 Lei Liu
Lei Liu Logan M. Locascio
Logan M. Locascio Sylvain Doré
Sylvain Doré- 1Department of Anesthesiology, Center for Translational Research in Neurodegenerative Disease and McKnight Brain Institute, University of Florida, Gainesville, FL, United States
- 2Departments of Neurology, Psychiatry, Pharmaceutics, and Neuroscience, University of Florida, Gainesville, FL, United States
Ischemic stroke is one of the leading causes of death and long-term disability worldwide; however, effective clinical approaches are still limited. The transcriptional factor Nrf2 is a master regulator in cellular and organismal defense against endogenous and exogenous stressors by coordinating basal and stress-inducible activation of multiple cytoprotective genes. The Nrf2 network not only tightly controls redox homeostasis but also regulates multiple intermediary metabolic processes. Therefore, targeting Nrf2 has emerged as an attractive therapeutic strategy for the prevention and treatment of CNS diseases including stroke. Here, the current understanding of the Nrf2 regulatory network is critically examined to present evidence for the contribution of Nrf2 pathway in rodent ischemic stroke models. This review outlines the literature for Nrf2 studies in preclinical stroke and focuses on the in vivo evidence for the role of Nrf2 in primary and secondary brain injuries. The dynamic change and functional importance of Nrf2 signaling, as well as Nrf2 targeted intervention, are revealed in permanent, transient, and global cerebral ischemia models. In addition, key considerations, pitfalls, and future potentials for Nrf2 studies in preclinical stroke investigation are discussed.
Introduction
Ischemic stroke is one of the leading causes of death and long-term disability worldwide (Benjamin et al., 2018), an event which is more disabling than it is fatal. In the U.S. alone, ~795,000 people each year suffer a new or recurrent stroke, of which 87% are ischemic (Dirnagl et al., 2003; Feigin et al., 2017; Benjamin et al., 2018). However, effective clinical approaches are still limited. A successful therapeutic strategy for salvaging ischemic brain tissue and promoting functional outcomes is to quickly restore blood flow during acute ischemic stroke by introducing intravenous recombinant tissue plasminogen activator (rtPA), which has been available since 1996 (Prabhakaran et al., 2015; Romano and Sacco, 2015), in combination with thrombectomy. However, more than 95% of patients do not benefit from rtPA due to the narrow therapeutic window (4.5 h) and limited indications (Hacke et al., 2008; Fonarow et al., 2011; Sandercock et al., 2012; Emberson et al., 2014). In addition, there is no efficacious treatment that exhibits long-term recovery improvement. Therefore, development of new therapeutic strategies by targeting vital cellular components of the ischemic cascade is urgently needed.
The transcriptional factor Nrf2 is a major regulator of cellular and organismal defense mechanism against endogenous and exogenous stresses by coordinating basal and stress-inducible activation of multiple cytoprotective genes (Leonardo and Doré, 2011; Suzuki et al., 2013; Hayes and Dinkova-Kostova, 2014; Tonelli et al., 2017; Cuadrado et al., 2018; Yamamoto et al., 2018). Since its discovery in 1994 (Moi et al., 1994), Nrf2 biology has been extensively studied in understanding the structure, molecular mechanism, function, regulation of Nrf2 activity, downstream pathways and implications as a therapeutic target of diseases (Ma, 2013; Suzuki et al., 2013; Kumar et al., 2014; Cuadrado et al., 2018; Yamamoto et al., 2018). It is now recognized as a master regulator of redox homeostasis through the control of a wide array of target genes that share a common DNA sequence called the antioxidant response element (ARE) in the promoter region ensuring that its activity increases in response to redox perturbation, energy or nutrient fluxes, inflammation, toxicity, and disease conditions (Ma, 2013; Cuadrado et al., 2018; Yamamoto et al., 2018). The equivalent response element in mice and rats is called the electrophile response element (EpRE) (Friling et al., 1990; Wasserman and Fahl, 1997; Itoh et al., 2010; Yamamoto et al., 2018). Besides mediating antioxidant responses, Nrf2 contributes to the regulation of many primary and secondary metabolic processes (Hayes and Dinkova-Kostova, 2014; Tonelli et al., 2017). Consequently, targeting Nrf2 has emerged as an attractive therapeutic strategy for stroke prevention and reversal (Calkins et al., 2009; Leonardo and Doré, 2011; Wang Y. C. et al., 2011; Ma, 2013; Kumar et al., 2014; Tonelli et al., 2017).
This paper outlines the current understanding of the Nrf2 regulatory network and critically examined the recent evidence for the contribution of Nrf2 pathway in experimental ischemic stroke models. It includes extensive literature review for Nrf2 studies in experimental ischemic stroke mouse and rat models published by June 30, 2018 with a special focus on in vivo evidence for the role of Nrf2 in ischemic brain injury. We summarized the dynamic regulation of Nrf2 signaling, functional importance, and its targeted intervention in permanent, transient, and global cerebral ischemia preclinical models. Finally, we also assessed key considerations, pitfalls, and the potential for future Nrf2 studies in stroke investigation.
Overview of the Nrf2 Regulatory Network
A Brief History
Nrf2 is widely expressed in mammalian cells. In 1991, a study in the field of toxicology revealed that oxidative stress activates antioxidant genes through the antioxidant-response element (ARE), a cis-acting regulatory element that contributes to cellular defense in eukaryotes (Rushmore et al., 1991; Hayes and Dinkova-Kostova, 2014). In 1994, Nrf2 was firstly reported in the molecular biology field as an NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region (Moi et al., 1994; Raghunath et al., 2018). However, its biological function was unclear. In 1996, the first study on Nrf2 knockout mice showed that no overt abnormal phenotype was detected although the mice were susceptible to stresses (Chan et al., 1996). In 1997, a landmark study illustrated that the induction of two ARE-driven genes, glutathione S-transferase (GST) and NAD(P)H:quinone oxidoreductase-1 (NQO1), was abolished in the Nrf2 knockout mice by the phenolic antioxidant butylated hydroxyanisole (BHA), revealing that Nrf2 controls drug-metabolizing enzymes in vivo (Itoh et al., 1997). Recently, series of breakthrough studies have revealed that Nrf2 coordinately regulates a wide array of antioxidant response element/electrophile responsive element (ARE/EpRE)-driven genes that play critical roles in controlling endogenous resistance to various intrinsic and extrinsic stressors (Ma, 2013; Suzuki et al., 2013; Hayes and Dinkova-Kostova, 2014; Tonelli et al., 2017; Yamamoto et al., 2018).
Structure Domain of Nrf2
Nrf2 belongs to the cap'n'collar subfamily of the basic-region leucine zipper (CNC-bZIP) transcription factors. It is a modular protein composed of seven functional domains, Nrf2-ECH homology (Neh) domains 1–7, which have distinct functions (Figure 1A) (Chan et al., 1993; Katoh et al., 2005; Hayes and Dinkova-Kostova, 2014; Tonelli et al., 2017). The Neh1 domain contains the well-conserved CNC-bZIP region that heterodimerizes with small musculoaponeurotic fibrosarcoma oncogene (sMaf) proteins and binds ARE/EpRE sequence in DNA. This CNC-bZIP region, which can be found in several species, is vital for Nrf2 function. The N-terminal Neh2 is the domain by which Nrf2 binds to its primary negative regulator Keap1 through its low-affinity DLG and the high-affinity ETGE motifs. The C-terminal Neh3 region represents a transactivation domain that binds to chromo-ATPase/ helicase DNA-binding protein (CHD) 6 and activates transcription of Nrf2 target genes in concert with the Neh4 and Neh5 domains. The Neh4 and Neh5 regions of Nrf2 are transactivation domains that recruit the cAMP response element-binding protein (CREB)-binding protein (CBP) and/or receptor-associated coactivator (RAC) 3. The Neh6 domain, independent of Keap1, mediates the repression of Nrf2 stability with another negative regulator the dimeric β-transducin repeat-containing protein (β-TrCP), a substrate adaptor for the S-phase kinase-associated protein 1 (Skp1)–Cul1–Rbx1 core E3 complex, through DSGIS and DSAPGS motifs. The Neh7 domain is involved in the negative control of Nrf2 by physically binding to the retinoid X receptor (RXR).
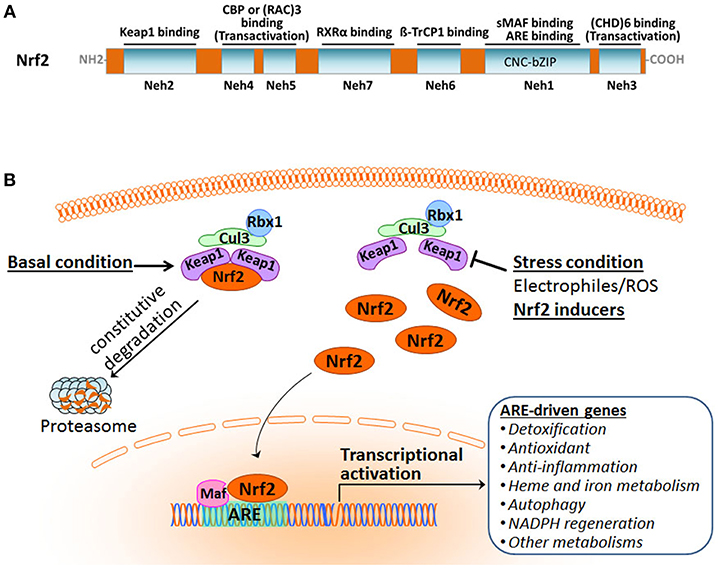
Figure 1. Overview of the Nrf2 pathway activation. (A) Functional domains of human Nrf2 protein. (B) The Keap1-dependent Nrf2 activation and response.
Overview of the Nrf2 Pathway
Organisms are equipped with a defense system to maintain homeostasis against constant intrinsic and extrinsic insults that result in the damage of nucleic acids, proteins, and membrane lipids. Nrf2 is a master regulator of the inducible cell defense system by controlling a broad range of cytoprotective genes (Hayes and Dinkova-Kostova, 2014; Suzuki and Yamamoto, 2015). Under basal homeostatic conditions, Nrf2 in the cytoplasm is predominately bound to the Kelch-like ECH-associating protein 1 (Keap1) through the Keap1–Cullin3 (Cul3)-Rbx1 ubiquitin E3 ligase complex and is constitutively degraded by the proteasome. Cul3 serves as a scaffolding protein that is bound to both Keap1 and Rbx1. As a result, Nrf2 abundance and activity are maintained at low levels (Figure 1B). Under stress conditions, Nrf2 protein is liberated from Keap1-mediated repression. The stabilized and accumulated Nrf2 translocates into the nucleus and, as a heterodimer with one of the small Maf proteins, binds to the ARE/EpRE in the promoter region of Nrf2 target genes, thus activating a wide array of cytoprotective genes (Ma, 2013; Kumar et al., 2014; Suzuki and Yamamoto, 2015; Taguchi and Yamamoto, 2017; Bellezza et al., 2018; Yamamoto et al., 2018).
It is becoming increasingly clear that Keap1 plays a central role in the regulatory mechanism of Nrf2. (1) Keap1 acts as a sensor of oxidative and electrophilic stresses for Nrf2 with a subcellular localization in the perinuclear cytoplasm (Suzuki and Yamamoto, 2017; Yamamoto et al., 2018). (2) Keap1 is a major repressor of Nrf2 through the activity of the Keap1–Cul3 complex. It is an adaptor protein for Cul3-dependent ubiquitination and specifically targets Nrf2 (Itoh et al., 1999, 2003; Kobayashi et al., 2004), thereby marking Nrf2 protein for rapid degradation by the ubiquitin–proteasome system. Under normal conditions, Nrf2 undergoes constitutive ubiquitination by the Keap1-Cul3 complex and resultant proteasomal degradation, leading to its low level of activity. Exposure to cellular insults like electrophiles or reactive oxygen species (ROS) brings activity of the E3 ubiquitin ligase Keap1-Cul3 complex to a halt, inhibiting Nrf2 ubiquitination and enabling Nrf2 protein stabilization and transcriptional activation. (3) Keap1 serves as a “floodgate” for Nrf2: Normally it functions to suppress Nrf2 nuclear translocation in response to cellular insults, the gene promotes translocation of Nrf2 into the nucleus to activate cytoprotective target gene expression. Keap1 has a relatively long half-life of 12 h compared to Nrf2's half-life of 20 min, contributing to its importance in the Nrf2 regulatory network.
Target Genes and Functions of Nrf2
The Nrf2-Maf complex binds to the specific promoter of its target genes. In the past decade, more than 200 target genes of Nrf2 have been identified through the analyses of gene expression profiling and chromatin immunoprecipitation (ChIP) (Suzuki et al., 2013; Kumar et al., 2014; Suzuki and Yamamoto, 2017; Rojo de la Vega et al., 2018). The Nrf2 target ARE-driven genes have been defined in encoding proteins for functional regulation of a wide range of biological processes including detoxification, antioxidation, anti-inflammation, NADPH regeneration, and intermediary metabolisms (Figure 1B). Nrf2 exerts multiple defense processes counteracting various stresses by the induction of these genes. Consequently, Nrf2 plays a fundamental role in maintaining the redox homeostasis of the cell.
Target Genes and Functions of Nrf2—Detoxification of Xenobiotics
Xenobiotics, including toxic chemicals and drugs, are detoxified by various drug-metabolizing enzymes and transporters. This detoxification mechanism is divided into three phases (Croom, 2012; Raghunath et al., 2018). Phase I enzymes mainly consist of cytochrome P450 oxidases that modify the xenobiotics through oxidation, reduction, or hydrolysis. Phase II conjugating enzymes include glutathione S-transferases, which catalyze the conjugation of reactive electrophile species with glutathione (GSH), attenuating the toxic potential of xenobiotics. Phase III transporters eliminate the GSH conjugates and offer protection against deleterious chemicals. It has been demonstrated that Nrf2 controls the expression of many drug-metabolizing enzymes [like distinct GST subunits and [NAD(P)H:quinone oxidoreductase 1) NQO1] and transporters [(drug-resistance-associated proteins (MRP)].
Target Genes and Functions of Nrf2—Antioxidant
A major function of Nrf2 is to resist oxidative stress, thereby maintaining redox homeostasis between reactive oxidants and endogenous antioxidant systems. Groups of Nrf2 targeted genes that regulate antioxidant defense and oxidant signaling have been defined. (1) Glutathione (GSH)-based: glutamate–cysteine ligase catalytic (GCLC) subunit and glutamate-cysteine ligase modifier (GCLM) control the entry of cystine into cells; glutathione peroxidase (GPX) 2 produces oxidized glutathione (GSSG) during the reduction of peroxides; and glutathione reductase (GSR) 1 reduces GSSG for maintenance of reduced intracellular GSH levels. (2) Thioredoxin (TXN)-based: Thioredoxin (TXN) 1, thioredoxin reductase (TXNRD) 1, and sulfiredoxin (SRXN) 1 reduce oxidized protein thiols. (3) Others: G6PDH and 6PGD reduce synthesis of NADPH, antioxidant protein Trx, stress response protein heme oxygenase 1 (HO1), and other proteins. Nrf2 coordinately regulates key components in the antioxidant system that precisely controls antioxidant defense at multiple levels, thus ensuring an adequate response to oxidants in time and space (Lin and Beal, 2006; Ma, 2013).
Target Genes and Functions of Nrf2—Anti-inflammation
Besides protecting against xenobiotic and oxidative insults, Nrf2 plays a role in anti-inflammation, which is supported by several recent findings (Kobayashi et al., 2016; Suzuki and Yamamoto, 2017). Several findings support the anti-inflammatory effect. The absence of Nrf2 leads to exacerbated inflammation in different murine models. Attenuated inflammation by Nrf2 is related to the inhibition of the nuclear factor-κB (NF-κB) pathway and proinflammatory cytokine level (Wardyn et al., 2015). Most recently, it was demonstrated that Nrf2 suppressed lipopolysaccharide (LPS)-induced inflammation by blocking the transcriptional upregulation of proinflammatory cytokines, including IL-6 and IL-1β. ChIP-seq and ChIP-qPCR analyses revealed that Nrf2 binds near the promoter region of these genes, and this Nrf2-mediated inhibition is independent of the Nrf2-binding motif and ROS level (Ahmed et al., 2017).
Target Genes and Functions of Nrf2—Heme and Iron Metabolism
Nrf2 also modulates the key components of heme and iron metabolism. HO1 catalyzes the cleavage of heme (which cannot be recycled) to form iron, carbon monoxide (CO), and biliverdin, which is immediately reduced to bilirubin (Doré and Snyder, 1999; Doré et al., 1999). HO1 is considered to be a stress protein that can be highly induced in a cell-specific manner by numberous conditions (Ewing et al., 1992). Iron is intimately linked to oxygen by acting as an essential nutrient cofactor in enzymes for oxygen transport, oxidative phosphorylation, and metabolite oxidation. However, excess labile iron—not bound to ferritin—would facilitate the formation of oxygen-derived free radicals capable of damaging biomolecules (Rouault, 2013; Kerins and Ooi, 2017). The biological utilization of iron is a tightly regulated process. Constitutive Nrf2 activation and subsequent deregulation of iron metabolism have been implicated in cancer development. Nrf2-mediated upregulation of HO1 or the iron storage protein ferritin can lead to enhanced proliferation and therapy resistance.
Target Genes and Functions of Nrf2—NADPH Regeneration and Other Metabolic Processes
NADPH is an essential cofactor for many drug-metabolizing enzymes and antioxidants. Nrf2 controls NADPH production by regulating key NADPH-generating enzymes, such as glucose-6-phosphate dehydrogenase (G6pd), 6-phosphogluconate dehydrogenase (Pgd), isocitrate dehydrogenase 1 (Idh1), and malic enzyme 1 (Leonardo and Doré, 2011; Dinkova-Kostova and Abramov, 2015). In addition, Nrf2 controls the expression of multiple metabolic enzymes, indicating its unique role between redox and intermediary metabolism. Nrf2 also regulates fatty acid oxidation and lipases that influence lipid metabolism and transcription factors (Hayes and Dinkova-Kostova, 2014).
Regulatory Mechanisms of Nrf2
Besides the predominant view that Nrf2 activity is mainly regulated at the protein stability level by Keap-mediated repression, accumulated evidence show that Nrf2 activity is tightly controlled at the transcriptional and post-translational levels during both basal and stressful conditions (Hayes and Dinkova-Kostova, 2014; Tonelli et al., 2017; Yamamoto et al., 2018). Given the central role of Nrf2 in cellular defense, the regulatory mechanisms of Nrf2 activity are intricate and multifaceted.
Regulatory Mechanisms of Nrf2–Transcriptional Regulation
Nrf2 mRNA is broadly expressed across different species (Ma, 2013; Tonelli et al., 2017; Yamamoto et al., 2018), and its basal levels vary within different organs. Nrf2 activity is at least partially regulated at the transcriptional level. (1) The rodent Nrf2 gene comprises two ARE-like sequences in its promoter region and, in response to electrophilic stimuli, appropriately autoregulates its own expression level in concert with sustaining the induction of ARE-driven genes, offering a positive feedback mechanism to augment the Nrf2 signal. In contrast, the ARE-like sequences in the human Nrf2 gene promoter region are reportedly associated with reduced expression of Nrf2, which enhances vulnerability to lung cancer. (2) The Nrf2 gene promoter comprises a binding site for NF-κB, which can be induced by environmental stress, injury or inflammation. Indeed, Nfe2l2 transcription level in human monocytes is indeed activated by LPS-induced inflammation. (3) It was reported that Nfe2l2 transcription in tumor cells can be amplified by the Notch signaling pathway and the phosphoinositide 3-kinase (PI3K)-Akt pathway, ultimately contributing to the increased expression of Nrf2.
Regulatory Mechanisms of Nrf2—Post-transcriptional Regulation
A microRNA (miRNA) is a small non-coding RNA molecule that functions in RNA silencing and post-transcriptional regulation of gene expression. At least eight miRNAs have been identified as direct modulators of Nrf2 expression at the transcriptomic level (Ayers et al., 2015). The miR-144 was identified to negatively regulate Nrf2 level in reticulocytes of homozygous sickle cell disease (HbSS) patients (Bryan et al., 2013; Kurinna and Werner, 2015) (140). Increased miR-144 is associated with the reduction of both Nrf2 level and GSH regeneration as well as impeded oxidative stress defense.
Regulatory Mechanisms of Nrf2—Post-translational Regulation in Nrf2 Protein Stability
Keap1-Dependent Repression Mechanism
(1) The prevailing view is that Keap1 is a primary repressor of Nrf2. Under basal conditions, Keap1 constantly targets Nrf2 for ubiquitination and proteasomal degradation, resulting in the disruption of Nrf2 protein stability and maintenance of Nrf2 signaling capacity at a very low level. In response to electrophiles or stressors, Nrf2 is liberated from the Keap1 repression (i.e., derepression) by inhibiting ubiquitylation and proteasomal degradation that allows newly-synthesized Nrf2 to be rapidly stabilized and activates transcriptional activation of cytoprotective genes. This view is supported by many reports of Keap1 knockout mice and Keap1 knockdown human cells that exhibited sufficient Nrf2 activity (Kensler et al., 2007; Yates et al., 2009; Bellezza et al., 2018; Yamamoto et al., 2018). Blockage of Keap1 expression by miR-141 or miR-200a triggers increased Nrf2 activity at the protein stabilization level. Transgenic complementation rescue assay is a comprehensive and powerful approach to delineate the in vivo functions of proteins (Motohashi et al., 2011; Katsuoka and Yamamoto, 2016). The esophagus lesions and resultant mortality in Keap1 knockout mice were fully retrieved by concurrent Nrf2 disruption, and aberrant phenotypes caused by Keap1 deficiency can also be rescued by transgene-derived Keap1. Epigenetic silencing of the Keap1 gene by hypermethylation of its promoter results in upregulation of Nrf2 in patients with gliomas and different cancers of breast, lung, prostate and colorectal (Tonelli et al., 2017). (2) The function of Keap1 within various electrophiles is related primarily to three critical cysteine residues: Cys151, Cys273, and Cys288 (Yamamoto et al., 2018). Keap1 is a thiol-rich protein with many cysteine residues and thus is sensitive to electrophiles (McMahon et al., 2010). Therefore, covalent modifications of the cysteine residues had been proposed to influence the function of Keap1, which releases Nrf2 (McMahon et al., 2010). Affirmatively, covalent binding of electrophiles to critical cysteine residues has been observed. Meanwhile, various lines of studies in cultured cells, zebrafish, and mice demonstrate that Keap1 functions as a sensor due to the prominent contribution of the cysteine residues. Multiple studies involving substitution of Cys151 strongly support its role as a main cysteine sensor by modulating the activity of the ubiquitin E3 ligase Keap1-Cul3 complex to respond to different electrophiles. (3) To activate Nrf2 by inhibiting Keap1 activity: Many ARE activity inducing agents are soft electrophiles that inhibit Keap1 by modifying Cys151, Cys226/Cys613, Cys-273/Cys-288, or Cys-434. Thus, the presence of multiple Cys-based sensors in Keap1 allows Nrf2 to be de-repressed in response to many xenobiotic stressors. (4) To activate Nrf2 by competitive inhibition of Keap1: p62, the first mammalian selective autophagy cargo receptor (Chu, 2018), is able to directly interact with the Nrf2- binding site on Keap1, a component of Cullin-3-type ubiquitin ligase for Nrf2 (Komatsu et al., 2010; Lau et al., 2010). Thus, an overproduction of p62 or a deficiency in autophagy competes with the interaction between Nrf2 and Keap1, resulting in Nrf2 stabilization and transcriptional activation of Nrf2 target genes. Additionally, p62 is a target gene of Nrf2 and creates a positive feedback loop by inducing ARE-driven gene transcription (Jain et al., 2010). Oxidative stress-triggered Nrf2 results in the induction and accumulation of p62 that in turn activates Nrf2 activation (Komatsu et al., 2010).
Keap1-Nondependent Repression Mechanism
(1) The Nrf2 is has been described to be suppressed by b-TrCP and GSK-3; in addition to the well-studied Keap1, E3 ubiquitin ligase adaptor β-TrCP is another negative repressor of Nrf2 stabilization (Rada et al., 2012). Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity (Chowdhry et al., 2013). Intraperitoneal injection of the GSK-3 inhibitor through the GSK-3/β-TrCP axis led to increased Nrf2 and HO1 levels in liver and hippocampus (Rada et al., 2012). Activation of the GSK3β/β-TrCP axis by gene knockdown of PHLPP1 in NRK52E cells enhanced Nrf2-responsive antioxidant enzymes HO1 and NQO1 (Mathur et al., 2018). (2) The Nrf2 is suppressed by CRIF1 and RNF4: CR6-interacting factor 1(CRIF1) physically interacts with both N- and C-terminal regions of Nrf2 and promotes Nrf2 ubiquitination and subsequent proteasome-mediated Nrf2 protein degradation (Kang et al., 2010). Crif1-knockdown BMMSCs caused increased oxidative stress and apoptosis after irradiation injury, partially due to a suppressed antioxidant response mediated by decreased Nrf2 nuclear translocation (Chen L. et al., 2017). RING finger protein 4 (Rnf4) mediates polyubiquitylation of polysumoylated Nrf2, leading to its subsequent degradation in promyelocytic leukemia-nuclear bodies (Malloy et al., 2013). Respiratory syncytial virus-induced Nrf2 degradation occurs in a SUMO-specific E3 ubiquitin ligase—RING finger protein 4 (Rnf4)-dependent manner.
Cerebral Ischemia And Relevant Preclinical Animal Models
Ischemic stroke is caused by cerebral arterial occlusion that leads to a critical reduction or loss of regional cerebral blood flow, and it occurs as a consequence of multiple vascular diseases, including cardioembolism, atherosclerosis, small vessel disease, and cryptogenic diseases (Adams and Biller, 2015; Mehndiratta et al., 2015). Interruption of the blood supply initiates complex spatial and temporal events involving hemodynamic, biochemical, and neurophysiologic alterations that ultimately lead to a pathological disturbance and a wide range of clinical symptoms (Lo et al., 2003; Iadecola and Anrather, 2011). The severity and temporal evolution of ischemic injury depend on the extent of cerebral blood flow (CBF), localization, duration of ischemia and coexisting systemic diseases among individual patients (Martini and Kent, 2007; Bang et al., 2008; Shen and Duong, 2008; Liebeskind, 2010).
Experimental ischemic stroke models are indispensable to our understanding of the events occurring in the ischemic and reperfused brains, enabling us to elucidate the pathophysiological mechanisms of ischemic brain injury and develop novel therapeutic treatments (Bosetti et al., 2017; Quinn et al., 2018). The vast majority of stroke models are carried in rats or mice because they present clear advantages of lower cost, resemblance to human cerebrovascular anatomy and physiology, and reproducibility of studies (Durukan and Tatlisumak, 2007; Durukan et al., 2008). In addition, genetic modification is usually applied in mice to illuminate molecular pathophysiological conditions like stroke. Cerebral ischemia in humans is divided into two categories: focal and global. Focal cerebral ischemia occurs when CBF is disrupted within a specific brain region, whereas global cerebral ischemia occurs when CBF is blocked throughout most or all of the brain. Given that ischemic stroke in humans occurs mostly in the territory of the middle cerebral artery (MCA), experimental focal cerebral ischemia models, including permanent and transient types, have served as the most widely used tool in the stroke research field (Dorr et al., 2007; Mehta et al., 2007). In focal cerebral ischemia, there is no blood flow at the infarct core, but there is usually a gradient of blood flow from the inner core to the neighboring ischemic area due to the collateral circulation. In global cerebral ischemia, global blood flow is completely stopped or remarkably reduced, depleting the energy supply and hindering cerebral metabolism and function. The reperfusion of blood flow effectively treats acute stroke; however, it can also exacerbate tissue damage and limit the recovery of function. Oxidative stress plays a key role in the pathogenesis of cerebral ischemia-reperfusion injury.
(1) Permanent cerebral ischemia (pdMCAO and pMCAO): Technically, the MCA can be selectively occluded at a distal or proximal site, referred to as permanent distal MCA occlusion (pdMCAO) or permanent (proximal) MCA occlusion (pMCAO) cerebral ischemia model, respectively. The pdMCAO model produces highly reproducible ischemic cortical lesions that are predominantly restricted to the barrel regions of cortex, inducing definable sensorimotor deficits that closely mimics ischemic stroke in humans. Thus, it is believed to be one of the most predictable and useful stroke models, allowing researchers to look at long-term recovery with high survival rates (Doyle and Buckwalter, 2014). Permanent cerebral ischemia can also be generated by the intraluminal suture method used in MCAO (pMCAO).
(2) Transient focal cerebral ischemia (tMCAO): This model is easy to perform in a controlled manner. The intraluminal suture MCAO model in rats and mice is the most frequently used model. This model exhibits reproducible MCA region infarctions that depend on the shape, size, and insertion length of the thread, allowing reperfusion by retracting the suture. MCAO generates ischemic cell death in the striatum and overlying the frontal, parietal, temporal, and portions of the occipital cortex. MCAO also precipitates variable damage in the thalamus, cervicomedullary junction, substantia nigra, and hypothalamus. Ischemic brain injury widely affects diverse brain regions and leads to complex motor, sensory, autonomic, and cognitive deficits (Carmichael, 2005). Usually ischemia must be induced for 60–120 min to obtain reproducible infarct volumes in transient focal ischemia models. In contrast, focal ischemia for more than 3 h precludes reversibility (Kasner and Grotta, 1998; Sicard and Fisher, 2009).
(3) Global cerebral ischemia (GCI): global cerebral ischemia (GCI) during cardiac arrest results in selective and delayed neuronal death of pyramidal neurons in the hippocampal CA1 region, similar to the situation in humans, and consequent cognitive decline (Traystman, 2003; Ostrowski et al., 2016).
Role Played by NRF2 During Cerebral Ischemia
In recent years, studies reported findings concerning the dynamic change of Nrf2 signaling, its functional importance, and its targeted intervention in cerebral ischemia. These findings provide insights into whether, when and how Nrf2 functions during brain injury. Accordingly, we mainly focused on the following questions. (1) What is the dynamic regulation of the Nrf2 signaling following cerebral ischemia? (2) Does the evidence from Nrf2−/− mice support the functional importance of Nrf2 during ischemic injury? (3) Whether Nrf2 induction is protective against ischemic injury and is facilitative for recovery? Specific focus is given to the in vivo evidence in different rodent cerebral ischemia models. Then, the pitfalls and concerns of current Nrf2 experimental ischemic stroke studies are discussed, which would be valuable for future studies.
The Dynamic Regulation of the NRF2 Signaling During Cerebral Ischemia
Normally, Nrf2 is largely localized in the cytoplasm and is maintained at a low basal level due to its binding affinity to Keap1. However, when cells are exposed to excessive oxidative stimuli during cerebral ischemia, Nrf2 is liberated from Keap1, translocates into the nucleus, and binds to the ARE sequence, thereby upregulating the expression of its target genes, which code cytoprotective proteins like anti-oxidative enzymes. In recent years, studies have provided substantial in vivo evidence of dynamic alternation of Nrf2 expression, as well as its target genes, and cellular and subcellular distribution of Nrf2 during different stages of cerebral ischemia. These findings, utilizing focal ischemia models with or without reperfusion in addition to global ischemia models, help us to identify the role of the Nrf2 regulatory network in the context of cerebral ischemia.
A number of studies with permanent cerebral ischemia models (pdMCAO and pMCAO) investigated the Nrf2/ARE pathway in response to ischemic insults (Table 1). Several permanent cerebral ischemia studies showed that, at 24 h after pMCAO, the protein expression levels of Nrf2 (total and nuclear) and its target antioxidant genes HO1 and SOD were upregulated in the ischemic cortex of mice or rats (Chang et al., 2013; Zhang J. et al., 2014; Zhao et al., 2014). The obvious increase of Nrf2 downstream antioxidant proteins can be sustained for at least 3 days following ischemia, revealed by the 1.8- to 3.6-fold increase in HO1, NQO1, SOD2, and GPx proteins after pdMCAO (Liu et al., 2018). The inflammatory factors IL-1β and IL-6 were dramatically increased in mouse brains for at least 48 h after pdMCAO (Clausen et al., 2017). These findings are supported by immunohistochemical analyses (Chen et al., 2012; Kao et al., 2013; Meng et al., 2014). Minimal Nrf2 and HO1 positive cells were detected in the sham group, indicating low baseline levels of Nrf2 and HO1 in the non-ischemic cortex areas, levels which increased significantly after pMCAO. Such signals were detected in neurons, astrocytes, and microglia, indicating concurrent expression of Nrf2 and HO1 in most cortical cells. On the contrary to above data, Nrf2 was reported to be significantly decreased at the mRNA level 6 h after pdMCAO, whereas the overall protein level was comparable to that under basal conditions (Clausen et al., 2017). A study reported a significantly decreased level of Nrf2 protein along with an increased level of antioxidant protein HO1 in rat cortex after pMCAO (Wang et al., 2018).
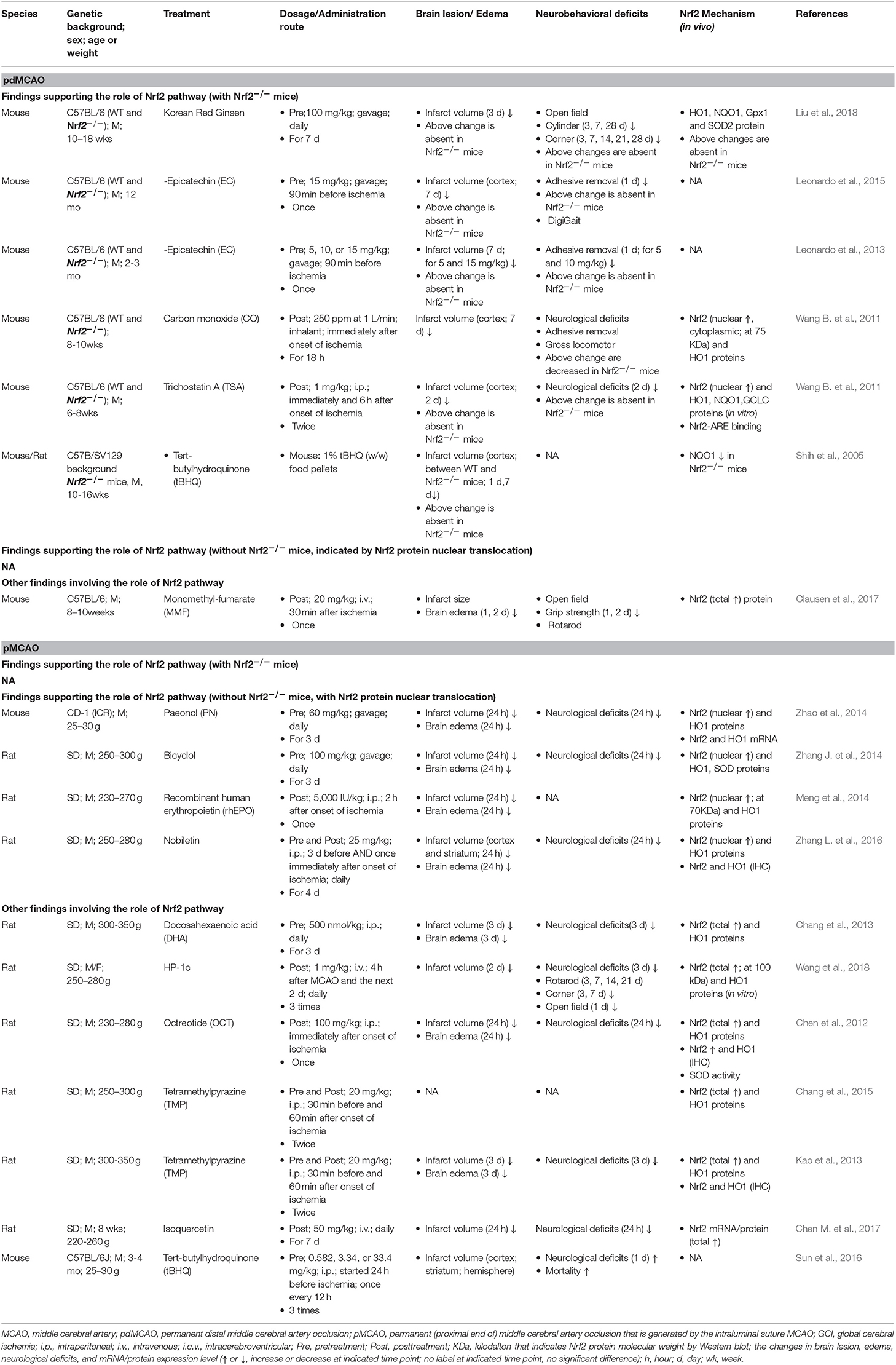
Table 1. Preclinical studies of Nrf2 in permanent cerebral ischemia models of mice and rats.
It appears that the majority of tMCAO studies support the activation of the Nrf2/ARE pathway in response to focal cerebral ischemia and perfusion (Tables 2, 3). Because the ischemia-reperfusion injury measured by infarct volume is widely accepted to be most severe at 24 h following tMCAO, most studies investigated the Nrf2 pathway by Western blot (WB) or immunohistological analyses at that time point, with some extending to earlier (2–8 h) or later stages (3–14 d). In these studies, the Nrf2 pathway was examined in different ischemic brain tissues including the cortex, hemisphere, hippocampus, striatum, and cerebellum. The researchers selected 1–2 h ischemia when designing these tMCAO ischemia-reperfusion animal models. We summarized these studies together in order to provide an overview of this field. Most studies measured total Nrf2 level in the ischemic cortex or brain at 24 h after tMCAO (1–2 h); studies subjects showed Nrf2 upregulation at the mRNA level (Li et al., 2013; Guo et al., 2014) and protein level (up to 2–3 fold higher) (Han et al., 2014; Li et al., 2015; Peng et al., 2015; Shi et al., 2015; Cai et al., 2017; Miao et al., 2018; Shang et al., 2018). Such an increase in Nrf2 protein might begin at 8 h (Cai et al., 2017) and be sustained over 3–14 days (Ding et al., 2015; Lin et al., 2016; Bai et al., 2017; Shang et al., 2018). Following nuclear translocation, Nrf2 protein accumulated in the nucleus (Li et al., 2013; Lv et al., 2017) and increased the Nrf2-binding activity to the ARE. Studies presented that the high nuclear Nrf2 protein level (Ding et al., 2015; An et al., 2018) and increased DNA binding activity of Nrf2 with the ARE can be observed, at 7 days after tMCAO (Li et al., 2014). The Nrf2 target antioxidant protein HO1 (up to 3 fold) (Li et al., 2013; Zhang M. et al., 2014; Hua et al., 2015; Peng et al., 2015; Lv et al., 2017; Yang et al., 2017), NQO1(Miao et al., 2018), GCLC and LCLM (Han et al., 2014; Shi et al., 2015) were also upregulated. Several reports showed that the higher expression level of HO1 can continue over 2–14 days (Zhang M. et al., 2014; Ding et al., 2015; Lin et al., 2016). At 24 h after tMCAO, the total and nuclear Nrf2 proteins levels in the ischemic hippocampus also implied significantly higher expression levels (Shi et al., 2015; Lou et al., 2016), which may continue for at least 2 days. In addition, it was reported by immunohistochemistry that, Nrf2 levels in the peri-infarct regions began to show a significant increase at 2 h with a peak at 8 h of reperfusion after 1 h tMCAO, and its target antioxidative proteins such as thioredoxin, glutathione, and HO1 showed significant increases at 24–72 h (Tanaka et al., 2011). Another study presented that Nrf2 immunoreactivity was detectable in the neurons, endothelial cells, astrocytes, and microglia in the peri-infarct area at 7 days after 1 h tMCAO (Shang et al., 2018). A luciferase mouse model, a Keap1-dependent oxidative stress detector, was employed to visualize the time-dependent Nrf2 expression from brain ischemia onset through 7 days after tMCAO (Takagi et al., 2014; Nakano et al., 2017). The in vivo optical signals of Nrf2 expression were not detected in the earliest stages but peaked at 24 h after ischemia. Such Nrf2 expression was mainly detected in in the penumbra area, largely localizing inside neurons and astrocytes (Srivastava et al., 2013; Takagi et al., 2014). By quantifying Nrf2 labeled with green fluorescent protein (GFP), researchers have found that Nrf2 and HO1 exhibited higher protein levels in the ischemic cortex at 4–48 h and striatum at 24 h after ischemia, which are consistent with the findings above. In contrast, some studies present the results that indicate suppression of the Nrf2/ARE pathway during tMCAO. At 24 h after tMCAO, the total (Wang L. et al., 2012; Pang et al., 2016; Wang et al., 2016, 2018; Chumboatong et al., 2017; Janyou et al., 2017) and nuclear (Wicha et al., 2017) Nrf2 proteins were found to be downregulated, and the same was observed with HO1 (Wang L. et al., 2012; Pang et al., 2016; Wang et al., 2016, 2018; Chumboatong et al., 2017; Wicha et al., 2017) and NQO1 (Janyou et al., 2017). On the contrary to above findings, several reports found that no change was detected in the markers above (Wu L. et al., 2013; Zhao et al., 2016; Wu G. et al., 2017; Zhang W. et al., 2018).
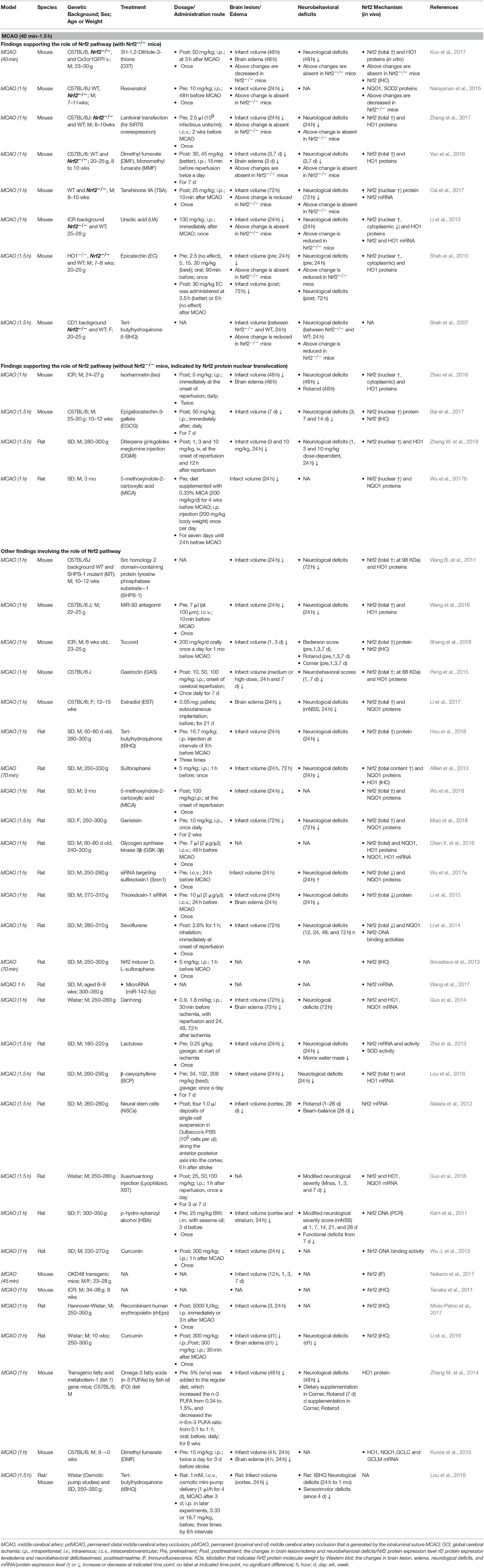
Table 2. Preclinical studies of Nrf2 in transient cerebral ischemia models of mice and rats (MCAO, 40 min-1.5 h).
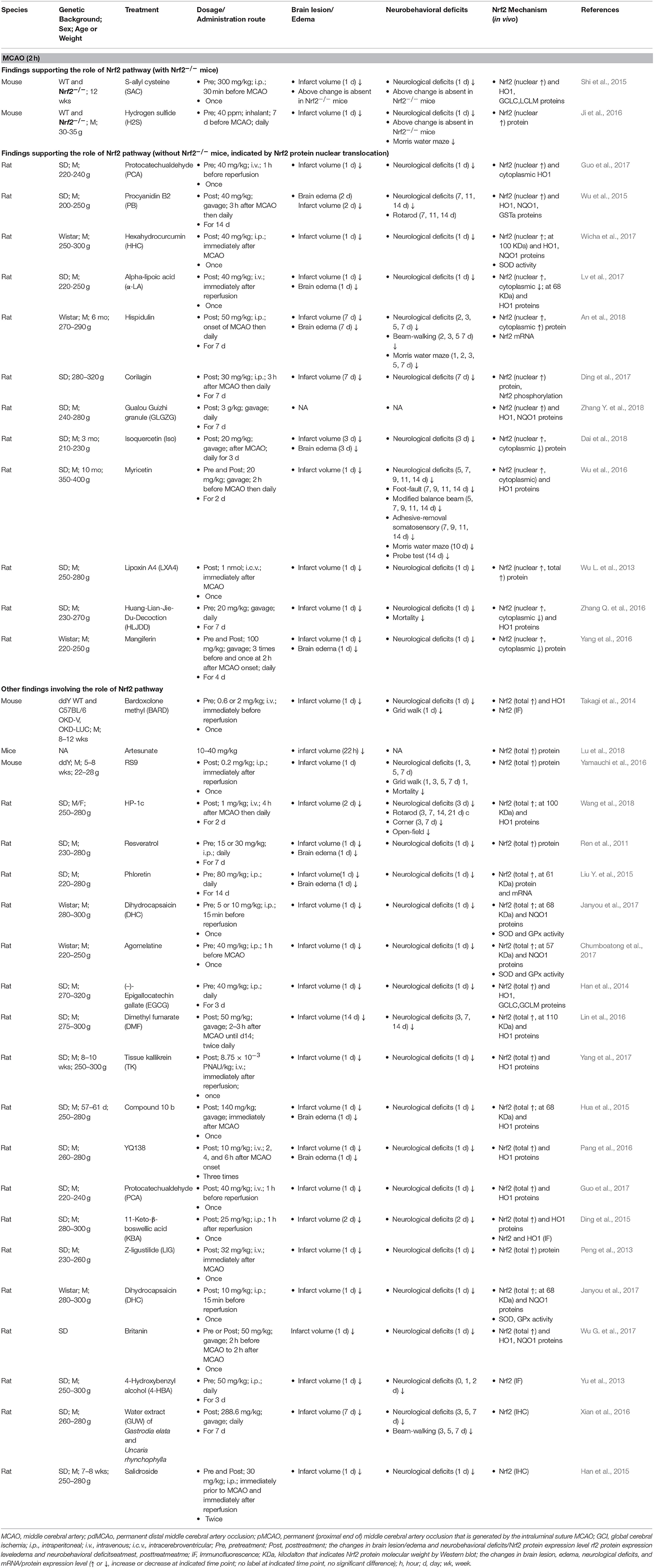
Table 3. Preclinical studies of Nrf2 in transient cerebral ischemia models of mice and rats (MCAO, 2 h).
The activation of the Nrf2/ARE pathway following global ischemia damage remains controversial (Table 4). This might be because of the various experimental factors like animal background, age, model, observation time points after ischemia, presence or absence of reperfusion, and site of the sample. At 24 h after GCI (2 vessel occlusion, 2VO), hippocampal Nrf2 content increased nearly 2-fold as determined by biochemical assay (Atef et al., 2018); striatal nuclear Nrf2 protein level and DNA binding activity of Nrf2 increased about 1.5-fold following reperfusion compared with the sham group; and there was notable upregulation in the detoxification and antioxidant proteins HO1, NQO1, GCLC, and GCLM (Ya et al., 2017). At 3 days after GCI (2VO), the total, cytoplasmic, and nuclear protein levels of Nrf2 were upregulated in the hippocampal CA1 region of rats although the increase in nuclear Nrf2 was not significant (Chen B. et al., 2016), a finding which is supported by another report (Lei et al., 2016). No change in HO1 protein level was detected at 3 days following brain injury (Chen B. et al., 2016). An immunofluorescence-based study showed that the Nrf2 level was slightly (but not significant) higher in the rats hippocampal CA1 area along with a decline of HO1 expression at 7 days after GCI (2VO) (Tulsulkar and Shah, 2013). In a rat GCI (4VO) model, activation of Nrf2 in the hippocampal CA1 region was examined over 3 days by four metrics including nuclear translocation of Nrf2, total Nrf2 protein level, DNA-binding of Nrf2, and induction of Nrf2 regulated proteins (Tu et al., 2015). The cytosolic but not nuclear Nrf2 protein was slightly upregulated from 12 to 72 h as evidenced by WB and immunostaining analyses, whereas the total Nrf2 protein level remained unchanged at 24 h. No change in the Nrf2 DNA binding activity was detected at 24 h and 72 h. The Nrf2-regulated antioxidant proteins HO1, NQO1, SOD2, and GPx1 were slightly altered within 72 h. These findings are supported by another report of GCI with less time of ischemia (Wang et al., 2013) that presented the upregulation of HO1 protein at 3 days. Lastly, it should be pointed out that the reports are inconclusive concerning the protein expression levels of Nrf2 with its target genes and subcellular distribution of Nrf2 following GCI. For example, hippocampal Nrf2 protein was reported to have not changed (Yang Y. et al., 2015), significantly decreased (Liu H. et al., 2015), or increased (Ashabi et al., 2015; Lee et al., 2015) following ischemia.
Table 4. Preclinical studies of Nrf2 in global cerebral ischemia models of mice and rats.
Functional Benefit of NRF2 In Cerebral Ischemia—in vivo Evidence From NRF2−/− Mice
Multiple lines of evidence demonstrate the beneficial contribution of Nrf2 in various pathological conditions, and most come from studies using Nrf2 knockout (Nrf2−/−) or knockdown animals. Reports showed that Nrf2−/− mice do not exhibit any overt abnormal phenotype regarding size, body weight, food intake, mobility, fertility, or other characteristics at baseline (Itoh et al., 1997; Wang et al., 2007; Wang Y. C. et al., 2011; Wang B. et al., 2012; Leonardo et al., 2013, 2015; Doré, 2015; Liu et al., 2018). In addition, Nrf2−/− mice exhibit similar cerebrovascular architecture in anastomosis, vascular physiology, and blood pH compared to their WT counterparts (Leonardo et al., 2015). Basal GST and NQO1 activities, but not GSH content, modestly but significantly decreased in multiple brain regions of Nrf2−/− mice compared to their Nrf2+/+ littermates, suggesting that the constitutive synthesis of brain GSH does not rely on Nrf2 function, at least in young adult mice (Shih et al., 2005).
However, in response to the various ischemic insults, it is becoming clear that Nrf2 plays a critical role in protection against ischemic brain damage (Tables 1–3). Indeed, older and recent studies with different cerebral ischemia models have revealed the functional benefit of Nrf2 on infarct volume, brain edema, and neurobehavioral deficits after ischemia. Following permanent ischemia injury without reperfusion, Nrf2 deficiency exacerbated the acute development of brain lesions, revealed by severe infarct volume at 3 days but not at 1 day after pdMCAO, and these mice exhibited relatively poor sensorimotor function over 28 days (Liu et al., 2018). At 2 days after pdMCAO, Nrf2−/− mice had significantly larger cortical infarct volume and more severe neurological deficits than WT controls (Wang B. et al., 2012). Another group showed that Nrf2 deficiency slightly (but not significantly) enlarged the cortical infarct size at 1 day and sustained considerably more damage at 7 day after pdMCAO (P < 0.01), which further supports the findings above (Shih et al., 2005). Moreover, it is well-known that reperfusion after a long period of ischemia can exacerbate brain damage. Following cerebral ischemia injury with reperfusion, several studies showed that Nrf2−/− mice exhibited aggravated acute brain damage in infarct volume and neurological deficits scores at 1 day after tMCAO with 1 h (Li et al., 2013; Yao et al., 2016; Zhang et al., 2017), 1.5 h (Shah et al., 2007), and 2 h (Shi et al., 2015) of ischemia. In these studies, no significant difference in blood pH, PaO2, PaCO2, or cerebral blood fluid (CBF) was observed before, during, or after ischemia-reperfusion between WT and Nrf2−/− at indicated time points as monitored by laser-Doppler flowmetry (Shah et al., 2010). In contrast, another contradictory report showed that no significant difference in infarct volume was observed between Nrf2−/− and WT mice 2 days after 1 h tMCAO (Narayanan et al., 2015). This discrepancy might be related to the different genetic background or age of mice or the experimental setting.
Nrf2 disruption depresses the capacity of the cellular antioxidant system and diminishes upregulation of its target cytoprotective proteins in response to ischemia (Tables 1–3). A study showed that, along with severe ischemic brain damage 3 days after pdMCAO, Nrf2 deficiency destroyed the ischemia-induced increase of antioxidant/detoxification proteins NQO1, HO1, SOD2, and Gpx1 (Liu et al., 2018). Oxidative stress and inflammation plays a key role in the pathological process of ischemia. Excessive production of ROS and reactive nitrogen species (RNS) contributes to the activation of inflammatory processes, further aggravating brain injury and functional deficits during or after stroke. Toll-like receptor 4 (TLR 4) initiates inflammatory processes and induces the expression of inflammatory elements IL-6, TNFα, and IL-1β through activation and nuclear translocation of NF-κB in cerebral ischemia/reperfusion injury (Arslan et al., 2010; Wang Y. C. et al., 2011). At 24 h after 1 h tMCAO, Nrf2 knockout upregulated the TLR4 and NF-κB proteins mediated by the inflammatory response in the ischemic cortex (Li et al., 2013).
Astrocytes interact closely with neurons to provide structural and functional support at multiple levels, including ion and water homeostasis, chemical signal transmission, blood flow regulation, immune and oxidative stress defense, and supply of metabolites (Liu et al., 2017). Given that the ARE-regulated genes are preferentially activated in glial cells, which have more effective detoxification and antioxidant capacities than neurons (Vargas and Johnson, 2009; Gan et al., 2012; Haskew-Layton et al., 2013), Nrf2 in glia cells can protect neurons from a wide array of insults like stroke (Vargas and Johnson, 2009). In response to ischemic insults, astrocytes undergo structural and functional changes, and interact with other glial cells to defend neurons (Magaki et al., 2018). Indeed, in the ischemic cortex, Nrf2 deficiency exacerbated reactive astrogliosis, dysfunction in glutamate metabolism, and water permeability within 3 days after pdMCAO. The spatiotemporal pattern of reactive astrogliosis correlated well with acute ischemic damage progression. Interestingly, the microglia activation was affected considerably under Nrf2 absence.
NRF2 Targeted Intervention In Ischemic Stroke—Advancements From Experimental Ischemic Stroke Model
Given the fundamental role of Nrf2 in redox homeostasis, many studies have presented the unique contribution of Nrf2 in neuroprotection against various diseases (Ma, 2013; Kumar et al., 2014; Sun et al., 2017; Cuadrado et al., 2018). For ischemic stroke, Nrf2/ARE activation is expected to trigger a cytoprotective response counteracting deleterious ischemic events. The beneficial effects of some candidate Nrf2 inducers were directly proved by the findings in Nrf2−/− mice. The typical findings were listed below (just to name a few), and more are also depicted in Tables 1–4.
Dimethyl Fumarate and Monomethyl Fumarate
Dimethyl fumarate (DMF) and its primary metabolite monomethyl fumarate (MMF) are two typical Nrf2 inducers. Several studies support their neuroprotective efficacy against ischemic brain injury through the activation of the Nrf2/HO1 pathway. DMF and MMF dramatically reduced infarct volume, brain edema, and neurological deficits over 7 days after tMCAO, along with the suppressed acute glial activation (Yao et al., 2016). Importantly, such evident protection was abolished in Nrf2−/− mice, indicating that the Nrf2 pathway is required for the benefit of DMF and MMF. During the late phase (7–14 d), DMF also acted as a potent immunomodulator, reducing the infiltration of neutrophils and T cells and the number of activated microglia and macrophages in the infarct region (Lin et al., 2016). Interestingly, DMF also attenuated brain edema formation during ischemia and stabilized the blood brain barrier (BBB) by preventing disruption of interendothelial tight junctions (Kunze et al., 2015). DMF protected against acute brain damage in edema volume and sensorimotor deficits after pdMCAO through anti-inflammatory cytokines (IL-10 and IL-12p70), further supporting the neuroprotective role of MMF (Clausen et al., 2017).
Sulforaphane
Sulforaphane (SFN), a typical Nrf2 activator, is a naturally occurring isothiocyanate found in cruciferous vegetables. Upregulation of Nrf2 by sulforaphane pretreatment was associated with increased HO1 expression in perivascular astrocytes in the peri-infarct regions and cerebral endothelium of the infarct core. BBB disruption, lesion progression, and neurological deficits were reduced after tMCAO. This indicates that the Nrf2 defense pathway in the cerebral microvasculature provides a novel therapeutic approach for preventing BBB breakdown and neurological dysfunction in stroke victims (Alfieri et al., 2013). There is a contradictory finding that sulforaphane treatment increased the overall mRNA levels of transcription of Nrf2, Hmox1, GCLC and GSTA4 but failed to reduce the infarct volume and motor deficits in a photothrombotic mouse model (Porritt et al., 2012).
Tert-Butylhydroquinone
Tert-butylhydroquinone (tBHQ) is a Nrf2 inducer widely used as a food additive. Nrf2 activation by tBHQ pretreatment reduced cortical damage and sensorimotor deficits from 24 h up to even 1 month after pdMCAO. Interestingly, larger infarcts were observed in Nrf2−/− mice 7 d after stroke, but not 24 h after stroke (Shih et al., 2005). This Nrf2-specific action of tBHQ in vivo was also validated in another study with tMCAO (Shah et al., 2007). A recent study showed that Nrf2 upregulation by tBHQ significantly reduced the expression of cytoplasmic thioredoxin interacting protein, nod-like receptor protein 3 (NLRP3) inflammasome, and downstream factors caspase-1, IL-1β, and IL-18, all of which contribute to the Nrf2-mediated neuroprotection against ischemic outcomes after tMCAO (Hou et al., 2018). Interestingly, there is a contradictory report that tBHQ-exacerbated stroke damage (Sun et al., 2016), revealed by increased post-stroke mortality and worsened outcomes after pMCAO.
MiR-93 Antagomir
MicroRNAs (miRNAs) play vital roles in regulating neuronal survival during cerebral ischemia/reperfusion injury. MiR-93, a direct negative modulator of Nrf2 expression at the transcriptomic level, serves as a potential therapeutic target for acute ischemic stroke. MiR-93 levels in the ischemic cortex of mice increased at 24 h and 48 h after tMCAO. MiR-93 antagomir treatment reduced infarction volume, neural apoptosis and neurological deficits through the Nrf2/HO1 antioxidant pathway (Wang et al., 2016).
Korean Red Ginseng
Korean Red Ginseng (Ginseng) is one of the most widely used herbal medicines with reported antioxidant and anti-inflammatory properties, displaying promising potential in neuroprotection. Ginseng pretreatment ameliorated short- and long-term sensorimotor deficits over 28 days, prevented the acute enlargement of lesion volume, attenuated reactive astroglial progression but not microglial activation, and enhanced the induction of Nrf2 target antioxidant proteins after pdMCAO in WT mice, an effect that was abolished in Nrf2−/− mice (Liu et al., 2018). Nrf2-dependent spatiotemporal reactive astrogliosis correlated well with acute ischemic damage progression. In contrast, Nrf2 deficiency exacerbated the ischemic conditions. This supported the conclusion that Ginseng pretreatment protects against acute sensorimotor deficits and promotes its long-term recovery after pdMCAO, at least partly, through Nrf2 activation. Attenuated reactive astrogliosis contributes to the Nrf2-depedent neuroprotection.
(–)-Epicatechin
The (–)-Epicatechin (EC) is especially abundant in cocoa, dark chocolate, and green tea, and it boosts antioxidant activity while supporting vascular function. EC-treated ischemic WT mice displayed a reduction of forelimb motor coordination impairments associated with reduced anatomical injury and microglia/macrophage activation in pMCAO and tMCAO models of adult WT mice, impairments which were abolished in tissues and neurons from Nrf2−/− mice (Shah et al., 2010). Interestingly, this neuroprotection was observed in 12 month-old WT and Nrf2−/− mice, indicating that age influences Nrf2 function (Leonardo et al., 2013, 2015).
Resveratrol
Resveratrol is a natural polyphenolic compound that is found in some dietary sources such as grapes, plums, and red wine. Trans-resveratrol pretreatment had neuroprotective effects on ischemic brain damage after tMCAO. This neuroprotective effect is likely exerted by upregulated expression of Nrf2, HO1 and SOD that ameliorates oxidative damage (Ren et al., 2011). Meanwhile, resveratrol preconditioning-mediated neuroprotection is reduced after tMCAO (Narayanan et al., 2015). WT and Nrf2−/− cortical mitochondria exhibited increased uncoupling and ROS production after resveratrol pre-treatments. Nrf2−/− astrocytes exhibited decreased mitochondrial antioxidant expression and were unable to upregulate cellular antioxidants.
Carbon Monoxide
Carbon monoxide (CO) is a gaseous second messenger produced when heme oxygenase enzymes catabolize heme. At low doses (Otterbein et al., 1999; Koehler and Traystman, 2002; Leffler et al., 2011), despite being traditionally viewed as a toxic agent (Cheng et al., 2012), CO displayed sustained neuroprotective efficacy against brain damage and progressive functional deficits via the Nrf2 pathway in tMCAO and pdMCAO models (Meffert et al., 1994; Maines, 1996; Alkadhi et al., 2001). Brain lesions in mice exposed to CO were 29.6 ± 12.6% smaller at 7 days after pdMCAO. Additionally, 18-h CO treatment led to Nrf2 dissociation from Keap1, nuclear translocation, increased activity of Nrf2 binding to the ARE sequence of the HO1 gene, and elevated HO1 expression from 6 to 48 h after CO exposure, the neuroprotection of which was abolished in Nrf2−/− mice (Wang B. et al., 2011).
Tetramethylpyrazine
Tetramethylpyrazine (TMP) is one of the main components of Ligusticum wallichii Franchat (Chuan Xiong) that has been used to treat neurovascular and cardiovascular diseases including stroke in traditional Chinese medicine. TMP has been found to protect against pdMCAO injury and reduce inflammation and ischemia-induced neutrophils through Nrf2/HO1 activation (Kao et al., 2013; Chang et al., 2015).
Curcumin
Curcumin is a phenolic pigment extracted from the rhizome of Curuma longa Linn, which has been widely used as medicine in many Asian countries. It has strong neuro-protective effects on neurological injuries, with virtually no toxicity, even at a high dose; though, its absorption-bioavailability is the main issue, and few groups are actively looking to test innovative formulation to circumvent this serious limitation. Curcumin significantly reduced infarct size, brain edema, neurological dysfunction, and oxidative stress levels in tMCAO rats, and this neuroprotective effect involves the Akt/Nrf2 and NF-κB pathways (Wu J. et al., 2013; Li et al., 2016).
Besides the candidate Nrf2 inducers above, many natural products, synthetic compounds and clinical drugs, such as ginkgo biloba (Tulsulkar and Shah, 2013), paeonol (Zhao et al., 2014), isoquercetin (Chen M. et al., 2017), tanshinone IIA (Cai et al., 2017), ursolic acid (Li et al., 2013), isorhamnetin (Zhao et al., 2016), docosahexaenoic acid (Chang et al., 2013), HP-1c (Wang et al., 2018), gastrodin (Peng et al., 2015), trichostatin A (Wang B. et al., 2012), bicyclol (Zhang J. et al., 2014), recombinant human erythropoietin (Meng et al., 2014), omega-3 fatty acids (Zhang M. et al., 2014), Tocovid (Shang et al., 2018), estradiol (Li et al., 2017), and more, have also been shown to benefit ischemic brain injury through mechanisms involving Nrf2.
Pitfalls and Considerations of Current NRF2 Experimental Ischemic Stroke Studies
Although most reports support the beneficial effect of Nrf2 activation by a multitude of variety of inducers against ischemic brain damage, many findings regarding the role of Nrf2 during ischemia or ischemic-reperfusion injury are contradictory. This could be attributed to a number of inconsistencies across studies, as briefly overview here. (1) Nrf2 antibody: Getting potent, selective and highly specific antibodies against the various species of Nrf2 has been an ongoing serious issue. Furthermore, what is the correct molecular weight of Nrf2 protein? The biologically relevant molecular weight of Nrf2 protein has been proven to be ~95–110 KDa, but not ~55–65 KDa, based on its 2-kb open reading frame or its tertiary structure (Lau et al., 2013; Kemmerer et al., 2015). Many reports presented the expression level of Nrf2 protein by Western blots at apparent different (or unspecified) molecular weights, which might be mainly responsible for the contradictory results among research groups, the lack of good negative controls to be run in parallel should be documented. Additionally, this issue may also contribute to the discrepancy regarding findings of Nrf2 in subcellular distribution and cell-type expression under normal and inducible conditions. (2) Questions remain as to which cell types would be most responsible for the neuroprotection associate with Nrf2. (3) Some reports also questioned the potential issues in the Nrf2 knockout lines and strains. (4) Furthermore, while many of these reported cytoprotective enzymes that are induced by Nrf2 activation, there are activate debates as to whether traditional freely available substrates are sufficiently high to explain the resulting beneficial properties of these metabolites/end-products. (5) While many of these compounds are assumed to be the bioactive entity, there are well-recognized issues on such drugs to be bioavailable and cross the blood brain barrier to then reach the target at a concentration that would be biologically relevant; thus, there is the possibility that indirect pathways are stimulated that would then indirectly activate the Nrf2 pathway. (6) Several of the studies have tested preventive medicine when give before ischemia while others have tested the therapeutic potential of the given drug concoction; though, one should not extrapolate conclusion of the universal protection until it is rigorously tested and independently validated. (7) Nrf2/ARE pathway: Is the Nrf2/ARE pathway activated or not? To validate that Nrf2 is activated, many reports based their conclusions on selected protein level measurements of total Nrf2 with or without its target markers like HO1 by WB. Based on the current understanding of Nrf2 activation, nucleus translocation of Nrf2 is essential to its activation, which is revealed by nuclear Nrf2 protein levels. Evidence that targets distinct components of the Nrf2/ARE pathway cascade will be expected. In addition, the application of pharmacological or genetic blockage of Nrf2 will be helpful to demonstrate whether Nrf2/ARE pathway activation is specific to the neuroprotection of the candidate Nrf2 inducer. (8) Ischemic brain tissue: Which part of the ischemic cerebral hemisphere was used for measurement? Despite variable ischemic damage to different brain regions (or together with reperfusion), various ischemic brain tissue like the cortex, ischemic region, and cerebral hemisphere was used for measurement, possibly leading to inconsistent results. These tissue samples such as the infarct core, peri-infarct, and healthy brain regions, could “dilute” and even mask the changes of markers interest during cerebral ischemia. The apparent alternation of Nrf2 expression happens inside the peri-infarct area. Accordingly, tissue from the same site of the peri-infarct area appears to be appropriate for the subsequent measurement that leads to the final conclusion. (9) Histological and neurobiological outcomes: Which marker and which time points are expected to have the functional benefit of a potent Nrf2 inducer? Most reports only selected infarct volume or neurological deficit score in the acute ischemic stage (1–3 days) without tracking long-term efficacy. As we know, ischemic insult induces severe motor, sensory, emotional, and cognitive deficits (Ferro et al., 2016), and long-term functional recovery is considered to be the goal of stroke intervention. More accurate and long-term histological and functional evaluations are expected for future studies. (10) Nrf2 activators: As mentioned above, a number of Nrf2 activators have been identified that appears to upregulate Nrf2/ARE pathway. However, thus far, only limited compounds including sulforaphane, CDDO-methyl ester, and DMF have consistently indicated sufficient pharmacokinetic and pharmacodynamic properties to act in brain (Lastres-Becker et al., 2016; Yamamoto et al., 2018). Future rigorous studies are needed to provide the necessary information concerning the clinical application prospect of Nrf2 targeted intervention.
This review focusses on Nrf2 activity in the context of ischemic stroke, though, over- or prolonged Nrf2 activation could potentially be problematic. For example, sustained activation of Nrf2 in drosophila would shorten its life expectancy (Tsakiri et al., 2013), and it would also promote the malignant transformation of human cells (Yang X. et al., 2015). It has also been reported that mutations in Nrf2 and Keap1 would promote cancer cell survival (Shibata et al., 2008); furthermore, many cancer cells would overexpress Nrf2 and this would be associated with cell resistance to cancer therapies (Taguchi and Yamamoto, 2017).
Conclusive Remarks And Perspective
Emerging evidence demonstrates that the Nrf2 network plays a crucial role in cellular adaption by controlling a wide range of cytoprotective proteins, counteracting distinct endogenous and exogenous insults while providing a promising optimal therapeutic target against various diseases from cancer to brain disorders. By using various ischemic stroke rodent models, recent preclinical studies provide direct in vivo evidence revealing the contribution of the Nrf2 pathway in ischemic stroke pathogenesis and neuroprotection. This review highlighted the promising potential of interventions targeting Nrf2, implying that the moderate activation of Nrf2 favors the attenuation of brain damage and long-term recovery from cerebral ischemia. Because the investigations of Nrf2 in stroke are still in the initial stages, future research is expected to elucidate the natural properties of Nrf2 in stroke leading to the development of novel drugs that target Nrf2.
Author Contributions
LL and SD conceived the study and designed the databases analysis. LL prepared the manuscript with input from SD. LL and LML searched databases, collected data, performed analyses, and prepared the tables and figure. All authors reviewed, discussed and approved the final manuscript.
Funding
This work was supported in part by the American Heart and Stroke Association Postdoctoral Fellowship 16POST31220032 (LL). Publication of this article was funded in part by the University of Florida Open Access Publishing Fund.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, H. P. Jr., and Biller, J. (2015). Classification of subtypes of ischemic stroke: history of the trial of org 10172 in acute stroke treatment classification. Stroke 46, e114–117. doi: 10.1161/STROKEAHA.114.007773
Ahmed, S. M., Luo, L., Namani, A., Wang, X. J., and Tang, X. (2017). Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 585–597. doi: 10.1016/j.bbadis.2016.11.005
Alfieri, A., Srivastava, S., Siow, R. C., Cash, D., Modo, M., Duchen, M. R., et al. (2013). Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 65, 1012–1022. doi: 10.1016/j.freeradbiomed.2013.08.190
Alkadhi, K. A., Al-Hijailan, R. S., Malik, K., and Hogan, Y. H. (2001). Retrograde carbon monoxide is required for induction of long-term potentiation in rat superior cervical ganglion. J. Neurosci. 21, 3515–3520. doi: 10.1523/JNEUROSCI.21-10-03515.2001
An, P., Wu, T., Yu, H., Fang, K., Ren, Z., and Tang, M. (2018). Hispidulin protects against focal cerebral ischemia reperfusion injury in rats. J. Mol. Neurosci. 65, 203–212. doi: 10.1007/s12031-018-1086-2
Arslan, F., Keogh, B., Mcguirk, P., and Parker, A. E. (2010). TLR2 and TLR4 in ischemia reperfusion injury. Mediators Inflamm. 2010:704202. doi: 10.1155/2010/704202
Ashabi, G., Khalaj, L., Khodagholi, F., Goudarzvand, M., and Sarkaki, A. (2015). Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 30, 747–754. doi: 10.1007/s11011-014-9632-2
Atef, R. M., Agha, A. M., Abdel-Rhaman, A. A., and Nassar, N. N. (2018). The ying and yang of adenosine A1 and A2A receptors on ERK1/2 activation in a rat model of global cerebral ischemia reperfusion injury. Mol. Neurobiol. 55, 1284–1298. doi: 10.1007/s12035-017-0401-1
Ayers, D., Baron, B., and Hunter, T. (2015). miRNA Influences in NRF2 pathway interactions within cancer models. J. Nucl. Acids 2015:143636. doi: 10.1155/2015/143636
Bai, Q., Lyu, Z., Yang, X., Pan, Z., Lou, J., and Dong, T. (2017). Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behav. Brain Res. 321, 79–86. doi: 10.1016/j.bbr.2016.12.037
Bang, O. Y., Saver, J. L., Buck, B. H., Alger, J. R., Starkman, S., Ovbiagele, B., et al. (2008). Impact of collateral flow on tissue fate in acute ischaemic stroke. J. Neurol Neuros. Psychiatry 79, 625–629. doi: 10.1136/jnnp.2007.132100
Bellezza, I., Giambanco, I., Minelli, A., and Donato, R. (2018). Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta 1865, 721–733. doi: 10.1016/j.bbamcr.2018.02.010
Benjamin, E. J., Virani, S. S., Callaway, C. W., Chamberlain, A. M., Chang, A. R., Cheng, S., et al. (2018). Heart disease and stroke statistics-2018 update: a report from the american heart association. Circulation 137, e67–e492. doi: 10.1161/CIR.0000000000000558
Bosetti, F., Koenig, J. I., Ayata, C., Back, S. A., Becker, K., Broderick, J. P., et al. (2017). Translational stroke research: vision and opportunities. Stroke 48, 2632–2637. doi: 10.1161/STROKEAHA.117.017112
Bryan, H. K., Olayanju, A., Goldring, C. E., and Park, B. K. (2013). The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 85, 705–717. doi: 10.1016/j.bcp.2012.11.016
Cai, M., Guo, Y., Wang, S., Wei, H., Sun, S., Zhao, G., et al. (2017). Tanshinone IIA elicits neuroprotective effect through activating the nuclear factor erythroid 2-related factor-dependent antioxidant response. Rejuvenation Res. 20, 286–297. doi: 10.1089/rej.2016.1912
Calkins, M. J., Johnson, D. A., Townsend, J. A., Vargas, M. R., Dowell, J. A., Williamson, T. P., et al. (2009). The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal. 11, 497–508. doi: 10.1089/ars.2008.2242
Carmichael, S. T. (2005). Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx 2, 396–409. doi: 10.1602/neurorx.2.3.396
Chan, J. Y., Han, X. L., and Kan, Y. W. (1993). Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc. Natl. Acad. Sci. U.S.A. 90, 11371–11375. doi: 10.1073/pnas.90.23.11371
Chan, K., Lu, R., Chang, J. C., and Kan, Y. W. (1996). NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U.S.A. 93, 13943–13948. doi: 10.1073/pnas.93.24.13943
Chang, C. Y., Kao, T. K., Chen, W. Y., Ou, Y. C., Li, J. R., Liao, S. L., et al. (2015). Tetramethylpyrazine inhibits neutrophil activation following permanent cerebral ischemia in rats. Biochem. Biophys. Res. Commun. 463, 421–427. doi: 10.1016/j.bbrc.2015.05.088
Chang, C. Y., Kuan, Y. H., Li, J. R., Chen, W. Y., Ou, Y. C., Pan, H. C., et al. (2013). Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 24, 2127–2137. doi: 10.1016/j.jnutbio.2013.08.004
Chen, B., Cao, H., Chen, L., Yang, X., Tian, X., Li, R., et al. (2016). Rifampicin attenuated global cerebral ischemia injury via activating the nuclear factor erythroid 2-related factor pathway. Front. Cell Neurosci. 10:273. doi: 10.3389/fncel.2016.00273
Chen, L., Ran, Q., Xiang, Y., Xiang, L., Li, F., Wu, J., et al. (2017). Co-Activation of PKC-delta by CRIF1 modulates oxidative stress in bone marrow multipotent mesenchymal stromal cells after irradiation by phosphorylating NRF2 Ser40. Theranostics 7, 2634–2648. doi: 10.7150/thno.17853
Chen, L., Wang, L., Zhang, X., Cui, L., Xing, Y., Dong, L., et al. (2012). The protection by octreotide against experimental ischemic stroke: up-regulated transcription factor Nrf2, HO-1 and down-regulated NF-kappaB expression. Brain Res. 1475, 80–87. doi: 10.1016/j.brainres.2012.07.052
Chen, M., Dai, L. H., Fei, A., Pan, S. M., and Wang, H. R. (2017). Isoquercetin activates the ERK1/2-Nrf2 pathway and protects against cerebral ischemia-reperfusion injury in vivo and in vitro. Exp. Ther. Med. 13, 1353–1359. doi: 10.3892/etm.2017.4093
Chen, X., Liu, Y., Zhu, J., Lei, S., Dong, Y., Li, L., et al. (2016). GSK-3beta downregulates Nrf2 in cultured cortical neurons and in a rat model of cerebral ischemia-reperfusion. Sci. Rep. 6:20196. doi: 10.1038/srep20196
Cheng, Y., Thomas, A., Mardini, F., Bianchi, S. L., Tang, J. X., Peng, J., et al. (2012). Neurodevelopmental consequences of sub-clinical carbon monoxide exposure in newborn mice. PLoS ONE 7:e32029. doi: 10.1371/journal.pone.0032029
Chowdhry, S., Zhang, Y., Mcmahon, M., Sutherland, C., Cuadrado, A., and Hayes, J. D. (2013). Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32, 3765–3781. doi: 10.1038/onc.2012.388
Chu, C. T. (2018). Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol Dis. 122, 23–34. doi: 10.1016/j.nbd.2018.07.015
Chumboatong, W., Thummayot, S., Govitrapong, P., Tocharus, C., Jittiwat, J., and Tocharus, J. (2017). Neuroprotection of agomelatine against cerebral ischemia/reperfusion injury through an antiapoptotic pathway in rat. Neurochem. Int. 102, 114–122. doi: 10.1016/j.neuint.2016.12.011
Clausen, B. H., Lundberg, L., Yli-Karjanmaa, M., Martin, N. A., Svensson, M., Alfsen, M. Z., et al. (2017). Fumarate decreases edema volume and improves functional outcome after experimental stroke. Exp. Neurol. 295, 144–154. doi: 10.1016/j.expneurol.2017.06.011
Croom, E. (2012). Metabolism of xenobiotics of human environments. Prog. Mol. Biol. Transl. Sci. 112, 31–88. doi: 10.1016/B978-0-12-415813-9.00003-9
Cuadrado, A., Manda, G., Hassan, A., Alcaraz, M. J., Barbas, C., Daiber, A., et al. (2018). Transcription factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol. Rev. 70, 348–383. doi: 10.1124/pr.117.014753
Dai, Y., Zhang, H., Zhang, J., and Yan, M. (2018). Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-kappaB pathway. Chem. Biol. Interact. 284, 32–40. doi: 10.1016/j.cbi.2018.02.017
Ding, Y., Chen, M., Wang, M., Li, Y., and Wen, A. (2015). Posttreatment with 11-keto-beta-boswellic acid ameliorates cerebral ischemia-reperfusion injury: nrf2/ho-1 pathway as a potential mechanism. Mol. Neurobiol. 52, 1430–1439. doi: 10.1007/s12035-014-8929-9
Ding, Y., Ren, D., Xu, H., Liu, W., Liu, T., Li, L., et al. (2017). Antioxidant and pro-angiogenic effects of corilagin in rat cerebral ischemia via Nrf2 activation. Oncotarget 8, 114816–114828. doi: 10.18632/oncotarget.22023
Dinkova-Kostova, A. T., and Abramov, A. Y. (2015). The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 88, 179–188. doi: 10.1016/j.freeradbiomed.2015.04.036
Dirnagl, U., Simon, R. P., and Hallenbeck, J. M. (2003). Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 26, 248–254. doi: 10.1016/S0166-2236(03)00071-7
Doré, S. (2015). Neuroprotective effert of carbon monoxide and Nrf2 in cerebral ischemia. Springerplus 4:L44. doi: 10.1186/2193-1801-4-S1-L44
Doré, S., and Snyder, S. H. (1999). Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N.Y. Acad. Sci. 890, 167–172. doi: 10.1111/j.1749-6632.1999.tb07991.x
Doré, S., Takahashi, M., Ferris, C. D., Hester, L. D., Guastella, D., and Snyder, S. H. (1999). Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. U.S.A. 96, 2445–2450. doi: 10.1073/pnas.96.5.2445
Dorr, A., Sled, J. G., and Kabani, N. (2007). Three-dimensional cerebral vasculature of the CBA mouse brain: a magnetic resonance imaging and micro computed tomography study. Neuroimage 35, 1409–1423. doi: 10.1016/j.neuroimage.2006.12.040
Doyle, K. P., and Buckwalter, M. S. (2014). A mouse model of permanent focal ischemia: distal middle cerebral artery occlusion. Methods Mol. Biol. 1135, 103–110. doi: 10.1007/978-1-4939-0320-7_9
Durukan, A., Strbian, D., and Tatlisumak, T. (2008). Rodent models of ischemic stroke: a useful tool for stroke drug development. Curr. Pharm. Des. 14, 359–370. doi: 10.2174/138161208783497688
Durukan, A., and Tatlisumak, T. (2007). Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 87, 179–197. doi: 10.1016/j.pbb.2007.04.015
Emberson, J., Lees, K. R., Lyden, P., Blackwell, L., Albers, G., Bluhmki, E., et al. (2014). Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet 384, 1929–1935. doi: 10.1016/S0140-6736(14)60584-5
Ewing, J. F., Haber, S. N., and Maines, M. D. (1992). Normal and heat-induced patterns of expression of heme oxygenase-1 (HSP32) in rat brain: hyperthermia causes rapid induction of mRNA and protein. J. Neurochem. 58, 1140–1149. doi: 10.1111/j.1471-4159.1992.tb09373.x
Feigin, V. L., Norrving, B., and Mensah, G. A. (2017). Global burden of stroke. Circ. Res. 120, 439–448. doi: 10.1161/CIRCRESAHA.116.308413
Ferro, J. M., Caeiro, L., and Figueira, M. L. (2016). Neuropsychiatric sequelae of stroke. Nat. Rev. Neurol. 12, 269–280. doi: 10.1038/nrneurol.2016.46
Fonarow, G. C., Smith, E. E., Saver, J. L., Reeves, M. J., Bhatt, D. L., Grau-Sepulveda, M. V., et al. (2011). Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation 123, 750–758. doi: 10.1161/CIRCULATIONAHA.110.974675
Friling, R. S., Bensimon, A., Tichauer, Y., and Daniel, V. (1990). Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. U.S.A. 87, 6258–6262. doi: 10.1073/pnas.87.16.6258
Gan, L., Vargas, M. R., Johnson, D. A., and Johnson, J. A. (2012). Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 32, 17775–17787. doi: 10.1523/JNEUROSCI.3049-12.2012
Guo, C., Wang, S., Duan, J., Jia, N., Zhu, Y., Ding, Y., et al. (2017). Protocatechualdehyde protects against cerebral ischemia-reperfusion-induced oxidative injury via protein kinase cepsilon/Nrf2/HO-1 pathway. Mol. Neurobiol. 54, 833–845. doi: 10.1007/s12035-016-9690-z
Guo, H., Adah, D., James, P. B., Liu, Q., Li, G., Ahmadu, P., et al. (2018). Xueshuantong injection (Lyophilized) attenuates cerebral ischemia/reperfusion injury by the activation of Nrf2-VEGF pathway. Neurochem. Res. 43, 1096–1103. doi: 10.1007/s11064-018-2523-x
Guo, H., Li, M. J., Liu, Q. Q., Guo, L. L., Ma, M. M., Wang, S. X., et al. (2014). Danhong injection attenuates ischemia/reperfusion-induced brain damage which is associating with Nrf2 levels in vivo and in vitro. Neurochem. Res. 39, 1817–1824. doi: 10.1007/s11064-014-1384-1
Hacke, W., Kaste, M., Bluhmki, E., Brozman, M., Davalos, A., Guidetti, D., et al. (2008). Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 359, 1317–1329. doi: 10.1056/NEJMoa0804656
Han, J., Wang, M., Jing, X., Shi, H., Ren, M., and Lou, H. (2014). (-)-Epigallocatechin gallate protects against cerebral ischemia-induced oxidative stress via Nrf2/ARE signaling. Neurochem. Res. 39, 1292–1299. doi: 10.1007/s11064-014-1311-5
Han, J., Xiao, Q., Lin, Y. H., Zheng, Z. Z., He, Z. D., Hu, J., et al. (2015). Neuroprotective effects of salidroside on focal cerebral ischemia/reperfusion injury involve the nuclear erythroid 2-related factor 2 pathway. Neural. Regen. Res. 10, 1989–1996. doi: 10.4103/1673-5374.172317
Haskew-Layton, R. E., Payappilly, J. B., Xu, H., Bennett, S. A., and Ratan, R. R. (2013). 15-Deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) protects neurons from oxidative death via an Nrf2 astrocyte-specific mechanism independent of PPARgamma. J. Neurochem. 124, 536–547. doi: 10.1111/jnc.12107
Hayes, J. D., and Dinkova-Kostova, A. T. (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218. doi: 10.1016/j.tibs.2014.02.002
Hou, Y., Wang, Y., He, Q., Li, L., Xie, H., Zhao, Y., et al. (2018). Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 336, 32–39. doi: 10.1016/j.bbr.2017.06.027
Hua, K., Sheng, X., Li, T. T., Wang, L. N., Zhang, Y. H., Huang, Z. J., et al. (2015). The edaravone and 3-n-butylphthalide ring-opening derivative 10b effectively attenuates cerebral ischemia injury in rats. Acta Pharmacol. Sin. 36, 917–927. doi: 10.1038/aps.2015.31
Iadecola, C., and Anrather, J. (2011). Stroke research at a crossroad: asking the brain for directions. Nat. Neurosci. 14, 1363–1368. doi: 10.1038/nn.2953
Itoh, K., Chiba, T., Takahashi, S., Ishii, T., Igarashi, K., Katoh, Y., et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322. doi: 10.1006/bbrc.1997.6943
Itoh, K., Mimura, J., and Yamamoto, M. (2010). Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid. Redox Signal. 13, 1665–1678. doi: 10.1089/ars.2010.3222
Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., Igarashi, K., Engel, J. D., et al. (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes. Dev. 13, 76–86. doi: 10.1101/gad.13.1.76
Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., O'connor, T., and Yamamoto, M. (2003). Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391. doi: 10.1046/j.1365-2443.2003.00640.x
Jain, A., Lamark, T., Sjottem, E., Larsen, K. B., Awuh, J. A., Overvatn, A., et al. (2010). p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 285, 22576–22591. doi: 10.1074/jbc.M110.118976
Janyou, A., Wicha, P., Jittiwat, J., Suksamrarn, A., Tocharus, C., and Tocharus, J. (2017). Dihydrocapsaicin attenuates blood brain barrier and cerebral damage in focal cerebral ischemia/reperfusion via oxidative stress and inflammatory. Sci. Rep. 7:10556. doi: 10.1038/s41598-017-11181-5
Ji, K., Xue, L., Cheng, J., and Bai, Y. (2016). Preconditioning of H2S inhalation protects against cerebral ischemia/reperfusion injury by induction of HSP70 through PI3K/Akt/Nrf2 pathway. Brain Res. Bull. 121, 68–74. doi: 10.1016/j.brainresbull.2015.12.007
Kam, K. Y., Yu, S. J., Jeong, N., Hong, J. H., Jalin, A. M., Lee, S., et al. (2011). p-Hydroxybenzyl alcohol prevents brain injury and behavioral impairment by activating Nrf2, PDI, and neurotrophic factor genes in a rat model of brain ischemia. Mol. Cells 31, 209–215. doi: 10.1007/s10059-011-0028-4
Kang, H. J., Hong, Y. B., Kim, H. J., and Bae, I. (2010). CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2 (NRF2) protein stability by proteasome-mediated degradation. J. Biol. Chem. 285, 21258–21268. doi: 10.1074/jbc.M109.084590
Kao, T. K., Chang, C. Y., Ou, Y. C., Chen, W. Y., Kuan, Y. H., Pan, H. C., et al. (2013). Tetramethylpyrazine reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. Exp. Neurol. 247, 188–201. doi: 10.1016/j.expneurol.2013.04.010
Kasner, S. E., and Grotta, J. C. (1998). Ischemic stroke. Neurol. Clin. 16, 355–372. doi: 10.1016/S0733-8619(05)70068-2
Katoh, Y., Iida, K., Kang, M. I., Kobayashi, A., Mizukami, M., Tong, K. I., et al. (2005). Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 433, 342–350. doi: 10.1016/j.abb.2004.10.012
Katsuoka, F., and Yamamoto, M. (2016). Small Maf proteins (MafF, MafG, MafK): history, structure and function. Gene 586, 197–205. doi: 10.1016/j.gene.2016.03.058
Kemmerer, Z. A., Ader, N. R., Mulroy, S. S., and Eggler, A. L. (2015). Comparison of human Nrf2 antibodies: a tale of two proteins. Toxicol. Lett. 238, 83–89. doi: 10.1016/j.toxlet.2015.07.004
Kensler, T. W., Wakabayashi, N., and Biswal, S. (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
Kerins, M. J., and Ooi, A. (2017). The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid Redox Signal 29, 1756–1773. doi: 10.1089/ars.2017.7176
Kobayashi, A., Kang, M. I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., et al. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004
Kobayashi, E. H., Suzuki, T., Funayama, R., Nagashima, T., Hayashi, M., Sekine, H., et al. (2016). Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 7:11624. doi: 10.1038/ncomms11624
Koehler, R. C., and Traystman, R. J. (2002). Cerebrovascular effects of carbon monoxide. Antioxid. Redox Signal. 4, 279–290. doi: 10.1089/152308602753666334
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell. Biol. 12, 213–223. doi: 10.1038/ncb2021
Kumar, H., Kim, I. S., More, S. V., Kim, B. W., and Choi, D. K. (2014). Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 31, 109–139. doi: 10.1039/C3NP70065H
Kunze, R., Urrutia, A., Hoffmann, A., Liu, H., Helluy, X., Pham, M., et al. (2015). Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp. Neurol. 266, 99–111. doi: 10.1016/j.expneurol.2015.02.022
Kuo, P. C., Yu, I. C., Scofield, B. A., Brown, D. A., Curfman, E. T., Paraiso, H. C., et al. (2017). 3H-1,2-Dithiole-3-thione as a novel therapeutic agent for the treatment of ischemic stroke through Nrf2 defense pathway. Brain Behav. Immun. 62, 180–192. doi: 10.1016/j.bbi.2017.01.018
Kurinna, S., and Werner, S. (2015). NRF2 and microRNAs: new but awaited relations. Biochem. Soc. Trans. 43, 595–601. doi: 10.1042/BST20140317
Lastres-Becker, I., Garcia-Yague, A. J., Scannevin, R. H., Casarejos, M. J., Kugler, S., Rabano, A., et al. (2016). Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in parkinson's disease. Antioxid. Redox Signal. 25, 61–77. doi: 10.1089/ars.2015.6549
Lau, A., Tian, W., Whitman, S. A., and Zhang, D. D. (2013). The predicted molecular weight of Nrf2: it is what it is not. Antioxid. Redox Signal. 18, 91–93. doi: 10.1089/ars.2012.4754
Lau, A., Wang, X. J., Zhao, F., Villeneuve, N. F., Wu, T., Jiang, T., et al. (2010). A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 30, 3275–3285. doi: 10.1128/MCB.00248-10
Lee, H., Park, Y. H., Jeon, Y. T., Hwang, J. W., Lim, Y. J., Kim, E., et al. (2015). Sevoflurane post-conditioning increases nuclear factor erythroid 2-related factor and haemoxygenase-1 expression via protein kinase C pathway in a rat model of transient global cerebral ischaemia. Br. J. Anaesth. 114, 307–318. doi: 10.1093/bja/aeu268
Leffler, C. W., Parfenova, H., and Jaggar, J. H. (2011). Carbon monoxide as an endogenous vascular modulator. Am. J. Physiol. Heart Circ. Physiol. 301, H1–H11. doi: 10.1152/ajpheart.00230.2011
Lei, X., Lei, L., Zhang, Z., and Cheng, Y. (2016). Neuroprotective effects of lycopene pretreatment on transient global cerebral ischemiareperfusion in rats: the role of the Nrf2/HO1 signaling pathway. Mol. Med. Rep. 13, 412–418. doi: 10.3892/mmr.2015.4534
Leonardo, C. C., Agrawal, M., Singh, N., Moore, J. R., Biswal, S., and Doré, S. (2013). Oral administration of the flavanol (-)-epicatechin bolsters endogenous protection against focal ischemia through the Nrf2 cytoprotective pathway. Eur. J. Neurosci. 38, 3659–3668. doi: 10.1111/ejn.12362
Leonardo, C. C., and Doré, S. (2011). Dietary flavonoids are neuroprotective through Nrf2-coordinated induction of endogenous cytoprotective proteins. Nutr. Neurosci. 14, 226–236. doi: 10.1179/1476830511Y.0000000013
Leonardo, C. C., Mendes, M., Ahmad, A. S., and Doré, S. (2015). Efficacy of prophylactic flavan-3-ol in permanent focal ischemia in 12-mo-old mice. Am. J. Physiol. Heart Circ. Physiol. 308, H583–591. doi: 10.1152/ajpheart.00239.2014
Li, B., Sun, J., Lv, G., Yu, Y., Wang, G., Xie, K., et al. (2014). Sevoflurane postconditioning attenuates cerebral ischemia-reperfusion injury via protein kinase B/nuclear factor-erythroid 2-related factor 2 pathway activation. Int. J. Dev. Neurosci. 38, 79–86. doi: 10.1016/j.ijdevneu.2014.08.005
Li, L., Chen, J., Sun, S., Zhao, J., Dong, X., and Wang, J. (2017). Effects of estradiol on autophagy and Nrf-2/ARE signals after cerebral ischemia. Cell. Physiol. Biochem. 41, 2027–2036. doi: 10.1159/000475433
Li, L., Zhang, X., Cui, L., Wang, L., Liu, H., Ji, H., et al. (2013). Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 1497, 32–39. doi: 10.1016/j.brainres.2012.12.032
Li, L., Zhu, K., Liu, Y., Wu, X., Wu, J., Zhao, Y., et al. (2015). Targeting thioredoxin-1 with siRNA exacerbates oxidative stress injury after cerebral ischemia/reperfusion in rats. Neuroscience 284, 815–823. doi: 10.1016/j.neuroscience.2014.10.066
Li, W., Suwanwela, N. C., and Patumraj, S. (2016). Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 106, 117–127. doi: 10.1016/j.mvr.2015.12.008
Liebeskind, D. S. (2010). Reperfusion for acute ischemic stroke: arterial revascularization and collateral therapeutics. Curr. Opin. Neurol. 23, 36–45. doi: 10.1097/WCO.0b013e328334da32
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Lin, R., Cai, J., Kostuk, E. W., Rosenwasser, R., and Iacovitti, L. (2016). Fumarate modulates the immune/inflammatory response and rescues nerve cells and neurological function after stroke in rats. J. Neuroinflammation 13:269. doi: 10.1186/s12974-016-0733-1
Liu, B., Teschemacher, A. G., and Kasparov, S. (2017). Astroglia as a cellular target for neuroprotection and treatment of neuro-psychiatric disorders. Glia 65, 1205–1226. doi: 10.1002/glia.23136
Liu, H., Zhang, J. J., Li, X., Yang, Y., Xie, X. F., and Hu, K. (2015). Post-occlusion administration of sodium butyrate attenuates cognitive impairment in a rat model of chronic cerebral hypoperfusion. Pharmacol. Biochem. Behav. 135, 53–59. doi: 10.1016/j.pbb.2015.05.012
Liu, L., Vollmer, M. K., Fernandez, V. M., Dweik, Y., Kim, H., and Doré, S. (2018). Korean red ginseng pretreatment protects against long-term sensorimotor deficits after ischemic stroke likely through Nrf2. Front. Cell. Neurosci. 12:74. doi: 10.3389/fncel.2018.00074
Liu, Y., Zhang, L., and Liang, J. (2015). Activation of the Nrf2 defense pathway contributes to neuroprotective effects of phloretin on oxidative stress injury after cerebral ischemia/reperfusion in rats. J. Neurol. Sci. 351, 88–92. doi: 10.1016/j.jns.2015.02.045
Lo, E. H., Dalkara, T., and Moskowitz, M. A. (2003). Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 4, 399–415. doi: 10.1038/nrn1106
Lou, J., Cao, G., Li, R., Liu, J., Dong, Z., and Xu, L. (2016). beta-Caryophyllene attenuates focal cerebral ischemia-reperfusion injury by Nrf2/HO-1 pathway in rats. Neurochem. Res. 41, 1291–1304. doi: 10.1007/s11064-016-1826-z
Lu, H., Wang, B., Cui, N., and Zhang, Y. (2018). Artesunate suppresses oxidative and inflammatory processes by activating Nrf2 and ROSdependent p38 MAPK and protects against cerebral ischemiareperfusion injury. Mol. Med. Rep. 17, 6639–6646. doi: 10.3892/mmr.2018.8666
Lv, C., Maharjan, S., Wang, Q., Sun, Y., Han, X., Wang, S., et al. (2017). alpha-lipoic acid promotes neurological recovery after ischemic stroke by activating the Nrf2/HO-1 pathway to attenuate oxidative damage. Cell. Physiol. Biochem. 43, 1273–1287. doi: 10.1159/000481840
Ma, Q. (2013). Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. doi: 10.1146/annurev-pharmtox-011112-140320
Magaki, S. D., Williams, C. K., and Vinters, H. V. (2018). Glial function (and dysfunction) in the normal and ischemic brain. Neuropharmacology 134, 218–225. doi: 10.1016/j.neuropharm.2017.11.009
Maines, M. (1996). Carbon monoxide and nitric oxide homology: differential modulation of heme oxygenases in brain and detection of protein and activity. Methods Enzymol. 268, 473–488. doi: 10.1016/S0076-6879(96)68049-5
Malloy, M. T., Mcintosh, D. J., Walters, T. S., Flores, A., Goodwin, J. S., and Arinze, I. J. (2013). Trafficking of the transcription factor Nrf2 to promyelocytic leukemia-nuclear bodies: implications for degradation of NRF2 in the nucleus. J. Biol. Chem. 288, 14569–14583. doi: 10.1074/jbc.M112.437392
Martini, S. R., and Kent, T. A. (2007). Hyperglycemia in acute ischemic stroke: a vascular perspective. J. Cereb. Blood Flow Metab. 27, 435–451. doi: 10.1038/sj.jcbfm.9600355
Mathur, A., Pandey, V. K., and Kakkar, P. (2018). Activation of GSK3beta/beta-TrCP axis via PHLPP1 exacerbates Nrf2 degradation leading to impairment in cell survival pathway during diabetic nephropathy. Free Radic. Biol. Med. 120, 414–424. doi: 10.1016/j.freeradbiomed.2018.04.550
McMahon, M., Lamont, D. J., Beattie, K. A., and Hayes, J. D. (2010). Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. U.S.A. 107, 18838–18843. doi: 10.1073/pnas.1007387107
Meffert, M. K., Haley, J. E., Schuman, E. M., Schulman, H., and Madison, D. V. (1994). Inhibition of hippocampal heme oxygenase, nitric oxide synthase, and long-term potentiation by metalloporphyrins. Neuron 13, 1225–1233. doi: 10.1016/0896-6273(94)90060-4
Mehndiratta, P., Chapman Smith, S., and Worrall, B. B. (2015). Etiologic stroke subtypes: updated definition and efficient workup strategies. Curr. Treat. Options Cardiovasc. Med. 17:357. doi: 10.1007/s11936-014-0357-7
Mehta, S. L., Manhas, N., and Raghubir, R. (2007). Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 54, 34–66. doi: 10.1016/j.brainresrev.2006.11.003
Meng, H., Guo, J., Wang, H., Yan, P., Niu, X., and Zhang, J. (2014). Erythropoietin activates Keap1-Nrf2/ARE pathway in rat brain after ischemia. Int. J. Neurosci. 124, 362–368. doi: 10.3109/00207454.2013.848439
Miao, Z. Y., Xia, X., Che, L., and Song, Y. T. (2018). Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of Nrf2 expression in ovariectomized rats. Neurol. Res. 40, 689–695. doi: 10.1080/01616412.2018.1462879
Moi, P., Chan, K., Asunis, I., Cao, A., and Kan, Y. W. (1994). Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U.S.A. 91, 9926–9930. doi: 10.1073/pnas.91.21.9926
Motohashi, H., Fujita, R., Takayama, M., Inoue, A., Katsuoka, F., Bresnick, E. H., et al. (2011). Molecular determinants for small Maf protein control of platelet production. Mol. Cell. Biol. 31, 151–162. doi: 10.1128/MCB.00798-10
Mrsic-Pelcic, J., Pilipovic, K., Pelcic, G., Vitezic, D., and Zupan, G. (2017). Decrease in oxidative stress parameters after post-ischaemic recombinant human erythropoietin administration in the hippocampus of rats exposed to focal cerebral ischaemia. Basic Clin. Pharmacol. Toxicol. 121, 453–464. doi: 10.1111/bcpt.12833
Nakano, Y., Yamashita, T., Li, Q., Sato, K., Ohta, Y., Morihara, R., et al. (2017). Time-dependent change of in vivo optical imaging of oxidative stress in a mouse stroke model. J. Neurosci. Res. 95, 2030–2039. doi: 10.1002/jnr.24047
Narayanan, S. V., Dave, K. R., Saul, I., and Perez-Pinzon, M. A. (2015). Resveratrol preconditioning protects against cerebral ischemic injury via nuclear erythroid 2-related factor 2. Stroke 46, 1626–1632. doi: 10.1161/STROKEAHA.115.008921
Ostrowski, R. P., Stepien, K., Pucko, E., and Matyja, E. (2016). Hyperbaric oxygen modalities are differentially effective in distinct brain ischemia models. Med. Gas. Res. 6, 39–47. doi: 10.4103/2045-9912.179344
Otterbein, L. E., Mantell, L. L., and Choi, A. M. (1999). Carbon monoxide provides protection against hyperoxic lung injury. Am. J. Physiol. 276, L688–L694. doi: 10.1152/ajplung.1999.276.4.L688
Pang, T., Wang, Y. J., Gao, Y. X., Xu, Y., Li, Q., Zhou, Y. B., et al. (2016). A novel GSK-3beta inhibitor YQ138 prevents neuronal injury induced by glutamate and brain ischemia through activation of the Nrf2 signaling pathway. Acta Pharmacol. Sin. 37, 741–752. doi: 10.1038/aps.2016.3
Peng, B., Zhao, P., Lu, Y. P., Chen, M. M., Sun, H., Wu, X. M., et al. (2013). Z-ligustilide activates the Nrf2/HO-1 pathway and protects against cerebral ischemia-reperfusion injury in vivo and in vitro. Brain Res. 1520, 168–177. doi: 10.1016/j.brainres.2013.05.009
Peng, Z., Wang, S., Chen, G., Cai, M., Liu, R., Deng, J., et al. (2015). Gastrodin alleviates cerebral ischemic damage in mice by improving anti-oxidant and anti-inflammation activities and inhibiting apoptosis pathway. Neurochem. Res. 40, 661–673. doi: 10.1007/s11064-015-1513-5
Porritt, M. J., Andersson, H. C., Hou, L., Nilsson, A., Pekna, M., Pekny, M., et al. (2012). Photothrombosis-induced infarction of the mouse cerebral cortex is not affected by the Nrf2-activator sulforaphane. PLoS ONE 7:e41090. doi: 10.1371/journal.pone.0041090
Prabhakaran, S., Ruff, I., and Bernstein, R. A. (2015). Acute stroke intervention: a systematic review. JAMA 313, 1451–1462. doi: 10.1001/jama.2015.3058
Quinn, T. J., Elliott, E., and Langhorne, P. (2018). Cognitive and mood assessment tools for use in stroke. Stroke 49, 483–490. doi: 10.1161/STROKEAHA.117.016994
Rada, P., Rojo, A. I., Evrard-Todeschi, N., Innamorato, N. G., Cotte, A., Jaworski, T., et al. (2012). Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell. Biol. 32, 3486–3499. doi: 10.1128/MCB.00180-12
Raghunath, A., Sundarraj, K., Nagarajan, R., Arfuso, F., Bian, J., Kumar, A. P., et al. (2018). Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biol. 17, 297–314. doi: 10.1016/j.redox.2018.05.002
Ren, J., Fan, C., Chen, N., Huang, J., and Yang, Q. (2011). Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem. Res. 36, 2352–2362. doi: 10.1007/s11064-011-0561-8
Rojo de la Vega, M., Chapman, E., and Zhang, D. D. (2018). NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43. doi: 10.1016/j.ccell.2018.03.022
Romano, J. G., and Sacco, R. L. (2015). Decade in review-stroke: progress in acute ischaemic stroke treatment and prevention. Nat. Rev. Neurol. 11, 619–621. doi: 10.1038/nrneurol.2015.199
Rouault, T. A. (2013). Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat. Rev. Neurosci. 14, 551–564. doi: 10.1038/nrn3453
Rushmore, T. H., Morton, M. R., and Pickett, C. B. (1991). The antioxidant responsive element. activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 266, 11632–11639.
Sakata, H., Niizuma, K., Yoshioka, H., Kim, G. S., Jung, J. E., Katsu, M., et al. (2012). Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J. Neurosci. 32, 3462–3473. doi: 10.1523/JNEUROSCI.5686-11.2012
Sandercock, P., Wardlaw, J. M., Lindley, R. I., Dennis, M., Cohen, G., Murray, G., et al. (2012). The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet 379, 2352–2363. doi: 10.1016/S0140-6736(12)60768-5
Shah, Z. A., Li, R. C., Ahmad, A. S., Kensler, T. W., Yamamoto, M., Biswal, S., et al. (2010). The flavanol (-)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J. Cereb. Blood Flow Metab. 30, 1951–1961. doi: 10.1038/jcbfm.2010.53
Shah, Z. A., Li, R. C., Thimmulappa, R. K., Kensler, T. W., Yamamoto, M., Biswal, S., et al. (2007). Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience 147, 53–59. doi: 10.1016/j.neuroscience.2007.02.066
Shang, J., Yan, H., Jiao, Y., Ohta, Y., Liu, X., Li, X., et al. (2018). Therapeutic effects of pretreatment with tocovid on oxidative stress in postischemic mice brain. J. Stroke Cerebrovasc. Dis. 27, 2096–2105. doi: 10.1016/j.jstrokecerebrovasdis.2018.03.012
Shen, Q., and Duong, T. Q. (2008). Quantitative prediction of ischemic stroke tissue fate. NMR Biomed. 21, 839–848. doi: 10.1002/nbm.1264
Shi, H., Jing, X., Wei, X., Perez, R. G., Ren, M., Zhang, X., et al. (2015). S-allyl cysteine activates the Nrf2-dependent antioxidant response and protects neurons against ischemic injury in vitro and in vivo. J. Neurochem. 133, 298–308. doi: 10.1111/jnc.12986
Shibata, T., Ohta, T., Tong, K. I., Kokubu, A., Odogawa, R., Tsuta, K., et al. (2008). Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. U.S.A. 105, 13568–13573. doi: 10.1073/pnas.0806268105
Shih, A. Y., Li, P., and Murphy, T. H. (2005). A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J. Neurosci. 25, 10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005
Sicard, K. M., and Fisher, M. (2009). Animal models of focal brain ischemia. Exp. Transl. Stroke Med. 1:7. doi: 10.1186/2040-7378-1-7
Srivastava, S., Alfieri, A., Siow, R. C., Mann, G. E., and Fraser, P. A. (2013). Temporal and spatial distribution of Nrf2 in rat brain following stroke: quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique. J. Physiol. 591, 3525–3538. doi: 10.1113/jphysiol.2013.257964
Sun, J., Hu, H., Ren, X., and Simpkins, J. W. (2016). Tert-butylhydroquinone compromises survival in murine experimental stroke. Neurotoxicol. Teratol. 54, 15–21. doi: 10.1016/j.ntt.2016.01.004
Sun, Y., Yang, T., Leak, R. K., Chen, J., and Zhang, F. (2017). Preventive and protective roles of dietary Nrf2 activators against central nervous system diseases. CNS Neurol. Disord. Drug Targets 16, 326–338. doi: 10.2174/1871527316666170102120211
Suzuki, T., Motohashi, H., and Yamamoto, M. (2013). Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 34, 340–346. doi: 10.1016/j.tips.2013.04.005
Suzuki, T., and Yamamoto, M. (2015). Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 88, 93–100. doi: 10.1016/j.freeradbiomed.2015.06.006
Suzuki, T., and Yamamoto, M. (2017). Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 292, 16817–16824. doi: 10.1074/jbc.R117.800169
Taguchi, K., and Yamamoto, M. (2017). The KEAP1-NRF2 system in cancer. Front. Oncol. 7:85. doi: 10.3389/fonc.2017.00085
Takagi, T., Kitashoji, A., Iwawaki, T., Tsuruma, K., Shimazawa, M., Yoshimura, S., et al. (2014). Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 72, 124–133. doi: 10.1016/j.freeradbiomed.2014.04.009
Tanaka, N., Ikeda, Y., Ohta, Y., Deguchi, K., Tian, F., Shang, J., et al. (2011). Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 1370, 246–253. doi: 10.1016/j.brainres.2010.11.010
Tonelli, C., Chio, I. I. C., and Tuveson, D. A. (2017). Transcriptional Regulation by Nrf2. Antioxid Redox Signal. doi: 10.1089/ars.2017.7342
Traystman, R. J. (2003). Animal models of focal and global cerebral ischemia. ILAR J. 44, 85–95. doi: 10.1093/ilar.44.2.85
Tsakiri, E. N., Sykiotis, G. P., Papassideri, I. S., Terpos, E., Dimopoulos, M. A., Gorgoulis, V. G., et al. (2013). Proteasome dysfunction in drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 12, 802–813. doi: 10.1111/acel.12111
Tu, J., Zhang, X., Zhu, Y., Dai, Y., Li, N., Yang, F., et al. (2015). Cell-permeable peptide targeting the Nrf2-Keap1 interaction: a potential novel therapy for global cerebral ischemia. J. Neurosci. 35, 14727–14739. doi: 10.1523/JNEUROSCI.1304-15.2015
Tulsulkar, J., and Shah, Z. A. (2013). Ginkgo biloba prevents transient global ischemia-induced delayed hippocampal neuronal death through antioxidant and anti-inflammatory mechanism. Neurochem. Int. 62, 189–197. doi: 10.1016/j.neuint.2012.11.017
Vargas, M. R., and Johnson, J. A. (2009). The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 11:e17. doi: 10.1017/S1462399409001094
Wang, B., Cao, W., Biswal, S., and Doré, S. (2011). Carbon monoxide-activated Nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke 42, 2605–2610. doi: 10.1161/STROKEAHA.110.607101
Wang, B., Zhu, X., Kim, Y., Li, J., Huang, S., Saleem, S., et al. (2012). Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 52, 928–936. doi: 10.1016/j.freeradbiomed.2011.12.006
Wang, J., Fields, J., Zhao, C., Langer, J., Thimmulappa, R. K., Kensler, T. W., et al. (2007). Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic. Biol. Med. 43, 408–414. doi: 10.1016/j.freeradbiomed.2007.04.020
Wang, L., Lu, Y., Deng, S., Zhang, Y., Yang, L., Guan, Y., et al. (2012). SHPS-1 deficiency induces robust neuroprotection against experimental stroke by attenuating oxidative stress. J. Neurochem. 122, 834–843. doi: 10.1111/j.1471-4159.2012.07818.x
Wang, N., Zhang, L., Lu, Y., Zhang, M., Zhang, Z., Wang, K., et al. (2017). Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating Nrf2/ARE signaling pathway. Biomed. Pharmacother. 89, 1187–1195. doi: 10.1016/j.biopha.2017.03.011
Wang, P., Liang, X., Lu, Y., Zhao, X., and Liang, J. (2016). MicroRNA-93 downregulation ameliorates cerebral ischemic injury through the Nrf2/HO-1 defense pathway. Neurochem. Res. 41, 2627–2635. doi: 10.1007/s11064-016-1975-0
Wang, R., Tu, J., Zhang, Q., Zhang, X., Zhu, Y., Ma, W., et al. (2013). Genistein attenuates ischemic oxidative damage and behavioral deficits via eNOS/Nrf2/HO-1 signaling. Hippocampus 23, 634–647. doi: 10.1002/hipo.22126
Wang, Y., Huang, Y., Xu, Y., Ruan, W., Wang, H., Zhang, Y., et al. (2018). A dual AMPK/Nrf2 activator reduces brain inflammation after stroke by enhancing microglia M2 polarization. Antioxid. Redox Signal. 28, 141–163. doi: 10.1089/ars.2017.7003
Wang, Y. C., Lin, S., and Yang, Q. W. (2011). Toll-like receptors in cerebral ischemic inflammatory injury. J. Neuroinflammation 8:134. doi: 10.1186/1742-2094-8-134
Wardyn, J. D., Ponsford, A. H., and Sanderson, C. M. (2015). Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 43, 621–626. doi: 10.1042/BST20150014
Wasserman, W. W., and Fahl, W. E. (1997). Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. U.S.A. 94, 5361–5366. doi: 10.1073/pnas.94.10.5361
Wicha, P., Tocharus, J., Janyou, A., Jittiwat, J., Changtam, C., Suksamrarn, A., et al. (2017). Hexahydrocurcumin protects against cerebral ischemia/reperfusion injury, attenuates inflammation, and improves antioxidant defenses in a rat stroke model. PLoS ONE 12:e0189211. doi: 10.1371/journal.pone.0189211
Wu, G., Zhu, L., Yuan, X., Chen, H., Xiong, R., Zhang, S., et al. (2017). Britanin ameliorates cerebral ischemia-reperfusion injury by inducing the Nrf2 protective pathway. Antioxid. Redox Signal. 27, 754–768. doi: 10.1089/ars.2016.6885
Wu, J., Chen, Y., Yu, S., Li, L., Zhao, X., Li, Q., et al. (2017a). Neuroprotective effects of sulfiredoxin-1 during cerebral ischemia/reperfusion oxidative stress injury in rats. Brain Res. Bull. 132, 99–108. doi: 10.1016/j.brainresbull.2017.05.012
Wu, J., Jin, Z., Yang, X., and Yan, L. J. (2018). Post-ischemic administration of 5-methoxyindole-2-carboxylic acid at the onset of reperfusion affords neuroprotection against stroke injury by preserving mitochondrial function and attenuating oxidative stress. Biochem. Biophys. Res. Commun. 497, 444–450. doi: 10.1016/j.bbrc.2018.02.106
Wu, J., Li, Q., Wang, X., Yu, S., Li, L., Wu, X., et al. (2013). Neuroprotection by curcumin in ischemic brain injury involves the Akt/Nrf2 pathway. PLoS ONE 8:e59843. doi: 10.1371/journal.pone.0059843
Wu, J., Li, R., Li, W., Ren, M., Thangthaeng, N., Sumien, N., et al. (2017b). Administration of 5-methoxyindole-2-carboxylic acid that potentially targets mitochondrial dihydrolipoamide dehydrogenase confers cerebral preconditioning against ischemic stroke injury. Free Radic. Biol. Med. 113, 244–254. doi: 10.1016/j.freeradbiomed.2017.10.008
Wu, L., Liu, Z. J., Miao, S., Zou, L. B., Cai, L., Wu, P., et al. (2013). Lipoxin A4 ameliorates cerebral ischaemia/reperfusion injury through upregulation of nuclear factor erythroid 2-related factor 2. Neurol. Res. 35, 968–975. doi: 10.1179/1743132813Y.0000000242
Wu, S., Yue, Y., Li, J., Li, Z., Li, X., Niu, Y., et al. (2015). Procyanidin B2 attenuates neurological deficits and blood-brain barrier disruption in a rat model of cerebral ischemia. Mol. Nutr. Food Res. 59, 1930–1941. doi: 10.1002/mnfr.201500181
Wu, S., Yue, Y., Peng, A., Zhang, L., Xiang, J., Cao, X., et al. (2016). Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 7, 2624–2634. doi: 10.1039/C6FO00419A
Xian, J. W., Choi, A. Y., Lau, C. B., Leung, W. N., Ng, C. F., and Chan, C. W. (2016). Gastrodia and uncaria (tianma gouteng) water extract exerts antioxidative and antiapoptotic effects against cerebral ischemia in vitro and in vivo. Chin. Med. 11:27. doi: 10.1186/s13020-016-0097-6
Ya, B. L., Li, H. F., Wang, H. Y., Wu, F., Xin, Q., Cheng, H. J., et al. (2017). 5-HMF attenuates striatum oxidative damage via Nrf2/ARE signaling pathway following transient global cerebral ischemia. Cell Stress Chaperones 22, 55–65. doi: 10.1007/s12192-016-0742-0
Yamamoto, M., Kensler, T. W., and Motohashi, H. (2018). The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169–1203. doi: 10.1152/physrev.00023.2017
Yamauchi, K., Nakano, Y., Imai, T., Takagi, T., Tsuruma, K., Shimazawa, M., et al. (2016). A novel nuclear factor erythroid 2-related factor 2 (Nrf2) activator RS9 attenuates brain injury after ischemia reperfusion in mice. Neuroscience 333, 302–310. doi: 10.1016/j.neuroscience.2016.07.035
Yang, J., Su, J., Wan, F., Yang, N., Jiang, H., Fang, M., et al. (2017). Tissue kallikrein protects against ischemic stroke by suppressing TLR4/NF-kappaB and activating Nrf2 signaling pathway in rats. Exp. Ther. Med. 14, 1163–1170. doi: 10.3892/etm.2017.4614
Yang, X., Wang, D., Ma, Y., Xu, X., Zhu, Z., Wang, X., et al. (2015). Continuous activation of Nrf2 and its target antioxidant enzymes leads to arsenite-induced malignant transformation of human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 289, 231–239. doi: 10.1016/j.taap.2015.09.020
Yang, Y., Zhang, J., Xiong, L., Deng, M., Wang, J., Xin, J., et al. (2015). Cognitive improvement induced by environment enrichment in chronic cerebral hypoperfusion rats: a result of upregulated endogenous neuroprotection? J. Mol. Neurosci. 56, 278–289. doi: 10.1007/s12031-015-0529-2
Yang, Z., Weian, C., Susu, H., and Hanmin, W. (2016). Protective effects of mangiferin on cerebral ischemia-reperfusion injury and its mechanisms. Eur. J. Pharmacol. 771, 145–151. doi: 10.1016/j.ejphar.2015.12.003
Yao, Y., Miao, W., Liu, Z., Han, W., Shi, K., Shen, Y., et al. (2016). Dimethyl fumarate and monomethyl fumarate promote post-ischemic recovery in mice. Transl. Stroke Res. 7, 535–547. doi: 10.1007/s12975-016-0496-0
Yates, M. S., Tran, Q. T., Dolan, P. M., Osburn, W. O., Shin, S., Mcculloch, C. C., et al. (2009). Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 30, 1024–1031. doi: 10.1093/carcin/bgp100
Yu, S., Zhao, J., Wang, X., Lei, S., Wu, X., Chen, Y., et al. (2013). 4-Hydroxybenzyl alcohol confers neuroprotection through up-regulation of antioxidant protein expression. Neurochem. Res. 38, 1501–1516. doi: 10.1007/s11064-013-1052-x
Zhai, X., Chen, X., Shi, J., Shi, D., Ye, Z., Liu, W., et al. (2013). Lactulose ameliorates cerebral ischemia-reperfusion injury in rats by inducing hydrogen by activating Nrf2 expression. Free Radic. Biol. Med. 65, 731–741. doi: 10.1016/j.freeradbiomed.2013.08.004
Zhang, J., Fu, B., Zhang, X., Zhang, L., Bai, X., Zhao, X., et al. (2014). Bicyclol upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. Bull. 100, 38–43. doi: 10.1016/j.brainresbull.2013.11.001
Zhang, L., Zhang, X., Zhang, C., Bai, X., Zhang, J., Zhao, X., et al. (2016). Nobiletin promotes antioxidant and anti-inflammatory responses and elicits protection against ischemic stroke in vivo. Brain Res. 1636, 130–141. doi: 10.1016/j.brainres.2016.02.013
Zhang, M., Wang, S., Mao, L., Leak, R. K., Shi, Y., Zhang, W., et al. (2014). Omega-3 fatty acids protect the brain against ischemic injury by activating Nrf2 and upregulating heme oxygenase 1. J. Neurosci. 34, 1903–1915. doi: 10.1523/JNEUROSCI.4043-13.2014
Zhang, Q., Wang, J., Zhang, C., Liao, S., Li, P., Xu, D., et al. (2016). The components of Huang-Lian-Jie-Du-Decoction act synergistically to exert protective effects in a rat ischemic stroke model. Oncotarget 7, 80872–80887. doi: 10.18632/oncotarget.12645
Zhang, W., Song, J. K., Yan, R., Li, L., Xiao, Z. Y., Zhou, W. X., et al. (2018). Diterpene ginkgolides protect against cerebral ischemia/reperfusion damage in rats by activating Nrf2 and CREB through PI3K/Akt signaling. Acta Pharmacol Sin. 39, 1259–1272. doi: 10.1038/aps.2017.149
Zhang, W., Wei, R., Zhang, L., Tan, Y., and Qian, C. (2017). Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 366, 95–104. doi: 10.1016/j.neuroscience.2017.09.035
Zhang, Y., Fan, L., Li, H., Wang, X., Xu, W., Chu, K., et al. (2018). Gualou guizhi granule protects against oxidative injury by activating Nrf2/ARE pathway in rats and PC12 cells. Neurochem. Res. 43, 1003–1009. doi: 10.1007/s11064-018-2507-x
Zhao, J. J., Song, J. Q., Pan, S. Y., and Wang, K. (2016). Treatment with isorhamnetin protects the brain against ischemic injury in mice. Neurochem. Res. 41, 1939–1948. doi: 10.1007/s11064-016-1904-2
Keywords: antioxidant response element, global cerebral ischemia, middle cerebral artery occlusion, permanent MCAO, preclinical models, reperfusion, stroke, transient MCAO
Citation: Liu L, Locascio LM and Doré S (2019) Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharmacol. 10:153. doi: 10.3389/fphar.2019.00153
Received: 26 October 2018; Accepted: 08 February 2019;
Published: 05 March 2019.
Edited by:
Javier Egea, Institute of Health Research of the University Hospital of La Princesa, SpainReviewed by:
Alexander A. Mongin, Albany Medical College, United StatesSiew Hua Gan, Monash University Malaysia, Malaysia
Copyright © 2019 Liu, Locascio and Doré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvain Doré, c2RvcmVAdWZsLmVkdQ==