 Yashika Bansal
Yashika Bansal Raghunath Singh
Raghunath Singh Ishwar Parhar
Ishwar Parhar Anurag Kuhad
Anurag Kuhad Tomoko Soga
Tomoko Soga- 1Pharmacology Research Lab, University Institute of Pharmaceutical Sciences UGC-Centre of Advanced Study, Panjab University, Chandigarh, India
- 2Brain Research Institute Monash Sunway, Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, Bandar Sunway, Malaysia
Depression is an incapacitating neuropsychiatric disorder. The serotonergic system in the brain plays an important role in the pathophysiology of depression. However, due to delayed and/or poor performance of selective serotonin reuptake inhibitors in treating depressive symptoms, the role of the serotonergic system in depression has been recently questioned further. Evidence from recent studies suggests that increased inflammation and oxidative stress may play significant roles in the pathophysiology of depression. The consequences of these factors can lead to the neuroprogression of depression, involving neurodegeneration, astrocytic apoptosis, reduced neurogenesis, reduced plasticity (neuronal and synaptic), and enhanced immunoreactivity. Specifically, increased proinflammatory cytokine levels have been shown to activate the kynurenine pathway, which causes increased production of quinolinic acid (QA, an N-Methyl-D-aspartate agonist) and decreases the synthesis of serotonin. QA exerts many deleterious effects on the brain via mechanisms including N-methyl-D-aspartate excitotoxicity, increased oxidative stress, astrocyte degeneration, and neuronal apoptosis. QA may also act directly as a pro-oxidant. Additionally, the nuclear translocation of antioxidant defense factors, such as nuclear factor (erythroid-derived 2)-like 2 (Nrf2), is downregulated in depression. Hence, in the present review, we discuss the role of QA in increasing oxidative stress in depression by modulating the nuclear translocation of nuclear factor (erythroid-derived 2)-like 2 and thus affecting the synthesis of antioxidant enzymes.
Introduction
Depression is a heterogeneous mood disorder characterized by mood alterations, anhedonia, low self-esteem, social withdrawal, feelings of guilt, idiopathic pain, loss of interest in enjoyable activities, and suicidal tendencies. According to the World Health Organization, more than 300 million people currently have depression and approximately 800,000 individuals with depression commit suicide every year. Suicide due to depression is the second leading cause of death among individuals 16–25 years of age (WHO, 2018).
Various antidepressants are available for the clinical treatment of depression, including typical antidepressants, atypical antidepressants, monoamine oxidase inhibitors, and selective serotonin reuptake inhibitors. These drugs’ mechanism of action aligns well with the monoamine theory of depression. This theory states that decreased levels of serotonin in the synaptic cleft in various brain regions correspond to increased activity of the monoamine degrading enzyme and decreased synthesis of serotonin by serotonergic neurons, ultimately leading to depression (Cowen and Browning, 2015).
Although advancements in medical research have contributed to increase depression treatment options, one-third of patients do not respond to conventional drug therapies. Given this, there is a pressing need to elucidate the pathophysiology of depression so that new pathways can be explored for the investigation of novel therapeutics. Emerging evidence from various clinical and preclinical studies has revealed that oxidative stress and increased activity of immune factor cascades play significant roles in the pathophysiology of depression (Liu et al., 2015; Kohler et al., 2016). Maes et al. were the first to report that abnormal activation of the immune system and hypothalamic-pituitary-adrenal axis hyperactivity occur in depression. Additionally, decreased serotonin has been implicated in depression as a consequence of cell-mediated immune activation leading to decrease availability of plasma L-tryptophan (a precursor for serotonin synthesis). Tryptophan is a common substrate for the kynurenine pathway (KP) and serotonin synthesis pathway (methoxyindole pathway). Activation of indoleamine 2,3-dioxygenase (IDO), a rate limiting enzyme of the KP in the brain and periphery, results in decreased tryptophan availability (Maes et al., 1991, 1993; Maes, 1993). Interferon (IFN)-γ is the major inducer of IDO while tumor necrosis factor-α, interleukin (IL)-6, IFN-β, and IFN-α also activate IDO to some extent. Many studies have found a positive link between neuroinflammation and IDO expression in neurodegenerative diseases and depression (Sapko et al., 2006; Kim et al., 2012; Dobos et al., 2015).
Tryptophan metabolism via the KP results in the production of a neurotoxin, quinolinic acid (QA), and a neuroprotective compound, kynurenic acid (KA). KA binds to the glutamate recognition site of the N-methyl-D-aspartate (NMDA) receptor and antagonizes it, while QA binds to the glycine site of the NMDA receptor with agonistic properties. Thus, KA prevents excitotoxicity induced by NMDA overstimulation. Kynurenine is the first metabolite of the KP and is catalyzed by IDO. IDO enhances the activity of the kynureninase enzyme, which catalyzes the breakdown of kynurenine to anthranilic acid. However, kynureninase also prevents the activity of kynurenine aminotransferase (KAT), which catalyzes the formation of neuroprotective KA from kynurenine. Eventually, the breakdown of kynurenine is linked to neurotoxic QA production and reduced KA production (Réus et al., 2015).
Imbalance in the levels of QA and KA has been reported in patients with major depressive disorder (MDD). Furthermore, increased levels of QA exert neurotoxic effects in the brain of patients with depression (explained in the next section) (Myint et al., 2012). Studies conducted by three different groups have shown that QA acts as a pro-oxidant and is associated with oxidative stress (Behan et al., 1999; Santamaría et al., 1999; Rossato et al., 2002). Although QA is an NMDA receptor agonist, QA-induced oxidative stress occurs in both NMDA-dependent and independent fashion and requires further exploration (Orlando et al., 2001; Behan and Stone, 2002; Guillemin, 2012).
A key factor crucial to combat increased oxidative stress is nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Nrf2 is a basic leucine zipper protein factor that acts as a master regulator of oxidative stress, maintains redox homeostasis, and provides protection against oxidative stress by transcribing various antioxidant enzymes. More specifically, studies have also shown downregulation of Nrf2 in depression and that Nrf2 activators, such as sulforaphane and its precursor glucoraphanin, exert antidepressive-like effects in depression (Martín-de-Saavedra et al., 2013; Yao et al., 2016).
In the present review, we discuss the role of QA, which might act as a pro-oxidant by impeding Nrf2 activity, an antioxidant protein implicated in clinical depression. Research on these two factors and their role in depression has led to emerging insight into the neuroprogression theory of depression and potential novel pharmacotherapeutics for its treatment.
The KP and Glial Cells in Depression
The KP is a metabolic pathway of tryptophan both in the periphery and central nervous system (CNS). In the periphery, 90% of tryptophan is found in the unbound form while 10% is bound to albumin. Only the free form of tryptophan can be transported through the blood-brain barrier (Jones et al., 2013). Tryptophan is metabolized to kynurenine by tryptophan 2,3-dioxygenase or IDO, a rate limiting enzyme of the KP. Tryptophan 2,3-dioxygenase catalyzes tryptophan catabolism in the liver and contributes to the peripheral levels of tryptophan, whereas IDO catalyzes tryptophan metabolism extrahepatically. In inflammatory conditions, IDO is induced by proinflammatory cytokines and shifts tryptophan metabolism to kynurenine. Kynurenine is further metabolized via three branches to KA, anthranilic acid, and QA by the enzymatic activity of KAT, kynureninase, and kynurenine monooxygenase, respectively. As shown in Figure 1, kynurenine is metabolized to KA through branch 1, to anthranilic acid through branch 2, and to 3-hydroxy kynurenine through branch 3. 3-Hydroxykynurenine is further metabolized to 3-hydroxyanthranilic acid in the presence of kynureninase. Finally, 3-hydroxyanthranilic acid is metabolized to QA in the presence of 3-hydroxyanthranilate 3,4-dioxygenase. Anthranilic acid formed via branch 2 is readily metabolized to 3-hydrocyanthranilic acid through non-specific hydroxylase, which further contributes to the synthesis of QA (Lima, 1998).
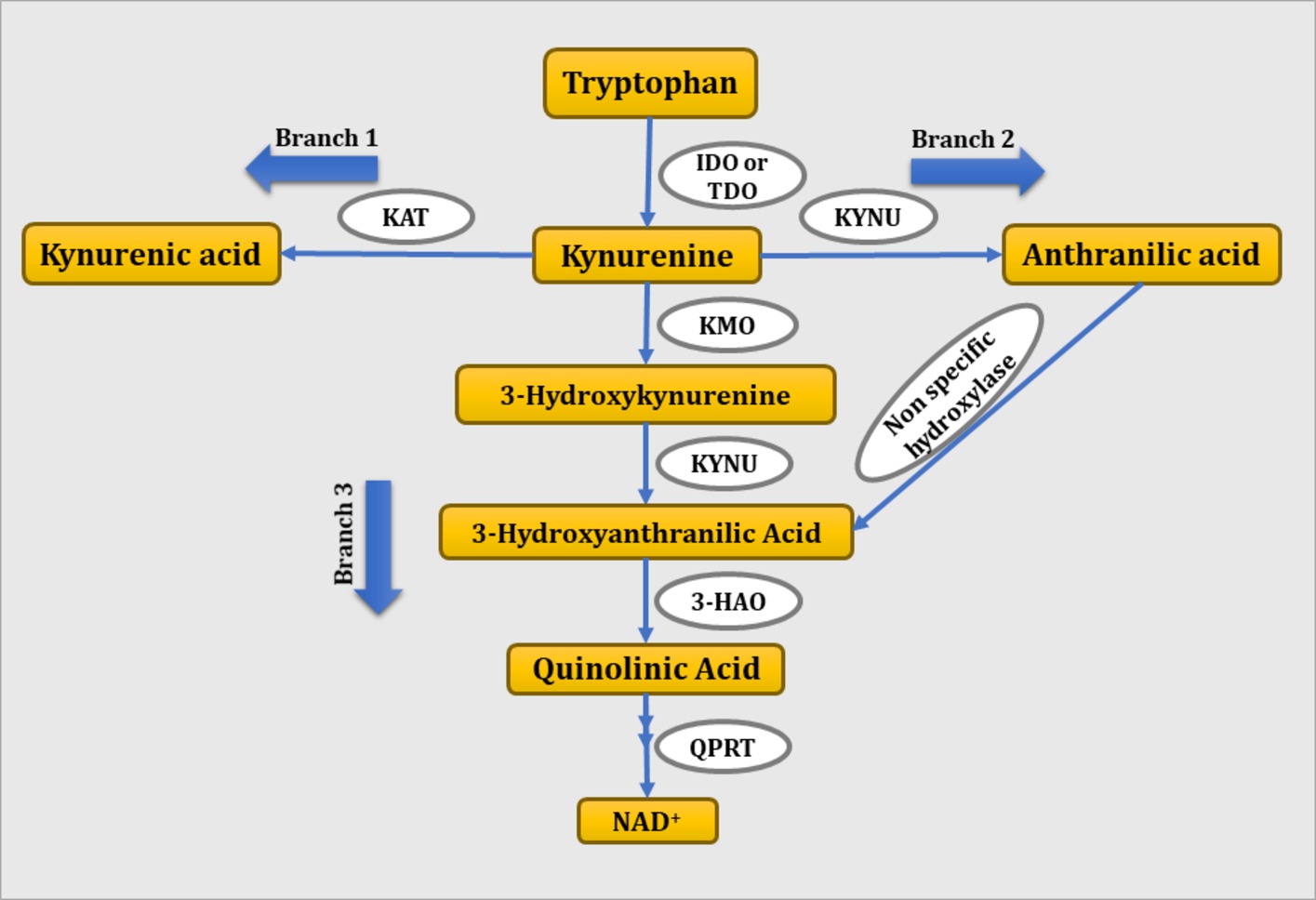
Figure 1. Schematic representation of tryptophan-kynurenine pathway. IDO, indoleamine 2,3-dioxygenase; TDO, tryptophan 2,3-dioxygenase; KMO, kynurenine monooxygenase; KYNU, kynureninase; 3-HAO, 3-hydroxyanthranilate 3,4-dioxygenase; QPRT: quinolinate phosphoribosyl transferase.
Glial cells, i.e., astrocytes and microglia, play a significant role in the development and proper function of the adult brain. Astrocytes are crucial for the formation and maturation of synapses, receptor trafficking, control of the homeostasis of ions and energy metabolites, and clearance of neurotransmitters for maintenance of the neuronal microenvironment (Araque et al., 2014; Dallérac and Rouach, 2016). Astrocytes and microglia have been found to play a potential role in inflammatory and neurodegenerative diseases as they act as both source and target of various inflammatory cytokines. Increased astrogliosis and microgliosis have been observed in several neurological and neurodegenerative disorders such as CNS injury, brain tumors, Huntington’s disease, stroke, epilepsy, Parkinson’s disease, or Alzheimer’s disease, whereas no astrogliosis takes place in depression. Postmortem studies have revealed considerably reduced number and packing density of astrocytes in subjects with depression compared to age-matched normal controls (Cotter et al., 2002; Gittins and Harrison, 2011). In addition, immunohistochemical analysis of glial fibrillary acidic protein, an astrocytic marker, revealed a significantly reduced area covered by glial fibrillary acidic protein-positive cell bodies and processes in various brain regions of young patients with depression compared to a control group (Müller et al., 2001; Fatemi et al., 2004; Miguel-Hidalgo et al., 2010; Gittins and Harrison, 2011; Chandley et al., 2013). In a recent preclinical study by Mendoza et al. (2018), reduced number and complexity of astrocytes in the dentate gyrus region of the hippocampus was seen in a restraint stress mouse model of depression. Contrary to this, animal studies have shown a clear increase in microglial activity in depressed rodents. Mice subjected to lipopolysaccharide (LPS)-induced microglial activation showed depressive-like behaviors (Henry et al., 2008). In a clinical study by Setiawan et al. (2015), significant elevation in the brain translocator protein density, a marker of microglial activation and neuroinflammation, was found in the prefrontal cortex, anterior angulate cortex, thalamus, hippocampus, dorsal putamen, and ventral striatum in patients with MDD compared to healthy controls. The mechanism by which activated microglia induce depression is through activation of microglial IDO and a further signaling pathway, i.e., the KP. Psychological stress, increased glucocorticoid levels, increased inflammatory cytokines, mainly IFN-γ but also tumor necrosis factor-α, IL-6, and their inducers such as LPS, activate microglial IDO (Kiank et al., 2010; Dantzer et al., 2011; Vaváková et al., 2015). Microglial KP-mediated depression is supported by clinical studies where IFN-α-induced immunotherapy increased peripheral and central KP metabolites. The severity of depressive-like symptoms is positively correlated with increased tryptophan metabolism (Capuron et al., 2003; Raison et al., 2010; Savitz, 2017). Additionally, postmortem studies of patients with unipolar depression have shown elevated number of QA-positive microglia in the subgenual anterior cingulate gyrus and anterior mid-cingulate cortex (Steiner et al., 2011). 1-Methlytryptophan (an inhibitor of IDO) and ketamine (an NMDA antagonist) significantly decreased depressive-like behavior by inhibiting QA-mediated NMDA signaling in rodents in an LPS-induced depression model (Aan Het Rot et al., 2012; Dobos et al., 2015). These findings revealed the prominent role of the KP in astrocytes and microglia in depression (Figure 2).
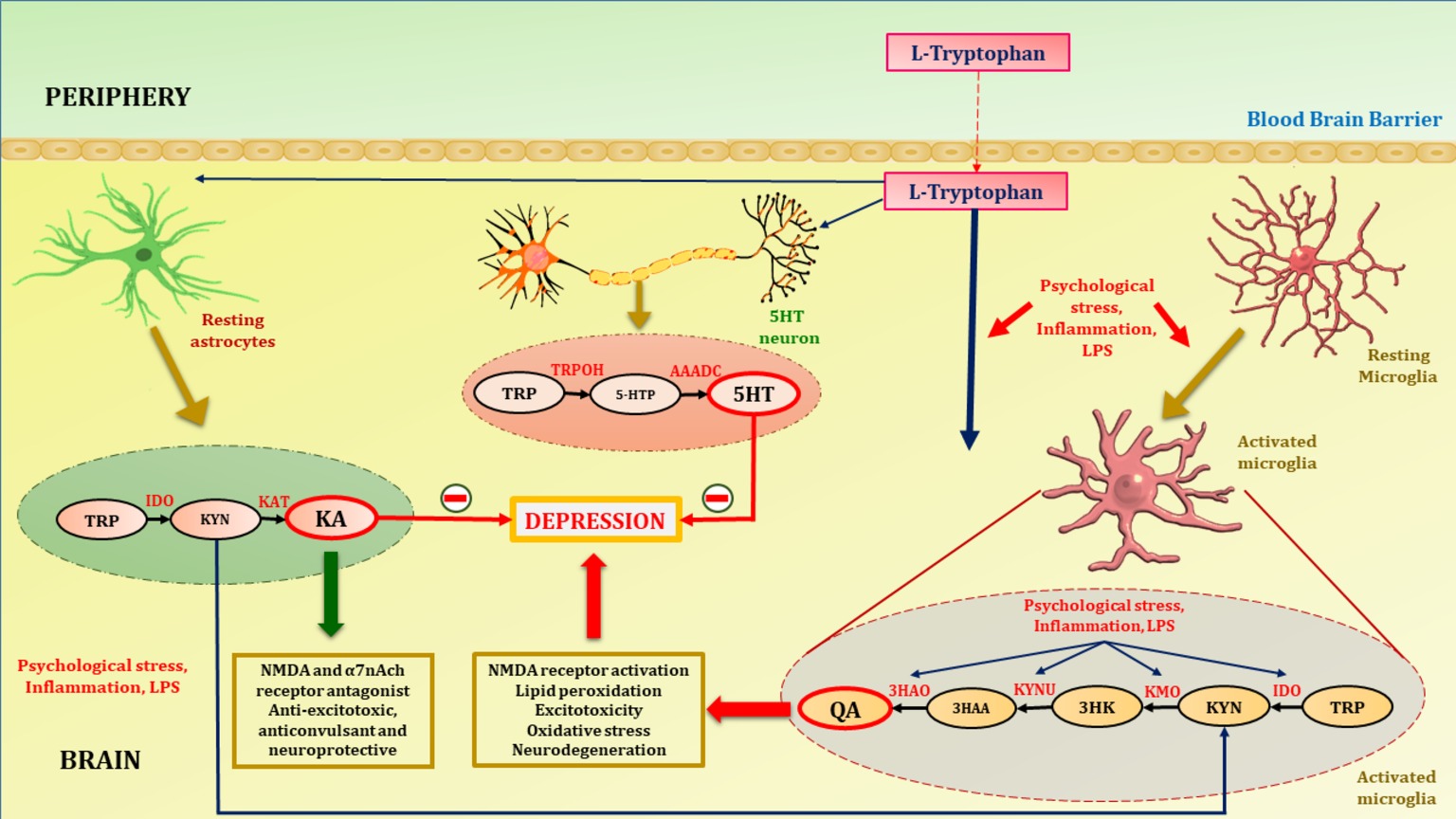
Figure 2. Tryptophan metabolism in astrocytes, microglia and 5HT neuron. Tryptophan, precursor of serotonin is transported to brain with the aid of non-specific competitive L-type amino acid transporters. At homeostatic conditions tryptophan is metabolized to KA in astrocytes and 5HT in serotonergic neurons. KA is an NMDA and α7nAch receptor antagonist, hence acts as anti-excitotoxic and anticonvulsant, thus provide neuroprotection. Increased activation of inflammatory cascades either by psychological stress and LPS activates microglia (microgliosis). Increased inflammation due to psychological stress or LPS activates the enzymes of neurotoxic branch of kynurenine pathway in microglia. This leads to increased production of QA. Psychological stress and activation of inflammatory cascades diverts the metabolism of tryptophan towards QA. This shift hampers the neuroprotection provided by KA and decreases synthesis of 5HT in serotonergic neurons thus contribute to the depression pathophysiology. TRP, tryptophan; IDO, indoleamine-2,3-dioxygenase; KAT, kynurenine aminotransferase; TRPOH, tryptophan hydroxylase; AAADC, aromatic L-amino acid decarboxylase; 5HT, serotonin; 5HTP, 5-hydroxytryptophan; QA, quinolinic acid; KA, kynurenic acid; KYN, kynurenine; 3HK, 3-hydroxykynurenine; 3HAA, 3-hydroxyanthranilic acid; KMO, kynurenine monooxygenase; KYNU, kynureninase; 3HAO, 3-hydroxyanthranilate 3,4-dioxygenase.
Quinolinic Acid in Depression
QA is a neurotoxic compound involved in the neuroprogression of depression. Neuroprogression is a term inclusive of the various stages of neurodegeneration including apoptosis, reduced neurogenesis, reduced plasticity (neuronal and synaptic), and increased immunoreactivity (Berk et al., 2011). Various dynamics are involved in the neuroprogression of depression, including disturbances in the serotonergic system, increased inflammation and cell-mediated immunity, oxidative and nitrosative stress, and neurotoxic compound production (Vaváková et al., 2015; Ruiz et al., 2018). QA is one such neurotoxic compound that acts as an NMDA receptor agonist and elicits excitotoxic damage via glutamatergic activation of neurons and astrocytes (Zhou et al., 2013).
The serotonin theory of depression asserts that “depression is a mood disorder characterized by decreased levels of monoamines, i.e., serotonin in the brain as a consequence of the increased activity of monoamine degrading enzyme, i.e., MAO or reduced serotonin synthesis” (Delgado, 2000). The reduced synthesis of serotonin accounts for the activation of the KP, as a consequence of increased inflammatory cascades in the brain (Ball et al., 2007). Critically, the fate of tryptophan depends on proinflammatory factor levels in the brain. During stress, the levels of circulating and brain proinflammatory cytokines (IL-6, IL-β, tumor necrosis factor-α, and IFN-γ) increase (Lanquillon et al., 2000; Kaestner et al., 2005; Thomas et al., 2005). This increase activates IDO and thus shifts tryptophan metabolism toward the tryptophan-KP (Leonard, 2010). Under basal conditions, kynurenine is metabolized predominantly to KA in astrocytes, the glia cells responsible for maintenance of homeostasis within the brain. KAT catalyzes the synthesis of KA from kynurenine and is not responsive to increased inflammation as are other enzymes of the KP. KAT is predominantly expressed in astrocytes (Rossi et al., 2008). During neuroinflammation, kynurenine metabolism shifts toward QA synthesis in microglia due to microglial activation (Delgado, 2000). Increased kynurenine levels and kynurenine monooxygenase expression are associated with neuroinflammation, characterized by activation of microglia and inflammatory cascades in the CNS. This shift from the methoxyindole pathway toward the KP increases the levels of the deleterious metabolite, QA, in the brain during increased neuroinflammation (Yan et al., 2015).
IDO-directed activity of pro-inflammatory cytokines (IL-6, IFN-α, and IFN-γ) can outpace KA synthesis by inhibiting KAT. A clinical study by Savitz et al. revealed reduced KA/QA levels in the cerebrospinal fluid (CSF) of patients with depression, MDD, and remitted patients with MDD compared to healthy individuals (Savitz et al., 2015). Additional studies have also shown a negative correlation between hippocampal and amygdala volume and increased QA levels in patients with bipolar disorder (Savitz et al., 2015). Another study reported that QA and IL-6 levels in the CSF are positively correlated with suicide attempts in patients with MDD. Clinical and postmortem studies with individuals who had attempted to commit suicide have shown clear increase in the levels of QA in the brain (Erhardt et al., 2013; Brundin et al., 2016). In a preclinical study by Laugeray et al. (2011), tryptophan was found to be metabolized to QA in the amygdala and striatum but not in the cingulate cortex. Furthermore, peripheral catabolism of tryptophan to kynurenine was found to increase in a murine model of depression, resulting in lower availability of plasma tryptophan to the brain (Laugeray et al., 2011).
Given the relationship between QA and depression, the next question that arises is how QA serves as an oxidative stress modulator. QA is not only excitotoxic but also a modulator of oxidative stress (Kubicova et al., 2013). Specifically, QA increases reactive oxygen species (ROS) by increasing the excitotoxicity of the NMDA receptor (Guillemin, 2012; Schwarcz et al., 2012). QA-induced toxicity can be mediated through both NMDA-dependent and independent mechanisms. In line with this, studies have shown that at pathological concentrations (i.e., 10–40 μM), QA forms complexes with iron (Fe) via the Fenton reaction and thus increases ROS levels (Stipek et al., 1997; Iwahashi et al., 1999; Müller et al., 2007; Kubicova et al., 2013). In a study by Goda et al. (1996), it was found that the QA-Fe2+ complex causes cell death via hydroxyl ion-induced DNA chain breakage and lipid peroxidation. QA also enhances free radical production by inducing nitric oxide synthase activity in neurons and astrocytes and also increases poly(ADP-ribose) polymerase and extracellular lactate dehydrogenase activities and impairs mitochondrial function (Braidy et al., 2010; Pérez-De La Cruz et al., 2010). QA has also been found to impair the activity of endogenous antioxidant enzymes. In a study by Rodríguez-Martínez et al. (2000), intrastriatal injection of QA reduced the activity of reduced glutathione and cytosolic copper/zinc superoxide dismutase, whereas it increased the levels of oxidized glutathione. Melatonin and deprenyl, potent free radical scavengers, prevented QA-induced oxidative stress via NMDA receptor-independent action and also elevated antioxidant enzyme levels (Behan et al., 1999; Antunes Wilhelm et al., 2013). Increased brain microglial density was reported by a postmortem study with individuals who had attempted to commit suicide (Steiner et al., 2008). In addition, microglial QA levels have also been shown to be upregulated in the brain of patients with depression (Steiner et al., 2011). In their study with patients with depression, Meier et al. found a positive correlation between a decreased KA/QA ratio in the CSF and reductions in gray matter volume in the rostral and subgenual anterior cingulate cortex in the medial prefrontal cortex (Meier et al., 2016). Patients with bipolar disorder were found to have decreased KA levels in the hippocampus and amygdala compared with healthy controls (Savitz et al., 2015). A recent meta-analysis by Ogyu et al. (2018) revealed increased QA levels and decreased KA and kynurenine levels in patients with depression. Additionally, at pathological concentrations (i.e., 10–40 μM), QA reduces metabolic and physical buffering of neurons by instigating astrocytic apoptosis and thus hampering protection to neurons from ROS and inflammatory processes that lead to neurodegeneration (Grant and Kapoor, 1998). Given this, constraining the activity of QA may reduce the neuroprogression of depression and thus serve as a potential target for its pharmacotherapeutic treatment.
Nrf2 in Depression
Nrf2, also known as NFE2L2, is a redox-sensitive transcription factor. It is a basic leucine zipper protein factor that belongs to the cap “n” collar subfamily. It plays an important role in the body’s endogenous antioxidant defense system by maintaining the intracellular redox homeostasis that when dysregulated, triggers oxidative stress (Moi et al., 1994). Nrf2 transcribes various antioxidant enzymes to protect against oxidative stress induced by injury and/or inflammation. Under physiological conditions, Nrf2 is retained in the cytosol in a dormant form via tethering to Kelch-like ECH associated protein-1 (Keap-1), an adaptor for the cullin-3/Rbx complex. Keap-1, in association with cullin-3/Rbx, causes ubiquitination and finally causes proteasomal degradation of Nrf2. Given this, Nrf2 is highly unstable at physiological conditions due to its negative regulation by Keap-1 (Dhakshinamoorthy and Jaiswal, 2001). In inflammatory and oxidative stress conditions, Nrf2 translocates to the nucleus after dissociating from Keap-1 and binds to antioxidant response element, also referred to as electrophile responsive element. Transcription of electrophile responsive element target genes that encode phase-II antioxidant proteins such as GCLC, NOQ1, and HO-1; detoxifying enzymes, antiapoptotic proteins, and proteasomes are then enhanced (Figure 3; Niture et al., 2014).
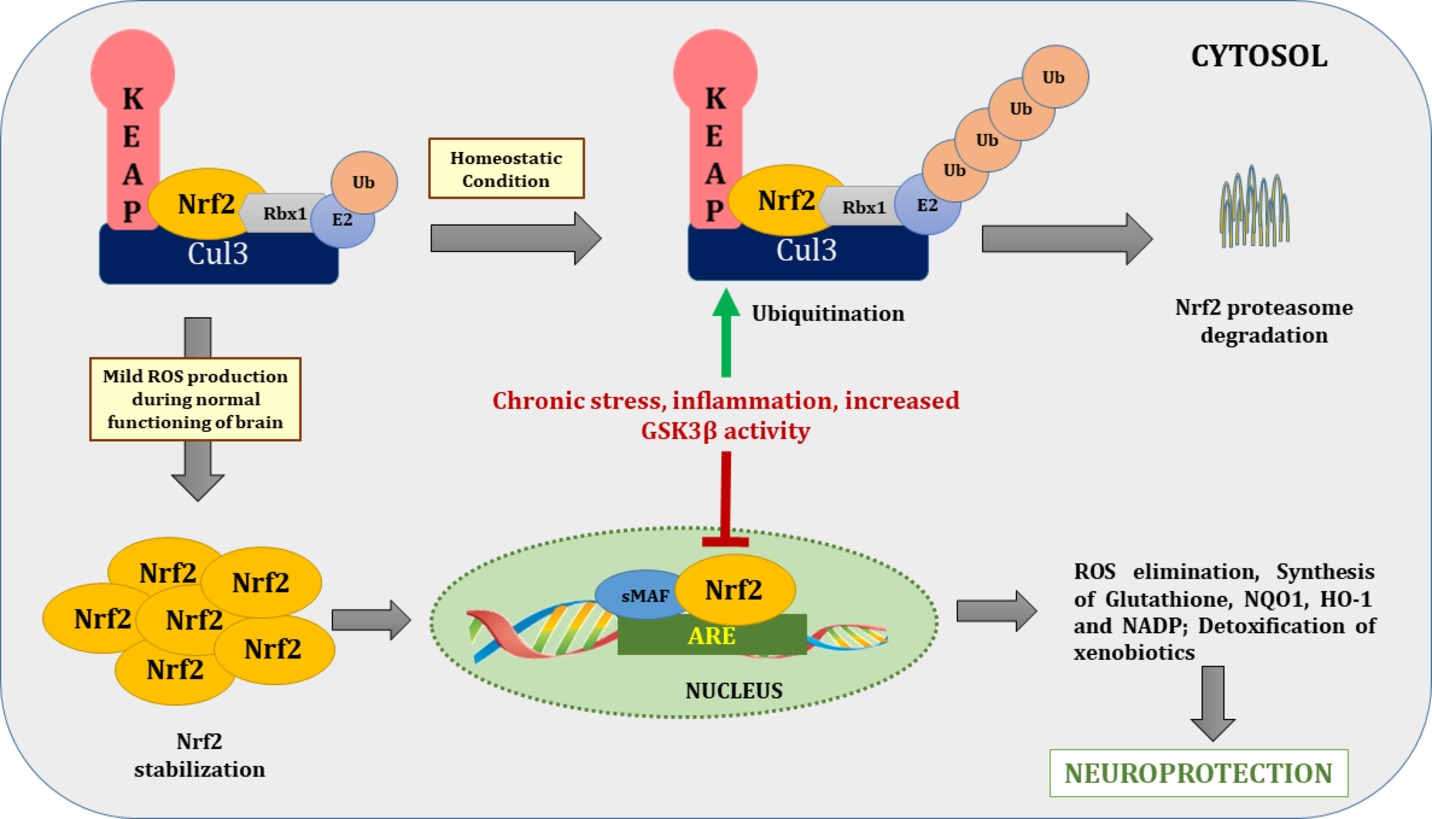
Figure 3. Nrf2/ARE pathway during basal and stressed conditions. During homeostatic conditions Nrf2 remains in tethering with Keap1 and Cul3-Rbx1-E2 ligase and undergoes proteasome degradation through ubiquitination. In normal brain functioning, mild ROS production dissociates Nrf2 from Keap1 and translocate it to the nucleus. After nuclear translocation, it binds with ARE to transcribe genes of various antioxidants enzymes to combat deleterious effects of ROS. On contrary during chronic stress, increased activity of inflammatory cascades and GSK-3β increased Nrf2 proteasome degradation via ubiquitination. Nrf2, nuclear factor (erythroid-derived 2)-like 2; Keap1, Kelch-like ECH associated protein-1; Cul-3, cullin 3; ROS, reactive oxygen species; ARE, antioxidant response element; Rbx1, ring box-1; E2, E2 ubiquitin-conjugating enzyme; Ub, ubiquitin.
Of special relevance here, studies have shown that downregulation of Nrf2 occurs in depression. In a recent study by Bouvier et al. (2016), rats that were exposed to persistently stressful conditions became progressively more vulnerable to depression due to persistent oxidative stress. This oxidative stress was found to be caused by diminished nuclear translocation of Nrf2, which prevented the transcription of antioxidants and detoxifying enzymes. In an in vitro study by Rojo et al. (2008), GSK-3β downregulated Nrf2 in neuronal cultures, providing a possible mechanism of inability to combat oxidative stress under persistent oxidant exposure (Figure 3).
The neurotrophic hypothesis of depression proposes that depression is also accompanied by decreased neurotrophic support, which is primarily linked to the brain-derived neurotrophic factor (BDNF) protein. A recent study reported that BDNF-deficient mice were more susceptible to stress-induced oxidative damage, indicating a direct link between oxidative stress and BDNF levels (Hacioglu et al., 2016). Bouvier et al. (2016) reported a positive correlation between BDNF and Nrf2 levels in rodents vulnerable to depression. In a study by Mendez-David et al. (2015), a positive correlation between BDNF and Nrf2 in the hippocampus of corticosterone-treated depressed rats was established. Besides depression, reduced levels of Nrf2 have further been noted in hippocampal astrocytes in patients with Alzheimer’s disease (Ramsey et al., 2007). Furthermore, when Nrf2 knockout mice were treated with corticosterone, they exhibited significant changes in prefrontal cortex neurotransmitter levels, including serotonin, glutamate, and dopamine, compared to wild-type controls (Mendez-David et al., 2015).
In addition to its role in neurotransmission and depression, Nrf2 is also linked to neuroprotection. Nrf2 protects neurons from the deleterious effects of oxidative and nitrosative stress and inflammatory cytokines by regulating astrocytic enzymes integral to glutathione redox signaling (Habas et al., 2013; Baxter and Hardingham, 2016). In a recent study by Yao et al. (2016), mice exhibiting depression-like behavior were found to have decreased levels of Keap-1 and Nrf2 protein in the prefrontal cortex, CA3, and dentate gyrus compared to controls. The authors also found that pre-treatment with dietary sulforaphane and 0.1% glucoraphanin (Nrf2 modulators) during the juvenile and adolescent stages prevented emergence of depression-like behavioral phenotypes in animals exposed to repeated social defeat stress (Yao et al., 2016). Additionally, Freitas et al. (2016) found that agmatine, an endogenous metabolite of L-arginine, protects corticosterone-induced apoptotic cell death and ROS production in hippocampal neuronal cells via induction of Nrf2. This group further evaluated the antidepressant potential of agmatine in a corticosterone mouse model of depression. Agmatine showed antidepressant properties in Nrf2(+/+) mice via induction of Nrf2 and BDNF and was unable to reverse the depression-like effect in Nrf2 knockout (Nrf2(−/−)) mice. Agmatine also prevented morphological changes in astrocytes and microglia in the CA1 region of the hippocampus of Nrf2(−/−) mice (Freitas et al., 2016).
Nrf2 is predominantly expressed in astrocytes. In studies by Calkins et al. (2004, 2010), specific Nrf2 activation in astrocytes provided protection against malonate and 3-nitropropionic acid induced mitochondrial complex II inhibition-mediated neurotoxicity both in vitro and in vivo. Additionally, the presence of tert-butyl hydroquinone (tBHQ; an Nrf2 activator) was unable to buffer against oxidative stress in pure neuronal cultures, whereas in a mixed culture of astrocytes and neurons, tBHQ caused Nrf2 activation and provided protection from oxidative stress (Bell et al., 2015). In another complimentary study conducted by (Kraft, 2004), similar in vitro results were reported wherein the pharmacological activation of mixed cultures by tBHQ protected against oxidative stress, whereas specific inhibition of Nrf2 in astrocytes reduced the protection. Although Nrf2 is predominantly expressed in astrocytes, overactivation of Nrf2 in astrocytes is protective in various neurogenerative diseases such as Parkinson’s disease (Chen et al., 2009), amyotrophic lateral sclerosis (Vargas et al., 2008), Huntington’s disease (Shih et al., 2005), and multiple sclerosis (Draheim et al., 2016). In another study, overactivation of neuronal Nrf2 was found to be protective in Alzheimer’s disease (Vargas et al., 2013), suggesting potential neural involvement. Collectively, these findings indicate that astrocytic activation of Nrf2 may be a potent pharmacotherapeutic target in various CNS diseases.
New findings on the role of Nrf2 in various CNS diseases have shown that Nrf2 is also important in the regulation of neuroinflammation that arises as a consequence of oxidative stress in the brain. In a study by Martín-de-Saavedra et al. (2013), Nrf2 was shown to reverse depression symptoms via an anti-inflammatory mechanism. This study also found that depletion of Nrf2 caused inflammation-induced depressive symptoms that were reversed by treatment with an anti-inflammatory drug, rofecoxib. Furthermore, this study also found that sulforaphane treatment in an LPS-induced inflammatory mouse model of depression increased nuclear translocation of Nrf2 and led to antidepressant-like effects (Martín-de-Saavedra et al., 2013). Also in the same study, Nrf2(−/−) animals displayed a reduction in BDNF expression in the hippocampus, demonstrating a prospective relationship between the neurotropic factor and Nrf2. Suggesting a potential mechanism for these effects of Nrf2, in a recent study by Kobayashi et al. (2016), Nrf2 was found to be an upstream regulator of cytokine production via inhibiting transcriptional upregulation of proinflammatory cytokine genes and thus decreased the levels of proinflammatory cytokines. Thus, Nrf2 exerts anti-inflammatory and antioxidant effects on biological systems. Collectively, the studies discussed here strongly suggest an essential role for Nrf2 in depression, pointing at the induction of Nrf2 as a potential target in the investigation of novel antidepressant drugs.
Interactions Between QA and Nrf2 in Depression
As suggested by the studies outlined previously, both QA and Nrf2 play substantial, and probably opposite, roles in the etiology of depression. While QA has been shown to contribute to depression, Nrf2 likely protects against depression. Given this, it is important to understand whether QA and Nrf2 are directly associated. To date, only two known in vitro studies have examined the direct relationship between QA and Nrf2 (Tasset et al., 2010; Colín-González et al., 2014). Suggesting a potential mechanism for this, Tasset et al. (2010) reported that exposure to QA abolished nuclear translocation of Nrf2 in rat striatal tissues. Nuclear translocation of Nrf2 in QA-treated striatal slices was noted after tBHQ (a Nrf2 modulator) treatment (Tasset et al., 2010). This indicates that QA abolishes Nrf2 translocation in the nucleus.
An additional study by Colín-González et al. (2014) reported that striatal slices from Nrf2(−/−) animals were more vulnerable to the oxidative damage caused by QA than were wild-type tissues. In mouse striatum slices, QA treatment after 1 h was found to stimulate Nrf2 nuclear translocation, while after 3 h, it was found to decrease Nrf2 translocation and phase II enzymatic activity and increase lipid peroxidation. Enhanced Nrf2 nuclear translocation may thus represent a compensatory mechanism for QA-induced oxidative stress, while downregulation of Nrf2 might contribute to cellular oxidative damage. Further, Nrf2(−/−) animals were found to be less responsive to QA-induced toxicity compared to wild-type animals (Colín-González et al., 2014). In a recent study by Ferreira et al. (2018), KA prevented QA-induced oxidative imbalance, mitochondrial dysfunction, and decreased Nrf2 levels in striatal slices. The NADPH inhibitor, apocynin, prevented QA-induced decrease in antioxidant levels such as glutathione, γ-GCL, and GPx activities and Nrf2 mRNA levels which is involved in the maintenance of antioxidant levels (Cruz-Álvarez et al., 2017).
Apart from NMDA-induced excitotoxicity by QA, studies have also shown that QA exerts toxic effects by increasing the oxidant-to-antioxidant ratio. Specifically, QA alters the ratio of reduced glutathione to oxidized glutathione and hinders the activity of other antioxidants, such as Cu and zinc-superoxide dismutase (Rodríguez-Martínez et al., 2000). However, Nrf2 protects against these pro-oxidant effects by increasing the transcription of various cytoprotective antioxidant genes such as GSH. For example, Harvey et al. (2009) demonstrated that Nrf2 is protective against oxidative stress by maintaining the glutathione redox state via transcriptional regulation of glutathione reductase. As noted previously, pathological concentrations of QA can cause astrocytic apoptosis, limiting the role of these cells in protecting neurons from ROS and other pro-inflammatory processes, thus causing neurodegeneration (Grant and Kapoor, 1998). In sum, Nrf2 protects against the deleterious effects of oxidative and nitrosative stress and inflammatory cytokines on neurons by cytoprotection of astrocytes via regulation of enzymes belonging to the glutathione redox system (Habas et al., 2013; Baxter and Hardingham, 2016). Based on the aforementioned studies, there may also be a direct link between QA and Nrf2, wherein QA constrains Nrf2 nuclear translocation. Further examining this process may serve as a viable potential target for the development of pharmacological treatments for depression (Figure 4).
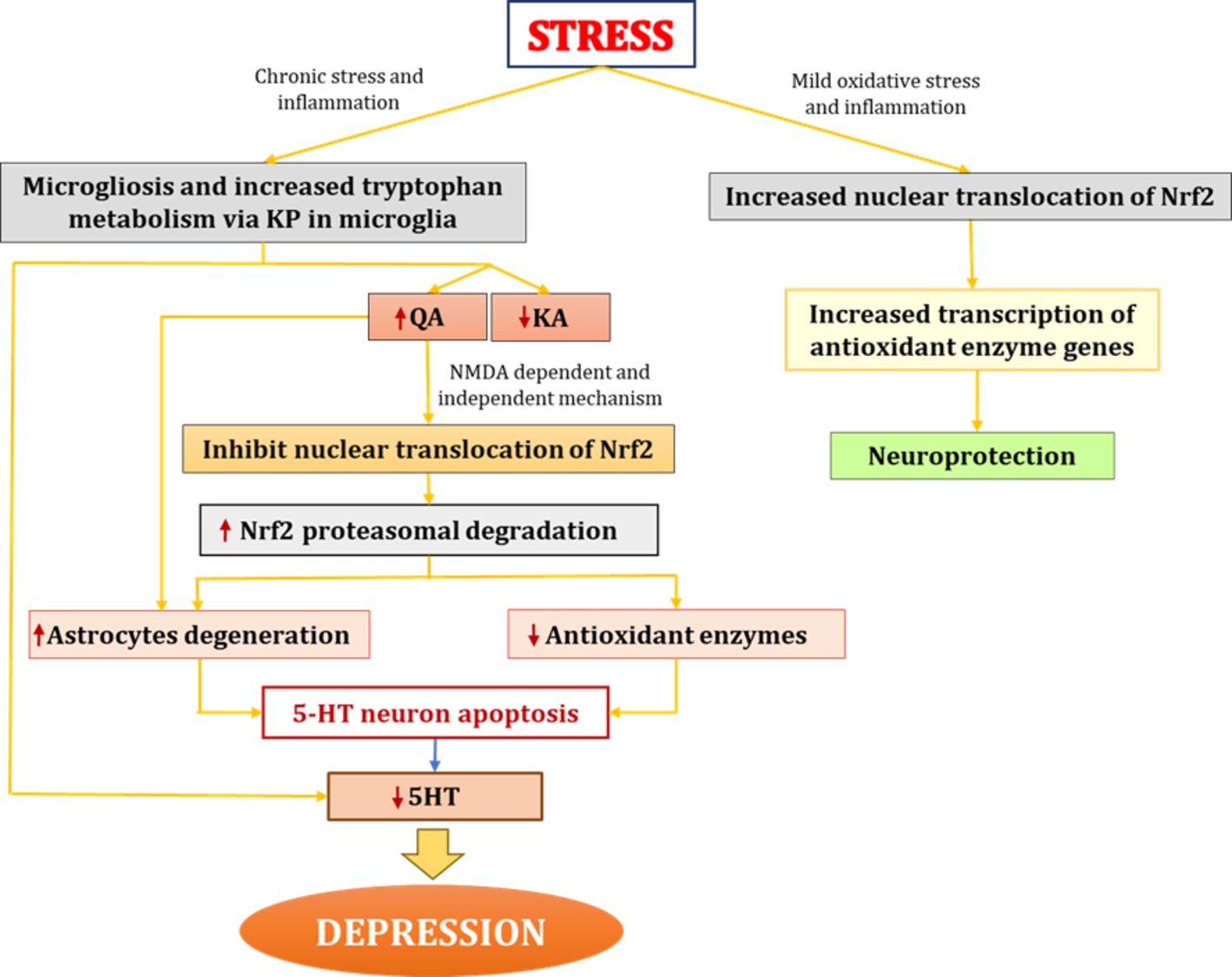
Figure 4. Interaction between QA and Nrf2 in depression. In chronic stress conditions, increased proinflammatory cytokines lead to microgliosis. Increased proinflammatory cytokines activate IDO (enzyme catalyzing first rate-limiting step of KP) and shift tryptophan metabolism from serotonin synthesis to KP in microglia. Elevated levels of QA further increases oxidative stress through NMDA agonistic activity and secondly might be through directly inhibiting nuclear translocation of Nrf2 and hence causes increased proteasome degradation of Nrf2 which ultimately led to decreased antioxidant levels and increased oxidative stress. QA also causes degeneration of astrocytes thus hampering protection and nutritional support to neurons. All this together lead to degeneration of 5HT neurons and decreased 5HT synthesis which results in depression. On the other hand mild oxidative stress stabilizes Nrf2 and increases Nrf2 transcribed antioxidant enzymes. IDO, indoleamine-2,3-dioxygenase; KP, kynurenine pathway; QA, quinolinic acid; KA, kynurenic acid; Nrf2, nuclear factor (erythroid-derived 2)-like 2; 5HT, serotonin.
Concluding Remarks
A growing body of preclinical and postmortem evidence has revealed that depression is often accompanied by hippocampal and prefrontal cortex neuronal atrophy. Decreased neuronal activity and plasticity across multiple brain regions have also been noted in clinical and modeled depression. Collectively, these morphological changes and altered neuronal functions may be due to increased oxidative stress, which contributes to neuronal and glial atrophy in depression. Specifically, the role of QA in mediating these changes and the broader pathophysiology of depression has become a subject of clinical and preclinical studies. A decreased KA/QA ratio has been noted in the CSF of remittent patients with depression. Moreover, QA is an endogenous toxin that also acts as a pro-oxidant and leads to the neuroprogression of depression. Furthermore, QA is linked to the activity of Nrf2, an endogenous antioxidant transcription factor. While the contributions of Nrf2 to CNS diseases have been understudied, it has been reported that Nrf2 is both a potent antioxidant transcription factor and also has anti-inflammatory effects that may be responsible for its antidepressant properties. Hence, further study of the associations among QA, Nrf2, and oxidative stress may provide novel insight into the neuroprogression and etiology of depression.
Author Contributions
YB wrote this review. RS and AK helped editing a draft review. IP and TS designed the flow of review paper, contributed to English editing and made scientific comments.
Funding
IBRO/APRC exchange fellowship 2017 from the International Brain Research Organization and DST inspire fellowship from Department of Science and Technology (DST), Delhi, India was awarded to YB.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aan Het Rot, M., Zarate, C. A., Charney, D. S., and Mathew, S. J. (2012). Ketamine for depression: where do we go from here? Biol. Psychiatry 72, 537–547. doi: 10.1016/j.biopsych.2012.05.003
Antunes Wilhelm, E., Ricardo Jesse, C., Folharini Bortolatto, C., and Wayne Nogueira, C. (2013). Correlations between behavioural and oxidative parameters in a rat quinolinic acid model of Huntington’s disease: protective effect of melatonin. Eur. J. Pharmacol. 701, 65–72. doi: 10.1016/j.ejphar.2013.01.007
Araque, A., Carmignoto, G., Haydon, P. G., Oliet, S. H. R., Robitaille, R., and Volterra, A. (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. doi: 10.1016/j.neuron.2014.02.007
Ball, H. J., Sanchez-Perez, A., Weiser, S., Austin, C. J. D., Astelbauer, F., Miu, J., et al. (2007). Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 396, 203–213. doi: 10.1016/j.gene.2007.04.010
Baxter, P. S., and Hardingham, G. E. (2016). Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 100, 147–152. doi: 10.1016/j.freeradbiomed.2016.06.027
Behan, W. M., McDonald, M., Darlington, L. G., and Stone, T. W. (1999). Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: protection by melatonin and deprenyl. Br. J. Pharmacol. 128, 1754–1760. doi: 10.1038/sj.bjp.0702940
Behan, W. M. H., and Stone, T. W. (2002). Enhanced neuronal damage by co-administration of quinolinic acid and free radicals, and protection by adenosine A2A receptor antagonists. Br. J. Pharmacol. 135, 1435–1442. doi: 10.1038/sj.bjp.0704613
Bell, K. F. S., Al-Mubarak, B., Martel, M. A., McKay, S., Wheelan, N., Hasel, P., et al. (2015). Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 6, 1–15. doi: 10.1038/ncomms8066
Berk, M., Kapczinski, F., Andreazza, A. C., Dean, O. M., Giorlando, F., Maes, M., et al. (2011). Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 35, 804–817. doi: 10.1016/j.neubiorev.2010.10.001
Bouvier, E., Brouillard, F., Molet, J., Claverie, D., Cabungcal, J. H., Cresto, N., et al. (2016). Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol. Psychiatry. 22, 1701–1713. doi: 10.1038/mp.2016.144
Braidy, N., Grant, R., Adams, S., and Guillemin, G. J. (2010). Neuroprotective effects of naturally occurring polyphenols on quinolinic acid-induced excitotoxicity in human neurons. FEBS J. 277, 368–382. doi: 10.1111/j.1742-4658.2009.07487.x
Brundin, L., Sellgren, C. M., Lim, C. K., Grit, J., Pålsson, E., Landén, M., et al. (2016). An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Transl. Psychiatry 6, 1–9. doi: 10.1038/tp.2016.133
Calkins, M. J., Jakel, R. J., Johnson, D. A., Chan, K., Kan, Y. W., and Johnson, J. A. (2004). Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc. Natl. Acad. Sci. USA 102, 244–249. doi: 10.1073/pnas.0408487101
Calkins, M. J., Vargas, M. R., Johnson, D. A., and Johnson, J. A. (2010). Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol. Sci. 115, 557–568. doi: 10.1093/toxsci/kfq072
Capuron, L., Neurauter, G., Musselman, D. L., Lawson, D. H., Nemeroff, C. B., Fuchs, D., et al. (2003). Interferon-alpha-induced changes in tryptophan metabolism: relationship to depression and paroxetine treatment. Biol. Psychiatry 54, 906–914. doi: 10.1016/S0006-3223(03)00173-2
Chandley, M. J., Szebeni, K., Szebeni, A., Crawford, J., Stockmeier, C. A., Turecki, G., et al. (2013). Gene expression deficits in pontine locus coeruleus astrocytes in men with major depressive disorder. J. Psychiatry Neurosci. 38, 276–284. doi: 10.1503/jpn.120110
Chen, P.-C., Vargas, M. R., Pani, A. K., Smeyne, R. J., Johnson, D. A., Kan, Y. W., et al. (2009). Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 106, 2933–2938. doi: 10.1073/pnas.0813361106
Colín-González, A. L., Luna-López, A., Königsberg, M., Ali, S. F., Pedraza-Chaverrí, J., and Santamaría, A. (2014). Early modulation of the transcription factor Nrf2 in rodent striatal slices by quinolinic acid, a toxic metabolite of the kynurenine pathway. Neuroscience 260, 130–139. doi: 10.1016/j.neuroscience.2013.12.025
Cotter, D., Mackay, D., Chana, G., Beasley, C., Landau, S., and Everall, I. P. (2002). Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 12, 386–394. doi: 10.1093/cercor/12.4.386
Cowen, P. J., and Browning, M. (2015). What has serotonin to do with depression? World Psychiatry 14, 158–160. doi: 10.1002/wps.20229
Cruz-Álvarez, S., Santana-Martínez, R., Avila-Chávez, E., Barrera-Oviedo, D., Hernández-Pando, R., Pedraza-Chaverri, J., et al. (2017). Apocynin protects against neurological damage induced by quinolinic acid by an increase in glutathione synthesis and Nrf2 levels. Neuroscience 350, 65–74. doi: 10.1016/j.neuroscience.2017.03.011
Dallérac, G., and Rouach, N. (2016). Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 144, 48–67. doi: 10.1016/j.pneurobio.2016.01.003
Dantzer, R., O’Connor, J. C., Lawson, M. A., and Kelley, K. W. (2011). Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinology 36, 426–436. doi: 10.1016/j.psyneuen.2010.09.012
Delgado, P. L. (2000). Depression: the case for a monoamine deficiency. J. Clin. Psychiatry 61(Suppl. 6), 7–11.
Dhakshinamoorthy, S., and Jaiswal, A. K. (2001). Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 20, 3906–3917. doi: 10.1038/sj.onc.1204506
Dobos, N., de Vries, E. F. J., Kema, I. P., Patas, K., Prins, M., Nijholt, I. M., et al. (2015). “The role of indoleamine 2,3-dioxygenase in a mouse model of neuroinflammation-induced depression” in Handbook of depression in Alzheimer’s disease (IOS Press), 163–174. doi: 10.3233/978-1-61499-542-5-163
Draheim, T., Liessem, A., Scheld, M., Wilms, F., Weißflog, M., Denecke, B., et al. (2016). Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia 64, 2219–2230. doi: 10.1002/glia.23058
Erhardt, S., Lim, C. K., Linderholm, K. R., Janelidze, S., Lindqvist, D., Samuelsson, M., et al. (2013). Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 38, 743–752. doi: 10.1038/npp.2012.248
Fatemi, S. H., Laurence, J. A., Araghi-Niknam, M., Stary, J. M., Schulz, S. C., Lee, S., et al. (2004). Glial fibrillary acidic protein is reduced in cerebellum of subjects with major depression, but not schizophrenia. Schizophr. Res. 69, 317–323. doi: 10.1016/j.schres.2003.08.014
Ferreira, F. S., Biasibetti-Brendler, H., Pierozan, P., Schmitz, F., Bertó, C. G., Prezzi, C. A., et al. (2018). Kynurenic acid restores Nrf2 levels and prevents quinolinic acid-induced toxicity in rat striatal slices. Mol. Neurobiol. 55, 1–12. doi: 10.1007/s12035-018-1003-2
Freitas, A. E., Egea, J., Buendia, I., Gómez-Rangel, V., Parada, E., Navarro, E., et al. (2016). Agmatine, by improving neuroplasticity markers and inducing Nrf2, prevents corticosterone-induced depressive-like behavior in mice. Mol. Neurobiol. 53, 3030–3045. doi: 10.1007/s12035-015-9182-6
Gittins, R. A., and Harrison, P. J. (2011). A morphometric study of glia and neurons in the anterior cingulate cortex in mood disorder. J. Affect. Disord. 133, 328–332. doi: 10.1016/j.jad.2011.03.042
Goda, K., Kishimoto, R., Shimizu, S., Hamane, Y., and Ueda, M. (1996). Quinolinic acid and active oxygens. Possible contribution of active oxygens during cell death in the brain. Adv. Exp. Med. Biol. 398, 247–254.
Grant, R. S., and Kapoor, V. (1998). Murine glial cells regenerate NAD, after peroxide-induced depletion, using either nicotinic acid, nicotinamide, or quinolinic acid as substrates. J. Neurochem. 70, 1759–1763. doi: 10.1046/j.1471-4159.1998.70041759.x
Guillemin, G. J. (2012). Quinolinic acid, the inescapable neurotoxin. FEBS J. 279, 1356–1365. doi: 10.1111/j.1742-4658.2012.08485.x
Habas, A., Hahn, J., Wang, X., and Margeta, M. (2013). Neuronal activity regulates astrocytic Nrf2 signaling. Proc. Natl. Acad. Sci. USA 110, 18291–18296. doi: 10.1073/pnas.1208764110
Hacioglu, G., Senturk, A., Ince, I., and Alver, A. (2016). Assessment of oxidative stress parameters of brain-derived neurotrophic factor heterozygous mice in acute stress model. Iran. J. Basic Med. Sci. 19, 388–393. doi: 10.22038/ijbms.2016.6810
Harvey, C. J., Thimmulappa, R. K., Singh, A., Blake, D. J., Ling, G., Wakabayashi, N., et al. (2009). Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 46, 443–453. doi: 10.1016/j.freeradbiomed.2008.10.040
Henry, C. J., Huang, Y., Wynne, A., Hanke, M., Himler, J., Bailey, M. T., et al. (2008). Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J. Neuroinflammation 5, 1–14. doi: 10.1186/1742-2094-5-15
Iwahashi, H., Kawamori, H., and Fukushima, K. (1999). Quinolinic acid, α-picolinic acid, fusaric acid, and 2,6-pyridinedicarboxylic acid enhance the Fenton reaction in phosphate buffer. Chem. Biol. Interact. 118, 201–215. doi: 10.1016/S0009-2797(99)00080-0
Jones, S. P., Guillemin, G. J., and Brew, B. J. (2013). The Kynurenine pathway in stem cell biology. Int. J. Tryptophan Res. 6, 57–66. doi: 10.4137/IJTR.S12626
Kaestner, F., Hettich, M., Peters, M., Sibrowski, W., Hetzel, G., Ponath, G., et al. (2005). Different activation patterns of proinflammatory cytokines in melancholic and non-melancholic major depression are associated with HPA axis activity. J. Affect. Disord. 87, 305–311. doi: 10.1016/j.jad.2005.03.012
Kiank, C., Zeden, J. P., Drude, S., Domanska, G., Fusch, G., Otten, W., et al. (2010). Psychological stress-induced, IDO1-dependent tryptophan catabolism: implications on immunosuppression in mice and humans. PLoS One 5, 1–15. doi: 10.1371/journal.pone.0011825
Kim, H., Chen, L., Lim, G., Sung, B., Wang, S., McCabe, M. F., et al. (2012). Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Invest. 122, 2940–2954. doi: 10.1172/JCI61884
Kobayashi, E. H., Suzuki, T., Funayama, R., Nagashima, T., Hayashi, M., Sekine, H., et al. (2016). Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 7, 1–14. doi: 10.1038/ncomms11624
Kohler, O., Krogh, J., Mors, O., and Eriksen Benros, M. (2016). Inflammation in depression and the potential for anti-inflammatory treatment. Curr. Neuropharmacol. 14, 732–742. doi: 10.2174/1570159X14666151208113700
Kraft, A. D. (2004). Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 24, 1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004
Kubicova, L., Hadacek, F., and Chobot, V. (2013). Quinolinic acid: neurotoxin or oxidative stress modulator? Int. J. Mol. Sci. 14, 21328–21338. doi: 10.3390/ijms141121328
Lanquillon, S. (2000). Cytokine Production and Treatment Response in Major Depressive Disorder. Neuropsychopharmacology 22, 370–379. doi: 10.1016/S0893-133X(99)00134-7
Laugeray, A., Launay, J. M., Callebert, J., Surget, A., Belzung, C., and Barone, P. R. (2011). Evidence for a key role of the peripheral kynurenine pathway in the modulation of anxiety- and depression-like behaviours in mice: focus on individual differences. Pharmacol. Biochem. Behav. 98, 161–168. doi: 10.1016/j.pbb.2010.12.008
Leonard, B. E. (2010). “The concept of depression as a dysfunction of the immune system” in Depression: From Psychopathology to Pharmacotherapy 53–71. doi: 10.1159/000319504
Lima, W. S. (1998). Seasonal infection pattern of gastrointestinal nematodes of beef cattle in Minas Gerais State–Brazil. Vet. Parasitol. 74, 203–214. doi: 10.1016/S0304-4017(97)00164-7
Liu, T., Zhong, S., Liao, X., Chen, J., He, T., Lai, S., et al. (2015). A meta-analysis of oxidative stress markers in depression. PLoS One 10. doi: 10.1371/journal.pone.0138904
Maes, M. (1993). A review on the acute phase response in major depression. Rev. Neurosci. 4, 407–416. doi: 10.1515/REVNEURO.1993.4.4.407
Maes, M., Bosmans, E., Suy, E., Vandervorst, C., DeJonckheere, C., and Raus, J. (1991). Depression-related disturbances in mitogen-induced lymphocyte responses and interleukin-1β and soluble interleukin-2 receptor production. Acta Psychiatr. Scand. 84, 379–386. doi: 10.1111/j.1600-0447.1991.tb03163.x
Maes, M., Scharpé, S., Meltzer, H. Y., Bosmans, E., Suy, E., Calabrese, J., et al. (1993). Relationships between interleukin-6 activity, acute phase proteins, and function of the hypothalamic-pituitary-adrenal axis in severe depression. Psychiatry Res. 49, 11–27. doi: 10.1016/0165-1781(93)90027-E
Martín-de-Saavedra, M. D., Budni, J., Cunha, M. P., Gómez-Rangel, V., Lorrio, S., del Barrio, L., et al. (2013). Nrf2 participates in depressive disorders through an anti-inflammatory mechanism. Psychoneuroendocrinology 38, 2010–2022. doi: 10.1016/j.psyneuen.2013.03.020
Meier, T. B., Drevets, W. C., Wurfel, B. E., Ford, B. N., Morris, H. M., Victor, T. A., et al. (2016). Relationship between neurotoxic kynurenine metabolites and reductions in right medial prefrontal cortical thickness in major depressive disorder. Brain Behav. Immun. 53, 39–48. doi: 10.1016/j.bbi.2015.11.003
Mendez-David, I., Tritschler, L., El Ali, Z., Damiens, M. H., Pallardy, M., David, D. J., et al. (2015). Nrf2-signaling and BDNF: a new target for the antidepressant-like activity of chronic fluoxetine treatment in a mouse model of anxiety/depression. Neurosci. Lett. 597, 121–126. doi: 10.1016/j.neulet.2015.04.036
Mendoza, C., Perez-Urrutia, N., Alvarez-Ricartes, N., Barreto, G. E., Pérez-Ordás, R., Iarkov, A., et al. (2018). Cotinine plus krill oil decreased depressive behavior, and increased astrocytes survival in the hippocampus of mice subjected to restraint stress. Front. Neurosci. 12, 1–11. doi: 10.3389/fnins.2018.00952
Miguel-Hidalgo, J. J., Waltzer, R., Whittom, A. A., Austin, M. C., Rajkowska, G., and Stockmeier, C. A. (2010). Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J. Affect. Disord. 127, 230–240. doi: 10.1016/j.jad.2010.06.003
Moi, P., Chan, K., Asunis, I., Cao, A., and Kan, Y. W. (1994). Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 91, 9926–9930. doi: 10.1073/pnas.91.21.9926
Müller, A. C., Dairam, A., Limson, J. L., and Daya, S. (2007). Mechanisms by which acyclovir reduces the oxidative neurotoxicity and biosynthesis of quinolinic acid. Life Sci. 80, 918–925. doi: 10.1016/j.lfs.2006.11.031
Müller, M. B., Lucassen, P. J., Yassouridis, A., Hoogendijk, W. J. G., Holsboer, F., and Swaab, D. F. (2001). Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur. J. Neurosci. 14, 1603–1612. doi: 10.1046/j.0953-816X.2001.01784.x
Myint, A. M., Schwarz, M. J., and Müller, N. (2012). The role of the kynurenine metabolism in major depression. J. Neural Transm. 119, 245–251. doi: 10.1007/s00702-011-0741-3
Niture, S. K., Khatri, R., and Jaiswal, A. K. (2014). Regulation of Nrf2–an update. Free Radic. Biol. Med. 66, 36–44. doi: 10.1016/j.freeradbiomed.2013.02.008
Ogyu, K., Kubo, K., Noda, Y., Iwata, Y., Tsugawa, S., Omura, Y., et al. (2018). Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. doi: 10.1016/j.neubiorev.2018.03.023
Orlando, L. R., Alsdorf, S. A., Penney, J. B., and Young, A. B. (2001). The role of group I and group II metabotropic glutamate receptors in modulation of striatal NMDA and quinolinic acid toxicity. Exp. Neurol. 167, 196–204. doi: 10.1006/exnr.2000.7542
Pérez-De La Cruz, V., Elinos-Calderón, D., Carrillo-Mora, P., Silva-Adaya, D., Konigsberg, M., Morán, J., et al. (2010). Time-course correlation of early toxic events in three models of striatal damage: modulation by proteases inhibition. Neurochem. Int. 56, 834–842. doi: 10.1016/j.neuint.2010.03.008
Raison, C. L., Dantzer, R., Kelley, K. W., Lawson, M. A., Woolwine, B. J., Vogt, G., et al. (2010). CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-α: relationship to CNS immune responses and depression. Mol. Psychiatry 15, 393–403. doi: 10.1038/mp.2009.116
Ramsey, C. P., Glass, C. A., Montgomery, M. B., Lindl, K. A., Ritson, G. P., Chia, L. A., et al. (2007). Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 66, 75–85. doi: 10.1097/nen.0b013e31802d6da9
Réus, G. Z., Jansen, K., Titus, S., Carvalho, A. F., Gabbay, V., and Quevedo, J. (2015). Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: evidences from animal and human studies. J. Psychiatr. Res. 68, 316–328. doi: 10.1016/j.jpsychires.2015.05.007
Rodríguez-Martínez, E., Camacho, A., Maldonado, P. D., Pedraza-Chaverrí, J., Santamaría, D., Galván-Arzate, S., et al. (2000). Effect of quinolinic acid on endogenous antioxidants in rat corpus striatum. Brain Res. 858, 436–439. doi: 10.1016/S0006-8993(99)02474-9
Rojo, A. I., De Sagarra, M. R., and Cuadrado, A. (2008). GSK-3β down-regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 105, 192–202. doi: 10.1111/j.1471-4159.2007.05124.x
Rossato, J. I., Ketzer, L. A., Centurião, F. B., Silva, S. J. N., Lüdtke, D. S., Zeni, G., et al. (2002). Antioxidant properties of new chalcogenides against lipid peroxidation in rat brain. Neurochem. Res. 27, 297–303. doi: 10.1023/A:1014907228580
Rossi, F., Schwarcz, R., and Rizzi, M. (2008). Curiosity to kill the KAT (kynurenine aminotransferase): structural insights into brain kynurenic acid synthesis. Curr. Opin. Struct. Biol. 18, 748–755. doi: 10.1016/j.sbi.2008.09.009
Ruiz, N. A. L., Del Angel, D. S., Olguin, H. J., and Silva, M. L. (2018). Neuroprogression: the hidden mechanism of depression. Neuropsychiatr. Dis. Treat. 14, 2837–2845. doi: 10.2147/NDT.S177973
Santamaría, D., Espinoza-González, V., Ríos, C., and Santamaría, A. (1999). Nomega-nitro-L-arginine, a nitric oxide synthase inhibitor, antagonizes quinolinic acid-induced neurotoxicity and oxidative stress in rat striatal slices. Neurochem. Res. 24, 843–848. doi: 10.1023/A:1020949812581
Sapko, M. T., Guidetti, P., Yu, P., Tagle, D. A., Pellicciari, R., and Schwarcz, R. (2006). Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: implications for Huntington’s disease. Exp. Neurol. 197, 31–40. doi: 10.1016/j.expneurol.2005.07.004
Savitz, J. (2017). “Role of kynurenine metabolism pathway activation in major depressive disorders” in Current topics in behavioral neurosciences, 249–268. doi: 10.1007/7854_2016_12
Savitz, J., Dantzer, R., Wurfel, B. E., Victor, T. A., Ford, B. N., Bodurka, J., et al. (2015). Neuroprotective kynurenine metabolite indices are abnormally reduced and positively associated with hippocampal and amygdalar volume in bipolar disorder. Psychoneuroendocrinology 52, 200–211. doi: 10.1016/j.psyneuen.2014.11.015
Savitz, J., Drevets, W. C., Wurfel, B. E., Ford, B. N., Bellgowan, P. S. F., Victor, T. A., et al. (2015). Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav. Immun. 46, 55–59. doi: 10.1016/j.bbi.2015.02.007
Schwarcz, R., Bruno, J. P., Muchowski, P. J., and Wu, H. Q. (2012). Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477. doi: 10.1038/nrn3257
Setiawan, E., Wilson, A. A., Mizrahi, R., Rusjan, P. M., Miler, L., Rajkowska, G., et al. (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72, 268–275. doi: 10.1001/jamapsychiatry.2014.2427
Shih, A. Y., Imbeault, S., Barakauskas, V., Erb, H., Jiang, L., Li, P., et al. (2005). Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J. Biol. Chem. 280, 22925–22936. doi: 10.1074/jbc.M414635200
Steiner, J., Bielau, H., Brisch, R., Danos, P., Ullrich, O., Mawrin, C., et al. (2008). Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J. Psychiatr. Res. 42, 151–157. doi: 10.1016/j.jpsychires.2006.10.013
Steiner, J., Walter, M., Gos, T., Guillemin, G. J., Bernstein, H. G., Sarnyai, Z., et al. (2011). Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflammation 8, 1–9. doi: 10.1186/1742-2094-8-94
Stipek, S., Stastny, F., Platenik, J., Crkovska, J., and Zima, T. (1997). The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem. Int. 30, 233–237. doi: 10.1016/S0197-0186(97)90002-4
Tasset, I., Pérez-De La Cruz, V., Elinos-Calderón, D., Carrillo-Mora, P., González-Herrera, I. G., Luna-López, A., et al. (2010). Protective effect of tert-butylhydroquinone on the quinolinic-acid-induced toxicity in rat striatal slices: role of the Nrf2-antioxidant response element pathway. Neurosignals 18, 24–31. doi: 10.1159/000243650
Thomas, A. J., Davis, S., Morris, C., Jackson, E., Harrison, R., and O’Brien, J. T. (2005). Increase in interleukin-1beta in late-life depression. Am. J. Psychiatry 162, 175–177. doi: 10.1176/appi.ajp.162.1.175
Vargas, M. R., Burton, N. C., Gan, L., Johnson, D. A., Schäfer, M., Werner, S., et al. (2013). Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS-linked mutant hSOD1 mouse models. PLoS One 8. doi: 10.1371/journal.pone.0056625
Vargas, M. R., Johnson, D. A., Sirkis, D. W., Messing, A., and Johnson, J. A. (2008). Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 28, 13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008
Vaváková, M., Ďuračková, Z., and Trebatická, J. (2015). Markers of oxidative stress and neuroprogression in depression disorder. Oxidative Med. Cell. Longev. 2015, 1–12. doi: 10.1155/2015/898393
WHO (2018). Depression. Available at: http://www.who.int/news-room/fact-sheets/detail/depression [Accessed March 8, 2019].
Yan, E. B., Frugier, T., Lim, C. K., Heng, B., Sundaram, G., Tan, M., et al. (2015). Activation of the kynurenine pathway and increased production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J. Neuroinflammation 12, 1–17. doi: 10.1186/s12974-015-0328-2
Yao, W., Zhang, J. C., Ishima, T., Dong, C., Yang, C., Ren, Q., et al. (2016). Role of Keap1-Nrf2 signaling in depression and dietary intake of glucoraphanin confers stress resilience in mice. Sci. Rep. 6, 1–13. doi: 10.1038/srep30659
Keywords: oxidative stress, depression, serotonin, quinolinic acid, tryptophan, Nrf2
Citation: Bansal Y, Singh R, Parhar I, Kuhad A and Soga T (2019) Quinolinic Acid and Nuclear Factor Erythroid 2-Related Factor 2 in Depression: Role in Neuroprogression. Front. Pharmacol. 10:452. doi: 10.3389/fphar.2019.00452
Edited by:
Javier Egea, Institute of Health Research of the University Hospital of La Princesa, SpainReviewed by:
Jessica Roos, Universitätsklinikum Frankfurt, GermanySatish Ramalingam, SRM Institute of Science and Technology, India
Copyright © 2019 Bansal, Singh, Parhar, Kuhad and Soga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoko Soga, dG9tb2tvLnNvZ2FAbW9uYXNoLmVkdQ==