 Edijs Vavers
Edijs Vavers Liga Zvejniece
Liga Zvejniece Tangui Maurice2
Tangui Maurice2- 1Laboratory of Pharmaceutical Pharmacology, Latvian Institute of Organic Synthesis, Riga, Latvia
- 2MMDN, University of Montpellier, INSERM, EPHE, UMR-S1198, Montpellier, France
Allosteric modulators of sigma-1 receptor (Sig1R) are described as compounds that can increase the activity of some Sig1R ligands that compete with (+)-pentazocine, one of the classic prototypical ligands that binds to the orthosteric Sig1R binding site. Sig1R is an endoplasmic reticulum membrane protein that, in addition to its promiscuous high-affinity ligand binding, has been shown to have chaperone activity. Different experimental approaches have been used to describe and validate the activity of allosteric modulators of Sig1R. Sig1R-modulatory activity was first found for phenytoin, an anticonvulsant drug that primarily acts by blocking the voltage-gated sodium channels. Accumulating evidence suggests that allosteric Sig1R modulators affect processes involved in the pathophysiology of depression, memory and cognition disorders as well as convulsions. This review will focus on the description of selective and non-selective allosteric modulators of Sig1R, including molecular structure properties and pharmacological activity both in vitro and in vivo, with the aim of providing the latest overview from compound discovery approaches to eventual clinical applications. In this review, the possible mechanisms of action will be discussed, and future challenges in the development of novel compounds will be addressed.
Introduction
The International Union of Basic and Clinical Pharmacology included sigma receptor in its list of receptors only in 2013 as a ligand-regulated non-opioid intracellular receptor (Alexander et al., 2013). Two pharmacologically distinct subtypes of sigma receptor, namely, sigma-1 receptor (Sig1R) and sigma-2 receptor (Sig2R), have been identified (Hellewell and Bowen, 1990; Quirion et al., 1992; Hellewell et al., 1994). Sig2R is known also as endoplasmic reticulum-resident transmembrane protein TMEM97 (Alon et al., 2017) involved in the regulation of cholesterol homeostasis and cell differentiation (Bartz et al., 2009; Haller et al., 2012). Number of selective Sig1R and Sig2R ligands have been described confirming significant differences in the pharmacological regulation of these subtypes. Thus, far no allosteric modulators of Sig2R have been reported.
Sig1R is an integral membrane-bound protein that is found in the nuclear membrane and endoplasmic reticulum and mitochondria-associated membrane (Mori et al., 2013; Mavlyutov et al., 2015; Su et al., 2016). Sig1R is expressed in both the CNS and peripheral tissues (Su and Junien, 1994). Sig1R is widely distributed in the brain, and it concentrates in specific areas involved in memory, emotion and sensory and motor functions (Alonso et al., 2000; Cobos et al., 2008). Sig1R, as described by its functional nature, is a chaperone protein and a unique cell protein modulator [reviewed in (Su et al., 2016)] that can amplify or reduce the signaling initiated when interacting with target proteins (Hayashi and Su, 2007; Zamanillo et al., 2012; Rodríguez-Muñoz et al., 2015). Therefore, Sig1R demonstrates properties that can be attributed to both chaperone proteins and receptors. However, the notion that allosteric modulators of Sig1R are identified is an additional argument for the “receptor” view of Sig1R.
Allosteric regulation is the regulation of protein activity by binding an effector molecule at a site other than the orthosteric or active site of a protein (Figure 1). The binding of allosteric modulators to a target protein induces a conformational change in the protein structure and changes the activity of orthosteric ligands (Figure 1). Allosteric modulators can be positive or negative effectors (PAMs or NAMs, respectively). PAMs increase the activity of the ligand, while NAMs block it (Figure 1). To date phenytoin, some benzazepine derivatives and stereoisomers of methylphenylpiracetam have been reported as PAMs of Sig1R while NAMs of Sig1R have not yet been described. The definition of allosteric Sig1R modulators might be artificial due to a lack of information on the orthosteric binding site for Sig1R. It is thought that the orthosteric binding site for Sig1R ligands is the binding site of (+)-SKF-10,047 and (+)-pentazocine, benzomorphan compounds that were the first identified to bind to Sig1R with high affinity and selectivity (Martin et al., 1976; Su, 1982). Thus, allosteric modulators of Sig1R are described as compounds that can increase the activity of Sig1R ligands that compete with [3H](+)-pentazocine for binding to Sig1R.
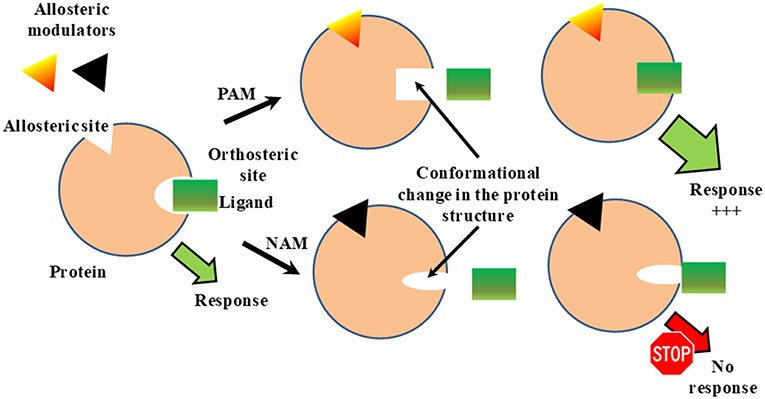
Figure 1. Classical model for allosteric regulation of receptor. PAM, positive allosteric modulation; NAM, negative allosteric modulation. This Figure has been modified from Vavers (2017).
At the moment, two compounds, the Sig1R and muscarinic receptor mixed agonist ANAVEX™ 2-73 (ANAVEX Life Sciences, ClinicalTrials.gov Identifier: NCT02244541) and the Sig1R antagonist E-52862 (ESTEVE, EudraCT number: 2012-000400-14) are being tested in clinical trials for the treatment of Alzheimer's disease and neuropathic pain, respectively, thus justifying the importance of Sig1R as a valid molecular target for clinical applications. Compared to widely used agonists and antagonists, allosteric modulators are less studied but are highly promising due to their advantageous clinical applications.
In recent years, significant progress has been achieved in the discovery, optimization and preclinical development of allosteric Sig1R modulators. These compounds can provide new advances in developing novel drugs, drug leads and research tools for Sig1R and have potential utility for the treatment of multiple human disorders. Accumulating evidence suggests that allosteric Sig1R modulators affect processes involved in the pathophysiology of depression, memory and cognition disorders as well as convulsions, and thus can provide novel strategies for the treatment of neurological disorders.
This review summarizes the literature and data on all known Sig1R allosteric modulators, including discovery and development, in vitro and in vivo pharmacological activities and discussion on allosteric regulation of Sig1R.
Discovery of Allosteric Sig1R Modulators
The first evidence indicating that a compound demonstrates allosteric activity on Sig1R came from radioligand binding studies. The first drug discovered as an allosteric modulator of Sig1R was phenytoin (diphenylhydantoin, Figure 2). Phenytoin has been used in clinical practice as an anti-convulsant since 1930 (Merritt and Putnam, 1984; Yaari et al., 1986). The anti-convulsant mechanism of phenytoin is the selective blockage of neuronal voltage-dependent sodium channels (Yaari et al., 1986). Over the course of competition binding studies, it was shown that phenytoin can increase the binding of [3H]dextromethorphan ([3H]DM) (Craviso and Musacchio, 1983) and [3H](+)-3-(3-hydroxyphenyl)-N-propylpiperidine ([3H](+)-3-PPP) in the guinea pig brain (Musacchio et al., 1989). These results were the first showing that phenytoin allosterically modulated the binding of prototypic sigma site ligands. Very rapidly, phenytoin sensitivity was even considered an intrinsic characteristic of the sigma-1 subtype of sigma sites, not shared by sigma-2 (Quirion et al., 1992). Sig1R were indeed defined mainly through their high-affinity sites for the dextrorotatory isomers of benzomorphans and their sensitivity to phenytoin. Moreover, similar to phenytoin, ropizine (SC-13504), an anti-convulsant benzhydryl piperazine (Figure 2), induced a marked concentration-dependent increase in the binding of [3H]DM (Musacchio et al., 1988) and [3H](+)-3-PPP (Musacchio et al., 1989). It was shown that the non-narcotic anti-tussive noscapine can dose-dependently potentiate the binding of [3H]DM in guinea pig brainstem homogenate (Craviso and Musacchio, 1983). In addition, hydrastine demonstrated similar activity on the binding of [3H]DM (Craviso and Musacchio, 1983). However, to date, noscapine and hydrastine have not been demonstrated to modulate the binding of more selective Sig1R ligands such as [3H](+)-pentazocine and are considered only putative allosteric Sig1R modulators.
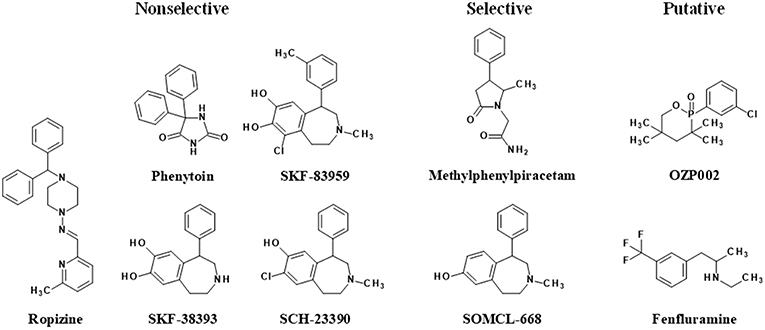
Figure 2. Allosteric modulators of Sig1R. Allostericity was not yet demonstrated for OZP002 and fenfluramine, only the Sig1R modulatory effect, and must therefore be considered as putative PAMs.
Later, compound SR31747A (N-cyclohexyl-N-ethyl-3-(3-chloro-4-cyclohexylphenyl)propen-2-ylamine hydrochloride) was also proposed to act as an allosteric modulator of peripheral sigma binding sites (Paul et al., 1994). Although SR31747A modulated the activity of Sig1R ligands in vivo and in vitro, the use of radiolabeled [3H]SR31747A demonstrated that SR31747A binds specifically, saturably and reversibly to rat spleen membranes and human lymphocytes at a single class of high-affinity sites, which were clearly different from the [3H](+)-pentazocine and [3H](+)-3-PPP binding sites (Paul et al., 1994). A few years later, the purified amino acid sequence of the [3H]SR31747A binding site was found to be a nuclear membrane protein related to a fungal C8-C7 sterol isomerase; additionally, this protein, which was called SR31747A-binding protein, was encoded by the ERG2 gene (Jbilo et al., 1997). The exact molecular mechanism of SR31747A has not been fully described thus far, but SR31747A is currently not considered an allosteric Sig1R modulator.
In 2013, more than two decades after the discovery of the positive allosteric Sig1R modulatory activity of phenytoin, it was shown that the atypical dopamine D1 receptor agonist SKF83959 (3-methyl-6-chloro-7,8-hydroxy-1-[3-methylphenyl]-2,3,4,5-tetrahydro-1H-3-benzazepine) and its analogs SKF38393 and SCH23390 act as allosteric Sig1R modulators (Guo et al., 2013; Figure 2). These compounds could enhance the binding activity of [3H](+)-pentazocine in brain and/or liver tissues, shift the saturation curve toward the left, and decrease the dissociation rate in binding kinetic analysis (Guo et al., 2013). At the same time another allosteric Sig1R modulator, methylphenylpiracetam (2-(5-methyl-2-oxo-4-phenyl-pyrrolidin-1-yl)-acetamide), was described (Veinberg et al., 2013; Figure 2). The activity of E1R, a 4R,5S-isomer of methylphenylpiracetam, was profiled using commercially available screening assays, which showed that the compound selectively modulates Sig1R activity but does not affect the other investigated neuronal receptors and ion channels (Zvejniece et al., 2014). The following detailed in vitro studies, in which more selective Sig1R ligands were used, provided solid evidence that E1R is a PAM of Sig1R (Zvejniece et al., 2014). Sig1R is the only molecular target described thus far that accounts for the pharmacological activity of E1R. Therefore, E1R is considered the first known selective allosteric Sig1R modulator. Later, several chemical derivatives of SKF83959 were synthesized to find a selective Sig1R allosteric modulator and to exclude the potential involvement of other receptors. One of these newly synthesized compounds, called SOMCL-668 (Figure 2), did not exhibit affinity for human dopamine D1, D2, D3, serotonin 5-HT1A, or 5-HT2A receptors (Zhang et al., 2014) but did show potent allosteric modulating activity at Sig1R (Guo et al., 2015). Therefore, SOMCL-668 has also been proposed as a selective allosteric Sig1R modulator.
Recently, a novel oxazaphosphinane compound derived from hydroxybupropion, was described (Volle et al., 2010). (±)-2-(3-chlorophenyl)-3,3,5,5-tetramethyl-2-oxo-[1,4,2]-oxazaphosphinane (OZP002) did not inhibit [3H](+)-pentazocine binding to Sig1R, as did bupropion or hydroxybupropion (Ritz and George, 1997), but rather moderately increased it. However, the drug did potentiate Sig1R agonist-induced antidepressant and anti-amnesic effects in wild-type mice but not in Sig1R-knock-out animals, and the effects were prevented by a Sig1R antagonist NE-100 suggesting a Sig1R positive modulatory effect (Maurice et al., 2017). Finally, fenfluramine (N-ethyl-α-methyl-3-(trifluoromethyl)-benzeneethanamine), a potent serotonin releaser activating multiple 5-HT receptor subtypes (Fuller et al., 1988), has been described for its positive modulatory activity at Sig1R (Maurice et al., 2018). Beyond serotonin, fenfluramine also binds Sig1R with high nanomolar affinity for Sig1R. However, in functional assays, fenfluramine potentiated the (+)-SKF-10,047-induced increase in the twitch contraction amplitude and the Sig1R/binding immunoglobulin protein (BiP) dissociation induced by the Sig1R agonist PRE-084, suggesting a positive modulatory action at Sig1R (Maurice et al., 2018).
The list of identified or suspected Sig1R positive modulators is presently being extended, and they present effective pharmacological activity that is closely related in vivo to that of orthosteric agonists. However, the different chemical structures of the identified compounds suggest a complex mode of action and clear specificities among the drugs.
The Pharmacological Activities of Allosteric Sig1R Modulators
Phenytoin
The allosteric modulation of sigma recognition sites by phenytoin has been demonstrated by the ability of phenytoin to stimulate the binding of various tritiated Sig1R agonists (Musacchio et al., 1988; McCann and Su, 1991; Chaki et al., 1996; Cobos et al., 2005), to slow dissociation from sigma sites and to shift sigma sites from a low-affinity state to a high-affinity state (DeHaven-Hudkins et al., 1993). A detailed comparison of the effects of phenytoin on the binding of [3H](+)-pentazocine and [3H]NE-100 using saturation and kinetics assays showed that phenytoin acts as PAM and can negatively modulate the binding of [3H]NE-100 by decreasing the specific binding and increasing the dissociation rate from Sig1R (Cobos et al., 2006), demonstrating significant allosteric effects on the Sig1R ligand binding to Sig1R.
Phenytoin has been used in the clinic against various types of epileptiform seizures for more than 80 years (Santulli et al., 2016). The mechanism by which phenytoin exerts its anti-seizure activity is primarily related to the inhibition of voltage-gated sodium channels (Tunnicliff, 1996). Phenytoin has been reported to reduce increases in extracellular K+ concentration in neuroblastoma cells (Nobile and Lagostena, 1998) and inhibit both Na+ (Rush and Elliott, 1997) and T-type Ca2+ currents in isolated dorsal root ganglia (Todorovic and Lingle, 1998). Phenytoin was shown to block the decrease in extracellular Ca2+ concentration effected by repetitive stimulation of pyramidal cells in hippocampal slices (Yaari et al., 1986). In rat cerebral synaptosomes phenytoin has been shown to increase the activity of Na+-K+-ATPase (Festoff and Appel, 1968). Phenytoin is known to block Na+ influx through sodium channels and the binding of [3H]phenytoin to these channels is inhibited by sodium channel blockers (Tunnicliff, 1996). It has been shown that Sig1R is involved in the regulation of sodium channels (Johannessen et al., 2009), however it has not been clearly demonstrated that the effects of phenytoin on sodium channels are Sig1R specific, and the in vivo pharmacological effects of phenytoin are not Sig1R related. Therefore, phenytoin is a non-selective allosteric modulator of Sig1R.
Ropizine (SC-13504)
Ropizine was shown to increase the binding of [3H]DM and [3H](+)-3-PPP in guinea pig brains (Musacchio et al., 1988, 1989). The allosteric effects of ropizine on [3H]DM binding are fully apparent at 10-fold lower concentrations than those of phenytoin (Musacchio et al., 1988). Ropizine has been shown to possess anti-convulsant activity in mice (Craig, 1967), cats (Edmonds and Stark, 1974) and dogs (Edmonds et al., 1978). It has been shown that ropizine is similar in efficacy to phenytoin in maximal electroshock-induced seizures (Novack et al., 1979), while it has limited anti-convulsant activity against chemically induced seizures (Edmonds et al., 1978, 1979). Ropizine is more active and potent than phenytoin in antagonizing the after discharges produced by cortical or hippocampal stimulation in cats (Joy and Edmonds, 1974). Data in the literature about the mechanism of action of ropizine are scarce (Table 1). It has been shown that ropizine inhibits magnesium-dependent ATPase activity in rat brain synaptosomes (Gilbert and Wyllie, 1976). Direct involvement of Sig1R in this pharmacological activity has not been demonstrated, and thus, ropizine is considered a non-selective allosteric modulator of Sig1R.
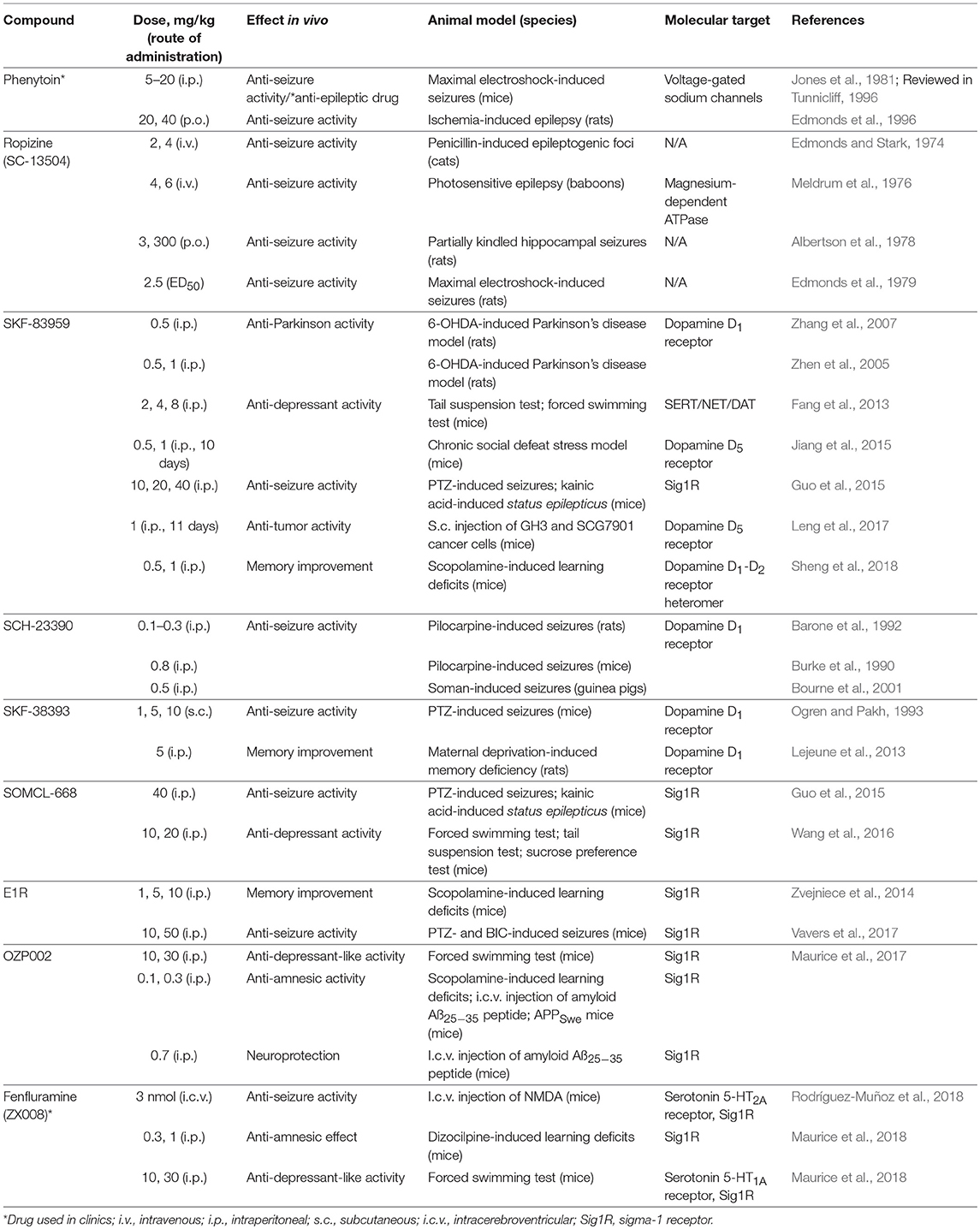
Table 1. Summary of in vivo effects of allosteric Sig1R modulators.
Methylphenylpiracetam (E1R)
The Sig1R site was the only site that E1R was discovered to target in in vitro pharmacological profiling assays, which included a number of ion channels, G protein-coupled receptors and CNS transporters (Zvejniece et al., 2014). The selected in vitro assays revealed that E1R did not bind directly to Sig1R but rather acted as a PAM of the receptor and enhanced the binding of an unselective sigma receptor radioligand [3H]DTG. E1R potentiated the contractions of rat vasa deferentia in the presence of the selective Sig1R agonist PRE-084 but not in the presence of the Sig2R agonist PB-28 (Zvejniece et al., 2014). In addition, E1R enhanced the effect of PRE-084 on the BDK-induced [Ca2+]i increase, thus confirming its Sig1R positive allosteric modulatory effect in vitro.
E1R successfully alleviated scopolamine-induced cognitive impairment in mice, as assessed using passive avoidance and spontaneous alternation (Y-maze) tests. The effects of E1R were antagonized by the selective Sig1R antagonist NE-100, thus confirming the Sig1R modulatory activity of E1R in vivo. E1R demonstrated dose-dependent anti-convulsant effects on pentylenetetrazole (PTZ)- and bicuculline (BIC)-induced seizures at doses of 10 and 50 mg/kg (Vavers et al., 2017; Tables 1, 3). To verify that Sig1R was involved in the anti-convulsant activity of E1R, the selective Sig1R antagonist NE-100 was used. The administration of NE-100 (5 mg/kg) before E1R (10 mg/kg) significantly restored the tonic seizure threshold to the basal level and therefore showed that the anti-seizure effect of E1R was mediated through Sig1R activity (Vavers et al., 2017). The pharmacological activity of E1R demonstrates that it is a selective PAM of Sig1R.
Benzazepine Derivatives
SKF83959, SCH23390, SKF38393
SKF83959 is an atypical D1 agonist (Downes and Waddington, 1993; Deveney and Waddington, 1995). Previously, it was shown that SKF83959 also exerted many D1 receptor-independent pharmacological effects. For example, SKF83959 suppressed excitatory synaptic transmission and voltage-activated Na+ current in rat hippocampus (Chu et al., 2011), inhibited the delayed rectifier potassium channel in primary culture neurons (Chen et al., 2009), and promoted the spontaneous release of glutamate in the rat somatosensory cortical neurons (Chu et al., 2010). The activity of SKF83959 was related to Sig1R based on the pharmacological activity of SKF83959 and similarities of the pharmacophore with some other Sig1R ligands. SKF-83959 increased the binding of [3H](+)-pentazocine to Sig1R in the brain and liver tissues and showed that SKF-83959 is PAM of Sig1R (Guo et al., 2013). SKF83959 has been shown to inhibit the generation of intracellular reactive oxygen species and the expression of tumor necrosis factor α, interleukin-1β, and cytokine-inducible nitric oxide synthase in lipopolysaccharide-stimulated mouse brain microglial BV2 cells (Wu et al., 2015). These effects of SKF83959 were blocked by Sig1R antagonists BD-1047 and BD1063 (Wu et al., 2015). In the same study, it was shown that in a [3H](+)-pentazocine binding assay, SKF83959 enhanced the binding activity of dehydroepiandrosterone (DHEA), a neurosteroid acting as a Sig1R agonist, by shifting the DHEA binding curve to the left. In addition, SKF83959 enhanced the anti-inflammatory effect of exogenous DHEA in a synergistic manner, which was dependent on Sig1R (Wu et al., 2015), thus showing that the anti-inflammatory effects of SKF83959 are due to positive allosteric Sig1R modulator activity in cells.
The anti-convulsant effects of SKF83959 at doses of 20 and 40 mg/kg in PTZ- and kainic acid-induced seizures were demonstrated to be mediated by modulating Sig1R (Guo et al., 2015, Tables 1, 3). The anti-convulsant effects of SKF83959 were blocked by the selective Sig1R antagonist BD-1047 at a dose of 1 mg/kg (Guo et al., 2015). SKF83959 has also demonstrated significant anti-Parkinson, anti-depressant, anti-tumor and memory-improving activity, which is associated with activity at dopamine receptors and regulation of dopamine reuptake (summarized in Table 1). In addition, it has been shown that SKF83959 inhibits voltage-gated sodium channels in cultured striatal neurons via the D1-like receptor-phosphatidylinositol-PKC pathway (Ma et al., 2015) and inhibits dopamine-sensitive adenylyl cyclase activity (Downes and Waddington, 1993), which are responsible for some additional in vivo activities not related to Sig1R.
SKF83959 analogs, such as SCH22390 and SKF38393, also have shown similar allosteric effects on Sig1R in liver tissue using [3H](+)-pentazocine (Guo et al., 2013). It was shown that SCH23390 affords protection against pinacolylmethylphosphonofluoridate-evoked electrical and motor seizure activity by inhibiting D1 receptor (Bourne and Fosbraey, 2000) but, similar to SKF83959, does not demonstrate anti-seizure activity in a PTZ-induced seizure model (Guo et al., 2015). SCH23390 is considered a highly potent D1 antagonist (Hyttel, 1983) and demonstrates moderately high affinity for the 5-HT2 and 5-HT1C receptors as well (Hicks et al., 1984). In contrast, SKF38393 is a selective D1 agonist (Molloy and Waddington, 1984), and the in vivo pharmacological activity of SKF38393 is D1 receptor dependent (Table 1). Overall, SKF83959 and its analogs SCH22390 and SKF38393 are non-selective allosteric modulators of Sig1R.
SOMCL-668
SOMCL-668 has demonstrated selective allosteric modulating activity on Sig1R (Guo et al., 2015). In the [3H](+)-pentazocine binding assay, SOMCL-668 shifted the saturation curve toward the left and significantly decreased the dissociation constant of the radioligand (Guo et al., 2015). SOMCL-668 synergized the effect of (+)-SKF-10,047 on Sig1R dissociation from binding immunoglobulin protein, which was further confirmed by immunoprecipitation and the plasma membrane translocation assay (Wang et al., 2016). SOMCL-668 increased the phosphorylation of glycogen synthase kinase-3β (GSK3β) on the Ser9 epitope and potentiated the agonist (+)-SKF-10,047-increased phosphorylation of (Ser9)-GSK3β in the hippocampus of mice and in hippocampal neuronal HT-22 cells, which was abolished by pretreatment with the Sig1R antagonist BD-1047 or by the knockdown of Sig1R in HT-22 cells (Wang et al., 2016). SOMCL-668 also significantly potentiated (+)-SKF-10,047-stimulated neurite growth and the production of BDNF (Wang et al., 2016), and the effect was Sig1R dependent.
The selective Sig1R allosteric modulator SOMCL-668 has been tested for its potential anti-depressant activity (Wang et al., 2016). SOMCL-668 at doses of 10 and 20 mg/kg significantly decreased the immobility time of mice in the forced-swimming and tail suspension tests, and these effects were blocked by the BD-1047 (Wang et al., 2016). In addition, daily administration of SOMCL-668 at a dose of 10 mg/kg for 1 week significantly reversed the decrease in the sucrose preference in the chronic mild stress model in mice (Wang et al., 2016). SOMCL-668 at a dose of 40 mg/kg also raised the seizure threshold in the maximal electroshock seizure test and prolonged the latencies to the clonus and generalized tonic-clonic convulsions as well as the survival time in the PTZ-induced seizure model (Guo et al., 2015). In kainic acid-induced status epilepticus SOMCL-668 prolonged the latency to seizure, lowered the average severity of seizure and shortened the duration of seizure (Guo et al., 2015). Moreover, the Sig1R antagonist BD-1047 at a dose of 1 mg/kg abolished the anti-seizure activity of SOMCL-668, indicating that its effect was Sig1R dependent (Guo et al., 2015; Table 1). Only Sig1R has been demonstrated to be involved in the pharmacological activities of SOMCL-668; therefore SOMCL-668 is considered a selective PAM of Sig1R.
OZP002
The oxazaphosphinane compound did not inhibit [3H](+)-pentazocine binding to sigma sites, but it did induce Sig1R receptor-like effects in vivo and potentiated the behavioral efficacy of Sig1R agonists. OZP002 had an antidepressant effect in the forced swimming test at 10 mg/kg and potentiated the effect of the Sig1R agonist igmesine at 5 mg/kg. These effects were blocked by coinjection of the Sig1R antagonist NE-100 or were absent in Sig1R-knock-out mice (Maurice et al., 2017). OZP002 prevented scopolamine-induced learning and memory impairments (spontaneous alternation or passive avoidance) at 0.1–0.3 mg/kg, and these effects were blocked not only by NE-100 but also, interestingly, by the α7 nicotinic acetylcholine receptor antagonist methyllycaconitine (Maurice et al., 2017). Moreover, the compound was analyzed for its neuroprotective activity in a pharmacological model of Alzheimer's disease, induced in mice by intracerebroventricular injection of oligomerized amyloid Aß25−35 peptide, and in APPSwe mice after a 2-months treatment. OZP002 prevented learning deficits in both models and decreased the expression levels of several markers of apoptosis, neuroinflammation and oxidative stress (Maurice et al., 2017). The drug therefore clearly acted as a Sig1R positive modulator and showed in vivo pharmacological activity closely related to that of Sig1R agonists, combining pharmacological efficacy, selectivity and therapeutic safety.
Fenfluramine (ZX008)
Fenfluramine has recently been shown in 2 phase 3 studies to have anti-convulsant activity in a childhood epilepsy condition, Dravet syndrome (Polster, 2018). It acts as a 5-HT agonist by interacting with 5-HT1D, 5-HT2A, and 5-HT2C receptors (Sourbron et al., 2016). The drug also interacts with Sig1R at nanomolar concentrations (Martin et al., 2017). In the Sig1R/BiP dissociation assay, it did not promote Sig1R/BiP dissociation but rather potentiated the effect of the Sig1R agonist PRE-084, suggesting a positive modulatory activity. The (+)-isomer of fenfluramine but not (-)-fenfluramine attenuated dizocilpine-induced deficits in a similar manner as PRE-084 (Maurice et al., 2018). It must be noted that the racemate and individual isomers of fenfluramine-related compound norfenfluramine did not affect dizocilpine-amnesia but rather prevented the PRE-084 effect, suggesting Sig1R antagonism. Combination between low doses of fenfluramine or (+)-fenfluramine and PRE-084 followed by calculation of combination indexes (Zhao et al., 2010; Maurice, 2016) showed that most combinations led to synergistic effects. Fenfluramine, as well as its active isomer (+)-fenfluramine, behaved in vitro and in vivo as Sig1R positive modulators. The drug is therefore a mixed 5-HT releaser/5-HT agonist and Sig1R modulator. Moreover, as the neuromodulatory role of Sig1R on neurotransmitter release, receptor activation and regulation of numerous ionophores is well-described, Sig1R modulation must be considered in the potential mechanism of action of its anti-convulsant activity demonstrated for Dravet syndrome (Polster, 2018).
Comparison of the Effects of Allosteric Sig1R Modulators
Stereochemistry of Allosteric Sig1R Modulators
A number of Sig1R ligands contains chiral centers in their molecular structures, which indicates that compounds are optically active and that there are possibly different stereoisomers of the same molecule, which also corresponds to different pharmacological activities for the same molecules. Geometric isomers of ropizine in cis and trans forms are possible, but the molecule of ropizine is not optically active. Additionally, phenytoin is not an optically active compound due to the lack of chiral atoms in the molecular structure. Methylphenylpiracetam is a complex molecule in terms of its stereochemistry. There are two chiral centers in the molecular structure of methylphenylpiracetam; therefore, it is possible to isolate four individual stereoisomers, which are denoted E1R, T1R, E1S, and T1S (reviewed in Veinberg et al., 2015; Figure 3).
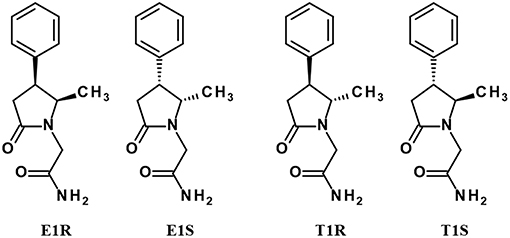
Figure 3. Stereoisomers of methylphenylpiracetam.
SKF83959 is a racemate that consists of the R-(+)- and S-(–)-enantiomers MCL-202 and MCL-201, respectively (Desai et al., 2007). The R-(+)- and S-(–)-enantiomers are also possible for SKF38393, SCH23390, and SOMCL-668 (Figure 4).
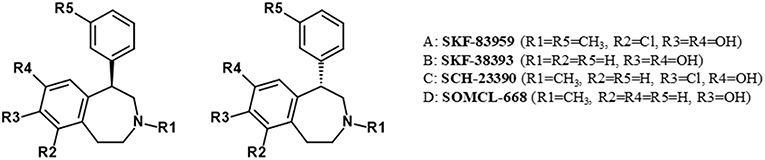
Figure 4. Enantiomers of benzazepine analogs.
All stereoisomers of methylphenylpiracetam are known to act as PAMs of Sig1R (Veinberg et al., 2013). In an ex vivo test the enantiomers with the R-configuration at the C-4 chiral center in the 2-pyrrolidone ring (E1R and T1R) were found to be more effective PAMs of Sig1R than their optical antipodes (Veinberg et al., 2013), showing that some stereo selectivity is possible also for allosteric Sig1R modulators.
The Sig1R modulatory activity of changes in the activity of Sig1R ligands induced by different enantiomers of benzazepine derivatives has not been compared. It would be interesting to compare the allosteric modulatory activity on Sig1R regarding the stereochemical properties of benzazepine derivatives. For example, it has been shown that the D1 receptor binding site favors the R-enantiomers of the D1 receptor-selective benzazepines over the corresponding S-enantiomers (Kebabian, 1988), which is true for both the D1 agonist SKF38393 and the D1 antagonist SCH23390 (Kebabian, 1988). In addition, the stereo selectivity of the R-isomer of SCH23390 for blockade of D1 receptors in vivo has been demonstrated (Ongini et al., 1987). In the case of benzazepine derivatives, Sig1R selectivity over dopamine receptors could depend on the pharmacological activity of individual isomers. However, this dependence has not been demonstrated so far.
The structure-activity relationship for fenfluramine is rather clear since the positive modulator activity of the drug originates from only the dextrorotatory isomer (+)-fenfluramine. In all behavioral responses analyzed, the racemate behaved as (+)-fenfluramine, suggesting chiral binding of the drug to the yet unidentified, modulatory binding site on Sig1R.
In vitro Effects
No screening assay is available for allosteric modulators of Sig1R. Therefore, different in vitro assays and in vivo pharmacological approaches have been used to describe and validate the activity of allosteric Sig1R modulators. For comparison, the Sig1R-related in vitro activities of allosteric Sig1R modulators are summarized in Table 2.
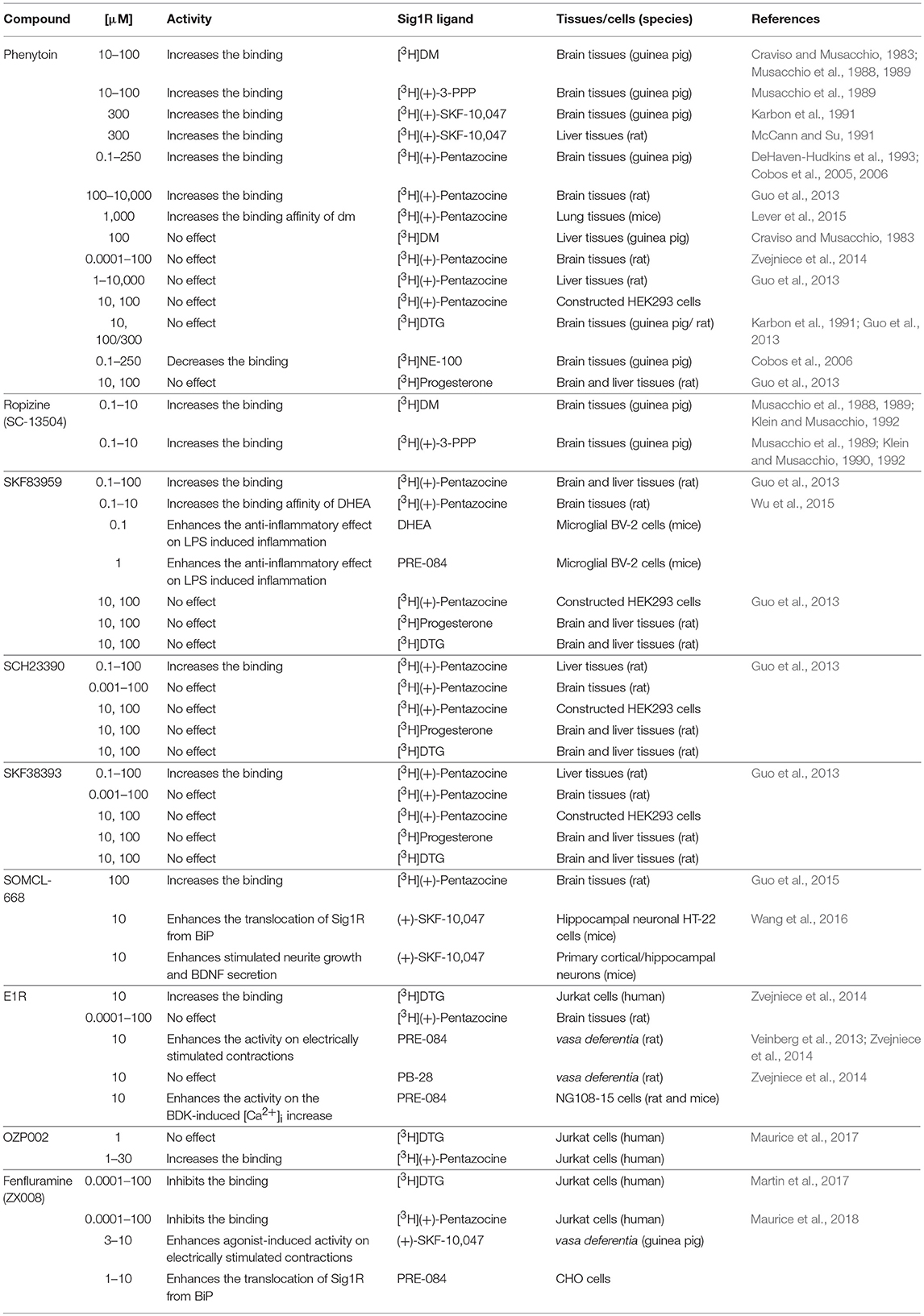
Table 2. The comparison of in vitro allosteric effects of Sig1R modulators.
The allosteric modulatory activity of phenytoin has previously been described in rat and guinea pig brains (DeHaven-Hudkins et al., 1993; Cobos et al., 2006; Guo et al., 2013), rat livers (McCann and Su, 1991), and mice lung tissues (Lever et al., 2015). In turn, it has been shown that phenytoin could modulate Sig1R ligand binding in rat brain tissue but not in rat liver tissue (Guo et al., 2013, Table 2). For example, SCH23390 and SKF38393 modulated the binding of [3H](+)-pentazocine only in liver tissues, while no detectable effects were observed in brain tissues (Guo et al., 2013). In the same study, SKF83959 was shown to allosterically modulate the binding of [3H](+)-pentazocine in both rat brain and liver tissues (Guo et al., 2013). The different effects of these Sig1R allosteric modulators in rat and guinea pig brain tissues have been described previously and could be explained by the variation in the size of the binding site between the two species (Klein and Musacchio, 1992). It seems that not only different species but also different tissues from the same animal species can respond differently to allosteric Sig1R modulators. SKF83959 and its analogs failed to change the binding activity of [3H](+)-pentazocine at Sig1R in human embryonic kidney (HEK)293 cells that stably expressed the Sig1R (Guo et al., 2013). The lack of binding modulatory activity of [3H](+)-pentazocine in HEK293 cells was also observed for phenytoin (Guo et al., 2013). Although [3H](+)-pentazocine displayed similar affinity for Sig1R in transfected HEK293 cells and rat brain tissues, the absence of allosteric modulation of Sig1R in the constructed system in vitro could be attributed to differences in Sig1R structure, cellular contents, or auxiliary proteins (Guo et al., 2013), which should be taken into account when studying mechanisms of Sig1R in vitro.
Above described issues rise number of questions in the understanding of molecular mechanisms of allosteric Sig1R modulators. Why allosteric modulatory activity of compounds has been observed in one type of tissues and/or animal species but not in the others? Is that because of the structure differences between compounds? Could it be because of different Sig1R structures, conformation states or even tissue specific Sig1R subtypes? Are there species specific Sig1R? It should be noted that some observed allosteric effects on Sig1R were shown after administration of allosteric modulator in high concentrations (Table 2). Thereby, how Sig1R-specific is the activity of allosteric modulators? Are there other proteins involved in the activity of compounds? What is the influence of endogenous compounds? Future studies must be focused to answer these questions. Nevertheless, it is clear that allosteric Sig1R modulators could be used as pharmacological tools to increase the global understanding of the physiological function of Sig1R and its interaction with ligands. Also the Sig1R-related in vivo activities of allosteric Sig1R modulators are very promising to reach clinical advantages.
Anti-seizure Activity
All allosteric Sig1R modulators demonstrate anti-seizure activity. However, the anti-seizure activity is not associated directly with Sig1R for most of allosteric Sig1R modulators (Table 3).
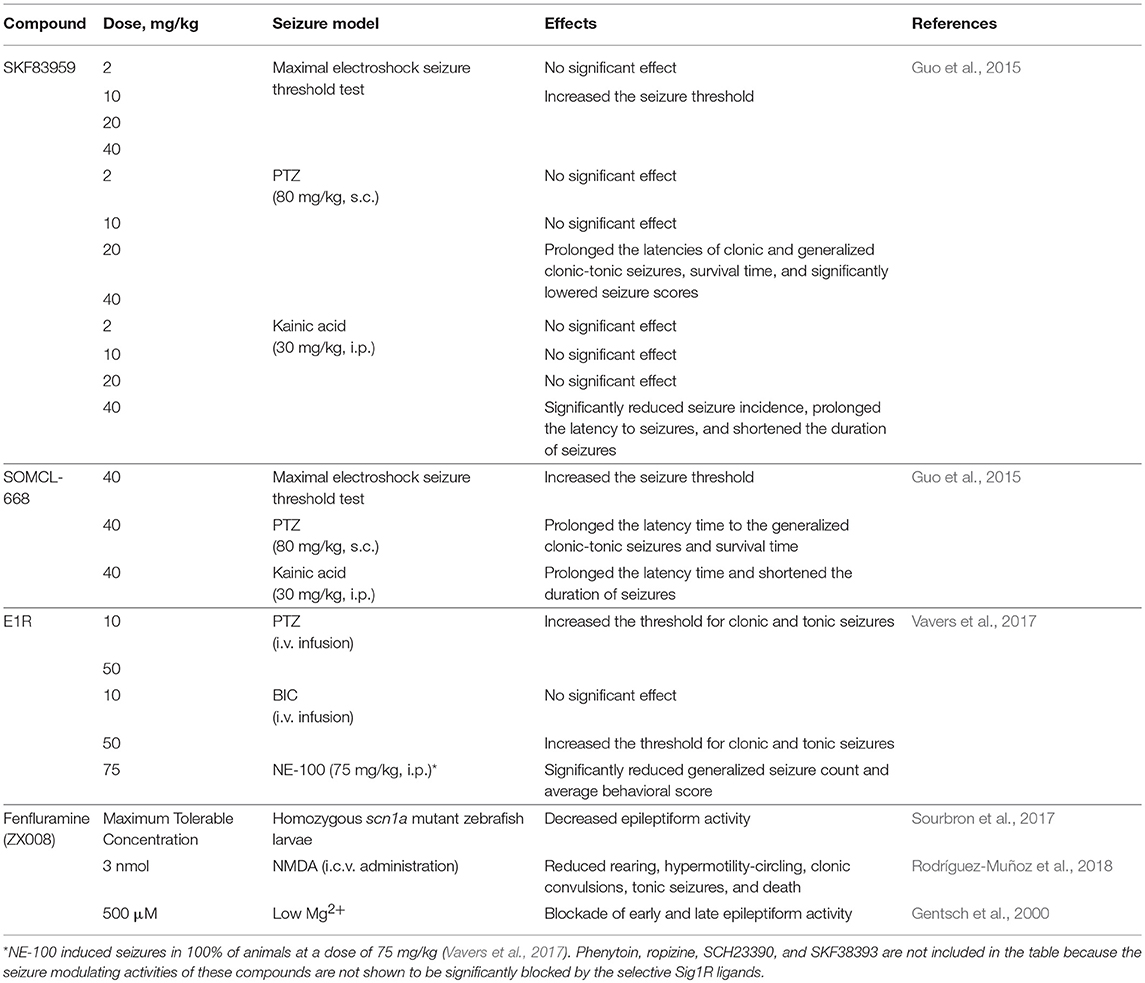
Table 3. Sig1R-dependent anti-seizure activity of allosteric Sig1R modulators.
It was demonstrated that the anti-seizure effect of E1R, SOMCL-668, and SKF83959 was blocked by the Sig1R selective antagonists NE-100 and BD-1047 (Guo et al., 2015; Vavers et al., 2017), while the anti-seizure effect of phenytoin was not blocked by BD-1047 (Guo et al., 2015). Moreover, anti-seizure effect of phenytoin is related to the inhibition of voltage-gated sodium channels (Tunnicliff, 1996). At the same time, it was shown that SCH23390, an analog of SKF83959, does not affect PTZ-induced seizures (Guo et al., 2015). Rather, SCH23390 has been shown to modulate seizures evoked by the subcutaneous administration of the cholinesterase inhibitor pinacolylmethylphosphonofluoridate and this activity was demonstrated to be D1 receptor dependent (Bourne et al., 2001). Current knowledge indicates that not all PAMs of Sig1R possess anti-seizure activities and that the anti-seizure effects are not always regulated by Sig1R.
Memory Improvement
Among all the positive allosteric Sig1R modulators described, E1R, OZP002, and fenfluramine showed Sig1R-dependent memory-improving effects (Zvejniece et al., 2014; Maurice et al., 2017, 2018). E1R, however, is the only modulator showing dose-dependent memory-improving activity in drug-naïve animals (Zvejniece et al., 2014). E1R and OZP002 successfully alleviated scopolamine-induced cognitive impairment in mice, as assessed using passive avoidance and spontaneous alternation tests (Zvejniece et al., 2014; Maurice et al., 2017). The effects were antagonized by the selective Sig1R antagonist NE-100, thus confirming the Sig1R modulatory activity of E1R in vivo. Moreover, fenfluramine showed anti-amnesic effects in mice treated with the non-competitive NMDA receptor antagonist dizocilpine (Maurice et al., 2018). This activity has long been described for Sig1R agonists (Maurice et al., 1994a,b) and is usually considered a potential activity test confirming the Sig1R agonist or antagonist activity of selective drugs. Finally, OZP002 showed anti-amnesic effects against learning deficits induced by amyloid Aβ25−35 peptide, a pharmacological model of symptomatic efficacy in Alzheimer's diease (Maurice et al., 1998). Some memory improvement activities have also been demonstrated for benzazepines SKF83959 and SKF38393. For example, administration of SKF83959 significantly improved scopolamine-induced memory impairments in the passive avoidance task, spontaneous alternation test, and place learning task in the Morris water maze task in mice (Sheng et al., 2018; Table 1). SKF38393 has been shown to improve temporal order memory performance in maternally deprived rats (Lejeune et al., 2013; Table 1). However, the effects of SKF83959 and SKF38393 on memory are dopamine receptor-mediated, and the involvement of Sig1R in the memory improvement of these compounds has not been demonstrated. In contrast to E1R treatment, treatment with phenytoin triggered memory impairment during the passive avoidance task (Reeta et al., 2009). Treatment of epilepsy with phenytoin has been shown to induce learning and memory deficits in patients as well (Mishra and Goel, 2015). Sig1R-related memory-improving activity could be specific for selective allosteric Sig1R modulators such as E1R and SOMCL-668. However, it has not been demonstrated that SOMCL-668 improves memory and cognition through a Sig1R-related pathway. Therefore, to date, the Sig1R-related memory-improving activity is specific only for E1R, OZP002, and fenfluramine.
Anti-depressant Activity
To date, SOMCL-668, OZP002 and fenfluramine have shown anti-depressant activity, which has been demonstrated to be Sig1R specific. The anti-depressant activity of SKF-83959 was shown to be regulated through other mechanisms (Table 1). It has been described that stereoisomers of methylphenylpiracetam, including E1R, did not induce any significant effects on the depressive condition in mice (Vavers et al., 2015). In the case of phenytoin, major depression as a complication of phenytoin intoxication has been described (Levkovitch et al., 1993). Therefore, anti-depressant activity seems to be shared by but not specific to allosteric modulators of Sig1R.
Allosteric Modulation of Sig1R
There are no clearly defined molecular mechanisms that could fully describe the function of Sig1R and the activity of Sig1R ligands. Thus, it is more difficult to describe the activity of allosteric modulators, which do not compete with orthosteric Sig1R ligands for binding in the active site of Sig1R but rather enhance the activity of Sig1R agonists. The crystal structure of Sig1R shows that the ligand-binding domain in the protein is highly conserved, and how ligands enter and exit this site remains unclear (Schmidt et al., 2016). The binding site for allosteric Sig1R modulators is probably located outside the orthosteric ligand-binding domain. Since allosteric modulators are compounds that induce a conformational change within the protein structure, PAMs should reorganize the Sig1R protein in a way that would allow agonists to freely enter the ligand-binding site. It has been discussed previously that phenytoin might induce a conformational change in the Sig1R and thus enhance the affinity of the orthosteric ligand [3H](+)-pentazocine for its binding site on Sig1R (Cobos et al., 2006), which fits classic description of the activity of allosteric modulators (Figure 6). However, it is not clear how allosteric modulators of Sig1R can distinguish between agonists and antagonists and then selectively enhance the activity of agonists, even though the agonists and antagonists sometimes contain the same structural moieties.
Classical models for allosteric modulation might not be attributed to allosteric modulation of Sig1R. Several previous experiments support the conclusion that in ligand binding, Sig1R functions as an oligomer (Schuster et al., 1995; Chu et al., 2013; Gromek et al., 2014). Therefore, oligomerization is a key functional property of the Sig1R that may be linked to ligand efficacy [Schmidt et al., 2016; reviewed in Chu and Ruoho (2016)] and could explain how ligands can regulate the activity of Sig1R (Figure 6). Sig1R ligands may regulate the activity of the receptor interaction with client proteins by altering the oligomeric/monomeric receptor ratio and favoring the oligomeric states (Mishra et al., 2015). It has been shown that the Sig1R agonist (+)-pentazocine increased the relative ratio of dimers and monomers, while the inhibitor haloperidol increased the incidence of higher oligomeric forms (Chu and Ruoho, 2016). This observation indicates that higher oligomeric forms of Sig1R might be functionally inactive (Figure 5A). Since all Sig1R allosteric modulators known thus far are PAMs and enhance the activity of Sig1R agonists, they might modulate Sig1R by stabilizing the agonist state of the receptor, providing an increase in the dimeric (Figure 5B) and/or monomeric (Figure 5C) protein forms.
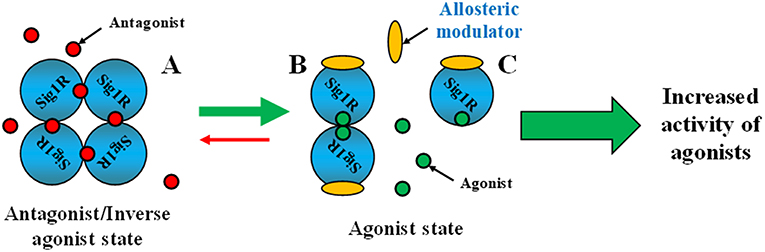
Figure 5. Stabilization of the agonist state of Sig1R by PAMs. A model showing possible mechanisms of Sig1R ligand activity. (A) Oligomeric form. For the agonist state, (B,C) represent the dimeric and monomeric forms of Sig1R, respectively. This Figure has been modified from (Chu and Ruoho, 2016).
Sig1R has already been described as a chaperone that modulates other receptor systems through heteromeric protein-protein interactions (Navarro et al., 2013; Pabba, 2013; Moreno et al., 2014; Su et al., 2016). Therefore, it is possible that heteromeric complexes formed by Sig1R and its target proteins could be regulated by allosteric modulators of Sig1R (Figure 6). Allosteric Sig1R modulators could serve as a bridge between Sig1R and its target proteins. The “molecular glue” concept was first discovered by observing the mechanism of action of natural products that promote immunomodulatory ternary complex formation (Che et al., 2018), and recently, some compounds of synthetic origin have been reported to induce novel protein-protein interactions [reviewed in (Che et al., 2018)]. The “molecular glue” principle could be attributed not only to heteromeric protein-protein interactions but also to homomeric Sig1R interactions and could explain how allosteric modulators could increase the number of Sig1R in the agonist state conformation, thus increasing the activity of Sig1R agonists.
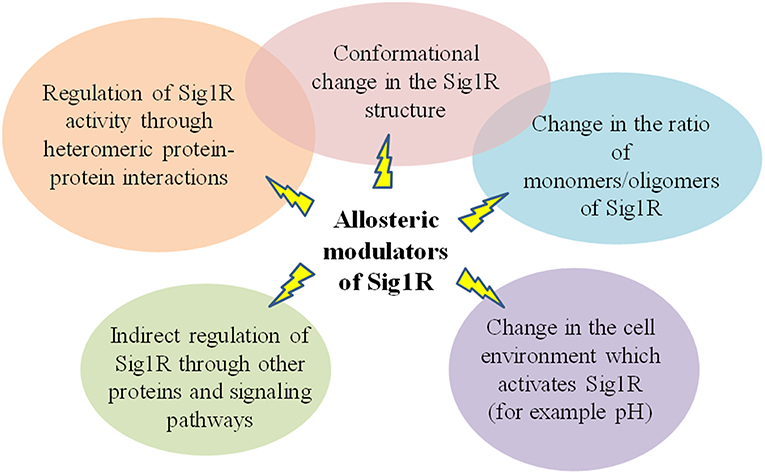
Figure 6. Summary of possible mechanisms of allosteric modulators of Sig1R.
It has been shown that compounds that bind to allosteric site of protein may activate the receptor in the absence of agonist (Schwartz and Holst, 2007). This condition, known as allosteric agonism, could explain the direct pharmacological effects of allosteric modulators of Sig1R observed both in vitro and in vivo (Zvejniece et al., 2014; Guo et al., 2015; Wang et al., 2016; Maurice et al., 2017, 2018). “Superagnonism” or ago-allosteric modulation also could be one of the possible descriptions of the modulatory activity of allosteric Sig1R ligands. An ago-allosteric modulator acts as both an agonist and an enhancer of agonist potency and provides “superagonism,” which would result in an efficacy >100% (Schwartz and Holst, 2007). One example of ago-allosteric modulatory activity could be the published in vitro effects of E1R (Zvejniece et al., 2014). It was shown that both selective Sig1R agonist PRE-084 and allosteric modulator E1R increased the BDK-induced [Ca2+]i increase, while the combination of both compounds resulted in an even more pronounced cellular response (Zvejniece et al., 2014). The similar “superagonism” could also be attributed to SOMCL-668. Both the allosteric Sig1R modulator SOMCL-668 and Sig1R agonist (+)-SKF-10,047 increased P(Ser9)-GSK-3β in hippocampal neuronal HT-22 cells, while the (+)-SKF-10,047-increased phosphorylation was potentiated by SOMCL-668 (Wang et al., 2016). While “superagonism” could be attributed only to some in vitro effects of E1R and SOMCL-668, it cannot be concluded that allosteric Sig1R modulators are superagonists. In addition, the direct pharmacological effects of allosteric Sig1R modulators could be explained by the presence of a possible endogenous agonists of Sig1R. N,N-dimethyltryptamine is considered as one of the most active endogenous Sig1R agonists identified thus far (Fontanilla et al., 2009; Su et al., 2009). Very recently, it was shown that also choline acts as an endogenous ligand (Brailoiu et al., 2019). However, to date no study has demonstrated the enhancement of endogenous agonist activity by known allosteric modulators of Sig1R. The acidity or alkalinity of a studied system is another issue that should be considered when evaluating possible mechanisms of allosteric Sig1R modulators (Figure 6) because it has been shown that pH by itself changes the activity of Sig1R ligands. Binding of ligands to Sig1R is pH dependent and is enhanced at higher pH (Largent et al., 1987). [3H](+)-Pentazocine binding to C6 glioma cell membranes was progressively increased at pH = 8.0 compared to buffer at pH = 7.0 and pH = 7.4 (Vilner et al., 1995). In addition, it was discovered that the pH of the medium markedly affected the activity of the Sig1R compounds in C6 glioma cells (Vilner et al., 1995). Increasing the pH from the range 7.2–7.4 to 8.3–8.5 caused a marked leftward shift in the dose curves for all active Sig1R compounds, while lowering the pH generally decreased the ability of sigma compounds to change cell morphology (Vilner et al., 1995). As it is known, all of the allosteric Sig1R modulators induce the same effect, which means an increase in the binding and activity of Sig1R ligands. Can allosteric modulators of Sig1R change the intracellular pH? Interestingly, it was previously demonstrated that a phenytoin-induced increase in the binding affinity of [3H]DM to brain homogenate is more marked at pH = 7.4 than at pH = 8.3 (Musacchio et al., 1988). Phenytoin is a weak acid with a pKa of approximately pH = 8.3. Therefore, at pH = 7.4 the concentration of unionized phenytoin is almost doubled, which may explain why phenytoin is much more potent at pH 7.4 (Musacchio et al., 1988). This finding demonstrates that pH plays a significant role in the process that determines the interaction between Sig1R ligands and receptors. Protonation or deprotonation of amino acids due to different pH could explain the change in the structure of Sig1R and therefore the change in the binding affinity of ligands to Sig1R. What is the role of allosteric modulators in these processes? In a number of structure-activity relationship studies, only agonists or antagonists have been used for evaluation of possible ligand-protein interactions. Additionally, no NMR data are available where allosteric modulators have been used to describe their molecular interaction with Sig1R. Most likely, three or more component systems (allosteric modulator-Sig1R-agonist and/or antagonist) must be applied to understand the interaction with Sig1R at the protein level. Detailed studies on allosteric Sig1R modulators at the protein level would be necessary to increase the global understanding of the interaction with the receptor and activity of Sig1R ligands.
Key Issues
Characterization of Binding Site
The binding site(s) for Sig1R allosteric modulators is(are) not identified. The binding dynamics in the Sig1R for allosteric compounds must be confirmed and characterized.
Screening System
No screening assay is available for allosteric modulators of Sig1R and must be developed and validated. Notably, modulation of the agonist-induced dissociation of Sig1R from BiP in vitro must be confirmed with all already identified modulators.
Stereoselectivity
The use of enantiomerically pure compounds is necessary to provide detailed evaluation of the in vitro and in vivo effects of allosteric Sig1R modulators. Comparison of all allosteric Sig1R modulators in the same experimental setup should be performed.
In vivo Proof of the Sig1R-Related Mechanism of Action
There is a lack of in vivo studies combining Sig1R agonists and allosteric Sig1R modulators to confirm the allosteric modulatory activity of a compound at Sig1R. In vitro concentrations and therapeutic doses in vivo should be comparable.
Conclusion
Allosteric pharmacology, that is, the design of drugs targeting sites topographically different from the orthosteric ligand binding site, is innovative approach to drug discovery. Allosteric modulation of Sig1R is an emerging and important target for designing novel drugs. A number of issues must be answered regarding the mechanisms of action and possible clinical applications of allosteric Sig1R modulators. More detailed classification of Sig1R ligands is inevitably necessary, and the development of new screening assay methods focused on ligand characterization is highly important to advance the understanding of the role of Sig1R and its allosteric modulators. Nevertheless, during the last 6 years, a large step forward in the understanding of allosteric modulation of Sig1R has been made. The first selective allosteric Sig1R modulators have been identified, which offer a basis for the discovery of novel and selective Sig1R compounds and increase knowledge of the impact of Sig1R regulation in living organisms, thereby providing novel treatment possibilities for various CNS-related diseases.
Author Contributions
EV and LZ wrote the manuscript in consultation with TM and MD. TM provided additional information on OZP002 and fenfluramine. All authors discussed the data and commented on the manuscript. MD supervised the manuscript.
Funding
This review article was supported by the European Regional Development Fund Project No. 1.1.1.1/16/A/292.
Conflict of Interest Statement
TM is co-inventor on the patent (PCT/EP2017/060129) describing OZP002 and received consultancy fees and research contracts from Zogenix Inc. The company had no role in the writing of this review.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Albertson, T. E., Peterson, S. L., and Stark, L. G. (1978). Effects of phenobarbital and SC-13504 on partially kindled hippocampal seizures in rats. Exp. Neurol. 61, 270–280. doi: 10.1016/0014-4886(78)90246-7
Alexander, S. P. H., Benson, H. E., Faccenda, E., Pawson, A. J., Sharman, J. L., McGrath, J. C., et al. (2013). The concise guide to pharmacology 2013/14: overview. Br. J. Pharmacol. 170, 1449–1458. doi: 10.1111/bph.12444
Alon, A., Schmidt, H. R., Wood, M. D., Sahn, J. J., Martin, S. F., and Kruse, A. C. (2017). Identification of the gene that codes for the σ2 receptor. Proc. Natl. Acad. Sci. U. S. A. 114, 7160–7165. doi: 10.1073/pnas.1705154114
Alonso, G., Phan, V., Guillemain, I., Saunier, M., Legrand, A., Anoal, M., et al. (2000). Immunocytochemical localization of the sigma1 receptor in the adult rat central nervous system. Neuroscience 97, 155–170. doi: 10.1016/S0306-4522(00)00014-2
Barone, P., Palma, V., de Bartolomeis, A., Cicarelli, G., and Campanella, G. (1992). Dopaminergic regulation of epileptic activity. Neurochem. Int. 20(Suppl.) 245S−249S. doi: 10.1016/0197-0186(92)90246-N
Bartz, F., Kern, L., Erz, D., Zhu, M., Gilbert, D., Meinhof, T., et al. (2009). Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 10, 63–75. doi: 10.1016/j.cmet.2009.05.009
Bourne, J. A., and Fosbraey, P. (2000). Novel method of monitoring electroencephalography at the site of microdialysis during chemically evoked seizures in a freely moving animal. J. Neurosci. Methods 99, 85–90. doi: 10.1016/S0165-0270(00)00217-X
Bourne, J. A., Fosbraey, P., and Halliday, J. (2001). SCH 23390 affords protection against soman-evoked seizures in the freely moving guinea-pig: a concomitant neurochemical, electrophysiological and behavioural study. Neuropharmacology 40, 279–288. doi: 10.1016/S0028-3908(00)00136-2
Brailoiu, E., Chakraborty, S., Brailoiu, G. C., Zhao, P., Barr, J. L., Ilies, M. A., et al. (2019). Choline is an intracellular messenger linking extracellular stimuli to IP(3)-evoked Ca2+ signals through sigma-1 receptors. Cell Rep. 26, 330–337.e4. doi: 10.1016/j.celrep.2018.12.051
Burke, K., Chandler, C. J., Starr, B. S., and Starr, M. S. (1990). Seizure promotion and protection by D-1 and D-2 dopaminergic drugs in the mouse. Pharmacol. Biochem. Behav. 36, 729–733. doi: 10.1016/0091-3057(90)90068-S
Chaki, S., Okuyama, S., Ogawa, S., Tanaka, M., Muramatsu, M., Nakazato, A., et al. (1996). Solubilization and characterization of binding sites for [3H]NE-100, a novel and potent sigma 1 ligand, from guinea pig brain. Life Sci. 59, 1331–1340. doi: 10.1016/0024-3205(96)00458-4
Che, Y., Gilbert, A. M., Shanmugasundaram, V., and Noe, M. C. (2018). Inducing protein-protein interactions with molecular glues. Bioorg. Med. Chem. Lett. 28, 2585–2592. doi: 10.1016/j.bmcl.2018.04.046
Chen, X.-Q., Zhang, J., Neumeyer, J. L., Jin, G.-Z., Hu, G.-Y., Zhang, A., et al. (2009). Arylbenzazepines are potent modulators for the delayed rectifier K+ channel: a potential mechanism for their neuroprotective effects. PLoS ONE 4:e5811. doi: 10.1371/journal.pone.0005811
Chu, H.-Y., Wu, Q., Zhou, S., Cao, X., Zhang, A., Jin, G.-Z., et al. (2011). SKF83959 suppresses excitatory synaptic transmission in rat hippocampus via a dopamine receptor-independent mechanism. J. Neurosci. Res. 89, 1259–1266. doi: 10.1002/jnr.22653
Chu, H.-Y., Yang, Z., Zhao, B., Jin, G.-Z., Hu, G.-Y., and Zhen, X. (2010). Activation of phosphatidylinositol-linked D1-like receptors increases spontaneous glutamate release in rat somatosensory cortical neurons in vitro. Brain Res. 1343, 20–27. doi: 10.1016/j.brainres.2010.04.043
Chu, U. B., Ramachandran, S., Hajipour, A. R., and Ruoho, A. E. (2013). Photoaffinity labeling of the sigma-1 receptor with N-[3-(4-nitrophenyl)propyl]-N-dodecylamine: evidence of receptor dimers. Biochemistry 52, 859–868. doi: 10.1021/bi301517u
Chu, U. B., and Ruoho, A. E. (2016). Biochemical pharmacology of the sigma-1 receptor. Mol. Pharmacol. 89, 142–153. doi: 10.1124/mol.115.101170
Cobos, E., Entrena, J., Nieto, F., Cendan, C., and Pozo, E. (2008). Pharmacology and therapeutic potential of sigma1 receptor ligands. Curr. Neuropharmacol. 6, 344–366. doi: 10.2174/157015908787386113
Cobos, E. J., Baeyens, J. M., and Del Pozo, E. (2005). Phenytoin differentially modulates the affinity of agonist and antagonist ligands for σ1 receptors of guinea pig brain. Synapse 55, 192–195. doi: 10.1002/syn.20103
Cobos, E. J., Lucena, G., Baeyens, J. M., and Del Pozo, E. (2006). Differences in the allosteric modulation by phenytoin of the binding properties of the sigma1 ligands [3H](+)-pentazocine and [3H]NE-100. Synapse 59, 152–161. doi: 10.1002/syn.20230
Craig, C. R. (1967). Anticonvulsant properties of some benzhydryl piperazine and benzhydryl piperidine compounds. Arch. Int. Pharmacodyn. Ther. 165, 328–336.
Craviso, G. L., and Musacchio, J. M. (1983). High-affinity dextromethorphan binding sites in guinea pig brain. II. Competition experiments. Mol. Pharmacol. 23, 629–640.
DeHaven-Hudkins, D. L., Ford-Rice, F. Y., Allen, J. T., and Hudkins, R. L. (1993). Allosteric modulation of ligand binding to [3H](+)pentazocine-defined sigma recognition sites by phenytoin. Life Sci. 53, 41–48. doi: 10.1016/0024-3205(93)90609-7
Desai, R. I., Neumeyer, J. L., Paronis, C. A., Nguyen, P., and Bergman, J. (2007). Behavioral effects of the R-(+)- and S-(-)-enantiomers of the dopamine D(1)-like partial receptor agonist SKF 83959 in monkeys. Eur. J. Pharmacol. 558, 98–106. doi: 10.1016/j.ejphar.2006.11.042
Deveney, A. M., and Waddington, J. L. (1995). Pharmacological characterization of behavioural responses to SKF 83959 in relation to “D1-like” dopamine receptors not linked to adenylyl cyclase. Br. J. Pharmacol. 116, 2120–2126.
Downes, R. P., and Waddington, J. L. (1993). Grooming and vacuous chewing induced by SKF 83959, an agonist of dopamine “D1-like” receptors that inhibits dopamine-sensitive adenylyl cyclase. Eur. J. Pharmacol. 234, 135–136.
Edmonds, H. L., Bellin, S. I., Chen, F. C., and Hegreberg, G. A. (1978). Anticonvulsant properties of ropizine in epileptic and nonepileptic beagle dogs. Epilepsia 19, 139–146. doi: 10.1111/j.1528-1157.1978.tb05024.x
Edmonds, H. L., Jiang, Y. D., Zhang, P. Y., and Shank, R. P. (1996). Anticonvulsant activity of topiramate and phenytoin in a rat model of ischemia-induced epilepsy. Life Sci. 59, PL127–PL131. doi: 10.1016/0024-3205(96)00379-7
Edmonds, H. L., and Stark, L. G. (1974). Penicillin-induced epileptogenic foci—II: the anticonvulsant and neuropharmacological effects of SC-13504 in the cat. Neuropharmacology 13, 269–277. doi: 10.1016/0028-3908(74)90077-X
Edmonds, H. L., Stark, L. G., Stark, D. M., McCormack, C. R., Sylvester, D. M., and Bellin, S. I. (1979). The anticonvulsant activity of ropizine in the rat. J. Pharmacol. Exp. Ther. 208, 236–242.
Fang, X., Guo, L., Jia, J., Jin, G., Zhao, B., Zheng, Y., et al. (2013). SKF83959 is a novel triple reuptake inhibitor that elicits anti-depressant activity. Acta Pharmacol. Sin. 34, 1149–1155. doi: 10.1038/aps.2013.66
Festoff, B. W., and Appel, S. H. (1968). Effect of diphenylhydantoin on synaptosome sodium-potassium-ATPase. J. Clin. Invest. 47, 2752–2758. doi: 10.1172/JCI105956
Fontanilla, D., Johannessen, M., Hajipour, A. R., Cozzi, N. V., Jackson, M. B., and Ruoho, A. E. (2009). The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323, 934–937. doi: 10.1126/science.1166127
Fuller, R. W., Snoddy, H. D., and Robertson, D. W. (1988). Mechanisms of effects of d-fenfluramine on brain serotonin metabolism in rats: uptake inhibition versus release. Pharmacol. Biochem. Behav. 30, 715–721. doi: 10.1016/0091-3057(88)90089-5
Gentsch, K., Heinemann, U., Schmitz, B., and Behr, J. (2000). Fenfluramine blocks low-Mg2+-induced epileptiform activity in rat entorhinal cortex. Epilepsia 41, 925–928. doi: 10.1111/j.1528-1157.2000.tb00273.x
Gilbert, J. C., and Wyllie, M. G. (1976). Effects of anticonvulsant and convulsant drugs on the ATPase activities of synaptosomes and their components. Br. J. Pharmacol. 56, 49–57. doi: 10.1111/j.1476-5381.1976.tb06958.x
Gromek, K. A., Suchy, F. P., Meddaugh, H. R., Wrobel, R. L., LaPointe, L. M., Chu, U. B., et al. (2014). The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 289, 20333–20344. doi: 10.1074/jbc.M113.537993
Guo, L., Chen, Y., Zhao, R., Wang, G., Friedman, E., Zhang, A., et al. (2015). Allosteric modulation of sigma-1 receptors elicits anti-seizure activities. Br. J. Pharmacol. 172, 4052–4065. doi: 10.1111/bph.13195
Guo, L., Zhao, J., Jin, G., Zhao, B., Wang, G., Zhang, A., et al. (2013). SKF83959 is a potent allosteric modulator of sigma-1 receptor. Mol. Pharmacol. 83, 577–586. doi: 10.1124/mol.112.083840
Haller, J. L., Panyutin, I., Chaudhry, A., Zeng, C., Mach, R. H., and Frank, J. A. (2012). Sigma-2 receptor as potential indicator of stem cell differentiation. Mol. Imaging Biol. 14, 325–335. doi: 10.1007/s11307-011-0493-3
Hayashi, T., and Su, T.-P. (2007). Sigma-1 receptor chaperones at the ER- mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Hellewell, S. B., and Bowen, W. D. (1990). A sigma-like binding site in rat pheochromocytoma (PC12) cells: decreased affinity for (+)-benzomorphans and lower molecular weight suggest a different sigma receptor form from that of guinea pig brain. Brain Res. 527, 244–253. doi: 10.1016/0006-8993(90)91143-5
Hellewell, S. B., Bruce, A., Feinstein, G., Orringer, J., Williams, W., and Bowen, W. D. (1994). Rat liver and kidney contain high densities of sigma 1 and sigma 2 receptors: characterization by ligand binding and photoaffinity labeling. Eur. J. Pharmacol. 268, 9–18. doi: 10.1016/0922-4106(94)90115-5
Hicks, P. E., Schoemaker, H., and Langer, S. Z. (1984). 5HT-receptor antagonist properties of SCH23390 in vascular smooth muscle and brain. Eur. J. Pharmacol. 105, 339–342. doi: 10.1016/0014-2999(84)90628-9
Hyttel, J. (1983). SCH 23390 - the first selective dopamine D-1 antagonist. Eur. J. Pharmacol. 91, 153–154. doi: 10.1016/0014-2999(83)90381-3
Jbilo, O., Vidal, H., Paul, R., De Nys, N., Bensaid, M., Silve, S., et al. (1997). Purification and characterization of the human SR 31747A-binding protein. A nuclear membrane protein related to yeast sterol isomerase. J. Biol. Chem. 272, 27107–27115. doi: 10.1074/jbc.272.43.27107
Jiang, B., Wang, F., Yang, S., Fang, P., Deng, Z.-F., Xiao, J.-L., et al. (2015). SKF83959 produces antidepressant effects in a chronic social defeat stress model of depression through BDNF-TrkB pathway. Int. J. Neuropsychopharmacol. 18:pyu096. doi: 10.1093/ijnp/pyu096
Johannessen, M., Ramachandran, S., Riemer, L., Ramos-Serrano, A., Ruoho, A. E., and Jackson, M. B. (2009). Voltage-gated sodium channel modulation by σ-receptors in cardiac myocytes and heterologous systems. Am. J. Physiol. Physiol. 296, C1049–C1057. doi: 10.1152/ajpcell.00431.2008
Jones, G. L., Amato, R. J., Wimbish, G. H., and Peyton, G. A. (1981). Comparison of anticonvulsant potencies of cyheptamide, carbamazepine, and phenytoin. J. Pharm. Sci. 70, 618–620. doi: 10.1002/jps.2600700611
Joy, R. M., and Edmonds, H. L. (1974). The antagonism of hippocampal and cortical afterdischarge by SC-13504, a new anticonvulsant. Neuropharmacology 13, 145–157. doi: 10.1016/0028-3908(74)90102-6
Karbon, E. W., Naper, K., and Pontecorvo, M. J. (1991). [3H]DTG and [3H](+)-3-PPP label pharmacologically distinct sigma binding sites in guinea pig brain membranes. Eur. J. Pharmacol. 193, 21–27. doi: 10.1016/0014-2999(91)90195-V
Kebabian, J. W. (1988). “The D-1 dopamine receptor,” in Central D1 Dopamine Receptors, ed M. J. Goldstein (New York, NY: Springer), 19–31.
Klein, M., and Musacchio, J. M. (1990). Computer-assisted analysis of dextromethorphan and (+)-3-(-3-hydroxyphenyl)-N-(1-propyl)piperidine binding sites in rat brain. Allosteric effects of ropizine. Life Sci. 47, 1625–1634. doi: 10.1016/0024-3205(90)90367-Z
Klein, M., and Musacchio, J. M. (1992). High-affinity dextromethorphan and (+)-3-(-3-hydroxyphenyl)-N-(1-propyl)piperidine binding sites in rat brain. Allosteric effects of ropizine. J. Pharmacol. Exp. Ther. 260, 990–999.
Largent, B. L., Wikström, H., Gundlach, A. L., and Snyder, S. H. (1987). Structural determinants of sigma receptor affinity. Mol. Pharmacol. 32, 772–784.
Lejeune, S., Dourmap, N., Martres, M.-P., Giros, B., Daugé, V., and Naudon, L. (2013). The dopamine D1 receptor agonist SKF 38393 improves temporal order memory performance in maternally deprived rats. Neurobiol. Learn. Mem. 106, 268–273. doi: 10.1016/j.nlm.2013.10.005
Leng, Z. G., Lin, S. J., Wu, Z. R., Guo, Y. H., Cai, L., Shang, H. B., et al. (2017). Activation of DRD5 (dopamine receptor D5) inhibits tumor growth by autophagic cell death. Autophagy 13, 1404–1419. doi: 10.1080/15548627.2017.1328347
Lever, J. R., Litton, T. P., and Fergason-Cantrell, E. A. (2015). Characterization of pulmonary sigma receptors by radioligand binding. Eur. J. Pharmacol. 762, 118–126. doi: 10.1016/j.ejphar.2015.05.026
Levkovitch, Y., Abramovitch, Y., and Nizan, I. (1993). [Dilantin toxicity as a possible cause of major depression]. Harefuah 124, 762–4, 795.
Ma, J., Long, L.-H., Hu, Z.-L., Zhang, H., Han, J., Ni, L., et al. (2015). Activation of D1-like receptor-dependent phosphatidylinositol signal pathway by SKF83959 inhibits voltage-gated sodium channels in cultured striatal neurons. Brain Res. 1615, 71–79. doi: 10.1016/j.brainres.2015.04.030
Martin, P., Gammaitoni, A., Farfel, G., Boyd, B., and Galer, B. (2017). 663. An examination of the mechanism of action of fenfluramine in dravet syndrome: a look beyond serotonin. Biol. Psychiatry 81, S268–S269. doi: 10.1016/j.biopsych.2017.02.1072
Martin, W. R., Eades, C. G., Thompson, J. A., Huppler, R. E., and Gilbert, P. E. (1976). The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 197, 517–532.
Maurice, T. (2016). Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav. Brain Res. 296, 270–278. doi: 10.1016/j.bbr.2015.09.020
Maurice, T., Farfel, G., Boyd, B., Gammaitoni, A., Galer, B., and Martin, P. (2018). Fenfluramine is a sigma-1 receptor positive modulator in mice. Soc. Neurosci. Abstr. 692.
Maurice, T., Hiramatsu, M., Itoh, J., Kameyama, T., Hasegawa, T., and Nabeshima, T. (1994a). Behavioral evidence for a modulating role of sigma ligands in memory processes. I. Attenuation of dizocilpine (MK-801)-induced amnesia. Brain Res. 647, 44–56. doi: 10.1016/0006-8993(94)91397-8
Maurice, T., Su, T. P., Parish, D. W., Nabeshima, T., and Privat, A. (1994b). PRE-084, a sigma selective PCP derivative, attenuates MK-801-induced impairment of learning in mice. Pharmacol. Biochem. Behav. 49, 859–869. doi: 10.1016/0091-3057(94)90235-6
Maurice, T., Su, T. P., and Privat, A. (1998). Sigma1 receptor agonists and neurosteroids attenuate B25-35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience 83, 413–428. doi: 10.1016/S0306-4522(97)00405-3
Maurice, T., Volle, J. N., Strehaiano, M., Pereira, C., Laborde, C., Virieux, D., et al. (2017). A novel phoshinolactone compound, OZP002, positive modulator of σ1 receptors, is neuroprotective in non-transgenic and transgenic mouse models of Alzheimer's disease. Soc. Neurosc. Abstr. 127.
Mavlyutov, T. A., Epstein, M., and Guo, L.-W. (2015). Subcellular localization of the sigma-1 receptor in retinal neurons - an electron microscopy study. Sci. Rep. 5:10689. doi: 10.1038/srep10689
McCann, D. J., and Su, T. P. (1991). Solubilization and characterization of haloperidol-sensitive (+)-[3H]SKF-10,047 binding sites (sigma sites) from rat liver membranes. J. Pharmacol. Exp. Ther. 257, 547–554.
Meldrum, B. S., Smyth, M. R., Franklin-Smyth, W., and Clifford, J. M. (1976). The relationship between the anticonvulsant properties of SC-13504 and its plasma levels, measured by polarography, in baboons with photosensitive epilepsy. Psychopharmacology 51, 59–64. doi: 10.1007/BF00426322
Merritt, H. H., and Putnam, T. J. (1984). Landmark article Sept 17, 1938: Sodium diphenyl hydantoinate in the treatment of convulsive disorders. JAMA 251, 1062–1067.
Mishra, A., and Goel, R. K. (2015). Comparative behavioral and neurochemical analysis of phenytoin and valproate treatment on epilepsy induced learning and memory deficit: search for add on therapy. Metab. Brain Dis. 30, 951–958. doi: 10.1007/s11011-015-9650-8
Mishra, A. K., Mavlyutov, T., Singh, D. R., Biener, G., Yang, J., Oliver, J. A., et al. (2015). The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 466, 263–271. doi: 10.1042/BJ20141321
Molloy, A. G., and Waddington, J. L. (1984). Dopaminergic behaviour stereospecific promoted by the D1 agonist R-SKF 38393 and selectively blocked by the D1 antagonist SCH 23390. Psychopharmacology 82, 409–410. doi: 10.1007/BF00427697
Moreno, E., Moreno-Delgado, D., Navarro, G., Hoffmann, H. M., Fuentes, S., Rosell-Vilar, S., et al. (2014). Cocaine disrupts histamine H3 receptor modulation of dopamine D1 receptor signaling: σ1-D1-H3 receptor complexes as key targets for reducing cocaine's effects. J. Neurosci. 34, 3545–3558. doi: 10.1523/JNEUROSCI.4147-13.2014
Mori, T., Hayashi, T., Hayashi, E., and Su, T.-P. (2013). Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 8:e76941. doi: 10.1371/journal.pone.0076941
Musacchio, J. M., Klein, M., and Paturzo, J. J. (1989). Effects of dextromethorphan site ligands and allosteric modifiers on the binding of (+)-[3H]3-(-3-hydroxyphenyl)-N-(1-propyl)piperidine. Mol. Pharmacol. 35, 1–5.
Musacchio, J. M., Klein, M., and Santiago, L. J. (1988). High affinity dextromethorphan binding sites in guinea pig brain: further characterization and allosteric interactions. J. Pharmacol. Exp. Ther. 247, 424–431.
Navarro, G., Moreno, E., Bonaventura, J., Brugarolas, M., Farré, D., Aguinaga, D., et al. (2013). Cocaine inhibits dopamine D2 receptor signaling via sigma-1-D2 receptor heteromers. PLoS ONE 8:e61245. doi: 10.1371/journal.pone.0061245
Nobile, M., and Lagostena, L. (1998). A discriminant block among K+ channel types by phenytoin in neuroblastoma cells. Br. J. Pharmacol. 124, 1698–1702. doi: 10.1038/sj.bjp.0702035
Novack, G. D., Stark, L. G., and Peterson, S. L. (1979). Anticonvulsant effects of benzhydryl piperazines on maximal electroshock seizures in rats. J. Pharmacol. Exp. Ther. 208, 480–484.
Ogren, S. O., and Pakh, B. (1993). Effects of dopamine D1 and D2 receptor agonists and antagonists on seizures induced by chemoconvulsants in mice. Pharmacol. Toxicol. 72, 213–220. doi: 10.1111/j.1600-0773.1993.tb01639.x
Ongini, E., Caporali, M. G., Massotti, M., and Sagratella, S. (1987). SCH 23390 and its S-enantiomer stereoselectively prevent EEG and behavioral activation induced by dopamine agonists in the rabbit. Pharmacol. Biochem. Behav. 26, 715–718. doi: 10.1016/0091-3057(87)90602-2
Pabba, M. (2013). The essential roles of protein–protein interaction in sigma-1 receptor functions. Front. Cell. Neurosci. 7:50. doi: 10.3389/fncel.2013.00050
Paul, R., Lavastre, S., Floutard, D., Floutard, R., Canat, X., Casellas, P., et al. (1994). Allosteric modulation of peripheral sigma binding sites by a new selective ligand: SR 31747. J. Neuroimmunol. 52, 183–192. doi: 10.1016/0165-5728(94)90112-0
Polster, T. (2018). Individualized treatment approaches: fenfluramine, a novel antiepileptic medication for the treatment of seizures in Dravet syndrome. Epilepsy Behav. doi: 10.1016/j.yebeh.2018.08.021 [Epub ahead of print].
Quirion, R., Bowen, W. D., Itzhak, Y., Junien, J. L., Musacchio, J. M., Rothman, R. B., et al. (1992). A proposal for the classification of sigma binding sites. Trends Pharmacol. Sci. 13, 85–86. doi: 10.1016/0165-6147(92)90030-A
Reeta, K. H., Mehla, J., and Gupta, Y. K. (2009). Curcumin is protective against phenytoin-induced cognitive impairment and oxidative stress in rats. Brain Res. 1301, 52–60. doi: 10.1016/j.brainres.2009.09.027
Ritz, M. C., and George, F. R. (1997). Cocaine toxicity: concurrent influence of dopaminergic, muscarinic and sigma receptors in mediating cocaine-induced lethality. Psychopharmacology 129, 311–321. doi: 10.1007/s002130050198
Rodríguez-Muñoz, M., Sánchez-Blázquez, P., and Garzón, J. (2018). Fenfluramine diminishes NMDA receptor-mediated seizures via its mixed activity at serotonin 5HT2A and type 1 sigma receptors. Oncotarget 9, 23373–23389. doi: 10.18632/oncotarget.25169
Rodríguez-Muñoz, M., Sánchez-Blázquez, P., Herrero-Labrador, R., Martínez-Murillo, R., Merlos, M., Vela, J. M., et al. (2015). The σ1 receptor engages the redox-regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control. Antioxid. Redox Signal. 22, 799–818. doi: 10.1089/ars.2014.5993
Rush, A. M., and Elliott, J. R. (1997). Phenytoin and carbamazepine: differential inhibition of sodium currents in small cells from adult rat dorsal root ganglia. Neurosci. Lett. 226, 95–98. doi: 10.1016/S0304-3940(97)00258-9
Santulli, L., Coppola, A., Balestrini, S., and Striano, S. (2016). The challenges of treating epilepsy with 25 antiepileptic drugs. Pharmacol. Res. 107, 211–219. doi: 10.1016/j.phrs.2016.03.016
Schmidt, H. R., Zheng, S., Gurpinar, E., Koehl, A., Manglik, A., and Kruse, A. C. (2016). Crystal structure of the human σ1 receptor. Nature 532, 527–530. doi: 10.1038/nature17391
Schuster, D. I., Arnold, F. J., and Murphy, R. B. (1995). Purification, pharmacological characterization and photoaffinity labeling of sigma receptors from rat and bovine brain. Brain Res. 670, 14–28. doi: 10.1016/0006-8993(94)01123-Y
Schwartz, T. W., and Holst, B. (2007). Allosteric enhancers, allosteric agonists and ago-allosteric modulators: where do they bind and how do they act? Trends Pharmacol. Sci. 28, 366–373. doi: 10.1016/j.tips.2007.06.008
Sheng, G., Zhang, J., Gao, S., Gu, Y., Jiang, B., and Gao, Q. (2018). SKF83959 has protective effects in the scopolamine model of dementia. Biol. Pharm. Bull. 41, 427–434. doi: 10.1248/bpb.b17-00877
Sourbron, J., Schneider, H., Kecskés, A., Liu, Y., Buening, E. M., Lagae, L., et al. (2016). serotonergic modulation as effective treatment for dravet syndrome in a zebrafish mutant model. ACS Chem. Neurosci. 7, 588–598. doi: 10.1021/acschemneuro.5b00342
Sourbron, J., Smolders, I., de Witte, P., and Lagae, L. (2017). Pharmacological analysis of the anti-epileptic mechanisms of fenfluramine in scn1a mutant zebrafish. Front. Pharmacol. 8:191. doi: 10.3389/fphar.2017.00191
Su, T.-P., Hayashi, T., and Vaupel, D. B. (2009). When the endogenous hallucinogenic trace amine N,N-dimethyltryptamine meets the sigma-1 receptor. Sci. Signal. 2:pe12. doi: 10.1126/scisignal.261pe12
Su, T.-P., Su, T.-C., Nakamura, Y., and Tsai, S.-Y. (2016). The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol. Sci. 37, 262–278. doi: 10.1016/j.tips.2016.01.003
Su, T. P. (1982). Evidence for sigma opioid receptor: binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea-pig brain. J. Pharmacol. Exp. Ther. 223, 284–290.
Su, T. P., and Junien, J. L. (1994). “Sigma receptors in the central nervous system and the periphery,” in Sigma Receptors, ed Y. Itzhak (London: Academic Press), 21–44.
Todorovic, S. M., and Lingle, C. J. (1998). Pharmacological properties of T-type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. J. Neurophysiol. 79, 240–252. doi: 10.1152/jn.1998.79.1.240
Tunnicliff, G. (1996). Basis of the antiseizure action of phenytoin. Gen. Pharmacol. 27, 1091–1097. doi: 10.1016/S0306-3623(96)00062-6
Vavers, E. (2017). Discovery of E1R: A Novel Positive Allosteric Modulator of Sigma-1 Receptor. Ph.D. thesis. Riga Stradins University, Riga.
Vavers, E., Svalbe, B., Lauberte, L., Stonans, I., Misane, I., Dambrova, M., et al. (2017). The activity of selective sigma-1 receptor ligands in seizure models in vivo. Behav. Brain Res. 328, 13–18. doi: 10.1016/j.bbr.2017.04.008
Vavers, E., Zvejniece, L., Veinberg, G., Svalbe, B., Domracheva, I., Vilskersts, R., et al. (2015). Novel positive allosteric modulators of sigma-1 receptor. Springerplus 4:P51. doi: 10.1186/2193-1801-4-S1-P51
Veinberg, G., Vavers, E., Orlova, N., Kuznecovs, J., Domracheva, I., Vorona, M., et al. (2015). Stereochemistry of phenylpiracetam and its methyl derivative: improvement of the pharmacological profile. Chem. Heterocycl. Compd. 51, 601–606. doi: 10.1007/s10593-015-1747-9
Veinberg, G., Vorona, M., Zvejniece, L., Vilskersts, R., Vavers, E., Liepinsh, E., et al. (2013). Synthesis and biological evaluation of 2-(5-methyl-4-phenyl-2- oxopyrrolidin-1-yl)-acetamide stereoisomers as novel positive allosteric modulators of sigma-1 receptor. Bioorganic Med. Chem. 21, 2764–2771. doi: 10.1016/j.bmc.2013.03.016
Vilner, B. J., de Costa, B. R., and Bowen, W. D. (1995). Cytotoxic effects of sigma ligands: sigma receptor-mediated alterations in cellular morphology and viability. J. Neurosci. 15, 117–134. doi: 10.1523/JNEUROSCI.15-01-00117.1995
Volle, J. N., Filippini, D., Krawczy, B., Kaloyanov, N., Van der Lee, A., Maurice, T., et al. (2010). Drug discovery: phosphinolactone, in vivo bioisostere of the lactol group. Org. Biomol. Chem. 8, 1438–1444. doi: 10.1039/b919345f
Wang, Y., Guo, L., Jiang, H.-F., Zheng, L.-T., Zhang, A., and Zhen, X.-C. (2016). Allosteric modulation of sigma-1 receptors elicits rapid antidepressant activity. CNS Neurosci. Ther. 22, 368–377. doi: 10.1111/cns.12502
Wu, Z., Li, L., Zheng, L.-T., Xu, Z., Guo, L., and Zhen, X. (2015). Allosteric modulation of sigma-1 receptors by SKF83959 inhibits microglia-mediated inflammation. J. Neurochem. 134, 904–914. doi: 10.1111/jnc.13182
Yaari, Y., Selzer, M. E., and Pincus, J. H. (1986). Phenytoin: mechanisms of its anticonvulsant action. Ann. Neurol. 20, 171–184. doi: 10.1002/ana.410200202
Zamanillo, D., Portillo-Salido, E., Vela, J. M., and Romero, L. (2012). “Sigma-1 receptor chaperone: pharmacology and therapeutic perspectives,” in Therapeutic Targets: Modulation, Inhibition, and Activation, eds L. M. Botana and M. Loza (Hoboken, NJ: Wiley), 225–278.
Zhang, H., Ma, L., Wang, F., Chen, J., and Zhen, X. (2007). Chronic SKF83959 induced less severe dyskinesia and attenuated L-DOPA-induced dyskinesia in 6-OHDA-lesioned rat model of Parkinson's disease. Neuropharmacology 53, 125–133. doi: 10.1016/j.neuropharm.2007.04.004
Zhang, J., Huang, J., Song, Z., Guo, L., Cai, W., Wang, Y., et al. (2014). Structural manipulation on the catecholic fragment of dopamine D(1) receptor agonist 1-phenyl-N-methyl-benzazepines. Eur. J. Med. Chem. 85, 16–26. doi: 10.1016/j.ejmech.2014.07.059
Zhao, L., Au, J. L.-S., and Wientjes, M. G. (2010). Comparison of methods for evaluating drug-drug interaction. Front. Biosci. 2, 241–249.
Zhen, X., Goswami, S., and Friedman, E. (2005). The role of the phosphatidyinositol-linked D dopamine receptor in the pharmacology of SKF83959. Pharmacol. Biochem. Behav. 80, 597–601. doi: 10.1016/j.pbb.2005.01.016
Keywords: sigma-1 receptor (Sig1R), allosteric modulator, phenytoin, E1R, SOMCL-668, SKF83959, OZP002, fenfluramine
Citation: Vavers E, Zvejniece L, Maurice T and Dambrova M (2019) Allosteric Modulators of Sigma-1 Receptor: A Review. Front. Pharmacol. 10:223. doi: 10.3389/fphar.2019.00223
Received: 14 December 2018; Accepted: 22 February 2019;
Published: 19 March 2019.
Edited by:
Salvatore Salomone, Università degli Studi di Catania, ItalyReviewed by:
Nadezhda A. German, Texas Tech University Health Sciences Center, United StatesPilar Sánchez-Blázquez, Spanish National Research Council (CSIC), Spain
Arnold Eino Ruoho, University of Wisconsin School of Medicine and Public Health, United States
Copyright © 2019 Vavers, Zvejniece, Maurice and Dambrova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edijs Vavers, ZWRpanMudmF2ZXJzQGZhcm0ub3NpLmx2