 Hu Li1
Hu Li1 Jian-Rui Li1
Jian-Rui Li1 Meng-Hao Huang1Jin-Hua Chen1Xiao-Qin Lv1Li-Li Zou1Jia-Li Tan1Biao Dong1
Meng-Hao Huang1Jin-Hua Chen1Xiao-Qin Lv1Li-Li Zou1Jia-Li Tan1Biao Dong1 Zong-Gen Peng1,2*Jian-Dong Jiang1,2,3*
Zong-Gen Peng1,2*Jian-Dong Jiang1,2,3*- 1Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 2Key Laboratory of Biotechnology of Antibiotics, The National Health and Family Planning Commission (NHFPC), Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 3State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
Treatment with direct-acting antivirals (DAAs) cures most patients infected with hepatitis C virus (HCV) in the real world. However, some patients, especially those with the underlying advanced liver disease, have a limited reduction of liver injury after achieving a sustained viral response (SVR). Bicyclol was widely used in clinics for the treatment of a variety of liver injuries but with an unknown mechanism for the treatment of hepatitis C. We investigated the anti-inflammatory effects and mechanisms of bicyclol in HCV-infected hepatocytes and further confirmed the putative results in a mouse hepatitis model induced by the coinjection of polyinosinic-polycytidylic acid [poly (I:C)] and D-galactosamine (D-GalN). The results showed that the activation of nuclear factor kappa B (NF-κB) and the subsequent increase of inflammatory factors were directly induced by HCV infection and were persistent after clearance of the virus in Huh7.5 cells. Bicyclol decreased the activation of NF-κB and the levels of inflammatory factors in HCV-infected hepatocytes by inhibiting the activation of the ROS-MAPK-NF-κB pathway, and the effect was synergistic with DAAs in HCV-infected hepatocytes. Bicyclol attenuated the ROS-MAPK-NF-κB axis via recovering mitochondrial function without a dependence on dihydronicotinamide adenine dinucleotide phosphate oxidase and superoxide dismutases. The anti-inflammatory effects and mechanism of bicyclol were verified in mouse hepatitis induced by the coinjection of poly(I:C)/D-GalN. Bicyclol directly ameliorates the chronic inflammation caused by HCV infection and might be used with DAAs or after DAA therapy for ultimately curing chronic hepatitis C.
Introduction
Hepatitis C, caused by infection of hepatitis C virus (HCV), is a worldwide prevailing disease. To date, direct-acting antivirals (DAAs) have substantially improved the sustained viral response (SVR) to above 90% (Jacobson et al., 2017). However, antiviral therapy does not drastically eliminate the liver disease progression, as evidenced by persistent progressive cirrhosis, liver failure, and HCC in a subset of patients who achieved an SVR (Conti et al., 2016; Reig et al., 2016; Jacobson et al., 2017; D’Ambrosio et al., 2018). A most recent cohort of prospective study followed up for 10 years even showed that post-SVR cirrhosis regression does not prevent HCC occurrence (D’Ambrosio et al., 2018). Actually, hepatitis C is associated with inflammation, oxidative stress, and metabolic disorders triggered by HCV infection, which creates the pro-oncogenic microenvironment and results in fibrogenesis, cirrhosis, and HCC (Koike and Miyoshi, 2006; Morgan et al., 2010; Lin et al., 2015; D’Ambrosio et al., 2018). Though early appropriate inflammatory responses are partially beneficial for protecting against pathogens, persistent and uncontrolled inflammatory responses have long-term and repeated malignant follow-up effects in chronic hepatitis C (Nakamoto and Kaneko, 2003; Koike and Miyoshi, 2006; Castello et al., 2010). Therefore, beyond merely achieving an SVR, the prevention of subsequent excessive inflammation, concurrently with other therapeutic regimens, is also imperative for ultimately eliminating hepatitis C (Koike and Miyoshi, 2006; Castello et al., 2010).
Bicyclol (4,4′-dimethoxy-5,6,5′,6′-bis (methylenedioxy)-2-hydroxymethyl-2′-methoxycarbonyl biphenyl) is an approved drug in China, which is used as a hepatoprotective and anti-inflammatory agent (Liu, 2009). Accumulating clinical evidence shows its substantial effects in various liver injuries, including viral hepatitis (Yao et al., 2002; Liu, 2009), with in vitro mechanisms related to multiple possible molecules, such as the Toll-like receptor, inosine 5′-monophosphate dehydrogenase II, heat shock protein 70/27, and heme oxygenase (Bao and Liu, 2009; Zhang et al., 2014, 2016). The anti-HCV activity of long-term use of bicyclol was also reported (Liu, 2009). However, unlike the single pathogen-associated molecular patterns or damage-associated molecular pattern signal, HCV infection-mediated inflammatory cytokine and chemokine storms in vitro and in vivo are acute and sophisticated, and there is still no research about the role and mechanism of bicyclol in hepatitis C. Here we analyzed the effects and mechanisms of bicyclol in HCV-infected hepatocytes and in mouse hepatitis induced by the coinjection of polyinosinic-polycytidylic acid [poly (I:C)] and D-galactosamine (D-GalN), which mimic the vigorous inflammatory state and activation of intracellular signaling pathways during HCV infection. We revealed that bicyclol attenuates liver inflammation induced by HCV infection via repressing the ROS-mediated activation of the MAPK/NF-κB signaling pathway.
Materials and Methods
Cells and Virus
The hepatocyte Huh7.5 cells and the plasmid pFL-J6/JFH/JC1, containing a full-length chimeric HCV cDNA, were kindly provided by the Vertex Pharmaceuticals Inc. (Boston, USA). The primary human hepatocytes (PHHs) were from the ScienCell Research Laboratories (San Diego, CA, USA). The HCV virus stock was prepared as previously described (Peng et al., 2011). For preparing the virus-free supernatants, the HCV stock was layered onto a 20% sucrose cushion and was ultracentrifuged at 250,000 g for 4 h at 4°C; the virus-free supernatants and DMEM-resuspended virus pellets under the cushion were collected. For preparing UV-inactivated HCV, the HCV stocks were irradiated by UV light at a dose of 20 mj/cm2 for 20 min.
Agents
Bicyclol was from the Beijing Union Pharmaceutical Company (Beijing, China) with purity over 99%. Anti-HCV positive drugs, such as sofosbuvir, simeprevir, and daclatasvir, were from the MedChemExpress (Princeton, NJ, USA). BAY11-7082 (Beyotime Biotechnology, Jiangsu, China), diphenyliodonium (DPI) (Sigma, St. Louis, MO, USA), N-acetylcysteine (Beyotime Biotechnology), SB203580 (CST, Beverly, MA, USA), SP600125 (CST), and U0126 (CST) were used in the experiments.
Cytotoxicity Assay
Huh7.5 cells were treated with drugs or the solvent control for 12 or 72 h. The cell viability was detected with an MTT assay and was calculated as described before (Peng et al., 2010).
Western Blot Assay
Western blot was performed as previously described (Peng et al., 2010). Briefly, after SDS-PAGE and transmembrane, the target proteins were accordingly probed with antibodies against β-actin (CST, cat#3700s), CuZn-SOD (CST, cat#2770), Mn-SOD (CST, cat#13141), cytochrome c (CST, cat#11940), Nox1 (Boster Biological Technology, cat#BA3335), Nox4 (Abcam, cat#ab133303), HCV Core (Abcam, cat#ab2740), HCV NS3 (Abcam, cat#ab13830), and phospho-specific or total p38, ERK, JNK, or NF-κB p65 (CST, cat#9910, #9926, and #3033). After an incubation with the corresponding HRP-conjugated secondary antibody, the signal of the target proteins was detected using the ChemiDo XRS gel imager system (Bio-Rad), with an enhanced chemiluminescence (ECL) kit (GE Healthcare Life Sciences, Pittsburgh, PA, USA) and was scanned with the Gelpro32 software. The ratio of the protein of interest to the internal control protein Actin was calculated and normalized as 1.00 for the control group.
Real-Time Quantitative Reverse Transcript PCR
HCV RNA was quantified with real-time quantitative reverse transcript PCR (qRT-PCR), and the result was calibrated with the internal control gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as previously reported (Peng et al., 2010). The mRNA of TNF-α, IL-6, and MIP-1β was amplified with specific primers (Table 1). Briefly, total RNA (1 μg) isolated from the cells or tissues was reverse transcribed into cDNA using the GoScript Reverse Transcription System (Promega) and was quantified with the GoTaq qPCR Master Mix (Promega) according to the manufacturer’s protocol. The relative mRNA amounts were calculated by the comparative Ct method after normalizing against the amount of GAPDH mRNA.

Table 1. The primers used in qRT-PCR.
Luciferase Reporter Assay
Huh7.5 cells were cotransfected with the plasmid pNF-κB-Luc (Promega), expressing firefly luciferase, and the pRL-SV40 vector (Promega), expressing Renilla luciferase, in a 10:1 mass ratio using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). After 24 h of transfection, the cells were pretreated with different inhibitors for 12 h and were then treated with or without 200 μM of H2O2 for another 2 h. The luciferase activity of the cell lysate was determined with a dual-luciferase reporter assay kit (Beyotime Biotechnology) according to the manufacturer’s protocol. The relative luciferase activities were calculated by the ratio of the intensity of firefly luminescence to the intensity of the reference Renilla luminescence.
Detection of Reactive Oxygen Species in the Cells and Liver Tissues
The treated cells were incubated with 5 μM dihydroethidium (DHE) (Beyotime Biotechnology) for 30 min at 37°C. After being washed, the cells were resuspended in PBS. The superoxide level was measured by a flow cytometer (Sanchez-Reus et al., 2005). The ROS assay in the liver tissues was performed as previously described (Wang et al., 2014). After fixing and then dehydrating at 4°C, the tissues were embedded using optimal cutting temperature compound (OCT) and were cut into sections of 5 μm thickness. The sections were washed and then incubated with DHE (50 μM) for 60 min followed by washing with PBS three times and were then mounted with an antifade mountant (Invitrogen). The red fluorescence signal was acquired with a fluorescence microscope, and the ROS accumulation was measured and statistically analyzed in three random vision fields by Image-Pro plus 6.0.
Animal Experiments
The male C57BL/6 mice (20.0 ± 2.0 g) were from the Beijing HuaFuKang Biological Technology Co. Ltd. The mice were randomly divided into five groups, with six mice in each group and were daily administered intragastrically with bicyclol (75, 150, and 300 mg/kg) dissolved in 0.5% carboxymethyl cellulose sodium (CMC-Na) or solvent control for 7 days. The mice were injected intravenously with polyinosinic-polycytidylic acid sodium (poly(I:C), InvivoGen) dissolved in pyrogen-free saline at a dose of 75 μg/kg and were simultaneously intraperitoneally injected with D-galactosamine (D-GalN, Sigma) at a dose of 500 mg/kg. The control group was injected intravenously and intraperitoneally with pyrogen-free saline. Sixteen hours after the injection, the mice were sacrificed, and the blood and liver tissues were collected. The blood samples were centrifuged, and the serum was collected. The liver tissues were stored at −80°C for total RNA and protein extraction or were embedded in 10% formaldehyde and 4% paraformaldehyde for hematoxylin-eosin (H&E) or DHE staining, respectively. Formalin-fixed and paraffin-embedded liver tissues were also used for the monocytes, macrophages and Kupffer cell marker CD68 immunohistochemical (IHC) staining. The distribution of ROS was detected through comparing the paraformaldehyde fixed liver tissue slice in white light, nucleus stained with DAPI, and ROS stained with DHE. Serum alanine transaminase (ALT) and aspartate transaminase (AST) were detected using ALT and AST assay kits (Nanjing Jiancheng Bioengineering Institute, China), respectively. Liver cytokine (TNF-α, IL-6, and MIP-1β) mRNA and protein levels were quantified with qRT-PCR and ELISA kits (USCN Life Science Inc., Wuhan, China), respectively. Animal experiments were conducted following the National Guidelines for Housing and Care of Laboratory Animals and were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee.
Isolation of Cytoplasmic and Mitochondrial Proteins
Mitochondria were isolated using a mitochondria isolation kit (Beyotime Biotechnology). Briefly, the cells were homogenized and centrifuged at 600 g for 10 min at 4°C to remove the nuclei. The supernatants were centrifuged again at 11,000 g for 10 min at 4°C to obtain crude mitochondria pellets. The cytosolic proteins were obtained by centrifuging the supernatants for another 10 min at 12,000 g. The proteins were boiled for western blot assay after determining the concentration using a BCA protein assay kit (Thermo).
Cellular Mitochondrial Membrane Potential Measurement
The mitochondrial membrane potential was measured by flow cytometry using Rho123 (Sigma) staining, as described previously (Wang et al., 2014). Briefly, the cells were incubated with Rho123 (0.5 μg/mL) at 37°C for 25 min and then were harvested and washed twice with PBS, and a total of 10,000 events per sample was measured by a flow cytometer. The fluorescent signal intensity was analyzed with BD CellQuest Pro software.
Measurement of Cellular NADPH Oxidase and CuZn/Mn-SOD Activity
The cells were collected and lysed, and the dihydronicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity was measured using an NADPH oxidase activity assay kit (Genmed Scientifics Inc., Shanghai, China), which was detected by an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA). The CuZn/Mn-SOD activity was measured by the CuZn/Mn-SOD assay kit (Beyotime Biotechnology). The relative activity was normalized to the protein contents and presented as a percent of the control.
Statistical Analysis
The data are presented as the mean ± standard deviation of over three independent experiments. Analysis of variance (ANOVA) followed by Student-Newman-Keuls (SNK) post hoc tests were performed to compare the different parameters between the groups using SPSS17.0. The value of statistical significance was set at p < 0.05.
Results
NF-κB-Mediated Inflammatory Response Is Directly Induced by HCV Infection and Is Persistent Even After Clearance of the Virus in Huh7.5 Cells
HCV infection increases the level of intracellular inflammatory cytokines and chemokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and chemokine macrophage inflammatory protein 1 beta (MIP-1β) in vivo and in vitro (Zeremski et al., 2007; Lin et al., 2014), and nuclear factor kappa B (NF-κB) activation is a common upstream event that causes the increase. We first verified whether HCV infection directly induced an inflammatory response in an NF-κB-dependent manner in vitro. Huh7.5 cells were infected with HCV for 0, 3, and 7 days, and the amount of phosphorylated NF-κB (p-NF-κB) was increased in a time-dependent manner without changing the total NF-κB amount (Figure 1A). Intracellular TNF-α, IL-6, and MIP-1β mRNA levels were also increased (Figure 1B) and were positively related to the HCV viral loads (Figure 1A,B). Therefore, our results verified that HCV infection induced NF-κB activation and the subsequent liver inflammatory response in Huh7.5 cells, which was consistent with previous reports (Lin et al., 2010, 2011, 2014).
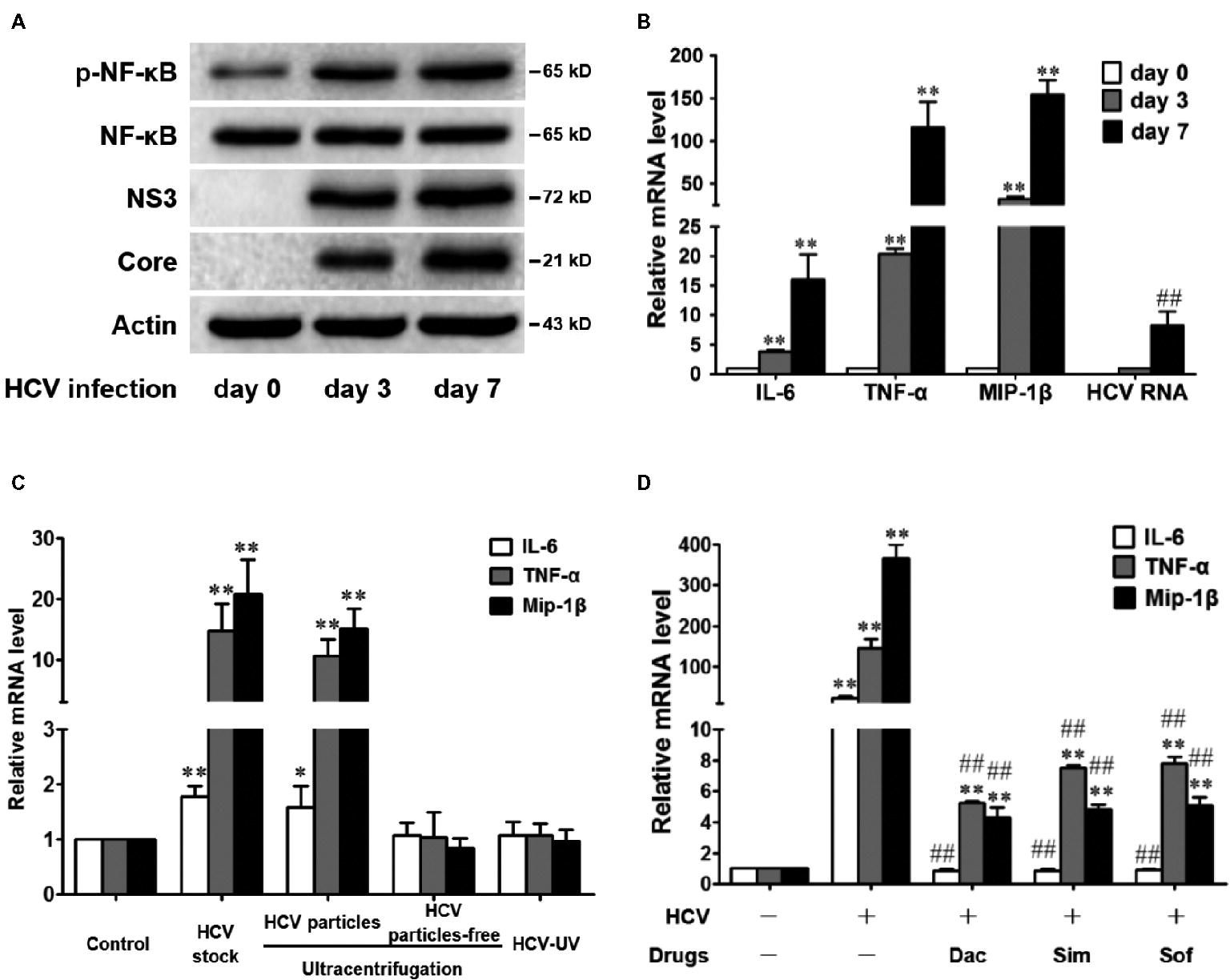
Figure 1. HCV infection induces NF-κB activation and subsequently increases the levels of inflammatory factors in Huh7.5 Cells. (A,B) Huh7.5 cells were infected with HCV (MOI = 1). At 0, 3, and 7 days, intracellular protein and RNA were detected by western blot (A) and qRT-PCR (B), respectively. (C) Huh7.5 cells were infected with infectious HCV (HCV particles obtained by ultracentrifugation or HCV stock, MOI = 2), UV-inactivated HCV (HCV-UV), and virus-free supernatants, and intracellular RNA was detected by qRT-PCR at 48 h. (D) Huh7.5 cells, 3 days post infection (MOI = 1), were treated with daclatasvir (Dac, 100 pM), simeprevir (Sim, 0.2 μM), or sofosbuvir (Sof, 0.5 μM) for 8 days, and the intracellular RNA was detected by qRT-PCR. The protein bands show the results of a representative experiment. The experiments were performed at least in triplicate, and each value represents the mean ± SD. ANOVA analysis with the SNK method was used in (B–D). *p < 0.05, **p < 0.01 versus the solvent control; #P < 0.05, ##P < 0.01 versus the HCV-infected control group.
To corroborate that the increased inflammatory response was mediated by HCV infection or by the increased inflammatory mediators in the supernatants, we compared the role of infectious HCV (HCV stock or HCV particles obtained by ultracentrifugation), UV-inactivated HCV (HCV-UV), and the virus-free supernatants. The results showed that infectious HCV increased TNF-α, IL-6, and MIP-1β mRNA levels, while the culture supernatants with HCV particle-free or inactivated HCV were unable to show this upregulation (Figure 1C), suggesting that the inflammatory response was exclusively activated by the HCV infection but not the inflammatory mediator in our infectious system.
In the clinic, aggravated liver disease progress after the clearance of HCV is still persistent (Lin et al., 2015; Jacobson et al., 2017). Therefore, we examined the inflammatory state after virus eradication in vitro. After the HCV-infected Huh7.5 cells were treated with DAAs for 8 days, HCV RNAs were undetectable (data not shown). However, the TNF-α and MIP-1β mRNA levels were still significantly higher than those in the control cells (Figure 1D), although the IL-6 mRNA decreased to the basal level. The results agree with previous reports in the clinic that anti-HCV therapy does not completely cure HCV-induced inflammatory diseases (Koike and Miyoshi, 2006; Morgan et al., 2010; Lin et al., 2015; D’Ambrosio et al., 2018).
Bicyclol Decreases the NF-κB Activation and Inflammatory Response in HCV-Infected Hepatocytes
Clinical and experimental data show that bicyclol has anti-inflammatory and hepatoprotective effects, and long-term use of bicyclol inhibits HCV replication via a new mechanism that is different from that of DAAs (Guo et al., 2003; Liu, 2009). To verify bicyclol’s anti-inflammatory and anti-HCV effects, Huh7.5 cells were infected with HCV and were simultaneously treated with bicyclol or DAAs (daclatasvir, simeprevir, and sofosbuvir) for 72 h. The results showed that both the bicyclol and DAA treatments decreased the levels of HCV RNA and proteins (Figure 2A) and the inflammatory factor TNF-α, IL-6, and MIP-1β mRNA levels (Figure 2B) without cytotoxicity (Figure 2C). However, bicyclol was more effective than the DAAs, with regard to the anti-inflammatory effects, under identical anti-HCV activities (Figure 2A,B).
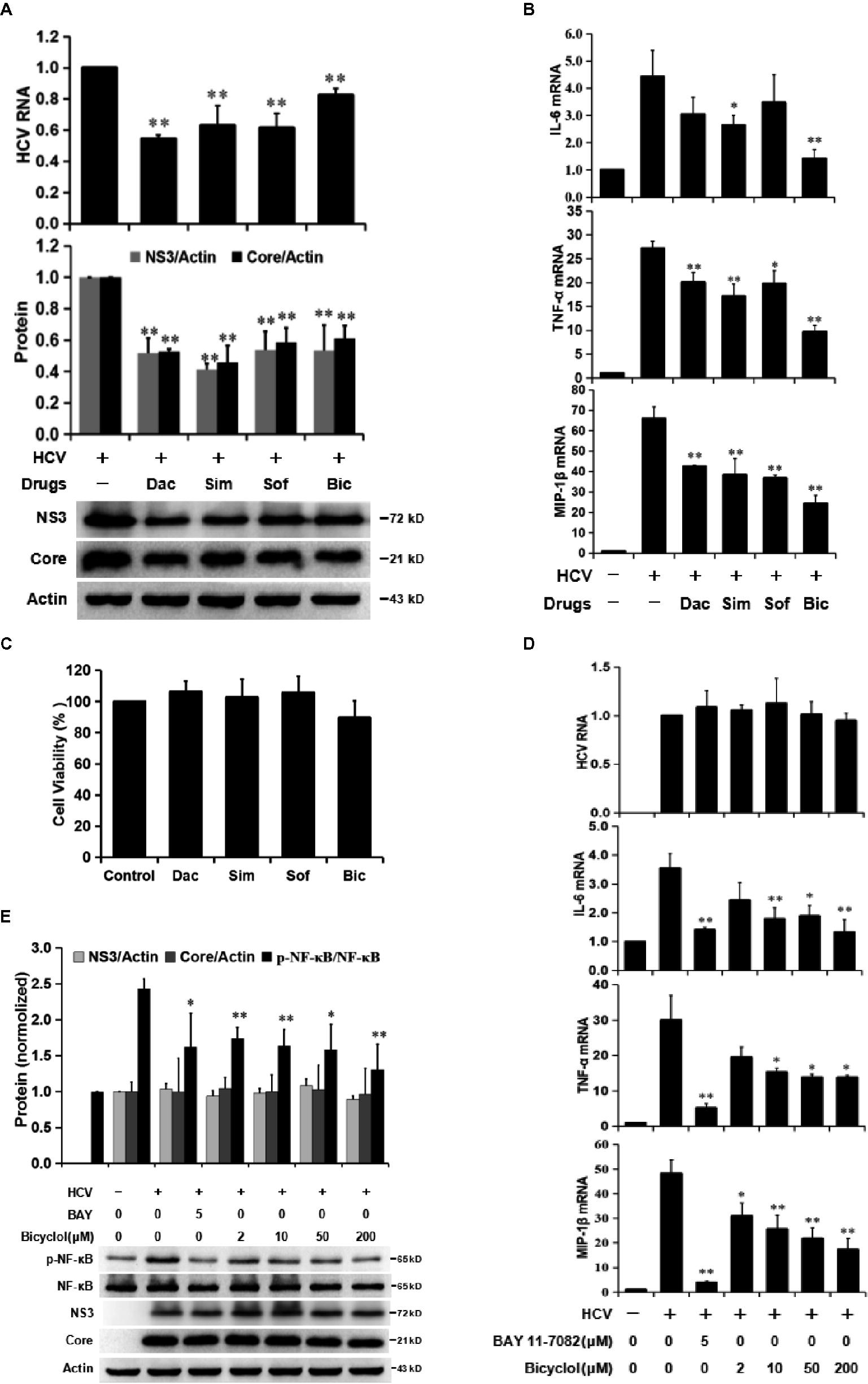
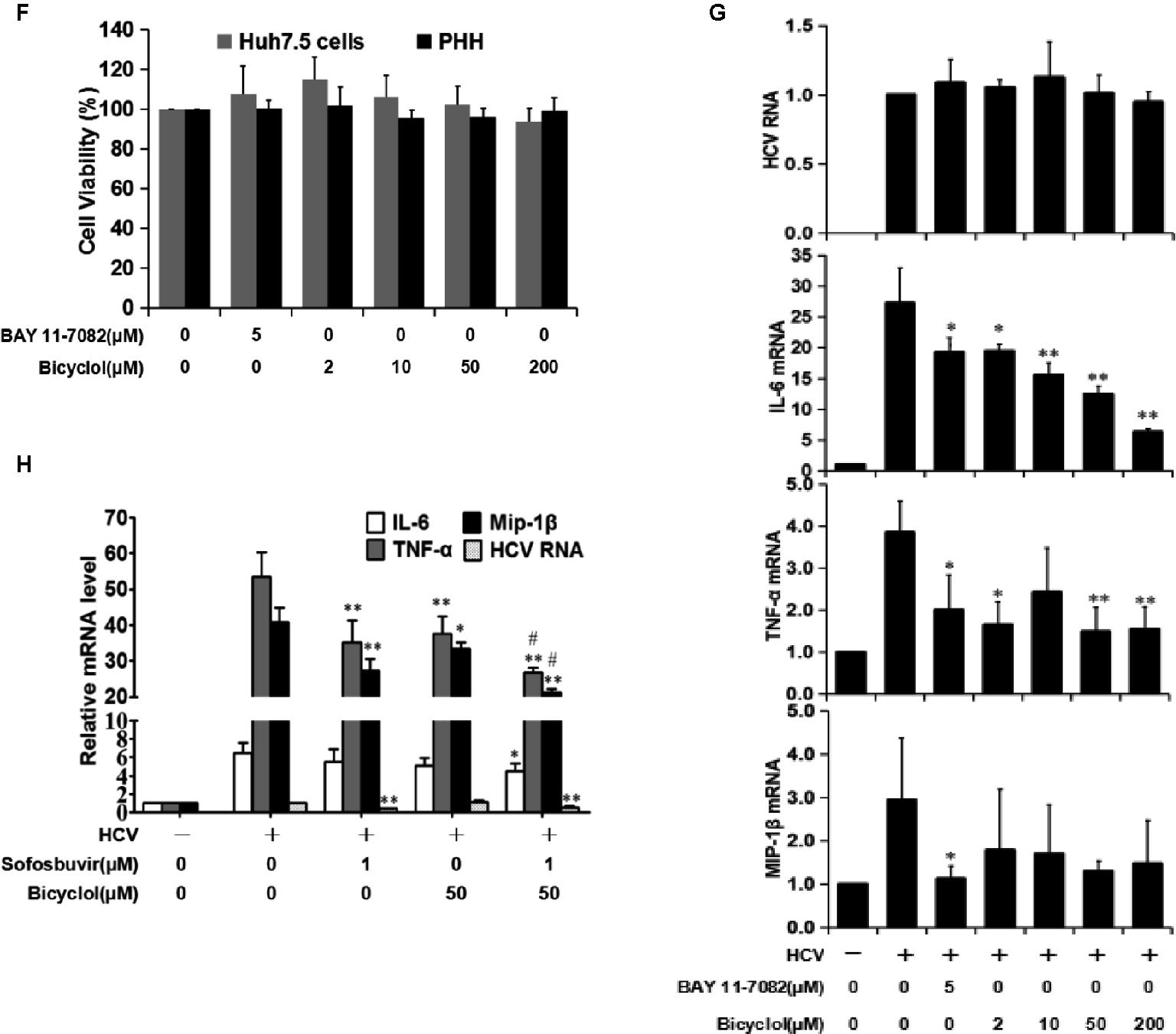
Figure 2. Bicyclol decreases HCV-induced NF-κB activation and the levels of inflammatory factors in hepatocytes. (A–C) Huh7.5 cells infected with HCV (MOI = 1) were simultaneously treated with bicyclol (Bic, 100 μM) or DAAs (daclatasvir (Dac, 10 pM), simeprevir (Sim, 0.02 μM), and sofosbuvir (Sof, 0.1 μM)) for 72 h, and intracellular RNA and proteins were detected by qRT-PCR and western blot (A,B), respectively, and the cytotoxicity was detected by MTT assay (C). (D–F) HCV-infected Huh7.5 cells were starved for 4 h and were then treated with the NF-κB inhibitor BAY11-7082 or bicyclol for 12 h; the intracellular RNA and proteins were detected by qRT-PCR (D) and western blot (E), respectively, and the cytotoxicity was detected by MTT assay (F). (F–G) Primary human hepatocytes (PHHs), 4 days post-HCV infection (MOI = 2), were treated with bicyclol for 12 h, and the intracellular RNAs were detected by qRT-PCR (G), and the cytotoxicity was detected by MTT assay (F). (H) Huh7.5 cells, 6 days post infection with HCV (MOI = 0.2), were treated with sofosbuvir (1 μM) and/or bicyclol (50 μM) for 24 h, and the intracellular RNA was detected by qRT-PCR. The protein bands show the results of a representative experiment. The experiments were performed at least in triplicate, and each value represents the mean ± SD. ANOVA analysis with the SNK method was used. *p < 0.05, **p < 0.01 versus the HCV-infected control group; #p < 0.05 versus the sofosbuvir-treated group.
To further distinguish bicyclol’s anti-inflammatory and anti-HCV effect, we examined whether bicyclol reduces the inflammatory response in HCV-infected Huh7.5 cells within 12 h of treatment. The results showed that TNF-α, IL-6, and MIP-1β mRNA levels were decreased (Figure 2D), and HCV-induced p-NF-κB activation was inhibited by bicyclol (Figure 2E) without changing the HCV RNA and protein levels (Figure 2D,E). Certainly, BAY11-7082, an NF-κB inhibitor, was effective in this model (Figure 2D,E) without cytotoxicity (Figure 2F). The results were similar in HCV-infected primary human hepatocytes (Figure 2G) and were without cytotoxicity (Figure 2F). Furthermore, the combined use of bicyclol with sofosbuvir synergistically reduced the inflammatory factors but not the HCV RNA level in HCV-infected Huh7.5 cells within 24 h of treatment (Figure 2H). These results suggest that bicyclol has a potential anti-inflammatory effect during HCV infection, which is at least partly not due to its anti-HCV effects.
Bicyclol Attenuates HCV-Induced Activation of NF-κB Through the Inhibition of the ROS-MAPK-NF-κB Pathway in Huh7.5 Cells
NF-κB activation is commonly related to the activation of its upstream mitogen-activated protein kinases (MAPKs), including p38 MAPK, extracellular signal-regulated kinase (ERK), and Jun N-terminal kinase (JNK), and the upper-stream reactive oxygen species (ROS) (Lin et al., 2010; Hsu et al., 2013). Two types of ROS, superoxide anion (O2•−) and secondary generated hydrogen peroxide (H2O2), are induced by HCV protein expression in replicon models and infectious system (Gong et al., 2001; Paracha et al., 2013) and, finally, contribute to the development of liver disease via NF-κB activation (Tardif et al., 2005; Lin et al., 2010). Previous reports show that bicyclol has a potent hepatocyte-protective effect partly by decreasing oxidative stress (Liu, 2009), and therefore, we assessed the role of bicyclol in the ROS-MAPK-NF-κB axis during HCV infection. HCV-infected Huh7.5 cells were pretreated with the ROS inhibitor diphenyliodonium (DPI) or bicyclol for 10 h and were then treated with or without H2O2 for another 2 h. The results showed that bicyclol decreased HCV infection induced cellular O2•- levels (Figure 3A) and inhibited the phosphorylation of p38, ERK, JNK, and NF-κB (Figure 3B), with unchanged amounts of HCV protein and total p38, ERK, JNK, and NF-κB (Figure 3B). The effect of bicyclol was similar with that of the ROS inhibitor DPI (Figure 3A,B), which was neutralized by exogenous H2O2 (Figure 3A,B). These results suggest that the HCV infection-activated ROS-MAPK-NF-κB pathway might be interrupted by bicyclol.
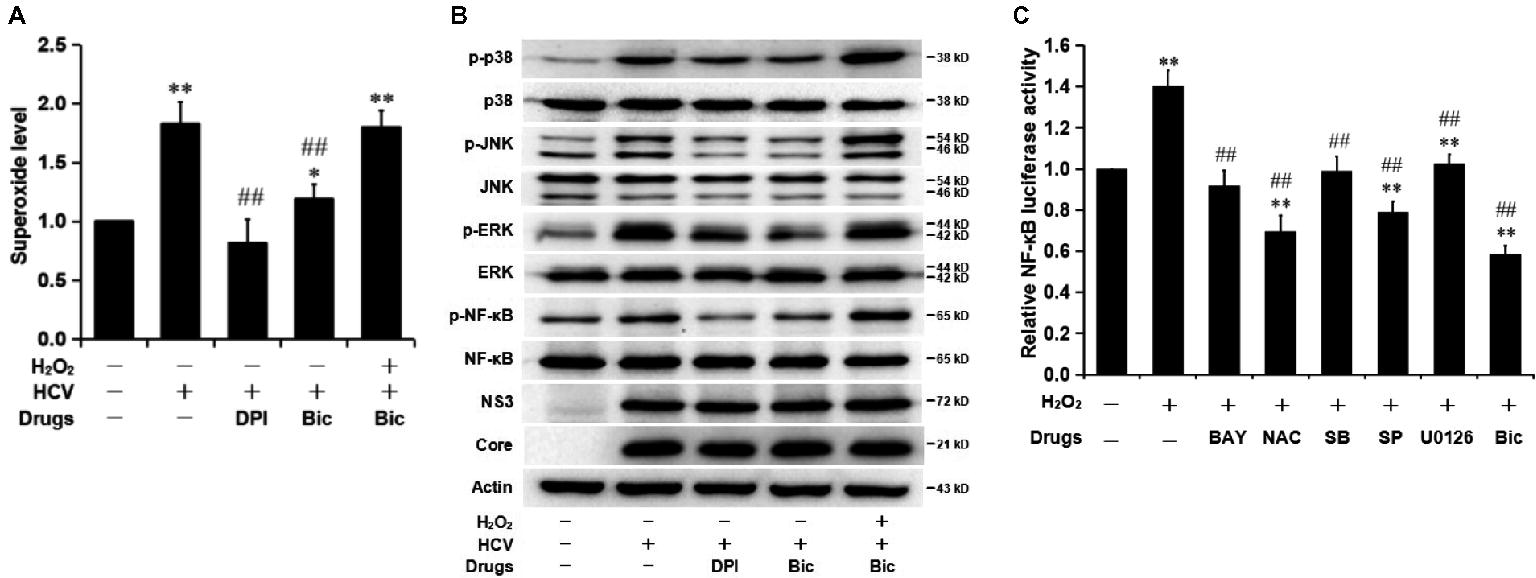
Figure 3. Bicyclol inhibits the HCV-induced activation of NF-κB by inhibiting the activation of the ROS-MAPK-NF-κB pathway in Huh7.5 cells. (A,B) Naive and HCV-infected (MOI = 1) Huh7.5 cells were serum starved for 4 h and pretreated with the ROS inhibitor DPI (20 μM) and bicyclol (Bic, 200 μM) for 10 h and were then treated with or without H2O2 (200 μM) for another 2 h; the intracellular superoxide levels (A) and phospho-specific and total p38/ERK/JNK and NF-κB amounts (B) were detected. (C) Huh7.5 cells were cotransfected with the plasmid pNF-κB-Luc expressing firefly luciferase and the control plasmid, pRL-SV40 vector, expressing Renilla luciferase in a 10:1 mass ratio. After 24 h of transfection, the cells were pretreated with the NF-κB inhibitor BAY11-7082 (BAY, 5 μM), ROS scavenger NAC (10 mM), p38 inhibitor SB203580 (SB, 10 μM), JNK inhibitor SP600125 (SP, 10 μM), ERK inhibitor U0126 (10 μM), or bicyclol (Bic, 200 μM) for 10 h and were then treated with or without H2O2 (200 μM) for another 2 h. The relative luciferase activity was measured. The experiments were performed at least in triplicate, and each value represents the mean ± SD. ANOVA analysis with the SNK method was used. *p < 0.05, **p < 0.01 versus the solvent control; #p < 0.05, ##p < 0.01 versus the HCV- or H2O2-treated group.
To further investigate the inactive role of bicyclol in the ROS- and MAPK-mediated NF-κB activation, we applied H2O2 to mimic the induced oxidative stress state by HCV infection. Huh7.5 cells, cotransfected with the plasmid pNF-κB-Luc and control plasmid pRL-SV40, were treated with the NF-κB inhibitor BAY11-7082, ROS scavenger N-acetylcysteine (NAC), p38 inhibitor SB203580, JNK inhibitor SP600125, ERK inhibitor U0126, or bicyclol for 10 h and were then stimulated with H2O2 for another 2 h. Predictably, we observed H2O2-induced NF-κB activation (Figure 3C), and the NF-κB activity was decreased by all those inhibitors or bicyclol (Figure 3C). These data further verified that bicyclol inhibits the HCV-induced inflammatory response through the ROS-MAPK-NF-κB pathway.
Bicyclol Decreases the Level of HCV-Induced ROS Through Restoration of Mitochondrial Function Without a Dependence on NADPH Oxidase and Superoxide Dismutases
Superoxide anion is mainly derived from mitochondrial electron transport chain and cytoplasmic production after dihydronicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation (Brault et al., 2013). Mitochondrial depolarization is widely reported to enhance the generation of O2•- (Budd et al., 1997). Therefore, we detected the effect of bicyclol on the mitochondrial transmembrane potential (ΔΨm). HCV-infected Huh7.5 cells were treated with bicyclol for 12 h, and ΔΨm and O2•− were detected. The results showed that HCV infection decreased ΔΨm (shown by Rho123 staining) and increased the superoxide anion level (shown by DHE staining) (Figure 4A), which is consistent with a previous report (Deng et al., 2008). Bicyclol reversed these changes in a dose-dependent manner (Figure 4A), suggesting that bicyclol might decrease the level of HCV-induced ROS by restoring HCV-decreased ΔΨm in HCV-infected cells.
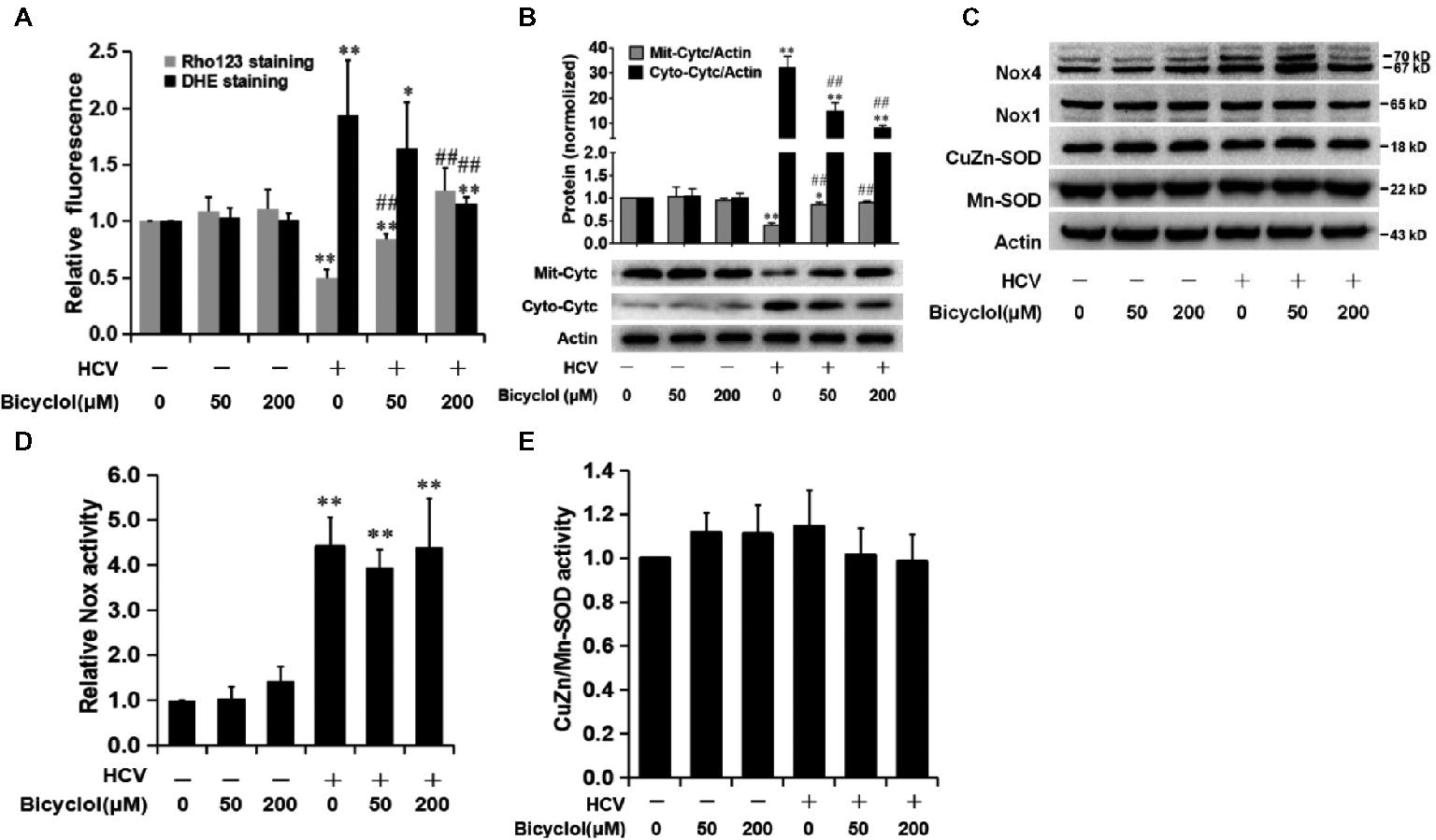
Figure 4. Bicyclol decreases the level of HCV-induced ROS through the restoration of mitochondrial function but is independent of NADPH oxidase and superoxide dismutases. Naive and HCV-infected Huh7.5 cells were treated with bicyclol for 12 h. The cells were stained by Rho123 (0.5 μg/mL) for 25 min to measure the mitochondrial membrane potential or stained by 5 μM dihydroethidium (DHE) for 30 min to measure the superoxide level using a flow cytometer (A). Cytosolic and mitochondrial proteins were extracted, and cytochrome c was detected by western blot (B). Total cellular protein was detected by western blot (C), and the activities of Nox (D) and CuZn/Mn-SOD (E) were detected with assay kits. The protein bands show the result of a representative experiment. The experiments were performed at least in triplicate, and each value represents the mean ± SD. ANOVA analysis with the SNK method was used. *p < 0.05, **p < 0.01 versus the solvent control; #p < 0.05, ##p < 0.01 versus the HCV-infected control group.
The decreased ΔΨm leads to mitochondrial dysfunction, which is characterized by cytochrome c release from mitochondria into cellular cytosol (Deng et al., 2008). Our results showed that mitochondrial cytochrome c was decreased (Figure 4B), while cytosol cytochrome c was increased (Figure 4B) after HCV infection. We also observed a decreased release of cytochrome c after treatment with 50 μM bicyclol for 12 h (Figure 4B), with no effect in naive Huh7.5 cells (Figure 4B), suggesting that the bicyclol decreasing the level of HCV-induced ROS might be through the restoration of mitochondrial function.
NADPH oxidase (Nox) catalyzes the transfer of electrons from NAD(P) H to O2 and thus produces O2•−. The degradation of O2•− into H2O2 relies on mitochondrial or cytosolic superoxide dismutases (Mn-SOD or CuZn-SOD). Previous studies showed that among the seven Nox enzymes (Nox1-5, Duox1, and Duox2), Nox1 and Nox4 proteins might be increased in HCV-infected or HCV core protein expressed cells (Boudreau et al., 2009; de Mochel et al., 2010), while the effect of HCV infection on Mn-SOD or CuZn-SOD was controversial (Abdalla et al., 2005; Lee et al., 2016). Our results showed that HCV only slightly increased the expression of Nox4, but not Nox1 (Figure 4C), while Nox activities were significantly increased after HCV infection (Figure 4D). However, the increased expressions and activities of the Nox were not changed after bicyclol treatment (Figure 4C,D). Meanwhile, the expression and activity of Mn-SOD and CuZn-SOD were not influenced by HCV infection or by bicyclol treatment (Figure 4C,E). These data hint that the decreased level of HCV-induced superoxide anion by bicyclol is independent of NADPH oxidases and superoxide dismutases.
Bicyclol Reduces Liver Injury and Inflammation via Attenuating the Activation of the ROS-MAPK-NF-κB Pathway in Hepatitis Mice
Simultaneous injection of poly(I:C) and D-GalN induces serious liver damage in mice, and thus, this rodent animal model is used to evaluate the protective role of the candidates (Huang et al., 2016). The levels of ALT and AST in the serum were increased after the coinjection of poly(I:C)/D-GalN (Figure 5A), suggesting that the liver was injured. However, the transaminase level was markedly decreased in a dose-dependent manner by an intragastric administration of bicyclol (Figure 5A). The mRNA (Figure 5B) and protein levels (Figure 5C) of liver tissues TNF-α, IL-6, and MIP-1β were significantly increased in the poly(I:C)/D-GalN group (Figure 5B,C), while bicyclol decreased the inflammation in a dose-dependent manner (Figure 5B,C). Histopathological findings also revealed that poly(I:C)/D-GalN induced spotted necrosis and inflammatory infiltration in the liver, and bicyclol alleviated these pathologic changes (Figure 5D, HE staining and CD68 IHC staining). These data suggest that bicyclol improves poly(I:C)/D-GalN-induced hepatitis in mice, which is consistent with the clinical data (Yao et al., 2002; Liu, 2009).
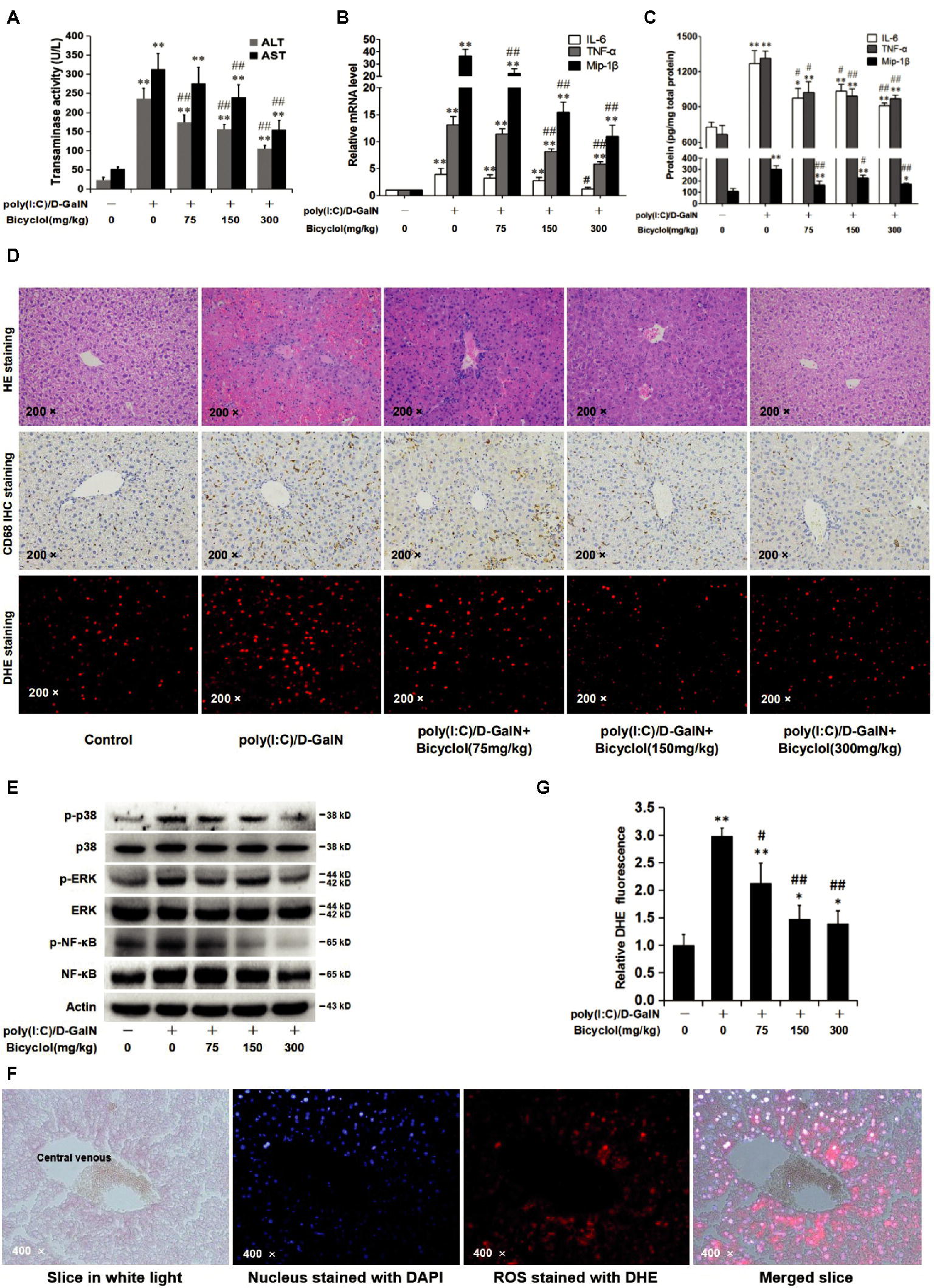
Figure 5. Bicyclol reduces liver injury and inflammation via attenuating the activation of the ROS-MAPK-NF-κB pathway in hepatitis mice. Mice (n = 6 for each group) were intragastrically administered with bicyclol daily or a solvent control for 7 days, and then, the mice were intravenously injected with poly(I:C) at a dose of 75 μg/kg or a solvent control. Simultaneously, the mice were intraperitoneally injected with D-GalN at a dose of 500 mg/kg or a solvent control. Sixteen hours after the injection, the serum was collected for the ALT and AST assays (A), and the liver tissues were prepared for RNA detection (B), ELISA assay (C), H&E and CD68 IHC staining (D), and western blot (E). The level of ROS in the liver tissues was detected using the fluorescent probe DHE (D, DHE staining), and the distribution of ROS was detected through comparing the paraformaldehyde fixed liver tissue slice in white light, nucleus stained with DAPI, and ROS stained with DHE (F). ROS accumulation was measured and analyzed in three random vision fields by Image-Pro plus 6.0 (F). ANOVA analysis with the SNK method was used. *p < 0.05, **p < 0.01 versus the control group; #p < 0.05, ##p < 0.01 versus the poly(I:C)/D-Gal treated group.
D-GalN or poly(I:C) triggers ROS production, MAPK pathway activation, and inflammatory response in vitro and in vivo (Dejager and Libert, 2008; Liu et al., 2008; Lin et al., 2009). We detected changes in the ROS-MAPK-NF-κB axis in the mouse liver. As shown in Figure 5E, poly(I:C)/D-GalN increased phosphorylated-MAPK (p-p38 and p-ERK) levels, while the p-JNK was not detectable, which might be due to the low level of p-JNK in this model. In the bicyclol-treated group, the levels of p-p38, p-ERK, and downstream p-NF-κB were obviously downregulated (Figure 5E). The distribution of liver cells (nucleus stained with DAPI) and ROS (stained with DHE) also demonstrated that the ROS was mainly distributed in hepatocytes (Figure 5F) and bicyclol decreased the liver ROS accumulation triggered by poly(I:C)/D-GalN in a dose-dependent manner (Figure 5D, DHE staining and 5G). The ROS is distributed mainly in hepatocytes These results suggest that bicyclol ameliorates poly(I:C)/D-GalN-induced liver injury and inflammatory factor release, at least partly, by inhibiting ROS generation and the subsequent activation of the MAPK/NF-κB pathway. The conclusion from the hepatitis mice model agreed with that from the HCV-infected cells (Figure 3).
Discussion
Bicyclol was widely used in China for the treatment of various types of liver injury, including hepatitis B and C (Guo et al., 2003; Liu, 2009). Its mechanism involves mitochondrial protection, antioxidant stress, anti-apoptosis, or/and toll-like receptor pathway inhibition (Liu, 2009; Zhang et al., 2016). However, the anti-inflammatory mechanism of bicyclol in hepatitis C remains to be clarified. Once infected with HCV, ~80% of individuals develop chronic hepatitis C naturally (Leone and Rizzetto, 2005). HCV triggers the occurrence of oxidative stress and the overproduction of inflammatory factors and, thus, facilitates disease progression and even leads to HCC (Nakamoto and Kaneko, 2003). In this study, we demonstrated that bicyclol significantly inhibited the inflammatory response in HCV-infected hepatocytes and improved liver injury in hepatitis mice, and the combined use of bicyclol with antivirals produced a synergistic anti-inflammatory effect. Detailed mechanism studies showed that bicyclol decreased the level of HCV-induced ROS by recovering the mitochondrial function independent of NADPH oxidase and superoxide dismutases and, thus, inhibited the HCV-induced inflammation by blocking the ROS-MAPK-NF-κB pathway (Figure 6). Moreover, after HCV clearance by treatment with DAAs, the increased level of inflammatory factors is still persistent (Figure 1D), suggesting that the restoration of liver injury is delayed following HCV elimination. This may be a reason that persistent disease progress is still persistent after finishing antiviral therapy in the clinic (Conti et al., 2016; Reig et al., 2016; Jacobson et al., 2017), and thus, the anti-inflammatory therapy regimen is still very important for those patients.
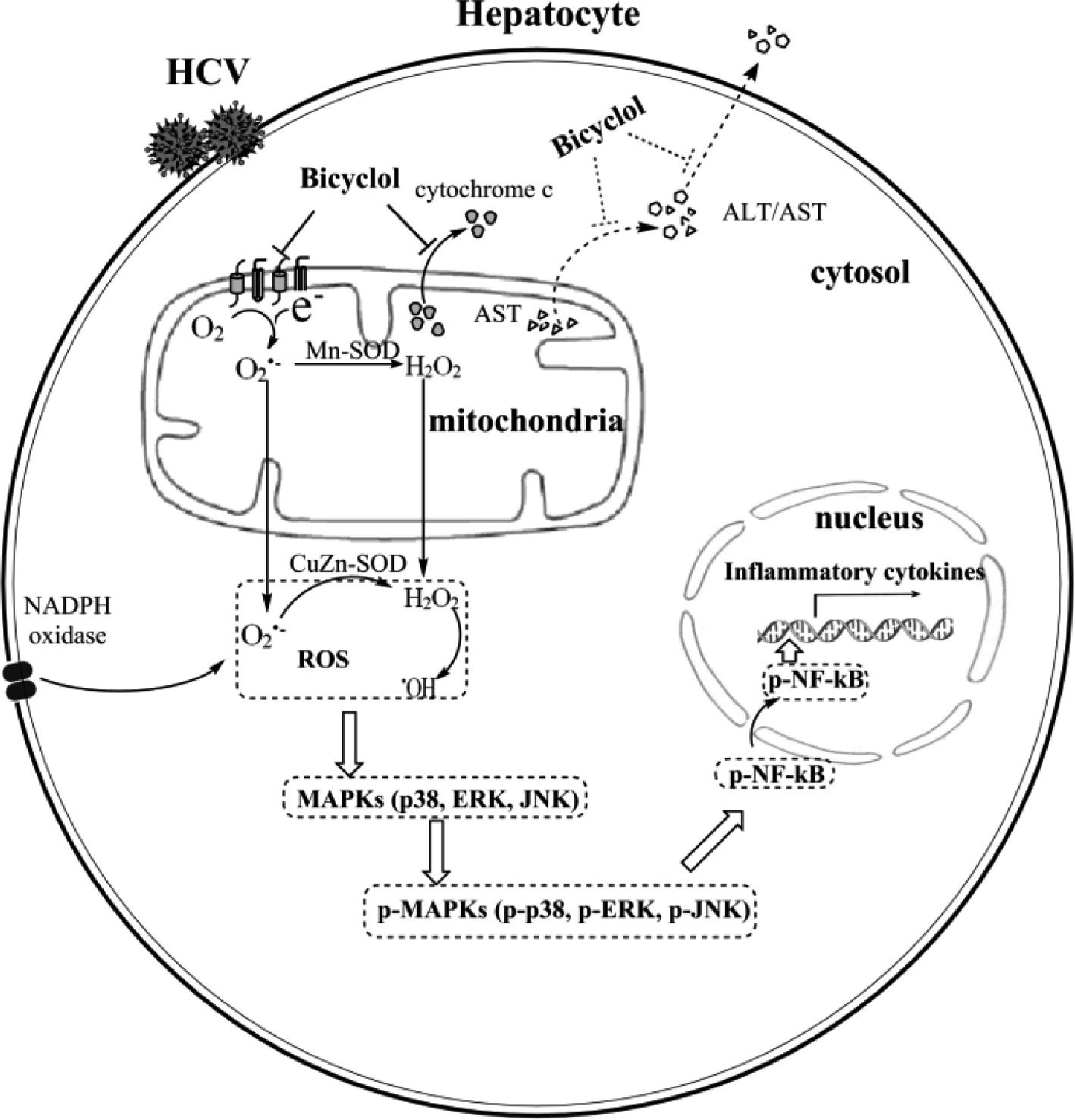
Figure 6. Schematic diagram of the anti-inflammatory mechanism of bicyclol. Bicyclol reverses the HCV-disturbed mitochondrial transmembrane potential, without altering the expressions and activities of NADPH oxidase and superoxide dismutases, decreases the cellular ROS levels, and inhibits the subsequent MAPK and NF-κB activation. The overall anti-inflammation and hepatoprotection effects of bicyclol are characterized by the decreased inflammatory cytokine production and the mitochondrial cytochrome c release to the cytosol and/or the ALT/AST release into the extracellular matrix. The dotted line shows the potential mechanism not verified in our study.
Clinical evidence suggests that the long-term application of bicyclol might, to some extent, decrease the HCV load (Guo et al., 2003; Liu, 2009). Our results demonstrated that bicyclol’s anti-inflammatory effect was distinguished from its anti-HCV roles (Figure 2), although its anti-HCV role might contribute to its anti-inflammatory effect. Transcription factors, such as NF-κB, AP-1, and STAT3, regulate the production of proinflammatory cytokines and chemokines during HCV infection, among which NF-κB plays a central role in the inflammatory response (Gong et al., 2001; Hassan et al., 2007). Therefore, we focused on the NF-κB-mediated inflammation response during HCV infection. Our data demonstrated that the inflammation inducer is HCV itself but not the inflammatory mediators in the culture supernatants (Figure 1C). A report showed that HCV infection induced the nuclear accumulation of NF-κB p65 and the subsequent activation of inflammatory response in TLR3-reconstituted hepatocytes (Huh7.5-TLR3 cells) but not in Huh7.5 cells (Li et al., 2012). However, Huh7.5 cells are a cell line with deficient TLR3 expression (Sumpter et al., 2005; Prescott et al., 2007), and we still detected the inflammatory factor storm and an increased level of p-NF-κB, with an unchanged level of total NF-κB (Figure 1A,B) during HCV infection, which was consistent with previous reports (Lin et al., 2010, 2011).
MAPK (p38, ERK, and JNK) phosphorylation is also widely reported to activate downstream of the IKK-IKB-NF-κB pathway (Lin et al., 2010; Das and Ghosh, 2017). NF-κB activation, induced by HCV, is accompanied by increased oxidative stress, which is characterized by enhanced intracellular ROS levels and the subsequent activation of the MAPK pathway (Koike and Miyoshi, 2006; Fujinaga et al., 2011). Additionally, the ROS-MAPK-NF-κB axis is also reported in other inflammatory models induced by LPS, poly(I:C), and virus (Liu et al., 2008; Lin et al., 2010; Hsu et al., 2013; Li et al., 2016). Our data from the HCV-infected Huh7.5 cells are consistent with these studies (Figure 3). Bicyclol inhibited the activation of the HCV-induced MAPK pathway, leading to decreased p-NF-κB levels (Figure 3B), and this effect might be through decreasing cellular superoxide levels (Figure 3A), which was confirmed in the NF-κB-Luc dual luciferase reporter system (Figure 3C). This might be the reason that bicyclol is efficacious in various hepatitis types because oxidative stress universally exists in patients.
Oxidative stress is generally induced during HCV infection, and the mitochondria are the main source of cellular superoxide (Brault et al., 2013). Our result showed that bicyclol decreased the HCV-induced ROS level by restoring the HCV-reduced mitochondrial transmembrane potential (Figure 4A), which further decreased abnormally the cytochrome c release into the cellular cytosol (Figure 4B). HCV-induced ROS generation was also reported to improve Nox1 and Nox4 expression in HCV-infected Huh7 cells or in HCV RNA-transfected HepG2 cells (Deng et al., 2008; Boudreau et al., 2009; de Mochel et al., 2010). However, in our infectious system, HCV infection only slightly increased the expression of Nox4 but not Nox1 (Figure 4C), while Nox activity was significantly increased after HCV infection (Figure 4D). This phenomenon might be due to the different hepatocytes and HCV infection systems, as HCV cDNA-transfected HepG2 cells displayed a basal level of Nox1 mRNA expression, and only a modest Nox4 mRNA level change (<10%) was detected in the JFH-AM2 HCV clone-infected Huh7 cells (Boudreau et al., 2009). Previous studies also suggest that Nox-dependent innate immune responses are less robust in the Huh7.5 cell line (Boudreau et al., 2009). In addition, Mn-SOD or CuZn-SOD, which degrades superoxide anion into H2O2, also affects cellular superoxide accumulation, and the effect on Mn-SOD or CuZn-SOD after HCV infection is controversial (Abdalla et al., 2005; Lee et al., 2016). Our result showed that HCV infection and bicyclol treatment had no effect on their expression and activities (Figure 4C,E). These data suggest that bicyclol decreases the level of HCV-induced ROS by restoring mitochondrial function without a dependence on NADPH oxidase and superoxide dismutases. However, the detailed mechanisms still need to be clarified.
Persistent intrahepatic inflammation and hepatocellular apoptosis lead to liver injury and even to HCC (Nakamoto and Kaneko, 2003). Poly(I:C), a synthetic analog of viral double-stranded RNA, is commonly used to study the RNA viral infection-induced immune response (Takahashi et al., 2006; Liu et al., 2008). It also induces a MAPK-dependent expression of proinflammatory cytokines and chemokines in cells (Liu et al., 2008). D-GalN is a hepatotoxic agent, which induces liver damage that closely resembles human viral hepatitis (Decker and Keppler, 1972). Previous reports suggest that D-GalN triggers liver ROS production, activates the MAPK signaling pathway in rats, and facilitates mitochondrial apoptosis to contribute to producing oxidative stress and inflammation in the liver (Quintero et al., 2002; Lin et al., 2009; Ganai et al., 2015). Coinjection of D-GalN and poly(I:C) synergistically mediates the severe liver injury (Dejager and Libert, 2008). Bicyclol significantly decreased poly(I:C)/D-GalN-induced liver injury and inflammatory response by reducing the ROS accumulation and the subsequent phosphorylation levels of p38, ERK, and NF-κB in this model (Figure 5), which was similar with the changes in the HCV-infected hepatocytes (Figure 3). Besides, the total NF-κB level in this mice hepatitis model also increased, which we speculated is caused by poly(I:C)/D-GalN induced damage-associated molecular patterns in vivo. The effect of bicyclol in our liver injury model is consistent with its hepatoprotection and anti-inflammation roles in other chemical and immunological liver injury mouse models (Liu, 2009). However, our data could not exclude the potentially direct role of bicyclol for blocking inflammatory factor triggered canonical NF-κB activation, and there needs more evidence for further confirmation.
Conclusion
The present study reported that bicyclol attenuated the inflammation in HCV-infected hepatocytes and ameliorated liver injury in mouse hepatitis induced by the coinjection of poly(I:C) and D-GalN. The detailed mechanism showed that bicyclol decreased the level of HCV-induced ROS by recovering mitochondrial function without a dependence on NADPH oxidase and superoxide dismutases and, thus, inhibited HCV-induced excessive inflammation by blocking the activation of the ROS-MAPK-NF-κB pathway. Because this pathologic pathway is universal in a variety of hepatitis types and inflammation still exists in HCV-infected patients after they achieve an SVR, bicyclol might contribute to eventually eliminating hepatitis C.
Author Contributions
HL designed and performed the experiments, analyzed the data and wrote the manuscript. J-RL and M-HH performed the experiments and analyzed the data. J-HC, X-QL, L-LZ, J-LT, and BD performed the experiments. J-DJ and Z-GP oversaw the project, designed the experiments, analyzed the data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work was supported by the National Mega-Project for “R&D for Innovative drugs,” Ministry of Science and Technology, China (2017ZX09101003-003-010), CAMS Innovation Fund for Medical Sciences (2017-I2M-3-012), and the National Natural Science Foundation, China (81621064).
References
Abdalla, M. Y., Ahmad, I. M., Spitz, D. R., Schmidt, W. N., and Britigan, B. E. (2005). Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J. Med. Virol. 76, 489–497. doi: 10.1002/jmv.20388
Bao, X. Q., and Liu, G. T. (2009). Induction of overexpression of the 27- and 70-kDa heat shock proteins by bicyclol attenuates concanavalin A-induced liver injury through suppression of nuclear factor-kappa B in mice. Mol. Pharmacol. 75, 1180–1188. doi: 10.1124/mol.108.053280
Boudreau, H. E., Emerson, S. U., Korzeniowska, A., Jendrysik, M. A., and Leto, T. L. (2009). Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: a new contributor to HCV-induced oxidative stress. J. Virol. 83, 12934–12946. doi: 10.1128/JVI.01059-09
Brault, C., Levy, P. L., and Bartosch, B. (2013). Hepatitis C virus-induced mitochondrial dysfunctions. Viruses 5, 954–980. doi: 10.3390/v5030954
Budd, S. L., Castilho, R. F., and Nicholls, D. G. (1997). Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 415, 21–24. doi: 10.1016/S0014-5793(97)01088-0
Castello, G., Costantini, S., and Scala, S. (2010). Targeting the inflammation in HCV-associated hepatocellular carcinoma: a role in the prevention and treatment. J. Transl. Med. 8, 109. doi: 10.1186/1479-5876-8-109
Conti, F., Buonfiglioli, F., Scuteri, A., Crespi, C., Bolondi, L., Caraceni, P., et al. (2016). Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 65, 727–733. doi: 10.1016/j.jhep.2016.06.015
D’Ambrosio, R., Aghemo, A., Rumi, M. G., Degasperi, E., Sangiovanni, A., Maggioni, M., et al. (2018). Persistence of hepatocellular carcinoma risk in hepatitis C patients with a response to IFN and cirrhosis regression. Liver Int. 38, 1459–1467. doi: 10.1111/liv.13707
Das, K., and Ghosh, M. (2017). Structured DAG oil ameliorates renal injury in streptozotocin-induced diabetic rats through inhibition of NF-kappa B and activation of Nrf2 pathway. Food Chem. Toxicol. 100, 225–238. doi: 10.1016/j.fct.2016.12.033
de Mochel, N. S., Seronello, S., Wang, S. H., Ito, C., Zheng, J. X., Liang, T. J., et al. (2010). Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 52, 47–59. doi: 10.1002/hep.23671
Decker, K., and Keppler, D. (1972). Galactosamine induced liver injury. Prog. Liver Dis. 4, 183–199.
Dejager, L., and Libert, C. (2008). Tumor necrosis factor alpha mediates the lethal hepatotoxic effects of poly(I:C) in D-galactosamine-sensitized mice. Cytokine 42, 55–61. doi: 10.1016/j.cyto.2008.01.014
Deng, L., Adachi, T., Kitayama, K., Bungyoku, Y., Kitazawa, S., Ishido, S., et al. (2008). Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J. Virol. 82, 10375–10385. doi: 10.1128/JVI.00395-08
Fujinaga, H., Tsutsumi, T., Yotsuyanagi, H., Moriya, K., and Koike, K. (2011). Hepatocarcinogenesis in hepatitis C: HCV shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species. Oncology 81(Suppl 1), 11–17. doi: 10.1159/000333253
Ganai, A. A., Khan, A. A., Malik, Z. A., and Farooqi, H. (2015). Genistein modulates the expression of NF-kappaB and MAPK (p-38 and ERK1/2), thereby attenuating d-galactosamine induced fulminant hepatic failure in Wistar rats. Toxicol. Appl. Pharmacol. 283, 139–146. doi: 10.1016/j.taap.2015.01.012
Gong, G., Waris, G., Tanveer, R., and Siddiqui, A. (2001). Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. U. S. A. 98, 9599–9604. doi: 10.1073/pnas.171311298
Guo, Q., Zhang, Y. J., Wang, J., Su, C. L., and Chang, X. (2003). Evaluation of clinical usage of bicyclol in the treatment of chronic HCV. Evaluat. Anal. Drug Use Hospitals China 3, 337–339. doi: 10.14009/j.issn.1672-2124.2003.06.005
Hassan, M., Selimovic, D., Ghozlan, H., and Abdel-Kader, O. (2007). Induction of high-molecular-weight (HMW) tumor necrosis factor (TNF) alpha by hepatitis C virus (HCV) non-structural protein 3 (NS3) in liver cells is AP-1 and NF-kappaB-dependent activation. Cell. Signal. 19, 301–311. doi: 10.1016/j.cellsig.2006.07.002
Hsu, C. C., Lien, J. C., Chang, C. W., Chang, C. H., Kuo, S. C., and Huang, T. F. (2013). Yuwen02f1 suppresses LPS-induced endotoxemia and adjuvant-induced arthritis primarily through blockade of ROS formation, NFkB and MAPK activation. Biochem. Pharmacol. 85, 385–395. doi: 10.1016/j.bcp.2012.11.002
Huang, Y. Q., Li, P. Y., Wang, J. B., Zhou, H. Q., Yang, Z. R., Yang, R. C., et al. (2016). Inhibition of Sophocarpine on Poly I: C/D-GalN-induced immunological liver injury in mice. Front. Pharmacol. 7, 256. doi: 10.3389/fphar.2016.00256
Jacobson, I. M., Lim, J. K., and Fried, M. W. (2017). American Gastroenterological Association Institute clinical practice update-expert review: care of patients who have achieved a sustained virologic response after antiviral therapy for chronic hepatitis C infection. Gastroenterology 152, 1578–1587. doi: 10.1053/j.gastro.2017.03.018
Koike, K., and Miyoshi, H. (2006). Oxidative stress and hepatitis C viral infection. Hepatol. Res. 34, 65–73. doi: 10.1016/j.hepres.2005.11.001
Lee, S. A., Kim, J., Sim, J., Kim, S. G., Kook, Y. H., Park, C. G., et al. (2016). A telomerase-derived peptide regulates reactive oxygen species and hepatitis C virus RNA replication in HCV-infected cells via heat shock protein 90. Biochem. Biophys. Res. Commun. 471, 156–162. doi: 10.1016/j.bbrc.2016.01.160
Leone, N., and Rizzetto, M. (2005). Natural history of hepatitis C virus infection: from chronic hepatitis to cirrhosis, to hepatocellular carcinoma. Minerva Gastroenterol. Dietol. 51, 31–46.
Li, K., Li, N. L., Wei, D., Pfeffer, S. R., Fan, M., and Pfeffer, L. M. (2012). Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55, 666–675. doi: 10.1002/hep.24763
Li, Y. X., Ren, Y. L., Fu, H. J., Zou, L., Yang, Y., and Chen, Z. (2016). Hepatitis B virus middle protein enhances IL-6 production via p38 MAPK/NF-kappaB pathways in an ER stress-dependent manner. PLoS One 11:e0159089. doi: 10.1371/journal.pone.0159089
Lin, B. R., Yu, C. J., Chen, W. C., Lee, H. S., Chang, H. M., Lee, Y. C., et al. (2009). Green tea extract supplement reduces D-galactosamine-induced acute liver injury by inhibition of apoptotic and proinflammatory signaling. J. Biomed. Sci. 16,35. doi: 10.1186/1423-0127-16-35
Lin, M. V., King, L. Y., and Chung, R. T. (2015). Hepatitis C virus-associated cancer. Annu. Rev. Pathol. 10, 345–370. doi: 10.1146/annurev-pathol-012414-040323
Lin, R. J., Chu, J. S., Chien, H. L., Tseng, C. H., Ko, P. C., Mei, Y. Y., et al. (2014). MCPIP1 suppresses hepatitis C virus replication and negatively regulates virus-induced proinflammatory cytokine responses. J. Immunol. 193, 4159–4168. doi: 10.4049/jimmunol.1400337
Lin, W., Tsai, W. L., Shao, R. X., Wu, G., Peng, L. F., Barlow, L. L., et al. (2010). Hepatitis C virus regulates transforming growth factor beta 1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 138, 2509–2518. doi: doi: 10.1053/j.gastro.2010.03.008
Lin, W., Wu, G., Li, S., Weinberg, E. M., Kumthip, K., Peng, L. F., et al. (2011). HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFkappaB. J. Biol. Chem. 286, 2665–2674. doi: 10.1074/jbc.M110.168286
Liu, G. T. (2009). Bicyclol: a novel drug for treating chronic viral hepatitis B and C. Med. Chem. 5, 29–43. doi: 10.2174/157340609787049316
Liu, Y., Kimura, K., Yanai, R., Chikama, T., and Nishida, T. (2008). Cytokine, chemokine, and adhesion molecule expression mediated by MAPKs in human corneal fibroblasts exposed to poly(I:C). Invest. Ophthalmol. Vis. Sci. 49, 3336–3344. doi: 10.1167/iovs.07-0972
Morgan, T. R., Ghany, M. G., Kim, H. Y., Snow, K. K., Shiffman, M. L., De Santo, J. L., et al. (2010). Outcome of sustained virological responders with histologically advanced chronic hepatitis C. Hepatology 52, 833–844. doi: 10.1002/hep.23744
Nakamoto, Y., and Kaneko, S. (2003). Mechanisms of viral hepatitis induced liver injury. Curr. Mol. Med. 3, 537–544. doi: 10.2174/1566524033479591
Paracha, U. Z., Fatima, K., Alqahtani, M., Chaudhary, A., Abuzenadah, A., Damanhouri, G., et al. (2013). Oxidative stress and hepatitis C virus. Virol. J. 10:251. doi: 10.1186/1743-422X-10-251
Peng, Z. G., Fan, B., Du, N. N., Wang, Y. P., Gao, L. M., Li, Y. H., et al. (2010). Small molecular compounds that inhibit hepatitis C virus replication through destabilizing heat shock cognate 70 messenger RNA. Hepatology 52, 845–853. doi: 10.1002/hep.23766
Peng, Z. G., Zhao, Z. Y., Li, Y. P., Wang, Y. P., Hao, L. H., Fan, B., et al. (2011). Host apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology 53, 1080–1089. doi: 10.1002/hep.24160
Prescott, J. B., Hall, P. R., Bondu-Hawkins, V. S., Ye, C., and Hjelle, B. (2007). Early innate immune responses to Sin Nombre hantavirus occur independently of IFN regulatory factor 3, characterized pattern recognition receptors, and viral entry. J. Immunol. 179, 1796–1802. doi: 10.4049/jimmunol.179.3.1796
Quintero, A., Pedraza, C. A., Siendones, E., Kamal ElSaid, A. M., Colell, A., Garcia-Ruiz, C., et al. (2002). PGE1 protection against apoptosis induced by D-galactosamine is not related to the modulation of intracellular free radical production in primary culture of rat hepatocytes. Free Radic. Res. 36, 345–355. doi: 10.1080/10715760290019372
Reig, M., Marino, Z., Perello, C., Inarrairaegui, M., Ribeiro, A., Lens, S., et al. (2016). Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 65, 719–726. doi: 10.1016/j.jhep.2016.04.008
Sanchez-Reus, M. I., Peinado, I. I., Molina-Jimenez, M. F., and Benedi, J. (2005). Fraxetin prevents rotenone-induced apoptosis by induction of endogenous glutathione in human neuroblastoma cells. Neurosci. Res. 53, 48–56. doi: 10.1016/j.neures.2005.05.009
Sumpter, R.Jr., Loo, Y. M., Foy, E., Li, K., Yoneyama, M., Fujita, T., et al. (2005). Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79, 2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005
Takahashi, N., Yamada, T., Narita, N., and Fujieda, S. (2006). Double-stranded RNA induces production of RANTES and IL-8 by human nasal fibroblasts. Clin. Immunol. 118, 51–58. doi: 10.1016/j.clim.2005.09.001
Tardif, K. D., Waris, G., and Siddiqui, A. (2005). Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 13, 159–163. doi: 10.1016/j.tim.2005.02.004
Wang, C., Chen, K., Xia, Y., Dai, W., Wang, F., Shen, M., et al. (2014). N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One 9:e108855. doi: 10.1371/journal.pone.0108855
Yao, G., Ji, Y., Zhou, X., Wang, Q., and Wu, W. (2002). Treatment of chronic hepatitis C by bicyclum, a randomized double-blind placebo controlled trial. Zhonghua Yi Xue Za Zhi 82, 958–960. doi: 10.3760/j:issn:0376-2491.2002.14.008
Zeremski, M., Petrovic, L. M., and Talal, A. H. (2007). The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J. Viral Hepat. 14, 675–687. doi: 10.1111/j.1365-2893.2006.00838.x
Zhang, J., Fu, B., Zhang, X., Zhang, L., Bai, X., Zhao, X., et al. (2014). Bicyclol upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. Bull. 100, 38–43. doi: 10.1016/j.brainresbull.2013.11.001
Keywords: bicyclol, hepatitis C virus, anti-inflammatory therapy, inflammatory factor, oxidative stress
Citation: Li H, Li J-R, Huang M-H, Chen J-H, Lv X-Q, Zou L-L, Tan J-L, Dong B, Peng Z-G and Jiang J-D (2018) Bicyclol Attenuates Liver Inflammation Induced by Infection of Hepatitis C Virus via Repressing ROS-Mediated Activation of MAPK/NF-κB Signaling Pathway. Front. Pharmacol. 9:1438. doi: 10.3389/fphar.2018.01438
Edited by:
Ying Yu, Shanghai Institutes for Biological Sciences (CAS), ChinaCopyright © 2018 Li, Li, Huang, Chen, Lv, Zou, Tan, Dong, Peng and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zong-Gen Peng, cHVtY3B6Z0AxMjYuY29t; Jian-Dong Jiang, amlhbmcuamRvbmdAMTYzLmNvbQ==