 129 | Rat NDG | Cat. current | Barann et al., 2002 |
| 5-HT3 | Receptor | CP55940 | Syn | Antag | 94 | Rat NDG | Cat. current | Fan, 1995 |
| 5-HT3 | Receptor | CP55940 | Syn | NCAnt | 648 | Rat NDG | Cat. current | Barann et al., 2002 |
| 5-HT3 | Receptor | JWH-015 | Syn | NCAnt | 147 | Rat NDG | Cat. current | Barann et al., 2002 |
| 5-HT3 | Receptor | LY320135 | Syn | NCAnt | 523 | Rat NDG | Cat. current | Barann et al., 2002 |
| 5-HT3 | Receptor | THC | Phyto | NCAnt | 38.4 | Rat NDG | Cat. current | Barann et al., 2002 |
| 5-HT3 | Receptor | WIN | Syn | Antag | 310 | Rat NDG | Cat. current | Fan, 1995 |
| 5-HT3 | Receptor | WIN | Syn | NCAnt | 104 | Rat NDG | Cat. current | Barann et al., 2002 |
| A2A | Receptor | CBD | Phyto | Agon | NA | EOC-20 | Cell prolif. | Carrier et al., 2006 |
| AMPA | Receptor | AEA | Endo | Antag | 160–240 | X. oocytes | Cat. current | Akinshola et al., 1999 |
| ANA trans | Transporter | CBD | Phyto | Antag | 25,300 | leukemia Cx | ANA uptake | De Petrocellis et al., 2011 |
| ANA trans | Transporter | CBG | Phyto | Antag | 11,300 | leukemia Cx | ANA uptake | De Petrocellis et al., 2011 |
| DAGLα | Enzyme | THCA | Phyto | Antag | 27,300 | COS-7 | 2-AG met | De Petrocellis et al., 2011 |
| δOP | Receptor | SR141716A | Syn | NCAnt | NA | CB1 KO | GTPγS | Zádor et al., 2014 |
| ENT1 | Transporter | CBD | Phyto | Antag | ~250 | EOC-20 | Nucleoside | Carrier et al., 2006 |
| ENT1 | Transporter | THC | Phyto | Antag | ~50 | EOC-20 | Nucleoside | Carrier et al., 2006 |
| FAAH | Enzyme | CBD | Phyto | Antag | 15,200 | Rat brain | ANA met | De Petrocellis et al., 2011 |
| GABAA B2 | Receptor | 2-AG | Endo | Agon | 1,100 | X. oocytes | Cl− current | Sigel et al., 2011 |
| GABAA B2 | Receptor | 2-AG | Endo | Antag | NA | HEK293 | Cl− current | Golovko et al., 2014 |
| GABAA B2 | Receptor | CP55940 | Syn | Antag | NA | HEK293 | Cl− current | Golovko et al., 2014 |
| Glycine | Receptor | 2-AG | Endo | Antag | NA | HCX neurons | Cl− current | Lozovaya et al., 2005 |
| Glycine | Receptor | AEA | Endo | Agon | 230–318 | Rat VTA | Cl− current | Hejazi et al., 2006 |
| Glycine | Receptor | AEA | Endo | Antag | 300 | HCX neurons | Cl− current | Lozovaya et al., 2005 |
| Glycine | Receptor | THC | Phyto | Agon | 115 | Rat VTA | Cl− current | Hejazi et al., 2006 |
| Glycine | Receptor | WIN | Syn | Antag | 300 | HCX neurons | Cl− current | Lozovaya et al., 2005 |
| Glycine α1 | Receptor | AEA | Endo | Antag | 38 | HEK293 | Cl− current | Yang et al., 2008 |
| Glycine α2 | Receptor | HU210 | Syn | Antag | 90 | HEK293 | Cl− current | Yang et al., 2008 |
| Glycine α2 | Receptor | HU308 | Syn | Antag | 1,130 | HEK293 | Cl− current | Yang et al., 2008 |
| Glycine α2 | Receptor | NAGly | Endo | Antag | 3,030 | HEK293 | Cl− current | Yang et al., 2008 |
| Glycine α2 | Receptor | WIN | Syn | Antag | 220 | HEK293 | Cl− current | Yang et al., 2008 |
| GLYT1a | Transporter | AEA | Endo | Agon | 30,000 | X. oocytes | Gly trans | Pearlman et al., 2003 |
| GPR119 | Receptor | OEA | Endo | Agon | 3,200 | Yeast | LacZ | Overton et al., 2006 |
| GPR119 | Receptor | PEA | Endo | Agon | >10,000 | Yeast | LacZ | Overton et al., 2006 |
| GPR18 | Receptor | AbCBD | Syn | Agon | ~0.3 μg/kg | RVLM | BP | Penumarti and Abdel-Rahman, 2014b |
| GPR18 | Receptor | AbCBD | Syn | Agon | 836 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR18 | Receptor | ACPA | Syn | Agon | 1,350 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR18 | Receptor | AM251 | Syn | Antag | 9,640 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR18 | Receptor | AEA | Endo | Agon | 383 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR18 | Receptor | CBD | Phyto | Antag | 5,110 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR18 | Receptor | NAGly | Endo | Agon | NA | CHO | Ca++ | Kohno et al., 2006 |
| GPR18 | Receptor | NAGly | Endo | Agon | ~30 | CHO | Cyclase inhib. | Kohno et al., 2006 |
| GPR18 | Receptor | NAGly | Endo | Agon | 45 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR18 | Receptor | O-1602 | Syn | Agon | 65 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR18 | Receptor | O-1918 | Syn | Antag | ~0.4 μg/kg | RVLM | BP | Penumarti and Abdel-Rahman, 2014b |
| GPR18 | Receptor | THC | Phyto | Agon | 960 | HEC-1B | MAPK act | McHugh et al., 2012 |
| GPR35 | Receptor | 2A-LPA | Endo | Agon | ~100 | HEK293 | Ca++ | Oka et al., 2010 |
| GPR55 | Receptor | 2-AG | Endo | Agon | 3 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | AbCBD | Phyto | Agon | 2,500 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | AM251 | Syn | Agon | 39 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | AEA | Endo | Agon | 18 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | CBD | Phyto | Antag | NA | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | CP55940 | Syn | Agon | 5 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | HU210 | Syn | Agon | 26 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | LPI | Endo | Agon | 200 | HEK293 | ERK | Oka et al., 2007 |
| GPR55 | Receptor | Noladin Ether | Endo | Agon | 10 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | O-1602 | Syn | Agon | 13 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | OEA | Endo | Agon | 440 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | PEA | Endo | Agon | 4 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | THC | Phyto | Agon | 8 | HEK293 | GTPγS | Ryberg et al., 2007 |
| GPR55 | Receptor | Virodhamine | Endo | Agon | 12 | HEK293 | GTPγS | Ryberg et al., 2007 |
| κOP | Receptor | SR141716A | Syn | InvAg | NA | CB1 KO | GTPγS | Zádor et al., 2015 |
| MAGL | Enzyme | CBG | Phyto | Antag | 95,700 | COS | 2-AG met | De Petrocellis et al., 2011 |
| MAGL | Enzyme | THCA | Phyto | Antag | 46,000 | COS | 2-AG met | De Petrocellis et al., 2011 |
| μOP | Receptor | Noladin Ether | Endo | NCAnt | NA | CB1 KO | GTPγS | Zádor et al., 2012 |
| μOP | Receptor | SR141716A | Syn | Antag | NA | CB1 KO | GTPγS | Cinar and Szücs, 2009 |
| μOP | Receptor | THC | Phyto | Agon | NA | Rat in vivo | Hot plate | Tulunay et al., 1981 |
| Na+ channel | Ion Channel | Ajulemic acid | Syn | Antag | ~3,000 | HEK293 | Na+ current | Foadi et al., 2014 |
| nAChR a4β2 | Receptor | AEA | Endo | Antag | NA | SH-EP1 | Na+ current | Spivak et al., 2007 |
| nAChR α7 | Receptor | 2-AG | Endo | Antag | 118 | X. oocytes | Na+ current | Oz et al., 2004 |
| nAChR α7 | Receptor | AEA | Endo | Antag | 30 | X. oocytes | Na+ current | Oz et al., 2003 |
| nAChR α7 | Receptor | CBD | Phyto | Antag | 11,300 | X. oocytes | Na+ current | Mahgoub et al., 2013 |
| nAChR α7 | Receptor | MA | Syn | Antag | ~1 μmol/kg | Anesth. Rat | HR | Baranowska et al., 2008 |
| NCX1 | Transporter | AEA | Endo | Antag | 4,700 | Rat myocytes | Na+/Ca++ | Kury et al., 2014 |
| NMDA | Receptor | AEA | Endo | Agon | NA | Rat ICV | BP | Malinowska et al., 2010 |
| NMDA | Receptor | AEA | Endo | Agon | NA | Rat HCX | Ca++ current | Yang et al., 2014 |
| TRPA1 | Ion Channel | AM251 | Syn | Agon | 10,000 | CHO | [Ca++]i | Patil et al., 2011 |
| TRPA1 | Ion Channel | CBD | Phyto | Agon | 110 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPA1 | Ion Channel | CBG | Phyto | Agon | 700 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPA1 | Ion Channel | THC | Phyto | Agon | 230 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPM8 | Ion Channel | CBD | Phyto | Antag | 60 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPM8 | Ion Channel | CBG | Phyto | Antag | 160 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPM8 | Ion Channel | THC | Phyto | Antag | 160 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPV1 | Ion Channel | AM251 | Syn | Agon | 10,000 | CHO | [Ca++]i | Patil et al., 2011 |
| TRPV1 | Ion Channel | AM630 | Syn | Agon | 10,000 | CHO | [Ca++]i | Patil et al., 2011 |
| TRPV1 | Ion Channel | CBD | Phyto | Agon | 1,000 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPV1 | Ion Channel | CBG | Phyto | Agon | 1,300 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPV2 | Ion Channel | CBD | Phyto | Agon | 1,250 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
| TRPV2 | Ion Channel | THC | Phyto | Agon | 650 | HEK293 | [Ca++]i | De Petrocellis et al., 2011 |
Summary of CNS-relevant non-cannabinoid receptor targets.
ICSS, intracranial self stimulation; HR, heart rate; BP, blood pressure; MAPK act, MAPK activation; Cat. current, Cation current; AEA, arachidonoyl ethanolamide or anandamide; MA, methanandamide; CBD, cannabidiol; CBG, cannabigerol; Agon, agonist; NCAnt, non-competitive antagonist; Antag, antagonist; NDG, nodose ganglion; Cx, culture; Lig. displ., displacement of ligand binding; MFB, medial forebran bundle.
Effects of cannabinoids and the cannabinoid receptors on cancer
Both CB1 and CB2 receptors, and their endogenous ligands, are expressed in a variety of peripheral organs, including; the GI tract, liver, bone, reproductive system, skin, and the immune system. Peripheral endocannabinoid signaling also regulates many aspects of human pathophysiology, including cancer. Cannabinoid agonists are used as palliative therapy for chemotherapy-induced nausea and vomiting, and they may also be beneficial in the treatment of cancer-related pain. Evidence from both in vitro and in vivo studies established the efficacy of these compounds in reducing tumor growth and proliferation (Ladin et al., 2016). Much of this efficacy is attributable to interaction with targets other than the classic CB1/CB2 receptors. Therefore, we focus on how off-receptor targets mediate cannabinoid effects on cancer. The cancer-related off-receptor targets reviewed here, and the cannabinoid ligands that interact with them, are summarized in Table 3.
Table 3
| Cancer type | Cell line | Drug | Target | Activity | Cellular response | Assay | Reference |
|---|---|---|---|---|---|---|---|
| Colon and colorectal cancer | HT29 | AEA | FAAH | Substrate | Reduced cell death | Adherent cell count | Patsos et al., 2005 |
| HCA7 | AEA | COX-2 | Substrate | Cell death | Adherent cell count | Patsos et al., 2005 | |
| HCT116 Caco-2 | CBG | TRPM8 | Antagonist | Apoptosis | • Caspase 3/7 activity • DNA fragmentation | Borrelli et al., 2014 | |
| SW480 HT29 | WIN | Phosphatase | Increased expression, Activation | Apoptosis | • PARP cleavage • TUNEL | Sreevalsan and Safe, 2013 | |
| SW480 | WIN | Phosphatase | Increased expression | Reduced proliferation, Apoptosis | • Cell number • PARP cleavage • Caspase 3 | Sreevalsan et al., 2011 | |
| Brain tumor: | |||||||
| Glioma | Ge227 Ge258 U87 U251 | AEA | TRPV1 | Agonist | Apoptosis | DNA fragmentation | Contassot et al., 2004b |
| U87 | CBD | TRPV2 | Agonist, Increased expression | Increased chemotherapeutic sensitivity, Inhibition of cell migration | • MTT (cell viability) • Chemotaxis (cell migration) | Nabissi et al., 2013 | |
| H4 | MA | COX-2 | Increased expression | Apoptosis | • Caspase • PARP cleavage | Eichele et al., 2006 | |
| GSC patient derived | CBD | TRPV2 | Agonist, Increased expression | Increased differentiation, Autophagy, Reduced proliferation | • Flow (differentiation) • LC3I, LC3II (autophagy) • MTT (proliferation) | Nabissi et al., 2015 | |
| Neuroblastoma | N1EE-115 | AEA | FAAH | Substrate | Reduced cell death | • MTT | Hamtiaux et al., 2011 |
| Prostate | LNCaP | WIN | Phosphatase | Increased expression | Reduced proliferation, Apoptosis | • Cell number • PARP cleavage • Caspase 3 | Sreevalsan et al., 2011 |
| Non-melanoma skin cancer | JWF2 | AEA | COX-2 | Substrate | Apoptosis | • PARP cleavage • Caspase 3 | Kuc et al., 2012 |
| JWF2 | AEA | FAAH | Substrate | Reduced apoptosis | • PARP cleavage • Caspase 3 • TUNEL | Kuc et al., 2012 | |
| Lung cancer | A549 H460 | CBD | PPAR⋎ COX-2 | Activation of PPAR⋎ & COX-2, Increased COX-2 expression | Apoptosis, Reduced tumor growth (xenograft) | DNA fragmentation | Ramer et al., 2013 |
| Murine lung cancer | L1C2 | MA | COX-2 | Expression and activity | Increased tumor growth | Tumor volume | Gardner et al., 2003 |
| HeLa | MA | PPAR⋎ COX-2 | Activation of PPAR⋎ & COX-2, Increased COX-2 expression | Apoptosis | DNA fragmentation | Eichele et al., 2009 | |
| Cervical cancer | HeLa Caski CC299 | AEA | TRPV1 | Agonist | Apoptosis | • subG0/G1 • Caspase 7 cleavage | Contassot et al., 2004a |
| Cholangiocarcinoma | Mc-ChA-1 | AEA | GPR55 | Activation | Apoptosis | Annexin V | DeMorrow et al., 2007 |
| Multiple myeloma | RPMI, U266 | CBD | TRPV2 | Agonist | Necrosis, Cell death, Increased therapeutic sensitivity | • Propidium iodide (necrosis) • MTT (cell death) | Morelli et al., 2014 |
Summary of cancer-relevant non-cannabinoid receptor targets.
AEA, arachidonoyl ethanolamide or anandamide; FAAH, fatty acid amide hydrolase; CBG, cannabigerol; TRPM8, transient receptor potential cation channel subfamily M member 8; WIN, WIN55212-2; CBD, cannabidiol; MA, methanandamide; PPAR⋎, peroxisome proliferator-activated receptor gamma; COX-2, cyclooxygenase-2; TRPV1, TRPV2, transient receptor potential cation channel subfamily V members 1 and 2; GPR55, G protein-coupled receptor 55; MTT, 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide; PARP, Poly (ADP-ribose) polymerase; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Non-CB1/CB2 receptor targets
Deorphanized G-protein-coupled receptors (GPCRs) in CNS and cancer
Four former orphan GPCRs (GPR55, GPR18, GPR119, and GPR35) have now been clearly established to transduce effects of a subset of both naturally-occurring and synthetic cannabinoid compounds. The available data demonstrate that cannabinoids directly activate these four GPCRs in the CNS, and they also activate GPR55 in cancer.
GPR55 in CNS
The most well-characterized of the deorphanized GPCRs, GPR55 (Ryberg et al., 2007), is expressed most highly in brain, adrenal gland, and digestive tract (although at reportedly low levels relative to that of CB1). When expressed in HEK293 cells, specific binding of the synthetic CB1 agonist CP55940 is observed, but not of the CB1/CB2 synthetic agonist WIN55212-2. Interestingly, a very small amount of specific GPR55 binding was observed using 50 nM of the CB1-selective antagonist/inverse agonist SR141716A (SR).
Using the same HEK293 cell line for GTPγS functional assays, the endogenous agonist 2-arachidonylglyerol (2-AG) potently activates GPR55 (EC50 = 3 nM), although virodhamine (EC50 = 12 nM) is a more complete agonist with about 50% greater efficacy. Notably, in a separate assay of intracellular calcium release, 2-AG and virodhamine showed no GPR55 agonism, suggesting this receptor is subject to agonist-dependent functional selectivity (Lauckner et al., 2008). Other potent agonists discovered in this initial screen of GPR55 efficacy include; palmitoylethanolamide (PEA), CP55940, Δ9-tetrahydrocannabinol (THC), noladin ether, and anandamide with EC50s of 4, 5, 8, 10, and 18 nM respectively, and efficacies similar to that of 2-AG. Interestingly, the non-psychoactive phytocannabinoid cannabidiol (CBD) was found to antagonize GPR55 activity at physiologically relevant concentrations.
GPR55 primarily couples through the relatively obscure Gα13. This interesting G-protein activates a cascade involving RhoA that, among other effects, ultimately alters actin polymerization and stability, implicating GPR55 signaling in processes related to neuronal morphology (Worzfeld et al., 2008). This potential role is further supported by effects to promote neurite retraction (Obara et al., 2011). More recently, evidence in transfected HEK293 cells indicates that signaling through Gq also occurs (Lauckner et al., 2008).
In ERK activation assays with GPR55-expressing HEK293 cells, lysophosphatidylinositol (LPI) is implicated as an important endogenous agonist (Oka et al., 2007). Notably, 2-AG and virodhamine were not effective in stimulating ERK in transfected HEK293 cells, possibly indicating ligand-dependent functional selectivity for different signal transduction pathways. LPI was also found to dose-dependently increase intracellular calcium in these cells with an EC50 ~ 1 μM, an effect possibly related to GPR55 activation of Gq. System-dependent efficacy of GPR55 signaling may also involve interaction with other receptors including CB1. GPR55 and CB1 have been shown to heterodimerize within rat and macaque striatum, possibly related to a role in motor behaviors. When co-expressed in HEK293 cells, CB1 and GPR55 antagonize respective agonist effects to promote ERK1/2 activation (Martínez-Pinilla et al., 2014).
The physiological significance of GPR55 expression in brain remains an open question; although, some evidence indicates a role in controlling ingestive behaviors (reviewed by Liu et al., 2015). A role in promoting appetite makes sense as it has been reported that GPR55 regulates peripheral metabolism and energy homoeostasis (reviewed by Simcocks et al., 2014). Deletion of GPR55 in mice has subtle effects on motor coordination, but it has shown no significant effect on several learning and memory tests, in contrast to the role of CB1 signaling, and despite dense GPR55 expression in hippocampus, striatum, and cortex (Wu et al., 2013). Spinal cord expression of GPR55 is upregulated in a chronic constriction injury model of neuropathic pain; this suggests that it may mediate some of the analgesic effects of N-arachidonoyl-serotonin (AA-5-HT, Malek et al., 2016). GPR55 activation by some agonists increases calcium release from intraneuronal stores and inhibits M-type potassium current, both tending to promote neuronal activity (Lauckner et al., 2008). These effects may be important in the context of CBD's ability to antagonize the receptor, as this non-psychoactive phytocannabinoid has recently been shown effective for the treatment of Dravet syndrome, a formerly intractable form of childhood epilepsy (Devinsky et al., 2017). Whether the efficacy of CBD in this syndrome involves GPR55 antagonism, or perhaps, involves some combination of the many other established targets of this drug (see Table 2) remains to be resolved. Other evidence for potential roles for GPR55 signaling includes: distinct expression in rod cells of primate retina suggesting a role in low-light vision (Bouskila et al., 2013) and; expression in microglia that, following LPI activation, acts to protect hippocampal neurons from excitotoxicity (Kallendrusch et al., 2013).
GPR55 in cancer
Despite the numerous studies illuminating the physiological functions of GPR55 in the CNS described above (section GPR55 in CNS), limited information is available about the role of this receptor in cancer. GPR55 is expressed in different cancer cell types raising the possibility that it may be a target for cancer chemotherapeutic agent development (reviewed by Falasca and Ferro, 2016). Much of the research conducted thus far indicates that GPR55 promotes tumor formation. GPR55 expression was examined in human tumor biopsy samples from breast, pancreatic, and glioblastoma patients (Andradas et al., 2011). In this study, high levels of GPR55 were strongly correlated with the aggressiveness of the malignancy. Additional evidence for GPR55-promotion of malignancy includes; (1) elevated levels of its endogenous ligand lysophosphatidylinositol (LPI) in ovarian cancers (Xiao et al., 2001) and; (2) exogenous LPI promotion of proliferation and migration of various cancer cell lines (Ford et al., 2010; Piñeiro et al., 2011). However, when GPR55 is activated by the endocannabinoid, AEA, cholangiocarcinoma cell survival is inhibited. AEA induces cholangiocarcinoma cell apoptosis by recruiting Fas death receptors into lipid rafts and activating the JNK signaling pathway (DeMorrow et al., 2007; Huang et al., 2011). Genetic disruption of GPR55 receptor expression using shRNA blocked the antiproliferative effects of AEA in vitro and in vivo (Huang et al., 2011). Of note, AEA-induced cell death was not reversed in the presence of CB1- (SR141716A) or CB2- (SR144528) selective antagonists although each cannabinoid receptor was expressed (DeMorrow et al., 2007). These findings suggest that AEA elicits antiproliferative effects on cholangiocarcinoma that are GPR55-dependent and CB receptor-independent. Other deorphanized GPCRs, including GPR18 and GPR35, have been identified as modulators of tumorigenesis (Okumura et al., 2004; Qin et al., 2011); however, the role of cannabinoid ligands in these systems is unclear. Examination of the deorphanized cannabinoid receptors in cancer will provide greater insight into their impact on tumor development and progression. In addition, determining whether classical cannabinoids mediate their effects through the deorphanized receptors will shed light on mechanisms of their antitumor activity.
GPR18 in CNS
Interestingly, microglia migration is stimulated by activation of a second formerly orphan cannabinoid-related receptor, GPR18 (McHugh et al., 2010). This Gi/o-coupled receptor was originally reported to be highly-expressed in testes and spleen (Gantz et al., 1997); yet, it clearly is also functional within brainstem (Penumarti and Abdel-Rahman, 2014b). GPR18 is activated by N-arachidonoyl glycine (NAGly, Kohno et al., 2006) that is a product of anandamide metabolism (Aneetha et al., 2009; Bradshaw et al., 2009). It is also activated by abnormal cannabidiol (abn-CBD) that, when infused into regions of the brainstem, causes reduction in blood pressure in a manner that involves nitric oxide synthase and adiponectin signaling (Penumarti and Abdel-Rahman, 2014a). As with GPR55 discussed above (section GPR55 in CNS), GPR18 receptor expression is upregulated following spinal cord injury; this suggests that it may mediate some of the analgesic effects of N-arachidonoyl-serotonin (AA-5-HT, Malek et al., 2016). Notably, the principal active constituent of Cannabis, THC, is a potent agonist of GPR18 when expressed in HEK293 cells (McHugh et al., 2012). The lesser Cannabis constituent, CBD, along with synthetic CB agonists CP55940 and WIN55212-2, have no activity at physiologically-relevant concentrations. Potent GPR18 activation by THC and NAGly has been suggested to underlie efficacy to resolve inflammatory pain (Burstein et al., 2011; McHugh et al., 2014; Crowe et al., 2015). Some controversy about GPR18 signal transduction was raised when NAGly failed to inhibit Ca++ currents following heterologous expression and agonist stimulation in rat superior cervical ganglion neurons (Lu et al., 2013). Whether this is a special case of system-dependent functional selectivity, or an indication that NAGly modulates neuronal activity via other receptor systems, remains an open question.
GPR119 in CNS
A third relevant G-protein-coupled receptor, GPR119, has been deorphanized. In humans, GPR119 was initially reported to be expressed only outside of CNS, most notably in pancreas, suggesting a peripheral metabolic function (Fredriksson et al., 2003). A possible CNS expression difference between human and rodent species is suggested in a patent application that indicates GPR119 is also expressed at high density in rat and mouse brain (Bonini et al., 2002). Mouse CNS expression is confirmed by high-affinity GPR119 binding and potent efficacy to reduce seizure-related hippocampal activity (Scott et al., 2014). At micromolar concentrations, the endogenous peroxisome proliferator-activated receptor alpha (PPARα) agonist oleoylethanolamide (OEA) activates GPR119, while anandamide appears to have partial efficacy in yeast cells expressing the receptor (Overton et al., 2006). Notably, 2-AG, CP55940, WIN55212-2, methanandamide, and JWH-133 did not activate GPR119. Supporting CNS activity, and a role in controlling ingestive behaviors, OEA and a synthetic GPR119 agonist with similar efficacy, but four-fold greater potency, PSN632408, were found to reduce food consumption and body weight in Sprague-Dawley rats (Overton et al., 2006). More recent evidence suggests that PSN632408 may lack selectivity for GPR119 in peripheral tissues (Ning et al., 2008). Despite evidence of GPR119 CNS activity, this receptor is being most closely studied in as a target for treating diabetes and other metabolic disorders (Ansarullah et al., 2013; Cornall et al., 2013, 2015).
GPR35 in CNS
A final Gi/o-coupled deorphanized receptor, GPR35, has been reported to transduce effects of 2-arachidonyl lysophosphatidic acid (2A-LPA) and kynurenic acid, both of which are present at relevant concentrations within neuronal tissues. 2A-LPA is a product of metabolism of the principal brain endocannabinoid, 2-arachidonyl glycerol (2-AG, Oka et al., 2010). Kynurenic acid is the product of tryptophan metabolism, and is most well-characterized as an endogenous antagonist of receptors for the principal excitatory neurotransmitter, NMDA (Stone et al., 2013). A very recent structure-activity study has led to identification of several potent GPR35 agonists and three structures critical for efficacy (Abdalhameed et al., 2017). GPR35 is expressed most predominantly in the gastrointestinal tract and in leukocytes (Wang et al., 2006). Low-level expression of GPR35 in brain reduces the likelihood that it significantly modulates neuronal activity within this region of the CNS. Within spinal cord, on the other hand, convincing evidence demonstrates functionally-relevant expression of GPR35, with distinct enrichment within murine dorsal horn ganglia (Cosi et al., 2011). In an acetic acid-induced pain assay, both kynurenic acid, and the phosphodiesterase inhibitor, zaprinast, were found to produce significant analgesia with EC50s of about 100 and 1 mg/kg, respectively. Note zaprinast is an effective agonist of GPR35 with a particularly high potency at rat vs. human forms of the receptor (Taniguchi et al., 2006). Homology between GPR35, and other LPA receptors, may suggest that 2A-LPA is more relevant than kynurenic acid as an endogenous modulator (Zhao and Abood, 2013). Clearly, more needs to be learned and understood about GPR35 signaling.
Opioid receptors in CNS
Evidence implicating interaction between cannabinoid and opioid signaling systems began to accumulate early, and before specific cannabinoid receptors were unequivocally identified. For example, it was well-established by the 1980s that THC effectively reduces symptoms of naloxone-precipitated opioid withdrawal (Hine et al., 1975). Similarly, the analgesic efficacy of THC was known to depend upon μ-opioid receptors (μOP, Cox et al., 2015), as use of an irreversible antagonist (chlornaltrexamine, that alkylates and destroys activity of μOPs) reduces efficacy of both morphine and THC in the hotplate assay (Tulunay et al., 1981). Using the μOP-selective radioligand, [3H]-dihydromorphine, it was found that THC non-competitively reduces the density of binding sites in brain membranes via an unknown mechanism (Vaysse et al., 1987). More recently, it has been demonstrated that augmentation of endocannabinoid signaling using the monoacylglycerol lipase (MAGL) inhibitor JZL184 (MAGL is an enzyme responsible for degrading the endocannabinoid 2-AG) has efficacy similar to THC in reducing opioid withdrawal symptoms. This suggests MAGL as a potential target for managing opioid addiction (Ramesh et al., 2011).
More progress has been made since development of CB1/CB2 receptor-deficient mice and availability of selective antagonists. Interestingly, when using CB1 knockout mice with the CB1-selective cannabinoid antagonist/inverse agonist SR, it was found that SR effectively reduces both basal levels of G-protein activation and the ability of the μOP peptide agonist, DAMGO, to stimulate GTPγS binding (Cinar and Szücs, 2009). These results were obtained using mouse cortical membranes (where μOP and CB1 are normally both distinctly expressed), and there was no difference in efficacy across tissue obtained from wild type and CB1 knockout animals, clearly demonstrating a non-CB1-related mechanism for SR's inverse agonist activity (although not excluding the possibility that CB1 constitutive activity drives inverse agonism in other systems). Additional experiments using CB1 and μOP transfected CHO cells revealed that micromolar concentrations of SR are able to displace ligands bound to μOPs, demonstrating an interesting direct SR-μOP interaction.
The nature of this SR-μOP interaction was further explored by demonstrating that both SR and the 2-AG-related endocannabinoid noladin ether are both able to potently antagonize DAMGO-stimulated GTPγS binding in membranes taken from wildtype animals (Zádor et al., 2012). In membranes from CB1 deficient mice, both SR and a combination of SR and noladin ether reduced the efficacy of DAMGO to stimulate GTPγS binding, without altering potency, suggesting μOP-CB1 interaction, and non-competitive antagonism of μOP in the absence of CB1. These studies employing DAMGO, relevant to μOP activity, were extended to employ the δ-opioid receptor (δOP) selective peptide agonist DPDPE. In CHO cells expressing δOP, SR reduced basal GTPγS activation with an EC50 ~ 1 μM. SR also non-competitively antagonized DPDPE stimulation of GTPγS binding to an extent similar to that achieved by the competitive δOP antagonist, naltrindole (Zádor et al., 2014). Completing evaluation of SR interaction with the three primary opioid receptor subtypes, the same group demonstrated SR displaces binding of the κ-opioid receptor (κOP) agonist U-69,593 from membranes prepared from transfected CHO cell membranes. Using these transfected cells in vitro, SR was found to inhibit basal levels of κOP activation in a manner antagonized by the κ-selective antagonist, nor-BNI, suggesting that SR is a κOP inverse agonist. In both CB1 deficient and wildtype mice, systemic pretreatment of animals with a low, 0.1 mg/kg dosage of SR decreased efficacy of the κOP peptide agonist, dynorphin A to activate GTPγS binding in preparations of brain membranes (Zádor et al., 2015). The magnitude of efficacy reductions following SR pretreatment was similar across CB1 knockouts and wildtype animals, and the potency was not altered, suggesting that SR acts to effectively reduce κOP density. Behavioral tests demonstrated an anxiolytic effect of low-dose (0.1 mg/kg subcutaneous) SR treatments, perhaps contrasting with established dysphoric effects of higher doses in other systems, including humans.
Related to morphine dependence, is evidence indicating that chronic morphine treatment of rats results in upregulation of both CB1 protein and mRNA encoding the receptor (Jin et al., 2014). Morphine-induced CB1 expression was also associated with cytokine release, including IL-6, and, most notably, IL-1B, in various brain regions, including cortex, hippocampus, cerebellum, and brain stem. Coincidently, CB1 upregulation and cytokine release implicate both cannabinoid signaling and immune responsiveness in effects associated with opioid dependence. Interestingly, the microRNA let-7d and CB1 receptors reciprocally downregulate each other in transfected SH-SY5Y cell cultures (Chiarlone et al., 2016). Let-7d-expressing SH-SY5Y cells show decreased sensitivity to methanandamide and morphine in stimulating ERK phosphorylation and a high degree of cross-tolerance following chronic treatments with the cannabinoid- and opioid agonists. Perhaps related to this interaction is evidence for efficacy of the opioid antagonist naltrexone (currently indicated for managing alcoholism) in the treatment of Cannabis dependence (Haney et al., 2015).
Serotonin receptors in CNS
5-HT3 ligand-gated cation channels in CNS
As with other systems, evidence for cannabinoid modulation of serotonin signaling was obtained soon after CB1 receptors were identified. This evidence began to accumulate through studies of the rat nodose ganglion which conducts afferent transmission from GI, heart, and lung. Activity in this ganglion is increased by the 5-HT3 subtype of serotonin-gated cation channels. Using the synthetic agonists CP55940 and WIN, and the most well-established endocannabinoid at the time, anandamide, it was discovered that each of these agonists non-competitively antagonizes serotonergic activation of 5-HT3 receptors at nanomolar concentrations (Fan, 1995). The inability to modulate cannabinoid effects on 5-HT3 activity with guanyl nucleotides suggested a non-G-protein dependent mechanism, and possible direct interaction with the ion channel. This finding raised speculation that analgesic and antiemetic efficacy of cannabinoid agonists may, at least in part, be attributable to 5-HT3 antagonism.
Effects in the isolated tissue preparation described above were corroborated by results of experiments using a human 5-HT3A-expressing CHO cell line (Barann et al., 2002). In patch clamp studies it was found that a series of synthetic, endogenous, and phytocannabinoid agonists potently inhibited serotonin activation of 5-HT3 cation channels. Notably, the phytocannabinoid THC was the most potent compound evaluated with EC50 ~ 40 nM. Cannabinoid antagonism was not inhibited by the CB1-selective antagonist SR. Specific binding of a 5-HT3-selective radioligand wasn't displaced by the cannabinoids employed, suggesting interaction with an allosteric site that doesn't influence serotonin affinity.
5-HT1A GPCRs in CNS
The 5-HT1A subtype of serotonin receptors are notably associated with presynaptic distribution, where, similar to CB1 receptors, their activation reduces probability of neurotransmitter release. Although this type of presynaptic autoreceptor mechanism is well-characterized for 5-HT1A, the distribution of these receptors is wide, and it includes both excitatory and inhibitory terminals, making prediction of effects produced by modulators difficult.
Multiple lines of evidence suggest that the enigmatic non-psychotropic phytocannabinoid CBD exerts at least some of its actions through agonism of 5-HT1A autoreceptors (McPartland et al., 2015). First, CBD displaces specific binding of the 5-HT1A-selective radioligand [3H]8-OH-DPAT, activates GTPγS binding and inhibits adenylyl cyclase activity in heterologously expressing CHO cell cultures (Russo et al., 2005). Second, 5-HT1A receptor expression is upregulated under conditions of neuropathic pain in a manner that is reduced by cannabinoid agonism (Palazzo et al., 2006). Finally, microinfusion of CBD into the bed nucleus of the stria terminalis (BNST) dose-dependently reduces anxiety as measured by both elevated plus maze and Vogel conflict tests (Gomes et al., 2011).
These anxiolytic CBD responses are reversed by pretreatment with the 5-HT1A-selective agent WAY-100635 (WAY) that has been reported to be a neutral antagonist, although agonism at D4 dopamine receptors has also been reported (Chemel et al., 2006). Recently, WAY-reversible anxiolytic effects of CBD as measured in the elevated plus maze have been confirmed by others (Fogaça et al., 2014). CBD mitigation of acute restraint stress was also found, although, perhaps importantly, this effect was more resistant to WAY antagonism, possibly suggesting involvement of another target in stress vs. anxiety responses. Interestingly, anxiolytic effects of both CB1 and 5-HT1A agonists have been reported in a non-mammalian teleost fish, suggesting a conserved relationship between cannabinoid and serotonergic signaling in responding to stress (Connors et al., 2014).
Extending studies of CBD anxiolysis that are reversed by the 5-HT1A antagonist WAY, mitigation of nausea as measure in the rat gaping model (Rock et al., 2014), reduction of both neuropathic pain (Ward et al., 2014) and panicolytic effects (Twardowschy et al., 2013) have all been reported. In the context of drug abuse, CBD antagonizes effects of morphine to reduce threshold intracranial self-stimulation responding—a measure of drug reward. CBD antagonism of morphine reward was reversed by microinjection of WAY into dorsal raphe, further implicating 5-HT1A involvement (Katsidoni et al., 2013). More recently, CBD has been reported to have rapid-onset antidepressive efficacy that is dependent upon 5-HT1A activation (Linge et al., 2016).
Other serotonin receptors in CNS
Physiological interactions, where activity of one signaling system influences activity of another, have been documented to occur between cannabinoid and serotonergic signaling in models of chronic pain (Campos et al., 2013); epilepsy (Devinsky et al., 2014); stress-induced analgesia (Yesilyurt et al., 2015) and; tic disorders (Ceci et al., 2015). These studies reinforce the possibility of using inhibitors of endocannabinoid uptake and metabolism therapeutically.
Adenosine receptors in CNS
Adenosine signaling in CNS is largely terminated by reuptake, making inhibitors of the transporter indirect-acting adenosinergic agonists. Such agents have shown anti-inflammatory efficacy via inhibiting release of immune mediators like TNFα (Noji et al., 2002). Interestingly, the phytocannabinoid CBD, discussed above, in the context of 5-HT1A agonism (5-HT1A GPCRs in CNS), appears to inhibit immunosuppressive effects via direct interaction with, and antagonism of, an adenosine transporter (Carrier et al., 2006). CBD anti-inflammatory effects were absent in adenosine A2A knockout mice, and reversed by a selective antagonist, implicating indirect agonism of A2A receptors as the mechanism of CBD-mediated immunosuppression.
Antagonists of adenosine receptors are established psychomotor stimulants. In the context of movement disorders, A2A receptors are expressed at high levels within the inhibitory indirect dopaminergic pathway of vertebrate striatum. Interneurons in this region also robustly express presynaptic CB1 receptors that inhibit activity of the inhibitory pathway, resulting in an overall activation of movement-related striatal signaling. Experiments employing CB1 deficient mice found these animals to be resistant to the psychomotor stimulant effect of adenosine receptor antagonists (Lerner et al., 2010). Further investigation found that the A2A-selective antagonist, SCH442416, doubled the concentration of the endocannabinoid, 2-AG, within striatum, but not cortex. Increased 2-AG release was associated with decreased indirect pathway activity as measured electrophysiologically. These results demonstrate that the psychomotor efficacy of A2A antagonists involve, to some extent, indirect endocannabinoid agonism. More recently it has been discovered that CB1-mediated disruption of memory consolidation is mitigated by A2A antagonism, further elaborating the co-dependence of cannabinoid and adenosinergic signaling (Mouro et al., 2017).
Excitatory amino acid receptors in CNS
NMDA receptors in CNS
Given general sedative effects of cannabinoid agonists, it isn't surprising that these agents act, at many sites, to reduce release of the principal excitatory neurotransmitter, NMDA. This was found to clearly be the case in rat brain slice preparations for synthetic agonists (Shen et al., 1996) and both the endocannabinoid anandamide and phytocannabinoid THC. Inhibitory effects of THC and anandamide were reversed in the presence of both the CB1-selective antagonist SR and Gi inhibitor pertussis toxin implicating cannabinoid-receptor involvement (Hampson et al., 1998). But, in the presence of SR, anandamide potentiation of NMDA-induced currents were noted. This stimulatory effect of anandamide was also observed in Xenopus oocytes expressing NMDA receptor (NMDAR) subunits, suggesting a direct agonist interaction with these cation channel proteins.
With similarities to results of experiments done in slice preparations, additional evidence for anandamide interaction with NMDA receptors derives from studies of the complex cannabinoid modulation of blood pressure control in rats (Malinowska et al., 2010). In this system, it was discovered that anandamide produces a pressor effect in the presence of the CB1-selective antagonist, SR. This increase in pressure was partially reduced by the NMDA receptor-selective antagonist MK-801, suggesting a possible direct interaction with relevance to central control of blood pressure.
More recently, the interaction of anandamide and NMDAR activation has been more elaborately studied through a series of electrophysiology studies in rat hippocampal sections and dissected cells (Yang et al., 2014). These experiments demonstrated that CA1 pyramidal cell NMDAR activation by anandamide was reversed in the presence of the TRPV1 antagonist capsazepine, raising the possibility of vanilloid receptor involvement in the anandamide effects discussed above. 2-AG was also studied in these experiments and found to produce a similar potentiation of NMDAR activation, although distinct in that it was not blocked by TRPV1 antagonism. Interestingly, the efficacy of 2-AG was increased when delivered intracellularly, suggesting interaction with NMDAR or other protein domains inside the neuron.
It has long been suspected that Cannabis abuse increases risk of psychoses (Colizzi et al., 2016). In a mouse model of NMDAR antagonist-precipitated psychosis (MK-801), the CB1-selective antagonist AM 251 reduced behavioral symptoms, including hyperactivity (Kruk-Slomka et al., 2016). Interestingly, CB1-antagonism also improved psychosis-related memory impairments in this model, suggesting a possible role for endocannabinoid signaling in the memory deficits associated with schizophrenia and bipolar disorder.
AMPA receptors in CNS
An early Xenopus oocyte expression study examining sensitivity of various AMPA receptor subunit compositions demonstrated that high concentrations of anandamide (EC50 > 100 μM) effectively inhibit/s AMPA agonist-initiated currents (Akinshola et al., 1999). Anandamide inhibition was not reversed by SR, as expected in the CB1-deficient expression system, and similar efficacy was not observed for the synthetic agonist WIN. Enhancement of anandamide's effect was produced by addition of forskolin, a direct adenylyl cyclase activator, and partially reversed by cyclase inhibitors, implicating this enzyme in mediating the effect.
An interesting physiological interaction between endocannabinoid and AMPA receptor signaling has been reported in chicken embryo spinal cord (Gonzalez-Islas et al., 2012). In this system, a basal endocannabinoid tone inhibits activity of motor neurons which decreases spontaneous activity. Reversal of endocannabinoid tone via application of the CB1-selective antagonist, AM 251 resulted in an increase of spontaneous activity that, over 2 days, resulted in AMPA receptor desensitization, implicating endocannabinoid signaling in regulation of motor activity during development.
Metabotropic glutamate receptors in CNS
Physiological interaction of metabotropic glutamate receptors (mGluR) and endocannabinoid signaling has been reported in striatal and hippocampal slice preparations (Jung et al., 2005). In this system, activation of the mGluR5 receptor subtype resulted in release of the endocannabinoid 2-AG but not anandamide. This finding implicates 2-AG as the endocannabinoid involved in mediating AMPA receptor-induced plasticity important to processes of learning and memory.
Inhibitory amino acid receptors in CNS
GABA and glycine are amino acid transmitters that activate inhibitory chloride channels, and in the case of GABAB, Gi/o-coupled metabotropic receptors that activate inhibitory inwardly rectifying K+ flow. Overall sedative effects of cannabinoid agonists made positive regulation of these inhibitory amino acid transmitter systems seem likely, although clear evidence of interaction with GABAergic signaling systems has emerged only recently.
Gaba receptors in CNS
In the case of cannabinoid receptor-independent effects on GABAergic signaling, in hippocampal slice preparations it has been found that WIN55212-2 and anandamide (but not 2-AG) potentiate GABA release within rat dentate gyrus. This release promoted measurable inhibitory post-synaptic currents (IPSCs) measured by electrophysiology. The fact these WIN55212-2 and anandamide-promoted currents were resistant to both CB1- and CB2-selective antagonists in tissues taken from CB1 deficient mice, is evidence that they are the result of interaction with a yet uncharacterized target (Hofmann et al., 2011). Interestingly, the agonist specificity (WIN55212-2 and anandamide) of this IPSC effect is similar to that discovered earlier in GTPγS binding assays employing mouse tissue (Breivogel et al., 2001) suggesting that the same target is responsible.
In the context of behavior, 2-AG has been shown to reduce locomotor activity in CB1/CB2 double knockout mice (Sigel et al., 2011). Unfortunately only endocannabinoids were investigated in this study leaving open the question of WIN55212-2 activity that is implicated by earlier studies mentioned above. Hypermotility was observed in mice deficient for the GABAA B2 subunit. When heterologously expressed in Xenopus oocytes, it was found that electrophysiological effects of 2-AG were only observed in systems producing B2 protein, suggesting this subunit is required for 2-AG interaction with GABAA. B2/2-AG interaction has been further supported by modeling and studies of the effect of amino acid substitutions (Baur et al., 2013). This work has recently been extended to a slice preparation model that demonstrates 2-AG influences GABAA signaling under physiologically-relevant conditions (Golovko et al., 2014). This physiological modulation involves both synaptic and extrasynaptic populations of GABAA receptors enhancing the former and inhibiting the latter in a manner to potentiate overall GABAergic inhibition (Brickley and Mody, 2012).
Endocannabinoid regulation of spinal nociceptive vs. non-nociceptive transmission has been established in vertebrate species (Pernía-Andrade et al., 2009). This work has been extended to demonstrate that the endocannabinoid 2-AG produces similar effects in a non-cannabinoid receptor-expressing invertebrate species of leech (Higgins et al., 2013). The target of 2-AG in this animal appears to be a TRPV channel, as a selective antagonist blocked the effect. Interestingly, the 2-AG effect to potentiate non-nociceptive vs. nociceptive transmission was antagonized by the GABAA-selective agent, bicuculline, suggesting a potential TRPV-GABAergic relationship in this physiological system.
Glycine receptors in CNS
Evidence for direct interaction of the endocannabinoids anandamide and 2-AG and glycine receptors were first reported from electrophysiological studies employing primary neuronal cell cultures and hippocampal slice preparations (Lozovaya et al., 2005). These experiments demonstrated that anandamide, 2-AG and the synthetic agonist WIN55212-2 inhibit glycine receptor conductance post-synaptically, and accelerate desensitization. This effect contrasts with the retrograde, presynaptic endocannabinoid mechanism that has been better characterized to date. Glycine receptor inhibition was observed in the presence of CB1, CB2, and vanilloid receptor antagonists, suggesting a direct agonist mechanism.
Complicating the picture, in a different Xenopus oocyte expression system, anandamide agonism of glycine receptors was discovered (Hejazi et al., 2006). These studies extended the effect to include the phytocannabinoid, THC. In an additional heterologous expression system, using HEK293 cell cultures expressing various glycine receptor subunits, an even more complex picture emerged (Yang et al., 2008). In these studies, anandamide and HU210 potently activated α1 subunit-containing glycine receptors. In contrast, the synthetic agonists HU210, HU308, and WIN55212-2 each potently (EC50 38—3030 nM) inhibited the α2 subunit-mediated currents, while the endocannabinoid NAGly (discussed above as a GPR18 agonist) activated it at high concentration. Efficacy and potency of glycine receptor inhibition varied with differential subunit expression. These types of apparent conflicts in pharmacological literature are common and, as we see with glycine receptors, are often attributable to different physiological/expression systems employed (Urban et al., 2007).
Cholinergic receptors in CNS
Anecdotes have long suggested interaction between cannabinoid and nicotinic cholinergic signaling systems. This relationship is supported by behavioral (Pryor et al., 1978) and more recently biochemical evidence that demonstrates nicotine modulates multiple effects of the phytocannabinoid, THC (Valjent et al., 2002).
A direct mechanism underlying cannabinoid interaction with nicotinic receptors has been revealed through a series of Xenopus oocyte expression studies employing the endocannabinoid anandamide. The first of these found that anandamide potently inhibits nicotinic receptors comprised of α7 subunits (IC50 ~ 30 nM, Oz et al., 2003). This inhibition was not altered by CB1 or CB2 antagonists or cAMP modulating agents suggesting a direct nicotinic receptor interaction. Anandamide inhibition of α7-mediated currents didn't alter nicotine potency, only efficacy, indicating a non-competitive antagonist mechanism. The second study expanded the array of cannabinoid agonists employed in the system finding that only the endocannabinoids anandamide and 2-AG produce potent inhibition (Oz et al., 2004). Notably, THC, WIN, CP55940 did not. The final papers in this series found that the potency of inhibitory effects of both anandamide and ethanol are increased by their co-administration (Oz et al., 2005) and that anandamide potently and non-competitively also antagonizes receptors comprised of α4β2 subunits (EC50 ~ 50 nM, Spivak et al., 2007). Also, the phytocannabinoid CBD was found to inhibit nicotinic currents, albeit less potently (EC50 ~ 11 μM, Mahgoub et al., 2013).
More recently, these studies have been extended out of the oocyte system and into in vivo models. In the first of these, the non-hydrolysable analog of anandamide, methanandamide, was found to effectively antagonize nicotinic receptors present in cardiac post-ganglionic sympathetic neurons (Baranowska et al., 2008). In a second anesthetized rat model, nicotine infusion increased activity of dopaminergic neurons in the ventral tegmental area, a region important to reward-related release of dopamine in nucleus accumbens. Nicotine stimulation of this activity was found to be reversed by URB597, an indirect cannabinoid agonist that inhibits anandamide metabolism (Melis et al., 2008). This effect did not extend to the hydrolysis-resistant methanandamide, suggesting that a product of an alternate anandamide metabolic pathway is responsible. Interestingly, both oleoylethanolamine (OEA) and palmitoylethanolamide (PEA) were found to have efficacies similar to anandamide. As OEA and PEA are known agonists of peroxisome proliferator-activated receptor-α (PPARα) activation of this receptor was studied as the potential mechanism for nicotinic receptor inhibition. Results revealed that PPARα activation promotes kinase activity that increases nicotinic receptor phosphorylation and inactivation. Whether the efficacy of anandamide, perhaps through increasing availability of ethanolamine precursors, depends upon a similar PPARα-related mechanism remains an open question.
Nuclear receptors in cancer
Peroxisome proliferator-activated receptors (PPAR) in cancer
PPARs are a superfamily of nuclear receptors that play an important role in the regulation of lipid metabolism, glucose homeostasis, cell differentiation and tumorigenesis (Vamecq and Latruffe, 1999). PPAR transcription factors form heterodimers with retinoid X receptor (RXR) that then bind to peroxisome proliferator responsive elements (PPRE) and regulate transcription of target genes. PPARs are classified into three subtypes: PPARα, PPARδ, and PPAR⋎. In cancer cells, targeting PPAR⋎ with selective agonists inhibits cell proliferation, induces programmed cell death (apoptosis and autophagy) and promotes cell differentiation in multiple in vitro and in vivo studies (Campbell et al., 2008).
PPARγ in cancer
Cannabinoids increase PPAR⋎ transcriptional activity by diverse mechanisms (reviewed in O'Sullivan, 2007). Cannabinoids can bind and activate cell surface cannabinoid receptors which promotes MAPK signal transduction resulting in downstream PPARγ activation. Cannabinoid-induced PPARγ activation can also occur independent of cannabinoid receptors by direct and indirect mechanisms. Cannabinoids (e.g., ajulemic acid) and endocannabinoids (e.g., AEA and 2-AG) can directly bind, and increase the transcriptional activity of, PPARγ. Indirect PPARγ activation occurs as a consequence of the metabolism of endocannabinoids (2-AG, AEA) to PPARγ agonists or by increasing the synthesis of endogenous PPAR agonists (Burstein, 2005; O'Sullivan, 2007).
Several studies have shown that PPAR⋎ mediates the antitumor activity of cannabinoids independent of CB1 and CB2 receptors. In lung cancer cells, PPARγ activation was required for CBD-induced apoptosis (Ramer et al., 2013). This effect of CBD on PPARγ was mediated by production of the prostaglandins (PGs), PGD2 and 15deoxy, Δ12, 14 PGJ2 (15d-PGJ2) which are PPARγ activators. Prevention of PG synthesis with a selective inhibitor of COX-2 activity (NS-398) or siRNA directed toward COX-2 reduced CBD-induced PPARγ nuclear translocation and cell death. However, the use of CB1 (AM-251), CB2 (AM-630), or TPRV1 (capsazepine) antagonists did not alter CBD-mediated apoptotic cell death. Furthermore, in human cervical carcinoma cells treated with the non-hydrolysable AEA derivative, met-AEA, apoptosis was reliant upon the production of PGD2 and PGJ2 as well as the activation of PPARγ (Eichele et al., 2009). These findings indicate that cannabinoid-induced prostaglandin synthesis and the engagement of these prostaglandins with PPARγ represents an important pathway by which cannabinoids regulate cellular fate independent of the cannabinoid receptors.
Ion channels in CNS and cancer
Direct cannabinoid modulation of the activity of ionotropic receptors have been discussed according to ligand type in the section above. In terms of interaction with non-ligand gated ion channels, cannabinoid receptor-stimulated G-proteins have long been known to inhibit voltage-gated calcium channels and to activate inward-rectifying potassium channels, consistent with inhibition of transmitter release (Shen et al., 1996). In addition to ligand-gated channel effects, evidence for direct cannabinoid interaction with other non-ligand-gated ion channels has emerged and is discussed below.
Voltage-gated sodium channels in CNS
In the case of the synthetic cannabinoid, ajulemic acid that is structurally-related to THC, patch clamp studies of HEK293 and ND7/23 cells expressing various types of voltage-gated sodium channels demonstrated inhibition with potencies ranging from 1 to 10 μM (Foadi et al., 2014). These results suggest that the efficacy of ajulemic acid to reduce neuropathic pain may involve sodium channel inhibition.
Transient receptor potential (TRP) cation channels in CNS
TRP channels are a superfamily of cation channels located on the cell plasma membrane that control inward movement of monovalent or divalent cations notably including Ca++. These channels are largely sensory-related, and they are particularly important to perception of temperature and mechanical stimulation (Voets et al., 2005). TRP channels have already been mentioned above (in the context of anandamide interaction with NMDA receptors) (section NMDA Receptors in CNS). In addition to a role in mediating peripheral afferent signaling, TRP channels are physiologically relevant to the extraneuronal function of vascular smooth muscle (Fernandes et al., 2012) and are also expressed within CNS (Vennekens et al., 2012). Several members of this family may represent “ionotropic cannabinoid receptors” moving cannabinoid signaling into line with most other CNS signaling systems that include both metabotropic and ion channel targets (Akopian et al., 2009).
Several members of this family, including TRP ankyrin type 1 (TRPA1), the vanilloid receptors types 1 and 2 (TRPV1 and TRPV2), and TRP melastatin type 8 (TRPM8), have been identified through a remarkably ambitious and thorough screening study as targets of an array of compounds isolated from Cannabis (De Petrocellis et al., 2011). These phytocannabinoids include cannabichromene (CBC), CBD, cannabidiol acid (CBDA), cannabidivarin (CBDV), cannabidivarin acid (CBDVA), cannabigerol (CBG), cannabigerol acid (CBGA), cannabigivarin (CBGV), cannabinol (CBN), THC, THC acid (THCA), and tetrahydrocannabivarin acid (THCVA). Each of these compounds are effective agonists for TRP1A with EC50s ranging from 90 nM to 8.4 μM and a rank order of potency of CBC > CBD > CBN > CBDV > CBG > THCV > CBGV > THCA > CBDA > CBGA > THCVA. At TRPV1; CBD, CBG, CBN, CBDV, CBGV, and THCV have agonist activity with EC50 < 10 μM (the most potent compound being CBD with EC50 = 1 μM). At TRPV2, several of the compounds had agonist activity with a rank order of potency = THC > CBD > CBGV > CBG > THCV > CBDV > CBN (EC50 ranging from 650 nM to 19 μM). Finally, at TRPM8, each of the compounds were found to effectively antagonize icilin-stimulated Ca++ conductance with a rank order of potency = CBD > CBG > THCA > CBN > THCV > CBDV > CBGA > THCVA > CBGV > CBDA and IC50s ranging from 60 nM to 4.8 μM.
Interestingly, the synthetic cannabinoid compounds AM251 and AM630, that are CB1- and CB2-selective antagonists/inverse agonists, respectively, have been found to activate Ca++ channels in primary cultures of trigeminal nerve neurons (Patil et al., 2011). When evaluated in CHO cell culture systems heterologously expressing TRP1A and TRPV1 channels, it was found that both CB receptor antagonists have agonist activity at TRP1A but not TRPV1. These findings suggest that in vivo AM251 and AM630 may activate sensory neurons via TRP1A agonism.
Focus on TRPV1 in CNS
Also known as capsaicin receptors or vanilloid receptors, these promiscuous cation channels are both temperature- and pH-sensitive. They are also capable of ligand activation, notably by capsaicin (the active constituent of spicy peppers). Shortly after its isolation and identification as the first endogenous cannabinoid present in brain, anandamide was found to interact with a non-cannabinoid receptor target: TRPV1 (Zygmunt et al., 1999). The relationship between anandamide and other endocannabinoid signaling and TRPV1 channel activity has been the subject of excellent recent reviews (Dainese et al., 2010; Marzo and Petrocellis, 2010; Di Marzo and De Petrocellis, 2012), and so, only two notable recent additions, confirming the physiological relevance of TRPV1 agonism by anandamide to neuronal activity, will be presented here.
The first is an interesting recent study of the analgesic effects of the fatty acid amide hydrolase (FAAH) inhibitor: URB597. This compound reduces metabolism of the endocannabinoid, anandamide, thereby enhancing effects of endogenous release. Systemic administration of URB597 effectively reduces measures of neuropathic pain in the rat chronic constriction injury model (Starowicz et al., 2013). Analgesic efficacy of URB597 was maintained in the presence of the CB1-selective antagonist, AM251. However, pretreatment of animals with the potent TRPV1 antagonist, iodoresiniferatoxin, reversed effects of URB597. These important results reinforce the role of endocannabinoid signaling in pain sensation, and they demonstrate that agonism of the TRPV1 subtype of vanilloid receptors is important to this type of analgesic efficacy of anandamide.
A second recent study employed mouse hippocampal cultures of CA1 presynaptic CB1-expressing GABAergic neurons and post-synaptic pyramidal neurons. Activities of both neuron types were studied simultaneously via an ambitious paired patch clamping technique (Lee et al., 2015). As expected, post-synaptic depolarizations were associated with increased 2-AG release and diffusion, ultimately resulting in presynaptic CB1 activation. In the case of perisomatic input to pyramidal cells, a basal tone of 2-AG stimulation of presynaptic CB1 receptors was found to maintain a “set point” of GABA release. This set point was revealed by increased inhibition following application of JZL184, an inhibitor of MAGL. As MAGL is the principal enzyme responsible for degrading 2-AG, inhibitors like JZL184 act as indirect agonists. Interestingly, addition of PF3845, an inhibitor of the enzyme FAAH responsible for degrading anandamide, effectively reduced 2-AG tone, suggesting an antagonistic relationship between the two endocannabinoids in regulating GABAergic signaling in hippocampus. The indirect anandamide agonist effect of PF3845 was reversed in the presence of AMG9810, a selective TRPV1 inhibitor, demonstrating that anandamide effectively modulates basal 2-AG tone via activation of this post-synaptic cation channel, presumably via reduced activity of the enzyme responsible for 2-AG synthesis, DAGLα.
TRP cation channels in cancer
A growing body of evidence implicates members of the TRP family, including TRPV1, TRPV2, and TRPM8, in calcium-mediated signal transduction that regulates proliferation, migration, and metastasis of cancer cells (reviewed by Déliot and Constantin, 2015; Yee, 2015). As discussed previously, numerous cannabinoids bind to, and modify, the activity of TRP channels. We review TRP-mediated activity of several cannabinoids in cancer below.
Vanilloid receptor type 1 (TRPV1) in cancer
TRPV1 Ca++ channels are located on the plasma membrane and other subcellular organelles including the endoplasmic reticulum (ER). TRPV1 regulates intracellular Ca++ levels by controlling Ca++ movement across the plasma membrane and its release from the ER and sarcoplasmic reticulum (Gallego-Sandín et al., 2009). Establishing the relationship between TRPV1 expression levels, and characteristics such as the tumor grade (predicted aggressiveness of the tumor) or tumor stage (tumor size and degree of spread), aids in determining its potential role in cancer. It has been reported that TRPV1 expression increased with increasing tumor grade in prostate cancer biopsies (Czifra et al., 2009), but it was inversely correlated with tumor stage in bladder cancer (Lazzeri et al., 2005) suggesting that the relationship between TRPV1 expression and tumor behavior may be tumor-type specific. Other studies provide evidence that TRPV1 activation is necessary for cannabinoid-induced tumor death. The endocannabinoid, AEA, decreased the viability of cervical cancer cells that overexpressed TRPV1, CB1, and CB2. In these cells, the antiproliferative effect of AEA was counteracted by blockade of TRPV1 but not by antagonism of the CB1 or CB2 receptors (Contassot et al., 2004a). Similarly, AEA initiated TRPV1-dependent, CB receptor-independent apoptosis in human glioma cells (Contassot et al., 2004b). Based upon these interesting findings, further investigation should uncover roles of TRPV1 in cancer and identify other cannabinoid agonists that decrease cancer growth by targeting this pathway.
Vanilloid receptor type 2 (TRPV2) in cancer
The specific role of TRPV2 in carcinogenesis appears to differ according to tumor type. In urothelial carcinoma, TRPV2 expression increased with increasing tumor stage and grade (Caprodossi et al., 2008). However, in hepatocellular carcinoma, TRPV2 expression was lower in poorly differentiated (compared to well-differentiated) tumors (Liu et al., 2010). In glioblastoma multiforme, elevated TRPV2 expression correlated with increased patient survival (Alptekin et al., 2015) while increased TRPV2 expression was associated with poor survival in esophageal squamous cell carcinoma (Zhou et al., 2014). Although the aforementioned studies demonstrate that the role of TRPV2 in cancer progression is unclear, research that examined the impact of cannabinoids on this cation channel demonstrated that TRPV2 activation decreased tumor cell survival and sensitized tumor cells to clinically available chemotherapeutic agents. The phytocannabinoid, CBD, increases inward movement of Ca++, but it has low affinity interactions with CB1 and CB2 receptors (De Petrocellis et al., 2011; McPartland et al., 2015). In glioblastoma, CBD increased the plasma membrane expression of TRPV2 and prevented cell resistance to carmustine (BCNU), doxorubicin, and temozolmide (Nabissi et al., 2013). This sensitization to cytotoxic chemotherapeutics was prevented by siRNA-mediated disruption of TRPV2 expression. Consistent with this finding, the antiproliferative effects of CBD in stem-like glioma cells were reversed in the presence of the Ca++ channel blocker, ruthenium red (RR), and the selective TRPV2 blocker, tranilast (Nabissi et al., 2015). As anticipated, antagonists of CB1 (AM251) and CB2 (AM630) were not able to rescue cells from cell death (Nabissi et al., 2015). Moreover, the sensitivity of multiple myeloma cells to bortezomib was heightened by cell exposure to CBD in a TRPV2-dependent manner (Hashimoto et al., 1986). These findings suggest calcium mobilization regulated by TRPV2 is essential for CBD-mediated cell death. Further investigation of the activity of TRPV2 in cancer is needed to determine the feasibility of utilizing cannabinoids to target this cation channel as a therapeutic strategy.
Transporters in CNS
Transporters function to actively facilitate movement of molecules across membranes against concentration and osmotic gradients. Drugs that target transporters exert effects through altering distribution of other molecules that, in turn, alter membrane potential of excitable membranes and/or terminate action of transmitters that are regulated by uptake. With respect to cannabinoid signaling, evidence suggests that transport may contribute to anandamide signal termination, in addition to metabolism by FAAH (Nicolussi and Gertsch, 2015). The anandamide transporter remains a target for development of selective inhibitors (Nicolussi et al., 2014).
Adding to the promiscuous interaction of anandamide with non-CB1/CB2 targets is evidence for inhibition of a Na+/Ca++ exchange pump (Kury et al., 2014). This inhibition was of both influx and efflux, and was not altered by CB1- and CB2-selective antagonists, pertussis toxin or guanyl nucleotide analogs, consistent with a direct interaction with the transporter. Although this transporter is most relevant to cardiac function, similar transporters are important to CNS activity, and these results suggest potential anandamide neuronal efficacy related to ion transport.
Part of the cannabinoid effects on glycinergic signaling discussed above (section Glycine Receptors in CNS) may in part be attributable to interaction with glycine transporters. In the case of glycine transporter GLYT1a, arachidonic acid and anandamide have interesting opposing modulatory actions (Pearlman et al., 2003). Interestingly, evidence that upregulation of GLT-1, the transporter for the excitatory transmitter glutamate, in preventing cannabinoid dependence, is also emerging (Gunduz et al., 2011).
As discussed above in the context of adenosine (section Adenosine Receptors in CNS), evidence demonstrates that the phytocannabinoid CBD antagonizes an adenosine transporter, reducing inflammatory responses (Carrier et al., 2006). This activity was absent in adenosine A2A knockout mice, and reversed by a selective antagonist, implicating indirect A2A agonism, via inhibition of transporter-mediated signal termination, as the mechanism of CBD-mediated immunosuppression.
While not demonstrating a direct interaction, studies of effects of the synthetic CB1/CB2 agonist, WIN, have revealed that dosages effective in reducing locomotor activity (0.1–1 mg/kg) also decrease expression of dopamine transporters (Fanarioti et al., 2015). Transporter densities were measured by in situ [3H]-WIN35428 binding. Decreased levels were observed in several brain regions relevant to drug abuse including: nucleus accumbens core and shell; substantia nigra and; ventral tegmentum.
Enzymes in CNS and cancer
Endocannabinoid synthesis and metabolism in CNS
The elaborate study of phytocannabinoids discussed above in the context of TRP channels [section Transient Receptor Potential (TRP) Cation Channels in CNS] in CNS also included evaluation of the same series of compounds for effects on activity of enzymes related to endocannabinoid signaling—including those responsible for endocannabinoid production (DAGLα), metabolism [monoacylglycerol lipase (MAGL), fatty acid amide hydrolase (FAAH), and N-acylethanolamine acid amide hydrolase (NAAA)] and uptake (anandamide cellular uptake [ACU]). Results demonstrated that a few of the compounds inhibit DAGLα with a rank order of potency CBDV = CBDA > CBGA = THCA = CBDVA (EC50s ranged from 17 to 35 μM, De Petrocellis et al., 2011). Both CBC and CBG partially inhibit MAGL activity with efficacies of ~50% and IC50s = 50 and 95 μM, respectively. THCA is a more fully-effective MAGL inhibitor with IC50 = 46 μM. Only CBD was found to inhibit FAAH at concentrations under 50 μM (IC50 = 15 μM). Only CBDA inhibited NAAA, the enzyme responsible for degradation of the endocannabinoid-like compound N-palmitoylethanolamine (PEA), doing so with IC50 = 23 μM.
Fatty acid amide hydrolase (FAAH) in cancer
FAAH is a membrane-bound serine hydrolase that catabolizes naturally occurring fatty acid amides including AEA, OEA, and PEA to fatty acids plus ethanolamine (Cravatt et al., 1996, 2001). FAAH regulates the levels of its main cannabinoid substrate AEA, and it is overexpressed in different types of cancer. FAAH expression was elevated in lung adenocarcinoma compared to non-malignant respiratory epithelial cells (Ravi et al., 2014). In prostate cancer cells, FAAH was overexpressed, and the upregulated FAAH levels correlated with poor patient prognosis (Thors et al., 2010). In addition, AEA levels and FAAH expression and activity were elevated in human colorectal cancer tissue compared to non-tumor colon tissue (Chen et al., 2015). Consistent with findings demonstrating that FAAH is overexpressed in cancer, in vitro and in vivo studies revealed that the antitumor activity of endocannabinoids was increased by inhibiting FAAH activity. AEA-mediated death in HT29 colorectal cancer cells was enhanced by its co-administration with the FAAH inhibitor, MAFP (Patsos et al., 2005). Similarly, the apoptotic effect of AEA was increased by co-exposure to the FAAH inhibitor URB597 in non-melanoma skin cancer cells (Kuc et al., 2012). In addition, FAAH inhibition with URB597 prevented AEA degradation and increased its cytotoxicity in neuroblastoma cells (Hamtiaux et al., 2011). The reduction in cell viability caused by the co-administration of AEA and URB597 was found to be independent of CB1, TRPV1, PPAR-α, PPAR-γ, and GPR55 receptor activity (Hamtiaux et al., 2011). These findings suggest that increasing cellular levels of AEA, by decreasing FAAH activity, allows greater quantities of this endocannabinoid to interact with its molecular targets (thereby enhancing its antitumor activity).
Monoacylglycerol lipase (MAGL) in cancer
MAGL is a membrane-associated serine hydrolase that metabolizes monoacylglycerols (MAG) to free fatty acids (FFA) plus glycerol. MAGL is a prominent regulator of the levels of the endocannabinoid, 2-AG. MAGL was found to be elevated in aggressive cancer cells (compared to non-aggressive cells) (Nomura et al., 2010). MAGL increased FFA production to promote tumor growth, tumor cell migration, and tumor invasion. Inhibiting MAGL expression with shRNAs, or blocking its activity with JZL184, reduced tumor cell migration in a manner that was not dependent on the CB1 or CB2 receptors (Nomura et al., 2010). These data indicate that the tumor promoting metabolic products of 2-AG increase cancer cell survival. As such, examining MAGL, 2-AG and FFA levels in different tumor types, and identifying cellular targets of FFAs, will be an important step toward understanding whether this pathway can be exploited for therapeutic benefit.
Cycloxygenase-2 (COX-2) in cancer
COX-2 oxygenates arachidonic acid (AA) to prostaglandin H2 (PGH2) that is further metabolized by PG synthase-E, -F2α, and -D to PGE2, PGF2α, and PGD2, respectively (Rouzer and Marnett, 2009). PGD2 is then dehydrated to J-series PGs. Substantial evidence points to a role for COX-2 and its products, notably PGE2, in the growth of epithelial tumors of the colon, lung, breast, skin, and other organs (Jiao et al., 2014; Ma et al., 2015; Majumder et al., 2015; Kiraly et al., 2016). Pro-inflammatory COX-2 and PGE2 promote cell proliferation, angiogenesis, and cell migration by activating PGE2 receptors which increase oncogene and cytokine activity (Rundhaug et al., 2007, 2011). In contrast, cannabinoids possess anti-inflammatory activity (Burstein and Zurier, 2009). The mechanism by which cannabinoids suppress inflammatory cascades is a matter of debate; although, some studies suggest that cannabinoids block cyclooxygenase activity. Ruhaak et al. reported that several cannabinoids isolated from Cannabis sativa inhibited COX-2 activity and reduced prostaglandin generation (Ruhaak et al., 2011). Tetrahydrocannabinolic acid (THC-A), cannabigerol (CBG), and cannabigerolic acid (CBGA) inhibited the activity of COX-2 in a cell-free enzyme assay with IC50 values of 6.3 × 10−4, 2.7 × 10−4, and 2.0 × 10−4 M, respectively. However, in TNFα stimulated cancer cells, a significant reduction in COX-2 activity was not observed (Ruhaak et al., 2011). In a different study, CBDA was a potent and selective inhibitor of COX-2 activity with an IC50 value of 2.2 μM and an IC50 value of 20 μM for COX-1 (Takeda et al., 2008). These authors suggested that the inhibitory effect of cannabinoids on COX-2 activity was due to the presence of the salicylic acid moiety in the chemical structure of CBDA. Additional studies are needed to uncover the biological and clinical relevance of cannabinoid-mediated cyclooxygenase inhibition.
In contrast to the inhibitory effects of cannabinoids on COX-2 activity mentioned previously, other reports indicate that cannabinoids upregulate COX-2 expression. A series of studies by Hinz et al. demonstrated that cannabinoid-induced COX-2 expression increased the synthesis of arachidonic acid-derived prostaglandins that promoted cell death (Hinz et al., 2004a,b). In lung cancer cells, CBD increased COX-2 expression and the synthesis of D- and J- series prostaglandins which initiated tumor cell apoptosis (Ramer et al., 2013). Met-AEA also increased COX-2 expression in a manner that was dependent on lipid rafts, ceramide, and the activation of p38 and p42/44 MAPK (Ramer et al., 2003; Hinz et al., 2004a). However, Met-AEA-induced apoptosis was not reversed by CB1 (AM251), CB2 (AM630), or TRPV1 (capsazepine) receptor antagonists in human neuroglioma cells (Hinz et al., 2004b). It was also reported that Met-AEA increased ceramide synthesis, COX-2 expression and the production of proapoptotic PGD2 in human cervical carcinoma cells (Eichele et al., 2009). In this study, inhibiting the production of D-series PGs using siRNA directed against lipocalin-PGDS or siRNA against PPARγ prevented Met-AEA-induced apoptosis. A reversal in Met-AEA-induced apoptosis, however, was not observed in the presence of the receptor antagonists, AM251, AM630 or capsazepine (Eichele et al., 2009). In contrast, Garder et al. determined that Met-AEA increased lung tumor growth in vitro and in vivo (Gardner et al., 2003). This pro-tumorigenic effect was blocked by pharmacological inhibitors of COX-2, p38, and p42/44, but it was not affected by the CB1 (SR141716) and CB2 (SR144528) receptor antagonists (Gardner et al., 2003).
AEA is also a substrate of COX-2, and, as a result, is cytotoxic in tumor cells that overexpress COX-2. AEA has an unmodified arachidonate backbone, and it is therefore susceptible to oxidative metabolism by COX-2 to form prostaglandin-ethanolamines, also known as prostamides (PM) (Yu et al., 1997; Kozak et al., 2002; Soliman et al., 2016). The metabolic products of AEA, prostamides-E2, F2α, and -D2, do not bind to prostaglandin receptors, and they are metabolically stable relative to prostaglandins derived from arachidonic acid (Kozak et al., 2001; Matias et al., 2004). Recently, we demonstrated that AEA was also metabolized by COX-2 to novel J series prostamides that initiated tumor cell apoptosis (Kuc et al., 2012; Ladin et al., 2017). Because epithelial cancer cells typically overexpress COX-2, AEA was metabolized to pro-apoptotic J-series prostamides in tumor cells, but J-series prostamides were not detected in non-tumor cells which had low endogenous levels of COX-2 (Soliman et al., 2016). Blockade of AEA degradation with the FAAH inhibitor, URB597, increased J-series prostaglandin synthesis and apoptosis; however, inhibition of CB1, CB2, and TRPV-1 receptors using selective antagonists did not reverse this effect (Soliman and Van Dross, 2016). Furthermore, cell treatment with exogenous 15-deoxy, Δ12, 14 prostamide J2 (15d-PMJ2), the most abundant J-series product of AEA metabolism by COX-2, also caused cell death in vitro and in vivo (Ladin et al., 2017). Reports by Pastos et al. also demonstrated that COX-2 was required to induce death in cancer cells treated with AEA (Patsos et al., 2005, 2010). Prostamides E2 and D2 were noted to cause tumor cell death while blockade of COX-2 activity or expression partially reversed this antiproliferative effect. However, AEA-mediated cell death was not blocked by CB1, CB2, or TRPV1 receptor antagonists (Patsos et al., 2010). Thus, COX-2 is a critical regulator of CB receptor-independent cannabinoid activity in cancer cells. These studies demonstrate that cannabinoid metabolism and signal transduction is modulated by COX-2 and that COX-2 is also inhibited by molecules within the cannabinoid family. This implies that a complex relationship exists between cannabinoids, the endocannabinoid system, and cyclooxygenases in cancer and potentially in other organ systems.
Phosphatases in cancer
The restoration of phosphatase expression and activity is a desirable effect of chemotherapeutic agents, as these enzymes dephosphorylate kinases, and other pro-tumorigenic proteins, that are often constitutively active in cancer (Perrotti and Neviani, 2013). The anticancer activity of WIN55212-2 was reported to be mediated by phosphatases in a cannabinoid receptor-independent manner (Sreevalsan et al., 2011; Sreevalsan and Safe, 2013). WIN55212-2 increased the expression of several phosphatases including dual specificity phosphatases 1 and 10 (DUSP1, DUSP10) and protein tyrosine phosphatase, non-receptor type 6 (PTPN6) in LNCaP prostate cancer cells (Sreevalsan et al., 2011). WIN55212-2 also increased PTPN6 expression and protein phosphatase A2 (PPA2) activity in SW480 colon cancer cells (Sreevalsan et al., 2011; Sreevalsan and Safe, 2013) and reduced the expression of the pro-tumorigenic transcription factors, specificity protein 1, 3, and 4 (SP1, SP3, and SP4). The effect of WIN55212-2 on SP expression and apoptosis was dependent on phosphatase PPA2, a prominent serine-threonine phosphatase in eukaryotic cells that regulates the cell cycle and apoptosis. Specifically, it was determined that WIN55212-2 increased the activity of PPA2 and activation of the Sp repressor, ZBTB10, thereby inhibiting Sp expression through a pathway that was regulated by micro RNA-27a (miR-27a). However, this cytotoxic activity was not prevented by blockade of CB1 or CB2 receptor activity (Sreevalsan and Safe, 2013). These reports suggest that cannabinoid activity is modulated by phosphatases independent of the cannabinoid receptors. Phosphatases have significant impacts on cellular behavior, because these enzymes control kinase activity. Therefore, examination of the role of phosphatases in cannabinoid activity may allow these proteins to be targeted to improve the action of cannabinoids.
Other non-receptor targets in cancer
Lipid rafts and ceramide
Lipid rafts are glycoprotein microdomains enriched in cholesterol and sphingolipids. Lipid rafts serve as organization centers that promote interactions between proteins to aid in intracellular signal transduction (Simons and Ikonen, 1997). Ceramide is a membrane lipid that displaces cholesterol from lipid rafts, enhances membrane rigidity, stabilizes rafts and induces formation of large raft domains (“platforms”) in plasma membranes (Simons and Ikonen, 1997; Megha and London, 2004). It has been reported that the antitumor activity of cannabinoids is regulated by ceramide and lipid rafts which control CB1 and/or CB2-mediated signal transduction (Bari et al., 2005; Sarnataro et al., 2006). However, other studies demonstrate that ceramide and lipid rafts transmit lethal cannabinoid signals independent of CB1 and CB2. DeMorrow et al., found that AEA increased ceramide synthesis and localization of the death receptor/ligand, Fas/FasL, to lipid rafts leading to cholangiocarcinoma cell death (DeMorrow et al., 2007; Huang et al., 2011). In these studies, selective antagonists of CB1 and CB2 did not inhibit the cytotoxicity of AEA. In a different report, lipid raft disruptors completely attenuated AEA-mediated cell death in neuroblastoma cells (Hamtiaux et al., 2011). This cytotoxicity was not blocked by pharmacological antagonism of CB1, CB2, GPR55, TRPV1, or PPAR⋎. Similarly, the disruption of lipid rafts, but not the antagonism of CB1, CB2, or TRPV1 receptors, prevented AEA cytotoxicity in cutaneous melanoma cells (Adinolfi et al., 2013). In addition, Sarker and Maruyama found that disruption of lipid rafts blocked AEA-induced oxidative stress and apoptosis independent of the CB1, CB2, or TRPV1 receptors in different cell lines (Sarker and Maruyama, 2003). Consistent with these findings, Met-AEA-induced apoptosis was inhibited by the use of ceramide synthase inhibitors and lipid raft disruptors but not by CB1, CB2, or TRPV1 receptor antagonists in human neuroglioma and cervical carcinoma cells (Hinz et al., 2004b; Eichele et al., 2009). Moreover, WIN55212-2, caused lipid raft-mediated cell death in cultured melanoma cells in a CB1/CB2 receptor-independent manner (Scuderi et al., 2011). Collectively, these findings suggest that cannabinoids reduce tumor cell viability by modulating lipid rafts through cannabinoid receptor-dependent and -independent pathways.
Oxidative stress in cancer
Reactive oxygen species (ROS) are second messenger signal transduction molecules in eukaryotic cells. Low levels of ROS are needed for physiological processes such as immune defense against pathogens, mitogenic responses, and maintenance of cellular homeostasis (Finkel, 1998; Kim et al., 1998; Dröge, 2002). Physiologic levels of ROS are maintained by a balance between antioxidant and pro-oxidant systems in the cell. However, when antioxidant systems become overwhelmed, oxidative stress occurs. Cancer cells contain supraphysiological levels of ROS that activate signaling pathways which promote cell proliferation, survival, angiogenesis, and metastasis (Nourazarian et al., 2014). However, excessively high ROS levels causes oxidative damage to cellular proteins, lipids, and DNA that triggers cell cycle arrest and cell death (Kaur et al., 2014). Structurally distinct cannabinoids can inhibit tumor cell survival by generating high levels of ROS. CBD decreased glutathione (GSH) levels and increased GSH reductase and GSH peroxidase content thereby causing cytotoxic oxidative stress in glioma cells (Massi et al., 2006). The cytotoxicity of CBD was abrogated by the antioxidant, α-tocopherol; however, blockade of CB receptor activity or ceramide synthesis did not significantly alter CBD cytotoxicity (Massi et al., 2004). Moreover, CBD-induced oxidative stress and cell death occurred in tumorigenic U87 glioma cells but not in non-tumorigenic primary glial cells (Massi et al., 2004), suggesting ROS elicits tumor-selective cytotoxicity. Ligresti et al. also found that antioxidants (α-tocopherol and vitamin C), but not CB receptor antagonists, blocked the antiproliferative activity of CBD in breast adenocarcinoma cells (Ligresti et al., 2006). Similarly, Shrivastava et al. determined that CBD increased ROS production that initiated autophagy and apoptosis independent of CB1, CB2, or TRPV1 in breast cancer cells (Shrivastava et al., 2011).
In several reports, the endocannabinoid, AEA, induced ROS-dependent, CB receptor-independent cell death. Oxidative stress and apoptosis in AEA treated non-melanoma skin cancer cells were prevented by the antioxidant, N-acetyl cysteine (NAC) (Van Dross, 2009). Blockade of CB1 and CB2 receptor activity did not rescue cells from AEA-mediated oxidative stress or apoptosis (Soliman and Van Dross, 2016). Similarly, in colon cancer cells, AEA-induced cell death and apoptosis was reversed by the use of antioxidants but not by CB1 or CB2 receptor antagonists (Gustafsson et al., 2009; Soliman, 2015). These findings suggests that oxidative stress regulates cannabinoid activity through pathways that circumvent the cannabinoid receptors.
Endoplasmic reticulum (ER) stress in cancer
ER stress occurs when the capacity of the cell to fold proteins is exceeded by the protein folding load resulting in the accumulation of unfolded proteins in ER (the unfolded protein response, UPR) (Holcik and Sonenberg, 2005). The UPR is an integrated intracellular signal transduction pathway that is regulated by three ER-resident stress sensors: double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase (PERK), activating transcription factor-6 (ATF6), and inositol requiring kinase-1 (IRE1) (Lin et al., 2008; Chakrabarti et al., 2011). Activated PERK phosphorylates the α subunit of eukaryotic initiation factor 2α (eIF2α), resulting in a reduction in global translation with a preferential increase in the expression of activated transcription factor 4 (ATF4)-regulated genes (reviewed in Marciniak et al., 2006). Activation of PERK, ATF6, and IRE1 promotes cell survival by decreasing protein synthesis and by increasing protein folding and degradation to reestablish ER homeostasis. However, excessive or prolonged ER stress activates cell death pathways primarily by increasing expression of the transcription factor, C/EBP homologous protein 10 (CHOP10, also known as the growth arrest and DNA damage-inducible gene 153 [GADD153]) which transcribes pro-apoptotic genes (reviewed in Marciniak and Ron, 2006). Several studies have shown that the ER stress pathway mediates the anticancer activity of cannabinoids. The phytocannabinoid, THC, increased phosphorylation of eIF2α and the expression of the CHOP10 transcriptional product, TRB3, in hepatocellular carcinoma cells (HEPG2), human glioma cells, and RasV12/E1A-transformed mouse embryonic fibroblasts (MEF, Salazar et al., 2009, 2013; Vara et al., 2013). TRB3 is known to inhibit cancer cell proliferation by inactivating AKT/ mammalian target of the rapamycin complex 1 (mTORC) signal transduction (Ohoka et al., 2005). In human glioma cells treated with THC, it was determined that TRB3 was required for inhibition of Akt/mTORC1 signaling and the induction of autophagy and apoptosis (Salazar et al., 2009). Furthermore, unlike Trib3+/+ (also known as TRB3) MEF cells, Trib3−/− MEFs were resistant to cell death caused by THC (Salazar et al., 2013). Similar results were observed when the two cell types were injected subcutaneously in nude mice that were subsequently treated with THC. THC reduced the growth of Trib3+/+ xenograft tumors; however, this effect was not seen in mice engrafted with Trib3−/− cells (Salazar et al., 2013). WIN55212-2 also induced ER stress and increased the expression of CHOP10 and TRB3 in cancer cells (Wasik et al., 2011; Notaro et al., 2014; Pellerito et al., 2014). In addition, WIN55212-2 reduced the viability of osteosarcoma cells by initiating cytotoxic autophagy (Notaro et al., 2014). Genetic ablation of CHOP10 using siRNA prevented WIN-induced autophagic cell death. WIN55212-2 also caused autophagy and apoptosis in human colorectal cancer cells that was prevented by reducing the expression of CHOP10 with siRNA (Pellerito et al., 2014). Interestingly, Wasik et al. reported that WIN55212-2-induced autophagic cell death in Mantle cell lymphoma (MCL) cells occurred independent of the CB1 and CB2 receptors (Wasik et al., 2011). Consistent with these observations, the ER stress inhibitors, phenylbutyric acid (PBA) and salubrinal, prevented apoptosis in non-melanoma skin cancer cells treated with AEA. However, selective antagonism of the CB1 and CB2 receptor failed to inhibit AEA-induced ER stress or apoptosis (Soliman and Van Dross, 2016; Soliman et al., 2016). Structurally diverse cannabinoids initiate cytotoxic ER stress in cancer cells independent of the cannabinoid receptors. Because the ER stress pathway is a primary regulator of protein folding and synthesis in cells, understanding the impact of cannabinoids on the ER stress pathway is of vital importance for development of effective therapeutic agents.
Conclusions
Cannabinoids have emerged as prominent modulators of diverse physiological and pathological processes. As such, efforts are underway to examine the efficacy of cannabinoid agonist and antagonists as therapeutic agents (McPartland et al., 2014; Russo, 2016; Toguri et al., 2016; Zhou et al., 2016). Cannabinoids primarily exert their effects through the cannabinoid receptors (CB1 and CB2); however, other receptors and molecular targets are now known to be important for their activity. The endocannabinoid system (ECS), which is composed of cannabinoids, cannabinoid receptors, and molecules involved in cannabinoid synthesis, uptake, and degradation, was initially described in the CNS. As a consequence, mechanisms of cannabinoid activity in the CNS are well-characterized compared to what is known about cannabinoid activity in cancer. This review sought to describe the cannabinoid receptor-independent actions of cannabinoids in systems where cannabinoid activity was both well- and poorly-defined. The studies examined herein indicate that, in the CNS, the activity of cannabinoids could be regulated by ion channels, enzymes, transporters, and receptors other than CB1 and CB2. In cancer cells, the CB receptor-independent activity of cannabinoids was found to be mediated by receptors, ion channels, lipid rafts, enzymes, and cellular stressors. Interestingly, in both the CNS and in cancer, several components of the ECS were important for CB1/CB2 receptor-independent activity including GRP55, TRP channels, FAAH, and MAGL. The dependence of cannabinoid activity on these common targets in dissimilar systems reinforces the importance of the ECS in cannabinoid signaling and activity.
Studies described in this review also reveal cannabinoid targets that are unique to the CNS and cancer. In the CNS, cannabinoids generated different biological responses through modulation of opioid, serotonin, adenosine, amino acid, and cholinergic receptors. Opioid, serotonin, NMDA, and GABA receptors have been implicated in cannabinoid-mediated modulation of cancer pain and chemotherapy-induced emesis (Maccarrone et al., 2009; Gu et al., 2011; Khasabova et al., 2011; Bolognini et al., 2013; Higgins et al., 2013; Ward et al., 2014). Future investigations should determine the role of these receptors in cannabinoid-induced tumor cell death—particularly in CNS malignancies. Research in cancer cells also demonstrated that the activity of cannabinoids could be mediated by lipid rafts, ER stress, oxidative stress, and enzymes. The involvement of lipid rafts, ROS and enzymes (COX-2 and phosphatases) in CNS cannabinoid signaling have been described in a few studies (Cannich et al., 2004; Maccarrone et al., 2009; Oddi et al., 2012; Penumarti and Abdel-Rahman, 2014b; Colín-González et al., 2015); however, the impact of the ER stress pathway on cannabinoid activity in the CNS remains an open question.
The diversity of cellular targets for cannabinoid ligands helps to explain the wide range of physiological responses to cannabinoid drugs. This range of efficacies also suggests that opportunities for employing cannabinoid-based therapies have only just begun to be explored. More needs to be learned about CB1/CB2-independent targets of cannabinoids to identify off-receptor effects, mechanisms of tolerance/ resistance and to explain unanticipated outcomes. In addition, by examining common and distinct cannabinoid targets in different biological systems, unique mechanisms of drug action can be uncovered. Among these biological systems are, notably, animals more primitive than chordates that lack the classical cannabinoid receptors (Elphick, 2012). Despite absence of these receptors, there is significant literature documenting behavioral effects in these organisms (reviewed by Soderstrom, 2009). To the extent that some signaling systems are conserved across vertebrate and invertebrate species (e.g., serotonergic signaling, Tierney, 2001) these behavioral effects may involve subsets of the array of non-CB1/CB2 targets reviewed here. Thus, invertebrate species, without interfering classical cannabinoid receptors (and therefore representing “clean” systems), hold promise for studying mechanisms of off-receptor cannabinoid effects.
Statements
Author contributions
KS and ES contributed equally to this work. KS, ES, and RV contributed to the conception and design of the review, the acquisition of data, and the analysis and interpretation of the data. All authors participated in drafting and revising the manuscript. All authors approved of the final version of the submitted manuscript.
Funding
This work was supported by a Brody Brothers Endowment Grant to RV.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AbdalhameedM. M.ZhaoP.HurstD. P.ReggioP. H.AboodM. E.CroattM. P. (2017). Structure-activity relationships of benzothiazole GPR35 antagonists. Bioorg. Med. Chem. Lett.27, 612–615. 10.1016/j.bmcl.2016.12.012
2
AdinolfiB.RomaniniA.VanniA.MartinottiE.ChiccaA.FogliS.et al. (2013). Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol.718, 154–159. 10.1016/j.ejphar.2013.08.039
3
AkinsholaB. E.TaylorR. E.OgunseitanA. B.OnaiviE. S. (1999). Anandamide inhibition of recombinant AMPA receptor subunits in Xenopus oocytes is increased by forskolin and 8-bromo-cyclic AMP. Naunyn. Schmiedebergs Arch. Pharmacol.360, 242–248.
4
AkopianA. N.RuparelN. B.JeskeN. A.PatwardhanA.HargreavesK. M. (2009). Role of ionotropic cannabinoid receptors in peripheral antinociception and antihyperalgesia. Trends Pharmacol. Sci.30, 79–84. 10.1016/j.tips.2008.10.008
5
AlptekinM.ErogluS.TutarE.SencanS.GeyikM. A.UlasliM.et al. (2015). Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med.36, 9209–9213. 10.1007/s13277-015-3577-x
6
AndradasC.CaffarelM. M.Pérez-GómezE.SalazarM.LorenteM.VelascoG.et al. (2011). The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene30, 245–252. 10.1038/onc.2010.402
7
AneethaH.O'DellD. K.TanB.WalkerJ. M.HurleyT. D. (2009). Alcohol dehydrogenase-catalyzed in vitro oxidation of anandamide to N-arachidonoyl glycine, a lipid mediator: synthesis of N-acyl glycinals. Bioorg. Med. Chem. Lett.19, 237–241. 10.1016/j.bmcl.2008.10.087
8
AnsarullahLuY.HolsteinM.DeRuyterB.RabinovitchA.GuoZ. (2013). Stimulating β-cell regeneration by combining a GPR119 agonist with a DPP-IV inhibitor. PLoS ONE8:e53345. 10.1371/journal.pone.0053345
9
BarannM.MolderingsG.BrüssM.BönischH.UrbanB. W.GöthertM. (2002). Direct inhibition by cannabinoids of human 5-HT3A receptors: probable involvement of an allosteric modulatory site. Br. J. Pharmacol.137, 589–596. 10.1038/sj.bjp.0704829
10
BaranowskaU.GöthertM.RudźR.MalinowskaB. (2008). Methanandamide allosterically inhibits in vivo the function of peripheral nicotinic acetylcholine receptors containing the α7-subunit. J. Pharmacol. Exp. Ther.326, 912–919. 10.1124/jpet.108.140863
11
BariM.BattistaN.FezzaF.Finazzi-AgròA.MaccarroneM. (2005). Lipid rafts control signaling of type-1 cannabinoid receptors in neuronal cells. Implications for anandamide-induced apoptosis. J. Biol. Chem.280, 12212–12220. 10.1074/jbc.M411642200
12
BaurR.KielarM.RichterL.ErnstM.EckerG. F.SigelE. (2013). Molecular analysis of the site for 2-arachidonylglycerol (2-AG) on the β2 subunit of GABAA receptors. J. Neurochem.126, 29–36. 10.1111/jnc.12270
13
BegumS.EmaniN.CheungA.WilkinsO.DerS.HamelP. A. (2005). Cell-type-specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene24, 1860–1872. 10.1038/sj.onc.1208315
14
BifulcoM.LaezzaC.PortellaG.VitaleM.OrlandoP.PetrocellisL. D.et al. (2001). Control by the endogenous cannabinoid system of ras oncogene-dependent tumor growth. FASEB J.15, 2745–2747. 10.1096/fj.01-0320fje
15
BologniniD.RockE. M.ClunyN. L.CascioM. G.LimebeerC. L.DuncanM.et al. (2013). Cannabidiolic acid prevents vomiting in Suncus murinus and nausea-induced behaviour in rats by enhancing 5-HT1A receptor activation. Br. J. Pharmacol.168, 1456–1470. 10.1111/bph.12043
16
BoniniJ. A.BorowskyB. E.AdhamN.BoyleN.ThompsonT. O. (2002). Methods of Identifying Compounds that Bind to SNORF25 Receptors. Available online at: http://www.google.com/patents/US6468756 (Accessed July 8, 2015).
17
BorrelliF.PaganoE.RomanoB.PanzeraS.MaielloF.CoppolaD.et al. (2014). Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a Cannabis-derived non-psychotropic cannabinoid. Carcinogenesis35, 2787–2797. 10.1093/carcin/bgu205
18
BouskilaJ.JavadiP.CasanovaC.PtitoM.BouchardJ.-F. (2013). Rod photoreceptors express GPR55 in the adult vervet monkey retina. PLoS ONE8:e81080. 10.1371/journal.pone.0081080
19
BradshawH. B.RimmermanN.HuS. S.BentonV. M.StuartJ. M.MasudaK.et al. (2009). The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem.10:14. 10.1186/1471-2091-10-14
20
BreivogelC. S.GriffinG.Di MarzoV.MartinB. R. (2001). Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol. Pharmacol.60, 155–163. 10.1124/mol.60.1.155
21
BrickleyS. G.ModyI. (2012). Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron73, 23–34. 10.1016/j.neuron.2011.12.012
22
BursteinS. (2005). PPAR-gamma: a nuclear receptor with affinity for cannabinoids. Life Sci.77, 1674–1684. 10.1016/j.lfs.2005.05.039
23
BursteinS. H.ZurierR. B. (2009). Cannabinoids, endocannabinoids, and related analogs in inflammation. AAPS J.11, 109–119. 10.1208/s12248-009-9084-5
24
BursteinS. H.McQuainC. A.RossA. H.SalmonsenR. A.ZurierR. E. (2011). Resolution of inflammation by N-arachidonoylglycine. J. Cell. Biochem.112, 3227–3233. 10.1002/jcb.23245
25
Busquets GarciaA.Soria-GomezE.BellocchioL.MarsicanoG. (2016). Cannabinoid receptor type-1: breaking the dogmas. F1000Research5:99010.12688/f1000research.8245.1
26
CabralG. A.RabornE. S.GriffinL.DennisJ.Marciano-CabralF. (2008). CB2 receptors in the brain: role in central immune function. Br. J. Pharmacol.153, 240–251. 10.1038/sj.bjp.0707584
27
CampbellM. J.CarlbergC.KoefflerH. P. (2008). A role for the PPARgamma in cancer therapy. PPAR Res.2008:314974. 10.1155/2008/314974
28
CamposA. C.de Paula SoaresV.CarvalhoM. C.FerreiraF. R.VicenteM. A.BrandãoM. L.et al. (2013). Involvement of serotonin-mediated neurotransmission in the dorsal periaqueductal gray matter on cannabidiol chronic effects in panic-like responses in rats. Psychopharmacology226, 13–24. 10.1007/s00213-012-2878-7
29
CannichA.WotjakC. T.KamprathK.HermannH.LutzB.MarsicanoG. (2004). CB1 cannabinoid receptors modulate kinase and phosphatase activity during extinction of conditioned fear in mice. Learn. Mem. Cold Spring Harb.11, 625–632. 10.1101/lm.77904
30
CaprodossiS.LucciariniR.AmantiniC.NabissiM.CanesinG.BallariniP.et al. (2008). Transient receptor potential vanilloid type 2 (TRPV2) expression in normal urothelium and in urothelial carcinoma of human bladder: correlation with the pathologic stage. Eur. Urol.54, 612–620. 10.1016/j.eururo.2007.10.016
31
CarrierE. J.AuchampachJ. A.HillardC. J. (2006). Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc. Natl. Acad. Sci. U.S.A.103, 7895–7900. 10.1073/pnas.0511232103
32
CeciC.Proietti OnoriM.MacrìS.LaviolaG. (2015). Interaction between the endocannabinoid and serotonergic system in the exhibition of head twitch response in four mouse strains. Neurotox. Res.27, 275–283. 10.1007/s12640-014-9510-z
33
ChakrabartiA.ChenA. W.VarnerJ. D. (2011). A review of the mammalian unfolded protein response. Biotechnol. Bioeng.108, 2777–2793. 10.1002/bit.23282
34
ChemelB. R.RothB. L.ArmbrusterB.WattsV. J.NicholsD. E. (2006). WAY-100635 is a potent dopamine D4 receptor agonist. Psychopharmacology188, 244–251. 10.1007/s00213-006-0490-4
35
ChenL.ChenH.LiY.LiL.QiuY.RenJ. (2015). Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol. Rep.34, 447–454. 10.3892/or.2015.3973
36
ChiarloneA.BörnerC.Martín-GómezL.Jiménez-GonzálezA.García-ConcejoA.García-BermejoM. L.et al. (2016). MicroRNA let-7d is a target of cannabinoid CB1 receptor and controls cannabinoid signaling. Neuropharmacology108, 345–352. 10.1016/j.neuropharm.2016.05.007
37
CinarR.SzücsM. (2009). CB1 receptor-independent actions of SR141716 on G-protein signaling: coapplication with the mu-opioid agonist Tyr-D-Ala-Gly-(NMe)Phe-Gly-ol unmasks novel, pertussis toxin-insensitive opioid signaling in mu-opioid receptor-Chinese hamster ovary cells. J. Pharmacol. Exp. Ther.330, 567–574. 10.1124/jpet.109.152710
38
Colín-GonzálezA. L.Paz-LoyolaA. L.SerratosI. N.SeminottiB.RibeiroC. A. J.LeipnitzG.et al. (2015). The effect of WIN 55,212-2 suggests a cannabinoid-sensitive component in the early toxicity induced by organic acids accumulating in glutaric acidemia type I and in related disorders of propionate metabolism in rat brain synaptosomes. Neuroscience310, 578–588. 10.1016/j.neuroscience.2015.09.043
39
ColizziM.McGuireP.PertweeR. G.BhattacharyyaS. (2016). Effect of cannabis on glutamate signalling in the brain: a systematic review of human and animal evidence. Neurosci. Biobehav. Rev.64, 359–381. 10.1016/j.neubiorev.2016.03.010
40
ConnorsK. A.ValentiT. W.LawlessK.SackermanJ.OnaiviE. S.BrooksB. W.et al. (2014). Similar anxiolytic effects of agonists targeting serotonin 5-HT1A or cannabinoid CB receptors on zebrafish behavior in novel environments. Aquat. Toxicol. Amst. Neth.151, 105–113. 10.1016/j.aquatox.2013.12.005
41
ContassotE.TenanM.SchnürigerV.PelteM.-F.DietrichP.-Y. (2004a). Arachidonyl ethanolamide induces apoptosis of uterine cervix cancer cells via aberrantly expressed vanilloid receptor-1. Gynecol. Oncol.93, 182–188. 10.1016/j.ygyno.2003.12.040
42
ContassotE.WilmotteR.TenanM.BelkouchM.-C.SchnürigerV.de TriboletN.et al. (2004b). Arachidonylethanolamide induces apoptosis of human glioma cells through vanilloid receptor-1. J. Neuropathol. Exp. Neurol.63, 956–963.
43
CornallL. M.HryciwD. H.MathaiM. L.McAinchA. J. (2015). Direct activation of the proposed anti-diabetic receptor, GPR119 in cardiomyoblasts decreases markers of muscle metabolic activity. Mol. Cell. Endocrinol.402, 72–85. 10.1016/j.mce.2015.01.006
44
CornallL. M.MathaiM. L.HryciwD. H.SimcocksA. C.O'BrienP. E.WentworthJ. M.et al. (2013). GPR119 regulates genetic markers of fatty acid oxidation in cultured skeletal muscle myotubes. Mol. Cell. Endocrinol.365, 108–118. 10.1016/j.mce.2012.10.003
45
CosiC.MannaioniG.CozziA.CarlàV.SiliM.CavoneL.et al. (2011). G-protein coupled receptor 35 (GPR35) activation and inflammatory pain: studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology60, 1227–1231. 10.1016/j.neuropharm.2010.11.014
46
CoxB. M.ChristieM. J.DeviL.TollL.TraynorJ. R. (2015). Challenges for opioid receptor nomenclature: IUPHAR Review 9. Br. J. Pharmacol.172, 317–323. 10.1111/bph.12612
47
CravattB. F.DemarestK.PatricelliM. P.BraceyM. H.GiangD. K.MartinB. R.et al. (2001). Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U.S.A.98, 9371–9376. 10.1073/pnas.161191698
48
CravattB. F.GiangD. K.MayfieldS. P.BogerD. L.LernerR. A.GilulaN. B. (1996). Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature384, 83–87. 10.1038/384083a0
49
CroweM. S.LeishmanE.BanksM. L.GujjarR.MahadevanA.BradshawH. B.et al. (2015). Combined inhibition of monoacylglycerol lipase and cyclooxygenases synergistically reduces neuropathic pain in mice. Br. J. Pharmacol.172, 1700–1712. 10.1111/bph.13012
50
CzifraG.VargaA.NyesteK.MarincsákR.TóthB. I.KovácsI.et al. (2009). Increased expressions of cannabinoid receptor-1 and transient receptor potential vanilloid-1 in human prostate carcinoma. J. Cancer Res. Clin. Oncol.135, 507–514. 10.1007/s00432-008-0482-3
51
DaineseE.OddiS.MaccarroneM. (2010). Interaction of endocannabinoid receptors with biological membranes. Curr. Med. Chem.17, 1487–1499. 10.2174/092986710790980087
52
De PetrocellisL.Di MarzoV. (2010). Non-CB1, non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: focus on G-protein-coupled receptors and transient receptor potential channels. J. Neuroimmune Pharmacol.5, 103–121. 10.1007/s11481-009-9177-z
53
De PetrocellisL.LigrestiA.MorielloA. S.AllaràM.BisognoT.PetrosinoS.et al. (2011). Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol.163, 1479–1494. 10.1111/j.1476-5381.2010.01166.x
54
DéliotN.ConstantinB. (2015). Plasma membrane calcium channels in cancer: alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta1848, 2512–2522. 10.1016/j.bbamem.2015.06.009
55
DeMorrowS.GlaserS.FrancisH.VenterJ.VaculinB.VaculinS.et al. (2007). Opposing actions of endocannabinoids on cholangiocarcinoma growth RECRUITMENT OF Fas AND Fas LIGAND TO LIPID RAFTS. J. Biol. Chem.282, 13098–13113. 10.1074/jbc.M608238200
56
DevinskyO.CilioM. R.CrossH.Fernandez-RuizJ.FrenchJ.HillC.et al. (2014). Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia55, 791–802. 10.1111/epi.12631
57
DevinskyO.CrossJ. H.LauxL.MarshE.MillerI.NabboutR.et al. (2017). Trial of cannabidiol for drug-resistant seizures in the dravet syndrome. N. Engl. J. Med.376, 2011–2020. 10.1056/NEJMoa1611618
58
Di MarzoV.De PetrocellisL. D. (2012). Why do cannabinoid receptors have more than one endogenous ligand?Philos. Trans. R. Soc. Lond. B Biol. Sci.367, 3216–3228. 10.1098/rstb.2011.0382
59
DrögeW. (2002). Free radicals in the physiological control of cell function. Physiol. Rev.82, 47–95. 10.1152/physrev.00018.2001
60
EicheleK.RamerR.HinzB. (2009). R(+)-methanandamide-induced apoptosis of human cervical carcinoma cells involves a cyclooxygenase-2-dependent pathway. Pharm. Res.26, 346–355. 10.1007/s11095-008-9748-3
61
EicheleK.WeinzierlU.RamerR.BruneK.HinzB. (2006). R(+)-methanandamide elicits a cyclooxygenase-2-dependent mitochondrial apoptosis signaling pathway in human neuroglioma cells. Pharm. Res.23, 90–94. 10.1007/s11095-005-8815-2
62
ElphickM. R. (2012). The evolution and comparative neurobiology of endocannabinoid signalling. Philos. Trans. R. Soc. Lond. B. Biol. Sci.367, 3201–3215. 10.1098/rstb.2011.0394
63
FalascaM.FerroR. (2016). Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv. Biol. Regul.60, 88–93. 10.1016/j.jbior.2015.10.003
64
FanP. (1995). Cannabinoid agonists inhibit the activation of 5-HT3 receptors in rat nodose ganglion neurons. J. Neurophysiol.73, 907–910.
65
FanariotiE.MavrikakiM.PanagisG.MitsacosA.NomikosG. G.GiompresP. (2015). Behavioral and neurochemical changes in mesostriatal dopaminergic regions of the rat after chronic administration of the cannabinoid receptor agonist WIN55,212-2. Int. J. Neuropsychopharmacol.18:pyu097. 10.1093/ijnp/pyu097
66
FernandesE.FernandesM.KeebleJ. (2012). The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br. J. Pharmacol.166, 510–521. 10.1111/j.1476-5381.2012.01851.x
67
FinkelT. (1998). Oxygen radicals and signaling. Curr. Opin. Cell Biol.10, 248–253.
68
FoadiN.BergerC.PilawskiI.StoetzerC.KarstM.HaeselerG.et al. (2014). Inhibition of voltage-gated Na+ channels by the synthetic cannabinoid ajulemic acid. Anesth. Analg.118, 1238–1245. 10.1213/ANE.0000000000000188
69
FogaçaM. V.ReisF. M. C. V.CamposA. C.GuimarãesF. S. (2014). Effects of intra-prelimbic prefrontal cortex injection of cannabidiol on anxiety-like behavior: involvement of 5HT1A receptors and previous stressful experience. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol.24, 410–419. 10.1016/j.euroneuro.2013.10.012
70
FordL. A.RoelofsA. J.Anavi-GofferS.MowatL.SimpsonD. G.IrvingA. J.et al. (2010). A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br. J. Pharmacol.160, 762–771. 10.1111/j.1476-5381.2010.00743.x
71
FredrikssonR.HöglundP. J.GloriamD. E. I.LagerströmM. C.SchiöthH. B. (2003). Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett.554, 381–388. 10.1016/S0014-5793(03)01196-7
72
Gallego-SandínS.Rodríguez-GarcíaA.AlonsoM. T.García-SanchoJ. (2009). The endoplasmic reticulum of dorsal root ganglion neurons contains functional TRPV1 channels. J. Biol. Chem.284, 32591–32601. 10.1074/jbc.M109.019687
73
GantzI.MuraokaA.YangY. K.SamuelsonL. C.ZimmermanE. M.CookH.et al. (1997). Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics42, 462–466. 10.1006/geno.1997.4752
74
GardnerB.ZhuL. X.SharmaS.TashkinD. P.DubinettS. M. (2003). Methanandamide increases COX-2 expression and tumor growth in murine lung cancer. FASEB J.17, 2157–2159. 10.1096/fj.03-0254fje
75
GolovkoT.MinR.LozovayaN.FalconerC.YatsenkoN.TsintsadzeT.et al. (2014). Control of inhibition by the direct action of cannabinoids on GABAA receptors. Cereb. Cortex25, 2440–2455. 10.1093/cercor/bhu045
76
GomesF. V.ResstelL. B. M.GuimarãesF. S. (2011). The anxiolytic-like effects of cannabidiol injected into the bed nucleus of the stria terminalis are mediated by 5-HT1A receptors. Psychopharmacology213, 465–473. 10.1007/s00213-010-2036-z
77
Gonzalez-IslasC.Garcia-BereguiainM. A.WennerP. (2012). Tonic and transient endocannabinoid regulation of AMPAergic miniature postsynaptic currents and homeostatic plasticity in embryonic motor networks. J. Neurosci.32, 13597–13607. 10.1523/JNEUROSCI.1229-12.2012
78
GuX.MeiF.LiuY.ZhangR.ZhangJ.MaZ. (2011). Intrathecal administration of the cannabinoid 2 receptor agonist JWH015 can attenuate cancer pain and decrease mRNA expression of the 2B subunit of N-methyl-D-aspartic acid. Anesth. Analg.113, 405–411. 10.1213/ANE.0b013e31821d1062
79
GunduzO.OltuluC.UlugolA. (2011). Role of GLT-1 transporter activation in prevention of cannabinoid tolerance by the beta-lactam antibiotic, ceftriaxone, in mice. Pharmacol. Biochem. Behav.99, 100–103. 10.1016/j.pbb.2011.04.012
80
GustafssonS. B.LindgrenT.JonssonM.JacobssonS. O. P. (2009). Cannabinoid receptor-independent cytotoxic effects of cannabinoids in human colorectal carcinoma cells: synergism with 5-fluorouracil. Cancer Chemother. Pharmacol.63, 691–701. 10.1007/s00280-008-0788-5
81
HampsonA. J.BornheimL. M.ScanzianiM.YostC. S.GrayA. T.HansenB. M.et al. (1998). Dual effects of anandamide on NMDA receptor-mediated responses and neurotransmission. J. Neurochem.70, 671–676.
82
HamtiauxL.HansoulleL.DauguetN.MuccioliG. G.GallezB.LambertD. M. (2011). Increasing antiproliferative properties of endocannabinoids in N1E-115 neuroblastoma cells through inhibition of their metabolism. PLoS ONE6:e26823. 10.1371/journal.pone.0026823
83
HaneyM.RameshD.GlassA.PavlicovaM.BediG.CooperZ. D. (2015). Naltrexone maintenance decreases cannabis self-administration and subjective effects in daily cannabis smokers. Neuropsychopharmacology40, 2489–2498. 10.1038/npp.2015.108
84
HashimotoY.MaxamA. M.GreeneM. I. (1986). T-cell antigen-receptor genes in autoimmune mice. Proc. Natl. Acad. Sci. U.S.A.83, 7865–7869.
85
HejaziN.ZhouC.OzM.SunH.YeJ. H.ZhangL. (2006). Delta9-tetrahydrocannabinol and endogenous cannabinoid anandamide directly potentiate the function of glycine receptors. Mol. Pharmacol.69, 991–997. 10.1124/mol.105.019174
86
HigginsA.YuanS.WangY.BurrellB. D. (2013). Differential modulation of nociceptive versus non-nociceptive synapses by endocannabinoids. Mol. Pain9:26. 10.1186/1744-8069-9-26
87
HineB.FriedmanE.TorrelioM.GershonS. (1975). Morphine-dependent rats: blockade of precipitated abstinence by tetrahydrocannabinol. Science187, 443–445.
88
HinzB.RamerR.EicheleK.WeinzierlU.BruneK. (2004a). R(+)-methanandamide-induced cyclooxygenase-2 expression in H4 human neuroglioma cells: possible involvement of membrane lipid rafts. Biochem. Biophys. Res. Commun.324, 621–626. 10.1016/j.bbrc.2004.09.095
89
HinzB.RamerR.EicheleK.WeinzierlU.BruneK. (2004b). Up-regulation of cyclooxygenase-2 expression is involved in R(+)-methanandamide-induced apoptotic death of human neuroglioma cells. Mol. Pharmacol.66, 1643–1651. 10.1124/mol.104.002618
90
HofmannM. E.BhatiaC.FrazierC. J. (2011). Cannabinoid receptor agonists potentiate action potential-independent release of GABA in the dentate gyrus through a CB1 receptor-independent mechanism. J. Physiol.589, 3801–3821. 10.1113/jphysiol.2011.211482
91
HolcikM.SonenbergN. (2005). Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol.6, 318–327. 10.1038/nrm1618
92
HuangL.RamirezJ. C.FramptonG. A.GoldenL. E.QuinnM. A.PaeH. Y.et al. (2011). Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab. Invest.91, 1007–1017. 10.1038/labinvest.2011.62
93
JaneroD. R.ThakurG. A. (2016). Leveraging allostery to improve G protein-coupled receptor (GPCR)-directed therapeutics: cannabinoid receptor 1 as discovery target. Expert Opin. Drug Discov.11, 1223–1237. 10.1080/17460441.2016.1245289
94
JiaoJ.IshikawaT.-O.DumlaoD. S.NorrisP. C.MagyarC. E.MikulecC.et al. (2014). Targeted deletion and lipidomic analysis identify epithelial cell COX-2 as a major driver of chemically induced skin cancer. Mol. Cancer Res.12, 1677–1688. 10.1158/1541-7786.MCR-14-0397-T
95
JinL.PanL.GuoY.ZhengY.NieZ.ZhuR. (2014). Expression and localization of cannabinoid receptor 1 in rats' brain treated with acute and repeated morphine. Acta Neurobiol. Exp.74, 288–297.
96
JungK.-M.MangieriR.StapletonC.KimJ.FegleyD.WallaceM.et al. (2005). Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol. Pharmacol.68, 1196–1202. 10.1124/mol.105.013961
97
KallendruschS.KremzowS.NowickiM.GrabiecU.WinkelmannR.BenzA.et al. (2013). The G protein-coupled receptor 55 ligand l-α-lysophosphatidylinositol exerts microglia-dependent neuroprotection after excitotoxic lesion. Glia61, 1822–1831. 10.1002/glia.22560
98
KatsidoniV.AnagnostouI.PanagisG. (2013). Cannabidiol inhibits the reward-facilitating effect of morphine: involvement of 5-HT1A receptors in the dorsal raphe nucleus. Addict. Biol.18, 286–296. 10.1111/j.1369-1600.2012.00483.x
99
KaurR.KaurJ.MahajanJ.KumarR.AroraS. (2014). Oxidative stress–implications, source and its prevention. Environ. Sci. Pollut. Res. Int.21, 1599–1613. 10.1007/s11356-013-2251-3
100
KhasabovaI. A.GielissenJ.ChandiramaniA.Harding-RoseC.OdehD. A.SimoneD. A.et al. (2011). CB1 and CB2 receptor agonists promote analgesia through synergy in a murine model of tumor pain. Behav. Pharmacol.22, 607–616. 10.1097/FBP.0b013e3283474a6d
101
KhuranaL.MackieK.PiomelliD.KendallD. A. (2017). Modulation of CB1 cannabinoid receptor by allosteric ligands: pharmacology and therapeutic opportunities. Neuropharmacology124, 3–12. 10.1016/j.neuropharm.2017.05.018
102
KimK. S.TakedaK.SethiR.PracykJ. B.TanakaK.ZhouY. F.et al. (1998). Protection from reoxygenation injury by inhibition of rac1. J. Clin. Invest.101, 1821–1826. 10.1172/JCI1830
103
KiralyA. J.SolimanE.JenkinsA.Van DrossR. T. (2016). Apigenin inhibits COX-2, PGE2, and EP1 and also initiates terminal differentiation in the epidermis of tumor bearing mice. Prostaglandins Leukot. Essent. Fatty Acids104, 44–53. 10.1016/j.plefa.2015.11.006
104
KohnoM.HasegawaH.InoueA.MuraokaM.MiyazakiT.OkaK.et al. (2006). Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem. Biophys. Res. Commun.347, 827–832. 10.1016/j.bbrc.2006.06.175
105
KozakK. R.CrewsB. C.MorrowJ. D.WangL.-H.MaY. H.WeinanderR.et al. (2002). Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem.277, 44877–44885. 10.1074/jbc.M206788200
106
KozakK. R.CrewsB. C.RayJ. L.TaiH. H.MorrowJ. D.MarnettL. J. (2001). Metabolism of prostaglandin glycerol esters and prostaglandin ethanolamides in vitro and in vivo. J. Biol. Chem.276, 36993–36998. 10.1074/jbc.M105854200
107
KreitzerF. R.StellaN. (2009). The therapeutic potential of novel cannabinoid receptors. Pharmacol. Ther.122, 83–96. 10.1016/j.pharmthera.2009.01.005
108
Kruk-SlomkaM.BudzynskaB.SlomkaT.BanaszkiewiczI.BialaG. (2016). The influence of the CB1 receptor ligands on the schizophrenia-like effects in mice induced by MK-801. Neurotox. Res.30, 658–676. 10.1007/s12640-016-9662-0
109
KucC.JenkinsA.Van DrossR. T. (2012). Arachidonoyl ethanolamide (AEA)-induced apoptosis is mediated by J-series prostaglandins and is enhanced by fatty acid amide hydrolase (FAAH) blockade. Mol. Carcinog.51, 139–149. 10.1002/mc.20770
110
KuryL. T. A.YangK.-H. S.ThayyullathilF. T.RajeshM.AliR. M.ShubaY. M.et al. (2014). Effects of endogenous cannabinoid anandamide on cardiac Na+/Ca2+ exchanger. Cell Calcium55, 231–237. 10.1016/j.ceca.2014.02.017
111
LadinD. A.SolimanE.EscobedoR.FitzgeraldT. L.YangL. V.BurnsC.et al. (2017). Synthesis and evaluation of the novel prostamide, 15-Deoxy, Δ(12,14)-Prostamide J2, as a selective antitumor therapeutic. Mol. Cancer Ther.16, 838–849. 10.1158/1535-7163.MCT-16-0484
112
LadinD. A.SolimanE.GriffinL.Van DrossR. (2016). Preclinical and clinical assessment of cannabinoids as anti-cancer agents. Front. Pharmacol.7:361. 10.3389/fphar.2016.00361
113
LaucknerJ. E.JensenJ. B.ChenH.-Y.LuH.-C.HilleB.MackieK. (2008). GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. U.S.A.105, 2699–2704. 10.1073/pnas.0711278105
114
LazzeriM.VannucchiM. G.SpinelliM.BizzocoE.BenefortiP.TuriniD.et al. (2005). Transient receptor potential vanilloid type 1 (TRPV1) expression changes from normal urothelium to transitional cell carcinoma of human bladder. Eur. Urol.48, 691–698. 10.1016/j.eururo.2005.05.018
115
LeeS.-H.LedriM.TóthB.MarchionniI.HenstridgeC. M.DudokB.et al. (2015). Multiple forms of endocannabinoid and endovanilloid signaling regulate the tonic control of GABA release. J. Neurosci.35, 10039–10057. 10.1523/JNEUROSCI.4112-14.2015
116
LernerT. N.HorneE. A.StellaN.KreitzerA. C. (2010). Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J. Neurosci.30, 2160–2164. 10.1523/JNEUROSCI.5844-09.2010
117
LigrestiA.MorielloA. S.StarowiczK.MatiasI.PisantiS.De PetrocellisL.et al. (2006). Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther.318, 1375–1387. 10.1124/jpet.106.105247
118
LinJ. H.WalterP.YenT. S. B. (2008). Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol.3, 399–425. 10.1146/annurev.pathmechdis.3.121806.151434
119
LingeR.Jiménez-SánchezL.CampaL.Pilar-CuéllarF.VidalR.PazosA.et al. (2016). Cannabidiol induces rapid-acting antidepressant-like effects and enhances cortical 5-HT/glutamate neurotransmission: role of 5-HT1A receptors. Neuropharmacology103, 16–26. 10.1016/j.neuropharm.2015.12.017
120
LiuB.SongS.JonesP. M.PersaudS. J. (2015). GPR55: from orphan to metabolic regulator?Pharmacol. Ther.145, 35–42. 10.1016/j.pharmthera.2014.06.007
121
LiuG.XieC.SunF.XuX.YangY.ZhangT.et al. (2010). Clinical significance of transient receptor potential vanilloid 2 expression in human hepatocellular carcinoma. Cancer Genet. Cytogenet.197, 54–59. 10.1016/j.cancergencyto.2009.08.007
122
LozovayaN.YatsenkoN.BeketovA.TsintsadzeT.BurnashevN. (2005). Glycine receptors in CNS neurons as a target for nonretrograde action of cannabinoids. J. Neurosci.25, 7499–7506. 10.1523/JNEUROSCI.0977-05.2005
123
LuV. B.PuhlH. L.IkedaS. R. (2013). N-arachidonyl glycine does not activate G protein–coupled receptor 18 signaling via canonical pathways. Mol. Pharmacol.83, 267–282. 10.1124/mol.112.081182
124
MaX.AokiT.TsuruyamaT.NarumiyaS. (2015). Definition of prostaglandin E2-EP2 signals in the colon tumor microenvironment that amplify inflammation and tumor growth. Cancer Res.75, 2822–2832. 10.1158/0008-5472.CAN-15-0125
125
MaccarroneM.De ChiaraV.GasperiV.ViscomiM. T.RossiS.OddiS.et al. (2009). Lipid rafts regulate 2-arachidonoylglycerol metabolism and physiological activity in the striatum. J. Neurochem.109, 371–381. 10.1111/j.1471-4159.2009.05948.x
126
MahgoubM.Keun-HangS. Y.SydorenkoV.AshoorA.KabbaniN.Al KuryL.et al. (2013). Effects of cannabidiol on the function of α7-nicotinic acetylcholine receptors. Eur. J. Pharmacol.720, 310–319. 10.1016/j.ejphar.2013.10.011
127
MajumderM.LandmanE.LiuL.HessD.LalaP. K. (2015). COX-2 elevates oncogenic miR-526b in breast cancer by EP4 activation. Mol. Cancer Res.13, 1022–1033. 10.1158/1541-7786.MCR-14-0543
128
MalekN.KostrzewaM.MakuchW.PajakA.KucharczykM.PiscitelliF.et al. (2016). The multiplicity of spinal AA-5-HT anti-nociceptive action in a rat model of neuropathic pain. Pharmacol. Res.111, 251–263. 10.1016/j.phrs.2016.06.012
129
MalinowskaB.ZakrzeskaA.KurzC. M.GöthertM.KwolekG.WielgatP.et al. (2010). Involvement of central beta2-adrenergic, NMDA and thromboxane A2 receptors in the pressor effect of anandamide in rats. Naunyn. Schmiedebergs Arch. Pharmacol.381, 349–360. 10.1007/s00210-010-0497-6
130
MarciniakS. J.RonD. (2006). Endoplasmic reticulum stress signaling in disease. Physiol. Rev.86, 1133–1149. 10.1152/physrev.00015.2006
131
MarciniakS. J.Garcia-BonillaL.HuJ.HardingH. P.RonD. (2006). Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol.172, 201–209. 10.1083/jcb.200508099
132
Martínez-PinillaE.Reyes-ResinaI.Oñatibia-AstibiaA.ZamarbideM.RicobarazaA.NavarroG.et al. (2014). CB1 and GPR55 receptors are co-expressed and form heteromers in rat and monkey striatum. Exp. Neurol.261, 44–52. 10.1016/j.expneurol.2014.06.017
133
MarzoV.PetrocellisL. (2010). Endocannabinoids as regulators of transient receptor potential (TRP)channels: a further opportunity to develop new endocannabinoid-based therapeutic drugs. Curr. Med. Chem.17, 1430–1449. 10.2174/092986710790980078
134
MassiP.VaccaniA.BianchessiS.CostaB.MacchiP.ParolaroD. (2006). The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell. Mol. Life Sci.63, 2057–2066. 10.1007/s00018-006-6156-x
135
MassiP.VaccaniA.CerutiS.ColomboA.AbbracchioM. P.ParolaroD. (2004). Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J. Pharmacol. Exp. Ther.308, 838–845. 10.1124/jpet.103.061002
136
MatiasI.ChenJ.De PetrocellisL.BisognoT.LigrestiA.FezzaF.et al. (2004). Prostaglandin ethanolamides (prostamides): in vitro pharmacology and metabolism. J. Pharmacol. Exp. Ther.309, 745–757. 10.1124/jpet.103.061705
137
McHughD.HuS. S.RimmermanN.JuknatA.VogelZ.WalkerJ. M.et al. (2010). N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci.11:44. 10.1186/1471-2202-11-44
138
McHughD.PageJ.DunnE.BradshawH. B. (2012). Δ(9) -Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br. J. Pharmacol.165, 2414–2424. 10.1111/j.1476-5381.2011.01497.x
139
McHughD.RoskowskiD.XieS.BradshawH. B. (2014). Δ(9)-THC and N-arachidonoyl glycine regulate BV-2 microglial morphology and cytokine release plasticity: implications for signaling at GPR18. Front. Pharmacol.4:162. 10.3389/fphar.2013.00162
140
McPartlandJ. M.DuncanM.Di MarzoV.PertweeR. G. (2015). Are cannabidiol and Δ(9) -tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br. J. Pharmacol.172, 737–753. 10.1111/bph.12944
141
McPartlandJ. M.GuyG. W.Di MarzoV. (2014). Care and feeding of the endocannabinoid system: a systematic review of potential clinical interventions that upregulate the endocannabinoid system. PLoS ONE9:e89566. 10.1371/journal.pone.0089566
142
Megha and LondonE. (2004). Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J. Biol. Chem.279, 9997–10004. 10.1074/jbc.M309992200
143
MelisM.PillollaG.LuchicchiA.MuntoniA. L.YasarS.GoldbergS. R.et al. (2008). Endogenous fatty acid ethanolamides suppress nicotine-induced activation of mesolimbic dopamine neurons through nuclear receptors. J. Neurosci.28, 13985–13994. 10.1523/JNEUROSCI.3221-08.2008
144
MorelliM. B.OffidaniM.AlesianiF.DiscepoliG.LiberatiS.OlivieriA.et al. (2014). The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer134, 2534–2546. 10.1002/ijc.28591
145
MouroF. M.BatalhaV. L.FerreiraD. G.CoelhoJ. E.BaqiY.MüllerC. E.et al. (2017). Chronic and acute adenosine A2A receptor blockade prevents long-term episodic memory disruption caused by acute cannabinoid CB1 receptor activation. Neuropharmacology117, 316–327. 10.1016/j.neuropharm.2017.02.021
146
NabissiM.MorelliM. B.AmantiniC.LiberatiS.SantoniM.Ricci-VitianiL.et al. (2015). Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer137, 1855–1869. 10.1002/ijc.29573
147
NabissiM.MorelliM. B.SantoniM.SantoniG. (2013). Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis34, 48–57. 10.1093/carcin/bgs328
148
NguyenT.LiJ.-X.ThomasB. F.WileyJ. L.KenakinT. P.ZhangY. (2017). Allosteric modulation: an alternate approach targeting the cannabinoid CB1 receptor. Med. Res. Rev.37, 441–474. 10.1002/med.21418
149
NicolussiS.GertschJ. (2015). Chapter fourteen - endocannabinoid transport revisited, in Vitamins & Hormones Hormones and Transport Systems. ed. LitwackG. (Academic Press), 441–485. Available online at: http://www.sciencedirect.com/science/article/pii/S0083672914000247 (Accessed September 21, 2015).
150
NicolussiS.ChiccaA.RauM.RihsS.SoeberdtM.AbelsC.et al. (2014). Correlating FAAH and anandamide cellular uptake inhibition using N-alkylcarbamate inhibitors: from ultrapotent to hyperpotent. Biochem. Pharmacol.92, 669–689. 10.1016/j.bcp.2014.09.020
151
NingY.O'NeillK.LanH.PangL.ShanL. X.HawesB. E.et al. (2008). Endogenous and synthetic agonists of GPR119 differ in signalling pathways and their effects on insulin secretion in MIN6c4 insulinoma cells. Br. J. Pharmacol.155, 1056–1065. 10.1038/bjp.2008.337
152
NojiT.TakayamaM.MizutaniM.OkamuraY.TakaiH.KarasawaA.et al. (2002). KF24345, an adenosine uptake inhibitor, suppresses lipopolysaccharide-induced tumor necrosis factor-α production and leukopenia via endogenous adenosine in mice. J. Pharmacol. Exp. Ther.300, 200–205. 10.1124/jpet.300.1.200
153
NomuraD. K.LongJ. Z.NiessenS.HooverH. S.NgS.-W.CravattB. F. (2010). Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell140, 49–61. 10.1016/j.cell.2009.11.027
154
NotaroA.SabellaS.PelleritoO.Di FioreR.De BlasioA.VentoR.et al. (2014). Involvement of PAR-4 in cannabinoid-dependent sensitization of osteosarcoma cells to TRAIL-induced apoptosis. Int. J. Biol. Sci.10, 466–478. 10.7150/ijbs.8337
155
NourazarianA. R.KangariP.SalmaninejadA. (2014). Roles of oxidative stress in the development and progression of breast cancer. Asian Pac. J. Cancer Prev.15, 4745–4751. 10.7314/APJCP.2014.15.12.4745
156
O'SullivanS. E. (2007). Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol.152, 576–582. 10.1038/sj.bjp.0707423
157
ObaraY.UenoS.YanagihataY.NakahataN. (2011). Lysophosphatidylinositol causes neurite retraction via GPR55, G13 and RhoA in PC12 cells. PLoS ONE6:e24284. 10.1371/journal.pone.0024284
158
OddiS.DaineseE.SandifordS.FezzaF.LanutiM.ChiurchiùV.et al. (2012). Effects of palmitoylation of Cys(415) in helix 8 of the CB(1) cannabinoid receptor on membrane localization and signalling. Br. J. Pharmacol.165, 2635–2651. 10.1111/j.1476-5381.2011.01658.x
159
OhokaN.YoshiiS.HattoriT.OnozakiK.HayashiH. (2005). TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J.24, 1243–1255. 10.1038/sj.emboj.7600596
160
OkaS.NakajimaK.YamashitaA.KishimotoS.SugiuraT. (2007). Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun.362, 928–934. 10.1016/j.bbrc.2007.08.078
161
OkaS.OtaR.ShimaM.YamashitaA.SugiuraT. (2010). GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun.395, 232–237. 10.1016/j.bbrc.2010.03.169
162
OkumuraS.BabaH.KumadaT.NanmokuK.NakajimaH.NakaneY.et al. (2004). Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer Sci.95, 131–135. 10.1111/j.1349-7006.2004.tb03193.x
163
OvertonH. A.BabbsA. J.DoelS. M.FyfeM. C. T.GardnerL. S.GriffinG.et al. (2006). Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab.3, 167–175. 10.1016/j.cmet.2006.02.004
164
OzM.JacksonS. N.WoodsA. S.MoralesM.ZhangL. (2005). Additive effects of endogenous cannabinoid anandamide and ethanol on α7-nicotinic acetylcholine receptor-mediated responses in Xenopus Oocytes. J. Pharmacol. Exp. Ther.313, 1272–1280. 10.1124/jpet.104.081315
165
OzM.RavindranA.Diaz-RuizO.ZhangL.MoralesM. (2003). The endogenous cannabinoid anandamide inhibits α7 nicotinic acetylcholine receptor-mediated responses in Xenopus Oocytes. J. Pharmacol. Exp. Ther.306, 1003–1010. 10.1124/jpet.103.049981
166
OzM.ZhangL.RavindranA.MoralesM.LupicaC. R. (2004). Differential effects of endogenous and synthetic cannabinoids on alpha7-nicotinic acetylcholine receptor-mediated responses in Xenopus Oocytes. J. Pharmacol. Exp. Ther.310, 1152–1160. 10.1124/jpet.104.067751
167
PalazzoE.de NovellisV.PetrosinoS.MarabeseI.VitaD.GiordanoC.et al. (2006). Neuropathic pain and the endocannabinoid system in the dorsal raphe: pharmacological treatment and interactions with the serotonergic system. Eur. J. Neurosci.24, 2011–2020. 10.1111/j.1460-9568.2006.05086.x
168
PatilM.PatwardhanA.SalasM. M.HargreavesK. M.AkopianA. N. (2011). Cannabinoid receptor antagonists AM251 and AM630 activate TRPA1 in sensory neurons. Neuropharmacology61, 778–788. 10.1016/j.neuropharm.2011.05.024
169
PatsosH. A.GreenhoughA.HicksD. J.Al KharusiM.CollardT. J.LaneJ. D.et al. (2010). The endogenous cannabinoid, anandamide, induces COX-2-dependent cell death in apoptosis-resistant colon cancer cells. Int. J. Oncol.37, 187–193. 10.3892/ijo_00000666
170
PatsosH. A.HicksD. J.DobsonR. R. H.GreenhoughA.WoodmanN.LaneJ. D.et al. (2005). The endogenous cannabinoid, anandamide, induces cell death in colorectal carcinoma cells: a possible role for cyclooxygenase 2. Gut54, 1741–1750. 10.1136/gut.2005.073403
171
PearlmanR. J.AubreyK. R.VandenbergR. J. (2003). Arachidonic acid and anandamide have opposite modulatory actions at the glycine transporter, GLYT1a. J. Neurochem.84, 592–601. 10.1046/j.1471-4159.2003.01549.x
172
PelleritoO.NotaroA.SabellaS.De BlasioA.VentoR.CalvarusoG.et al. (2014). WIN induces apoptotic cell death in human colon cancer cells through a block of autophagic flux dependent on PPARγ down-regulation. Apoptosis Int. J. Program. Cell Death19, 1029–1042. 10.1007/s10495-014-0985-0
173
PenumartiA.Abdel-RahmanA. A. (2014a). Neuronal nitric oxide synthase-dependent elevation in adiponectin in the rostral ventrolateral medulla underlies g protein-coupled receptor 18-mediated hypotension in conscious rats. J. Pharmacol. Exp. Ther.351, 44–53. 10.1124/jpet.114.216036
174
PenumartiA.Abdel-RahmanA. A. (2014b). The novel endocannabinoid receptor GPR18 is expressed in the rostral ventrolateral medulla and exerts tonic restraining influence on blood pressure. J. Pharmacol. Exp. Ther.349, 29–38. 10.1124/jpet.113.209213
175
Pernía-AndradeA. J.KatoA.WitschiR.NyilasR.KatonaI.FreundT. F.et al. (2009). Spinal endocannabinoids and CB1 receptors mediate C-Fiber–induced heterosynaptic pain sensitization. Science325, 760–764. 10.1126/science.1171870
176
PerrottiD.NevianiP. (2013). Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol.14, e229–e238. 10.1016/S1470-2045(12)70558-2
177
PertweeR. G. (1997). Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther.74, 129–180. 10.1016/S0163-7258(97)82001-3
178
PertweeR. G. (2010). Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr. Med. Chem.17, 1360–1381. 10.2174/092986710790980050
179
PiñeiroR.MaffucciT.FalascaM. (2011). The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene30, 142–152. 10.1038/onc.2010.417
180
PryorG. T.LarsenF. F.HusainS.BraudeM. C. (1978). Interactions of Δ9-tetrahydrocannabinol with d-Amphetamine, cocaine, and nicotine in rats. Pharmacol. Biochem. Behav.8, 295–318. 10.1016/0091-3057(78)90320-9
181
QinY.VerdegaalE. M. E.SideriusM.BebelmanJ. P.SmitM. J.LeursR.et al. (2011). Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: the constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res.24, 207–218. 10.1111/j.1755-148X.2010.00781.x
182
RamerR.HeinemannK.MerkordJ.RohdeH.SalamonA.LinnebacherM.et al. (2013). COX-2 and PPAR-γ confer cannabidiol-induced apoptosis of human lung cancer cells. Mol. Cancer Ther.12, 69–82. 10.1158/1535-7163.MCT-12-0335
183
RamerR.WeinzierlU.SchwindB.BruneK.HinzB. (2003). Ceramide is involved in r(+)-methanandamide-induced cyclooxygenase-2 expression in human neuroglioma cells. Mol. Pharmacol.64, 1189–1198. 10.1124/mol.64.5.1189
184
RameshD.RossG. R.SchlosburgJ. E.OwensR. A.AbdullahR. A.KinseyS. G.et al. (2011). Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J. Pharmacol. Exp. Ther.339, 173–185. 10.1124/jpet.111.181370
185
RaviJ.SnehA.ShiloK.NasserM. W.GanjuR. K. (2014). FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget5, 2475–2486. 10.18632/oncotarget.1723
186
RockE. M.LimebeerC. L.NavaratnamR.StichtM. A.BonnerN.EngelandK.et al. (2014). A comparison of cannabidiolic acid with other treatments for anticipatory nausea using a rat model of contextually elicited conditioned gaping. Psychopharmacology231, 3207–3215. 10.1007/s00213-014-3498-1
187
RouzerC. A.MarnettL. J. (2009). Cyclooxygenases: structural and functional insights. J. Lipid Res.50(Suppl.), S29–S34. 10.1194/jlr.R800042-JLR200
188
RuhaakL. R.FelthJ.KarlssonP. C.RafterJ. J.VerpoorteR.BohlinL. (2011). Evaluation of the cyclooxygenase inhibiting effects of six major cannabinoids isolated from Cannabis sativa. Biol. Pharm. Bull.34, 774–778. 10.1248/bpb.34.774
189
RundhaugJ. E.MikulecC.PavoneA.FischerS. M. (2007). A role for cyclooxygenase-2 in ultraviolet light-induced skin carcinogenesis. Mol. Carcinog.46, 692–698. 10.1002/mc.20329
190
RundhaugJ. E.SimperM. S.SurhI.FischerS. M. (2011). The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev.30, 465–480. 10.1007/s10555-011-9317-9
191
RussoE. B. (2016). Current therapeutic cannabis controversies and clinical trial design issues. Front. Pharmacol.7:309. 10.3389/fphar.2016.00309
192
RussoE. B.BurnettA.HallB.ParkerK. K. (2005). Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res.30, 1037–1043. 10.1007/s11064-005-6978-1
193
RybergE.LarssonN.SjögrenS.HjorthS.HermanssonN.-O.LeonovaJ.et al. (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol.152, 1092–1101. 10.1038/sj.bjp.0707460
194
SalazarM.CarracedoA.SalanuevaI. J.Hernández-TiedraS.LorenteM.EgiaA.et al. (2009). Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Invest.119, 1359–1372. 10.1172/JCI37948
195
SalazarM.LorenteM.García-TaboadaE.Hernández-TiedraS.DavilaD.FrancisS. E.et al. (2013). The pseudokinase tribbles homologue-3 plays a crucial role in cannabinoid anticancer action. Biochim. Biophys. Acta1831, 1573–1578. 10.1016/j.bbalip.2013.03.014
196
SarfarazS.AdhamiV. M.SyedD. N.AfaqF.MukhtarH. (2008). Cannabinoids for cancer treatment: progress and promise. Cancer Res.68, 339–342. 10.1158/0008-5472.CAN-07-2785
197
SarkerK. P.MaruyamaI. (2003). Anandamide induces cell death independently of cannabinoid receptors or vanilloid receptor 1: possible involvement of lipid rafts. Cell. Mol. Life Sci.60, 1200–1208. 10.1007/s00018-003-3055-2
198
SarnataroD.PisantiS.SantoroA.GazzerroP.MalfitanoA. M.LaezzaC.et al. (2006). The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Mol. Pharmacol.70, 1298–1306. 10.1124/mol.106.025601
199
ScottJ. S.BowkerS. S.BrocklehurstK. J.BrownH. S.ClarkeD. S.EasterA.et al. (2014). Circumventing seizure activity in a series of g protein coupled receptor 119 (GPR119) agonists. J. Med. Chem.57, 8984–8998. 10.1021/jm5011012
200
ScuderiM. R.CantarellaG.ScolloM.LempereurL.PalumboM.Saccani-JottiG.et al. (2011). The antimitogenic effect of the cannabinoid receptor agonist WIN55212-2 on human melanoma cells is mediated by the membrane lipid raft. Cancer Lett.310, 240–249. 10.1016/j.canlet.2011.07.008
201
ShenM.PiserT. M.SeyboldV. S.ThayerS. A. (1996). Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J. Neurosci.16, 4322–4334.
202
ShevyrinV.MelkozerovV.EndresG. W.ShafranY.MorzherinY. (2016). On a new cannabinoid classification system: a sight on the illegal market of novel psychoactive substances. Cannabis Cannabinoid Res.1, 186–194. 10.1089/can.2016.0004
203
ShrivastavaA.KuzontkoskiP. M.GroopmanJ. E.PrasadA. (2011). Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther.10, 1161–1172. 10.1158/1535-7163.MCT-10-1100
204
SigelE.BaurR.RáczI.MarazziJ.SmartT. G.ZimmerA.et al. (2011). The major central endocannabinoid directly acts at GABAA receptors. Proc. Natl. Acad. Sci. U.S.A.108, 18150–18155. 10.1073/pnas.1113444108
205
SimcocksA. C.O'KeefeL.JenkinK. A.MathaiM. L.HryciwD. H.McAinchA. J. (2014). A potential role for GPR55 in the regulation of energy homeostasis. Drug Discov. Today19, 1145–1151. 10.1016/j.drudis.2013.12.005
206
SimonsK.IkonenE. (1997). Functional rafts in cell membranes. Nature387, 569–572. 10.1038/42408
207
SoderstromK. (2009). Lessons from nonmammalian species. Curr. Top. Behav. Neurosci.1, 173–198. 10.1007/978-3-540-88955-7_7
208
SoderstromK.JohnsonF. (2001). Zebra finch CB1 cannabinoid receptor: pharmacology and in vivo and in vitro effects of activation. J. Pharmacol. Exp. Ther.297, 189–197.
209
SoderstromK.LeidM.MooreF. L.MurrayT. F. (2000). Behaviroal, pharmacological, and molecular characterization of an amphibian cannabinoid receptor. J. Neurochem.75, 413–423. 10.1046/j.1471-4159.2000.0750413.x
210
SolimanE. (2015). Molecular Mechanism of Anandamide-Induced Apoptosis in Non-melanoma Skin Cancer. Available online at: http://thescholarship.ecu.edu/handle/10342/4959 (Accessed April 4, 2017).
211
SolimanE.Van DrossR. (2016). Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: receptor-independent endocannabinoid signaling. Mol. Carcinog.55, 1807–1821. 10.1002/mc.22429
212
SolimanE.HendersonK. L.DanellA. S.Van DrossR. (2016). Arachidonoyl-ethanolamide activates endoplasmic reticulum stress-apoptosis in tumorigenic keratinocytes: role of cyclooxygenase-2 and novel J-series prostamides. Mol. Carcinog.55, 117–130. 10.1002/mc.22257
213
SpivakC. E.LupicaC. R.OzM. (2007). The endocannabinoid anandamide inhibits the function of α4β2 nicotinic acetylcholine receptors. Mol. Pharmacol.72, 1024–1032. 10.1124/mol.107.036939
214
SreevalsanS.SafeS. (2013). The cannabinoid WIN 55,212-2 decreases specificity protein transcription factors and the oncogenic cap protein eIF4E in colon cancer cells. Mol. Cancer Ther.12, 2483–2493. 10.1158/1535-7163.MCT-13-0486
215
SreevalsanS.JosephS.JutooruI.ChadalapakaG.SafeS. H. (2011). Induction of apoptosis by cannabinoids in prostate and colon cancer cells is phosphatase dependent. Anticancer Res.31, 3799–3807.
216
StarowiczK.MakuchW.KorostynskiM.MalekN.SlezakM.ZychowskaM.et al. (2013). Full inhibition of spinal FAAH Leads to TRPV1-mediated analgesic effects in neuropathic rats and possible lipoxygenase-mediated remodeling of anandamide metabolism. PLoS ONE8:e60040. 10.1371/journal.pone.0060040
217
StoneT. W.StoyN.DarlingtonL. G. (2013). An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol. Sci.34, 136–143. 10.1016/j.tips.2012.09.006
218
TakedaS.MisawaK.YamamotoI.WatanabeK. (2008). Cannabidiolic acid as a selective cyclooxygenase-2 inhibitory component in cannabis. Drug Metab. Dispos. Biol. Fate Chem.36, 1917–1921. 10.1124/dmd.108.020909
219
TaniguchiY.Tonai-KachiH.ShinjoK. (2006). Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett.580, 5003–5008. 10.1016/j.febslet.2006.08.015
220
ThorsL.BerghA.PerssonE.HammarstenP.StattinP.EgevadL.et al. (2010). Fatty acid amide hydrolase in prostate cancer: association with disease severity and outcome, CB1 receptor expression and regulation by IL-4. PLoS ONE5:e12275. 10.1371/journal.pone.0012275
221
TierneyA. J. (2001). Structure and function of invertebrate 5-HT receptors: a review. Comp. Biochem. Physiol. A Mol. Integr. Physiol.128, 791–804. 10.1016/S1095-6433(00)00320-2
222
ToguriJ. T.CaldwellM.KellyM. E. M. (2016). Turning down the thermostat: modulating the endocannabinoid system in ocular inflammation and pain. Front. Pharmacol.7:304. 10.3389/fphar.2016.00304
223
TulunayF. C.AyhanI. H.PortogheseP. S.TakemoriA. E. (1981). Antagonism by chlornaltrexamine of some effects of Δ9-tetrahydrocannabinol in rats. Eur. J. Pharmacol.70, 219–224. 10.1016/0014-2999(81)90217-X
224
TwardowschyA.Castiblanco-UrbinaM. A.Uribe-MariñoA.BiagioniA. F.Salgado-RohnerC. J.de CrippaJ. A. S.et al. (2013). The role of 5-HT1A receptors in the anti-aversive effects of cannabidiol on panic attack-like behaviors evoked in the presence of the wild snake Epicrates cenchria crassus (Reptilia, Boidae). J. Psychopharmacol. Oxf. Engl.27, 1149–1159. 10.1177/0269881113493363
225
UrbanJ. D.ClarkeW. P.ZastrowM.von NicholsD. E.KobilkaB.WeinsteinH.et al. (2007). Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther.320, 1–13. 10.1124/jpet.106.104463
226
ValjentE.MitchellJ. M.BessonM.-J.CabocheJ.MaldonadoR. (2002). Behavioural and biochemical evidence for interactions between Δ9-tetrahydrocannabinol and nicotine. Br. J. Pharmacol.135, 564–578. 10.1038/sj.bjp.0704479
227
VamecqJ.LatruffeN. (1999). Medical significance of peroxisome proliferator-activated receptors. Lancet354, 141–148. 10.1016/S0140-6736(98)10364-1
228
Van DrossR. T. (2009). Metabolism of anandamide by COX-2 is necessary for endocannabinoid-induced cell death in tumorigenic keratinocytes. Mol. Carcinog.48, 724–732. 10.1002/mc.20515
229
VaraD.MorellC.Rodríguez-HencheN.Diaz-LaviadaI. (2013). Involvement of PPARγ in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis.4:e618. 10.1038/cddis.2013.141
230
VaysseP. J.GardnerE. L.ZukinR. S. (1987). Modulation of rat brain opioid receptors by cannabinoids. J. Pharmacol. Exp. Ther.241, 534–539.
231
VennekensR.MenigozA.NiliusB. (2012). TRPs in the Brain, in Reviews of Physiology, Biochemistry and Pharmacology, Vol. 163 Reviews of Physiology, Biochemistry and Pharmacology, eds. NiliusB.AmaraS. G.GudermannT.JahnR.LillR.OffermannsS.et al. (Berlin; Heidelberg: Springer), 27–64. Available online at: http://link.springer.com/chapter/10.1007/112_2012_8 (Accessed September 1, 2015).
232
VoetsT.TalaveraK.OwsianikG.NiliusB. (2005). Sensing with TRP channels. Nat. Chem. Biol.1, 85–92. 10.1038/nchembio0705-85
233
WangJ.SimonaviciusN.WuX.SwaminathG.ReaganJ.TianH.et al. (2006). Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem.281, 22021–22028. 10.1074/jbc.M603503200
234
WardS. J.McAllisterS. D.KawamuraR.MuraseR.NeelakantanH.WalkerE. A. (2014). Cannabidiol inhibits paclitaxel-induced neuropathic pain through 5-HT(1A) receptors without diminishing nervous system function or chemotherapy efficacy. Br. J. Pharmacol.171, 636–645. 10.1111/bph.12439
235
WasikA. M.AlmestrandS.WangX.HultenbyK.DacklandÅ.-L.AnderssonP.et al. (2011). WIN55,212-2 induces cytoplasmic vacuolation in apoptosis-resistant MCL cells. Cell Death Dis.2:e225. 10.1038/cddis.2011.106
236
WorzfeldT.WettschureckN.OffermannsS. (2008). G12/G13-mediated signalling in mammalian physiology and disease. Trends Pharmacol. Sci.29, 582–589. 10.1016/j.tips.2008.08.002
237
WuC.-S.ChenH.SunH.ZhuJ.JewC. P.Wager-MillerJ.et al. (2013). GPR55, a G-protein coupled receptor for lysophosphatidylinositol, plays a role in motor coordination. PLoS ONE8:e60314. 10.1371/journal.pone.0060314
238
XiaoY. J.SchwartzB.WashingtonM.KennedyA.WebsterK.BelinsonJ.et al. (2001). Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: comparison of the lysophospholipid contents in malignant vs nonmalignant ascitic fluids. Anal. Biochem.290, 302–313. 10.1006/abio.2001.5000
239
YangK.LeiG.XieY.-F.MacDonaldJ. F.JacksonM. F. (2014). Differential regulation of NMDAR and NMDAR-mediated metaplasticity by anandamide and 2-AG in the hippocampus. Hippocampus24, 1601–1614. 10.1002/hipo.22339
240
YangZ.AubreyK. R.AlroyI.HarveyR. J.VandenbergR. J.LynchJ. W. (2008). Subunit-specific modulation of glycine receptors by cannabinoids and N-arachidonyl-glycine. Biochem. Pharmacol.76, 1014–1023. 10.1016/j.bcp.2008.07.037
241
YeeN. S. (2015). Roles of TRPM8 ion channels in cancer: proliferation, survival, and invasion. Cancers7, 2134–2146. 10.3390/cancers7040882
242
YesilyurtO.SeyrekM.TasdemirS.KahramanS.DeveciM. S.KarakusE.et al. (2015). The critical role of spinal 5-HT7 receptors in opioid and non-opioid type stress-induced analgesia. Eur. J. Pharmacol.762, 402–410. 10.1016/j.ejphar.2015.04.020
243
YuM.IvesD.RameshaC. S. (1997). Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem.272, 21181–21186.
244
ZádorF.KocsisD.BorsodiA.BenyheS. (2014). Micromolar concentrations of rimonabant directly inhibits delta opioid receptor specific ligand binding and agonist-induced G-protein activity. Neurochem. Int.67, 14–22. 10.1016/j.neuint.2013.12.005
245
ZádorF.LénártN.CsibrányB.SánthaM.MolnárM.TukaB.et al. (2015). Low dosage of rimonabant leads to anxiolytic-like behavior via inhibiting expression levels and G-protein activity of kappa opioid receptors in a cannabinoid receptor independent manner. Neuropharmacology89, 298–307. 10.1016/j.neuropharm.2014.10.008
246
ZádorF.OtvösF.BenyheS.ZimmerA.PáldyE. (2012). Inhibition of forebrain μ-opioid receptor signaling by low concentrations of rimonabant does not require cannabinoid receptors and directly involves μ-opioid receptors. Neurochem. Int.61, 378–388. 10.1016/j.neuint.2012.05.015
247
ZhangH.-Y.GaoM.LiuQ.-R.BiG.-H.LiX.YangH.-J.et al. (2014). Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. U.S.A.111, E5007–E5015. 10.1073/pnas.1413210111
248
ZhaoP.AboodM. E. (2013). GPR55 and GPR35 and their relationship to cannabinoid and lysophospholipid receptors. Life Sci.92, 453–457. 10.1016/j.lfs.2012.06.039
249
ZhouJ.BurkovskiyI.YangH.SardinhaJ.LehmannC. (2016). CB2 and GPR55 receptors as therapeutic targets for systemic immune dysregulation. Front. Pharmacol.7:264. 10.3389/fphar.2016.00264
250
ZhouK.ZhangS.-S.YanY.ZhaoS. (2014). Overexpression of transient receptor potential vanilloid 2 is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Med. Oncol. Northwood Lond. Engl.31:17. 10.1007/s12032-014-0017-5
251
ZygmuntP. M.PeterssonJ.AnderssonD. A.ChuangH.SørgårdM.Di MarzoV.et al. (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature400, 452–457. 10.1038/22761
Summary
Keywords
cannabinoid, cannabinoid receptor-independent, central nervous system, Cancer, experimental therapeutics
Citation
Soderstrom K, Soliman E and Van Dross R (2017) Cannabinoids Modulate Neuronal Activity and Cancer by CB1 and CB2 Receptor-Independent Mechanisms. Front. Pharmacol. 8:720. doi: 10.3389/fphar.2017.00720
Received
24 May 2017
Accepted
25 September 2017
Published
10 October 2017
Volume
8 - 2017
Edited by
Vsevolod V. Gurevich, Vanderbilt University, United States
Reviewed by
Chris J. Van Koppen, ElexoPharm GmbH, Germany; Carmela Parenti, Università degli Studi di Catania, Italy
Updates
Copyright
© 2017 Soderstrom, Soliman and Van Dross.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rukiyah Van Dross vandrossr@ecu.edu
This article was submitted to Experimental Pharmacology and Drug Discovery, a section of the journal Frontiers in Pharmacology
†These authors have contributed equally to this work.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.