 Nicole El-Darzi
Nicole El-Darzi Artem Astafev
Artem Astafev Natalia Mast
Natalia Mast Aicha Saadane
Aicha Saadane Irina A. Pikuleva
Irina A. Pikuleva- Department of Ophthalmology and Visual Sciences, Case Western Reserve University, Cleveland, OH, United States
N,N-dimethyl-3β-hydroxycholenamide (DMHCA) is an experimental pharmaceutical and a steroidal liver X receptor (LXR) agonist, which does not induce undesired hepatic lipogenesis. Herein, DMHCA was evaluated for its retinal effects on normal C57BL/6J and Cyp27a1−/−Cyp46a1−/− mice; the latter having higher retinal total and esterified cholesterol in addition to retinal vascular abnormalities. Different doses and two formulations were used for DMHCA delivery either via drinking water (C57BL/6J mice) or by oral gavage (Cyp27a1−/−Cyp46a1−/− mice). The duration of treatment was 1 week for C57BL/6J mice and 2 or 4 weeks for Cyp27a1−/−Cyp46a1−/− mice. In both genotypes, the higher DMHCA doses (37–80 mg/kg of body weight/day) neither increased serum triglycerides nor serum cholesterol but altered the levels of retinal sterols. Total retinal cholesterol was decreased in the DMHCA-treated mice, mainly due to a decrease in retinal unesterified cholesterol. In addition, retinal levels of cholesterol precursors lanosterol, zymosterol, desmosterol, and lathosterol were changed in Cyp27a1−/−Cyp46a1−/− mice. In both genotypes, DMHCA effect on retinal expression of the LXR target genes was only moderate and gender-specific. Collectively, the data obtained provide evidence for a decrease in retinal cholesterol as a result of DMHCA acting in the retina as an enzyme inhibitor of cholesterol biosynthesis rather than a LXR transcriptional activator. Specifically, DMHCA appears to partially inhibit the cholesterol biosynthetic enzyme Δ24-dehydrocholesterol reductase rather than upregulate the expression of LXR target genes involved in reverse cholesterol transport. The identified DMHCA dosages, formulations, and routes of delivery as well as the observed effects on the retina should be considered in future studies using DMHCA as a potential therapeutic for age-related macular degeneration and diabetic retinopathy.
Introduction
The retina is a thin tissue that lines the back of the eye and responds to light by converting it into an electrical signal. The retina is very small: only 1204 mm2 in humans and 15.6 mm2 in mice (Remtulla and Hallett, 1985; Panda-Jonas et al., 1994), a size that makes pharmacological investigations of the retina challenging. In addition, the retina is separated from the systemic circulation by the blood–retinal barrier, which may limit drug delivery to the retina. Lowering of cholesterol in the retina is one of the approaches that are currently under investigation for treatment of age-related macular degeneration (Pikuleva and Curcio, 2014), a common eye disease leading to vision loss in the elderly of industrialized countries. Indeed, significant amounts of cholesterol are present in drusen and subretinal drusenoid deposits (Curcio et al., 2001, 2005; Oak et al., 2014), the two hallmarks of AMD, and AMD risk factors include variants of several cholesterol-related genes (ABCA1, APOE, CETP, LIPC, LPL, LRP6, and VLDLR) (Miller, 2013). The majority (72%) of retinal cholesterol (at least in mice) is provided by local biosynthesis, which proceeds via the Bloch and Kandutsch-Russell pathways (Figure 1); the remaining 28% of cholesterol are taken up from the systemic circulation (Saadane et al., 2014; Lin et al., 2016). Retinal cholesterol elimination proceeds via metabolism to oxysterols catalyzed by CYP27A1 and CYP46A1 enzymes as well as lipoprotein-mediated reverse cholesterol transport to the liver (Tserentsoodol et al., 2006; Curcio et al., 2011; Fujihara et al., 2014; Pikuleva and Curcio, 2014). Additionally, small amounts of cholesterol excess stay in the retina in the form of cholesterol esters (Bretillon et al., 2008; Saadane et al., 2016).
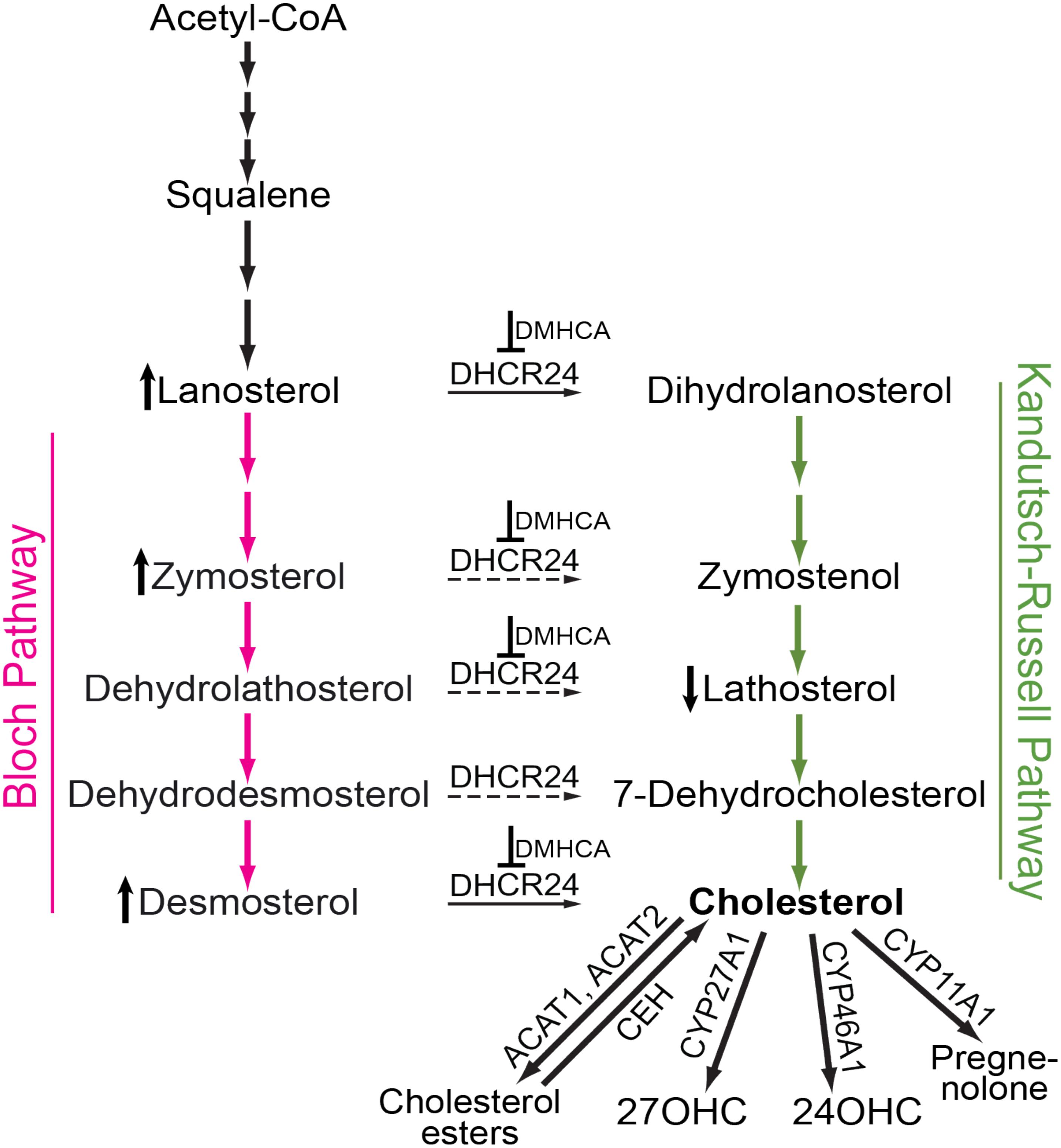
FIGURE 1. Schematic representation of cholesterol-related pathways of pertinence to the present work. Cholesterol biosynthesis starts from acetyl coenzyme A (Acetyl-CoA) and proceeds either via the Bloch (magenta arrows) or Kandutsch-Russell (green arrows) pathways. In certain tissues and cell types, there may be a switch from the Bloch to the Kandutsch-Russell pathway due to the action of Δ24-dehydrocholesterol reductase (DHCR24, solid and dashed arrows are the confirmed and potential points of the pathway crossover, respectively). Pharmacologic inhibition (for example with DMHCA, blunt-end arrows) may increase the levels of some of the DHCR24 substrates (e.g., lanosterol, zymosterol, and desmosterol, the vertical upward black arrow) and decrease the levels of some of the DHCR24 metabolites (e.g., lathosterol, the vertical downward black arrow), thus decreasing the levels of tissue cholesterol. If tissue cholesterol is in excess, it can be esterified by acyl-coenzyme A:cholesterol acyltransferases 1 and 2 (ACAT1 and ACAT2) and stored intracellularly in the form of lipid droplets. When cellular cholesterol levels are decreased, cholesterol esters are hydrolyzed to cholesterol by cholesterol ester hydrolases (CEH) with several enzymes being known to carry out this enzymatic reaction. Cholesterol excess can also be hydroxylated by CYP enzymes 27A1, 46A1, and 11A1 to yield 27-hydroxycholesterol (27OHC), 24-hydroxycholesterol (24OHC), and pregnenolone, respectively. These cholesterol derivatives either diffuse into the systemic circulation for delivery to the liver and subsequent degradation to bile acids or are metabolized locally at the site of production.
N,N-dimethyl-3β-hydroxycholenamide (DMHCA) is an experimental pharmaceutical and a synthetic ligand for LXRs (Janowski et al., 1999; Spencer et al., 2001), a family of transcription factors. LXRs act as cellular sterol sensors and are activated by desmosterol (a cholesterol precursor) and certain oxysterols (metabolites of cholesterol) (Janowski et al., 1996; Chen et al., 2007; Spann et al., 2012). The two isoforms of this family, LXRα and LXRβ (Alberti et al., 2000), bind the same ligands and either increase (gene transactivation) or decrease (gene transrepression) the expression of the target genes (Glass and Ogawa, 2006; Kalaany and Mangelsdorf, 2006; Zelcer and Tontonoz, 2006). Originally, LXRs were discovered as nuclear receptors that limit cholesterol content by upregulating the expression of the genes involved in reverse cholesterol transport (e.g., Abca1, Abcg1, and Apoe). Later, LXR target genes were identified in the pathways of fatty acid biosynthesis (e.g., Srebp1c, Acc1, Acc2, Fasn, and Scd1), cholesterol uptake (Idol), glucose metabolism (Glut4), neovascularization (Vegf), and immune/inflammatory responses (e.g., ArgII, Cox-2, iNos, and Il-6) (Calkin and Tontonoz, 2012). In the latter, the proinflammatory genes (e.g., Cox-2, iNos, and Il-6) were downregulated by LXRs in contrast to the upregulation of cholesterol-, fatty acid-, and glucose-related genes (Glass and Ogawa, 2006; Zelcer and Tontonoz, 2006). Both gene transactivation and transrepression represent LXR activities that are of therapeutic importance (Hong and Tontonoz, 2014), with the exception of the induction of fatty acid synthesis in the liver, which is reflected by an increase in serum triglyceride levels. Significant efforts are directed toward developing LXR ligands that lack the undesired upregulation of hepatic lipogenesis and bind LXRs in a tissue-, pathway-, and isoform-specific manner (Hong and Tontonoz, 2014).
N,N-dimethyl-3β-hydroxycholenamide was initially synthesized for a study of structural requirements for the LXR ligands (Janowski et al., 1999). Later, this compound was found to activate the tested LXR target genes in the liver, small intestine, and peritoneal macrophages of C57BL/6 mice while having only a minor effect on hepatic fatty acid biosynthesis and serum triglyceride levels (Quinet et al., 2004). Subsequent DMHCA evaluation on Apoe−/− mice confirmed the desired upregulation of the LXR target genes and a lack of undesired serum hypertriglyceridemia; this evaluation also documented the antiatherogenic properties of DMHCA (Kratzer et al., 2009). A favorable pharmacologic profile of DMHCA was established and prompted further investigations of this compound (Caldas et al., 2011; Patel et al., 2014; Yu et al., 2016; Muller et al., 2017). Unexpectedly, in addition to LXR binding, DMHCA was discovered to inhibit DHCR24 both in vitro and in vivo (Pfeifer et al., 2011). DHCR24 catalyzes Δ24 reduction of the side chain of cholesterol precursors during cholesterol biosynthesis (Figure 1) and links the Bloch and Kandutsch-Russell pathways (Zerenturk et al., 2013; Mitsche et al., 2015). DMHCA inhibition of DHCR24 opened new pharmacologic options including those for treating hepatitis C viral infection, certain forms of cancers, and atherosclerosis (Muller et al., 2017). Herein, we evaluated DMHCA for its effect on the retina in mice. We found that under the treatment paradigms employed, DMHCA partially inhibited DHCR24 in the retina and reduced retinal cholesterol but did not seem to upregulate the expression of the LXR target genes.
Materials and Methods
Materials
DMHCA, lanosterol, [26,26,26,27,27,27-2H6]lanosterol, zymosterol, [2,2,3,4,4,-2H5]zymosterol, lathosterol, and desmosterol were from Avanti Polar Lipids, Inc. (Alabaster, AL, United States). Cholesterol and [3H]cholesterol were from Steraloids, Inc. (Newport, RI, United States) and PerkinElmer (Waltham, MA, United States), respectively. [26,26,26,27,27,27-2H6]Desmosterol and [1,2,5,6α-2H4]lathosterol were from C/D/N Isotopes Inc. (Pointe-Claire, QC, Canada). Rodent chow (5P76 Prolab Isopro RMH 3000) was from T. R. Last Co. (Saxonburg, PA, United States). All other chemicals were from Sigma-Aldrich (St. Louis, MO, United States) unless otherwise indicated. Recombinant CYP27A1, CYP46A1, adrenodoxin reductase, adrenodoxin, and NADPH-CYP oxidoreductase were expressed and purified as described (Sagara et al., 1992, 1993; Hanna et al., 1998; Mast et al., 2006, 2012).
Animals
All animals were 4-month old. C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME, United States), and Cyp27a1−/−Cyp46a1−/− mice were generated as described (Saadane et al., 2014). Both genotypes were free of the Crblrd8 mutation. Mice were maintained on a standard 12-h light (approximately 10 lux)-dark cycle and were fed standard rodent chow and water ad libitum. Rodent chow contained only 0.02% cholesterol (w/w). All animal procedures were approved by the Case Western Reserve University Institutional Animal Care and Use Committee and have been carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health.
Pharmacologic Treatments
All experiments were initiated in the morning. C57BL/6J mice received DMHCA in drinking water. Cyp27a1−/−Cyp46a1−/− mice received DMHCA by oral gavage. For delivery in drinking water, DMHCA stocks (84.3, 16.9, and 33.7 mg/l) were prepared for groups 1–3, respectively, in aqueous 11% 2-hydroxypropyl-β-cyclodextrin (HPCD) and then were diluted 10-fold with drinking water. This DMHCA-containing water was provided to mice for 7 days ad libitum. The control group received 1.1% HPCD in drinking water. On day 7, mice were fasted overnight but had access to water and were sacrificed the following morning. For delivery by gavage, DMHCA stock (100 mg/ml) in ethanol was prepared and diluted 10-fold with aqueous 5% HPCD. Animals received from 0.17 to 0.23 ml of 10 mg/ml DMHCA in aqueous solution of 10% ethanol and 4.5% HPCD so that the administered dose of DMHCA was equivalent to 80 mg/kg of BW. Animals underwent daily gavage for 15 or 29 days. The control group received aqueous solution of 10% ethanol and 4.5% HPCD. On day 14 or 28, mice were fasted overnight and sacrificed the next morning 1 h after the 15th or 29th gavage administration.
Serum Analyses
Blood was withdrawn via cardiac puncture after mice were anesthetized via intraperitoneal injection of 80 mg/kg ketamine (Animal Health, Fort Dodge, IA, United States) mixed with 15 mg/kg xylazine (Akorn Inc., Lake Forest, IL, United States) in phosphate buffered saline, pH 7.4. The serum was isolated as described (Mast et al., 2010) and analyzed for total cholesterol and triglycerides by Marshfield Labs (Marshfield Clinic, Marshfield, WI, United States).
Tissue Isolation and Sterol Quantifications
Mouse liver and retinas were isolated and processed as described (Zheng et al., 2015). Sterol quantifications were performed by isotope dilution gas chromatography-mass spectroscopy using deuterated sterol analogs as internal standards (Mast et al., 2011). Both unesterified and total retinal cholesterol (the sum of unesterified and esterified cholesterol) were measured.
Quantitative Real Time PCR
Total RNA (1 μg) from the samples of individual livers or pooled retinas was isolated as described (Zheng et al., 2015) using the TRIzol Reagent (Life Technologies, Grand Island, NY, United States). This RNA was then converted to cDNA by SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. PCR reactions were performed in triplicate and were normalized to GAPDH. The primer sequences are shown in Table 1. Changes in relative mRNA level were calculated by the 2−ΔΔCt method (Pfaffl, 2001).

TABLE 1. Primer sequences for quantitative real-time PCR.
CYP27A1 and CYP46A1 Enzyme Assays
CYP27A1 and CYP46A1 were evaluated for cholesterol 27- and 24-hydroxylation, respectively. The conditions for each enzyme assay were as described (Mast et al., 2012; Lam et al., 2018). For each P450, the cholesterol concentration was equal to 0.5 Km for cholesterol (2.3 and 2.7 μM for CYP27A1 and CYP46A1, respectively) (Mast et al., 2008, 2015), and DMHCA concentrations were equal to 45 and 43 μM for CYP27A1 and CYP46A1, respectively. Cholesterol was added from a 1 mM stock in 4.5% aqueous HPCD, and DMHCA was added from an 8 mM stock in 11% HPCD. Control incubations contained 0.06% HPCD. Product formation was linear with time and P450 concentration. More than 75% of inhibition of the P450 activity in this enzyme assay is indicative of a strong inhibitor (Mast et al., 2012; Lam et al., 2018).
Statistics
The data represent either the mean ± SEM or the mean ± SD; the number of animals (n) is indicated in each figure or figure legend. Data were analyzed either by a two-tailed, unpaired Student’s t-test or by one-way ANOVA followed by Dunnett’s test for post hoc analysis using GraphPad Prism software (La Jolla, CA, United States). Statistical significance was defined as ∗P ≤ 0.05; ∗∗P ≤ 0.01; ∗∗∗P ≤ 0.001.
Results
Pilot Studies on C57BL/6J Mice
A control and three treatment groups (1–3) were used, each comprised of two female and three male mice housed in two separate cages based on gender. The intended DMHCA doses were 20, 40, and 80 mg/kg of BW for groups 1–3, respectively, assuming that an average C57BL/6J mouse weighs ∼25 g and drinks ∼5.8 ml of water per day (Bachmanov et al., 2002). Thus, different amounts of DMHCA were added to drinking water for groups 1–3 (“Materials and Methods” section). However, the measurements of the intake of DMHCA-containing water (Figure 2A) revealed that both female and male mice had a trend toward a decreased water intake with increasing DMHCA concentrations; the lowest water intake being by group 3 (Figure 2A). In contrast, there did not seem to be a dependence of mouse BW on the administered DMHCA dose, except a slight decrease in BW at the end of the treatment in both female and male mice (≤10% and not statistically significant in male mice, Figure 2B). These data are in agreement with previous studies showing that DMHCA treatment may slightly decrease mouse BW (Kratzer et al., 2009; Patel et al., 2014). Thus, most of the actual DMHCA doses were lower than the intended doses (Table 2).
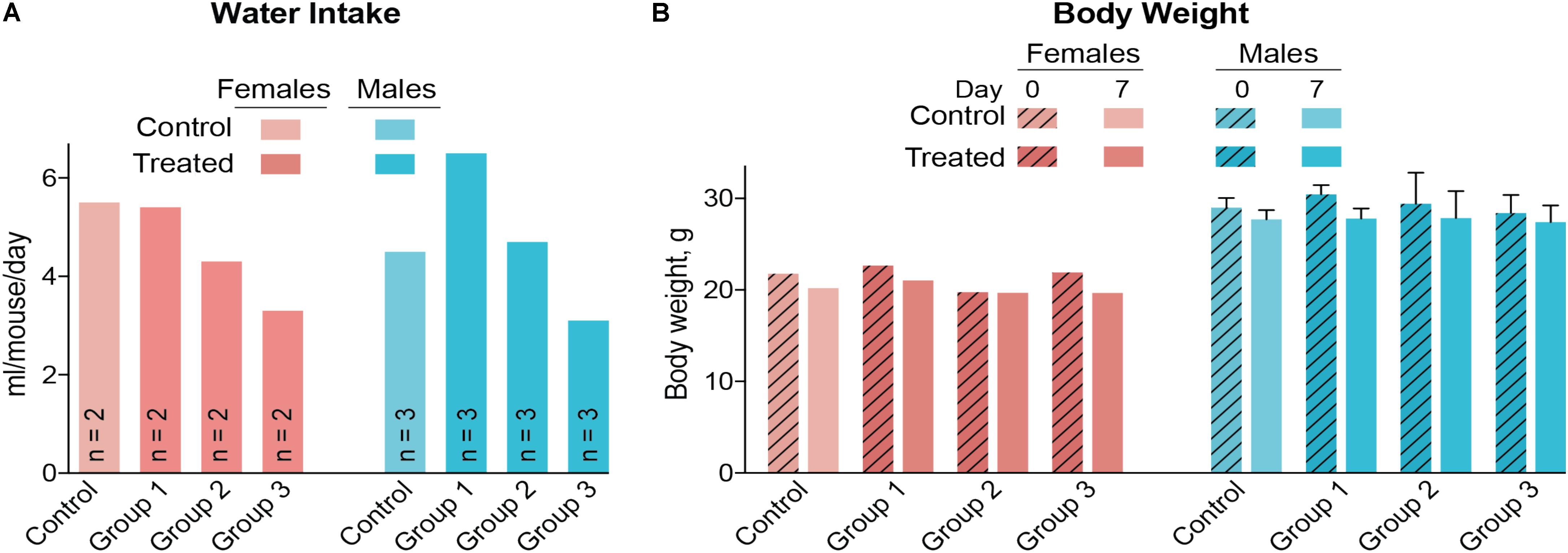
FIGURE 2. Water consumption (A) and body weight (B) of C57BL/6J mice treated with different DMHCA doses. The DMHCA doses are shown in Table 2. The duration of treatment was 7 days. DMHCA was delivered in drinking water containing 1.1% HPCD. The control group received drinking water containing 1.1% HPCD. The control and each dosing group was comprised of two female mice and three male mice with each gender housed in a separate cage. Water consumption was recorded at the end of treatment and divided by the number of mice per cage and duration of treatment. Results for the body weight measurements represent the mean for two female mice and the mean ± SD for three male mice.

TABLE 2. DMHCA doses delivered in drinking water to C57BL/6J mice.
Serum triglycerides and serum total cholesterol were measured in the vehicle-(control) and DMHCA-treated groups to confirm the lack of undesired liver lipogenesis (Figures 3A,B). No changes in the serum levels of these lipids were found, although we documented a higher interindividual variability in female mice than in male mice. Then, retinal cholesterol and retinal cholesterol precursors were quantified: desmosterol and lathosterol, which are sterol intermediates in the Bloch and Kandutsch-Russell pathways of cholesterol biosynthesis, respectively (Figure 1; Zerenturk et al., 2013). There was a trend toward an increased desmosterol levels and decreased lathosterol levels with increasing DMHCA doses (Figures 3C,D). Also, total cholesterol was decreased by 14% in group 3, which received the highest DMHCA dose (Figure 3E).
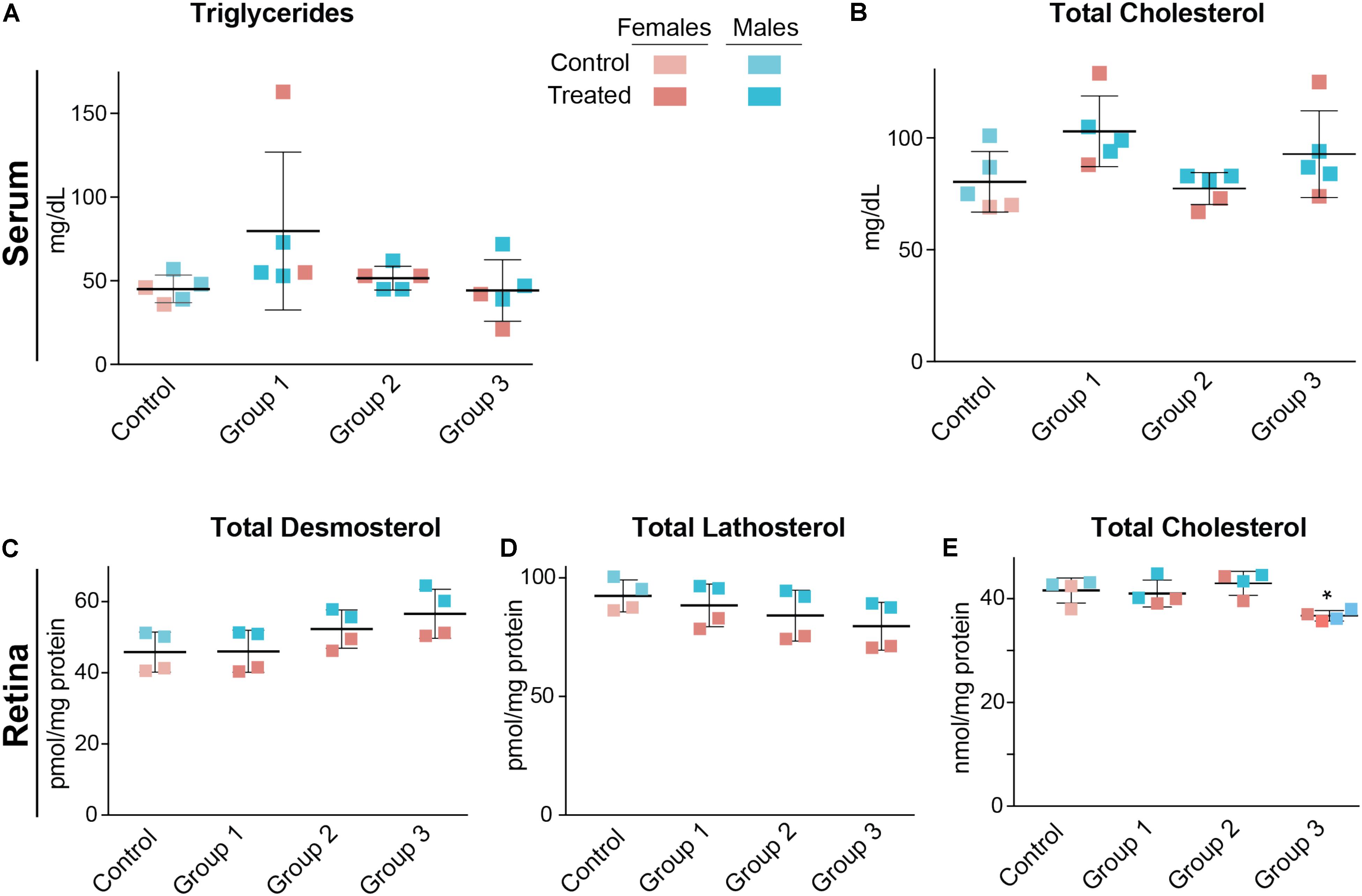
FIGURE 3. Serum (A,B) and retinal lipids (C–E) of C57BL/6J mice treated with different DMHCA doses. The DMHCA doses and treatment paradigm were the same as in Figure 2. The results are the mean ± SD of the measurements in the samples from individual animals of both genders. Asterisk is a significant change relative to the control group as assessed by one-way ANOVA followed by Dunnett’s test for post hoc analysis. ∗P ≤ 0.05.
Finally, gene expression was evaluated in the liver and retina. Hepatic expression of LXR target genes were mostly unchanged, except a paradoxical dose-dependent decrease in the Srebp1c expression (Figure 4A). Additionally, Acc1, Scd1, and Inos in group 3 had the largest extent of upregulation. Yet, serum triglyceride levels were not increased in DMHCA-treated mice (Figure 3A) suggesting that increased Srebp1c, Acc1, and Scd1 expression did not translate into increased liver lipogenesis. In the retina, a dose-dependent decrease seemed to occur only in the expression of pro-inflammatory genes Cox-2 and Il-6 with the remaining changes in gene expression did not seem to be dose-dependent (Figure 4B).
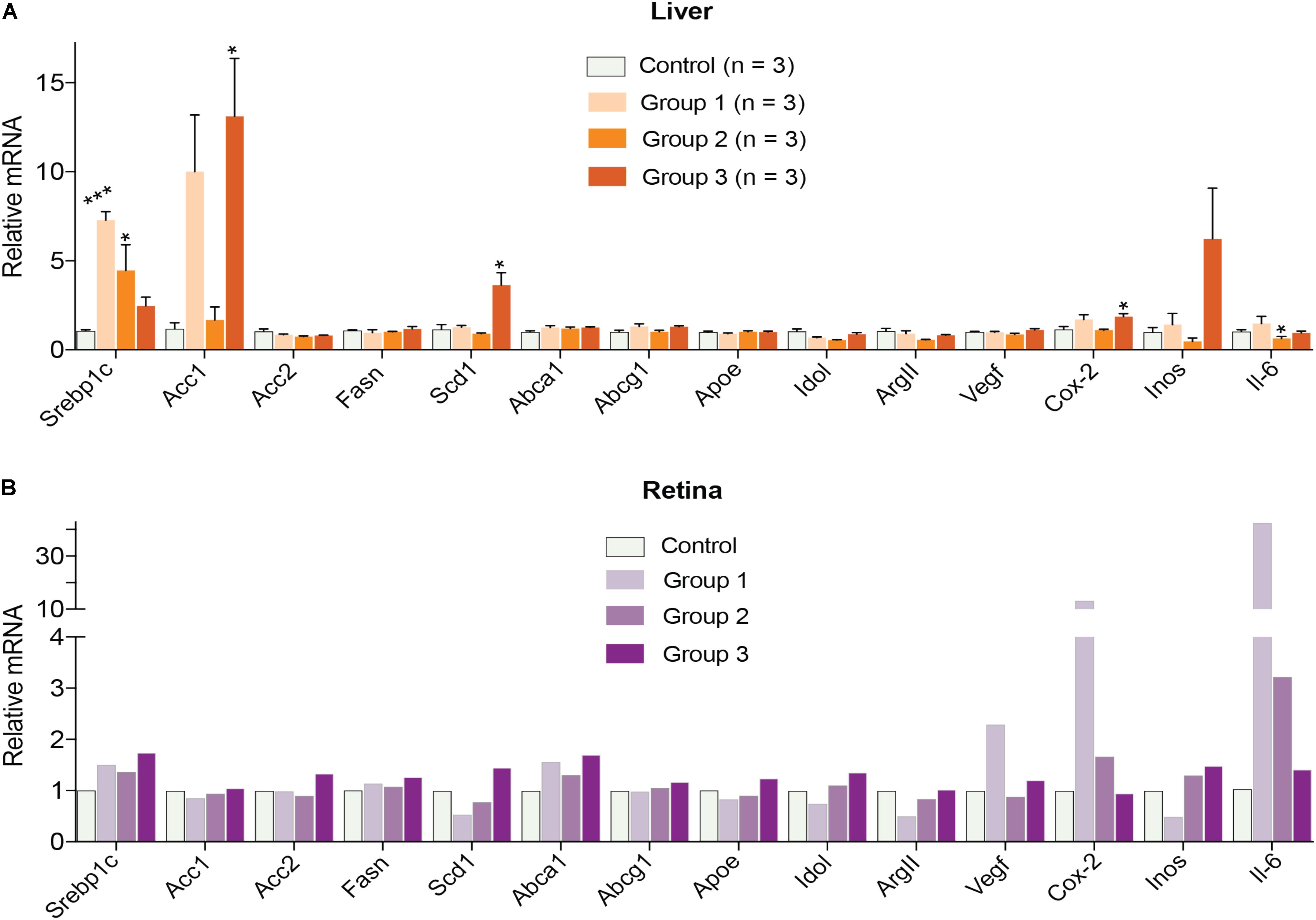
FIGURE 4. Hepatic (A) and retinal (B) gene expression in C57BL/6J mice treated with different DMHCA doses. The DMHCA doses and treatment paradigm were the same as in Figure 2. The results are the mean ± SEM of the duplicate measurements in the individual samples of the liver from two female one male mouse (A) or a pooled sample of five retinas from two female and three male mice (B). Asterisks are significant changes relative to the control group as assessed by one-way ANOVA followed by Dunnett’s test for post hoc analysis. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001.
Thus, the results of the pilot dose-dependence studies were encouraging. They confirmed a lack of hypertriglyceridemia in mice treated with DMHCA doses of up to 51 mg/kg BW. They identified the effective DMHCA doses (37 and 51 mg/kg BW for male and female mice, respectively) that led to a decrease in total retinal cholesterol. Accordingly, we decided to test DMHCA on Cyp27a1−/−Cyp46a1−/− mice who develop retinal lesions similar to those found in type 3 neovascular age-related macular degeneration and early stage diabetic retinopathy (Saadane et al., 2014). Of importance was that the total retinal cholesterol in Cyp27a1−/−Cyp46a1−/− mice is twice as high as in wild-type mice on the same C57BL/6J;129S6/SvEv background (Saadane et al., 2014), and retinal cholesterol excess in this genotype is stored in the form of cholesterol esters (Saadane et al., 2016). Because of such high and unusual retinal cholesterol content, we decided to: (1) increase the DMHCA dose to 80 mg/kg BW/day; (2) increase the treatment time to either 2 or 4 weeks; and (3) switch from drug delivery in drinking water to gavage to prevent animal dehydration and ensure that every animal in its respective treatment group receives the same DMHCA dose.
DMHCA Treatment of Cyp27a1−/−Cyp46a1−/− Mice
Both female and male mice were used for the 2-week treatment study, and only female mice were used in the 4-week treatment experiment due to the female gender having a higher interindividual variability in their serum lipid profile (Figures 3A,B). DMHCA (80 mg/kg BW/day) was formulated with aqueous 10% ethanol/4.5% HPCD and delivered orally by gavage. Average food intake was the same in vehicle-treated (control) and DMHCA-treated mice (Figure 5A), and average BWs were not significantly different by the end of either 2- or 4-week treatment with the exception of the control group in the 4-week experiment that had a small decrease in the BW (Figure 5B).
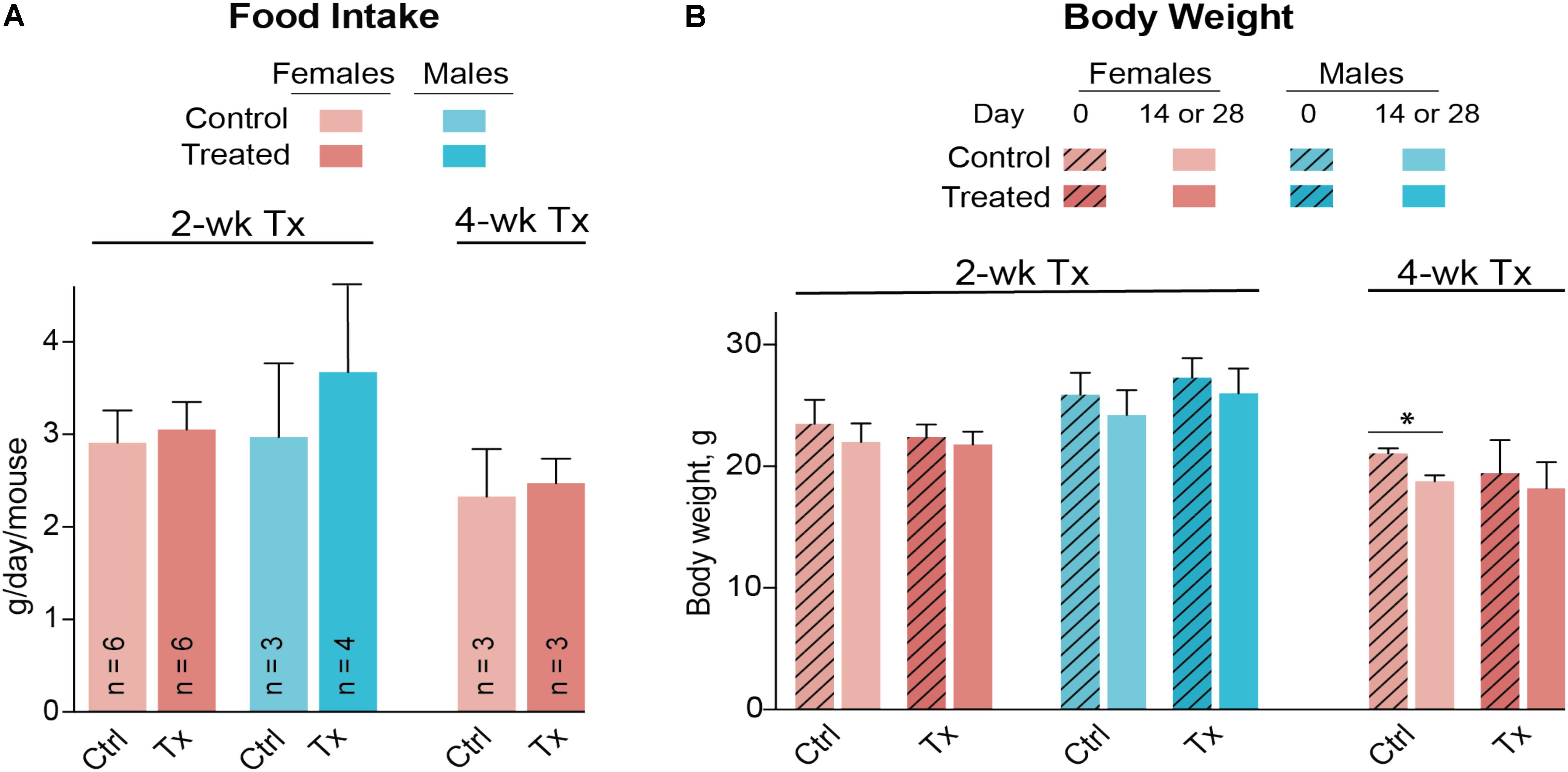
FIGURE 5. Food consumption (A) and body weight (B) of Cyp27a1−/−Cyp46a1−/− mice treated with DMHCA. DMHCA was formulated with aqueous solution of 10% ethanol and 4.5% HPCD and delivered by oral gavage at a 80 mg/kg BW/day dose for 2 or 4 weeks. The number of animals per group (n) is indicated in A and is the same in B. Each animal was housed in a separate cage; food consumption was recorded at the end of treatment and divided by duration of treatment. The results are the mean ± SD of the measurements in individual animals. Asterisks are significant changes relative to the control group as assessed by a two-tailed, unpaired Student’s t-test. ∗P ≤ 0.05. Ctrl, control; Tx, treatment.
Similar to C57BL/6J mice, Cyp27a1−/−Cyp46a1−/− mice did not show gender differences in the levels of serum triglycerides and total cholesterol (Figures 6A,B). Serum triglycerides were decreased by 35% after a 4-week treatment, whereas total serum cholesterol remained unchanged after 2 and 4 weeks of treatment. The most pronounced effect of DMHCA was on the levels of retinal sterols. The levels of total lanosterol, zymosterol, and desmosterol were increased by ≥70% in both the 2- and 4-week treatment groups as compared to the control groups (Figures 6C–E), whereas the lathosterol levels were decreased by up to 46% (Figure 6F). As compared to changes in retinal levels of cholesterol precursors, changes in the levels of total cholesterol in DMHCA-treated mice were smaller: only 15 and 17% in the 2- and 4-week treatment groups, respectively (Figure 6G). Remarkably, these decreases in total cholesterol mainly occurred due to a reduction in the levels of unesterified cholesterol with the levels of cholesterol esters being essentially unchanged.
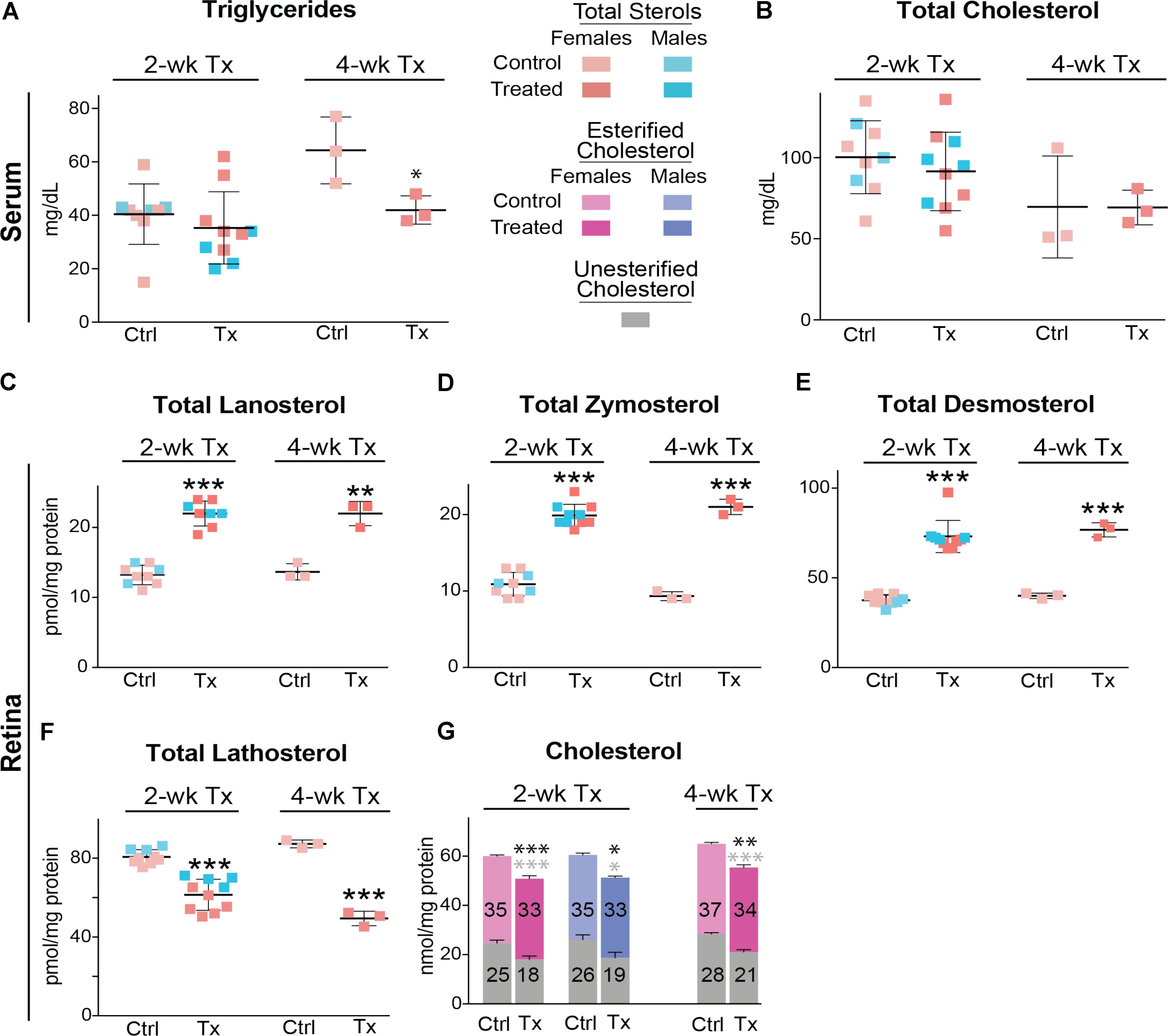
FIGURE 6. Serum (A,B) and retinal (C–G) lipids of Cyp27a1−/−Cyp46a1−/− mice treated with DMHCA. The treatment paradigm and number of animals per group were the same as in Figure 5. The results are the mean ± SD of the measurements in samples from individual animals of both genders. Numbers inside the bars in G represent the mean values for esterified and unesterified cholesterol; black and gray asterisks are significant changes relative to the control group for total and unesterified cholesterol, respectively, as assessed by a two-tailed, unpaired Student’s t-test. ∗P ≤ 0.05, ∗∗P ≤ 0.01, and ∗∗∗P ≤ 0.001. When female and male mice in a 2-week treatment were analyzed separately, statistical analyses did not reveal differences as compared to the group comprised of both genders. Ctrl, control; Tx, treatment.
Since the decrease in retinal cholesterol was similar in 2- and 4-week treated mice, the DMHCA effect on retinal gene expression was evaluated in 2-week treated mice. In the liver, DMHCA administration did not generally affect the expression of the studied genes (Figures 7A,B), except a 2-fold increase in the expression of Scd1 in female mice and a ≥2-fold decrease in the expression of Fasn and Inos in male mice. In the retina, however, 5 out of the 9 studied genes in female mice (Srepb1c, Fasn, Scd1, Abca1, and Vegf) and 3 out of the 7 genes in male mice (Fasn, Apoe, and Vegf) showed an altered expression after DMHCA treatment (Figures 7C,D). Yet, these changes were moderate (up to 40%) and gender-specific: there was a pattern of increased gene expression in female mice, while in male mice, gene expression was decreased.
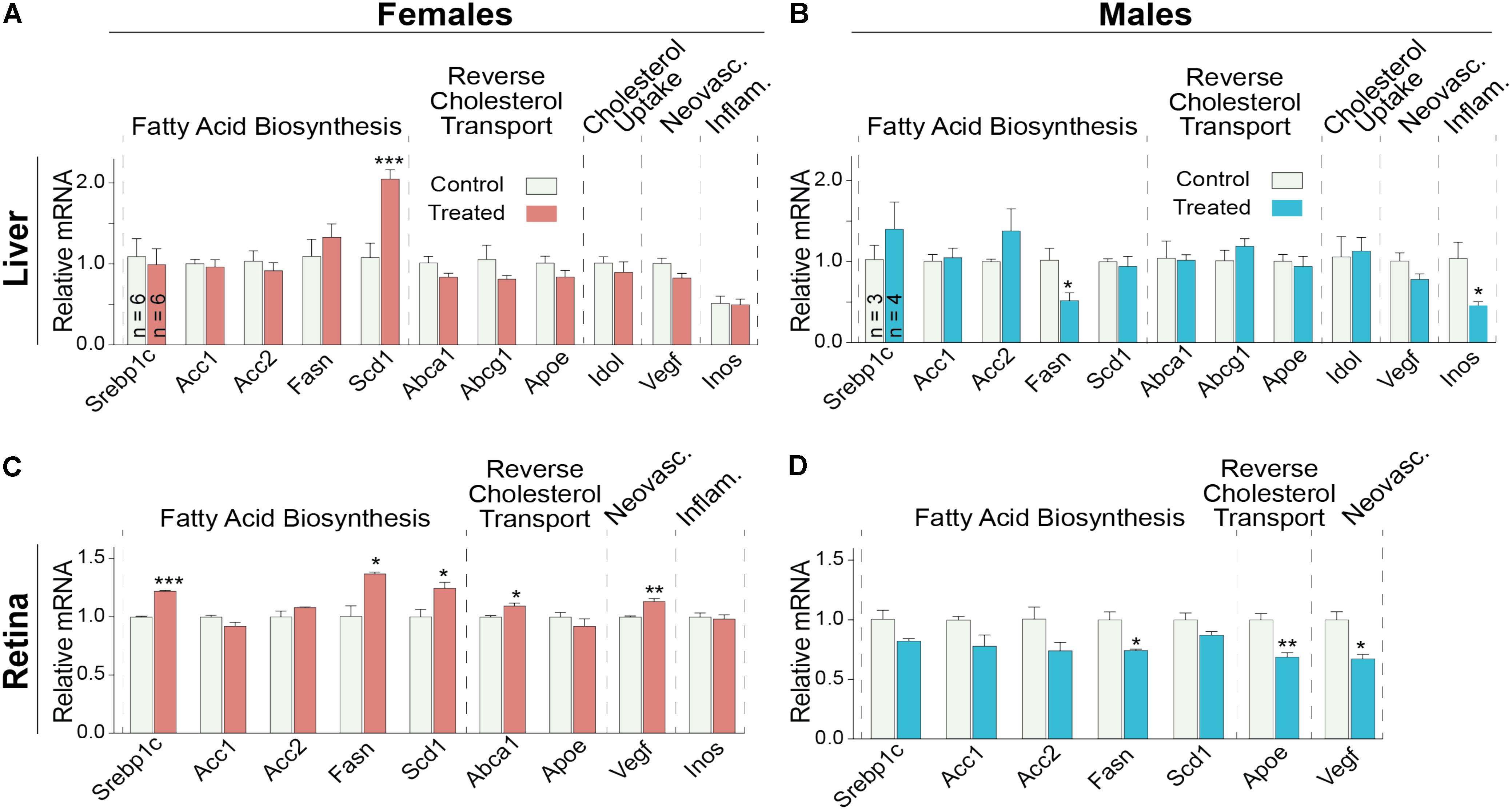
FIGURE 7. Hepatic (A,B) and retinal (C,D) gene expression in Cyp27a1−/−Cyp46a1−/− mice treated with DMHCA for 2 weeks. The treatment paradigm was the same as in Figure 4. (A,B) The results are the mean ± SEM of triplicate measurements in the individual samples of the liver from six female mice (A) and three to four male mice (B) or a pooled sample of individual retinas from six female mice (C) and three male mice (D). Asterisks are significant changes relative to the control group as assessed by a two-tailed, unpaired Student’s t-test. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001.
DMHCA Effect on CYP27A1 and CYP46A1
CYP27A1 and CYP46A1 are the two major cholesterol hydroxylases in the retina that utilize cholesterol as a substrate to produce different metabolites, 27-hydroxycholesterol and 24-hydroxycholesterol (Figure 1), respectively (Saadane et al., 2014). The levels of retinal cholesterol were different in C57BL/6J and Cyp27a1−/−Cyp46a1−/− mice, and mice of the two genotypes received different DMHCA doses in different formulations. Nevertheless, DMHCA reduced retinal cholesterol in both genotypes by the same extent, 14 and 15–17%. Such an effect could be due to DMHCA inhibition of CYP27A1 and/or CYP46A1 in C57BL/6J mice, and thus a lack of cholesterol removal by metabolism in the retina of both genotypes. Hence, we used purified recombinant CYP27A1 and CYP46A1 and tested DMHCA for >75% enzyme inhibition in the in vitro enzyme assay, an indication of a strong P450 inhibitor (Lam et al., 2018). Neither CYP27A1 nor CYP46A1 were inhibited by DMHCA in vitro (Table 3), thus suggesting that retinal metabolism of cholesterol does not contribute to retinal cholesterol reduction as a result of DMHCA treatment.

TABLE 3. Effect of DMHCA on in vitro cholesterol hydroxylation by CYP27A1 and CYP46A1.
Discussion
To the best of our knowledge, we have pioneered an in vivo study of retinal effects of DMHCA, an experimental steroidal LXR agonist. First, we conducted a pilot dose dependence study of DMHCA on a small group of mice from a common laboratory strain C57BL/6J (Figures 2–4). Then, we evaluated Cyp27a1−/−Cyp46a1−/− mice (Figures 5–7), which are on the C57BL/6J;129S6/SvEv background with total retinal cholesterol ∼1.5 times higher than that of wild-type mice on the C57BL/6J background. Retinal cholesterol excess in these animals is stored in the form of cholesterol esters, and their retinas have a number of pathologies (Saadane et al., 2014, 2016). Significant cholesterol esterification is unusual for the retina because normally, cholesterol esters are present in only very small amounts (up to 15%) both in mouse or human retina (Bretillon et al., 2008; Mast et al., 2011). Nevertheless, cholesterol esters are present in drusen, a hallmark of age-related macular degeneration, whereas subretinal drusenoid deposits, another hallmark of this disease, are mainly composed of unesterified cholesterol (Curcio et al., 2001, 2005; Oak et al., 2014). Hence, to be clinically relevant, evaluation of experimental pharmaceuticals for age-related macular degeneration should include the quantifications of different forms of retinal cholesterol: total, unesterified, and esterified cholesterol.
We found that at oral daily doses of 37 mg/kg BW given to male mice and 51 mg/kg BW given to female mice (Table 2), DMHCA decreased the levels of total retinal cholesterol in the C57BL/6J strain without affecting the levels of serum cholesterol (Figures 3B,E). Similarly, DMHCA (80 mg/kg BW/day) reduced retinal but not serum cholesterol in Cyp27a1−/−Cyp46a1−/− mice (Figures 6B,G). Collectively, these data suggest that DMHCA crosses the blood–retina barrier and reaches neural retina where it decreases cholesterol. Previously, DMHCA was shown to be readily absorbed in the intestine and distributed to different organs (Pfeifer et al., 2011). In C57BL/6 mice, the highest DMHCA concentration at an 80 mg/kg BW/day dose was found in the small intestine with lower drug concentrations in the kidney, heart, lung, and the liver (Pfeifer et al., 2011). In the brain, DMHCA was detected only in very small amounts and did not show the effects observed in the other organs (Pfeifer et al., 2011). Thus, unlike the blood–retina barrier, the blood–brain barrier seems to limit DMHCA availability to the brain. Conversely, in a mouse model of a traumatic brain injury, DMHCA (50 mg/kg BW) was reported to be well distributed in the brain and increase the expression of the LXR target Abca1, while decreasing the expression of pro-inflammatory Il-1β and Tnfα at the site of injury (Yu et al., 2016).
The retina is a neural tissue composed of both neurons and astrocytes. In mice, the main source of retinal cholesterol is local biosynthesis, which accounts for ∼72% of total retinal cholesterol (Lin et al., 2016). Retinal cholesterol biosynthesis proceeds via both, the Bloch and Kandutsch-Russell pathways because the metabolites from both pathways were detected in the retina in the same concentration range (Omarova et al., 2012). Desmosterol is a marker of cholesterol biosynthesis in astrocytes (Pfrieger and Ungerer, 2011) and is the last sterol intermediate in the Bloch pathway. Desmosterol is reduced by DHCR24 to yield cholesterol (Figure 1). The levels of this cholesterol precursor will increase, if DHCR24 is inhibited, because desmosterol will not be converted to cholesterol. Similarly, there may be an increase in the levels of lanosterol and zymosterol, the upstream desmosterol precursor and also the substrates for DHCR24. Lathosterol is a marker of cholesterol biosynthesis in neurons (Pfrieger and Ungerer, 2011) and is the penultimate sterol intermediate in the Kandutsch-Russell pathway (Figure 1). Lathosterol is a product of DHCR24 reduction of dehydrolathosterol, an intermediate in the Bloch pathway (Zerenturk et al., 2013; Mitsche et al., 2015). The levels of lathosterol will decrease, if DHCR24 is inhibited, because lathosterol will not be formed. Accordingly, an increase in retinal lanosterol, zymosterol, and desmosterol levels with a simultaneous decrease in retinal lathosterol levels (Figures 6C–F) suggest that treatment with an 80 mg/kg BW/day DMHCA dose partially inhibited DHCR24 and both pathways of retinal cholesterol biosynthesis. This interpretation is supported by previous studies on HepG2 cells treated with increasing DMHCA concentrations and supplemented with desmosterol as a substrate for DHCR24 (Pfeifer et al., 2011). Increasing DMHCA concentrations led to a decrease in cholesterol production by these HepG2 cells and an increase in desmosterol levels, thus providing evidence that DMHCA indeed inhibits DHCR24.
Retinal expression of genes involved in reverse cholesterol transport was either not increased or increased only moderately in DMHCA-treated mice (Figures 7C,D). Therefore, our sterol and mRNA quantifications collectively suggest that under the experimental conditions used, namely the DMHCA dose, formulation, route of delivery, and treatment time, this LXR agonist probably reduces retinal cholesterol (Figure 6G) via partial inhibition of DHCR24 rather than its transcriptional LXR activity controlling the expression of genes involved in reverse cholesterol transport. Moderate effects of DMHCA on retinal expression of LXR target genes (Figures 7C,D) are in agreement with experiments that directly compared the potency of DMHCA to T0901317 or GW3965, which are first generation non-steroidal LXR agonists that induce liver lipogenesis. When the same doses of DMHCA, T0901317, or GW3965 were used (50 or 80 mg/kg BW/day), T0901317 and GW3965 were more potent LXR agonists than DMHCA in the liver of C57BL/6 mice or in the C57BL/6 brain with traumatic injury (Quinet et al., 2004; Kratzer et al., 2009; Yu et al., 2016). We hypothesize that a higher, >100 mg/kg BW/day, DMHCA dose could possibly upregulate the reverse cholesterol transport genes in the retina upon oral DMHCA delivery.
Partial retinal DHCR24 inhibition by DMHCA revealed in the present work should not be viewed as a negative DMHCA effect on the retina and prevent further evaluations of DMHCA as a pharmaceutical for the treatment of retinal diseases. First, moderate accumulation of desmosterol does not seem to be toxic as indicated by apparently healthy heterozygous carriers of the DHCR24 inactivating mutations. These subjects have only moderately elevated levels of plasma desmosterol, and the levels of their serum cholesterol are normal (FitzPatrick et al., 1998; Andersson et al., 2002; Zolotushko et al., 2011). Second, DHCR24 inhibition may lead to partial inhibition of cellular cholesterol biosynthesis and thus decrease intracellular cholesterol independent of the upregulation of reverse cholesterol transport genes. Third, desmosterol is an LXR agonist, and accumulation of desmosterol in macrophage foam cells was shown to activate LXRs and suppresses inflammatory gene expression, while upregulating the expression of reverse cholesterol transport genes (Spann et al., 2012). A similar mechanism could be operative in some retinal cells, e.g., microglia/macrophages. However, because of a small number of these cells in the retina, which is composed of multiple cell types (Fliesler and Bretillon, 2010), this mechanism was not detected in the present work when total mRNA was isolated from the whole retina. Finally, beneficial effects of GW3965, a first generation LXR agonist, on early stages diabetic retinopathy were already established in a mouse model (Hazra et al., 2012). Therefore, evaluations of other LXR agonists, which do not induce hepatic lipogenesis and have unique pharmacologic profile, are required. DMHCA should be tested for the effects on the retina either at higher doses or using eye-specific routes of delivery (e.g., ophthalmic eye drops or intravitreal injections). A caveat to studies leading to higher retinal DMHCA concentrations is that they may increase retinal Dhcr24 expression via SREBP2 (Zerenturk et al., 2012), a sensor for intracellular cholesterol levels and a transcriptional activator of cellular cholesterol biosynthesis (Horton et al., 2002; Brown and Goldstein, 2009). Indeed, a decrease in retinal cholesterol in DMHCA-treated mice due to DHCR24 inhibition by DMHCA can potentially upregulate SREBP2 processing and thereby increase Dhcr24 expression. This increase may ultimately overcome DMHCA inhibition of DHCR24 and diminish retinal cholesterol lowering.
An interesting finding of the present work is an apparent lack of DMHCA effect on the levels of retinal cholesterol esters (Figure 6G), which may be generated when cholesterol is in excess in a cell and are rapidly hydrolyzed (Figure 1) when there is a decrease in the levels of cellular cholesterol (Chang et al., 2006). ACAT1, a cholesterol esterifying enzyme present in the retina (Saadane et al., 2016), was shown to be activated by cholesterol and oxysterols on the enzyme allosteric site (Zhang et al., 2003; Liu et al., 2005). Thus, it is plausible that DMHCA, a steroidal compound, could activate ACAT1 as well. Furthermore, the retina was also shown to express lysosomal acid lipase (Elner, 2002), one of the several enzymes with cholesterol ester hydrolyzing activities (Ghosh, 2012). Accordingly, unchanged cholesterol ester levels in DMHCA-treated mice could either be due to ACAT1 activation by DMHCA or DMHCA inhibition of a cholesterol ester hydrolase. The identification of the mechanism beyond the unchanged cholesterol ester levels in the retina of DMHCA-treated mice was beyond the scope of the present work. However, we may investigate this finding in the future because it may point us to an important enzyme that controls the levels of cholesterol esters in drusen, a hallmark of AMD.
In summary, the present work identified DMHCA dosages, formulations, and routes of delivery which reduce total retinal cholesterol without increasing serum triglyceride and cholesterol levels. Notably, a reduction in total retinal cholesterol was due to a reduction in unesterified cholesterol rather than esterified cholesterol. We also show that in Cyp27a1−/−Cyp46a1−/− mice, a genotype with a significantly increased retinal cholesterol, DMHCA administration altered retinal levels of cholesterol precursors lanosterol, zymosterol, desmosterol, and lathosterol. This result suggests a partial inhibition of retinal DHCR24. Retinal expression of the LXR target genes was affected only moderately in DMHCA-treated mice, thus providing evidence that at the doses tested and genotypes studied, DMHCA acts in the retina mainly as an enzyme inhibitor of cholesterol biosynthesis rather than a LXR agonist.
Author Contributions
NE-D, NM, and IP participated in research design. NE-D, AA, NM, AS, and ML conducted the experiments. NE-D, AA, NM, AS, ML, and IP performed the data analysis. IP wrote the manuscript.
Funding
This work was supported in part by National Institutes of Health Grant EY018383 and EY011373 to (IP) and EY025383. IP is a Carl F. Asseff Professor of Ophthalmology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Tomilade Adepoju for initial experiments on C57BL/6J mice and the Visual Sciences Research Center Core Facilities (supported by National Institutes of Health Grant EY011373) for assistance with mouse breeding (Heather Butler and Kathryn Franke) and animal genotyping (John Denker).
Abbreviations
ACAT1 and ACAT2, acyl-coenzyme A:cholesterol acyltransferases 1 and 2, respectively; BW, body weight; CYP, cytochrome P450; DHCR24, Δ24-dehydrocholesterol reductase; DMHCA, N,N-dimethyl-3β-hydroxycholenamide; HPCD, 2-hydroxypropyl-β-cyclodextrin; LXR, liver X receptor; qRT-PCR, quantitative real time PCR.
References
Alberti, S., Steffensen, K. R., and Gustafsson, J. A. (2000). Structural characterisation of the mouse nuclear oxysterol receptor genes LXRalpha and LXRbeta. Gene 243, 93–103.
Andersson, H. C., Kratz, L., and Kelley, R. (2002). Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am. J. Med. Genet. 113, 315–319.
Bachmanov, A. A., Reed, D. R., Beauchamp, G. K., and Tordoff, M. G. (2002). Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 32, 435–443.
Bretillon, L., Thuret, G., Gregoire, S., Acar, N., Joffre, C., Bron, A. M., et al. (2008). Lipid and fatty acid profile of the retina, retinal pigment epithelium/choroid, and the lacrimal gland, and associations with adipose tissue fatty acids in human subjects. Exp. Eye Res. 87, 521–528. doi: 10.1016/j.exer.2008.08.010
Brown, M. S., and Goldstein, J. L. (2009). Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J. Lipid Res. 50(Suppl.), S15–S27. doi: 10.1194/jlr.R800054-JLR200
Caldas, Y. A., Giral, H., Cortazar, M. A., Sutherland, E., Okamura, K., Blaine, J., et al. (2011). Liver X receptor-activating ligands modulate renal and intestinal sodium-phosphate transporters. Kidney Int. 80, 535–544. doi: 10.1038/ki.2011.159
Calkin, A. C., and Tontonoz, P. (2012). Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 13, 213–224. doi: 10.1038/nrm3312
Chang, T. Y., Chang, C. C., Ohgami, N., and Yamauchi, Y. (2006). Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22, 129–157.
Chen, W., Chen, G., Head, D. L., Mangelsdorf, D. J., and Russell, D. W. (2007). Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 5, 73–79.
Curcio, C. A., Johnson, M., Rudolf, M., and Huang, J. D. (2011). The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 95, 1638–1645. doi: 10.1136/bjophthalmol-2011-300344
Curcio, C. A., Millican, C. L., Bailey, T., and Kruth, H. S. (2001). Accumulation of cholesterol with age in human Bruch’s membrane. Invest. Ophthalmol. Vis. Sci. 42, 265–274.
Curcio, C. A., Presley, J. B., Malek, G., Medeiros, N. E., Avery, D. V., and Kruth, H. S. (2005). Esterified and unesterified cholesterol in drusen and basal deposits of eyes with age-related maculopathy. Exp. Eye Res. 81, 731–741.
Elner, V. M. (2002). Retinal pigment epithelial acid lipase activity and lipoprotein receptors: effects of dietary omega-3 fatty acids. Trans. Am. Ophthalmol. Soc. 100, 301–338.
FitzPatrick, D. R., Keeling, J. W., Evans, M. J., Kan, A. E., Bell, J. E., Porteous, M. E., et al. (1998). Clinical phenotype of desmosterolosis. Am. J. Med. Genet. 75, 145–152.
Fliesler, S. J., and Bretillon, L. (2010). The ins and outs of cholesterol in the vertebrate retina. J. Lipid Res. 51, 3399–3413. doi: 10.1194/jlr.R010538
Fujihara, M., Cano, M., and Handa, J. T. (2014). Mice that produce ApoB100 lipoproteins in the RPE do not develop drusen yet are still a valuable experimental system. Invest. Ophthalmol. Vis. Sci. 55, 7285–7295. doi: 10.1167/iovs.14-15195
Ghosh, S. (2012). Early steps in reverse cholesterol transport: cholesteryl ester hydrolase and other hydrolases. Curr. Opin. Endocrinol. Diabetes Obes. 19, 136–141. doi: 10.1097/MED.0b013e3283507836
Glass, C. K., and Ogawa, S. (2006). Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 6, 44–55.
Hanna, I. H., Teiber, J. F., Kokones, K. L., and Hollenberg, P. F. (1998). Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332.
Hazra, S., Rasheed, A., Bhatwadekar, A., Wang, X., Shaw, L. C., Patel, M., et al. (2012). Liver X receptor modulates diabetic retinopathy outcome in a mouse model of streptozotocin-induced diabetes. Diabetes Metab. Res. Rev. 61, 3270–3279. doi: 10.2337/db11-1596
Hong, C., and Tontonoz, P. (2014). Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discov. 13, 433–444. doi: 10.1038/nrd4280
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131.
Janowski, B. A., Grogan, M. J., Jones, S. A., Wisely, G. B., Kliewer, S. A., Corey, E. J., et al. (1999). Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl. Acad. Sci. U.S.A. 96, 266–271.
Janowski, B. A., Willy, P. J., Devi, T. R., Falck, J. R., and Mangelsdorf, D. J. (1996). An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383, 728–731.
Kalaany, N. Y., and Mangelsdorf, D. J. (2006). LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu. Rev. Physiol. 68, 159–191.
Kratzer, A., Buchebner, M., Pfeifer, T., Becker, T. M., Uray, G., Miyazaki, M., et al. (2009). Synthetic LXR agonist attenuates plaque formation in apoE-/- mice without inducing liver steatosis and hypertriglyceridemia. J. Lipid Res. 50, 312–326. doi: 10.1194/jlr.M800376-JLR200
Lam, M., Mast, N., and Pikuleva, I. A. (2018). Drugs and scaffold that inhibit cytochrome P450 27A1 in vitro and in vivo. Mol. Pharmacol. 93, 101–108. doi: 10.1124/mol.117.110742
Lin, J. B., Mast, N., Bederman, I. R., Li, Y., Brunengraber, H., Bjorkhem, I., et al. (2016). Cholesterol in mouse retina originates primarily from in situ de novo biosynthesis. J. Lipid Res. 57, 258–264. doi: 10.1194/jlr.M064469
Liu, J., Chang, C. C., Westover, E. J., Covey, D. F., and Chang, T. Y. (2005). Investigating the allosterism of acyl-CoA:cholesterol acyltransferase (ACAT) by using various sterols: in vitro and intact cell studies. Biochem. J. 391, 389–397.
Mast, N., Lin, J. B., and Pikuleva, I. A. (2015). Marketed drugs can inhibit cytochrome P450 27A1, a potential new target for breast cancer adjuvant therapy. Mol. Pharmacol. 88, 428–436. doi: 10.1124/mol.115.099598
Mast, N., Linger, M., Clark, M., Wiseman, J., Stout, C. D., and Pikuleva, I. A. (2012). In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol. Pharmacol. 82, 824–834. doi: 10.1124/mol.112.080424
Mast, N., Murtazina, D., Liu, H., Graham, S. E., Bjorkhem, I., Halpert, J. R., et al. (2006). Distinct binding of cholesterol and 5-cholestane-3 alpha,7 alpha,12 alpha-triol to cytochrome P450 27A1: evidence from modeling and site-directed mutagenesis studies. Biochemistry 45, 4396–4404.
Mast, N., Reem, R., Bederman, I., Huang, S., DiPatre, P. L., Bjorkhem, I., et al. (2011). Cholestenoic acid is an important elimination product of cholesterol in the retina: comparison of retinal cholesterol metabolism with that in the brain. Invest. Ophthalmol. Vis. Sci. 52, 594–603. doi: 10.1167/iovs.10-6021
Mast, N., Shafaati, M., Zaman, W., Zheng, W., Prusak, D., Wood, T., et al. (2010). Marked variability in hepatic expression of cytochromes CYP7A1 and CYP27A1 as compared to cerebral CYP46A1. Lessons from a dietary study with omega 3 fatty acids in hamsters. Biochim. Biophys. Acta 1801, 674–681. doi: 10.1016/j.bbalip.2010.03.005
Mast, N., White, M. A., Bjorkhem, I., Johnson, E. F., Stout, C. D., and Pikuleva, I. A. (2008). Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U.S.A. 105, 9546–9551.
Miller, J. W. (2013). Age-related macular degeneration revisited–piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am. J. Ophthalmol. 155, 1.e13–35.e13. doi: 10.1016/j.ajo.2012.10.018
Mitsche, M. A., McDonald, J. G., Hobbs, H. H., and Cohen, J. C. (2015). Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. eLife 4:e07999. doi: 10.7554/eLife.07999
Muller, C., Hemmers, S., Bartl, N., Plodek, A., Korner, A., Mirakaj, V., et al. (2017). New chemotype of selective and potent inhibitors of human delta 24-dehydrocholesterol reductase. Eur. J. Med. Chem. 140, 305–320. doi: 10.1016/j.ejmech.2017.08.011
Oak, A. S. W., Messinger, J. D., and Curcio, C. A. (2014). Subretinal drusenoid deposits: further characterization by lipid histochemistry. Retina 34, 825–826.
Omarova, S., Charvet, C. D., Reem, R. E., Mast, N., Zheng, W., Huang, S., et al. (2012). Abnormal vascularization in mouse retina with dysregulated retinal cholesterol homeostasis. J. Clin. Invest. 122, 3012–3023. doi: 10.1172/JCI63816
Panda-Jonas, S., Jonas, J. B., Jakobczyk, M., and Schneider, U. (1994). Retinal photoreceptor count, retinal surface area, and optic disc size in normal human eyes. Ophthalmology 101, 519–523.
Patel, M., Wang, X. X., Magomedova, L., John, R., Rasheed, A., Santamaria, H., et al. (2014). Liver X receptors preserve renal glomerular integrity under normoglycaemia and in diabetes in mice. Diabetologia 57, 435–446. doi: 10.1007/s00125-013-3095-6
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45.
Pfeifer, T., Buchebner, M., Chandak, P. G., Patankar, J., Kratzer, A., Obrowsky, S., et al. (2011). Synthetic LXR agonist suppresses endogenous cholesterol biosynthesis and efficiently lowers plasma cholesterol. Curr. Pharm. Biotechnol. 12, 285–292.
Pfrieger, F. W., and Ungerer, N. (2011). Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50, 357–371.
Pikuleva, I. A., and Curcio, C. A. (2014). Cholesterol in the retina: the best is yet to come. Prog. Retin. Eye Res. 41, 64–89. doi: 10.1016/j.preteyeres.2014.03.002
Quinet, E. M., Savio, D. A., Halpern, A. R., Chen, L., Miller, C. P., and Nambi, P. (2004). Gene-selective modulation by a synthetic oxysterol ligand of the liver X receptor. J. Lipid Res. 45, 1929–1942.
Remtulla, S., and Hallett, P. E. (1985). A schematic eye for the mouse, and comparisons with the rat. Vision Res. 25, 21–31.
Saadane, A., Mast, N., Charvet, C., Omarova, S., Zheng, W., Huang, S. S., et al. (2014). Retinal and non-ocular abnormalities in Cyp27a1(-/-) Cyp64a1(-/-) mice with dysfunctional metabolism of cholesterol. Am. J. Pathol. 184, 2403–2419. doi: 10.1016/j.ajpath.2014.05.024
Saadane, A., Mast, N., Dao, T., Ahmad, B., and Pikuleva, I. A. (2016). Retinal Hypercholesterolemia triggers cholesterol accumulation and esterification in photoreceptor cells. J. Biol. Chem. 291, 20427–20439. doi: 10.1074/jbc.M116.744656
Sagara, Y., Hara, T., Ariyasu, Y., Ando, F., Tokunaga, N., and Horiuchi, T. (1992). Direct expression in Escherichia coli and characterization of bovine adrenodoxins with modified amino-terminal regions. FEBS Lett. 300, 208–212.
Sagara, Y., Wada, A., Takata, Y., Waterman, M. R., Sekimizu, K., and Horiuchi, T. (1993). Direct expression of adrenodoxin reductase in Escherichia coli and the functional characterization. Biol. Pharm. Bull. 16, 627–630.
Spann, N. J., Garmire, L. X., McDonald, J. G., Myers, D. S., Milne, S. B., Shibata, N., et al. (2012). Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 151, 138–152. doi: 10.1016/j.cell.2012.06.054
Spencer, T. A., Li, D., Russel, J. S., Collins, J. L., Bledsoe, R. K., Consler, T. G., et al. (2001). Pharmacophore analysis of the nuclear oxysterol receptor LXRalpha. J. Med. Chem. 44, 886–897.
Tserentsoodol, N., Gordiyenko, N. V., Pascual, I., Lee, J. W., Fliesler, S. J., and Rodriguez, I. R. (2006). Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol. Vis. 12, 1319–1333.
Yu, S., Li, S., Henke, A., Muse, E. D., Cheng, B., Gustav, W., et al. (2016). Dissociated sterol-based liver X receptor agonists as therapeutics for chronic inflammatory diseases. FASEB J. 30, 2570–2579. doi: 10.1096/fj.201600244R
Zelcer, N., and Tontonoz, P. (2006). Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest. 116, 607–614.
Zerenturk, E. J., Sharpe, L. J., and Brown, A. J. (2012). Sterols regulate 3beta-hydroxysterol Delta24-reductase (DHCR24) via dual sterol regulatory elements: cooperative induction of key enzymes in lipid synthesis by sterol regulatory element binding proteins. Biochim. Biophys. Acta 1821, 1350–1360. doi: 10.1016/j.bbalip.2012.07.006
Zerenturk, E. J., Sharpe, L. J., Ikonen, E., and Brown, A. J. (2013). Desmosterol and DHCR24: unexpected new directions for a terminal step in cholesterol synthesis. Prog. Lipid Res. 52, 666–680. doi: 10.1016/j.plipres.2013.09.002
Zhang, Y., Yu, C., Liu, J., Spencer, T. A., Chang, C. C., and Chang, T. Y. (2003). Cholesterol is superior to 7-ketocholesterol or 7 alpha-hydroxycholesterol as an allosteric activator for acyl-coenzyme A:cholesterol acyltransferase 1. J. Biol. Chem. 278, 11642–11647.
Zheng, W., Mast, N., Saadane, A., and Pikuleva, I. A. (2015). Pathways of cholesterol homeostasis in mouse retina responsive to dietary and pharmacologic treatments. J. Lipid Res. 56, 81–97. doi: 10.1194/jlr.M053439
Keywords: N, N-dimethyl-3β-hydroxycholenamide, retina, cholesterol, Δ24-dehydrocholesterol, CYP27A1, CYP46A1, liver X receptor
Citation: El-Darzi N, Astafev A, Mast N, Saadane A, Lam M and Pikuleva IA (2018) N,N-Dimethyl-3β-hydroxycholenamide Reduces Retinal Cholesterol via Partial Inhibition of Retinal Cholesterol Biosynthesis Rather Than its Liver X Receptor Transcriptional Activity. Front. Pharmacol. 9:827. doi: 10.3389/fphar.2018.00827
Received: 30 March 2018; Accepted: 09 July 2018;
Published: 25 July 2018.
Edited by:
Vsevolod V. Gurevich, Vanderbilt University, United StatesReviewed by:
Marc Poirot, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceSpyridon Theofilopoulos, Swansea University, United Kingdom
Copyright © 2018 El-Darzi, Astafev, Mast, Saadane, Lam and Pikuleva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irina A. Pikuleva, aWFwOEBjYXNlLmVkdQ==