 Fawaz Alasmari
Fawaz Alasmari Charles R. Ashby Jr.
Charles R. Ashby Jr. Frank S. Hall
Frank S. Hall Youssef Sari
Youssef Sari Amit K. Tiwari
Amit K. Tiwari- 1Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia
- 2Department of Pharmacology and Experimental Therapeutics, College of Pharmacy and Pharmaceutical Sciences, University of Toledo, Toledo, OH, United States
- 3Pharmaceutical Sciences, College of Pharmacy, St. John's University Queens, New York, NY, United States
Introduction
Inflammation of neuronal tissue, or neuro-inflammation, is associated with neurological diseases, including Alzheimer's disease (AD) (Patel et al., 2005; Walters et al., 2016; Wang B. et al., 2016). The exact role of neuro-inflammation in AD remains uncertain as it may be a result of other causative factors in AD, but can subsequently contribute to the course of the disease, or be caused by other factors. Neuro-inflammation is significantly correlated with changes in the expression of brain proteins that regulate the transport or signaling pathways of endogenous and exogenous molecules (Tilleux and Hermans, 2007; Kim et al., 2015; Gao et al., 2017). ATP-binding cassette (ABC) proteins, such as ABCB1 (P-glycoprotein, P-gp), are highly expressed in the brain capillary endothelial cells of the blood - brain barrier (BBB) and limit the uptake of certain endogenous and exogenous compounds into the brain (Löscher and Potschka, 2005; Zhang et al., 2015). Several studies have reported alterations in the expression and functions of ABCB1 in AD models (Wijesuriya et al., 2010; van Assema et al., 2012). The formation of amyloid beta (Aβ) (a substrate of ABCB1) plaques in the brain is a histological hallmark associated with AD (Lee et al., 2004; Wildburger et al., 2017). The ABCB1 transporter removes Aβ from the brain into the circulatory system (Hartz et al., 2010; ElAli and Rivest, 2013). Thus, alterations in the expression or function of ABCB1 may affect the progression of AD. The role of ABCB1 in AD progression and treatment has been recently reviewed, elsewhere (Pahnke et al., 2014; Sita et al., 2017). However, the focus of this opinion article is to discuss the effects of neuro-inflammatory cytokines on ABCB1 function and their role in the pathogenesis of AD.
Alzheimer's Disease is Associated With Neuro-Inflammation
AD is the leading cause of dementia in the elderly and its prevalence has significantly increased over the last two decades (Reitz and Mayeux, 2014). Epidemiological studies indicated that more than 4.5 million people in the United States (U.S.) had AD in 2000 and this number may triple by 2050 (Hebert et al., 2003). Learning and memory impairments, as well as cognitive dysfunction, have been observed in animal models of AD (Dao et al., 2013; Webster et al., 2014; Xiao et al., 2017). Notably, AD is characterized by the formation of neurofibrillary tangles and amyloid beta (Aβ) plaques and the loss of cholinergic neurons in multiple brain regions (Paulson et al., 2008; Iba et al., 2013; Parent et al., 2013). The cleavage of amyloid precursor protein (APP) by the enzymes beta (β) secretase and gamma (γ) secretase produces Aβ in the brain (O'Brien and Wong, 2011). Studies indicate that Aβ causes neuro-inflammation through various signaling pathways (Liu et al., 2012; Parajuli et al., 2013). Aβ affects inflammatory signaling by activating toll-like receptor-2 (TLR-2) (Liu et al., 2012). Additionally, the incubation of human monocytes in vitro with Aβ (10 μM) for 30 min increases the mRNA expression of the pro-inflammatory cytokine IL-1β, while incubation for 48 h increases tumor necrosis factor-α (TNF-α) (Yates et al., 2000). This study also reported that concurrent incubation of mouse microglial cells with fibrillar Aβ (10 μM) and lipopolysaccharide (6.25, 12.5, or 25 ng/ml) for 48 h significantly increased the release of IL-1β and TNF-α compared to microglial cells that were incubated with only lipopolysaccharide. A prior study reported that nucleotide-binding and oligomerization domains, as well as caspase-1, are involved in oligomeric Aβ-induced interleukin-1β (IL-1β) processing (Parajuli et al., 2013). Further studies found that Aβ activates NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3)/caspase1 inflammasome signaling pathway, resulting in neuro-inflammation induction (Gold and El Khoury, 2015; Saresella et al., 2016). The mRNA and protein expression of the NLRP3 inflammasome was increased in monocytes in individuals with moderate or severe AD (Saresella et al., 2016). The activation of this pathway has been found to increase the production of active inflammatory cytokines such as IL-1β (Gold and El Khoury, 2015). This indicates that stimulating the NLRP3/caspase1 inflammasome/IL-1β cascade might affect ABCB1 function or expression in AD patients. Studies are warranted to explore the pharmacological role of this pathway in modulating ABCB1 in AD models. In addition, IL-1β was detected in the nucleus basalis (NB) 24 h following the injection of Aβ (4 μg/μL) into the NB of rats (Giovannini et al., 2002). This effect was associated with activation of microglia and p38 MAPK pathway. Conversely, the incubation of cortical glial cells with IL-1 (100 ng/ mL) for 14 h or IL-6 (50–200 ng/mL) for 6 h significantly increased the mRNA expression of APP (Del Bo et al., 1995). Pro-inflammatory cytokines, such as TNF-α or interferon-γ (IFN-γ), have been reported to increase the production of Aβ in astrocytes expressing APP (Yamamoto et al., 2007). TNF-α, IFN-γ, and IL-β1 have been shown to stimulate γ-secretase, thereby increasing Aβ levels (Liao et al., 2004). Moreover, the pre-incubation of neuroblastoma cells with 0.1 mM of ibuprofen for 12 h significantly reduced Aβ secretion induced by 24 h of incubation with TNF-α and IFN-γ (Blasko et al., 2001). These data suggest that targeting these signaling pathways stimulated by Aβ could provide a pharmacological strategy to attenuate neuro-inflammation associated with AD, potentially improving AD symptoms and slowing disease progression.
Role of Neuro-Inflammatory Cytokines in Neurodegenerative Diseases and Psychiatric Diseases, and Their Effect on ABCB1 Expression
The production of pro-inflammatory cytokines has been found in pre-clinical models of various neurodegenerative diseases. For example, transient focal ischemia was reported to be associated with an increase in the levels of TNF-α (Chu et al., 2007). Moreover, the concentrations of IL-1β, IFN-γ, and TNF-α are increased in the brains of animals following traumatic brain injury produced by Feeney's weight-drop model (Wei et al., 2012). Long-term exposure to drugs of abuse, such as ethanol, produces a significant increase in TNF-α concentrations in the hippocampus of rats (Alfonso-Loeches et al., 2010; Marshall et al., 2016b). The exposure of C57BL/6J mice and Wistar rats to ethanol significantly increases IL-1β mRNA expression and concentrations, respectively, in the brain compared to the control groups (Alfonso-Loeches et al., 2010; Marshall et al., 2016a). In addition, a significant increase in immunoreactive TNF-α in glial cells has been reported in the substantia nigra of Parkinson's disease (PD) patients compared to the control group (Boka et al., 1994). Furthermore, the levels of IL-1β and IL-6 are significantly increased in striatal dopaminergic neurons of PD patients (Mogi et al., 1994). IL-1β levels were increased in the frontal cortex and hippocampus of AD patients compared to individuals with vascular dementia and control subjects (Cacabelos et al., 1994). Importantly, the level of inflammatory cytokines was positively correlated with the level of Aβ in a mouse model of AD (Patel et al., 2005). These findings indicate that the levels of pro-inflammatory cytokines are increased in pre-clinical neurodegenerative and psychiatric diseases models, as well as the disease states they are purported to model.
Several studies investigated the effects of certain pro-inflammatory cytokines on the expression and function of ABCB1 (Evseenko et al., 2007; Iqbal et al., 2012; Walther et al., 2015). The mRNA and protein expression of Abcb1/ABCB1 were significantly decreased following incubation with TNF-α (30 ng/mL) for 24–72 h (Walther et al., 2015). Furthermore, incubation of cytotrophoblasts with TNF-α (20 ng/mL) or IL-1β (2 ng/mL) significantly decreased the expression of the Abcb1/ABCB1 mRNA and protein (Evseenko et al., 2007). ABCB1 function and mRNA levels in cultured guinea pig brain endothelial cells (harvested at postnatal day 14) were significantly decreased following incubation with TNF-α, IL-1β, or IL-6, at 3.3 × 103 or 103 pg/mL, for 24 h (Iqbal et al., 2012). Thus, neuro-inflammatory cytokines affect the expression and function of ABCB1, suggesting that neuro-inflammation in neurodegenerative diseases, including AD, may alter ABCB1 expression, although this remains to be determined.
The Impact of Modulating ABCB1 on the Progression of Alzheimer's Disease
Data suggest a relationship between neuro-inflammation, regulation of ABCB1 transporter, and Aβ clearance in the brain. The release of IL-1β, IL-6, and TNF-α are increased in brain micro-vessels compared to larger vessels of AD patients (Grammas and Ovase, 2001). Overall, IL-1 levels are also significantly increased in the brain of AD patients, compared to control subjects (Griffin et al., 1989). IL-6 levels in the cortex and hippocampus of AD patients are greater than those in control subjects (Bauer et al., 1991; Strauss et al., 1992). Since these cytokines, as discussed above, can regulate ABCB1 expression, an increase in pro-inflammatory cytokines in the brains of AD patients could reduce ABCB1 expression or function, contributing to the pathogenesis or progression of AD. A reduction in the expression of ABCB1 may lead to the accumulation of substances in the brain that promote inflammation or contribute to neurodegeneration in AD, most notably Aβ. Indeed, ABCB1 overexpression attenuated neurodegeneration in a mouse AD Model (Qosa et al., 2012; Durk et al., 2014), which is likely due to its transport of Aβ. In a mouse model of AD, increased Aβ is eliminated from the brain by ABCB1 (Bruckmann et al., 2017). St. John's Wort also significantly decreased the accumulation of Aβ in the brain, in part, by increasing the expression of the ABCB1 protein in mice (Brenn et al., 2014). These findings were further supported by studies reporting that 1α,25-dihydroxy-vitamin D3 significantly decreased the concentrations of Aβ in the cerebral cortex of mouse AD model (Durk et al., 2014) and increased ABCB1 activity and expression in brain capillaries of rats and mice, as well as in isolated endothelial cells of human micro-vessels (Chow et al., 2011; Durk et al., 2012). Rifampicin (20 mg/kg i.p., once daily for 3 weeks) had prophylactic efficacy against the development and progression of symptoms in a mouse AD model by decreasing Aβ levels via the upregulation of ABCB1 transporters in brain microvessels (Qosa et al., 2012). These findings were confirmed by previous studies showing that ABCB1 is critical in Aβ uptake across the BBB using ABCB1-knockout mice (Wang W. M. et al., 2016).
Thus, there are data indicating the involvement of the ABCB1 transporter in Aβ transport and targeting this transporter may attenuate the progression of AD. The treatment of mice with 5 mg/kg/day IP of oleocanthal, an anti-inflammatory compound, significantly increases the clearance of Aβ from the cerebral cortex of mice, in part, by upregulating ABCB1 transporters in brain micro-vessels (Qosa et al., 2015). This effect was associated with a decrease in IL-1β levels and a decrease in the activation of astrocytes. This indicates that neuro-inflammation may contribute to the accumulation of Aβ in the brain by decreasing the expression of ABCB1 transporters, which could contribute AD pathogenesis (Figure 1). Importantly, Aβ induced an increase in the release of pro-inflammatory cytokines (Yates et al., 2000; Liu et al., 2012; Parajuli et al., 2013), which may further reduce the expression of the ABCB1 transporter in a positive feedback loop that might contribute to the long-term trajectory of the illness. This hypothesis was supported by a previous study reporting that administration of Aβ-42 at 4 μg/h via subcutaneous transplanted ALZET pumps for 24 h significantly decreased the expression of Abcb1 mRNA at the BBB of 90-day old mice (Brenn et al., 2011). A recent study reported that the incubation of isolated rat brain capillaries with Aβ-40 (10 nM for 6 h) significantly decreased the expression and transport activity of the ABCB1 transporter and this effect was associated with a degradation of the ubiquitin-proteasome (UP) (Hartz et al., 2016). The inhibition of UP in the microglial cell line enhanced the secretion of TNF-α (Kwon et al., 2008). Therefore, a decrease in the ABCB1 transporter expression in the BBB may lead to deposition of Aβ in the brain, contributing to the progression of AD.
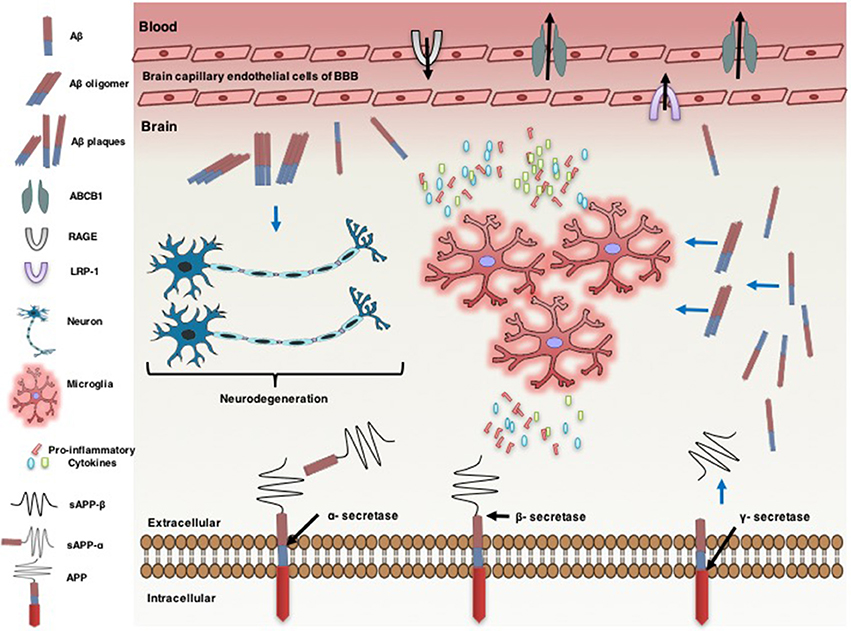
Figure 1. Neuro-inflammatory cytokines modulate ATP-binding cassette B1 (ABCB1), affecting the pathogenesis of Alzheimer's disease. Alzheimer's disease is associated with an increase in the formation of amyloid beta (Aβ) protein in the brain. Amyloid precursor protein (APP) generates Aβ through sequential proteolysis by beta (β) secretase and gamma (γ) secretase enzymes. Soluble APP-alpha (sAPP-α) and soluble APP-beta (sAPP-β) are produced through cleavage of APP by α-secretase and β-secretase, respectively. Aβ is transported across brain into the blood and vice versa by lipoprotein-related protein-1 (LRP-1) and the receptor for advanced glycation end products (RAGE), respectively. ABCB1 is also involved in uptake of Aβ from the brain into the circulatory system. However, the increase of Aβ plaque levels in the brain accelerates neuro-degenerations. The accumulation of Aβ leads to neuro-inflammation, characterized by activated microglia and the production of pro-inflammatory cytokines [e.g., interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α)]. Pro-inflammatory cytokines overexpression down regulates ABCB1 expression in the endothelial cells of brain capillary at the blood brain barrier (BBB). Pro-inflammatory cytokines increase the expression of APP. This effect is associated with a further increase in the accumulation of Aβ in the brain.
Conclusion and Future Directions
The accumulation of Aβ, a substrate of ABCB1, in the brain is associated with a decrease in the expression of ABCB1, which could affect pathogenesis of AD. We hypothesize that pro-inflammatory cytokines decrease the expression of ABCB1 in the endothelial cells of the BBB, reducing Aβ efflux, based on data from using preclinical models of AD. The upregulation of the ABCB1 transporter could decrease the accumulation of Aβ, thereby potentially attenuating the progression of AD. Future research is warranted to determine the precise role of neuro-inflammatory signaling pathways in regulating ABCB1 expression, and in the pathogenesis of AD, including cognitive effects. We suggest that compounds or treatments with dual actions, including anti-inflammatory actions and ABCB1 stimulatory effects, may have greater efficacy in reducing the progression of AD. Indeed, in a recent study, new compounds with dual actions for the treatment of AD symptoms were synthesized, although these compounds also targeted other pathways (Pang et al., 2017). The overexpression of ABCB1 may be pivotal in attenuating AD symptoms induced by the deposition of Aβ in the brain. Finally, reducing the accumulation of Aβ in the brain would also lead to a decrease in the levels of pro-inflammatory cytokines from microglia, limiting the down-regulatory effects of these cytokines on ABCB1, and interrupting the positive feedback loop between Aβ and neuro-inflammation that may be critical to disease progression in AD.
Author Contributions
FA, AT, and FH put in the idea and drafted the opinion letter. YS and CA helped provide valuable inputs and edited the opinion letter.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This opinion article was written during the period of funding supported by Award Number (R01AA019458 to YS), the University of Toledo start-up grant (F110760) to AT, and the University of Toledo Start-up grant (F110744) to FH. The authors would like to thank Rami M. Alzhrani for his technical assistance.
References
Alfonso-Loeches, S., Pascual-Lucas, M., Blanco, A. M., Sanchez-Vera, I., and Guerri, C. (2010). Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 30, 8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010
Bauer, J., Strauss, S., Schreiter-Gasser, U., Ganter, U., Schlegel, P., Witt, I., et al. (1991). Interleukin-6 and α-2-macroglobulin indicate an acute-phase state in Alzheimer's disease cortices. FEBS Lett. 285, 111–114. doi: 10.1016/0014-5793(91)80737-N
Blasko, I., Apochal, A., Boeck, G., Hartmann, T., Grubeck-Loebenstein, B., and Ransmayr, G. (2001). Ibuprofen decreases cytokine-induced amyloid beta production in neuronal cells. Neurobiol. Dis. 8, 1094–1101. doi: 10.1006/nbdi.2001.0451
Boka, G., Anglade, P., Wallach, D., Javoy-Agid, F., Agid, Y., and Hirsch, E. (1994). Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci. Lett. 172, 151–154. doi: 10.1016/0304-3940(94)90684-X
Brenn, A., Grube, M., Jedlitschky, G., Fischer, A., Strohmeier, B., Eiden, M., et al. (2014). St. John's Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer's disease mouse model—role of P-glycoprotein. Brain Pathol. 24, 18–24. doi: 10.1111/bpa.12069
Brenn, A., Grube, M., Peters, M., Fischer, A., Jedlitschky, G., Kroemer, H. K., et al. (2011). Beta-amyloid downregulates MDR1-P-glycoprotein (Abcb1) expression at the blood-brain barrier in mice. Int. J. Alzheimers dis. 2011:690121. doi: 10.4061/2011/690121
Bruckmann, S., Brenn, A., Grube, M., Niedrig, K., Holtfreter, S., von Bohlen und Halbach, O., et al. (2017). Lack of P-glycoprotein results in impairment of removal of beta-amyloid and increased intraparenchymal cerebral amyloid angiopathy after active immunization in a transgenic mouse model of Alzheimer's disease. Curr. Alzheimer Res. 14, 656–667. doi: 10.2174/1567205013666161201201227
Cacabelos, R., Alvarez, X. A., Fernández-Novoa, L., Franco, A., Mangues, R., Pellicer, A., et al. (1994). Brain interleukin-1 beta in Alzheimer's disease and vascular dementia. Methods Find. Exp. Clin. Pharmacol. 16, 141–151.
Chow, E. C., Durk, M. R., Cummins, C. L., and Pang, K. S. (2011). 1α, 25-Dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr (–/–) and fxr (+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J. Pharmacol. Exp. Ther. 337, 846–859. doi: 10.1124/jpet.111.179101
Chu, K., Lee, S.-T., Sinn, D.-I., Ko, S.-Y., Kim, E.-H., Kim, J.-M., et al. (2007). Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke 38, 177–182. doi: 10.1161/01.STR.0000252091.36912.65
Dao, A. T., Zagaar, M. A., Levine, A. T., Salim, S., Eriksen, J. L., and Alkadhi, K. A. (2013). Treadmill exercise prevents learning and memory impairment in Alzheimer's disease-like pathology. Curr. Alzheimer Res. 10, 507–515. doi: 10.2174/1567205011310050006
Del Bo, R., Angeretti, N., Lucca, E., De Simoni, M. G., and Forloni, G. (1995). Reciprocal control of inflammatory cytokines, IL-1 and IL-6, and β-amyloid production in cultures. Neurosci. Lett. 188, 70–74. doi: 10.1016/0304-3940(95)11384-9
Durk, M. R., Chan, G. N., Campos, C. R., Peart, J. C., Chow, E. C., Lee, E., et al. (2012). 1α, 25-Dihydroxyvitamin D3-liganded vitamin D receptor increases expression and transport activity of P-glycoprotein in isolated rat brain capillaries and human and rat brain microvessel endothelial cells. J. Neurochem. 123, 944–953. doi: 10.1111/jnc.12041
Durk, M. R., Han, K., Chow, E. C., Ahrens, R., Henderson, J. T., Fraser, P. E., et al. (2014). 1α, 25-Dihydroxyvitamin D3 reduces cerebral amyloid-β accumulation and improves cognition in mouse models of Alzheimer's disease. J. Neurosci. 34, 7091–7101. doi: 10.1523/JNEUROSCI.2711-13.2014
ElAli, A., and Rivest, S. (2013). The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer's disease. Front. Physiol. 4:45. doi: 10.3389/fphys.2013.00045
Evseenko, D. A., Paxton, J. W., and Keelan, J. A. (2007). Independent regulation of apical and basolateral drug transporter expression and function in placental trophoblasts by cytokines, steroids, and growth factors. Drug Metab. Dispos. 35, 595–601. doi: 10.1124/dmd.106.011478
Gao, R., Ji, M. H., Gao, D. P., Yang, R. H., Zhang, S. G., Yang, J. J., et al. (2017). Neuroinflammation-induced downregulation of hippocampacal neuregulin 1-ErbB4 signaling in the parvalbumin interneurons might contribute to cognitive impairment in a mouse model of sepsis-associated encephalopathy. Inflammation 40, 387–400. doi: 10.1007/s10753-016-0484-2
Giovannini, M. G., Scali, C., Prosperi, C., Bellucci, A., Vannucchi, M. G., Rosi, S., et al. (2002). β-Amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol. Dis. 11, 257–274. doi: 10.1006/nbdi.2002.0538
Gold, M., and El Khoury, J. (2015). β-amyloid, microglia, and the inflammasome in Alzheimer's disease. Semin. Immunopathol. 37, 607-611. doi: 10.1007/s00281-015-0518-0
Grammas, P., and Ovase, R. (2001). Inflammatory factors are elevated in brain microvessels in Alzheimer's disease. Neurobiol. Aging 22, 837–842. doi: 10.1016/S0197-4580(01)00276-7
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Nat. Acad. Sci. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Hartz, A. M., Miller, D. S., and Bauer, B. (2010). Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-β in a mouse model of Alzheimer's disease. Mol. Pharmacol. 77, 715–723. doi: 10.1124/mol.109.061754
Hartz, A. M., Zhong, Y., Wolf, A., LeVine, H., Miller, D. S., and Bauer, B. (2016). Aβ40 reduces P-glycoprotein at the blood–brain barrier through the ubiquitin–proteasome pathway. J. Neurosci. 36, 1930–1941. doi: 10.1523/JNEUROSCI.0350-15.2016
Hebert, L. E., Scherr, P. A., Bienias, J. L., Bennett, D. A., and Evans, D. A. (2003). Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch. Neurol. 60, 1119–1122. doi: 10.1001/archneur.60.8.1119
Iba, M., Guo, J. L., McBride, J. D., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J. Neurosci. 33, 1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013
Iqbal, M., Ho, H. L., Petropoulos, S., Moisiadis, V. G., Gibb, W., and Matthews, S. G. (2012). Pro-inflammatory cytokine regulation of P-glycoprotein in the developing blood-brain barrier. PLoS ONE 7:e43022. doi: 10.1371/journal.pone.0043022
Kim, W., Zekas, E., Lodge, R., Susan-Resiga, D., Marcinkiewicz, E., Essalmani, R., et al. (2015). Neuroinflammation-induced interactions between protease-activated receptor 1 and proprotein convertases in HIV-associated neurocognitive disorder. Mol. Cell. Biol. 35, 3684–3700. doi: 10.1128/MCB.00764-15
Kwon, S.-J., Ahn, T.-B., Yoon, M.-Y., and Jeon, B. S. (2008). BV-2 stimulation by lactacystin results in a strong inflammatory reaction and apoptotic neuronal death in SH-SY5Y cells. Brain Res. 1205, 116–121. doi: 10.1016/j.brainres.2008.02.030
Lee, S. P., Falangola, M. F., Nixon, R. A., Duff, K., and Helpern, J. A. (2004). Visualization of β-amyloid plaques in a transgenic mouse model of Alzheimer's disease using MR microscopy without contrast reagents. Magn. Reson. Med. 52, 538–544. doi: 10.1002/mrm.20196
Liao, Y.-F., Wang, B.-J., Cheng, H.-T., Kuo, L.-H., and Wolfe, M. S. (2004). Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 279, 49523–49532. doi: 10.1074/jbc.M402034200
Liu, S., Liu, Y., Hao, W., Wolf, L., Kiliaan, A. J., Penke, B., et al. (2012). TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 188, 1098–1107. doi: 10.4049/jimmunol.1101121
Löscher, W., and Potschka, H. (2005). Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2, 86–98. doi: 10.1602/neurorx.2.1.86
Marshall, S. A., Casachahua, J. D., Rinker, J. A., Blose, A. K., Lysle, D. T., and Thiele, T. E. (2016a). IL-1 receptor signaling in the basolateral amygdala modulates binge-like ethanol consumption in male C57BL/6J mice. Brain Behav. Immun. 51, 258–267. doi: 10.1016/j.bbi.2015.09.006
Marshall, S. A., Geil, C. R., and Nixon, K. (2016b). Prior binge ethanol exposure potentiates the microglial response in a model of alcohol-induced neurodegeneration. Brain Sci. 6:16. doi: 10.3390/brainsci6020016
Mogi, M., Harada, M., Kondo, T., Riederer, P., Inagaki, H., Minami, M., et al. (1994). Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 180, 147–150. doi: 10.1016/0304-3940(94)90508-8
O'Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Pahnke, J., Langer, O., and Krohn, M. (2014). Alzheimer's and ABC transporters—new opportunities for diagnostics and treatment. Neurobiol. Dis. 72, 54–60. doi: 10.1016/j.nbd.2014.04.001
Pang, X., Fu, H., Yang, S., Wang, L., Liu, A.-L., Wu, S., et al. (2017). Evaluation of novel dual acetyl-and butyrylcholinesterase inhibitors as potential Anti-Alzheimer's disease agents using pharmacophore, 3D-QSAR, and molecular docking approaches. Molecules 22:E1254. doi: 10.3390/molecules22081254
Parajuli, B., Sonobe, Y., Horiuchi, H., Takeuchi, H., Mizuno, T., and Suzumura, A. (2013). Oligomeric amyloid β induces IL-1β processing via production of ROS: implication in Alzheimer's disease. Cell Death Dis. 4:e975. doi: 10.1038/cddis.2013.503
Parent, M. J., Bedard, M.-A., Aliaga, A., Minuzzi, L., Mechawar, N., Soucy, J.-P., et al. (2013). Cholinergic depletion in Alzheimer's disease shown by [18F] FEOBV autoradiography. Int. J. Mol. Imaging 2013:205045. doi: 10.1155/2013/205045
Patel, N. S., Paris, D., Mathura, V., Quadros, A. N., Crawford, F. C., and Mullan, M. J. (2005). Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J. Neuroinflammation 2:9. doi: 10.1186/1742-2094-2-9
Paulson, J. B., Ramsden, M., Forster, C., Sherman, M. A., McGowan, E., and Ashe, K. H. (2008). Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer's disease. Am. J. Pathol. 173, 762–772. doi: 10.2353/ajpath.2008.080175
Qosa, H., Abuznait, A. H., Hill, R. A., and Kaddoumi, A. (2012). Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer's disease. J. Alzheimers Dis. 31, 151–165. doi: 10.3233/JAD-2012-120319
Qosa, H., Batarseh, Y. S., Mohyeldin, M. M., El Sayed, K. A., Keller, J. N., and Kaddoumi, A. (2015). Oleocanthal enhances amyloid-β clearance from the brains of tgswdi mice and in vitro across a human blood-brain barrier model. ACS Chem. Neurosci. 6, 1849–1859. doi: 10.1021/acschemneuro.5b00190
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
Saresella, M., La Rosa, F., Piancone, F., Zoppis, M., Marventano, I., Calabrese, E., et al. (2016). The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol. Neurodegener. 11:23. doi: 10.1186/s13024-016-0088-1
Sita, G., Hrelia, P., Tarozzi, A., and Morroni, F. (2017). P-glycoprotein (ABCB1) and oxidative stress: focus on Alzheimer's disease. Oxid. Med. Cell. Longev. 2017:7905486. doi: 10.1155/2017/7905486
Strauss, S., Bauer, J., Ganter, U., Jonas, U., Berger, M., and Volk, B. (1992). Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer's disease patients. Lab. Invest. 66, 223–230.
Tilleux, S., and Hermans, E. (2007). Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 85, 2059–2070. doi: 10.1002/jnr.21325
van Assema, D. M., Lubberink, M., Rizzu, P., van Swieten, J. C., Schuit, R. C., Eriksson, J., et al. (2012). Blood–brain barrier P-glycoprotein function in healthy subjects and Alzheimer's disease patients: effect of polymorphisms in the ABCB1 gene. EJNMMI Res. 2:57. doi: 10.1186/2191-219X-2-57
Walters, A., Phillips, E., Zheng, R., Biju, M., and Kuruvilla, T. (2016). Evidence for neuroinflammation in Alzheimer's disease. Prog. Neurol. Psychiatry 20, 25–31. doi: 10.1002/pnp.444
Walther, W., Kobelt, D., Bauer, L., Aumann, J., and Stein, U. (2015). Chemosensitization by diverging modulation by short-term and long-term TNF-α action on ABCB1 expression and NF-κB signaling in colon cancer. Int. J. Oncol. 47, 2276–2285. doi: 10.3892/ijo.2015.3189
Wang, B., Wang, X. Q., Yang, S. S., Liu, X., Feng, D. Y., Lu, F. N., et al. (2016). Neuroprotective effects of nitidine in Parkinson's disease models through inhibiting microglia activation: role of the Jak2–Stat3 pathway. RSC Adv. 6, 71328–71337. doi: 10.1039/C6RA11759G
Wang, W. M., Bodles-Brakhop, A. W., and Barger, S. (2016). A role for P-glycoprotein in clearance of Alzheimer amyloid β-peptide from the brain. Curr. Alzheimer Res. 13, 615–620. doi: 10.2174/1567205013666160314151012
Webster, S. J., Bachstetter, A. D., Nelson, P. T., Schmitt, F. A., and Van Eldik, L. J. (2014). Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 5:88. doi: 10.3389/fgene.2014.00088
Wei, J., Pan, X., Pei, Z., Wang, W., Qiu, W., Shi, Z., et al. (2012). The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J. Trauma Acute Care Surg. 73, 654–660. doi: 10.1097/TA.0b013e31825133c0
Wijesuriya, H. C., Bullock, J. Y., Faull, R. L., Hladky, S. B., and Barrand, M. A. (2010). ABC efflux transporters in brain vasculature of Alzheimer's subjects. Brain Res. 1358, 228–238. doi: 10.1016/j.brainres.2010.08.034
Wildburger, N. C., Esparza, T. J., LeDuc, R. D., Fellers, R. T., Thomas, P. M., Cairns, N. J., et al. (2017). Diversity of amyloid-beta proteoforms in the Alzheimer's disease brain. Sci. Rep. 7:9520. doi: 10.1038/s41598-017-10422-x
Xiao, M.-F., Xu, D., Craig, M. T., Pelkey, K. A., Chien, C.-C., Shi, Y., et al. (2017). NPTX2 and cognitive dysfunction in Alzheimer's Disease. Elife 6:e23798. doi: 10.7554/eLife.23798
Yamamoto, M., Kiyota, T., Horiba, M., Buescher, J. L., Walsh, S. M., Gendelman, H. E., et al. (2007). Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am. J. Pathol. 170, 680–692. doi: 10.2353/ajpath.2007.060378
Yates, S. L., Burgess, L. H., Kocsis-Angle, J., Antal, J. M., Dority, M. D., Embury, P. B., et al. (2000). Amyloid β and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J. Neurochem. 74, 1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x
Keywords: Alzheimer's disease, ABCB1, pro-inflammatory cytokines, amyloid beta, blood brain barrier
Citation: Alasmari F, Ashby CR Jr, Hall FS, Sari Y and Tiwari AK (2018) Modulation of the ATP-Binding Cassette B1 Transporter by Neuro-Inflammatory Cytokines: Role in the Pathogenesis of Alzheimer's Disease. Front. Pharmacol. 9:658. doi: 10.3389/fphar.2018.00658
Received: 02 April 2018; Accepted: 01 June 2018;
Published: 20 June 2018.
Edited by:
Francisco Lopez-Munoz, Universidad Camilo José Cela, SpainReviewed by:
Chung-Pu Wu, Chang Gung University, TaiwanAlejandro Romero, Complutense University of Madrid, Spain
Copyright © 2018 Alasmari, Ashby, Hall, Sari and Tiwari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youssef Sari, eW91c3NlZi5zYXJpQHV0b2xlZG8uZWR1
Amit K. Tiwari, YW1pdC50aXdhcmlAdXRvbGVkby5lZHU=