 Jaya Kumar
Jaya Kumar Amro Solaiman
Amro Solaiman Pasuk Mahakkanukrauh
Pasuk Mahakkanukrauh Rashidi Mohamed
Rashidi Mohamed Srijit Das
Srijit Das- 1Department of Physiology, Faculty of Medicine, Universiti Kebangsaan Malaysia Medical Centre, Kuala Lumpur, Malaysia
- 2Department of Anatomy, Universiti Kebangsaan Malaysia Medical Centre, Kuala Lumpur, Malaysia
- 3Department of Anatomy, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
- 4Excellence Centre in Forensic Osteology Research Center, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
- 5Department of Familty Medicine, Universiti Kebangsaan Malaysia Medical Centre, Kuala Lumpur, Malaysia
In the last several decades, sleep-related epilepsy has drawn considerable attention among epileptologists and neuroscientists in the interest of new paradigms of the disease etiology, pathogenesis and management. Sleep-related epilepsy is nocturnal seizures that manifest solely during the sleep state. Sleep comprises two distinct stages i.e., non-rapid eye movement (NREM) and rapid eye movement (REM) that alternate every 90 min with NREM preceding REM. Current findings indicate that the sleep-related epilepsy manifests predominantly during the synchronized stages of sleep; NREM over REM stage. Sleep related hypermotor epilepsy (SHE), benign partial epilepsy with centrotemporal spikes or benign rolandic epilepsy (BECTS), and Panayiotopoulos Syndrome (PS) are three of the most frequently implicated epilepsies occurring during the sleep state. Although some familial types are described, others are seemingly sporadic occurrences. In the present review, we aim to discuss the predominance of sleep-related epilepsy during NREM, established familial links to the pathogenesis of SHE, BECTS and PS, and highlight the present available pharmacotherapy options.
Introduction
Epilepsy is characterized by frequent and unpredictable disruptions of brain functions resulting in “epileptic seizures.” Epilepsy has a great impact on the quality of life through increased incidence of injury and death, unemployment rates, lower monthly incomes, higher household costs and high absenteeism at work and schools (Jennum et al., 2017; Trinka et al., 2018; Wibecan et al., 2018). An epileptic seizure is considered as a transient episode of signs or symptoms, including transitory confusion, staring speech, irrepressible jerking movements, loss of consciousness, psychic symptoms such as fear and anxiety, due to the abnormal synchronous neuronal activity of the brain. The International League Against Epilepsy (ILAE) published a recent clinical definition of epilepsy in which a patient with any of the following conditions is considered to be an epileptic i.e., (i) two or more unprovoked seizures within more than 24 h apart; (ii) one unprovoked seizure and a probability of further seizures similar to the general recurrence risk, occurring over the next 10 years; (iii) definite diagnosis of an epilepsy syndrome (Fisher et al., 2014). Genesis of epilepsy is attributed to various predispositions that include neurological, perceptive, psychological, and social factors, which could either stimulate or worsen the syndrome. In early 2017, the point prevalence of active epilepsy was found to be 6.38/1,000 individuals, while the lifetime prevalence was 7.60/1,000 persons. Meanwhile, the annual cumulative incidence of epilepsy was 67.77/100,000 persons and the incidence rate was 61.44/100,000 person-years. The active annual prevalence, prevalence during lifetime and the incidence of epilepsy were found to be higher in the developing countries (Fiest et al., 2017).
A systematic review revealed that epilepsies of unknown etiology had the highest prevalence compared to the epilepsies of known origin (Fiest et al., 2017). These were due to known underlying factors that cause seizures such as brain damage (Sizemore et al., 2018), metabolic diseases (Tumiene et al., 2018), infections (Bartolini et al., 2018), hemorrhagic stroke (Zhao et al., 2018), and gene mutations (Leonardi et al., 2018). These precipitating factors tilt the balance between excitatory and inhibitory neurotransmissions which has been established in different types of epilepsy. Physical and psychological comorbidities are usually accompanied with epilepsy, such as depression (Jamal-Omidi et al., 2018), sleep disorders (Castro et al., 2018), and body injuries (Mahler et al., 2018). Advanced cases may suffer from memory loss (Reyes et al., 2018), behavioral disorders (Jalihal et al., 2018), and disturbance of autonomic functions (Fialho et al., 2018). The rate of sudden death in epileptic patients was reported to be three times higher than non-epileptic individuals (Kothare and Trevathan, 2018; Pati et al., 2018).
Sleep deprivation is very common among the epileptic patients and lack of sleep could worsen the seizure expressions (Neto et al., 2016). In animal models, sleep deprivation was shown to heighten the propensity to seizures (McDermott et al., 2003). Sleep deprivation has been correlated with decline in various aspects of brain functional connectivity (Nilsonne et al., 2017). Generally, sleep deprivation is secondary to other factors such as illness, emotional or psychological stress, and alcohol use. Hence, lack of sleep alone may not be sufficient to cause seizures (Razavi and Fisher, 2017). A large body of literature on the effects of epilepsy on sleep and/or sleep-deprivation on the epileptic state has been collated (St Louis, 2011; Unterberger et al., 2015).
Sleep-related epilepsy represents nocturnal seizures that manifest solely during the sleep state (Tchopev et al., 2018). Approximately 12% epileptic patients are affected by sleep-related epilepsy with the majority suffering from focal epilepsy (Derry and Duncan, 2013; Losurdo et al., 2014). In a recent case report, focal epilepsies were anatomically linked to epileptogenic origins at the right frontal lobe, using white matter tractography MRI (Tchopev et al., 2018). In a separate study, ambulatory electroencephalogram (EEG) measurement in outpatient setting reported frontal lobe seizures to manifest more readily between 12 a.m. and 12 p.m., particularly around 6:30 a.m., whereas temporal lobe seizures expressed more frequently between 12 p.m. and 12 a.m., specifically around 8:50 p.m. (Pavlova et al., 2012). In addition to seizure onset, few seizures seem to propagate more readily during sleep, based on anatomical locus. Medial temporal lobe regions were shown more likely to manifest spike production or propagation during NREM sleep stage compared to other brain regions (Lambert et al., 2018). Sleep-related epilepsy is often misdiagnosed as sleep disorders (Tinuper and Bisulli, 2017), especially in cases where the seizures manifest exclusively during sleep. Over the past decade, the discovery of numerous pre-disposing genes and availability of advanced diagnostic tools have shed more light in understanding the nature of sleep-related epilepsy.
In the present review, we discuss sleep-related epilepsy with particular emphasis on three of the most frequently implicated epilepsies during the sleep state which include sleep related hypermotor epilepsy (SHE), benign partial epilepsy with centrotemporal spikes (BECTS), and Panayiotopoulos Syndrome (PS).
Expression of Seizures in NREM vs. REM Sleep Stages
In comparison with rapid eye movement (REM) sleep, the expression of focal seizure was 87 times more common in N1, 68 times more likely in N2, and 51 times more likely in N3. For generalized seizures, the seizure rate was 3.1 times higher in N1, 3.13 times higher in N2 and 6.59 times higher in N3 compared to the REM stage. Sleep-related epilepsies such as Benign Epilepsy of Childhood with rolandic spikes were common during the non-rapid eye movement (NREM) stages, especially during N3 (Ng and Pavlova, 2013), SHE was expressed more readily during N1/N2 (Nobili et al., 2014; Yeh and Schenck, 2014), and PS during N1 (Demirbilek and Dervent, 2004). Taken together, the existing literature suggests that sleep-related seizures are more likely to occur during the NREM stages of sleep.
NREM sleep is known as the state of neuronal synchronization, whereas REM as the most desynchronized sleep state. EEG findings suggest synchronization changes are more likely to take place during the transitions between the sleep states, rather than during the particular sleep states (Baghbani et al., 2018). In general, two types of synchronization exist; long-range (involves numerous brain regions) and local synchronization (involves adjacent neurons). During seizures, the long-range synchronization is impaired and local synchronization is enhanced as a result of the altered extracellular content of calcium (Ca2+) and potassium (K+) ions. In the pathogenesis of paroxysmal discharge, various predisposing factors (familial vs. sporadic) could alter the electrophysiological properties of numerous receptors, which may potentially decrease the extracellular level of Ca2+ and increase the extracellular content of K+, simultaneously (Amzica et al., 2002). Such changes inhibit synaptic transmission and propagation of action potential (Seigneur and Timofeev, 2011), which subsequently impair long-range synchronization and promote electrical coupling between cortical interneurons (Galarreta and Hestrin, 2001) and glial cells (Giaume and McCarthy, 1996). Long-range synchronization is also impaired during the slow-wave sleep (N3) (Ng and Pavlova, 2013). A preponderance of cortical slow oscillations takes place at this stage that results in a significant drop in the extracellular content of Ca2+, leading to high rates of synaptic failures (Steriade et al., 1993; Crochet et al., 2005). For instance, during the N3 stage, the mesenphalic reticular formation cholinergic neurons that allow transmission of impulses from the thalamus to the cortex are least active (less active during NREM stages) (Ng and Pavlova, 2013). As hypothesized by Timofeev et al. (2012) the significant drop in extracellular levels of Ca2+ during slow-wave sleep can promote the opening of hemichannels (Thimm et al., 2005) and electrical coupling between neighboring neurons (local synchrony) (Timofeev et al., 2012). Taken together, neuronal synchronization (local) along with pre-existing pro-epileptic conditions (such as channellopathies) seem to reinforce the predominance of seizure expressions during sleep state.
The seizure expressions during NREM stages are simplified in Table 1.
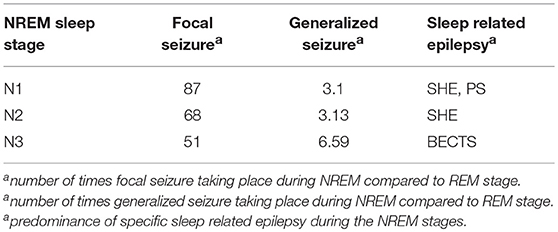
Table 1. Table showing the expression of seizures during NREM sleep.
Sleep Related Hypermotor Epilepsy
SHE, or previously known as the Nocturnal Frontal Lobe Epilepsy (NFLE) is a type of sleep-related epilepsy with frontal and extrafrontal regions as seizure onset zones that characterized by peculiar motor aspects of seizures (Tinuper et al., 2016; Vignatelli et al., 2017). The hallmark feature of this rare partial epilepsy is motor seizures that manifest almost exclusively during the NREM stage of sleep. The familial form of this epilepsy, also widely known as the autosomal dominant NFLE manifests between the age 8 and 12 years (Picard and Scheffer, 2012). SHE is the first of its kind to be associated with a causative agent, which are mutations of 3 subunit genes of nicotinic acetylcholine receptors (nAChRs) (Steinlein et al., 1995; Marini and Guerrini, 2007); reported in approximately 10% of the affected families (Heron et al., 2007).
The notion that SHE is a channelopathy is derived from early findings on mutations of nAChRs genes such as cholinergic receptor nicotinic alpha 2 subunit (CHRNA2), cholinergic receptor nicotinic alpha 4 subunit (CHRNA4) and cholinergic receptor nicotinic beta 2 subunit (CHRNB2) encoding for nAChRs containing subunits of α4β2 or heteromers with subunits of α2/β2/β4 (Figure 1) (Di Resta et al., 2010; Wallace and Bertrand, 2013; Becchetti et al., 2015). The anatomical projections of cholinergic neurons from pons and basal forebrain toward thalamus and cortex have been implicated in the regulation of sleep-wake cycle (Saper et al., 2010). Existing literature suggests increased acetylcholine release during wakefulness and REM sleep, whereas marked decrease during NREM sleep (Jones, 2008). Findings from mutant murine models of SHE (expressing B2-V287L) showed altered sleep pattern and development of spontaneous seizures during the slow wave sleep state (O'Neill et al., 2013). Molecular results show that mutated nAChRs could be hyperfunctional, and thus maintaining abnormal gamma-Aminobutyric acid-(GABA)ergic and glutamatergic neurotransmission, even with very minimal acetylcholine (Ach) available to bind to (Aracri et al., 2010). Hyperfunction of nAChRs also could be due to longer duration of Ach remaining in the synapse. In a recent study, a novel autosomal recessive phenotype of SHE was identified in a two-generation Australian family of Italian origins. Whole genome sequencing revealed mutations of proline rich membrane anchor 1 (PRIMA1) on chromosome 14 that encodes for PRIMA1 transmembrane protein, which anchors acetylcholinesterase at synapses for hydrolysis of acetylcholine. The authors went on to point out that perturbations in the cholinergic responses attributed to dysfunctional acetylcholinesterase could alter central and peripheral process of seizure expressions and likely to transform to SHE (Hildebrand et al., 2015). More recently, the occurrence of sleep-related epilepsy in SHE has been directly related to dysregulation of GABAergic neurotransmission. The reversal potential of GABAA-mediated inhibitory post-synaptic potential requires the movement of chloride ions (Cl−) in and out of the cells, which are mediated by co-transporters such as Na+/K+/Cl− co-transporter-1 (NKCC1) and K+/Cl− co-transporter-2 (KCC2) (Kaila et al., 2014). In a murine model of SHE (expressing B2-V287L), delayed surface expression of KCC2 was reported in layer V of prefrontal cortex (PFC), which is the most susceptible part of PFC to epileptiform activities. The delay was noticed during the first postnatal weeks, which led the authors to suggest that PFC is more prone to neuronal network-related pathologies such as epilepsy (Amadeo et al., 2018). Neural network in layer V of PFC is predominantly regulated by nAChRs expressing β2 subunit (Poorthuis et al., 2012). Despite mutation at B2-V287L, the cell surface expression of nAChRs remained the same (Manfredi et al., 2009). Thus, functional changes were thought to be attributable to expression of B2-V287L (Amadeo et al., 2018). Altered Ca2+ signals following hyperactivity of nAChRs could upregulate the surface expression of KCC2 as a compensatory mechanism to counterbalance the overactive neuronal network by increasing the Cl− turnover. Collectively, these lead to retardation of GABAergic switch in PFC and ultimately hyperexcitability (Amadeo et al., 2018).
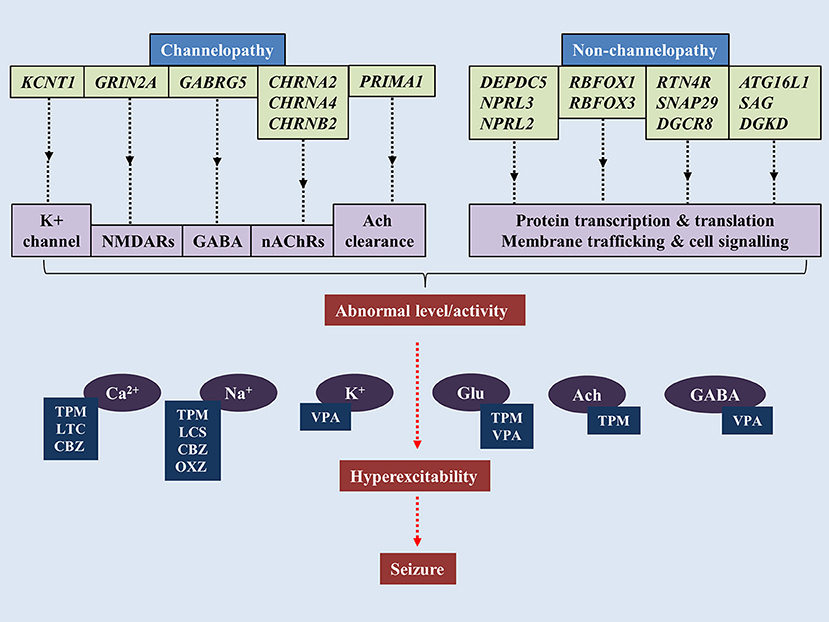
Figure 1. Pathogenesis of sleep-related epilepsy. Mutations of genes associated with channelopathy and non-channelopathy origin of sleep related epilepsy could disrupt the balance between inhibitory and excitatory neurotransmissions in central nervous system, leading to manifestation of seizure. Various anti-epileptic drugs alleviate seizure by restoring chemical balance in brain. TPM, topiramate; VPA, valproic acid; CBZ, carbamazepine; OXC, oxcarbazepine; LTC, levetiracetam; LCS, lacosamide.
In addition to nAChRs, mutations in KCNT1 (gene encoding for potassium-sodium activated channel subfamily T member 1) were also linked to a rather severe form of SHE and sporadic SHE. The patients found with these mutations are also presented with various psychiatric features and intellectual disabilities, which was dissimilar to the usual form of SHE. The onset age for KCNT1 mutation-related SHE was below the onset age of classical SHE and the penetrance of KCNT1 mutations are 100% (Heron et al., 2012) which is higher than classical SHE. KCNT1 encodes for KCNT1 channel subunit which binds with potassium-sodium activated subfamily T member 2 (KCNT2) to form the heterotetrameric complex of the channel. The KCNT1 mutations mostly affect the nicotinamide adenine dinucleotide interaction with C-terminal of the channel, which could disrupt the modulation of the channels' functions (Tamsett et al., 2009).
More recent findings have focused on the non-channelopathy-based pathogenesis of the SHE (Dibbens et al., 2013; Picard et al., 2014; Korenke et al., 2016). DEPDC5 (Disheveled, Eg-10 and Pleckstrin Domain containing proteins) is a gene that encodes a protein structurally-related to Gap Activity Toward Rags 1 (GATOR1), which is an important negative modulator of mammalian target of Rapamycin (mTOR) Complex 1 (mTORC1) that regulates various cell functions (Bar-Peled et al., 2013). DEPDC5 mutations have been implicated in familial temporal lobe epilepsy, SHE and familial focal epilepsy with variable loci (Dibbens et al., 2013; Ishida et al., 2013; Martin et al., 2014). Other genes expressing GATOR1 such as nitrogen permease regulator-like 2 (NPRL2) and nitrogen permease regulator-like 3 (NPRL3) were also linked to sporadic and familial form of epilepsy (Ricos et al., 2016). It was thought that mal-interaction between mTORC1 and GATOR1 may alter cortical neuroarchitecture as epileptic patients with NPRL3 mutations were presented with dysplastic brain lesions (Sim et al., 2016). Nevertheless, few patients despite experiencing seizures showed no anomalies in brain imaging, suggesting the structural changes could be microscopic or other unknown pathway could mediate the pathogenesis (Korenke et al., 2016). From a functional perspective, reduced negative modulation of mTORC1 leads to overactivity of the protein complex, which has been demonstrated in epileptic brains (Sha et al., 2012; Sosunov et al., 2012). In addition, inhibition of mTORC1 has been shown to block epileptogenesis (Huang et al., 2010). Thus, it was hypothesized that hyperactivated mTORC1 signaling leads to rhythmic increase in neuronal excitability (Cho, 2012).
Benign Childhood Epilepsy With Centrotemporal Spikes
BECTS, also known as Rolandic Epilepsy (RE) is the most common type of epilepsy syndrome in children. The typical onset age of BECTS is between 3 and 13 years, with spontaneous remission of seizures upon puberty (Berg et al., 2010). The hallmark feature of BECTS' EEG is high voltage spike and wave, mainly centrotemporal spikes. The seizures predominantly occur during NREM sleep and last for 1–3 min (Stephani, 2000).
Although initially described as idiopathic (Panayiotopoulos et al., 2008), several studies postulated a hereditary link to the disease (Vears et al., 2012; Shi et al., 2018). Numerous potential genes predisposing to BECTS were investigated to no avail (Neubauer et al., 1998; Strug et al., 2009; Pal et al., 2010). Proline-rich transmembrane protein gene (PRRT2) that was associated with paroxysmal kinesigenic dyskinesias (Chen W. J. et al., 2011) and RE (Dimassi et al., 2014), were screened in 9 cohorts of 53 sporadic patients and 250 controls in a mainland Chinese population. Genomic sequencing revealed no association between PRRT2 mutations and BECTS (Che et al., 2017). In a separate study, genes associated with epilepsy-aphasia spectrum, that encode for elongator acetyltransferase complex subunit 4 (ELP4) and sushi-repeat containing protein X-linked 2 (SRPX2) (Roll et al., 2006; Strug et al., 2009), were studied for their potential interactions in BECTS. The investigators utilized whole genome sequencing on 290 patients with European ancestry diagnosed with RE or atypical RE (ARE) in Germany, Canada and Austria and no pathological link between RE/ARE and ELP4 and SRPX2 genes were found (Reinthaler et al., 2014). The role of ELP4 in pathogenesis of BECTS was also downplayed by another study conducted in a Greek population (Gkampeta et al., 2014). The genetic risk factor in BECTS was identified in the gene that encodes for α2 subunit of N-methyl-D-aspartate receptors (NMDARs) (GRIN2A). A mutational analysis carried out in 2 independent cohorts of 359 patients identified heterozygous mutations in 27 of 359 subjects and described exon-disrupting microdeletions in 3 of 286 individuals (Lemke et al., 2013). More recently, an unbiased gene-burden analysis of 194 patients against 567 in-house and 33370 online ExAC controls showed only GRIN2A rare CSDD15, CADD15 + LOF, and LOF variants were more frequent in BECTS (Bobbili et al., 2018). NMDARs are glutamate-bound excitatory receptors with important roles in synaptic transmission and plasticity (Paoletti, 2011). Numerous animal models have implicated altered NMDARs functions in development of epilepsy (Frasca et al., 2011; Di Maio et al., 2012). Thus, mutations in GRIN2A are thought to affect the electrophysiological property of GluN2A subunit containing NMDARs (Lemke et al., 2013). In addition to GRIN2A, genes DEPDC5 (Lal et al., 2014), gamma-aminobutyric acid receptor subunit gamma-2 (GABRG2) (Reinthaler et al., 2015), RNA Binding Fox-1 Homolog 1 (RBFOX1), RNA Binding Fox-1 Homolog 3 (RBFOX3) (Lal et al., 2013) and KCNT1 (Shi et al., 2018) were also implicated in BECTS.
Persistent neuropsychiatric deficits in executive functions, intelligence and attention have been reported in BECTS patients despite spontaneous remission during the adolescence (Currie et al., 2018; Ofer et al., 2018). Myriad of neuroimaging studies associated the cognitive decline with abnormal cortical changes (Overvliet et al., 2013; Pardoe et al., 2013). Generally, cortical gray matter decreases over time from childhood to young adulthood (Shaw et al., 2006). However, in the BECTS population, the changes in cortical thickness were greater compared to the control group and there was also delay in reaching the normative values. This may explain the persistence of language problems in BECTS patients even after the remission (Pardoe et al., 2013). On the contrary, a more recent study precluded any direct relationship between centrotemporal spikes frequency and morphological changes in cortex of BECTS patients. However, one particular region, R pars opercularis showed thinner cortex in BECTS children relating the atypical cortical features with poor processing speed (Fujiwara et al., 2018). Several studies have shown BECTS to predominantly affect the left hemisphere of the brain, linking BECTS to language dysfunction seen in the children (Overvliet et al., 2013). This was further corroborated by the discovery of widespread white matter abnormalities confining to the left hemisphere of BECTS children with intellectual disabilities, particularly verbal IQ (Kim et al., 2014). In addition to intellectual disabilities, left hemisphere anomaly was also correlated with greater social fear among BECTS patients (Potegal et al., 2018). More recently, the network re-organization leading to cognitive dysfunctions in BECTS was thought to originate from right homologous brain areas. In treatment naïve, early-onset BECTS patients, escalated brain activity was seen in the right Broca's area during the early stage of the disease, suggesting the compensatory change to take place in the right hemisphere of the brain (Chen et al., 2018). Subcortical structures such as the basal ganglia, sensorimotor networks, and striato-cortical circuitry were also shown to have epilepsy-related functional connectivity with BECTS (Li et al., 2017).
Panayiotopoulos Syndrome
PS is a common idiopathic childhood epilepsy syndrome with predominant features of autonomic signs. The majority of the children were found to be in the age group of 1–14 years, with 4–5 years being more predominantly affected by PS (Caraballo et al., 2000). The most common symptoms exhibited by PS patients are full emetic triad (nausea, retching, vomiting), pallor, deviation in head and eyes, generalized seizures, ictal syncope (Yalçin and Toydemir, 2017), changes in thermoregulatory (Panayiotopoulos, 2005) and cardiorespiratory functions (Yamamoto et al., 2018). A very recent investigation on the incidence of PS recorded over 16 months in a population aged below 16 years old, reported 0.8/100,000 new cases. Similar study also recorded 6.1/100,000 new cases of BECTS within the same population, reporting 13 times higher prevalence of BECTS than PS (Weir et al., 2018).
The transient focal EEG abnormalities that are usually seen in the epilepsy, or also known as the “functional spikes” were initially thought to be confined to the occipital lobe in PS patients (Tsai et al., 2001). However, follow-up studies documented the functional spikes to shift to extra-occipital regions such as prefrontal and frontal (Kokkinos et al., 2010; Yoshinaga et al., 2010), parietal and lateral temporal lobe (Leal et al., 2008). This multifocal hyperexcitability nature of PS has led the researchers to correlate the affected cortical areas to the hypothalamic autonomic centers and the limbic system to the manifestation of transient hyperactive central autonomic network, which is the cardinal feature of PS (Ten Donkelaar and Horim, 2011). Generally, seizures in PS patients are sleep-related. Some studies have reported nearly all PS-associated seizures to take place during sleep (Caraballo et al., 2007), whereas some reported the 69.9% seizures to occur during sleep and 12.9% during awake (Specchio et al., 2010). More recent findings suggest the seziures are more likely to take place during awakenings (66.7%) (Yalçin and Toydemir, 2017). In sum, the constellation of findings suggests the PS-associated seizures to take place either during early awakening or sleeping hours.
The etiology of PS has been the subject of intense research, yet much of its pathophysiology has remained elusive. In 2007, a 12-year-old girl who presented with PS, was reported to carry a sporadic missense mutation in sodium voltage-gated channel alpha subunit 1 (SCN1A), the gene encodes a voltage-gated sodium channel that was implicated in the pathology of Dravet Syndrome (Grosso et al., 2007). Two years later, another study reported two siblings with PS to have the SCN1A mutation. However, the father of the children also shared the SCN1A mutation, and surprisingly never experienced a seizure. This suggests that SCN1A mutation may merely increase the susceptibility to an idiopathic focal epilepsy phenotype (Livingston et al., 2009). In another study, 2 monozygotic twins presented with PS were found to have no mutations in the SCN1A gene or the GABRG2 gene (another gene associated with Dravet Syndrome). Based on the early onset of PS in these patients and the severity of their symptoms, the authors concluded that mutations in the SCN1A gene may regulate the severity of the syndrome rather than the genesis of the disorder (Martín Del Valle et al., 2011). In a very recent study, a 6-year old girl diagnosed with PS was presented with a de novo 2.6 Mb deletion in 22q11.2 and an additional 172 kb duplication in 2q37.1 (Bertini et al., 2017). Deletion in 22q11.2 was associated with loss of genes involved in brain function and development, such as RTN4R (reticulon four receptor, NOGO RECEPTOR) (Pan et al., 2005; Ramasamy et al., 2014), SNAP29 (synaptosomal-associated protein 29) and gene responsible for biogenesis of micro-mRNA, especially in mammalian brain such as DGCR8 (microprocessor complex subunit 8) (Cheng et al., 2014). In addition to this, the 172 kb duplication in 2q37.1 was related to three genes, which include ATG16L1 (Autophagy 16-like 1), SAG (S-antigen; retina and pineal gland), and DGKD (diacylglycerol kinase delta). Among these genes, DGKD coding for a cytoplasmic enzyme that phosphorylates diacylglycerol to produce phosphatidic acid has been implicated in epilepsy (Leach et al., 2007).
Similar to BECTS, children suffering from PS also demonstrate cognitive deficits, particularly in global visual-motor integration, writing, reading, arithmetic skills, verbal and visual-spatial memory (Germanò et al., 2005). The cognitive abnormalities reported in some of the PS patients was a result of the propagation of the interictal activity to various brain regions, including the frontal (Germanò et al., 2005) and parietal lobes (Lopes et al., 2014). In concordance with this finding, a more recent study reported changes in volume of prefrontal lobe and prefrontal-to-frontal lobe volume ratio in 3 PS patients who presented with status epilepticus. Conversely, the non- status epilepticus PS patients possess cortical growth pattern similar to that of healthy controls, suggesting the manifestation of SE in PS may impair the cognitive behavior of some PS patients (Kanemura et al., 2015).
Pharmacotherapy for BECTS
The spontaneous remission of BECTS in adolescence has cast controversy over its treatment; in particular, as to whether or not to use anti-epileptic drugs (AEDs). A review of 110 recommendations from 96 published materials on BECTS revealed two-third of the findings to favor and one-thirds not to favor the use of AEDs. Most of those in favor with AEDs use, advocate for pharmacotherapy only in cases with early onset and multiple seizure expressions and also to limit the treatment to 1 year (Hughes, 2010). The need for AEDs was evident in some cases owing to the severity of the seizures, cognitive impairments and behavioral abnormalities that accompany the seizures in a large population of young children with BECTS (Kavros et al., 2008; Sarco et al., 2011; Samaitiene et al., 2012). In addition, there seem to be geographic differences in pharmacological management of BECTS. Sulthiamine (STM) was the most commonly prescribed AED in Austria and Germany (Gross-Selbeck, 1995). In the United States, the preferred AED for BECTS was carbamazepine (CBZ)/oxcarbazepine (OXZ) (Arzimanoglou and Wheless, 2007). European epileptologists prefer valproic acid (VPA) as the drug of choice for BECTS (Wheless et al., 2007).
There is a considerable amount of literature on VPA use in reducing electroclinical abnormalities (Gelisse et al., 1999; Xiao et al., 2014), increasing the threshold of motor evoked potentials (Nezu et al., 1997) and controlling epileptic negative myoclonus induced by other AEDs (Yang et al., 2008). In 2013, the ILAE recommended monotherapy with VPA for BECTS (level C evidence) (Glauser et al., 2013). Prolonged exposure to VPA was associated with weight gain in 40% of children (Corman et al., 1997). CBZ was shown to successfully treat BECTS populations in Japan (Oka et al., 2004), China (Ma and Chan, 2003), Greek (Gkampeta et al., 2015), and the United States (Wheless et al., 2005). Apart from reducing seizure incidences, CBZ also improved cognitive functions in BECTS patients by altering the epilepsy-induced changes in P300 event-related potential (Naganuma et al., 1994). One of the most undesirable side effects of CBZ was the drug-induced non-epileptic myoclonus and tic-like movements (Magaudda and Di Rosa, 2012). OXZ, a chemical twin of CBZ, was reported to normalize EEG, improved cognition and effectively controlled seizure expressions (Tzitiridou et al., 2005). It has a mild adverse effect profile such as headache and sedation (Coppola et al., 2007).
STM, a carbonic anhydrase inhibitor reduced spike and seizure frequencies (Wirrell et al., 2008), normalized EEG (Bast et al., 2003) with low seizure remission rate (91% success for 24 weeks of observation) (Borggraefe et al., 2013). Adverse effects such as impaired cognitive functions (Wirrell et al., 2008) and metabolic acidosis (Borggraefe et al., 2013) were associated with STM. On the other hand, LEV was shown to control seizures without (Bello-Espinosa and Roberts, 2003) and with minimum side effects (Verrotti et al., 2007). Few researchers reported that LEV improves BECTS-related impairments in auditory verbal memory and baseline auditory comprehension (Kossoff et al., 2007). The success rate of LEV on seizure remission was 81% as reported by Borggraefe et al. (2013). Adverse effects such as suicidal ideations (Borggraefe et al., 2013) and psychosis (Kossoff et al., 2001) were related to LEV use. Topiramate, a novel AED was shown to reduce epileptiform frequency and inhibit epileptiform discharges in BECTS patients. Its side effect includes anorexia, nausea, headache, and hypohidrosis / adiaphoresis (Liu et al., 2016).
Pharmacotherapy for Sleep Related Hypermotor Epilepsy
CBZ has been documented as the most commonly prescribed drug to manage SHE (Provini et al., 1999; Gambardella et al., 2000). Almost two-thirds of the SHE patients responded well to bedtime low doses (200–1,000 mg/kg) of CBZ. However, one third of the patients remained resistant to the drug (Provini et al., 2000). High blood brain barrier penetrance of CBZ (Shorvon, 2000) indicates the propensity of the AED to react with a variety of neuronal receptors. In agreement with this, CBZ was shown to reduce the action potential frequency of voltage-gated sodium channels (McLean and MacDonald, 1986; Schwarz and Grigat, 1989) which in turn could alter the neuronal excitability by impairing the glutamate release (Sitges et al., 2007) or potentiating (gamma-Aminobutyric acid) GABAA receptors (Zheng et al., 2009). More importantly, CBZ inhibits α4β2 and α2β4 subunits of nicotinic receptors (Di Resta et al., 2010) which have often been implicated in SHE. Therefore, it seems that inhibition of the nicotinic receptors by CBZ may suppress glutamate excitability and potentiate GABA activity in the thalamocortical system and hippocampus to attenuate hyperexcitability (Albuquerque et al., 2009; Aracri et al., 2010).
Many epileptologists reported better outcomes with OXZ in controlling nocturnal seizures (Raju et al., 2007; Romigi et al., 2008), even in patients unresponsive to CBZ and other AEDs (Raju et al., 2007). Similar to CBZ, OXZ block voltage-gated sodium channels (MacDonald and Rogawski, 2008), potentiate GABAA receptors (Zheng et al., 2009) and inhibits α2β4 subunits of nicotinic receptors (Di Resta et al., 2010). In addition to OXZ, Oldani and co-researchers found administration of topiramate (50–300 mg daily at bedtime) as add-on (3 patients) or monotheraphy (21 patients) to reduce nocturnal seizures in 62.5% of patients and six of the total patients were seizure free in the follow-up that ranged from 6 months to 6 years. The authors reported mild adverse events include weight loss (6 patients), paresthesias (3 patients), and speech dysfunction (2 patients) which disappeared within 3 months (Oldani et al., 2006). More recently, in a Taiwanese series of 10 case studies, CBZ, OXZ, and topiramate along with other AEDs effectively reduced nocturnal seizures by 75% and abolished diurnal attack by more than 90% without producing any adverse effects (Yeh and Schenck, 2017).
Claudio Liguori and colleagues administered lacosamide as add-on therapy in 2 SHE patients that were unresponsive to other AEDs. Addition of lacosomide (200 mg/day) to polytherapy (CBZ+TPM) and (OXZ+clonazepam) dramatically abolished the nocturnal seizure expressions and both patients were then continued on lacosomide monotherapy for 12 months and remained seizures free (Liguori et al., 2016). In a separate study, administration of LCM (300–600 mg/kg) to eight patients with refractory-SHE reduced the seizure frequency in 5 patients for more than 50 and 25% in one patient. The authors also reported mild and reversible adverse events in most of the patients, such as transient fatigue and diplopia. However, one patient was withdrawn from lacosomide for feeling continuously “spaced out” (Samarasekera et al., 2018).
Pharmacotherapy for Panayiotopoulos Syndrome
Most clinicians believe children with PS may not require prophylaxis therapy with AED (Panayiotopoulos, 2002, 2004). Nevertheless, 10–20% of PS patients face persistent autonomic status epilepticus that could last for days (García and Rubio, 2009) which place them at great risk of developing life threatening severe cardiorespiratory dysfunctions (Camfield and Camfield, 2005; Verrotti et al., 2005). In addition, there is a consensus among the epileptologists that AED therapy should be reserved for the patients with unusually frequent and severe form of seizures that could affect the quality of their lives (Ferrie et al., 2006). To date, there is no single monotherapy of any AED has been shown to be superior to the rest.
Garcia and Rubio have reported recurrence of seizures in some PS patients after 6 months of treatment with VPA. The same authors also showed LEV (1,000–2,000 mg/kg) that initially introduced as add-on therapy to these patients, and then as monotherapy, successfully reduced the occurrences of seizures and all the patients remained seizure free for 2–3 years (García and Rubio, 2009). In parallel to this finding, numerous studies have documented the potential advantages of LEV in pediatric epileptic cases owing to the AED's lack of interaction with other drugs, favorable elimination kinetics and significant protein binding ability (Leppik, 2001). In addition, LEV also lacks adverse effects such as weight gain, polycystic ovarian syndrome, hair loss, and rash that have been most frequently implicated in the use of VPA, CBZ and lamotrigine (Konishi et al., 1993; Barron et al., 2000). As mentioned earlier, long term use of LEV was associated with behavior-related adverse events (Kossoff et al., 2001; Borggraefe et al., 2013).
Rectal, buccal or intravenous (IV) benzodiazepines were commonly used to manage autonomic status epilepticus manifestations in PS (Ferrie et al., 2006). However, Lacroix et al. (2011) urged the clinicians to practice great caution over the use of BDZ to treat autonomic seizures. The authors reported severe respiratory depression following the benzodiazepines administration (diazepam 0.5–0.6 mg/kg, IR; lorazepam 0.05–0.06 mg/kg, IV) for seizures with autonomic manifestations in five patients. The authors went on to suggest that the use of other BDZ such as buccal midazalom, or a more autonomic tolerant AEDs such as VPA or LE for the acute management of autonomic seizures (Lacroix et al., 2011) should be considered.
Future Recommendations
The past decade has witnessed the birth of various AEDs. Despite their efficacy, they are not without severe adverse effects, especially in prolonged exposure to refractory epileptic patients. This calls for discovery of novel, more specific molecular-targeting pharmacotherapies. Therapeutic diets such as ketogenic diet and low-glycemic index diet were shown to be effective in treating drug-resistant epileptic patients (Pfeifer and Thiele, 2005; Neal et al., 2008). As well, adjunctive therapy with fenofibrate was shown to markedly reduce the seizure frequency in human and animal models of SHE (Puligheddu et al., 2017). Fenofibrates are agonists of peroxisome proliferator-activated receptor alpha (PPARα), which inhibits β2-containing nicotinic receptors by phosphorylating β2 (Melis et al., 2010; Puligheddu et al., 2013). In animal models of SHE, chronic diet with fenofibrates reduced the nicotine-induced spontaneous inhibitory postsynaptic current in pyramidal neurons of the frontal lobe. This subsequently attenuated the cholinergic overactivation and expressions of seizures (Puligheddu et al., 2017). Taken together, these findings warrant further investigation of the role of fenofibrate and PPARα in the pathogenesis of sleep-related epilepsy.
Identification of GRIN2A mutations in BECTS and other childhood epilepsies has thrown light on the role of GluN2A subunit-containing NMDARs in epilepsy (Lemke et al., 2013; Gao et al., 2017; Von Stülpnagel et al., 2017). Recent findings indicate that GRIN2A mutations prolong NMDARs' deactivation time, decrease the amplitude of current responses, reduce glutamate potency, reduce channel open probability and accentuate the sensitivity of NMDARs toward negative allosteric modulators (Gao et al., 2017; Sibarov et al., 2017). It is postulated that reduced NMDARs function may impair the inhibitory effects of GABAergic interneurons in the prefrontal cortex (Xi et al., 2009) and cerebral cortex (Bagasrawala et al., 2016); leading to epilepsy (Gao et al., 2017). In addition, NMDARs are also modulated by metabotropic glutamate receptor subtype 5 (mGlu5) (Chen H. H. et al., 2011). Negative modulation of mGlu5 has been promising in attenuating hyperexcitability of central nervous system (Kumar et al., 2013, 2016, 2017) and even reducing the spike-wave discharges in numerous animal models of epilepsy (McCool et al., 1998; Chapman et al., 2000). More studies are needed to further elucidate the functional link between mGlu5, GABA, and NMDARs in epilepsy. Discovery of mutations in DEPDC5, NPRL2, and NPRL3 that encode for GATOR1 (negative modulator of mTORC1) have pioneered the channelopathy-independent approach in understanding the pathological process of NFLE. To date, mTORC1 inhibitors have only been proven successful in treating epilepsy in tuberous sclerosis (Curatolo, 2015) and polyhydramnios megalencephaly symptomatic epilepsy (Parker et al., 2013). Nevertheless, the potential functional link between mutations of mTORC1-related genes and non-lesional focal epilepsy (Myers and Scheffer, 2017) merits future studies.
We summarized the pharmacotherapy of options for BECTS, PS and SHE in Tables 2–4, respectively. The mechanism of actions for the AEDs is listed in Table 5.
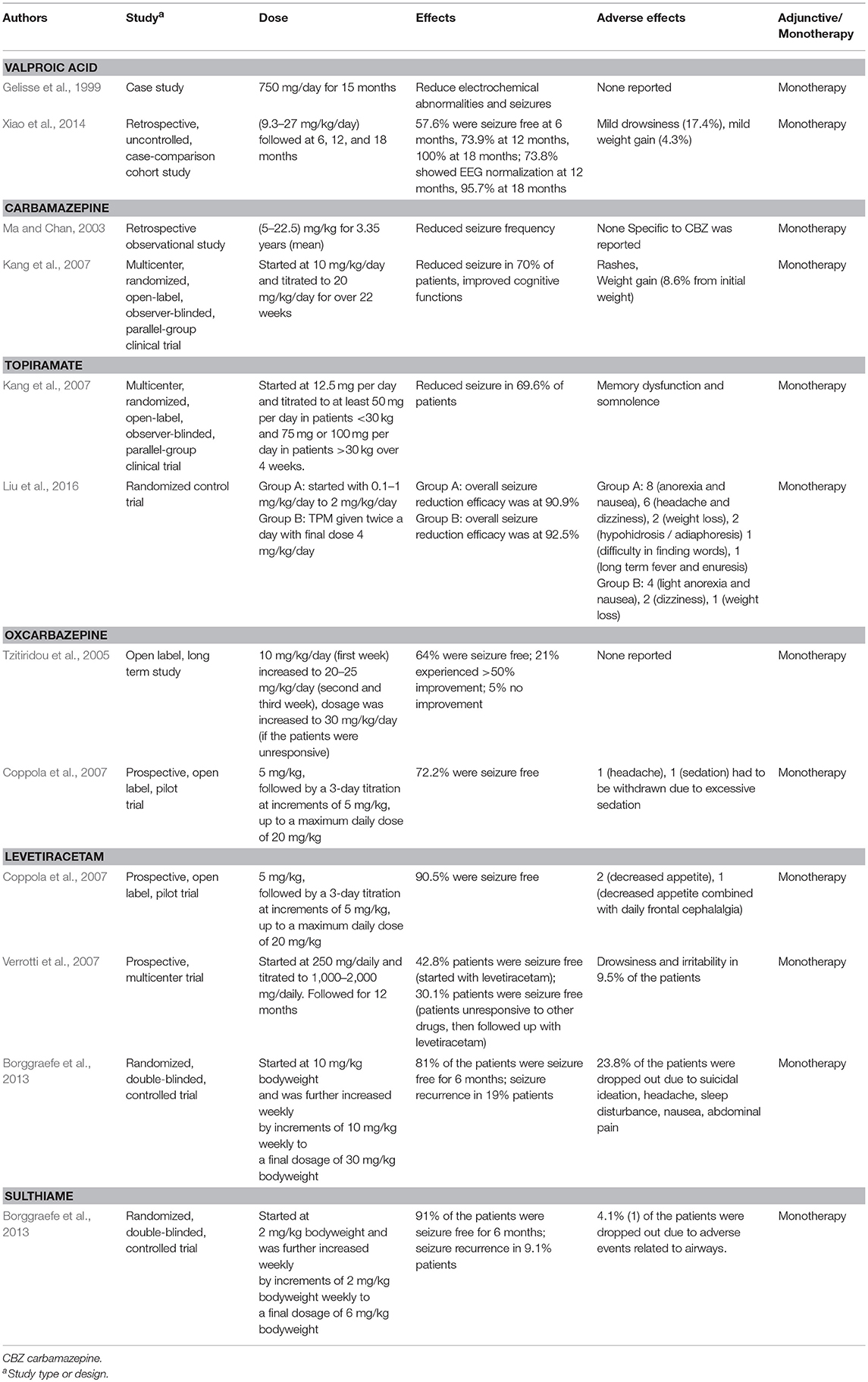
Table 2. Table showing pharmacotherapy options for BECTS.
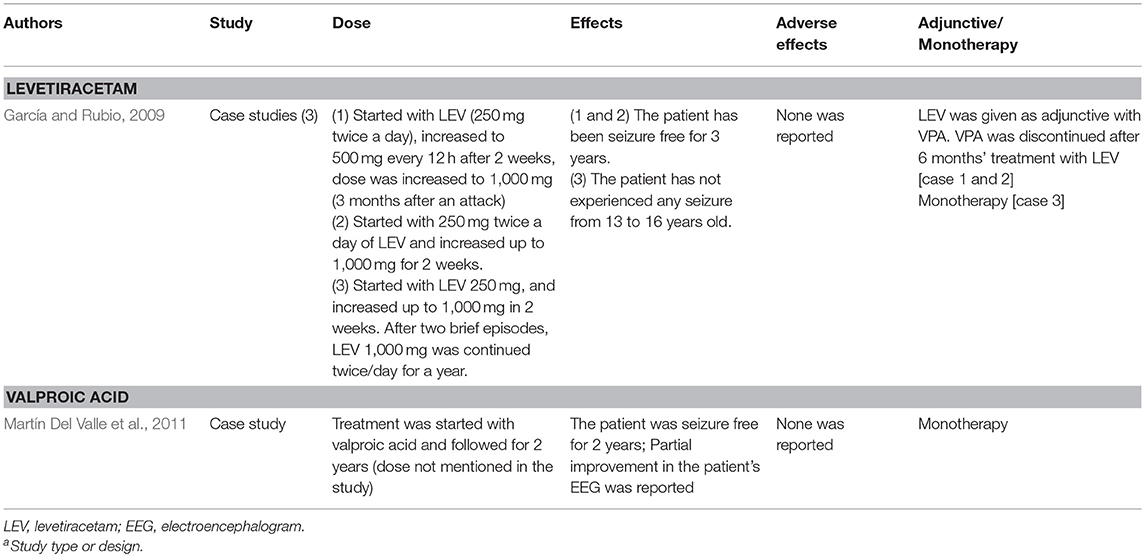
Table 3. Table showing pharmacotherapy options for Panayiotopoulos Syndrome.
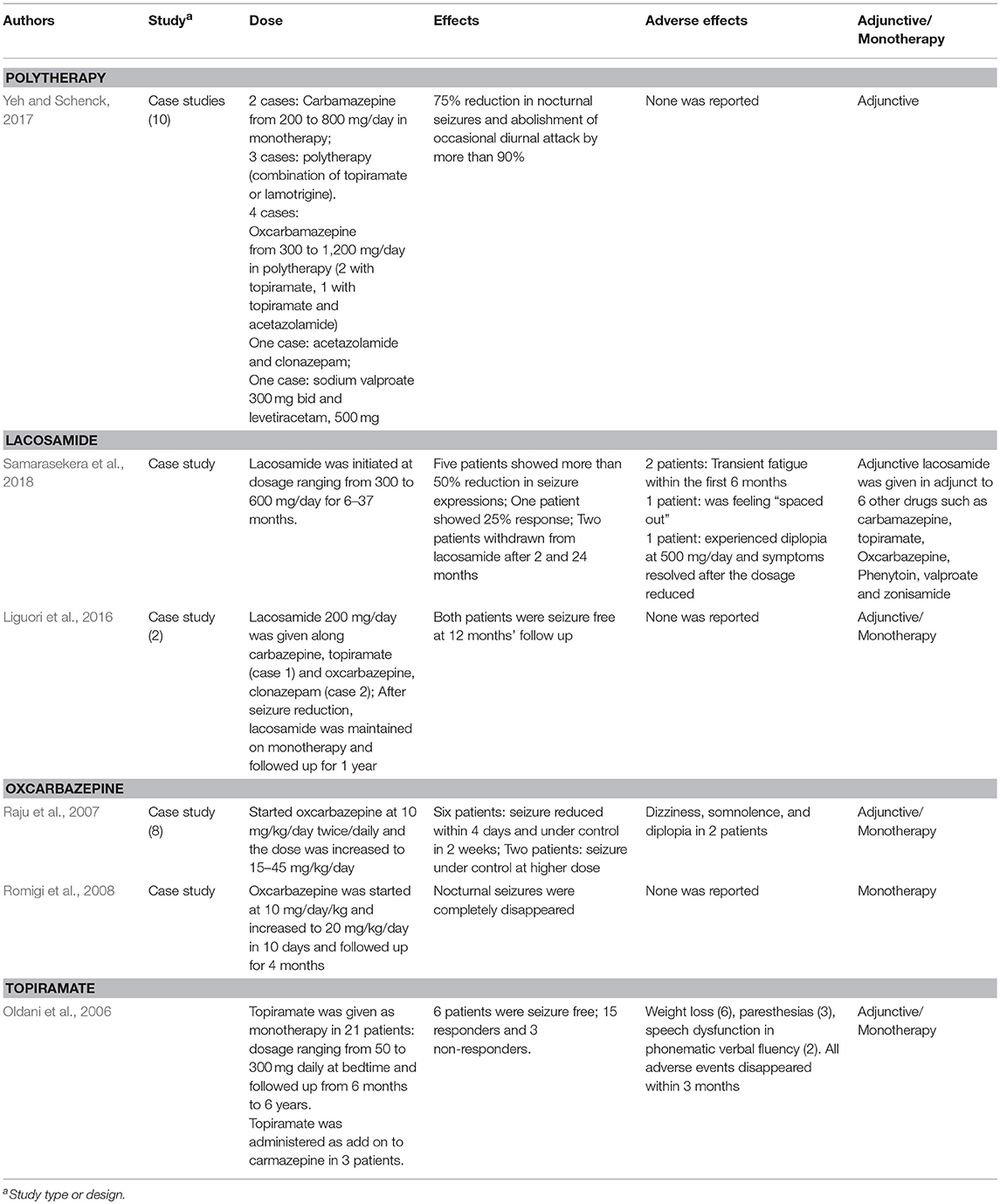
Table 4. Table showing pharmacotherapy options for sleep related hypermotor epilepsy.
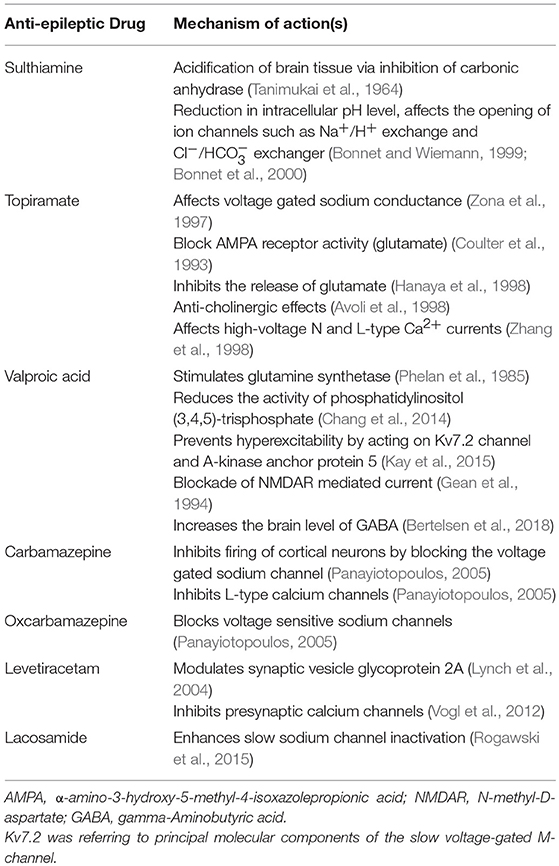
Table 5. Table showing the mechanism of actions for anti-epileptic drugs.
Author Contributions
JK, AS, PM, and SD conceived the idea. JK, AS, PM, SD, and RM wrote the manuscript. JK and SD prepared the tables. JK, PM, and SD edited the manuscript.
Funding
This study was supported by the FRGS/1/2016 /SKK 08/UKM/03/3.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Ernie for the critical review of the manuscript.
Abbreviations
ILAE, International League Against Epilepsy; SHE, sleep related hypermotor epilepsy; BECTS, benign partial epilepsy with centrotemporal spikes; PS, Panayiotopoulos Syndrome; REM, rapid eye movement; NREM, non-rapid eye movement; Ca2+, calcium; K+, potassium; NFLE, Nocturnal Frontal Lobe Epilepsy; nAChRs, nicotinic acetylcholine receptors; CHRNA2, cholinergic receptor nicotinic alpha 2 subunit; CHRNA4, cholinergic receptor nicotinic alpha 4 subunit; CHRNB2, cholinergic receptor nicotinic beta 2 subunit; GABA, gamma-Aminobutyric acid; Ach, acetylcholine; PRIMA1, proline rich membrane anchor 1; Cl−, chloride; NKCC1, Na+/K+/Cl− co-transporter-1; KCC2, K+/Cl− co-transporter-2; PFC, prefrontal cortex; KCNT1, potassium-sodium activated channel subfamily T member 1; KCNT2, potassium-sodium activated channel subfamily T member 1; DEPDC5, Disheveled, Eg-10 and Pleckstrin Domain containing proteins; GATOR1, Gap Activity Toward Rags 1; mTOR, mammalian target of Rapamycin; mTORC1, mTOR Complex 1; NPRL2, nitrogen permease regulator-like 2; NPRL3, nitrogen permease regulator-like 3; RE, rolandic epilepsy; PRRT2, proline-rich transmembrane protein gene; ELP4, elongator acetyltransferase complex subunit 4; SRPX2, sushi-repeat containing protein X-linked 2; ARE, atypical RE; NMDARs, N-methyl-D-aspartate receptors; GRIN2A, α2 subunit of N-methyl-D-aspartate receptors; GABRG2, gamma-aminobutyric acid receptor subunit gamma-2; RBFOX1, RNA Binding Fox-1 Homolog 1; RBFOX3, RNA Binding Fox-1 Homolog 3; SCN1A, sodium voltage-gated channel alpha subunit 1; RTN4R, reticulon four receptor, NOGO RECEPTOR; SNAP29, synaptosomal-associated protein 29; DGCR8, microprocessor complex subunit 8; ATG16L1, Autophagy 16-like 1; SAG, S-antigen; retina and pineal gland; DGKD, diacylglycerol kinase delta; STM, sulthiamine; CBZ, carbamazepine; OXZ, oxcarbazepine; VPA, valproic acid; LEV, levetiracetam; PPARα, peroxisome proliferator-activated receptor alpha.
References
Albuquerque, E. X., Pereira, E. F. R., Alkondon, M., and Rogers, S.W. (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120. doi: 10.1152/physrev.00015.2008
Amadeo, A., Coatti, A., Aracri, P., Ascagni, M., Iannantuoni, D., Modena, D., et al. (2018). Postnatal changes in K+/Cl− cotransporter-2 expression in the forebrain of Mice Bearing a Mutant Nicotinic Subunit Linked to Sleep-Related Epilepsy. Neuroscience 386, 91–107. doi: 10.1016/j.neuroscience.2018.06.030
Amzica, F., Massimini, M., and Manfridi, A. (2002). Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J. Neurosci. 22, 1042–1053. doi: 10.1523/JNEUROSCI.22-03-01042.2002
Aracri, P., Consonni, S., Morini, R., Perrella, M., Rodighiero, S., Amadeo, A., et al. (2010). Tonic modulation of GABA release by nicotinic acetylcholine receptors, in layer V of the murine prefrontal cortex. Cereb.Cortex 7, 1539–1555. doi: 10.1093/cercor/bhp214
Arzimanoglou, A., and Wheless, J. W. (2007). Children with epilepsy: are they the same on both sides of the Atlantic, and do the same treatments work? Epileptic. Disord. 9:351e352. doi: 10.1684/epd.2007.0137
Avoli, M., Palmieri, C., and Kawasaki, H. (1998). Topiramate depresses carbachol-induced depolarizing plateau potentials in the rat subiculum nl rltro. Epilepsia 39 (Suppl 6):43.
Bagasrawala, I., Memi, F.V, Radonjic, N., and Zecevic, N. (2016). N-methyl d-aspartate receptor expression patterns in the human fetal cerebral cortex. Cereb. Cortex 27, 5041–5053. doi: 10.1093/cercor/bhw289
Baghbani, S., Baril, J. F., and Ng, M. (2018). “EEG brain synchronization in epileptic patients during sleep,” in Canadian Medical and Biological Engineering Society Proceedings, Vol. 40 (Winnipeg, MN).
Bar-Peled, L., Chantranupong, L., Cherniack, A. D., Chen, W.W, Ottina, K. A., Grabiner, B. C., et al. (2013). A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106. doi: 10.1126/science.1232044
Barron, T. F., Hunt, S. L., Hoban, T. F., and Price, M. L. (2000). Lamotrigine monotherapy in children. Pediatr. Neurol. 23, 160–163. doi: 10.1016/S0887-8994(00)00162-4
Bartolini, L., Libbey, J. E., Ravizza, T., Fujinami, R. S., Jacobson, S., and Gaillard, W. D. (2018). Viral triggers and inflammatory mechanisms in pediatric epilepsy. Mol. Neurobiol. [Epub ahead of print].doi: 10.1007/s12035-018-1215-5
Bast, T., Völp, A., Wolf, C., and Rating, D. (2003). The influence of sulthiame on EEGin childrenwith benign childhood epilepsy with centrotemporal spikes (BECTS). Epilepsia 44, 215–220. doi: 10.1046/j.1528-1157.2003.51001.x
Becchetti, A., Aracri, P., Meneghini, S., Brusco, S., and Amadeo, A. (2015). The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front. Physiol. 6:22 doi: 10.3389/fphys.2015.00022
Bello–Espinosa, L. E., and Roberts, S. L. (2003). Levetiracetam for benign epilepsy of childhood with centrotemporal spikes�three cases. Seizure 12, 157–159. doi: 10.1016/S1059-1311(03)00004-9
Berg, A. T., Berkovic, S. F., Brodie, M. J., Buchhalter, J., Cross, J. H., et al. (2010). Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 51, 676–685. doi: 10.1111/j.1528-1167.2010.02522.x
Bertelsen, F., Landau, A. M., Vase, K. H., Jacobsen, J., Scheel-Krüger, J., and Møller, A. (2018). Acute in vivo effect of valproic acid on the GABAergic system in rat brain: A [11C] Ro15-4513 microPET study. Brain Res 1680, 110–114. doi: 10.1016/j.brainres.2017.12.018
Bertini, V., Valetto, A., Azzarà, A., Legitimo, A., Saggese, G., Consolini, R., et al. (2017). A case of 22q11 deletion syndrome (22q11DS) with a panayiotopoulos epileptic pattern: are additional copy-number variations a possible second Hit in modulating the 22q11DS phenotype? Front. pediatr. 5:48. doi: 10.3389/fped.2017.00048
Bobbili, D. R., Lal, D., May, P., Reinthaler, E. M., Jabbari, K., Thiele, H., et al. (2018). Exome-wide analysis of mutational burden in patients with typical and atypical Rolandic epilepsy. Eur. J. Hum. Genet. 26, 258–264. doi: 10.1038/s41431-017-0034-x
Bonnet, U., Leniger, T., and Wiemann, M. (2000). Alteration of intracellular pH and activity of CA3-pyramidal cells in guinea pig hippocampal slices by inhibition of transmembrane acid extrusion. Brain Res. 872, 116–124. doi: 10.1016/S0006-8993(00)02350-7
Bonnet, U., and Wiemann, M. (1999). Ammonium prepulse: effects on intracellular pH and bioelectric activity of CA3-neurones in guinea-pig hippocampal slices. Brain Res.840, 16–22. doi: 10.1016/S0006-8993(99)01687-X
Borggraefe, I., Bonfert, M., Bast, T., Neubauer, B. A., Schotten, K. J., Maßmann, K., et al. (2013). Levetiracetam vs. sulthiame in benign epilepsy with centrotemporal spikes in childhood: a double-blinded, randomized, controlled trial (German HEAD Study). Eur. J. Paediatr. Neurol. 17, 507–514. doi: 10.1016/j.ejpn.2013.03.014
Camfield, P., and Camfield, C. (2005). Sudden unexpected death in people with epilepsy: a pediatric perspective. Semin. Pediatr. Neurol. 12, 10–14. doi: 10.1016/j.spen.2004.11.005
Caraballo, R., Cers'iosimo, R., and Fejerman, N. (2007). Panayiotopoulos syndrome: aprospective study of 192 patients. Epilepsia 48, 1054–1061. doi: 10.1111/j.1528-1167.2007.01085.x
Caraballo, R., Cersosimo, R., Medina, C., and Fejerman, N. (2000). Panayiotopoulos- type benign childhood occipital epilepsy: a prospective study. Neurology 55, 1096–1100. doi: 10.1212/WNL.55.8.1096
Castro, S. C. C., Baroni, G. V., Martins, W. A., Palmini, A. L. F., and Bisol, L. W. (2018). Suicide risk, temperament traits, and sleep quality in patients with refractory epilepsy. Epilepsy Behav. 80, 254–258. doi: 10.1016/j.yebeh.2018.01.022
Chang, P., Walker, M. C., and Williams, R. S. (2014). Seizure-induced reduction in PIP3 levels contributes to seizure-activity and is rescued by valproic acid. Neurobiol. Dis. 62, 296–306. doi: 10.1016/j.nbd.2013.10.017
Chapman, A. G., Nanan, K., Williams, M., and Meldrum, B. S. (2000). Anticonvulsant activity of two metabotropic glutamate group I antagonists selective for the mGlu5 receptor: 2-methyl-6-(phenylethynyl)- pyridine (MPEP), and (E)-6-methyl-2-styryl-pyridine. Neuropharmacology 39, 1567–1574. doi: 10.1016/S0028-3908(99)00242-7
Che, X. Q., Sun, Z. F., Mao, X., Xia, K., Yan, X. X., Jiang, H., et al. (2017). Mutation screening of the PRRT2 gene for benign epilepsy with centrotemporal spikes in Chinese mainland population. Int. J. Neurosci. 127, 10–13. doi: 10.3109/00207454.2015.1136886
Chen, H. H., Liao, P. F., and Chan, M. H. (2011). mGluR5 positive modulators both potentiate activation and restore inhibition in NMDA receptors by PKC dependent pathway. J. Biomed. Sci. 18:19. doi: 10.1186/1423-0127-18-19
Chen, S., Fang, J., An, D., Xiao, F., Chen, D., Chen, T., et al. (2018). The focal alteration and causal connectivity in children with new-onset benign epilepsy with centrotemporal spikes. Sci. Rep. 8:5689. doi: 10.1038/s41598-018-23336-z
Chen, W. J., Lin, Y., Xiong, Z. Q., Wei, W., Ni, W., Tan, G. H., et al. (2011). Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet. 43, 1252–1255. doi: 10.1038/ng.1008
Cheng, T. L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W. H., Xu, W., et al. (2014). MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 28, 547–560. doi: 10.1016/j.devcel.2014.01.032
Cho, C. H. (2012). Molecular mechanism of circadian rhythmicity of seizures in temporal lobe epilepsy. Front. Cellular Neurosci. 6:55. doi: 10.3389/fncel.2012.00055
Coppola, G., Franzoni, E., Verrotti, A., Garone, C., Sarajlija, J., Operto, F. F., et al. (2007). Levetiracetam or oxcarbazepine as monotherapy in newly diagnosed benign epilepsy of childhood with centrotemporal spikes (BECTS): an open-label, parallel group trial. Brain. Dev. 29, 281–284. doi: 10.1016/j.braindev.2006.09.008
Corman, C. L., Leung, N. M., and Guberman, A. H. (1997). Weight gain in epileptic patients during treatment with valproic acid: a retrospective study. Can. J. Neurol. Sci. 24, 240–4. doi: 10.1017/S0317167100021879
Coulter, D. A., Sombati, S., and Delorenzo, R. J. (1993). Selective effects of topiramate on sustained repetitive firing and spontaneous bursting in cultured hippocampal neurons. Epilepsia 34 (Suppl. 2), 123.
Crochet, S., Chauvette, S., Boucetta, S., and Timofeev, I. (2005). Modulation of synaptic transmission in neocortex by network activities. Eur. J. Neurosci. 21, 1030–1044. doi: 10.1111/j.1460-9568.2005.03932.x
Curatolo, P. (2015). Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr.Neurol. 52, 281–289. doi: 10.1016/j.pediatrneurol.2014.10.028
Currie, N. K., Lew, A. R., Palmer, T. M., Basu, H., De Goede, C., Iyer, A., et al. (2018). Reading comprehension difficulties in children with rolandic epilepsy. Dev. Med. Child Neurol. 60, 275–282. doi: 10.1111/dmcn.13628
Demirbilek, V., and Dervent, A. (2004). Panayiotopoulos syndrome: video-EEG illustration of a typical seizure. Epileptic. Disord. 6, 121–4.
Derry, C. P., and Duncan, S. (2013). Sleep and epilepsy. Epilepsy. Behav. 26, 394–404. doi: 10.1016/j.yebeh.2012.10.033
Di Maio, R., Mastroberardino, P. G., Hu, X., Montero, L. M., and Greenamyre, J. T. (2012). Thiol oxidation and altered NR2B/NMDA receptor functions in in vitro and in vivo pilocarpine models: implications for epileptogenesis. Neurobiol. Dis. 49, 87–98. doi: 10.1016/j.nbd.2012.07.013
Di Resta, C., Ambrosi, P., Curia, G., and Becchetti, A. (2010). Effect of carbamazepine and oxcarbazepine on wild-type and mutant neuronal nicotinic acetylcholine receptors linked to nocturnal frontal lobe epilepsy. Eur. J. Pharmacol. 643, 13–20. doi: 10.1016/j.ejphar.2010.05.063
Dibbens, L.M., de Vries, B., Donatello, S., Heron, S. E., Hodgson, B. L., Chintawar, S., et al. (2013). Mutations inDEPDC5 cause familial focal epilepsy with variable foci. Nat. Genet. 45, 546–551. doi: 10.1038/ng.2599
Dimassi, S., Labalme, A., Lesca, G., Rudolf, G., Bruneau, N., Hirsch, E., Arzimanoglou, A., et al. (2014). A subset of genomic alterations detected in rolandic epilepsies contains candidate or known epilepsy genes including GRIN2A and PRRT2. Epilepsia 55, 370–378. doi: 10.1111/epi.12502
Ferrie, C., Caraballo, R., Covanis, A., Demirbilek, V., Dervent, A., Kivity, S., et al. (2006). Panayiotopoulos syndrome: a consensus view. Dev. Med. Child. Neurol. 48, 236–240. doi: 10.1017/S0012162206000508
Fialho, G. L., Wolf, P., Walz, R., and Lin, K. (2018). Increased cardiac stiffness is associated with autonomic dysfunction in patients with temporal lobe epilepsy. Epilepsia 59, e85–e90. doi: 10.1111/epi.14084
Fiest, K. M., Sauro, K. M., Wiebe, S., Patten, S. B., Kwon, C.-S., Dykeman, J., et al. (2017). Prevalence and incidence of epilepsy. A systematic review and meta-analysis of international studies. Neurology. 88, 296–303. doi: 10.1212/WNL.0000000000003509
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–482. doi: 10.1111/epi.12550
Frasca, A., Aalbers, M., Frigerio, F., Fiordaliso, F., Salio, M., Gobbi, M., et al. (2011). Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol. Dis. 43, 507–515. doi: 10.1016/j.nbd.2011.04.024
Fujiwara, H., Tenney, J., Kadis, D. S., Byars, A., Altaye, M., Spencer, C., Vannest, J., et al. (2018). Cortical morphology, epileptiform discharges, and neuropsychological performance in BECTS. Acta Neurol. Scand. [Epub ahead of print].doi: 10.1111/ane.12997
Galarreta, M., and Hestrin, S. (2001). Spike transmission and synchrony detection in networks of GABAergic interneurons. Science 292, 2295–2299. doi: 10.1126/science.1061395
Gambardella, A., Annesi, G., De Fusco, M., Patrignani, A., Aguglia, U., Annesi, F., et al. (2000). A new locus for autosomal dominant nocturnal frontal lobe epilepsy maps to chromosome 1. Neurology 55, 1467–1471. doi: 10.1212/WNL.55.10.1467
Gao, K., Tankovic, A., Zhang, Y., Kusumoto, H., Zhang, J., Chen, W., et al. (2017). A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS ONE. 12:e0170818. doi: 10.1371/journal.pone.0170818
García, C., and Rubio, G. (2009). Efficacy and safety of levetiracetam in the treatment of Panayiotopoulos syndrome. Epilepsy. Res. 85, 318–320. doi: 10.1016/j.eplepsyres.2009.03.024
Gean, P. W., Huang, C. C., and Tsai, J. J. (1994). Valproic acid suppresses the synaptic response mediated by the NMDA receptors in rat amygdalar slices. Brain Res. Bull. 33, 333–336. doi: 10.1016/0361-9230(94)90202-X
Gelisse, P., Genton, P., Raybaud, C., Thiry, A., and Pincemaille, O. (1999). Benign childhood epilepsy with centrotemporal spikes and hippocampal atrophy. Epilepsia 40, 1312–1315. doi: 10.1111/j.1528-1157.1999.tb00864.x
Germanò, E., Gagliano, A., Magazu, A., Sferro, C., Calarese, T., Mannarino, E., et al. (2005). Benign childhood epilepsy with occipital paroxysms: neuropsychological findings. Epilepsy. Res. 64, 137-−150. doi: 10.1016/j.eplepsyres.2005.03.004
Giaume, C., and McCarthy, K. D. (1996). Control of gap-junctional communication in astrocytic networks. Trends. Neurosci. 19, 319–325. doi: 10.1016/0166-2236(96)10046-1
Gkampeta, A., Fidani, L., Clarimon, J., Kalinderi, K., Katopodi, T., Zafeiriou, D., et al. (2014). Association of brain-derived neurotrophic factor (BDNF) and elongator protein complex 4 (ELP4) polymorphisms with benign epilepsy with centrotemporal spikes in a Greek population. Epilepsy. Res. 108, 1734–1739. doi: 10.1016/j.eplepsyres.2014.09.005
Gkampeta, A., Fidani, L., Zafeiriou, D., and Pavlou, E. (2015). Benign epilepsy with centrotemporal spikes: relationship between type of seizures and response to medication in a Greek population. J. Neurosci. Rural. Pract. 6, 545–548. doi: 10.4103/0976-3147.165420
Glauser, T., Ben-Menachem, E., Bourgeois, B., Cnaan, A., Guerreiro, C., Kalviainen, R., et al. (2013). Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 54, 551–563. doi: 10.1111/epi.12074
Grosso, S., Orrico, A., Galli, L., Di Bartolo, R., Sorrentino, V., and Balestri, P. (2007). SCN1A mutation associated with atypical Panayiotopoulos syndrome. Neurology 69, 609–611. doi: 10.1212/01.wnl.0000266666.10404.53
Gross-Selbeck, G. (1995). Treatment of “benign” partial epilepsies of childhood, including atypical forms. Neuropediatrics 26, 45–50.
Hanaya, R., Sasa, M., Ujihara, H., Ishihara, K., Serikawa, T., Iida, K., et al. (1998). Suppression by topiramate of epileptiform burst discharges in hippocampal CA3 neurons of spontaneously epileptic rat in vitro. Brain Res. 789, 274–282. doi: 10.1016/S0006-8993(98)00116-4
Heron, S. E., Scheffer, I. E., Berkovic, S. F., Dibbens, L. M., and Mulley, J.C. (2007). Channelopathies in idiopathic epilepsy. Neurotherapeutics 4, 295–304. doi: 10.1016/j.nurt.2007.01.009
Heron, S. E., Smith, K. R., Bahlo, M., Nobili, L., Kahana, E., Licchetta, L., et al. (2012). Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 44, 1188–1190. doi: 10.1038/ng.2440
Hildebrand, M. S., Tankard, R., Gazina, E. V., Damiano, J. A., Lawrence, K. M., Dahl, H. H., et al. (2015). PRIMA1 mutation: a new cause of nocturnal frontal lobe epilepsy. Ann. Clin. Transl. Neurol. 2, 821–830. doi: 10.1002/acn3.224
Huang, X., Zhang, H., Yang, J., Wu, J., McMahon, J., Lin, Y., et al. (2010). Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol. Dis. 40, 193–199. doi: 10.1016/j.nbd.2010.05.024
Hughes, J. R. (2010). Benign epilepsy of childhood with centrotemporal spikes (BECTS): to treat or not to treat, that is the question. Epilepsy. Behav. 19, 197–203. doi: 10.1016/j.yebeh.2010.07.018
Ishida, S., Picard, F., Rudolf, G., No,é, E., Achaz, G., Thomas, P., et al. (2013). Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat. Genet. 45, 552–555. doi: 10.1038/ng.2601
Jalihal, V., Shankar, R., Henley, W., Parrett, M., Tittensor, P., McLean, B. N., et al. (2018). Eslicarbazepine acetate as a replacement for levetiracetam in people with epilepsy developing behavioral adverse events. Epilepsy Behav. 80, 365–369. doi: 10.1016/j.yebeh.2018.01.020
Jamal-Omidi, S., Collins, C., Fulchiero, E., Liu, H., Colon-Zimmermann, K., Fuentes-Casiano, E., et al. (2018). Assessing depression severity with a self-rated vs. rater-administered instrument in patients with epilepsy. Epilepsy Behav. 85, 52–57. doi: 10.1016/j.yebeh.2018.05.018
Jennum, P., Sabers, A., Christensen, J., Ibsen, R., and Kjellberg, J. (2017). Welfare consequences for people with epilepsy and their partners: a matched nationwide study in Denmark. Seizure 49, 17–24. doi: 10.1016/j.seizure.2017.05.007
Jones, B. E. (2008). Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann. N. Y. Acad. Sci. 1129, 26–34. doi: 10.1196/annals.1417.026
Kaila, K., Ruusuvuori, E., Seja, P., Voipio, J., and Puskarjov, M. (2014). GABA actions and ionic plasticity in epilepsy. Curr. Opin. Neurobiol. 26, 34–41. doi: 10.1016/j.conb.2013.11.004
Kanemura, H., Sano, F., Ohyama, T., Aoyagi, K., Sugita, K., and Aihara, M. (2015). Sequential prefrontal lobe volume changes and cognitive dysfunctions in children with Panayiotopoulos syndrome presenting with status epilepticus. Epilepsy. Res. 112, 122–129. doi: 10.1016/j.eplepsyres.2015.02.019
Kang, H. C., Eun, B. L., Wu Lee, C., Ku Moon, H., Kim, J. S., Wook Kim, D., et al. (2007). The effects on cognitive function and behavioral problems of topiramate compared to carbamazepine as monotherapy for children with benign rolandic epilepsy. Epilepsia 48, 1716–1723. doi: 10.1111/j.1528-1167.2007.01160.x
Kavros, P. M., Clarke, T., Strug, L. J., Halperin, J. M., Dorta, N. J., and Pal, D. K. (2008). Attention impairment in rolandic epilepsy: systematic review. Epilepsia. 49, 1570–1580. doi: 10.1111/j.1528-1167.2008.01610.x
Kay, H. Y., Greene, D. L., Kang, S., Kosenko, A., and Hoshi, N. (2015). M-current preservation contributes to anticonvulsant effects of valproic acid. J. Clin. Invest. 125, 3904–3914. doi: 10.1172/JCI79727
Kim, S. E., Lee, J. H., Chung, H. K., Lim, S. M., and Lee, H. W. (2014). Alterations in white matter microstructures and cognitive dysfunctions in benign childhood epilepsy with centrotemporal spikes. Eur.J.Neurol. 21, 708–717. doi: 10.1111/ene.12301
Kokkinos, V., Koutroumanidis, M., Tsatsou, K., Koupparis, A., Tsiptsios, D., and Panayiotopoulos, C. P. (2010). Multifocal spatiotemporal distribution of interictal spikes in Panayiotopoulos syndrome. Clin. Neurophysiol. 121, 859–869. doi: 10.1016/j.clinph.2010.01.019
Konishi, T., Naganuma, Y., Hongo, K., Murakami, M., Yamatani, M., and Okada, T. (1993). Carbamazepine-induced skin rash in children with epilepsy. Eur. J. Pediatr. 152, 605–608. doi: 10.1007/BF01954091
Korenke, G. C., Eggert, M., Thiele, H., Nürnberg, P., Sander, T., and Steinlein, O. K. (2016), Nocturnal frontal lobe epilepsy caused by a mutation in the GATOR1 complex gene NPRL3. Epilepsia 57, e60–e63. doi: 10.1111/epi.13307
Kossoff, E. H., Bergey, G. K., Freeman, J. M., and Vining, E. P. (2001). Levetiracetam psychosis in children with epilepsy. Epilepsia 42, 1611–1613. doi: 10.1046/j.1528-1157.2001.32101.x
Kossoff, E. H., Los, J. G., and Boatman, D. F. (2007). A pilot study transitioning children onto levetiracetam monotherapy to improve language dysfunction associated with benign rolandic epilepsy. Epilepsy Behav. 11, 514–517. doi: 10.1016/j.yebeh.2007.07.011
Kothare, S. V., and Trevathan, E. (2018). Sudden death risk among children with epilepsy. Neurology 91, 57–58. doi: 10.1212/WNL.0000000000005763
Kumar, J., Hapidin, H., Bee, Y. T. G., and Ismail, Z. (2013). Effects of the mGluR5 antagonist MPEP on ethanol withdrawal induced anxiety-like syndrome in rats. Behav. Brain. Funct. 9:43. doi: 10.1186/1744-9081-9-43
Kumar, J., Hapidin, H., Bee, Y. T. G., and Ismail, Z. (2016). Effects of acute ethanol administration on ethanol withdrawal induced anxiety-like syndrome in rats: a biochemical study. Alcohol 50, 9–17. doi: 10.1016/j.alcohol.2015.10.001
Kumar, J., Ismail, Z., Hatta, N. H., Baharuddin, N., Hapidin, H., Get, B.ee, et al. (2017). Alcohol Addiction- Metabotropic Glutamate Receptor Subtype 5 And Its Ligands: How They All Come Together Curr. Drug.Targets 19, 907–915. doi: 10.2174/1389450118666170511144302
Lacroix, L., Fluss, J., Gervaix, A., and Korff, C. M. (2011). Benzodiazepines in the acute management of seizures with autonomic manifestations: anticipate complications! Epilepsia 52, e156–e159. doi: 10.1111/j.1528-1167.2011.03201.x
Lal, D., Reinthaler, E. M., Altmüller, J., Toliat, M. R., Thiele, H., Nürnberg, P., Sander, T., et al. (2013). RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS ONE 8:e73323. doi: 10.1371/journal.pone.0073323
Lal, D., Reinthaler, E. M., Schubert, J., Muhle, H., Riesch, E., Kluger, G., Hahn, A., et al. (2014). DEPDC5 mutations in genetic focal epilepsies of childhood. Ann. Neurol. 75, 788–792. doi: 10.1002/ana.24127
Lambert, I., Roehri, N., Giusiano, B., Carron, R., Wendling, F., Benar, C., et al. (2018). Brain regions and epileptogenicity influence epileptic interictal spike production and propagation during NREM sleep in comparison with wakefulness. Epilepsia 59, 235–243. doi: 10.1111/epi.13958
Leach, N. T., Sun, Y., Michaud, S., Zheng, Y., Ligon, K. L., Ligon, A. H., et al. (2007). Disruption of diacylglycerol kinase delta (DGKD) associated with seizures in humans and mice. Am. J. Hum. Genet. 80, 792–799. doi: 10.1086/513019
Leal, A. J., Ferreira, J. C., Dias, A. I., and Calado, E. (2008). Origin of frontal lobe spikes in the early onset benign occipital lobe epilepsy (Panayiotopoulos syndrome). Clin. Neurophysiol. 119, 1985–1991. doi: 10.1016/j.clinph.2008.04.299
Lemke, J. R., Lal, D., Reinthaler, E. M., Steiner, I., Nothnagel, M., Alber, M., et al. (2013). Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 45, 1067–1072. doi: 10.1038/ng.2728
Leonardi, E., Dazzo, E., Aspromonte, M. C., Tabaro, F., Pascarelli, S., Tosatto, S. C., et al. (2018). CNTNAP2 mutations and autosomal dominant epilepsy with auditory features. Epilepsy Res. 139, 51–53. doi: 10.1016/j.eplepsyres.2017.11.006
Leppik, I. E. (2001). The place of levetiracetam in the treatment of epilepsy. Epilepsia 42, 44-−45. doi: 10.1046/j.1528-1157.2001.0420s4044.x
Li, R., Ji, G. J., Yu, Y., Yu, Y., Ding, M. P., and Tang, Y. L. (2017). Epileptic discharge related functional connectivity within and between networks in benign epilepsy with centrotemporal spikes. Int. J. Neural. Syst. 27:1750018. doi: 10.1142/S0129065717500186
Liguori, C., Romigi, A., Placidi, F., Sarpa, M. G., Mercuri, N. B., and Izzi, F. (2016). Effective treatment of nocturnal frontal lobe epilepsy with lacosamide: a report of two cases. Sleep. Med. 23, 121–122. doi: 10.1016/j.sleep.2015.09.028
Liu, C., Song, M., and Wang, J. (2016). Nightly oral administration of topiramate for benign childhood epilepsy with centrotemporal spikes. Childs. Nerv. Sys. 32, 839–843. doi: 10.1007/s00381-016-3043-0
Livingston, J. H., Cross, J. H., Mclellan, A., Birch, R., and Zuberi, S. M. (2009). A novel inherited mutation in the voltage sensor region of SCN1A is associated with Panayiotopoulos syndrome in siblings and generalized epilepsy with febrile seizures plus. J. Child. Neurol. 24, 503–508. doi: 10.1177/0883073808324537
Lopes, R., Simões, M. R., and Leal, A. J. (2014). Neuropsychological abnormalities in children with the Panayiotopoulos syndrome point to parietal lobe dysfunction. Epilepsy. Behav. 31, 50–55. doi: 10.1016/j.yebeh.2013.11.013
Losurdo, A., Proserpio, P., Cardinale, F., Gozzo, F., Tassi, L., Mai, R., et al. (2014). Drug-resistant focasleep related epilepsy: results and predictors of surgical outcome. Epilepsy Res. 108, 953–962. doi: 10.1016/j.eplepsyres.2014.02.016
Lynch, B. A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S. M., Matagne, A., et al. (2004). The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA. 101, 9861–9866. doi: 10.1073/pnas.0308208101
Ma, C. K., and Chan, K. Y. (2003). Benign childhood epilepsy with centrotemporal spikes: a study of 50 Chinese children. Brain. Dev. 25, 390–395. doi: 10.1016/S0387-7604(03)00003-2
MacDonald, R. L., and Rogawski, M. A. (2008). “Cellular effects of antiepileptic drugs,” in Epilepsy. A Comprehensive Textbook. eds J. Engel Jr., T. A. Pedley (Philadelphia: Lippincott Williams and Wilkins).
Magaudda, A., and Di Rosa, G. (2012). Carbamazepine-induced non-epileptic myoclonus and tic-like movements. Epileptic Disord. 14:172–173. doi: 10.1684/epd.2012.0504
Mahler, B., Carlsson, S., Andersson, T., and Tomson, T. (2018). Risk for injuries and accidents in epilepsy: a prospective population-based cohort study. Neurology 90, e779–e789 doi: 10.1212/WNL.0000000000005035
Manfredi, I., Zani, A. D., Rampoldi, L., Pegorini, S., Bernascone, I., Moretti, M., et al. (2009). Expression of mutant β2 nicotinic receptors during development is crucial for epileptogenesis. Hum. Mol. Genet. 18, 1075–1088. doi: 10.1093/hmg/ddp004
Marini, C., and Guerrini, R. (2007). The role of the nicotinic acetylcholine receptors in sleep-related epilepsy. Biochem Pharmacol. 74:1308–1314. doi: 10.1016/j.bcp.2007.06.030
Martín Del Valle, F., Díaz Negrillo, A., Ares Mateos, G., Sanz Santaeufemia, F. J., Del Rosal Rabes, T., and González-Valcárcel Sánchez-Puelles, F. J. (2011). Panayiotopoulos syndrome: probable genetic origin, but not in SCN1A. Eur. J. Paediatr. Neurol. 15, 155–157. doi: 10.1016/j.ejpn.2010.08.002
Martin, C., Meloche, C., Rioux, M. F., Nguyen, D. K., Carmant, L., Andermann, E., et al. (2014). A recurrent mutation in DEPDC5 predisposes to focal epilepsies in the French- Canadian population. Clin. Genet. 86, 570–574. doi: 10.1111/cge.12311
McCool, B. A., Pin, J. P., Harpold, M. M., Brust, P. F., Stauderman, K. A., and Lovinger, D.M. (1998). Rat group I metabotropic glutamate receptors inhibit neuronal Ca2+ channels via multiple signal transduction pathways in HEK 293 cells. J. Neurophysiol. 79, 379–391. doi: 10.1152/jn.1998.79.1.379
McDermott, C. M., LaHoste, G. J., Chen, C., Musto, A., Bazan, N. G., and Magee, J. C. (2003). Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J. Neurosci. 23, 9687–9695. doi: 10.1523/JNEUROSCI.23-29-09687.2003
McLean, M. J., and MacDonald, R. L. (1986). Carbamazepine and 10–11 epoxicarbamazepine produce use- and voltage-dependent limitation of rapidly firing action potentials of mouse central neurons in cell culture. J. Pharmacol. Exp. Ther. 238, 727–738.
Melis, M., Carta, S., Fattore, L., Tolu, S., Yasar, S., Goldberg, S. R., et al. (2010). Peroxisome proliferator-activated receptors-alpha modulate dopamine cell activity through nicotinic receptors. Biol. Psychiatry 68, 256–264. doi: 10.1016/j.biopsych.2010.04.016
Myers, K. A., and Scheffer, I. E. (2017). DEPDC5 as a potential therapeutic target for epilepsy. Expert. Opin.Ther. Targets 21, 591–600. doi: 10.1080/14728222.2017.1316715
Naganuma, Y., Konishi, T., Hongou, K., Murakami, M., Yamatani, M., Yagi, S., et al. (1994). Auditory event-related potentials in benign childhood epilepsy with centrotemporal spike: the effects of carbamazepine. Clin. Electroencephalogr. 25, 8–12. doi: 10.1177/155005949402500105
Neal, E. G., Chaffe, H. M., Schwartz, R. H., Lawson, M., Edwards, N., Fitzsimmons, G., et al. (2008). The ketogenic diet in the treatment of epilepsy in children: a randomised, controlled trial. Lancet.Neurol. 7, 500–506. doi: 10.1016/S1474-4422(08)70092-9
Neto, K.F, Noschang, R., and Nunes, M.L. (2016). The relationship between epilepsy, sleep disorders and attention deficit hyperactivity disorder (ADHD) in children: a review of literature. Sleep. Science. 9, 158–163. doi: 10.1016/j.slsci.2016.09.003
Neubauer, B. A., Fiedler, B., Himmelein, B., Kämpfer, F., Lässker, U., Schwabe, G., et al. (1998). Centrotemporal spikes in families with rolandic epilepsy: linkage to chromosome 15q14. Neurology 51, 1608–1612. doi: 10.1212/WNL.51.6.1608
Nezu, A., Kimura, S., Ohtsuki, N., and Tanaka, M. (1997). Transcranial magnetic stimulation in benign childhood epilepsy with centro-temporal spikes. Brain. Dev. 19, 134–137. doi: 10.1016/S0387-7604(96)00497-4
Ng, M., and Pavlova, M. (2013). Why are seizures rare in rapid eye movement sleep? review of the frequency of seizures in different sleep stages. Epilepsy Res. Treat. 2013:932790. doi: 10.1155/2013/932790
Nilsonne, G., Tamm, S., Schwarz, J., Almeida, R., Fischer, H., Kecklund, G., et al. (2017). Intrinsic brain connectivity after partial sleep deprivation in young and older adults: results from the Stockholm Sleepy Brain study. Sci. Rep. 7:9422. doi: 10.1038/s41598-017-09744-7
Nobili, L., Proserpio, P., Combi, R., Provini, F., Plazzi, G., Bisulli, F., et al. (2014). Nocturnal frontal lobe epilepsy. Curr. Neurol. Neurosci. Rep. 14:424. doi: 10.1007/s11910-013-0424-6
Ofer, I., Jacobs, J., Jaiser, N., Akin, B., Hennig, J., Schulze-Bonhage, A., et al. (2018). Cognitive and behavioral comorbidities in Rolandic epilepsy and their relation with default mode network's functional connectivity and organization. Epilepsy Behav. 78, 179–186. doi: 10.1016/j.yebeh.2017.10.013
Oka, E., Murakami, N., Ogino, T., Kobayashi, K., Ohmori, I., Akiyama, T., et al. (2004). Initiation of treatment and selection of antiepileptic drugs in childhood epilepsy. Epilepsia 45, 17–19. doi: 10.1111/j.0013-9580.2004.458004.x
Oldani, A., Manconi, M., Zucconi, M., Martinelli, C., Ferini-Strambi, L., et al. (2006). Topiramate treatment for nocturnal frontal lobe epilepsy. Seizure 15, 649–652. doi: 10.1016/j.seizure.2006.07.002
O'Neill, H. C., Laverty, D. C., Patzlaff, N. E., Cohen, B. N., Fonck, C., McKinney, S., et al. (2013). Mice expressing the ADNFLE valine 287 leucine mutation of the β2 nicotinic acetylcholine receptor subunit display increased sensitivity to acute nicotine administration and altered presynaptic nicotinic receptor function. Pharmacol. Biochem. Behav. 103, 603–621. doi: 10.1016/j.pbb.2012.10.014
Overvliet, G. M., Besseling, R. M., Jansen, J. F., van der Kruijs, S. J., Vles, J. S., Hofman, P. A., et al. (2013). Early onset of cortical thinning in children with rolandic epilepsy. Neuroimage. Clin. 2, 434–439. doi: 10.1016/j.nicl.2013.03.008
Pal, D. K., Li, W., Clarke, T., Lieberman, P., and Strug, L. J. (2010). Pleiotropic effects of the 11p13 locus on developmental verbal dyspraxia and EEG centrotemporal sharp waves. Genes. Brain. Behav. 9, 1004–1012. doi: 10.1111/j.1601-183X.2010.00648.x
Pan, P. Y., Cai, Q., Lin, L., Lu, P. H., Duan, S., and Sheng, Z. H. (2005). SNAP-29-mediated modulation of synaptic transmission in cultured hippocampal neurons. J. Biol. Chem. 280, 25769–25779. doi: 10.1074/jbc.M502356200
Panayiotopoulos, C. P. (2002). Panayiotopoulos Syndrome: A Common and Benign Childhood Epileptic Syndrome. London, United Kingdom: John Libbey & Company.
Panayiotopoulos, C. P. (2004). Autonomic seizures and autonomic status epilepticus peculiar to childhood: diagnosis and management. Epilepsy. Behav. 5, 286–295. doi: 10.1016/j.yebeh.2004.01.013
Panayiotopoulos, C. P. (ed.). (2005). “Benign childhood focal seizures and related epileptic syndromes,” in The Epilepsies: Seizures, Syndromes and Management (Bladon Medical Publishing), 223–269.
Panayiotopoulos, C. P. (2005). The Epilepsies: Seizures, Syndromes and Management. Oxfordshire (UK): Bladon Medical Publishing;. Chapter 14, Pharmacopoeia of Prophylactic Antiepileptic Drugs. Available Online at https://www.ncbi.nlm.nih.gov/books/NBK2597/
Panayiotopoulos, C. P., Michael, M., Sanders, S., Valeta, T., and Koutroumanidis, M. (2008). Benign childhood focal epilepsies: assessment of established and newly recognized syndromes, Brain 131, 2264–2286. doi: 10.1093/brain/awn162
Paoletti, P. (2011). Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 33, 1351–1365. doi: 10.1111/j.1460-9568.2011.07628.x
Pardoe, H. R., Berg, A. T., Archer, J. S., Fulbright, R. K., and Jackson, G. D. (2013). A neurodevelopmental basis for BECTS: evidence from structural MRI. Epilepsy Res. 105, 133–139. doi: 10.1016/j.eplepsyres.2012.11.008
Parker, W. E., Orlova, K. A., Parker, W. H., Birnbaum, J. F., Krymskaya, V. P., Goncharov, D. A., et al. (2013). Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci. Transl. Med. 5:182ra53. doi: 10.1126/scitranslmed.3005271
Pati, S., Edwards, L., Riley, K., and DeWolfe, J. (2018). Chronic ambulatory electrocorticography findings preceding sudden death in epilepsy. Neurol. Clin. Pract. 8, e1–e3. doi: 10.1212/CPJ.0000000000000450
Pavlova, M. K., Lee, J. W., Yilmaz, F., and Dworetzky, B. A. (2012). Diurnal pattern of seizures outside the hospital: is there a time of circadian vulnerability?. Neurology 78, 1488–1492. doi: 10.1212/WNL.0b013e3182553c23
Pfeifer, H. H., and Thiele, E. A. (2005). Low-glycemic-index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 65, 1810–1812. doi: 10.1212/01.wnl.0000187071.24292.9e
Phelan, P., Regan, C., Kitty, C., and Dunne, A. (1985). Sodium valproate stimulates the pa*iculate form of glutamine synthetase in rat brain. Neuropharmacology 24, 895–902.
Picard, F., Makrythanasis, P., Navarro, V., Ishida, S., de Bellescize, J., Ville, D., et al. (2014). DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 82, 2101–2106. doi: 10.1212/WNL.0000000000000488
Picard, F., and Scheffer, I. E. (2012). “Genetically determined focal epilepsies,”in Epileptic Syndromes in Infancy, Childhood and Adolescence, eds M. Bureau, P. Genton, C. Drave, et al. 5th Ed. (Montrouge: John Libbey Eurotext) 349–361.
Poorthuis, R. B., Bloem, B., Schak, B., Wester, J., de Kock, C. P., and Mansvelder, H. D. (2012). Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cerebr. Cortex 23, 148–161. doi: 10.1093/cercor/bhr390
Potegal, M., Drewel, E. H., and MacDonald, J. T. (2018). Tantrums, emotion reactions and their EEG correlates in childhood benign rolandic epilepsy vs. complex partial seizures: exploratory observations. Front. Behav. Neurosci. 12:40. doi: 10.3389/fnbeh.2018.00040
Provini, F., Plazzi, G., Montagna, P., and Lugaresi, E. (2000). The wide clinical spectrum of nocturnal frontal lobe epilepsy. Sleep.Med. Rev. 4, 375–386. doi: 10.1053/smrv.2000.0109
Provini, F., Plazzi, G., Tinuper, P., Vandi, S., Lugaresi, E., and Montagna, P. (1999). Nocturnal frontal lobe epilepsy: a clinical and polygraphic overview of 100 consecutive cases. Brain 122, 1017–1031. doi: 10.1093/brain/122.6.1017
Puligheddu, M., Melis, M., Pillolla, G., Milioli, G., Parrino, L., Terzano, G. M., et al. (2017). Rationale for an adjunctive therapy with fenofibrate in pharmacoresistant nocturnal frontal lobe epilepsy. Epilepsia 58, 1762–1770. doi: 10.1111/epi.13863
Puligheddu, M., Pillolla, G., Melis, M., Lecca, S., Marrosu, F., De Montis, M. G., et al. (2013). PPAR-alpha agonists as novel antiepileptic drugs: preclinical findings. PLoS ONE 8:e64541. doi: 10.1371/journal.pone.0064541
Raju, G. P., Sarco, D. P., Poduri, A., Riviello, J. J., Bergin, A. M., and Takeoka, M. (2007). Oxcarbazepine in children with nocturnal frontal-lobe epilepsy. Pediatr. Neurol. 37, 345–349. doi: 10.1016/j.pediatrneurol.2007.06.013
Ramasamy, S., Yu, F., Hong Yu, Y., Srivats, H., Dawe, G. S., and Ahmed, S. (2014). NogoR1 and PirB signaling stimulates neural stem cell survival and proliferation. Stem Cells 32, 1636–1648. doi: 10.1002/stem.1645
Razavi, B., and Fisher, R. S. (2017). Sleep and Epilepsy. In Sleep and Neurologic Disease Available online at: https://medicinainternaelsalvador.com/wp-content/uploads/2017/08/7.-Sleep-and-Epilepsy.pdf (Last accessed on 13.4.2018). 129–140
Reinthaler, E. M., Dejanovic, B., Lal, D., Semtner, M., Merkler, Y., Reinhold, A., et al. (2015). Rare variants in γ-aminobutyric acid type A receptor genes in rolandic epilepsy and related syndromes. Ann. Neurol. 77, 972–986. doi: 10.1002/ana.24395
Reinthaler, E. M., Lal, D., Jurkowski, W., Feucht, M., Steinböck, H., Gruber-Sedlmayr, U., et al. (2014). Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia 55, e89–e93. doi: 10.1111/epi.12712
Reyes, A., Holden, H. M., Chang, Y. H. A., Uttarwar, V. S., Sheppard, D. P., DeFord, N. E., et al. (2018). Impaired spatial pattern separation performance in temporal lobe epilepsy is associated with visuospatial memory deficits and hippocampal volume loss. Neuropsychologia 111, 209–215. doi: 10.1016/j.neuropsychologia.2018.02.009
Ricos, M. G., Hodgson, B. L., Pippucci, T., Saidin, A., Ong, Y. S., Heron, S. E., et al. (2016), Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann. Neurol. 79, 120–131. doi: 10.1002/ana.24547.
Rogawski, M. A., Tofighy, A., White, H. S., Matagne, A., and Wolff, C. (2015). Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res. 110, 189–205. doi: 10.1016/j.eplepsyres.2014.11.021
Roll, P., Rudolf, G., Pereira, S., Royer, B., Scheffer, I. E., Massacrier, A., et al. (2006). SRPX2 mutations in disorders of language cortex and cognition. Hum. Mol. Genet. 15, 1195–1207. doi: 10.1093/hmg/ddl035
Romigi, A., Marciani, M. G., Placidi, F., Pisani, L. R., Izzi, F., Torelli, F., et al. (2008). Oxcarbazepine in nocturnal frontal-lobe epilepsy: a further interesting report. Pediatr. Neurol. 39:298. doi: 10.1016/j.pediatrneurol.2008.07.015
Samaitiene, R., Norkuniene, J., Jurkevičiene, G., and Grikiniene, J. (2012). Behavioral problems in children with benign childhood epilepsy with centrotemporal spikes treated and untreated with antiepileptic drugs. Medicina (Kaunas). 48, 338–344.
Samarasekera, S. R., Berkovic, S. F., and Scheffer, I. E. (2018). A case series of lacosamide as adjunctive therapy in refractory sleep-related hypermotor epilepsy (previously nocturnal frontal lobe epilepsy). J.Sleep. Res. 27:e12669. doi: 10.1111/jsr.12669
Saper, C. B., Fuller, P. M., Pedersen, N. P., Lu, J., and Scammell, T. E. (2010). Sleep state switching. Neuron 68, 1023–1042. doi: 10.1016/j.neuron.2010.11.032
Sarco, D. P., Boyer, K., Lundy-Krigbaum, S. M., Takeoka, M., Jensen, F., Gregas, M., et al. (2011). Benign rolandic epileptiform discharges are associated with mood and behavior problems. Epilepsy Behav. 22, 298–303. doi: 10.1016/j.yebeh.2011.06.023
Schwarz, J. R., and Grigat, G. (1989). Phenytoin and carbamazepine: potential- and frequencydependent block of Na currents in mammalian myelinated nerve fibers. Epilepsia 30, 286–294. doi: 10.1111/j.1528-1157.1989.tb05300.x
Seigneur, J., and Timofeev, I. (2011). Synaptic impairment induced by paroxysmal ionic conditions in neocortex. Epilepsia 52, 132–139. doi: 10.1111/j.1528-1167.2010.02784.x
Sha, L. Z., Xing, X. L., Zhang, D., Yao, Y., Dou, W. C., Jin, L. R., et al. (2012). Mapping the spatio-temporal pattern of the mammalian target of rapamycin (mTOR) activa- tion in temporal lobe epilepsy. PLoS ONE 7:e39152. doi: 10.1371/journal.pone.0039152
Shaw, P., Greenstein, D., Lerch, J., Clasen, L., Lenroot, R., Gogtay, N., et al. (2006). Intellectual ability and cortical development in children and adolescents. Nature 440, 676–679. doi: 10.1038/nature04513
Shi, X., Chen, J., Lu, Q., He, F., Zhou, J., Zhou, S. Zou, L., et al. (2018). Whole-exome sequencing revealing de novo heterozygous variant of KCNT1 in a twin discordant for benign epilepsy with centrotemporal spikes. J. Paediatr. Child Health 54, 709–710. doi: 10.1111/jpc.13939
Sibarov, D. A., Bruneau, N., Antonov, S. M., Szepetowski, P., Burnashev, N., and Giniatullin, R. (2017). Functional properties of human NMDA receptors associated with epilepsy-related mutations of GluN2A subunit. Front. Cell. Neurosci. 11:155. doi: 10.3389/fncel.2017.00155
Sim, J. C., Scerri, T., Fanjul-Fernández, M., Riseley, J. R., Gillies, G., Pope, K., et al. (2016). Familial cortical dysplasia caused by mutation in the mTOR regulator NPRL3. Ann. Neurol. 79, 132–137. doi: 10.1002/ana.24502
Sitges, M., Chiu, L. M., Guarneros, A., and Nekrassov, V. (2007). Effects of carbamazepine, phenytoin, lamotrigine, oxcarbazepine, topiramate and vinpocetine on Na+ channel-mediated release of [3H] glutamate in hippocampal nerve endings. Neuropharmacology 52, 854–862. doi: 10.1016/j.neuropharm.2007.08.016
Sizemore, G., Lucke-Wold, B., Rosen, C., Simpkins, J. W., Bhatia, S., and Sun, D. (2018). Temporal lobe epilepsy, stroke, and traumatic brain injury: mechanisms of hyperpolarized, depolarized, and flow-through ion channels utilized as tri-coordinate biomarkers of electrophysiologic dysfunction. OBM Neurobiol. 2:009. doi: 10.21926/obm.neurobiol.1802009
Sosunov, A. A., Wu, X., McGovern, R. A., Coughlin, D. G., Mikell, C. B., Goodman, R.R., et al. (2012). The mTOR pathway is activated in glial cells in mesial temporal Sclerosis. Epilepsia 53, 78–86. doi: 10.1111/j.1528-1167.2012.03478.x
Specchio, N., Trivisano, M., Di Ciommo, V., et al. (2010). Panayiotopoulos syndrome:a clinical, EEG, and neuropsychological study of 93 consecutive patients. Epilepsia 51, 2098–2107. doi: 10.1111/j.1528-1167.2010.02639.x
St Louis, E. K. (2011). Sleep and epilepsy: strange bedfellows no more. Minerva. Pneumol. 50, 159–176.
Steinlein, O. K., Mulley, J. C., Propping, P., Wallace, R. H., Phillips, H. A., Sutherland, G. R., et al. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 11, 201–203. doi: 10.1038/ng1095-201
Stephani, U. (2000). Typical semiology of benign childhood epilepsy with centrotemporal spikes (BCECTS). Epileptic. Disord. 2, S3–S4.
Steriade, M., McCormick, D. A., and Sejnowski, T. J. (1993).Thalamocortical oscillations in the sleeping and aroused brain. Science. 262, 679–685. doi: 10.1126/science.8235588
Strug, L. J., Clarke, T., Chiang, T., Chien, M., Baskurt, Z., Li, W., et al. (2009). Centrotemporal sharp wave EEG trait in rolandic epilepsy maps to Elongator Protein Complex 4 (ELP4). Eur. J. Hum. Genet. 17, 1171–1181. doi: 10.1038/ejhg.2008.267
Tamsett, T. J., Picchione, K. E., and Bhattacharjee, A. (2009). NAD+ activates KNa channels in dorsal root ganglion neurons. J. Neurosci. 29, 5127–5134. doi: 10.1523/JNEUROSCI.0859-09.2009
Tanimukai, H., Nishimura, T., and Sano, I. (1964). N-(4'sulfamylphenyl)-butansultam (1–4) (Ospolot) als Carboanhydratase-Inhibitor im Gehirn. Klin. Wochenschr. 42, 918–920.
Tchopev, Z. N., Yeh, P. H., Morgan, G. W., Meyer, E., Wolf, J. M., Ollinger, J. M., et al. (2018). Acquired sleep-related hypermotor epilepsy with disrupted white matter tracts assessed by multishell diffusion magnetic resonance imaging. Front. Neurol. 9:6. doi: 10.3389/fneur.2018.00006
Ten Donkelaar, H. J., and Horim, A. (2011). “The hypothalamus and hypothalamohypophysial systems,” in Clinical Neuroanatomy, Brain Circuitry and Its Disorders, ed H. J. ten Donkelaar ed. (Berlin: Springer-Verlag). doi: 10.1007/978-3-642-19134-3_13
Thimm, J., Mechler, A., Lin, H., Rhee, S., and Lal, R. (2005). Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. J. Biol. Chem. 280, 10646–10654. doi: 10.1074/jbc.M412749200
Timofeev, I., Bazhenov, M., Seigneur, J., and Sejnowski, T. (2012). “Neuronal synchronization and thalamocortical rhythms in sleep, wake and epilepsy,”in Source: Jasper's Basic Mechanisms of the Epilepsies [Internet]. eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, A. V. Delgado-Escueta 4th Ed. (Bethesda, MD: National Center for Biotechnology Information (US)). doi: 10.1093/med/9780199746545.003.0013
Tinuper, P., and Bisulli, F. (2017). From nocturnal frontal lobe epilepsy to sleep-related hypermotor epilepsy: a 35-year diagnostic challenge. Seizure 44, 87–92. doi: 10.1016/j.seizure.2016.11.023
Tinuper, P., Bisulli, F., Cross, J. H., Hesdorffer, D., Kahane, P., Nobili, L., et al. (2016). Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology 86, 1210–1212. doi: 10.1212/WNL.0000000000002666
Trinka, E., Kwan, P., Lee, B., and Dash, A. (2018). Epilepsy in Asia: Disease burden, management barriers, and challenges. Epilepsia. doi: 10.1111/epi.14458
Tsai, M. L., Lo, H. Y., and Chaou, W. T. (2001). Clinical and electroencephalographic findings in early and late onset benign childhood epilepsy with occipital paroxysms. Brain Dev. 23, 401–405. doi: 10.1016/S0387-7604(01)00246-7
Tumiene, B., Peterlin, B., Maver, A., and Utkus, A. (2018). Contemporary scope of inborn errors of metabolism involving epilepsy or seizures. Metabolic Brain Dis. [Epub ahead of print].doi: 10.1007/s11011-018-0288-1
Tzitiridou, M., Panou, T., Ramantani, G., Kambas, A., Spyroglou, K., and Panteliadis, C. (2005). Oxcarbazepine monotherapy in benign childhood epilepsy with centrotemporal spikes: a clinical and cognitive evaluation. Epilepsy. Behav. 7, 458–467. doi: 10.1016/j.yebeh.2005.07.012
Unterberger, I., Gabelia, D., Prieschl, M., Chea, K., Hofer, M., Högl, B., et al. (2015). Sleep disorders and circadian rhythm in epilepsy revisited: a prospective controlled study. Sleep. Med. 16, 237–242. doi: 10.1016/j.sleep.2014.09.021
Vears, D. F., Tsai, M. H., Sadleir, L. G., Grinton, B. E., Lillywhite, L. M., Carney, P. W., et al. (2012). Clinical genetic studies in benign childhood epilepsy with centrotemporal spikes. Epilepsia 53, 319–324. doi: 10.1111/j.1528-1167.2011.03368.x
Verrotti, A., Coppola, G., Manco, R., Ciambra, G., Iannetti, P., Grosso, S., et al. (2007). Levetiracetam monotherapy for children and adolescents with benign rolandic seizures. Seizure 16, 271–275. doi: 10.1016/j.seizure.2006.12.006
Verrotti, A., Salladini, C., Trotta, D., di Corcia, G., and Chiarelli, F. (2005). Ictal cardiorespiratory arrest in Panayiotopoulos sindrome. Neurology 64, 1816–1817. doi: 10.1212/01.WNL.0000162027.49577.1C
Vignatelli, L., Bisulli, F., Giovannini, G., Licchetta, L., Naldi, I., Mostacci, B., et al. (2017). Prevalence of sleep-related hypermotor epilepsy-formerly named nocturnal frontal lobe epilepsy-in the adult population of the Emilia-Romagna Region, Italy. Sleep 40:zsw041. doi: 10.1093/sleep/zsw041
Vogl, C., Mochida, S., Wolff, C., Whalley, B. J., and Stephens, G. J. (2012). The SV2A Ligand Levetiracetam Inhibits Presynaptic Ca2+ Channels Via an Intracellular Pathway. Molecular pharmacology, mol-111. doi: 10.1124/mol.111.076687
Von Stülpnagel, C., Ensslen, M., Møller, R. S., Pal, D. K., Masnada, S., Veggiotti, P., et al. (2017). Epilepsy in patients with GRIN2A alterations: Genetics, neurodevelopment, epileptic phenotype and response to anticonvulsive drugs. Eur. J.Paediatr. Neurol. 21, 530–541. doi: 10.1016/j.ejpn.2017.01.001
Wallace, T. L., and Bertrand, D. (2013). Importance of the nicotinic acetylcholine receptor system in the prefrontal cortex. Biochem. pharmacol. 85, 1713–1720. doi: 10.1016/j.bcp.2013.04.001
Weir, E., Gibbs, J., and Appleton, R. (2018). Panayiotopoulos syndrome and benign partial epilepsy with centro-temporal spikes: a comparative incidence study. Seizure 57, 66–69. doi: 10.1016/j.seizure.2018.03.002
Wheless, J. W., Clarke, D. F., Arzimanoglou, A., and Carpenter, D. (2007). Treatment of pediatric epilepsy: european expert opinion. Epileptic Disord. 9:353–412. doi: 10.1684/epd.2007.0144
Wheless, J. W., Clarke, D. F, and Carpenter, D. (2005). Treatment of pediatric epilepsy: expert opinion. J. Child. Neurol. 20, S1–S56. doi: 10.1177/088307380502000101
Wibecan, L., Fink, G., Tshering, L., Bruno, V., Patenaude, B., Nirola, D. K., et al. (2018). Bhutan Epilepsy Project. The economic burden of epilepsy in Bhutan. Trop Med. Int. Health 23, 342–358. doi: 10.1111/tmi.13035
Wirrell, E., Sherman, E. M., Vanmastrigt, R., and Hamiwka, L. (2008). Deterioration in cognitive function with benign epilepsy of childhood with central temporal spikes treated with sulthiame. J. Child. Neurol. 23, 14–21. doi: 10.1177/0883073807307082
Xi, D., Keeler, B., Zhang, W., Houle, J. D., and Gao, W. J. (2009). NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J. Neurosci. Methods 176, 172–118. doi: 10.1016/j.jneumeth.2008.09.013
Xiao, F., An, D., Deng, H., Chen, S., Ren, J., and Zhou, D. (2014). Evaluation of levetiracetam and valproic acid as low-dose monotherapies for children with typical benign childhood epilepsy with centrotemporal spikes (BECTS). Seizure 23,756–761. doi: 10.1016/j.seizure.2014.06.006
Yalçin, A. D., and Toydemir, H. E. (2017). Panayiotopoulos Syndrome with a special emphasis of ictal semiologic features. Epilepsy Res. 131, 37–43. doi: 10.1016/j.eplepsyres.2017.02.006
Yamamoto, H., Naito, H., Osako, T., Tsukahara, K., Yamada, T., Yumoto, T., et al. (2018). Ictal cardiorespiratory arrest associated with status epilepticus in panayiotopoulos syndrome. Acta Med. Okayama 72, 297–300. doi: 10.18926/AMO/56076
Yang, Z. X., Liu, X. Y., Qin, J., Zhang, Y. H., Bao, X. H., Chang, X. Z., et al. (2008). Clinical and electrophysiologic studies on epileptic negative myoclonus in atypical benign partial epilepsy of childhood. Zhonghua. Er. Ke. Za. Zhi. 46, 885–890.
Yeh, S. B., and Schenck, C. H. (2014). Sporadic nocturnal frontal lobe epilepsy: a consecutive series of 8 cases. Sleep Sci. 7, 170–177. doi: 10.1016/j.slsci.2014.09.016
Yeh, S. B., and Schenck, C. H. (2017). Sporadic nocturnal frontal lobe epilepsy report on two cases and review of the first taiwanese series of 10 cases. Acta Neurol.Taiwan 26, 55–63.
Yoshinaga, H., Kobayashi, K., and Ohtsuka, Y. (2010). Characteristics of the synchronous occipital and frontopolar spike phenomenon in Panayiotopoulos syndrome. Brain. Dev. 32, 603–608. doi: 10.1016/j.braindev.2009.09.007
Zhang, X., Velumian, A. A., Jones, O. T., and Carlen, P. L. (1998). Topiramate reduces high-voltage activated Ca:“ currents in CAI pyramidal neurons m rllro. Epilepsia 39(Suppl. 6), 44.
Zhao, Y., Li, X., Zhang, K., Tong, T., and Cui, R. (2018). The progress of epilepsy after stroke. Curr. Neuropharmacol. 16, 71–78. doi: 10.2174/1570159X15666170613083253
Zheng, T., Clarke, A. L., Morris, M. J., Reid, C.A, Petrou, S., and O'Brien, T.J. (2009). Oxcarbazepine, not its active metabolite, potentiates GABAA activation and aggravates absence seizures. Epilepsia 50, 83–87. doi: 10.1111/j.1528-1167.2008.01759.x
Keywords: epilepsy, sleep, seizure, SHE, BECTS, PS, rolandic, panayiotopoulos
Citation: Kumar J, Solaiman A, Mahakkanukrauh P, Mohamed R and Das S (2018) Sleep Related Epilepsy and Pharmacotherapy: An Insight. Front. Pharmacol. 9:1088. doi: 10.3389/fphar.2018.01088
Received: 16 April 2018; Accepted: 07 September 2018;
Published: 27 September 2018.
Edited by:
Ayanabha Chakraborti, University of Alabama at Birmingham, United StatesReviewed by:
Nasiara Karim, University of Malakand, PakistanMuthuraju Sangu, Universiti Sains Malaysia Health Campus, Malaysia
Copyright © 2018 Kumar, Solaiman, Mahakkanukrauh, Mohamed and Das. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Srijit Das, ZHJzcmlqaXRAZ21haWwuY29t