 Robert M. Rapoport
Robert M. Rapoport Manoocher Soleimani
Manoocher Soleimani- 1Department of Pharmacology and Systems Physiology, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 2Research Service, Veterans Affairs Medical Center, Cincinnati, OH, United States
- 3Department of Medicine, University of Cincinnati College of Medicine, Cincinnati, OH, United States
Thiazide diuretic (TZD)-mediated chronic reduction of arterial pressure is thought to occur through decreased total peripheral vascular resistance. Further, the decreased peripheral vascular resistance is accomplished through TZD activation of an extrarenal target, resulting in inhibition of vascular constriction. However, despite greater than five decades of investigation, little progress has been made into the identification of the TZD extrarenal target. Proposed mechanisms range from direct inhibition of constrictor and activation of relaxant signaling pathways in the vascular smooth muscle to indirect inhibition through decreased neurogenic and hormonal regulatory pathways. Surprisingly, particularly in view of this lack of progress, comprehensive reviews of the subject are absent. Moreover, even though it is well recognized that 1) several types of hypertension are insensitive to TZD reduction of arterial pressure and, further, TZD fail to reduce arterial pressure in normotensive subjects and animals, and 2) different mechanisms underlie acute and chronic TZD, findings derived from these models and parameters remain largely undifferentiated. This review 1) comprehensively describes findings associated with TZD reduction of arterial pressure; 2) differentiates between observations in TZD-sensitive and TZD-insensitive hypertension, normotensive subjects/animals, and acute and chronic effects of TZD; 3) critically evaluates proposed TZD extrarenal targets; 4) proposes guiding parameters for relevant investigations into extrarenal TZD target identification; and 5) proposes a working model for TZD chronic reduction of arterial pressure through vascular dilation.
Introduction
Thiazide diuretics (TZD) are one of the most widely prescribed therapeutic agents for treatment of hypertension and are also used in the treatment of heart failure and stroke (Olde Engberink et al., 2015). However, the mechanism underlying chronic reduction of arterial pressure to TZD remains unclear despite investigations over greater than five decades. It is widely agreed that decreased total peripheral vascular resistance underlies TZD chronic pressure reduction (Table 1a). Moreover, an extrarenal target for TZD action has been proposed (Table 1a). However, there is little consensus on the extrarenal TZD target responsible for the chronic reduction of arterial pressure. Proposals range widely from indirect mechanisms, in which TZD inhibit regulatory sites upstream from the vasculature, to TZD direct inhibition of vascular constriction (Table 1a).

Table 1 Text citations1.
Likely contributing to the wide range of proposed TZD targets for chronic reduction of arterial pressure and, furthermore, largely overlooked, are the use of the following:
1) Hypertensive models that are sensitive and insensitive to TZD arterial pressure reduction. In this regard, TZD reduce arterial pressure in some hypertensive patients, while TZD lack efficacy in others, i.e., “responders” and “nonresponders,” respectively (Table 1b). Indeed, as great as 20% of hypertensive patients are resistant to TZD arterial pressure reduction (Dudenbostel et al., 2017). Similarly, models of hypertension can be sensitive (1 kidney/DOCA-salt, 1 kidney/DOCA, DOCA-salt, angiotensin II-salt, spontaneous hypertensive rats, dietary salt in rats, NO synthase inhibitor, cyp1a1-Ren2 hypertensive rats, capsaicin-high salt, and low renin hypertension) or insensitive (renal, e.g., following renal artery constriction; neurogenic/sympathetic nervous system, e.g., following aortic depressor plus sinus nerve ligation and vagi-aortic nerve ligation, increased dietary salt in mice, angiotensin II, and hyperaldosteronism) to TZD arterial pressure reduction (Table 1c). Indeed, overall predictors of greater responsiveness to TZD include lower levels of plasma renin and urine aldosterone (Chapman et al., 2002). Clearly, the efficacy of TZD and other antihypertensive agents to reduce arterial pressure depends upon the target sites in the different types of hypertension (Gong et al., 2012).
2) Normotensive subjects and animals. TZD fail to reduce arterial pressure in normotensive subjects/animals (Table 1d; Figure 1).
3) Acute TZD challenge. Contrasting mechanisms underlie TZD acute and chronic reduction of arterial pressure, with the former renal mediated, caused by diuresis and accompanying decreased plasma volume, and the latter in which arterial pressure reduction and plasma volume depletion are dissociated (Table 1a; Figure 1).
4) Supra-therapeutic dose/concentration of TZD. At therapeutic dose, TZD selectively inhibit the Na+/Cl− cotransporter (NCC; SLC12A3; Sinning et al., 2017). NCC, which is expressed on the apical membrane of distal convoluted tubule cells, is the renal target responsible for TZD diuresis (Table 1e). However, at supra-therapeutic dose, TZD also inhibit the a) Na+-dependent chloride–bicarbonate exchanger (NDCBE; SLC4A8; Table 1f). NDCBE, which is located on the basolateral membrane of medullary collecting duct cells, regulates cell pH (Xu et al., 2018) and b) carbonic anhydrase isozymes (Swenson, 2014). Carbonic anhydrase isoenzymes are located in the renal proximal tubule and intercalated cells, where they mediate bicarbonate transport, as well as extrarenally (Swenson, 2014). Also, a number of in vivo studies on the effects of TZD on arterial blood pressure and a large majority of in vitro studies of the effects of TZD on the vasculature generally utilized supra-therapeutic TZD concentrations [(Na+/Cl−Cotransporter (NCC; SLC1283), In Vivo TZD on Vascular Contractility Determined In Vitro].
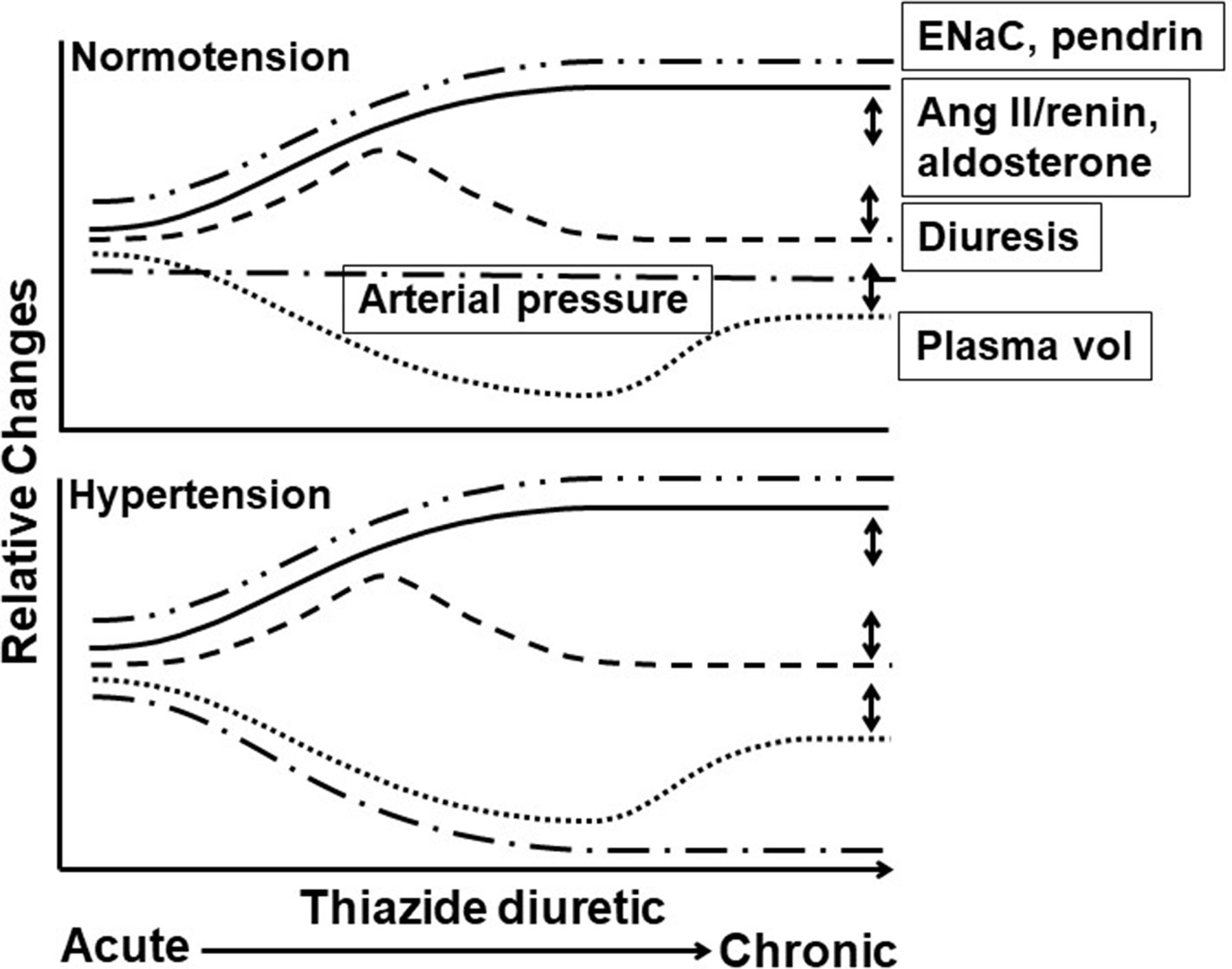
Figure 1 Time course of thiazide diuretic effects on arterial pressure, plasma volume, diuresis, and levels of angiotensin II/renin, ENaC, and pendrin in hypertension in responders and normotension. Acute through chronic time course of thiazide diuretic (TZD) effects in normotension (top panel) and hypertension (bottom panel) on relative changes in arterial pressure (__.__), plasma volume (…), renin activity/angiotenisn II and aldosterone plasma levels (_____), diuresis (----), and epithelial sodium channel (ENaC) and pendrin expression (__..). Features of the time course include (1) plasma volume: decreased with acute TZD and partial return to pre-TZD level with chronic TZD, with similar magnitude and time course in normotension and hypertension; (2) arterial pressure: reduced with acute and chronic TZD in hypertension and unaffected in normotension despite similar changes in plasma volume and diuresis; (3) renin activity/angiotensin II and aldosterone plasma levels: increased with acute and chronic TZD. Angiotensin II/renin and aldosterone levels in normotension with TZD have not been measured (to our knowledge) and, therefore, are speculated based upon the decreased plasma volume; and (4) ENaC and pendrin expression: increased with acute and chronic TZD, with similar magnitude and time course in normotension and hypertension. Increased ENaC and pendrin expression likely compensate for the Double-headed arrows indicate variable effects. TZD-induced diuresis mediated through NCC inhibition. Only single time points for ENaC and pendrin have been reported, and thus, the time course is speculative. Double-headed arrows indicate variable effects. See text for additional details.
This review 1) comprehensively describes findings related to TZD reduction of arterial pressure; 2) differentiates between observations derived from TZD-sensitive and TZD-insensitive hypertensive models, normotensive subjects/animals, acute and chronic TZD, and TZD dose/concentration; 3) proposes guiding parameters for clinically relevant, extrarenal TZD target identification; 4) critically evaluates proposed TZD extrarenal targets; and 5) proposes a working model for TZD chronic reduction of arterial pressure through dilation of the vasculature.
(1) Dose/Concentration of TZD
The use of supra-therapeutic doses/concentrations in in vivo and ex vivo studies undoubtedly contributes to the lack of clarity regarding the mechanism of TZD chronic reduction of arterial pressure. In order to clarify this potential impact, it is important to initially establish the values which constitute supra-therapeutic TZD doses/concentrations.
Therapeutic TZD Doses/Concentrations
TZD doses exceeded those required for maximal efficacy in early years of TZD use (Tamargo et al., 2014). Subsequently, TZD doses were substantially decreased in order to lower the incidence of side effects, with the overwhelming majority of TZD doses established at less than p.o. 50 mg/day (approximately 0.65 mg/kg/day; Musini et al., 2014; Tamargo et al., 2014; Vongpatanasin, 2014).
Bioavailability of TZD varies from 65% to 95% (Tamargo et al., 2014). Treatment of hypertension with p.o. hydrochlorothiazide 7.5–25 mg/day (approximately 0.1–0.3 mg/kg/day) resulted in a median plasma concentration of 0.26 µM (Sigaroudi et al., 2018). Consistent with the plasma hydrochlorothiazide level derived at these doses (Sigaroudi et al., 2018), treatment of hypertension and congestive heart failure with p.o. 75 mg/day (approximately 1 mg/kg/day) and 50 mg/day (approximately 0.7 mg/kg/day), respectively, yielded peak blood levels of approximately 1–3 µM (Beermann and Groschinsky‐Grind, 1977; Beermann and Groschinsky‐Grind, 1978; Beermann and Groschinsky‐Grind, 1979).
(2) Diuresis and Plasma Volume
Fundamental to TZD reduction of arterial pressure and, thus, elucidation of the TZD extrarenal target, is increased diuresis and resultant decreased plasma volume. Characteristics of TZD-induced diuresis and decreased plasma volume include differentiation into acute and chronic phases and associated underlying mechanisms.
Responders and TZD-Sensitive Hypertension Models
Acute TZD reduction of arterial pressure correlates with diuresis and the associated decreased plasma volume in hypertensive humans and animals (Table 1g; Figure 1). Exceptions to this correlation are TZD pressure reduction prior to decreased plasma volume (Gifford et al., 1961; Greene et al., 1961; Lockett and Nicholas, 1968).
Plasma volume repletion reversed acute TZD reduction of arterial pressure, consistent with the correlation between acute arterial pressure reduction and diuresis (Figure 1). Specifically, after TZD treatment for 6 days, dextran infusion increased arterial pressure as well as the cardiac index and reduced total peripheral vascular resistance in humans (Dustan et al., 1959). However, after TZD treatment for 7 days, in which the plasma volume decrease was maintained at the initial reduced level, plasma and saline infusion did not alter arterial pressure (Hollander et al., 1960). Also, in barbiturate-anesthetized 1 kidney/DOCA hypertensive rats challenged with TZD for 5–8 days, hypotonic saline and dextrose infusion did not reverse the reduced arterial pressure, although hypertonic saline infusion increased systolic pressure, albeit not diastolic pressure (Daniel, 1962).
In contrast to acute TZD, chronic TZD reduction of arterial pressure appears dissociated from diuresis and decreased plasma volume (Figure 1). With respect to diuresis, TZD chronic reduction in arterial pressure is maintained while diuresis is minimized in hypertensive humans (Table 1h). TZD also did not alter serum Na+ (Table 1h). Additionally, water and Na+ balance returned to normal during TZD challenge of hypertensive high Na+ diet/angiotensin II rats and spontaneously hypertensive rats, while reduced arterial pressure was maintained (Ballew and Fink, 2001; Jessup et al., 2008). Also consistent with the dissociation between diuresis and chronic TZD reduction of arterial pressure is the lesser diuretic efficacy of TZD than inhibitors of the luminal Na–K–Cl cotransporter (NKCC2) located in the thick ascending limb of Henle (loop diuretics), i.e., the NCC and NKCC2 regulate approximately 7% and 25% of Na+ absorption, respectively (Glover and O’Shaughnessy, 2013), while the antihypertensive efficacy of TZD was greater than that of NKCC2 inhibitors (Anderson et al., 1971; Araoye et al., 1978; Holland et al., 1979). Contributing to the minimal diuresis is increased expression of epithelial Na+ channel (ENaC) and Na+ anion exchanger, pendrin (Na et al., 2003; Alshahrani and Soleimani, 2017; Figure 1).
With respect to plasma volume, TZD-decreased plasma volume apparently returned to pre-TZD level despite continuous TZD for 1–12 months in hypertensive humans (Table 1i; Figure 1). Whether, in fact, plasma volume completely or only partially returned to pre-TZD level is unclear because a number of these studies demonstrated a tendency for reduced plasma volume. Moreover, the magnitude of plasma volume return to pre-TZD level may be overestimated by increased hemoglobin (Hilden et al., 1968; Leth, 1970). Along these lines, the TZD lowered plasma volume only partially returned at 2–3 months of TZD treatment (Winer, 1961; Hansen, 1968), and as determined at a single time point during TZD treatment, plasma volume was decreased at 12 and 24 months of TZD (Lund-Johansen, 1970; Tarazi et al., 1970; de Carvalho et al., 1977).
Also consistent with the only partial return of plasma volume with chronic TZD is the return of TZD-reduced arterial pressure following plasma volume expansion with dextran plus saline or glucose infusion at 2 weeks to 8 months of TZD treatment in hypertensive humans (Wilson and Freis, 1959). In partial agreement, following 2 weeks to 9 months of TZD treatment, dextran plus glucose infusion resulted in partial return of systolic pressure while the decreased diastolic pressure remained (Winer, 1961).
Additionally, plasma volume repletion with elevated dietary Na+ reversed TZD chronic reduction of arterial pressure in hypertensive humans (Wilkins et al., 1958). At 6 days post single TZD injection (i.v. 1 g of chlorothiazide), increased dietary Na+ of 140 mEq/day (1.8 g/day) from previously lowered 9 mEq/day (0.2 g/day), completely reversed the sustained reduction of arterial pressure to pre-TZD level (Wilkins et al., 1958). In partial agreement, greater amounts of dietary Na+ salt, i.e., 6–12 g/day (2.4–4.8 g/day Na+) only partially reversed TZD-mediated arterial pressure reduction, and complete reversal required 20 g/day (8 g/day Na+; Freis et al., 1958; Wilkins et al., 1958; Freis, 1959; Winer, 1961). Also, following withdrawal of TZD, plasma volume increases (Wilson and Freis, 1959; Tarazi et al., 1970).
Apparently inconsistent with the correlation between elevated Na+ and reversal of TZD-reduced arterial pressure, TZD reduction of arterial pressure remained despite prevention of Na+ loss by mineralocorticoid co-administration (Hollander et al., 1960). However, it is likely that TZD-decreased plasma volume was actually maintained because weight loss, an indirect measure of plasma volume, also remained following mineralocorticoid co-administration (Hollander et al., 1960).
Decreased Na+ excretion as the result of Na+-restricted diet (urinary excretion from 3.75 g Na+/day to 2 g Na+/day) which was associated with reduced arterial pressure and increased renin activity and aldosterone plasma level, also failed to prevent further arterial pressure reduction by TZD in the absence or presence of angiotensin receptor antagonists in humans (Priddle et al., 1970; Vogt et al., 2008; Wang et al., 2015b). The TZD arterial pressure reduction was still associated with plasma volume contraction because Na+-restricted diet did not prevent TZD-increased Na+ excretion (urine volume not reported) and futher increased plasma renin activity and aldosterone levels (Priddle et al., 1970; Vogt et al., 2008).
Summary and Conclusions: Responders and TZD-Sensitive Hypertension Models
Among the findings in support of an extrarenal, vascular mechanism mediating TZD chronic reduction of arterial pressure in hypertensive humans and animals is the maintained pressure reduction despite return of plasma volume towards pre-TZD levels (Figure 1). Overall, however, it appears that a remaining component of TZD acute decreased plasma volume is required for chronic TZD reduction in arterial pressure (Figures 1, 2). Thus, TZD chronic reduction of arterial pressure is dependent upon decreased plasma volume both as a trigger for the subsequent maintained arterial pressure reduction and for its ability to sustain the reduction (Figure 2).
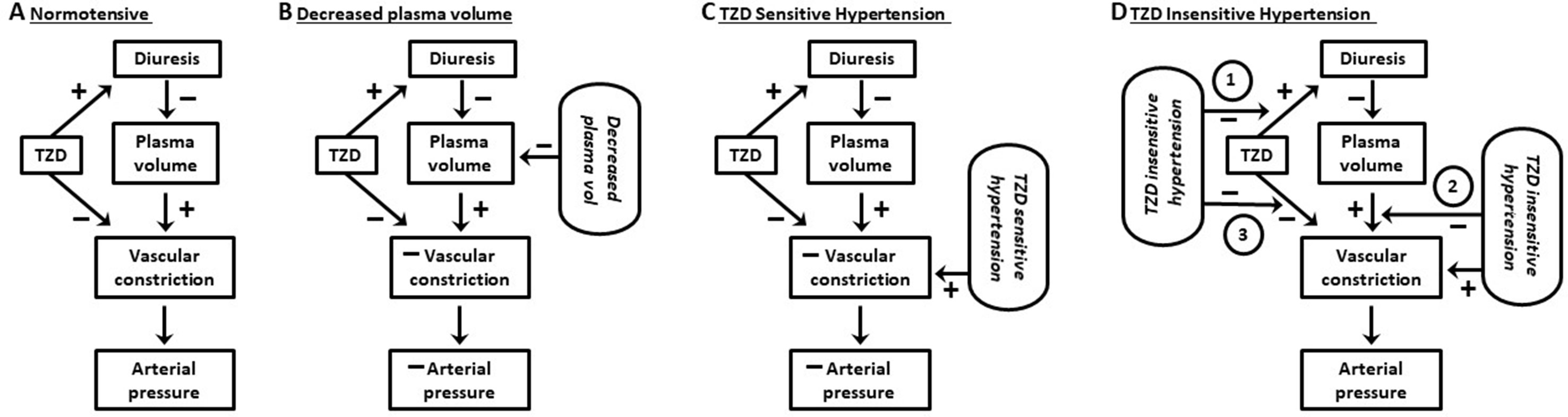
Figure 2 Working model of effects of thiazide diuretics on vascular constriction and arterial pressure.(A) Normotensive subjects: thiazide diuretics (TZD) induce diuresis but fail to reduce arterial pressure. The lack of pressure reduction results from a balance between vasoconstriction due to (1) signaling pathways activated in response to TZD decrease in plasma volume and (2) TZD direct inhibition of the vasoconstriction. (B) Normotensive subjects and decreased plasma Na+: vasoconstriction due to activation of compensatory regulatory pathways in response to decreased plasma Na+ by Na+-restricted diet plus TZD-induced diuresis is unable to mitigate the reduction in arterial pressure caused by TZD direct inhibition of the constriction. (C) TZD-sensitive hypertension: TZD reduce arterial pressure due to TZD direct inhibition of vasoconstriction overcoming constriction due to both plasma volume contraction (in response to decreased plasma volume) and hypertension. (D) TZD-insensitive hypertension: TZD fail to reduce arterial pressure due to inhibition of diuresis, plasma volume contraction, and/or prevention of TZD inhibition of vascular constriction (circled 1, 2, and 3, respectively). See text for additional details.
Normotensive Humans and Animals
Acute and chronic TZD failed to reduce arterial pressure in normotensive humans and animals despite the diuresis and decreased plasma volume (Table 1j; Figure 1). A supra-therapeutic TZD dose was required to reduce arterial pressure in dogs (Preziosi et al., 1959). Lack of TZD reduction of arterial pressure was not due to decreased diuresis because TZD caused similar magnitudes of changes in Na+ and K+ per body weight, as well as extracellular fluid volume and serum osmolarity, in normotensive subjects and hypertensive patients (Freis et al., 1960; Hollander et al., 1960; Figure 1). Diuresis due to NKCC2 inhibitors was also greater than diuresis due to TZD in normotensive subjects, while NKCC2 inhibitors still failed to reduce arterial pressure (Joubert et al., 1968).
In apparent contrast to the lack of TZD reduction of arterial pressure in normotensive humans and animals (Table 1j; Figure 1), in hypertensive humans. TZD and Na+-restricted diet elicited a steady reduction of arterial pressure (Priddle et al., 1970). Na+-restricted diet did not prevent TZD diuresis and natriuresis in rats (Hofmann and Sagartz, 1970) and, thus, were presumably also not prevented in humans.
Although TZD did not cause diuresis in normotensive mice, the absence of diuresis may actually reflect the relatively delayed assessment, i.e., day 3–4 post initial TZD (Alshahrani and Soleimani, 2017). This suggestion is supported by the return to baseline within 24 and 72 h of increased diuresis due to p.o. TZD q.d. (Fuchs et al., 1960; Claus-Walker et al., 1977) and 48 h p.o. TZD b.i.d. (Wilson and Freis, 1959). Also, ENaC and pendrin expression, which presumably compensates for TZD diuresis through increased Na+ reabsorption, increased on day 3 post initial TZD treatment (earliest time point assayed; Alshahrani and Soleimani, 2017; Figure 1). Along these lines, diuresis increased 2 h post a single i.v. injection of TZD in rats and ENaC activity remained unchanged (Frindt et al., 2017).
Summary and Conclusions: Normotensive Humans and Animals
TZD failed to reduce arterial pressure in normotensive humans and animals despite similar magnitudes of diuresis and plasma volume decrease as in hypertensive humans and animals (Figures 1, 2). The lack of TZD reduction of arterial pressure suggests a balance between compensatory contraction of the plasma volume and TZD mitigation of the contracted volume through inhibition of vasoconstriction (Figure 2). However, this balance is disrupted by further plasma volume contraction, which can occur, e.g., with increased Na+ loss, resulting in TZD reduction of arterial pressure (Figure 2).
Responders versus Nonresponders
TZD-mediated increased diuresis and plasma volume reduction, and body weight loss were greater in responders than nonresponders (de Carvalho et al., 1977; Freis et al., 1988; Table 2). Also, TZD failed to decrease plasma volume in a study limited to nonresponders (Svendsen et al., 1983). The greater contracted plasma volume of responders was suggested to underlie increased TZD reduction of total peripheral vascular resistance (de Carvalho et al., 1977). Consistent with the lesser magnitude of plasma volume contraction in nonresponders is the lack of angiotensin II and aldosterone plasma level elevation (Svendsen et al., 1983; Constrictor Pathways In Vivo, Endogenous Activation; Table 2). The lesser decrease in plasma volume in nonresponders than responders was not associated with lesser decreases in serum K+ (de Carvalho et al., 1977; Svendsen et al., 1983).
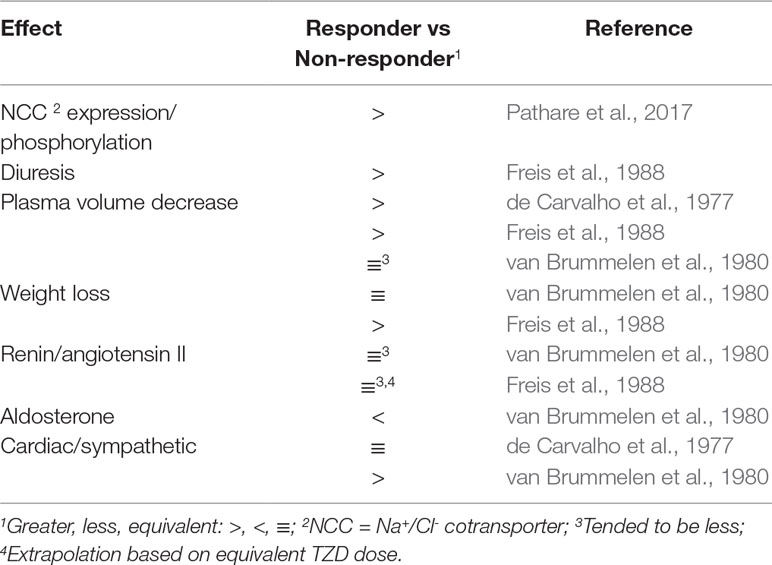
Table 2 Effects of Thiazide Diuretics in Responders and Nonresponders on Physiological and Biochemical Parameters in Hypertension.
In contrast, there was a tendency for increased plasma volume contraction in chronic TZD-treated nonresponders than responders (van Brummelen et al., 1980; Table 2). It was suggested that the tendency for greater plasma volume contraction in nonresponders, which was associated with a tendency for greater elevated plasma levels of angiotensin II (Constrictor Pathways In Vivo, Endogenous Activation; Table 2), resulted in increased peripheral vascular resistance (van Brummelen et al., 1980) and, thus, diminished TZD-decreased arterial pressure. On the other hand, body weight was not different in responders and nonresponders (van Brummelen et al., 1980; Table 1).
Summary and Conclusions: Responders versus Nonresponders
The dependency of TZD reduction of arterial pressure upon relaxation of vasoconstriction suggests that the inability of TZD to reduce arterial pressure in nonresponders may reflect the lack of compensatory vasoconstriction (Table 2; Figure 2). That is, the inability of TZD to elicit diuresis results in lack of decreased plasma volume and, therefore, absence of compensatory vasoconstriction (Table 2; Figure 2). This conclusion is supported by the lack of TZD arterial pressure reduction in normotensive humans and animals despite increased diuresis (above). Additionally, lack of diuresis in some types of hypertension associated with nonresponders could result from prevention of diuresis (Table 2; Figure 2).
Alternatively, TZD may induce diuresis in TZD-insensitive hypertension with different etiologies. Thus, the absence of arterial pressure reduction could result from inhibitory effects post plasma volume depletion (Figure 2). Additional studies of the effects of TZD in nonresponders with different hypertensive etiologies are warranted, including the mechanism underlying TZD reduction of arterial pressure in nonresponders with co-administration of other antihypertensive agents (Sander and Giles, 2011; Tamargo et al., 2014).
(3) Na+/Cl− Cotransporter (NCC; SLC12A3)
Insight into the mechanism underlying TZD chronic reduction of arterial pressure may be derived 1) through the effects of TZD on the activity of NCC, the renal target for TZD diuresis (Table 1e), and the relationship between NCC activity and arterial pressure in responders and nonresponders; 2) in NCC knockout and NCC/pendrin double-knockout mice (Schultheis et al., 1998; Loffing et al., 2004; Soleimani et al., 2012; Alshahrani et al., 2017a); and 3) in humans with dysfunctional NCC, i.e., Gitelman’s syndrome (Table 1l);.
NCC Activity Determinations
NCC activity determinations include increased NCC expression and phosphorylation, with the latter an indicator of NCC trafficking (Glover and O’Shaughnessy, 2013; Pathare et al., 2017). These two measurements may be unrelated because phosphorylation can be dissociated from increased NCC expression (Frindt et al., 2017). TZD binding to NCC, determined by high-affinity binding with 3H-metolazone (Beaumont et al., 1988), has also been used as an indirect measure of NCC expression and, thus, activity (Chen et al., 1990; Morsing et al., 1991). However, even though TZD increased NCC binding, transport was reduced (Morsing et al., 1991). Additional potential complications with binding studies include the inability to account for NCC isoforms along with selective isoform phosphorylation (Tutakhel et al., 2016; Pathare et al., 2017), as well as the influence of Na+ and Cl− concentrations (Beaumont et al., 1988; Tran et al., 1990).
Responders
In responders, chronic TZD increased NCC expression and NCC phosphorylation, as assayed in urinary vesicular exosomes (Pathare et al., 2017).
Normotensive Animals
Acute and chronic TZD increased NCC expression and phosphorylation in whole kidney and, in some studies, distal convoluted tubules from normotensive rats and mice (Northern and Western blot, in situ hybridization, and immunohistochemistry; Loffing et al., 1996; Na et al., 2003; Nijenhuis et al., 2005; Frindt et al., 2017). TZD also increased 3H-metolazone high-affinity binding to NCC in kidney membrane fractions from normotensive rats (Chen et al., 1990; Morsing et al., 1991).
Enhanced delivery of Na+ to the distal collecting duct likely underlies the increased NCC expression (Pathare et al., 2017) because furosemide, a diuretic that acts through inhibition of NKCC2, also increased NCC expression and binding in rat kidney (Chen et al., 1990; Na et al., 2003). Also, angiotensin receptor antagonists failed to increase NCC expression and phosphorylation even though arterial pressure was reduced to a similar magnitude as TZD (Pathare et al., 2017).
Responders versus Nonresponders
TZD-induced NCC expression and phosphorylation were greater in responders than nonresponders, as determined in urinary vesicular exosomes (Pathare et al., 2017; Table 1).
Summary and Conclusions: Responders, Normotensive Animals, and Responders versus Nonresponders
Increased NCC expression and phosphorylation due to TZD are consistent with the greater amount of diuresis in responders than nonresponders (Freis et al., 1988; Table 2; Figure 2; Diuresis and Plasma Volume). In normotensive animals, there is also a positive relationship between TZD-mediated NCC expression/phosphorylation and diuresis (Diuresis and Plasma Volume). NCC activation is restricted to agents that increase Na+ delivery to the distal collecting duct.
NCC Knockout and NCC/Pendrin Double-Knockout Mice
NCC knockout mice demonstrated changes similar to those observed in Gitelman’s syndrome, in which NCC is dysfunctional (below), including hypomagnesemia, hypocalciuria, and increased renin expression (determined in the kidney; Schultheis et al., 1998; Loffing et al., 2004; Alshahrani et al., 2017a). It should be noted, however, that TZD increased NCC expression and that NCC expression is decreased in Gitelman’s syndrome (Joo et al., 2007; Isobe et al., 2013; Yang et al., 2013; Frindt et al., 2017). This difference in NCC expression with TZD and Gitelman’s syndrome may reflect different mechanisms whereby TZD inhibit NCC activity (Payne, 2012) and of NCC dysfunction in Gitelman’s syndrome (Riveira-Munoz et al., 2007a, Riveira-Munoz et al., 2007b; Wang et al., 2015a).
Plasma bicarbonate levels were also elevated in NCC knockout mice, indicative of underlying, compensated alkalosis (Loffing et al., 2004). Additionally, aldosterone plasma levels were elevated (Loffing et al., 2004), although elevated aldosterone plasma levels were not consistently observed (Schultheis et al., 1998).
Basal arterial pressure: Basal arterial pressure was not significantly decreased in NCC knockout mice, although there was a tendency for decreased arterial pressure (Schultheis et al., 1998). The absence of hypotension in NCC knockout mice was attributed to compensation of mildly decreased intravascular volume (Schultheis et al., 1998; Loffing et al., 2004; Alshahrani et al., 2017a). Lack of hypotension in NCC knockout mice (Schultheis et al., 1998; Loffing et al., 2004; Alshahrani et al., 2017a), but hypotension in 50% of Gitelman’s syndrome patients (Cruz et al., 2001; below), may reflect differences in Na+ handling in mice and humans. This suggestion is supported by the arterial pressure reduction with Na+-restricted diet still containing 77.5% of normal in humans (Priddle et al., 1970), but the absence of arterial pressure reduction by more severe Na+-restricted diet of 0.01% and 0.1% in wild-type mice (Schultheis et al., 1998; Alshahrani et al., 2017a).
In contrast, arterial pressure was lower in NCC knockout mice with Na+-restricted diet (0.01% or 0.1%; Schultheis et al., 1998; Alshahrani et al., 2017a). Thus, it appears that compensatory vasoconstrictor pathways were unable to fully normalize the lower arterial pressure in NCC knockout mice subjected to Na+-restricted diet despite further increased renin expression (Schultheis et al., 1998; Alshahrani et al., 2017a). Similarly, NCC/pendrin double-knockout mice, which demonstrated severe diuresis and natriuresis along with even greater renin expression, was associated with further reduced arterial pressure (Soleimani et al., 2012).
TZD on arterial pressure: Hydrochlorothiazide failed to reduce arterial pressure in NCC knockout mice, despite s.c. administration at the supra-therapeutic dose of 20 mg/kg, i.e., approximately 30-fold greater than the therapeutic dose (Alshahrani et al., 2017a; Dose/Concentration of TZD). In fact, 20 mg/kg of hydrochlorothiazide would result in a plasma concentration of approximately 50 µM (Dose/Concentration of TZD). Lack of hydrochlorothiazide reduced arterial pressure in NCC knockout mice may reflect conditions similar to those associated with normotensive subjects and animals (Figure 2). That is, vasoconstriction available for TZD relaxation is minimal due to the relatively small magnitude of decreased intravascular volume and, thus, minimal compensatory vasoconstriction (Figure 2).
In contrast, hydrochlorothiazide (20 mg/kg) reduced arterial pressure in NCC knockout mice subjected to Na+-restricted diet (Alshahrani et al., 2017a). Moreover, in NCC/pendrin double-knockout mice, hydrochlorothiazide elicited an even greater reduction in arterial pressure than in NCC knockout mice subjected to Na+-restricted diet (Alshahrani et al., 2017a). Thus, the magnitude of hydrochlorothiazide arterial pressure reduction correlated with greater underlying vasoconstriction as the result of increased intravascular volume depletion (Soleimani et al., 2012; Alshahrani et al., 2017a; Figure 2). Consistent with this suggestion is that prevention of intravascular volume depletion with increased dietary Na+ from 1% to 7% abolished both the lowered basal arterial pressure and TZD reduction of arterial pressure in NCC/pendrin double-knockout mice (Alshahrani et al., 2017a).
It was suggested that TZD reduction of arterial pressure in the absence of NCC provided evidence for an extrarenal TZD target (Alshahrani et al., 2017a). Further, hydrochlorothiazide arterial pressure reduction does not appear mediated by decreased plasma volume, due to decreased plasma Na+ (Alshahrani et al., 2017a). This conclusion is based upon the finding that urine production was actually decreased in NCC/pendrin double-knockout mice (Alshahrani et al., 2017a). The decreased urine production was attributed to lowered kidney perfusion with reduced arterial pressure (Alshahrani et al., 2017a). Also contributing to the decreased urine production was lowered water intake (Alshahrani et al., 2017a). Inexplicably, food intake was greatly decreased by hydrochlorothiazide (Alshahrani et al., 2017a).
Measurements of plasma Na+ would further test whether decreased plasma volume through lowered plasma Na+ was involved in hydrochlorothiazide reduction of arterial pressure in NCC knockout mice subjected to Na+-restricted diet and NCC/pendrin double-knockout mice (Alshahrani et al., 2017a). In this regard, hydrochlorothiazide increased Na+ excretion in NCC knockout mice (Leviel et al., 2010; Eladari and Chambrey, 2011). Furthermore, the increased Na+ excretion by hydrochlorothiazide appears mediated through inhibition of the Na+-driven chloride–bicarbonate exchanger (NDCBE; SLC4A8e; Ware et al., 2017; Nadal et al., 2018; Xu et al., 2018) because increased Na+ excretion, at least at 50 mg/kg i.p. hydrochlorothiazide, was absent in NCC/NDCBE double-knockout mice (Leviel et al., 2010; Eladari and Chambrey, 2011; Eladari and Hübner, 2011; Sinning et al., 2017).
It should also be considered that the hydrochlorothiazide dose that reduced arterial pressure in the absence of NCC (Alshahrani et al., 2017a) yields a plasma concentration in the range that inhibits NDCBE. That is, hydrochlorothiazide doses that increased Na+ excretion in NCC knockout mice, i.p. hydrochlorothiazide 4 and 50 mg/kg (Leviel et al., 2010; Eladari and Chambrey, 2011), correspond to plasma concentrations of at least 10 and 100 µM, respectively (Dose/Concentration of TZD). In fact, the hydrochlorothiazide concentration used in the metabolic studies in NCC/pendrin double-knockout mice, 40 mg/kg, would yield a plasma concentration of approximately 100 µM (Alshahrani et al., 2017a; Dose/Concentration of TZD). Further, 100 µM hydrochlorothiazide inhibited NDCBE in perfused renal cortical collecting ducts of mice (Leviel et al., 2010). In comparison, the IC50 for metolazone inhibition of Na+ uptake by NCC was 2 or 0.3 µM (depending upon the report; Sabath et al., 2004; Moreno et al., 2006). Also, essentially complete inhibition of NCC by metolazone occurred at 10 µM (Sabath et al., 2004; Moreno et al., 2006).
On the other hand, 10 mg/kg i.p. hydrochlorothiazide was selective for NCC as demonstrated in STE20/SPS1-related proline/alanine-rich kinase (SPAK)-dead knock-in mice (Glover and O’Shaughnessy, 2013). Specifically, in SPAK-dead knock-in mice, which demonstrated decreased NCC expression and phosphorylation leading to overall decreased NCC activity, hydrochlorothiazide saliuresis was absent (Rafiqi et al., 2010; Glover and O’Shaughnessy, 2013). Although NKCC2 expression and phosphorylation were also decreased in SPAK-dead knock-in mice (Rafiqi et al., 2010; Glover and O’Shaughnessy, 2013), saliuresis to the loop diuretic, furosemide, was not decreased (Rafiqi et al., 2010; Glover and O’Shaughnessy, 2013). However, whether NDCBE function is also decreased in SPAK-dead knock-in mice is not known (to our knowledge).
Although a lowered IC50 for TZD inhibition due to increased NDCBE expression following NCC knockout (Eladari and Chambrey, 2011) could occur, depending upon the presence of NDCBE spare transporters, a lowered IC50 does not appear to occur at least in Gitelman’s syndrome. In support of this conclusion is the decreased Na+ and Cl− excretion with a single therapeutic dose of hydrochlorothiazide (p.o. 50 mg; Colussi et al., 1997; Peng et al., 2017; below).
While NDCBE-mediated TZD reduction of arterial pressure in NCC knockout mice subjected to Na+-restricted diet and NCC/pendrin double-knockout mice (Alshahrani et al., 2017a) may occur through hyponatremia, it should also be considered that hyponatremia may not accompany increased Na+ excretion. However, TZD-induced hyponatremia has been associated with decreased NCC (Channavajjhala et al., 2018). In fact, hyponatremia was observed in approximately one of seven TZD-treated patients (Glover and Clayton, 2012; Burst et al., 2017). Further, in patients with demonstrated hyponatremia treated with hydrochlorothiazide plus amiloride, challenge with 50 mg of hydrochlorothiazide, albeit combined with 5 mg of amiloride, caused hyponatremia (Friedman et al., 1989).
A major caveat, however, with respect to the potential functional role of NDCBE in Na+ handling and, therefore, possible high dose TZD-induced saliuresis and/or natriuresis, is the location of NDCBE within the kidney. Specifically, it was suggested that NDCBE was located in β-intercalated cells of the cortical collecting duct, consistent with saliuersis/natriuresis regulation (Ware et al., 2017; Nadal et al., 2018). In direct contrast, we were unable to locate NDCBE in cortical collecting duct β-intercalated cells (Xu et al., 2018). Moreover, NDCBE was restricted to the basolateral membrane of the medullary collecting duct (Xu et al., 2018), thereby questioning the potential role of NDCBE in Na+ handling.
Summary and Conclusions: NCC Knockout and NCC/Pendrin Double-Knockout Mice
TZD reduction of arterial pressure in NCC knockout mice subjected to Na+-restricted diet, but not NCC knockout mice with normal Na+ diet, and an even greater pressure reduction in NCC/pendrin double-knockout mice (Alshahrani et al., 2017a) are consistent with the dependency of TZD reduction of arterial pressure on decreased plasma volume per se and associated compensatory vasoconstriction (Diuresis and Plasma Volume; Figure 2). Thus, the considerable diuresis associated with less reduction of arterial pressure with NKCC2 inhibitors compared with TZD (Wilson and Freis, 1959; Anderson et al., 1971; Araoye et al., 1978; Holland et al., 1979; Diuresis and Plasma Volume) would reflect unopposed compensatory vasoconstriction with the NKCC2 inhibitors.
However, while TZD-reduced arterial pressure in NCC knockout mice with Na+-restricted diet and an even greater pressure reduction in NCC/pendrin double-knockout mice are suggestive of an extrarenal TZD target, an involvement of hyponatremia due to NDCBE inhibition cannot be completely eliminated because of the use of supra-therapeutic TZD dose (Alshahrani et al., 2017a). In fact, lack of TZD reduction of arterial pressure in Gitelman’s syndrome (below) at least indirectly supports the possibility of NDCBE inhibition in TZD reduction of arterial pressure. Overall, however, considering that NDCBE is apparently located on the basolateral membrane of medullary collecting duct (Xu et al., 2018) rather than in β-intercalated cells of the cortical collecting duct (Ware et al., 2017; Nadal et al., 2018), the function of NDCBE in response to hydrochlorothiazide needs to be revisited.
Gitelman’s Syndrome
Gitelman’s syndrome is an autosomal recessive malady attributed to dysfunctional NCC, largely due to missense mutations (Riveira-Munoz et al., 2007a; Riveira-Munoz et al., 2007b; Wang et al., 2015a). These mutations result in undetectable or reduced NCC amounts in urinary exosomes (Joo et al., 2007; Isobe et al., 2013; Yang et al., 2013). Consistent with NCC dysfunction in Gitelman’s syndrome is the lesser increase in urinary excretion of Na+ and Cl− to hydrochlorothiazide (single, therapeutic dose of p.o. 50 mg; Table 1k; Dose/Concentration of TZD). In contrast, urinary excretion of Na+ and Cl− increased in response to furosemide (loop diuretic; i.v. bolus infusion of 40 mg in Colussi et al., 1997 and i.m. 20 mg in Peng et al., 2016).
The phenotype associated with Gitelman’s syndrome includes hypomagnesemia, hypocalciuria, hypochloremia, and hypokalemic alkalosis (Table 1I; mimicked by NCC knockout mice; Schultheis et al., 1998; Alshahrani et al., 2017a; above). Vascular volume depletion is also associated with Gitelman’s syndrome, which results in elevated plasma renin levels/activity and aldosterone levels (Tsukamoto et al., 1995; Simon et al., 1996). Hypotension occurs in approximately 50% of patients with Gitelman’s syndrome (62% and 12% of patients and control subjects, respectively; Cruz et al., 2001). Based on measurements in neutrophils and monocytes from patients with Gitelman’s syndrome, the hypotension and absence of hypertension despite elevated plasma renin levels/activity and aldosterone levels were attributed to up-regulation/down-regulation of signaling pathways that would result in decreased vasoconstriction and basal vascular tone (Calò et al., 1998, Calò et al., 1999; Calò, 2006). These signaling pathways include decreased inositol trisphosphate release of intracellular Ca2+, decreased activation of protein kinase C, increased nitric oxide formation due to increased endothelial nitric oxide synthase expression, inhibition of oxidation signaling mechanisms (which mediate vasoconstriction to angiotensin II and other constrictors), decreased G-protein coupling through reduced αq expression, decreased agonist activation of Rho kinase through down-regulation of RhoA/Rho kinase signaling, and increased heme-oxygenase-1 expression (Calò et al., 1998, Calò et al., 1999; Calò, 2006).
However, possibly inconsistent with the predicted decreased agonist constriction and/or basal vascular tone in patients with Gitelman’s syndrome (Calò et al., 1998, Calò et al., 1999; Calò, 2006) are 1) autonomic function was preserved in hypotensive, Gitelman’s syndrome patients (Sartori et al., 2007). Autonomic function was determined indirectly by reflex tests including the Valsalva maneuver, cold pressor, hand grip, and hyperventilation and by norepinephrine and arginine vasopressin plasma levels in response to the reflex tests (Sartori et al., 2007). And 2) basal blood flow in the forearm of two hypotensive patients with Gitelman’s syndrome did not differ from basal blood flow in normotensive and hypertensive subjects (Pickkers et al., 1998).
Also, possibly inconsistent with the potential extrarenal TZD target in NCC knockout mice subjected to Na+-restricted diet and in NCC/pendrin double-knockout mice (Alshahrani et al., 2017a), in two patients with Gitelman’s syndrome with 8 and 25 µg hydrochlorothiazide/min/dL forearm infusion (plasma hydrochlorothiazide concentrations of 2.7 and 11.0 µM, respectively), blood flow did not increase (Pickkers et al., 1998). Similarly, while 75 µg hydrochlorothiazide/min/dl infusion (38 µM of hydrochlorothiazide) increased forearm blood flow in Gitelman’s syndrome patients, the magnitude of increase was not different from the increased blood flow in normotensive and hypertensive subjects (Pickkers et al., 1998). The increased blood flow to 75 µg hydrochlorothiazide infusion likely resulted from non-specific effects to the approximately 20-fold greater than therapeutic plasma levels of hydrochlorothiazide (Pickkers et al., 1998; Dose/Concentration of TZD).
Additionally, based upon the acute hydrochlorothiazide reduction of arterial pressure in NCC knockout mice subjected to Na+-restricted diet and in NCC/pendrin double-knockout mice (Alshahrani et al., 2017a; above), TZD may have been predicted to reduce arterial pressure in patients with Gitelman’s syndrome. However, hydrochlorothiazide infusion into the forearm also did not reduce arterial pressure in the two Gitelman’s syndrome patients (as well as the normotensive and hypertensive subjects; Pickkers et al., 1998). The limited duration of hydrochlorothiazide infusion, i.e., 5 min, however, may have been insufficient to determine whether arterial pressure was actually reduced (Pickkers et al., 1998). Arterial pressure was also not reduced following diagnostic challenge with p.o. 50 mg of hydrochlorothiazide in Gitelman’s syndrome patients (L Chen and G Colussi, personal communications), with heart rate also remaining unchanged (G Colussi, personal communication). Lack of hydrochlorothiazide arterial pressure reduction in patients with Gitelman’s syndrome appeared independent of basal arterial pressure, as hydrochlorothiazide failed to reduce arterial pressure in Gitelman’s syndrome patients with low-normal mean arterial blood pressures, ranging at basal from 73 to 96 mmHg (n = 5; G Colussi, personal communication).
On the other hand, plasma volume depletion in patients with Gitelman’s syndrome may be insufficient to demonstrate TZD reduction of arterial pressure, consistent with the inability of TZD to reduce arterial pressure in NCC knockout mice with normal Na+ diet (Alshahrani et al., 2017a). However, significant plasma volume depletion likely occurs in Gitelman’s syndrome based on the elevated plasma renin levels/activity and aldosterone levels (Tsukamoto et al., 1995; Simon et al., 1996). Further, treatment of Gitelman’s syndrome, a condition associated with high Na+ intake, includes ad libitum dietary Na+ (Blanchard et al., 2017). Despite the increased Na+ intake, the intake apparently is insufficient to fully compensate for the decreased plasma volume (Diuresis and Plasma Volume) and, therefore the contribution of the decreased plasma volume to the hypotension.
Summary and Conclusions: Gitelman’s Syndrome
Compensation of contracted plasma volume in Gitelman’s syndrome appears to occur through elevated aldosterone and renin and possibly other vasoconstrictor pathways (Simon et al., 1996). Hypotension associated with Gitelman’s syndrome, along with lack of hypertension despite increased renin activity and aldosterone plasma levels (Simon et al., 1996), suggest both an incomplete compensation and a prevention of increased constriction due to reduced vascular contractility (Calò et al., 1998, Calò et al., 1999; Calò, 2006). However, unaltered autonomic nervous system regulation of arterial blood pressure and absence of changes in basal and TZD-mediated forearm blood flow in patients with Gitelman’s syndrome (Pickkers et al., 1998; Sartori et al., 2007) do not appear to support a mechanism whereby up-regulation or down-regulation of these non-selective vascular inhibitory and excitatory signaling pathways, respectively, underlies hypotension and inhibition of anticipated elevated arterial pressure due to apparent activation of the renin–angiotensin II–aldosterone pathway (Calò et al., 1998, Calò et al., 1999; Calò, 2006).
Thus, it is tempting to conclude that selective inhibition of the renin–angiotensin II–aldosterone pathway underlies the compromised vascular constriction. This conclusion appears consistent with lower plasma renin and urine aldosterone levels as predictors of greater TZD efficacy of arterial pressure reduction (Chapman et al., 2002; Diuresis and Plasma Volume). A further speculation is that alterations in blood electrolytes and pH associated with Gitelman’s syndrome, i.e., hypomagnesemia, hypocalciuria, hypochloremia, and hypokalemic alkalosis (Table 1l), underlie possible compromised renin–angiotensin II–aldosterone pathway. However, it appears that these conditions do not decrease the formation of angiotensin II (Tsukamoto et al., 1995).
The absence of TZD reduction of arterial pressure in Gitelman’s syndrome is not supportive of an extrarenal TZD target suggested by TZD reduction of arterial pressure in NCC knockout mice subjected to Na+-restricted diet and in NCC/pendrin double-knockout mice (Alshahrani et al., 2017a). However, this apparent lack of support could be explained if intravascular volume contraction is less in Gitelman’s syndrome than in knockout mice models (above).
(4) Constrictor Pathways In Vivo
(a) Endogenous Activation
An additional assessment of contracted plasma volume in response to TZD is determination of compensatory constrictor pathways. Moreover, TZD may inhibit the compensatory vasoconstriction, eliciting reduced vascular tone and resulting in only partial compensation of the arterial pressure reduction. Additionally, these same constrictor pathways may be activated in some types of hypertension. Thus, determination of the effects of TZD on these constrictor pathways is fundamental to the identification of the TZD extrarenal target. The involvement of constrictor pathways can be assessed in part through determinations of endogenous levels of humoral and neurogenic constrictor factors and effects of TZD on constriction by these factors. Major compensatory constrictor pathways include the renin–angiotensin II–aldosterone and autonomic nervous systems. Overall, however, the relevance of plasma renin activity and angiotensin II plasma levels is unclear due to the importance of intrarenal versus systemic levels of these factors (Reudelhuber, 2013).
Renin–Angiotensin II–Aldosterone System
Responders and TZD-Sensitive Hypertension Models
A varied relationship exists between TZD-induced changes in renin activity/angiotensin II levels in plasma and reduction in arterial pressure (Figure 1). In hypertensive patients, chronic TZD-mediated increased renin activity/angiotensin II levels were maintained, partially maintained, or returned to levels not significantly different from pre-TZD levels (Table 1m).
However, conclusions that chronic TZD failed to elevate renin activity/angiotensin II levels are complicated by the underlying varied relationship between hypertension and plasma renin activity (Alderman et al., 2004; Atlas, 2007). An additional complication is the inclusion of a significant number of patients on additional antihypertensive medications, although acute TZD increased renin activity despite inclusion of multi-antihypertensive-treated patients (Bourgoignie et al., 1968). The detection of increased renin activity with acute but not chronic TZD may reflect a greater increase in the former (Bourgoignie et al., 1968). In the additional study that demonstrated a lack of increased renin activity/angiotensin II levels with chronic TZD, the patient population could be divided into those with and those without elevated plasma angiotensin II levels (Xiao et al., 2013). Arterial pressure was not reported, and, thus, possible association between TZD arterial pressure reduction and plasma angiotensin II levels remained unexamined (Xiao et al., 2013). Similar to elevated renin activity/angiotensin II levels in response to TZD, Na+-restricted diet also increased renin expression/angiotensin II levels in humans (Luft et al., 1991; Pimenta et al., 2009).
TZD-induced changes in aldosterone plasma levels generally correlated with increased renin activity/angiotensin II plasma levels (Figure 1). This correlation is based, however, upon a limited number of measurements of aldosterone plasma levels with and, for that matter, without renin activity/angiotensin II plasma levels in response to TZD. In fact, aldosterone levels tended to be elevated and were not significantly elevated when renin activity/angiotensin II levels were increased at 5 weeks and 3 months TZD treatment in humans, respectively (Maxwell and Gross, 1979; Lijnen et al., 1981). Further, renin activity/angiotensin II levels were at baseline at 2 months TZD treatment (Bourgoignie et al., 1968; Xiao et al., 2013). Similarly, aldosterone levels in the urine were not elevated following 3 days to 2 months of treatment with TZD (Gifford et al., 1961).
Enhanced TZD reduction of arterial pressure occurred with angiotensin receptor antagonists and angiotensin-converting enzyme inhibitor in human and spontaneously hypertensive rat (Table 1p), as well as with a mineralocorticoid receptor antagonist in human (Hollander et al., 1960).
Normotensive Humans and Animals
Renin activity/angiotensin II and aldosterone plasma levels in normotensive humans in response to TZD are largely unreported. Consistent with TZD-increased renin activity/angiotensin II levels, juxtaglomerular granularity increased following 9 weeks’ treatment with TZD in rat (Tobian et al., 1962). Na+-restricted diet also increased renin expression/angiotensin II levels in humans and NCC knockout mice and elevated Na+ diet decreased angiotensin II levels in humans (Genest et al., 1964; Alshahrani et al., 2017a).
Based upon the similar changes in plasma volume in responders and normotensive humans (Freis et al., 1960; Hollander et al., 1960; Diuresis and Plasma Volume), it is predicted that TZD increased renin activity/angiotensin II levels, as well as aldosterone levels, will be similar to those of responders (Figure 1). Na+-restricted diet increased aldosterone levels in humans (Genest et al., 1964).
Responders versus Nonresponders
TZD tended to increase renin activity/angiotensin II and aldosterone plasma levels to a greater magnitude in nonresponders than responders (van Brummelen et al., 1980; Freis et al., 1988; Table 2). It was suggested that the lack of arterial pressure reduction was due to compensatory angiotensin II vasoconstriction (van Brummelen et al., 1980; Freis et al., 1988). In contrast, TZD failure to increase renin activity and aldosterone levels in nonresponders was also observed (Svendsen et al., 1983), presumably due to underlying compensatory constrictor mechanisms.
Autonomic Nervous System
Responders and TZD-Sensitive Hypertension Models
Plasma levels of norepinephrine remained elevated with chronic TZD, consistent with increased sympathetic activity (Lake et al., 1979). Also consistent with the increased norepinephrine plasma levels (Lake et al., 1979), chronic TZD in hypertensive humans increased cardiac output/rate, although increased cardiac output/rate was also not observed (Table 1n). However, sympatholytic procedures and agents employed in the past to reduce arterial pressure, i.e., ganglionic blockers, reserpine, and sympathectomy, enhanced or did not prevent TZD arterial pressure reduction (Table 1o).
Normotensive Humans and Animals
As in some responders and TZD-sensitive hypertension models (Table 1n), heart rate was unaltered with chronic TZD in dogs (Jandhyala et al., 1972). Reserpine enhanced the arterial pressure reduction due to supra-therapeutic TZD dose in dogs, which would tend not to support the involvement of the sympathetic nervous system (Preziosi et al., 1959). In fact, TZD-lowered catecholamine levels in a number of tissues in dogs and mice suggest TZD-decreased sympathetic activity (Preziosi et al., 1961).
Responders versus Nonresponders
Similar responses to the Valsalva maneuver of TZD-treated responders and nonresponders do not support a role for autonomic nervous system activity and cardiac output in the differential reduction of arterial pressure (de Carvalho et al., 1977; Table 2). Resting cardiac output remained unchanged in responders and nonresponders (de Carvalho et al., 1977), presumably due to compensatory mechanisms. In partial contrast, while acute TZD decreased cardiac output in responders and nonresponders, with chronic TZD cardiac output returned to pre-TZD levels in responders but not in nonresponders (van Brummelen et al., 1980).
Summary and Conclusions: Endogenous Activation
Increased renin activity/angiotensin II and norepinephrine plasma levels in hypertensive humans and animals are consistent with a remaining component of decreased plasma volume in response to chronic TZD (Figure 1). Thus, TZD-reduced arterial pressure, while due to inhibition of compensatory angiotensin II and norepinephrine vasoconstriction as well as presumably other compensatory constrictor pathways, is also mitigated by further compensatory vasoconstriction (Figure 2). Additionally, increased renin activity/angiotensin II plasma levels by Na+-restricted diet suggest that similar constrictor pathways mediate the compensatory vasoconstriction due to plasma volume reduction in response to lowered Na+ and chronic TZD plasma volume decrease. Furthermore, the lesser magnitude of elevated renin activity/angiotensin II plasma levels in nonresponders (Table 2) is consistent with the lack of plasma volume decrease and resultant absence of arterial pressure reduction.
It should be noted, however, that increased renin activity/angiotensin II levels in response to chronic TZD were not consistently observed in hypertensive humans and animals (Figure 1). Furthermore, renin activity/angiotensin II levels were not consistently greater in responders than nonresponders (Table 2). Clearly, the limited number and contrasting findings of renin activity/angiotensin II levels, as well as aldosterone levels, warrant additional studies.
The enhancement or lack of prevention of TZD arterial pressure reduction by angiotensin receptor antagonists and angiotensin-converting enzyme inhibitor (Table 1p), mineralocorticoid receptor antagonists (Hollander et al., 1960), and ganglionic blockers, reserpine, and sympathectomy (Table 1o) appears inconsistent with TZD reduction of arterial pressure through inhibition of the renin–angiotensin–aldosterone and sympathetic nervous systems. Alternatively, TZD arterial pressure reduction would be enhanced by agents and procedures that disrupt the renin–angiotensin–aldosterone and sympathetic nervous system if TZD only partially inhibit activation of these systems.
The inability of TZD to reduce arterial pressure in neurogenic, i.e., sympathetic nervous system hypertension (Table 1c), also suggests that TZD inhibition of catecholamine-induced vasoconstriction is not involved in the reduction of arterial pressure. On the other hand, hypertension associated with high but not low renin activity/angiotensin II plasma levels is resistant to TZD reduction of arterial pressure (Chapman et al., 2002; Table 1c). The efficacy of TZD to reduce arterial pressure depending upon magnitude of renin–angiotensin II pathway activation may, by analogy, suggest that hypertension associated with high sympathetic nervous system activity is insensitive to arterial pressure reduction.
(b) Exogenous Agonist
TZD inhibition of the compensatory contracted plasma volume as well as constriction underlying certain types of hypertension may involve direct inhibition of vascular constriction. This assessment can be achieved in part through the effects of TZD on exogenous agonist pressor responses.
Numerous findings describe the effects of acute TZD on agonist-elevated and agonist-reduced vascular bed and mean arterial pressure (Table 1q). However, based upon the close correlation between acute TZD reduction of arterial pressure and diuresis, the extrarenal-mediated TZD reduction of arterial pressure is most likely absent during acute TZD (Diuresis and Plasma Volume). Thus, findings presented are limited to effects of chronic TZD on pressor responses (Table 3).
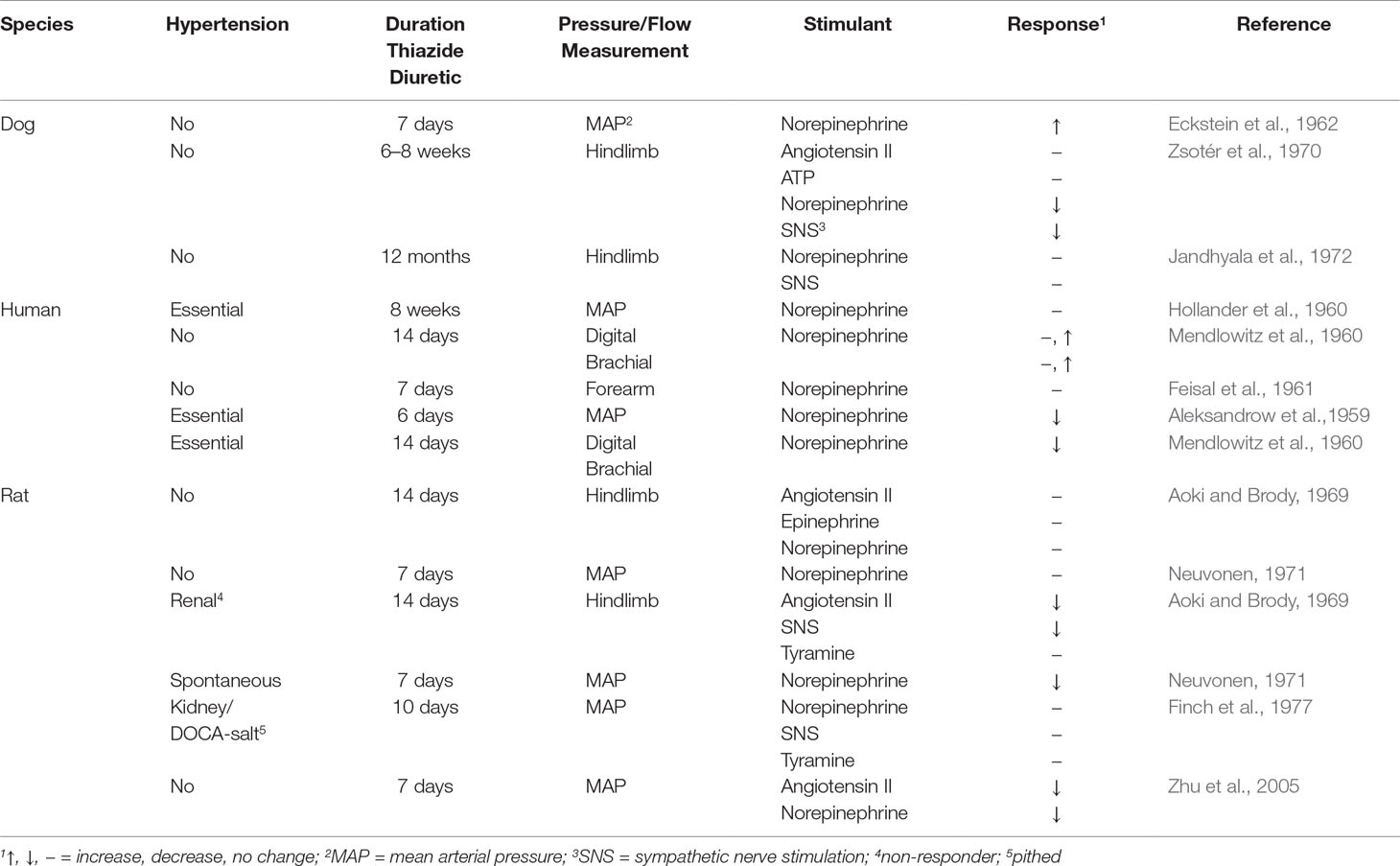
Table 3 Effect of Chronic Thiazide Diuretics on Agonist- and Sympathetic Nerve Stimulation-Elevated Arterial Pressure in Normotension and Hypertension.
Responders and TZD-Sensitive Hypertension Models
In hypertensive patients treated with TZD, norepinephrine pressor responses remained unaltered (Hollander et al., 1960; Table 3). In contrast, TZD reduced the norepinephrine increased digital, brachial, and mean arterial pressure (Aleksandrow et al., 1959; Mendlowitz et al., 1960; Table 3) and increased forearm basal flow at supra-therapeutic dose (Pickkers et al., 1998). TZD reduction of mean arterial pressure was independent of hypovolemia because similar magnitudes of TZD reduction of mean arterial pressure occurred in hypovolemic and euvolemic patients (Mendlowitz et al., 1960). On the other hand, the magnitude of TZD reduction of the norepinephrine pressor response exceeded the magnitude of arterial pressure reduction (Mendlowitz et al., 1960).
Although not challenged with agonist, venoconstriction due to deep breathing and the Valsalva maneuver decreased in TZD-treated hypertensive patients (Ogilvie and Schlieper, 1970). These findings are consistent with TZD lowering of neurogenic activity (Ogilvie and Schlieper, 1970).
The norepinephrine pressor response in spontaneously hypertensive rats was also decreased by TZD (Neuvonen, 1971; Table 3). In contrast, TZD did not decrease pressor responses to norepinephrine, tyramine, and sympathetic nerve stimulation in 1 kidney/DOCA-salt hypertensive rats (Finch et al., 1977; Table 3). The differential TZD reduction of agonist pressor response depending upon the agonist (Table 3; Finch et al., 1977; Neuvonen, 1971) is consistent with the dependency of TZD reduction of hypertension upon the mechanism underlying the hypertension (Gong et al., 2012).
Normotensive Humans and Animals
In normotensive humans, TZD decreased norepinephrine-elevated digital and brachial pressure, although norepinephrine did not consistently elevate pressure (Mendlowitz et al., 1960). Similarly, norepinephrine decreased forearm blood flow, and TZD increased the flow in Responders and Nonresponders (i.e., reduced the pressure; Feisal et al., 1961; Table 3). The TZD-increased flow was attributed to increased cardiac output, possibly as the result of decreased cardioregulatory reflexes (Feisal et al., 1961).
In support of this suggestion, TZD treatment increased cardiac output and caused lesser decreased heart rate as compared to untreated dogs (Eckstein et al., 1962; Table 3). TZD also further increased norepinephrine elevated mean arterial pressure (Eckstein et al., 1962; Table 3). Thus, it was suggested that desensitization of carotid sinus and baroreceptor reflexes was responsible for the norepinephrine-induced increased heart rate and cardiac output with chronic TZD, resulting in further elevation of mean arterial pressure (Eckstein et al., 1962; Table 3). However, a role for decreased cardiac output in chronic TZD reduction of arterial pressure has been discounted (Central Vasomotor Centers).
In dog hindlimb, TZD did not reduce the elevated pressure due to norepinephrine, epinephrine, angiotensin II, and/or sympathetic nerve stimulation (Aoki and Brody, 1969; Jandhyala et al., 1972; Table 3). However, tonic, non-sympathetic, neurogenic tone was decreased by chronic TZD (Jandhyala et al., 1972; Table 3). In contrast, also in dog hindlimb, TZD reduced the elevated pressure due to norepinephrine and sympathetic nerve stimulation, but not to angiotensin II and ATP (Zsotér et al., 1970; Table 3). However, whether TZD actually reduced norepinephrine elevated mean arterial pressure in the dogs was not reported (Zsoter et al., 1907). TZD was without effect (Neuvonen, 1971) or reduced the norepinephrine, as well as the angiotensin II pressor response in rats (Zhu et al., 2005; Table 3). Thus, similar to the different efficacies of TZD to reduce arterial pressure depending upon the type of hypertension (Table 1b and c), there is varied TZD decrease in agonist pressor response.
Responders versus Nonresponders
TZD reduced elevated pressure to sympathetic nerve stimulation, angiotensin II, and norepinephrine, but not to tyramine, in the perfused hindlimb of renal hypertensive rats (Aoki and Brody, 1969; Table 3). The relevancy of the reduced pressor responses is unclear because TZD did not decrease mean arterial pressure in renal hypertensive rats (Aoki and Brody, 1969; Table 3).
Summary and Conclusions: Exogenous Agonist
Varied TZD reduction of norepinephrine-elevated arterial pressure, along with lack of correlation between these reductions and chronic TZD-reduced mean arterial pressure, casts doubt upon the involvement of possible decreased norepinephrine pressor response in arterial pressure reduction. Few, if any, studies determined TZD effects on pressor responses to agonists other than norepinephrine.
(5) Vascular Contractility In Vitro
An additional assessment of TZD inhibition of increased tone due to plasma volume reduction and associated with hypertension is TZD inhibition in the isolated vasculature. The direct inhibition of increased tone can be determined by the effects of TZD a) in vivo on subsequently isolated vasculature and b) in vitro on agonist constriction.
(a) In Vivo TZD on Vascular Contractility Determined In Vitro
Responders and TZD-Sensitive Hypertension Models
In femoral artery from hypertensive rats due to nitric oxide synthase inhibitor, a 7-week challenge with hydrochlorothiazide, which lowered the elevated arterial pressure, failed to increase acetylcholine-induced relaxation in isolated femoral artery (Sládková et al., 2007; Table 4).
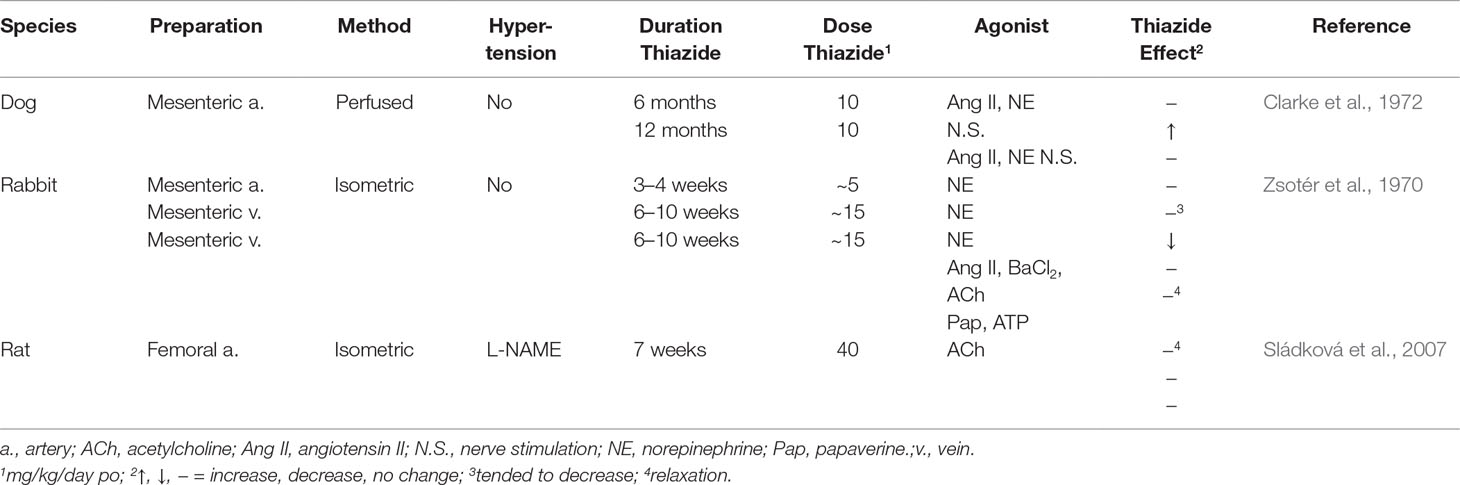
Table 4 Effect of Thiazide Diuretics In Vivo on Vascular Constriction In Vitro.
Normotensive Animals
In mesenteric arterial bed isolated from normotensive dogs, a 10-month challenge with hydrochlorothiazide failed to reduce norepinephrine-, angiotensin II-, and nerve stimulation-elevated perfusion pressure (Clarke et al., 1972; Table 4). In possible contrast, in rabbits challenged with hydrochlorothiazide for 6–10 weeks, constriction of mesenteric artery and vein to norepinephrine tended to decrease and decreased, respectively (Zsotér et al., 1970; Table 4). However, the significance of the reduced norepinephrine constriction is not clear because the vein is a capacitance vessel (Zsotér et al., 1970; Table 4). Moreover, norepinephrine constriction was not decreased following up to 4 weeks of challenge with hydrochlorothiazide (Zsotér et al., 1970; Table 4).
(b) In Vitro TZD on Vascular Contractility
Numerous studies investigated TZD inhibition of vascular contractility in vitro (Tables 1r and 5). Moreover, a number of these studies, but not all, demonstrated TZD inhibition of constriction (Tables 1s and 5). Proposed mechanisms underlying the inhibition of vascular contractility include carbonic anhydrase inhibition, increased nitric oxide release, BKCa2+ channel activation, decreased extracellular/intracellular Na+, decreased sensitization to intracellular Ca2+, RhoA and Rho kinase down-regulation, and inhibition of voltage-dependent Ca2+ channel activation (Table 1s).
However, i) supra-therapeutic TZD dose (Dose/Concentration of TZD) was required in a majority of studies to demonstrate inhibition of vasoconstriction (Table 5). Even though rabbits challenged with hydrochlorothiazide for 7 weeks with the supra-therapeutic dose of 40 mg/kg/day failed to increase acetylcholine relaxation of isolated femoral artery, while incubation of rabbit isolated femoral artery with 100 µM hydrochlorothiazide enhanced acetylcholine relaxation (Sládková et al., 2007; Vascular Contractility In Vitro, In Vivo TZD on Vascular Contractility Determined In Vitro; Tables 4 and 5). Indeed, the requirement of supra-therapeutic concentrations in vitro is similar to findings in humans of TZD-increased forearm blood flow, which required at least 38 µM of hydrochlorothiazide (Pickkers and Hughes, 1995; Pickkers et al., 1998), and in dogs, which required 9–18 times the usual therapeutic dose of chlorothiazide (Overbeck and Haddy, 1960). Concentration–response (inhibition of constriction) curves to TZD were not performed in a number of investigations (Table 5). Thus, TZD potency in these investigations remains to be established. ii) The magnitude of TZD inhibition of constriction depended significantly upon the TZD (Afsar et al., 2016). iii) Conclusions regarding mechanism underlying the inhibition apparently depended upon the contractile agent and/or vessel as observed, e.g., with the role of BKCa2+ channel inhibition (Calder et al., 1992, Calder et al., 1993, Calder et al., 1994; Colas et al., 2000a; Table 5). iv) In vitro effects may differ from in vivo effects. For example, the proposal that TZD decreased constriction through BKCa2+ channel inhibition based upon in vitro findings (Colas et al., 2000a; Table 5) is not supported by observations that i.p. paxilline, a BKCa2+ channel blocker, and a BKCa2+ channel/NCC/pendrin in triple-knockout mice did not prevent acute, hydrochlorothiazide reduction of arterial pressure (Alshahrani et al., 2017a). And v) vessels were not obtained from animals chronically treated with TZD (Table 5).
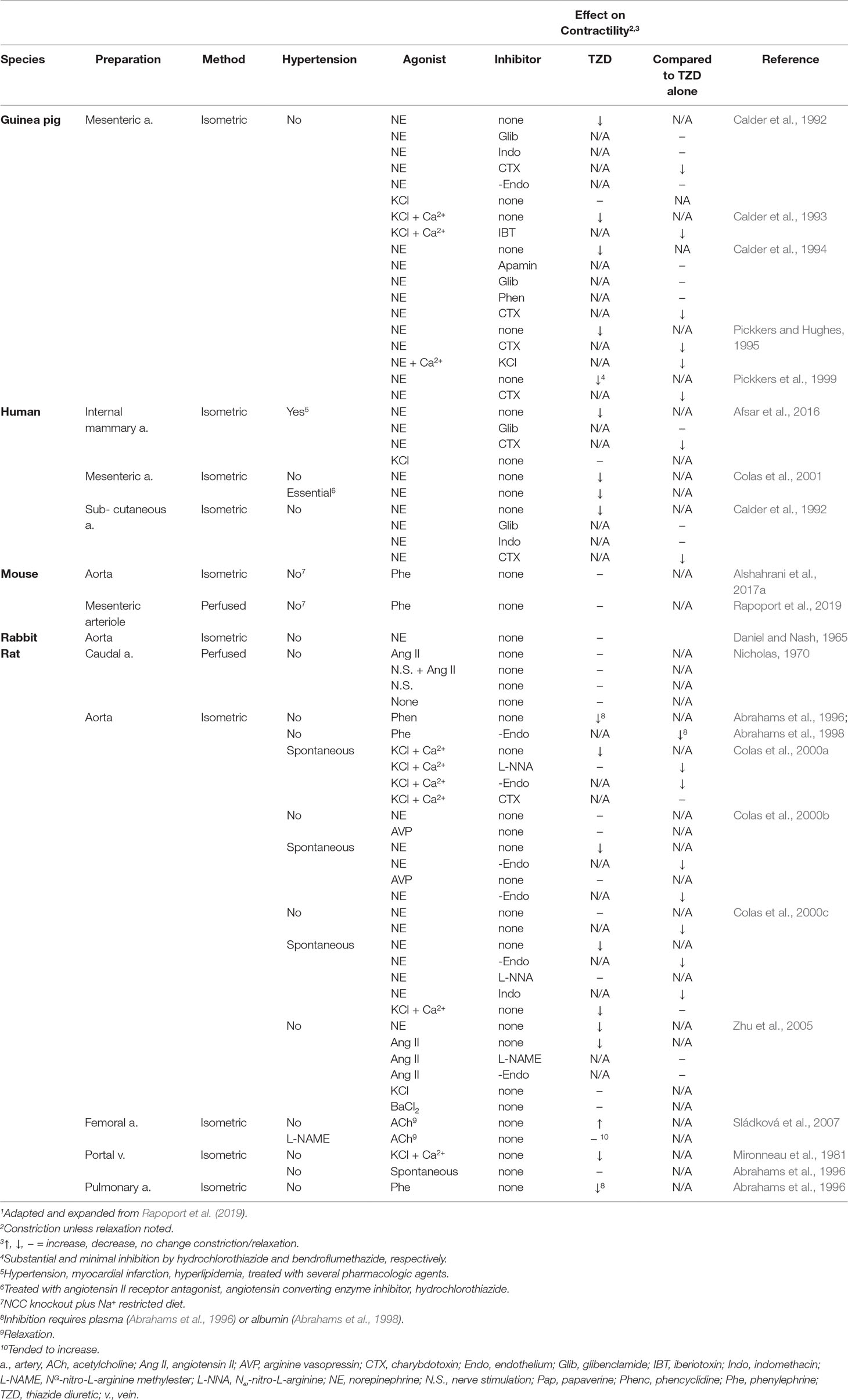
Table 5 Effect of Thiazide Diuretics In Vitro on Vascular Constriction1.
Generally consistent with the dependency of TZD reduction of arterial pressure on altered regulatory protein expression is the minimal and greatly reduced arterial pressure with acute infusion and with 3-day TZD treatment, respectively, in hypertensive, angiotensin II-salt rats (Ballew and Fink, 2001). Additionally, TZD inhibition of vasoconstriction was attributed to decreased expression of proteins that sensitize agonist constriction, RhoA and Rho kinase (Zhu et al., 2005; Table 4). This conclusion was based in large part on decreased expression of these proteins with TZD challenge of cultured vascular smooth muscle cells (Zhu et al., 2005). Consistent with these findings is the more recent demonstration that chronic hydrochlorothiazide reduced arterial pressure in the hypertensive 1 kidney/DOCA-salt rat and decreased the associated elevated Rho-kinase (as well as gene expression of markers of remodeling) in the aorta (Araos et al., 2016).
On the other hand, decreased Rho-kinase would be expected to inhibit and enhance agonist constriction and relaxation, respectively (Nakamura et al., 2003). However, 1) chronic TZD failed to consistently inhibit constriction and relaxation was not enhanced (Constrictor Pathways In Vivo, Exogenous Agonist; Vascular Contractility In Vitro, In Vivo TZD on Vascular Contractility Determined In Vitro; Tables 3 and 4); 2) while Rho-kinase inhibition reduces arterial pressure in rats with normal arterial pressure (Dhaliwal et al., 2007), chronic (and acute) TZD do not reduce arterial pressure in rat and other animals and in human (Introduction; Diuresis and Plasma Volume; Table 1j and d, Figure 1; unreported in Araos et al. (2016) is whether hydrochlorothiazide decreased mean arterial pressure in sham operated rats); 3) although hydrochlorothiazide in vitro at clinically relevant concentrations decreased Rho-kinase expression and agonist constriction, based upon the varied inhibitory effects of TZD on vasoconstriction in vitro as well as the use in many studies of supra-clinical TZD concentrations (Vascular Contractility In Vitro, In Vitro TZD on Vascular Contractility; Tables 1r, s, and 5), it is should be considered that inhibition of Rho-kinase activity by TZD is indirect and requires chronic exposure (Araos et al., 2016). In contrast, the Rho-kinase inhibitor, fasudil (Y27632), causes direct inhibition (Nakamura et al., 2003) and 4) intrarenal rather than systemic angiotensin II may be the important modulator of vascular contractility (Reudelhuber, 2013; Constrictor Pathways In Vivo, Endogenous Activation). Thus, the possible role of decreased Rho-kinase activity in chronic TZD reduction of arterial pressure remains unclear.
Summary and Conclusions: Vascular Contractility In Vitro
Lack of inhibition of agonist-induced constriction in vessels isolated from animals chronically challenged with TZD and in NCC knockout mice subject to Na+-restricted diet and NCC/pendrin double-knockout mice, despite TZD reduction of arterial pressure (Clarke et al., 1972; Sládková et al., 2007; Rapoport et al., 2019; Table 5), suggests that the condition(s) allowing TZD to reduce arterial pressure is absent in vitro. Thus, these findings (Clarke et al., 1972; Sládková et al., 2007; Alshahrani et al., 2017a; Rapoport et al., 2019; Table 5) do not support proposed mechanisms of TZD inhibition of vasoconstriction based upon in vitro challenge with TZD (Vascular Contractility In Vitro; Table 5). Indeed, the presence of the TZD target in vivo, but not in vitro, is consistent with reversal of TZD chronic reduction of arterial pressure with plasma volume repletion (Diuresis and Plasma Volume).
TZD-induced decreased vasoconstriction could occur through inhibition/activation of smooth muscle signaling pathways or requires up-regulation/down-regulation of these pathways. However, it is unlikely that expression of a potential TZD target at least within the vasculature is up-regulated/down-regulated because i) arteries isolated from animals chronically treated with TZD failed to demonstrate decreased constriction and/or increased relaxation (Zsotér et al., 1970; Clarke et al., 1972; Sládková et al., 2007; Table 3); ii) agonist pressor responses were not decreased with chronic TZD in a number of animal and human investigations (Table 3); iii) differences in phenylephrine constriction were not detected (independent of structural change) in vessels isolated from wild type and both NCC/pendrin double knockout and NCC knockout plus Na+ diet-restricted mice (Alshahrani et al., 2017a; Alshahrani et al., 2017b; Rapoport et al., 2019; Table 5); iv) hydrochlorothiazide, at the supra-therapeutic dose of 20 mg/kg, reduced arterial pressure within 1 h (shortest time period examined in unanesthetized mice) in NCC knockout mice subjected to Na+-restricted diet and NCC/pendrin knockout mice (Alshahrani et al., 2017a). It is unlikely that 1 h is sufficient time to significantly alter protein expression. However, a caveat to this conclusion is that hydrochlorothiazide at this dose may reduce arterial pressure through hyponatremia, rather than an extrarenal target (Alshahrani et al., 2017a; [Na+/Cl– Contransporter (NCC; SLC12A3)]); and v) chronic TZD in vivo with and without additional TZD challenge of vessels isolated from these animals did not inhibit agonist constriction (Zsotér et al., 1970; Clarke et al., 1972; Sládková et al., 2007; Table 4).
An array of mechanisms has been proposed for TZD direct inhibitory effects on vasoconstriction. However, the relevancy of these mechanisms is limited by the procedures adopted for these determinations (“b” above; In Vitro TZD on Vascular Contractility). Furthermore, with respect to overall considerations vis-à-vis measurements of vascular contractility—i.e., 1) the use of resistance type vessels rather than conduit vessels 2) the vessels should be derived from vascular beds associated with resistance, and 3) a relatively small change in contractility, which may or may not be detected depending upon measurement sensitivity, underlies a large change in blood flow, i.e., blood flow is proportional to the fourth power of the radius (Poiseulle’s law)—these considerations appear largely satisfied through recent video-imaging measurements of changes in diameter in mesenteric arterioles, i.e., resistance vessels (Rapoport et al., 2019). Moreover, the mesenteric vascular bed is relevant to determinations of peripheral vascular resistance and, thus, arterial blood pressure regulation (Heyndrickx et al., 1976; Christensen and Mulvany, 1993).
(6) Central Vasomotor Centers
In addition to peripheral actions of TZD, potential central effects of TZD need to be considered. The relevancy of central effects of TZD is unlikely because central access of TZD following peripheral administration is limited, as demonstrated in patients with various neurological diseases (Sigaroudi et al., 2018).
Responders and TZD-Sensitive Hypertension Models
TZD injection into the hypothalamus reduced arterial pressure in spontaneously hypertensive rats (Bergmann and Altshuler, 1996). However, in hypertensive patients, TZD was neutral with respect to central systolic pressure (Morgan et al., 2004; Mackenzie et al., 2009; Kwon et al., 2013; Manisty and Hughes, 2013).
Direct TZD application to the carotid artery sinus in the presence and absence of norepinephrine and epinephrine failed to reduce arterial pressure in dogs with acute hypertension due to vagi-aortic nerve denervation (Preziosi et al., 1959; Preziosi et al., 1961). Concomitant with the findings related to baroreceptor reflexes (Preziosi et al., 1959; Preziosi et al., 1961), while TZD increased the gain of renal nerve activity and heart rate in spontaneously hypertensive rats, the increase was non-selective in that similar findings were observed with several classes of anti-hypertensive agents (Kumagai et al., 1996).
Normotensive Animals
TZD did not lower arterial pressure when infused into the carotid artery in a dog preparation with ligated internal and external carotid arteries with and without carotid sinus denervation (Preziosi et al., 1961). Consistent with these findings TZD were without effect on isolated guinea pig and rabbit heart (Preziosi et al., 1959).
Summary and Conclusions: Central Vasomotor Centers
It is unlikely that TZD act centrally at vasomotor centers to decrease peripheral vascular resistance. Further, the relevance of determinations of TZD effects in normotensive animals is unclear because TZD do not lower arterial pressure in normotensive humans and animals (Diuresis and Plasma Volume).
(7) Other Signaling Pathways Based Upon Genetic Analysis
Hypertension Responders and Nonresponders
In terms of baseline protein expression in responders and nonresponders, responders had greater mRNA baseline levels of a number of proteins including vasodilator-stimulated phosphoprotein (VASP) and metabolites of the sphingolipid metabolic pathway (Turner et al., 2013; Shahin et al., 2017; Sá et al., 2018). It was suggested that increased baseline VASP expression and the sphingolipid metabolic pathway were responsible for TZD efficacy of arterial blood pressure reduction (Shahin et al., 2017).
Summary and Conclusions: Other Signaling Pathways Based Upon Genetic Analysis
The involvement of these pathways in TZD reduction of arterial pressure warrants additional investigation. Indeed, it remains unclear whereby, e.g., increased sphingolipid metabolism would result in greater efficacy of TZD reduction of arterial pressure (Shahin et al., 2017).
(8) Guiding Parameters
Suggested required parameters for relevant investigations into extrarenal TZD target identification include the following:
(a) Therapeutic TZD dose/concentration: In vivo doses should be in the therapeutic range (Dose/Concentration of TZD). Indeed, higher TZD doses elicit several effects not relevant to TZD mechanism of action at therapeutic dose (Na+/Cl−Contransporter [NCC; SLC12A3]). In vitro concentrations should reflect TZD plasma levels (Dose/Concentration of TZD and Vascular Contractility In Vitro).
(b) Chronic TZD: The extrarenal TZD target is exposed/present with chronic TZD (Diuresis and Plasma Volume and Vascular Contractility In Vitro). Thus, the relevancy of acute TZD challenge in vivo and in vitro is unclear.
(c) TZD effect on normal arterial pressure: TZD should not reduce arterial pressure in normotensive animals. This stipulation is derived from the lack of effect of TZD on arterial pressure in normotensive humans and animals (Diuresis and Plasma Volume). Thus, the relevancy of acute TZD challenge on vascular reactivity in normotensive animals and in vascular tissue from normotensive animals is unclear (Constrictor Pathways In Vivo; Vascular Contractility In Vitro).
(d) TZD and hypertension: TZD should reduce arterial pressure in hypertensive models (“responders” in humans; Diuresis and Plasma Volume). Moreover, due to the dependency of TZD reduction of arterial pressure on underlying hypertension, findings should be derived from hypertensive models and from tissues obtained from these models (Diuresis and Plasma Volume; Vascular Contractility In Vitro).
(e) TZD and NCC: The acute effects of TZD should be mediated through NCC inhibition ([Na+/Cl− Cotransporter (NCC; SLC12A3)]).
(f) Other diuretics: The TZD target should not be utilized by non-TZD, e.g., NKCC2 inhibitors (loop diuretics; Diuresis and Plasma Volume).
(g) TZD-insensitive hypertension (“nonresponders” in humans): The TZD target should not be exposed/present in hypertensive models insensitive to TZD arterial pressure reduction (Diuresis and Plasma Volume, [Na+/Cl− Cotransporter (NCC; SLC12A3)]; Constrictor Pathways In Vivo).
(9) Perspective
(a) TZD Target Site
It is unlikely that TZD selectively inhibit the presumably different TZD targets associated with each of the multiple types of hypertension sensitive to reduction by TZD (Table 1c; Introduction). For example, hypertension varies with respect to the magnitudes of sympathetic nerve activity and vascular contractile sensitivity (Table 1t). Thus, as illustrated in the working model (Figure 2), TZD reduction of arterial pressure is unrelated to direct inhibition of a specific constrictor pathway but rather through a common effect that inhibits constriction to multiple pathways. These multiple pathways underlie constriction (1) to TZD-decreased plasma volume (consistent with earlier proposals of Tapia et al., 1957; Freis et al., 1958; Dustan et al., 1959; Tarazi et al., 1970) and (2) responsible for the numerous TZD-sensitive types of hypertension (Table 1c; Introduction). Alternatively, a particular pathway sensitive to TZD inhibition, e.g., renin–angiotensin II, could underlie the multiple types of hypertension associated with responders.
In any case, the above proposal suggests that TZD reduction of arterial pressure is mediated by inhibition of underlying compensatory vasoconstrictor pathways (Figure 2). Indeed, removal of the compensatory constriction, through repletion of plasma volume by infusion with dextran plus saline/glucose or elevated dietary Na+, reversed TZD chronic reduction of arterial pressure (Freis et al., 1958; Wilkins et al., 1958; Wilson and Freis, 1959; Winer, 1961). Also consistent with a common TZD target is enhancement of agonist constriction with myogenic tone (Faber and Meininger, 1990).
It should also be noted that, despite the potential attractiveness of the suggestion that NCC serves as the extrarenal target underlying TZD chronic arterial pressure reduction based upon the lower arterial pressure in Gitelman’s syndrome (Calò, 2006), this suggestion is inconsistent with restriction of NCC expression to the kidney, i.e., as evidenced by lack of NCC expression and binding in the aorta, as well as the adrenal gland, brain, heart, intestine (large and small), liver, lung, pancreas, placenta, salivary gland, skeletal muscle, spleen, and testis in humans and/or rats (Beaumont et al., 1988; Hebert and Gamba, 1995; Mastroianni et al., 1996).
(b) TZD Arterial Pressure Reduction
As illustrated in the working model of Figures 2A–D:
(A) Normotensive subjects/animals: The proposed dependency of TZD arterial pressure reduction on inhibition of compensatory vasoconstrictor pathways (Guiding Parameters (a) Therapeutic TZD dose/concentration) suggests that the absence of TZD reduction of arterial pressure in normotensive subjects and animals (Diuresis and Plasma Volume) is due to complete mitigation by compensatory vasoconstriction of the TZD inhibition of constriction (Figure 2A).
(B) Decreased plasma volume: In contrast to the lack of TZD reduction of arterial pressure in normotensive subjects/animals, TZD reduced arterial pressure following enhanced plasma volume depletion, as observed in NCC knockout mice subjected to Na+-restricted diet and NCC/pendrin knockout mice (Alshahrani et al., 2017a). Thus, even greater compensatory vasoconstriction is required to limit the decrease in arterial pressure (Alshahrani et al., 2017a). However, a further decrease in plasma volume due to TZD cannot be fully compensated for by the already enhanced magnitude of compensatory vasoconstriction. The lack of complete mitigation of the plasma volume reduction, therefore, fails to fully compensate for TZD inhibition of vasoconstriction (Figure 2B). A caveat to this explanation is the possible mediation of TZD reduction of arterial pressure in NCC knockout mice subjected to Na+-restricted diet and NCC/pendrin knockout mice (Alshahrani et al., 2017a) through NDCBE inhibition ([Na+/Cl− Contransporter (NCC; SLC12A3)]).
(C) TZD-sensitive hypertension: Similar to TZD reduction in arterial pressure following enhanced plasma volume depletion (Alshahrani et al., 2017a), in TZD-sensitive hypertension, the magnitude of constriction due to hypertension plus decreased plasma volume in response to TZD cannot fully mitigate TZD inhibition of constriction (Figure 2C). Thus, TZD reduces the elevated arterial pressure (Figure 2C). This proposal further suggests, therefore, that reduction of arterial pressure in hypertension requires an underlying component of reduced plasma volume. In this regard, this suggestion is consistent with the reversal of the TZD lowered arterial pressure with plasma volume repletion (Diuresis and Plasma Volume). It should also be considered, however, that a particular pathway sensitive to TZD inhibition, e.g., renin–angiotensin II, could underlie the multiple types of hypertension associated with responders (Guiding Parameters (a) Therapeutic TZD dose/concentration).
(D) TZD-insensitive hypertension: Considering the suggested requirement for an underlying component of reduced plasma volume to lower elevated arterial pressure (C above), the inability to decrease arterial pressure in some types of hypertension may be due to prevention of contracted plasma volume (Figure 2D). The lack of contracted plasma volume is consistent with the greater TZD-increased diuresis, plasma volume reduction, and body weight loss in responders than nonresponders and the inability of TZD to decrease plasma volume in nonresponders (de Carvalho et al., 1977; Svendsen et al., 1983; Freis et al., 1988; Table 2; Diuresis and Plasma Volume). Alternatively, the constrictor pathways underlying TZD-insensitive hypertension may overcome the TZD inhibition of constriction (Figure 2D).
Clearly, the identity of the extrarenal site of TZD inhibition of vascular tone remains for further investigation. Moreover, this site is presumably active in Gitelman’s syndrome. TZD-insensitive hypertensive models should also assist in this identification through delineation of factors in common with, and different from, TZD-sensitive hypertensive models and unique to particular models. Additionally, several genetic signals associated with sensitivity to TZD reduction of arterial pressure have been identified (Melander et al., 2013; Shahin and Johnson, 2016; Sá et al., 2018). Whether these or other signals underlie TZD inhibition of vasoconstriction remains for further investigation. Identification of the TZD extrarenal target will provide the basis for conversion of nonresponder to responder and the development of novel antihypertensive therapies acting through this site.
Author Contributions
RR and MS conceived, RR wrote, and MS reviewed the article.
Funding
This study was supported by a Merit Review award from the Department of Veterans Affairs (5I01BX001000), funds from the Center on Genetics of Transport and Epithelial Biology at the University of Cincinnati, and grants from the Dialysis Clinic, Inc. and US Renal Care (MS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Glenn Doerman (University of Cincinnati) for the illustration and Drs. Limeng Chen (Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China) and Giacomo Colussi (ASST Grande Ospedale Metropolitano Niguarda, Milano, Italy), for the personal communications.
References
Abrahams, Z., Pang, M. Y., Lam, E. K., Wright, J. M. (1998). What is the plasma cofactor required by diuretics for direct vascular relaxant effect in vitro? J. Hypertens. 16, 801–809. doi: 10.1097/00004872-199816060-00011
Abrahams, Z., Tan, L. L., Pang, M. Y., Abrahams, B., Tan, M. M., Wright, J. M. (1996). Demonstration of an in vitro direct vascular relaxant effect of diuretics in the presence of plasma. J. Hypertens. 14, 381–388. doi: 10.1097/00004872-199603000-00016
Afsar, S., Hemsinli, D., Ozyazgan, S., Akkan, A. G., Arslan, C. (2016). The effects of potassium channels in human internal mammary artery. Pharmacology 9, 72–77. doi: 10.1159/000442528
Alderman, M. H., Cohen, H. W., Sealey, J. E., Laragh, J. H. (2004). Plasma renin activity levels in hypertensive persons: their wide range and lack of suppression in diabetic and in most elderly patients. Am. J. Hypertens. 17, 1–7.
Aleksandrow, D., Wysznacka, W., Gajewski, J. (1959). Influence of chlorothiazide upon arterial responsiveness to nor-epinephrine in hypertensive subjects. N. Engl. J. Med. 261, 1052–1055. doi: 10.1056/NEJM195911192612103
Alshahrani, S., Rapoport, R. M., Soleimani, M. (2017b). Vascular contractile reactivity in hypotension due to reduced renal reabsorption of Na(+) and restricted dietary Na(+). Naunyn. Schmiedebergs. Arch. Pharmacol. 390, 321–326. doi: 10.1007/s00210-017-1340-0
Alshahrani, S., Rapoport, R. M., Zahedi, K., Jiang, M., Nieman, M., Barone, S., et al. (2017a). The non-diuretic hypotensive effects of thiazides are enhanced during volume depletion states. PLoS One 12, e0181376. doi: 10.1371/journal.pone.0181376
Alshahrani, S., Soleimani, M. (2017). Ablation of the Cl–/HCO3– exchanger pendrin enhances hydrochlorothiazide-induced diuresis. Kidney Blood Press. Res. 42, 444–455. doi: 10.1159/000479296
Anderson, J., Godfrey, B. E., Hill, D. M., Munro-Faure, A. D., Sheldon, J. (1971). A comparison of the effects of hydrochlorothiazide and of frusemide in the treatment of hypertensive patients. Q. J. Med. 40, 541–560.
Aoki, V. S., Brody, M. J. (1969). The effect of thiazide on the sympathetic nervous system of hypertensive rats. Arch. Int. Pharmacodyn. Ther. 177, 423–434.
Araos, P., Mondaca, D., Jalil, J. E., Yañez, C., Novoa, U., Mora, I., et al. (2016). Diuretics prevent Rho-kinase activation and expression of profibrotic/oxidative genes in the hypertensive aortic wall. Ther. Adv. Cardiovasc. Dis. 10, 338–347. doi: 10.1177/1753944716666208
Araoye, M. A., Chang, M. Y., Khatri, I. M., Freis, E. D. (1978). Furosemide compared with hydrochlorothiazide. Long-term treatment of hypertension. JAMA 240, 1863–1866. doi: 10.1001/jama.1978.03290170045023
Ashek, A., Menzies, R. I., Mullins, L. J., Bellamy, C. O., Harmar, A. J., Kenyon, C. J., et al. (2012). Activation of thiazide-sensitive co-transport by angiotensin II in the cyp1a1-Ren2 hypertensive rat. PLoS One 7, e36311. doi: 10.1371/journal.pone.0036311
Atlas, S. A. (2007). The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 13 (8 Suppl B), 9–20.
Ballew, J. R., Fink, G. D. (2001). Characterization of the antihypertensive effect of a thiazide diuretic in angiotensin II-induced hypertension. J. Hypertens. 19, 1601–1606. doi: 10.1097/00004872-200109000-00012
Baum, T., Shropshire, A. T. (1967). Vasoconstriction induced by sympathetic stimulation during development of hypertension. Am. J. Physiol. 212, 1020–1024. doi: 10.1152/ajplegacy.1967.212.5.1020
Beaumont, K., Vaughn, D. A., Fanestil, D. D. (1988). Thiazide diuretic drug receptors in rat kidney: identification with [3H]metolazone. Proc. Natl. Acad. Sci. U.S.A. 85, 2311–2314. doi: 10.1073/pnas.85.7.2311
Beavers, W. R., Blackmore, W. P. (1958). Effect of chlorothiazide on vascular reactivity. Proc. Soc. Exp. Biol. Med. 98, 133–135. doi: 10.3181/00379727-98-23964
Beermann, B., Groschinsky-Grind, M. (1977). Pharmacokinetics of hydrochlorothiazide in man. Eur. J. Clin. Pharmacol. 12, 297–303. doi: 10.1007/BF00607430
Beermann, B., Groschinsky-grind, M. (1978). Antihypertensive effect of various doses of hydrochlorothiazide and its relation to the plasma level of the drug. Eur. J. Clin. Pharmacol. 13, 195–201. doi: 10.1007/BF00609982
Beermann, B., Groschinsky-Grind, M. (1979). Pharmacokinetics of hydrochlorothiazide in patients with congestive heart failure. Br. J. Clin. Pharmacol. 7, 579–583. doi: 10.1111/j.1365-2125.1979.tb04646.x
Bergmann, F., Altshuler, R. (1996). Blood pressure effects of hydrochlorothiazide, applied to the rat hypothalamus. J. Basic. Clin. Physiol. Pharmacol. 7, 331–339. doi: 10.1515/JBCPP.1996.7.4.331
Blanchard, A., Bockenhauer, D., Bolignano, D., Calò, L. A., Cosyns, E., Devuyst, O., et al. (2017). Gitelman syndrome: consensus and guidance from a Kidney Disease: improving Global Outcomes (KDIGO) Controversies Conference. Kid. Int. 91, 24–33. doi: 10.1016/j.kint.2016.09.046
Borghi, C., Omboni, S. (2014). Zofenopril plus hydrochlorothiazide combination in the treatment of hypertension: an update. Expert. Rev. Cardiovasc. Ther. 12, 1055–1065. doi: 10.1586/14779072.2014.946405
Bourgoignie, J. J., Catanzaro, F. J., Perry, H. M., Jr. (1968). Renin–angiotensin–aldosterone system during chronic thiazide therapy of benign hypertension. Circulation 37, 27–35. doi: 10.1161/01.CIR.37.1.27
Burst, V., Grundmann, F., Kubacki, T., Greenberg, A., Becker, I., Rudolf, D., et al. (2017). Thiazide-associated hyponatremia, report of the hyponatremia registry: an observational multicenter international study. Am. J. Nephrol 45, 420–430. doi: 10.1159/000471493
Calder, J. A., Schachter, M., Sever, P. S. (1992). Direct vascular actions of hydrochlorothiazide and indapamide in isolated small vessels. Eur. J. Pharmacol. 220, 19–26. doi: 10.1016/0014-2999(92)90006-P
Calder, J. A., Schachter, M., Sever, P. S. (1993). Ion channel involvement in the acute vascular effects of thiazide diuretics and related compounds. J. Pharmacol. Exp. Ther. 265, 1175–1180.
Calder, J. A., Schachter, M., Sever, P. S. (1994). Potassium channel opening properties of thiazide diuretics in isolated guinea pig resistance arteries. J. Cardiovasc Pharmacol. 24, 158–164. doi: 10.1097/00005344-199407000-00024
Calhoun, D. A. (2013). Hyperaldosteronism as a common cause of resistant hypertension. Annu. Rev. Med. 64, 233–247. doi: 10.1146/annurev-med-042711-135929
Calò, L., Davis, P. A., Milani, M., Cantaro, S., Antonello, A., Favaro, S., et al. (1999). Increased endothelial nitric oxide synthase mRNA level in Bartter’s and Gitelman’s syndrome. Relationship to vascular reactivity. Clin. Nephrol. 51, 12–17.
Calò, L., Sartore, G., Bassi, A., Basso, C., Bertocco, S., Marin, R., et al. (1998). Reduced susceptibility to oxidation of low-density lipoprotein in patients with overproduction of nitric oxide (Bartter’s and Gitelman’s syndrome). J. Hypertens. 16, 1001–1008. doi: 10.1097/00004872-199816070-00014
Calò, L. A. (2006). Vascular tone control in humans: insights from studies in Bartter’s/Gitelman’s syndromes. Kidney Int. 69, 963–966. doi: 10.1038/sj.ki.5000253
Channavajjhala, S. K., Bramley, R., Peltz, T., Oosthuyzen, W., Jia, W., Kinnear, S., et al. (2018). Urinary extracellular vesicle protein profiling and endogenous lithium clearance support excessive renal sodium wasting and water reabsorption in thiazide-induced hyponatremia. Kidney Int. Rep. 4, 139–147. doi: 10.1016/j.ekir.2018.09.011
Chapman, A. B., Schwartz, G. L., Boerwinkle, E., Turner, S. T. (2002). Predictors of antihypertensive response to a standard dose of hydrochlorothiazide for essential hypertension. Kidney Int. 61, 1047–1055. doi: 10.1046/j.1523-1755.2002.00200.x
Chen, Z. F., Vaughn, D. A., Beaumont, K., Fanestil, D. D. (1990). Effects of diuretic treatment and of dietary sodium on renal binding of 3H-metolazone. J. Am. Soc. Nephrol. 1, 91–98.
Christensen, K. L., Mulvany, M. J. (1993). Mesenteric arcade arteries contribute substantially to vascular resistance in conscious rats. J. Vasc. Res. 30, 73–79. doi: 10.1159/000158978
Clarke, D. E., Ertel, R. J., Adams, H. R., Buckley, J. P. (1972). Acute and chronic effects of hydrochlorothiazide on vascular adrenergic mechanisms. Eur. J. Pharmacol. 19, 380–384. doi: 10.1016/0014-2999(72)90105-7
Claus-Walker, J., Cardus, D., Griffith, D., Halstead, L. S. (1977). Metabolic effects of sodium restriction and thiazides in tetraplegic patients. Paraplegia 15, 3–10. doi: 10.1038/sc.1977.2
Colas, B., Colas, J. L., Masson, H., Slama, M., Collin, T., Andrejak, M. (2000a). Effect of methyclothiazide on the entry of calcium into vascular smooth muscle cells. Arch Mal Coeur Vaiss. 93, 901–904.
Colas, B., Collin, T., Safraou, F., Chatelain, D., Cordonnier, C., Henry, X., et al. (2001). Direct vascular actions of methyclothiazide in remodeled mesenteric arteries from hypertensive patients. Am. J. Hypertens. 14, 989–994. doi: 10.1016/S0895-7061(01)02158-6
Colas, B., Slama, M., Collin, T., Safar, M., Andrejak, M. (2000b). Mechanisms of methyclothiazide-induced inhibition of contractile responses in rat aorta. Eur. J. Pharmacol. 408, 63–67. doi: 10.1016/S0014-2999(00)00704-4
Colas, B., Slama, M., Masson, H., Colas, J. L., Collin, T., Arnould, M. L., et al. (2000c). Direct vascular actions of methyclothiazide and indapamide in aorta of spontaneously hypertensive rats. Fundam. Clin. Pharmacol. 14, 363–368. doi: 10.1111/j.1472-8206.2000.tb00417.x
Colussi, G., Bettinelli, A., Tedeschi, S., De Ferrari, M. E., Syrén, M. L., Borsa, N., et al. (2007). A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin. J. Am. Soc. Nephrol. 2, 454–460. doi: 10.2215/CJN.02950906
Colussi, G., Rombolà, G., Brunati, C., De Ferrari, M. E. (1997). Abnormal reabsorption of Na+/Cl– by the thiazide-inhibitable transporter of the distal convoluted tubule in Gitelman’s syndrome. Am. J. Nephrol. 17, 103–111. doi: 10.1159/000169082
Conway, J., Lauwers, P. (1960). Hemodynamic and hypotensive effects of long-term therapy with chlorothiazide. Circulation 21, 21–27. doi: 10.1161/01.CIR.21.1.21
Crosley, AP, Jr, Cullen, R. C., White, D., Freeman, J. F., Castillo, C. A., Rowe, G. G. (1960). Studies of the mechanism of action of chlorothiazide in cardiac and renal diseases. I. Acute effects on renal and systemic hemodynamics and metabolism. J. Lab. Clin. Med. 55, 182–190.
Cruz, D. N., Shaer, A. J., Bia, M. J., Lifton, R. P., Simon, D. B. (2001). Yale Gitelman’s and Bartter’s Syndrome Collaborative Study Group. Gitelman’s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 59, 710–717. doi: 10.1046/j.1523-1755.2001.059002710.x
Daniel, E. E., Nash, C. W. (1965). The effects of diuretic and non-diuretic benzothiadiazine and of structurally related diuretic drugs on active ion transport and contractility in smooth muscles. Arch. Int. Pharmacodyn. Ther. 158, 139–154.
Daniel, E. E. (1962). On the mechanism of antihypertensive action of hydrochlorothiazide in rats. Circ. Res. 11, 941–954. doi: 10.1161/01.RES.11.6.941
de Carvalho, J. G., Dunn, F. G., Lohmöller, G., Frohlich, E. D. (1977). Hemodynamic correlates of prolonged thiazide therapy: comparison of responders and nonresponders. Clin. Pharmacol. Ther. 22, 875–880. doi: 10.1002/cpt1977226875
Dhaliwal, J. S., Casey, D. B., Greco, A. J., Badejo, A. M., Jr., Gallen, T. B., Murthy, S. N., et al. (2007). Rho kinase and Ca2+ entry mediate increased pulmonary and systemic vascular resistance in L-NAME-treated rats. Am. J. Physiol. 293, L1306–L1313.
Dollery, C. T., Harington, M., Kaufman, G. (1959). The mode of action of chlorothiazide in hypertension: with special reference to potentiation of ganglion-blocking agents. Lancet 1, 1215–1218. doi: 10.1016/S0140-6736(59)90895-5
Duarte, J. D., Cooper-DeHoff, R. M. (2010). Mechanisms for blood pressure lowering and metabolic effects of thiazide and thiazide-like diuretics. Expert. Rev. Cardiovasc. Ther. 8, 793–802. doi: 10.1586/erc.10.27
Dudenbostel, T., Siddiqui, M., Gharpure, N., Calhoun, D. A. (2017). Refractory versus resistant hypertension: novel distinctive phenotypes. J. Nat. Sci. 3, e430.
Dustan, H. P., Cumming, G. R., Corcoran, A. C., Page, I. H. (1959). A mechanism of chlorothiazide-enhanced effectiveness of antihypertensive ganglioplegic drugs. Circulation 19, 360–365. doi: 10.1161/01.CIR.19.3.360
Eckstein, J. W., Abboud, F. M., Pereda, S. A. (1962). The effect of norepinephrine on cardiac output, arterial blood pressure, and heart rate in dogs treated with chlorothiazide. J. Clin. Invest. 41, 1578–1583. doi: 10.1172/JCI104615
Eladari, D., Chambrey, R. (2011). Identification of a novel target of thiazide diuretics. J. Nephrol. 24, 391–394. doi: 10.5301/JN.2011.8403
Eladari, D., Hübner, C. A. (2011). Novel mechanisms for NaCl reabsorption in the collecting duct. Curr. Opin. Nephrol. Hypertens. 20, 506–511. doi: 10.1097/MNH.0b013e3283486c4a
Ellison, D. H., Loffing, J. (2009). Thiazide effects and adverse effects: insights from molecular genetics. Hypertension 54, 196–202. doi: 10.1161/HYPERTENSIONAHA.109.129171
Faber, J. E., Meininger, G. A. (1990). Selective interaction of alpha-adrenoceptors with myogenic regulation of microvascular smooth muscle. Am. J. Physiol. 259, H1126–H1133. doi: 10.1152/ajpheart.1990.259.4.H1126
Feisal, K. A., Eckstein, J. W., Horsley, A. W., Keasling, H. H. (1961). Effects of chlorothiazide on forearm vascular responses to norepinephrine. J. Appl. Physiol. 16, 549–552. doi: 10.1152/jappl.1961.16.3.549
Finch, L., Hicks, P. E., Moore, R. A. (1977). Changes in vascular reactivity in experimental hypertensive animals following treatment with indapamide. J. Pharm. Pharmacol. 29, 739–743. doi: 10.1111/j.2042-7158.1977.tb11452.x
Friedman, S. M., Nakashima, M., Friedman, C. L. (1960). Relation of saluretic and hypotensive effects of hydrochlorothiazide in the rat. Am. J. Physiol. 198, 148–152. doi: 10.1152/ajplegacy.1960.198.1.148
Freis, E. D. (1959). Treatment of hypertension with chlorothiazide. J. Am. Med. Assoc. 169, 105–108. doi: 10.1001/jama.1959.03000190007002
Freis, E. D., Reda, D. J., Materson, B. J. (1988). Volume (weight) loss and blood pressure response following thiazide diuretics. Hypertension 12, 244–250. doi: 10.1161/01.HYP.12.3.244
Freis, E. D., Wanko, A., Schnaper, H. W., Frohlich, E. D. (1960). Mechanism of the altered blood pressure responsiveness produced by chlorothiazide. J. Clin. Invest. 39, 1277–1281. doi: 10.1172/JCI104143
Freis, E. D., Wanko, A., Wilson, I. M., Parrish, A. E. (1958). Chlorothiazide in hypertensive and normotensive patients. Ann. N. Y. Acad. Sci. 71, 450–455. doi: 10.1111/j.1749-6632.1958.tb46773.x
Friedman, E., Shadel, M., Halkin, H., Farfel, Z. (1989). Thiazide-induced hyponatremia. Reproducibility by single dose rechallenge and an analysis of pathogenesis. Ann. Intern. Med. 110, 24–30. doi: 10.7326/0003-4819-110-1-24
Frindt, G., Yang, L., Uchida, S., Weinstein, A. M., Palmer, L. G. (2017). Responses of distal nephron Na(+) transporters to acute volume depletion and hyperkalemia. Am. J Physiol 313, F62–F73. doi: 10.1152/ajprenal.00668.2016
Frohlich, E. D., Schnaper, H. W., Wilson, I. M., Freis, E. D. (1960). Hemodynamic alterations in hypertensive patients due to chlorothiazide. N. Engl. J. Med. 262, 1261–1263. doi: 10.1056/NEJM196006232622502
Fuchs, M., Moyer, J. H., Newman, B. E. (1960). Human clinical pharmacology of the newer diuretics: benzothiadiazine and phthalimidine. Ann. N. Y. Acad. Sci. 88, 795–808. doi: 10.1111/j.1749-6632.1960.tb20072.x
Genest, J., de Champlain, J., Veryat, R., Boucher, R., Tremblay, G. Y., Strong, C. G., et al. (1964). Role of the renin–angiotensin system in various physiological and pathological states. Hypertension 13, 97–116.
Gifford, RW, Jr, Mattox, V. R., Orvis, A. L., Sones, D. A., Rosevear, J. W. (1961). Effect of thiazide diuretics on plasma volume, body electrolytes, and excretion of aldosterone in hypertension. Circulation 24, 1197–1205. doi: 10.1161/01.CIR.24.5.1197
Gillenwater, J. Y., Scott, J. B., Frohlich, E. D. (1962). Effect of chlorothiazide on response of renal vascular bed to vasoactive substances. Circ. Res. 11, 283–286. doi: 10.1161/01.RES.11.2.283
Glover, M., Clayton, J. (2012). Thiazide-induced hyponatraemia: epidemiology and clues to pathogenesis. Cardiovasc. Ther. 30, e219–e226. doi: 10.1111/j.1755-5922.2011.00286.x
Glover, M., O’Shaughnessy, K. M. (2013). Molecular insights from dysregulation of the thiazide-sensitive WNK/SPAK/NCC pathway in the kidney: gordon syndrome and thiazide-induced hyponatraemia. Clin. Exp. Pharmacol. Physiol. 40, 876–884. doi: 10.1111/1440-1681.12115
Gong, Y., McDonough, C. W., Wang, Z., Hou, W., Cooper-DeHoff, R. M., Langaee, T. Y., et al. (2012). Hypertension susceptibility loci and blood pressure response to antihypertensives: results from the pharmacogenomic evaluation of antihypertensive responses study. Circ. Cardiovasc. Genet. 5, 686–691. doi: 10.1161/CIRCGENETICS.112.964080
Greene, M. A., Boltax, A. J., Niv, M., Rogow, E. (1963). The influence of chlorothiazide upon the cardiovascular response to a vasoconstrictor drug, phenylephrine. Am. J. Med. Sci. 246, 575–583. doi: 10.1097/00000441-196311000-00008
Greene, M. A., Boltax, A. J., Scherr, E. S. (1961). Acute effects of intravenous chlorothiazide upon cardiovascular hemodynamics. Am. Heart J. 62, 659–669. doi: 10.1016/0002-8703(61)90374-X
Greene, M. A., Boltax, A. J., Scherr, E. S., Niv, M. (1964). Mechanisms by which chlorothiazide potentiates the vasodepressor effect of ganglionic blocking agent (trimethaphan). Am. J. Med. 36, 87–95. doi: 10.1016/0002-9343(64)90151-2
Grossman, E., Peleg, E., Carroll, J., Shamiss, A., Rosenthal, T. (1994). Hemodynamic and humoral effects of the angiotensin II antagonist losartan in essential hypertension. Am. J. Hypertens. 7, 1041–1044. doi: 10.1093/ajh/7.12.1041
Hansen, J. (1968). Hydrochlorothiazide in the treatment of hypertension. The effects on blood volume, exchangeable sodium and blood pressure. Acta Med. Scand. 183, 317–321. doi: 10.1111/j.0954-6820.1968.tb10483.x
Hebert, S. C., Gamba, G. (1995). Molecular cloning of the renal diuretic-sensitive electroneutral sodium-(potassium)-chloride cotransporters. Proc. Assoc Am Physicians. 107, 76–80.
Heyndrickx, G. R., Boettcher, D. H., Vatner, S. F. (1976). Effects of angiotensin, vasopressin, and methoxamine on cardiac function and blood flow distribution in conscious dogs. Am. J. Physiol. 231, 1579–1587. doi: 10.1152/ajplegacy.1976.231.5.1579
Hilden, M., Leth, A., Hilden, T. (1968). High haemoglobin values during medical treatment of hypertension. Br. Med. J. 3, 163–165. doi: 10.1136/bmj.3.5611.163
Hofmann, L. M., Sagartz, J. W. (1970). Effects of hydrochlorothiazide, spironolactone and metyrapone on electrolyte excretion and zona glomerulosa width in the sodium depleted rat. Arch. Int. Pharmacodyn. Ther. 185, 76–84.
Holland, O. B., Gomez-Sanchez, C. E., Kuhnert, L. V., Poindexter, C., Pak, C. Y. (1979). Antihypertensive comparison of furosemide with hydrochlorothiazide for black patients. Arch. Intern. Med. 139, 1015–1021. doi: 10.1001/archinte.1979.03630460047016
Holland, O. B., von Kuhnert, L., Campbell, W. B., Anderson, R. J. (1983). Synergistic effect of captopril with hydrochlorothiazide for the treatment of low-renin hypertensive black patients. Hypertension 5, 235–239. doi: 10.1161/01.HYP.5.2.235
Hollander, W., Chobanian, A. V., Wilkins, R. W. (1960). The role of diuretics in the management of hypertension. Ann. N. Y. Acad. Sci. 88, 975–989. doi: 10.1111/j.1749-6632.1960.tb20089.x
Hughes, A. D. (2004). How do thiazide and thiazide-like diuretics lower blood pressure? J. Renin. Angiotensin. Aldosterone Syst. 5, 155–160. doi: 10.3317/jraas.2004.034
Isobe, K., Mori, T., Asano, T., Kawaguchi, H., Nonoyama, S., Kumagai, N., et al. (2013). Development of enzyme-linked immunosorbent assays for urinary thiazide-sensitive Na–Cl cotransporter measurement. Am. J. Physiol. 305, F1374–F1381. doi: 10.1152/ajprenal.00208.2013
Jandhyala, B. S., Cavero, I., Buckley, J. P. (1972). Effects of prolonged hydrochlorothiazide administration on neurogenic tone in the hind limb vasculature. Eur. J. Pharmacol. 17, 357–364. doi: 10.1016/0014-2999(72)90116-1
Jessup, J. A., Brosnihan, K. B., Gallagher, P. E., Chappell, M. C., Ferrario, C. M. (2008). Differential effect of low dose thiazides on the renin angiotensin system in genetically hypertensive and normotensive rats. J. Am. Soc. Hypertens. 2, 106–115. doi: 10.1016/j.jash.2007.10.005
Joo, K. W., Lee, J. W., Jang, H. R., Heo, N. J., Jeon, U. S., Oh, Y. K., et al. (2007). Reduced urinary excretion of thiazide sensitive Na–Cl cotransporter in Gitelman syndrome: preliminary data. Am. J. Kidney Dis. 50, 765–773. doi: 10.1053/j.ajkd.2007.07.022
Joubert, L., Radouco-Thomas, C., Loiselle, J. M., Radouco-Thomas, S., Turmel-Dorion, L., Warren, Y. (1968). A study in human pharmacology: evaluation of four diuretics and a placebo. Can. Med. Assoc. J. 99, 57–63.
Kumagai, K., Suzuki, H., Ichikawa, M., Jimbo, M., Nishizawa, M., Ryuzaki, M., et al. (1996). Comparison of early and late start of antihypertensive agents and baroreceptor reflexes. Hypertension 27, 209–218. doi: 10.1161/01.HYP.27.2.209
Kwon, B. J., Jang, S. W., Choi, K. Y., Kim, D. B., Cho, E. J., Ihm, S. H., et al. (2013). Comparison of the efficacy between hydrochlorothiazide and chlorthalidone on central aortic pressure when added on to candesartan in treatment-naïve patients of hypertension. Hypertens Res. 36, 79–84. doi: 10.1038/hr.2012.143
Lake, C. R., Ziegler, M. G., Coleman, M. D., Kopin, I. J. (1979). Hydrochlorothiazide-induced sympathetic hyperactivity in hypertensive patients. Clin. Pharmacol. Ther. 26, 428–432. doi: 10.1002/cpt1979264428
Lauwers, P., Conway, J. (1960). Effect of long-term treatment with chlorothiazide on body fluids, serum electrolytes, and exchangeable sodium in hypertensive patients. J. Lab. Clin. Med. 56, 401–408.
Leth, A. (1970). Changes in plasma and extracellular fluid volumes in patients with essential hypertension during long-term treatment with hydrochlorothiazide. Circulation 42, 479–485. doi: 10.1161/01.CIR.42.3.479
Leviel, F., Hübner, C. A., Houillier, P., Morla, L., El Moghrabi, S., Brideau, G., et al. (2010). The Na+-dependent chloride–bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J. Clin. Invest. 120, 1627–1635. doi: 10.1172/JCI40145
Li, J., Wang, D. H. (2007). Function and regulation of epithelial sodium transporters in the kidney of a salt-sensitive hypertensive rat model. J. Hypertens. 25, 1065–1072. doi: 10.1097/HJH.0b013e3280a8b87d
Lijnen, P., Fagard, R., Staessen, J., Amery, A. (1981). Effect of chronic diuretic treatment on the plasma renin–angiotensin–aldosterone system in essential hypertension. Br. J. Clin. Pharmacol. 12, 387–392. doi: 10.1111/j.1365-2125.1981.tb01231.x
Lockett, M. F., Nicholas, T. E. (1968). The effects of hydrochlorothiazide and frusemide on noradrenaline sensitivity and blood pressure of salt-loaded rats before and after nephrectomy. Br. J. Pharmacol. Chemother. 33, 136–144. doi: 10.1111/j.1476-5381.1968.tb00481.x
Loffing, J., Vallon, V., Loffing-Cueni, D., Aregger, F., Richter, K., Pietri, L., et al. (2004). Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman’s syndrome. J. Am. Soc. Nephrol. 15, 2276–2288. doi: 10.1097/01.ASN.0000138234.18569.63
Loffing, J., Loffing-Cueni, D., Hegyi, I., Kaplan, M. R., Hebert, S. C., Le Hir, M., et al. (1996). Thiazide treatment of rats provokes apoptosis in distal tubule cells. Kidney Int. 50, 1180–1190. doi: 10.1038/ki.1996.426
Luft, F. C., Fineberg, N. S., Weinberger, M. H. (1991). Long-term effect of nifedipine and hydrochlorothiazide on blood pressure and sodium homeostasis at varying levels of salt intake in mildly hypertensive patients. Am. J. Hypertens. 4, 752–760. doi: 10.1093/ajh/4.9.752
Lund-Johansen, P. (1970). Hemodynamic changes in long-term diuretic therapy of essential hypertension. A comparative study of chlorthalidone, polythiazide and hydrochlorothiazide. Acta Med. Scand. 187, 509–518. doi: 10.1111/j.0954-6820.1970.tb02977.x
Mackenzie, I. S., McEniery, C. M., Dhakam, Z., Brown, M. J., Cockcroft, J. R., Wilkinson, I. B. (2009). Comparison of the effects of antihypertensive agents on central blood pressure and arterial stiffness in isolated systolic hypertension. Hypertension 54, 409–413. doi: 10.1161/HYPERTENSIONAHA.109.133801
Manisty, C. H., Hughes, A. D. (2013). Meta-analysis of the comparative effects of different classes of antihypertensive agents on brachial and central systolic blood pressure, and augmentation index. Br. J. Clin. Pharmacol. 75, 79–92. doi: 10.1111/j.1365-2125.2012.04342.x
Mastroianni, N., De Fusco, M., Zollo, M., Arrigo, G., Zuffardi, O., Bettinelli, A., et al. (1996). Molecular cloning, expression pattern, and chromosomal localization of the human Na–Cl thiazide-sensitive cotransporter (SLC12A3). Genomics 35, 486–493. doi: 10.1006/geno.1996.0388
Maxwell, M. H., Gross, C. (1979). “Tricynafen and hydrochlorothiazide in essential hypertension. Effect of the renin–angiotensin II–aldosterone system and on electrolyte balance,” in Postgrad Med Commun. : A new class of diuretics with uricosuric activity (Symposium, Dallas, TX: Universtiy of Texas, Southwestern Medical School), Nov 1978; 1979, 43–50
McCubbin, J. W., Page, I. H. (1963). Neurogenic component of chronic renal hypertension. Science 139, 210–215. doi: 10.1126/science.139.3551.210
Meier, R., Gross, F., Tripod, J., Turrian, H. (1957). Pharmakologische charakterisirung von synthetischem hyprtensin. Experientia 13, 361–362. doi: 10.1007/BF02179167
Melander, O., Saarela, J., Ripatti, S., Wahlstrand, B., Manunta, P., Kontula, K., et al. (2013). Genomic association analysis of common variants influencing antihypertensive response to hydrochlorothiazide. Hypertension 62, 391–397. doi: 10.1161/HYPERTENSIONAHA.111.00436
Mendlowitz, M., Naftchi, N., Gitlow, S. E., Weinreb, H. L., Wolf, R. L. (1960). The effect of chlorothiazide and its congeners on the digital circulation in normotensive subjects and in patients with essential hypertensions. Ann. N. Y. Acad. Sci. 88, 964–974. doi: 10.1111/j.1749-6632.1960.tb20088.x
Mironneau, J., Savineau, J. P., Mironneau, C. (1981). Compared effects of indapamide, hydrochlorothiazide and chlorthalidone on electrical and mechanical activities in vascular smooth muscle. Eur. J. Pharmacol. 75, 109–113. doi: 10.1016/0014-2999(81)90068-6
Moreno, E., Cristóbal, P. S., Rivera, M., Vázquez, N., Bobadilla, N. A., Gamba, G. (2006). Affinity-defining domains in the Na–Cl cotransporter: a different location for Cl– and thiazide binding. J. Biol. Chem. 281, 17266–17275. doi: 10.1074/jbc.M602614200
Morgan, T., Lauri, J., Bertram, D., Anderson, A. (2004). Effect of different antihypertensive drug classes on central aortic pressure. Am. J. Hypertens. 17, 118–123. doi: 10.1016/j.amjhyper.2003.09.012
Morsing, P., Velázquez, H., Wright, F. S., Ellison, D. H. (1991). Adaptation of distal convoluted tubule of rats. II. Effects of chronic thiazide infusion. Am. J. Physiol. 261, F137–F143. doi: 10.1152/ajprenal.1991.261.1.F137
Musini, V. M., Nazer, M., Bassett, K., Wright, J. M. (2014). Blood pressure-lowering efficacy of monotherapy with thiazide diuretics for primary hypertension. Cochrane Database Syst. Rev. (5), CD003824. doi: 10.1002/14651858.CD003824.pub2
Na, K. Y., Oh, Y. K., Han, J. S., Joo, K. W., Lee, J. S., Earm, J. H., et al. (2003). Upregulation of Na+ transporter abundances in response to chronic thiazide or loop diuretic treatment in rats. Am. J. Physiol. 284, F133–F143. doi: 10.1152/ajprenal.00227.2002
Nadal, J., Channavajjhala, S. K., Jia, W., Clayton, J., Hall, I. P., Glover, M. (2018). Clinical and molecular features of thiazide-induced hyponatremia. Curr. Hypertens. Rep. 20, 31. doi: 10.1007/s11906-018-0826-6
Nakamura, A., Hayashi, K., Ozawa, Y., Fujiwara, K., Okubo, K., Kanda, T., et al. (2003). Vessel- and vasoconstrictor-dependent role of rho/rho-kinase in renal microvascular tone. J. Vasc. Res. 40, 244–251.
Neuvonen, P. J. (1971). Influence of diuretics and diazoxide on ions and vascular reactivity in normotensive and spontaneously hypertensive rats. Ann. Med. Exp. Biol. Fenn. 49, 109–119.
Nicholas, T. E. (1970). Potentiation of the effects of noradrenaline and of sympathetic stimulation of the perfused rat caudal artery by angiotensin. J. Pharm. Pharmacol. 22, 37–41. doi: 10.1111/j.2042-7158.1970.tb08381.x
Nicholas, T. E. (1971). Responses of mean arterial pressure to pressor agents and diuretics in renal hypertensive and salt hypertensive rats. Br. J. Pharmacol. 42, 179–192. doi: 10.1111/j.1476-5381.1971.tb07099.x
Nijenhuis, T., Vallon, V., van der Kemp, A. W., Loffing, J., Hoenderop, J. G., Bindels, R. J. (2005). Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J. Clin. Invest. 115, 1651–1658. doi: 10.1172/JCI24134
Ogilvie, R. I., Schlieper, E. (1970). The effect of hydrochlorothiazide on venous reactivity in hypertensive man. Clin. Pharmacol. Ther. 11, 589–594. doi: 10.1002/cpt1970114589
Olde Engberink, R. H., Frenkel, W. J., van den Bogaard, B., Brewster, L. M., Vogt, L., van den Born, B. J. (2015). Effects of thiazide-type and thiazide-like diuretics on cardiovascular events and mortality: systematic review and meta-analysis. Hypertension 65, 1033–1040. doi: 10.1161/HYPERTENSIONAHA.114.05122
Overbeck, W., Haddy, F. J. (1960). Acute effect of chlorothiazide upon vascular resistance in the dog forelimb. Clin. Res. 8, 189. doi: 10.1016/0002-9149(61)90131-X
Pathare, G., Tutakhel, O. A. Z., van der Wel, M. C., Shelton, L. M., Deinum, J., Lenders, J. W. M., et al. (2017). Hydrochlorothiazide treatment increases the abundance of the NaCl cotransporter in urinary extracellular vesicles of essential hypertensive patients. Am. J Physiol 312, F1063–F1107. doi: 10.1152/ajprenal.00644.2016
Payne, J. A. (2012). Molecular operation of the cation chloride cotransporters: ion binding and inhibitor interaction. Curr. Top Membr. 70, 215–237. doi: 10.1016/B978-0-12-394316-3.00006-5
Peng, X., Jiang, L., Chen, C., Qin, Y., Yuan, T., Wang, O., et al. (2017). Increased urinary prostaglandin E2 metabolite: a potential therapeutic target of Gitelman syndrome. PLoS One 12, e0180811. doi: 10.1371/journal.pone.0180811
Peng, X. Y., Jiang, L. P., Yuan, T., Yue, C., Zheng, K., Wang, O., et al. (2016). Value of chloride clearance test in differential diagnosis of Gitelman syndrome. Zhongguo. Yi. Xue. Ke. Xue. Yuan. Xue Bao. 38, 275–282.
Peng, X., Zhao, B., Zhang, L., Jiang, L., Yuan, T., Wang, Y., et al. (2018). Hydrochlorothiazide test as a tool in the diagnosis of Gitelman syndrome in Chinese patients. Front. Endocrinol. 9, 559. doi: 10.3389/fendo.2018.00559
Pickkers, P., Garcha, R. S., Schachter, M., Smits, P., Hughes, A. (1999). Inhibition of carbonic anhydrase accounts for the direct vascular effects of hydrochlorothiazide. Hypertension 33, 1043–1048. doi: 10.1161/01.HYP.33.4.1043
Pickkers, P., Hughes, A. D. (1995). Relaxation and decrease in [Ca2+] i by hydrochlorothiazide in guinea-pig isolated mesenteric arteries. Br. J. Pharmacol. 114, 703–707. doi: 10.1111/j.1476-5381.1995.tb17195.x
Pickkers, P., Hughes, A. D., Russel, F. G., Thien, T., Smits, P. (1998). Thiazide-induced vasodilation in humans is mediated by potassium channel activation. Hypertension 32, 1071–1076. doi: 10.1161/01.HYP.32.6.1071
Pimenta, E., Gaddam, K. K., Oparil, S., Aban, I., Husain, S., Dell’Italia, L. J., et al. (2009). Effects of dietary sodium reduction on blood pressure in subjects with resistant hypertension: results from a randomized trial. Hypertension 54, 475–481. doi: 10.1161/HYPERTENSIONAHA.109.131235
Pourafshar, N., Alshahrani, S., Karimi, A., Soleimani, M. (2018). Thiazide therapy in chronic kidney disease: renal and extra renal targets. Curr. Drug. Metab. 19, 1012–1020. doi: 10.2174/1389200219666180702104559
Preziosi, P., Bianchi, A., Loscalzo, B., De Schaepdyver, A. F. (1959). On the pharmacology of chlorothiazide, with special regard to its diuretic and anti-hypertensive effects. Arch. Int. Pharmacodyn. Ther. 118, 467–495.
Preziosi, P., De Schaepdryver, A. F., Marmo, E., Miele, E. (1961). On the mechanism of the anti-hypertensive effect of hydrochlorothiazide. Arch. Int. Pharmacodyn. Ther. 131, 209–229. doi: 10.1159/000219490
Priddle, W. W., Liu, S. F., Breithaupt, D. J. (1970). Management of hypertension—further sodium and potassium studies. J. Am. Geriatr. Soc 18, 861–92. doi: 10.1111/j.1532-5415.1970.tb02840.x
Rafiqi, F. H., Zuber, A. M., Glover, M., Richardson, C., Fleming, S., Jovanović, S., et al., (2010). Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med. 2, 63–75.
Rapoport, R. M., LeBlanc, A. J., Jason, E., Beare, J. E., Soleimani, M. (2019). Lack of thiazide diuretic inhibition of agonist constriction of mouse mesenteric arterioles ex vivo. Naunyn. Schmiedebergs. Arch. Pharmacol. 392, 117–121. doi: 10.1007/s00210-018-1590-5
Riveira-Munoz, E., Chang, Q., Godefroid, N., Hoenderop, J. G., Bindels, R. J., Dahan, K., et al. (2007a). Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J. Am. Soc. Nephrol. 18, 1271–1283. doi: 10.1681/ASN.2006101095
Riveira-Munoz, E., Chang, Q., Bindels, R. J., Devuyst, O. (2007b). Gitelman’s syndrome: towards genotype-phenotype correlations? Pediatr. Nephrol. 22, 326–332. doi: 10.1007/s00467-006-0321-1
Sá, A. C. C., Webb, A., Gong, Y., McDonough, C. W., Shahin, M. H., Datta, S., et al. (2018). Blood pressure signature genes and blood pressure response to thiazide diuretics: results from the PEAR and PEAR-2 studies. BMC Med. Genomics. 11, 55. doi: 10.1186/s12920-018-0370-x
Sabath, E., Meade, P., Berkman, J., de los Heros, P., Moreno, E., Bobadilla, N. A., et al. (2004). Pathophysiology of functional mutations of the thiazide-sensitive Na–Cl cotransporter in Gitelman disease. Am. J Physiol 287, F195–F203. doi: 10.1152/ajprenal.00044.2004
Sander, G. E., Giles, T. D. (2011). Resistant hypertension: concepts and approach to management. Curr. Hypertens. Rep. 13, 347–355. doi: 10.1007/s11906-011-0226-7
Sartori, M., Parotto, E., Bonso, E., Semplicini, A., Palatini, P., Pessina, A. C., et al. (2007). Autonomic nervous system function in chronic hypotension associated with Bartter and Gitelman syndromes. Am. J. Kidney Dis. 49, 330–335. doi: 10.1053/j.ajkd.2006.10.023
Schultheis, P. J., Lorenz, J. N., Meneton, P., Nieman, M. L., Riddle, T. M., Flagella, M., et al. (1998). Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl– cotransporter of the distal convoluted tubule. J. Biol. Chem. 273, 29150–29155. doi: 10.1074/jbc.273.44.29150
Shah, S., Khatri, I., Freis, E. D. (1978). Mechanism of antihypertensive effect of thiazide diuretics. Am. Heart J. 95, 611–618. doi: 10.1016/0002-8703(78)90303-4
Shahin, M. H., Gong, Y., Frye, R. F., Rotroff, D. M., Beitelshees, A. L., Baillie, R. A., et al. (2017). Sphingolipid metabolic pathway impacts thiazide diuretics blood pressure response: insights from genomics, metabolomics, and lipidomics. J. Am. Heart. Assoc. 7, e006656. doi: 10.1161/JAHA.117.006656
Shahin, M. H., Johnson, J. A. (2016). Mechanisms and pharmacogenetic signals underlying thiazide diuretics blood pressure response. Curr. Opin. Pharmacol. 27, 31–37. doi: 10.1016/j.coph.2016.01.005
Siddiqui, M., Dudenbostel, T., Calhoun, D. A. (2016). Resistant and refractory hypertension: antihypertensive treatment resistance vs treatment failure. Can. J. Cardiol. 32, 603–606. doi: 10.1016/j.cjca.2015.06.033
Sigaroudi, A., Kinzig, M., Wahl, O., Stelzer, C., Schroeter, M., Fuhr, U., et al. (2018). Quantification of hydrochlorothiazide and ramipril/ramiprilate in blood serum and cerebrospinal fluid: a pharmacokinetic assessment of central nervous system adverse effects. Pharmacology 102, 133–137. doi: 10.1159/000489999
Silah, J. G., Jones, R. E., Bashour, F. A., Kaplan, N. M. (1965). The effect of acute administration of chlorothiazide upon the pressor responsiveness to angiotensin and norepinephrine. Am. Heart J. 69, 301–305. doi: 10.1016/0002-8703(65)90269-3
Simon, D. B., Nelson-Williams, C., Bia, M. J., Ellison, D., Karet, F. E., Molina, A. M., et al. (1996). Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–Cl cotransporter. Nat. Genet. 12, 24–30. doi: 10.1038/ng0196-24
Sinning, A., Radionov, N., Trepiccione, F., López-Cayuqueo, K. I., Jayat, M., Baron, S., et al. (2017). Double knockout of the Na+-driven Cl–/HCO3– exchanger and Na+/Cl– cotransporter induces hypokalemia and volume depletion. J. Am. Soc. Nephrol. 28, 130–139. doi: 10.1681/ASN.2015070734
Sládková, M., Kojsová, S., Jendeková, L., Pechánová, O. (2007). Chronic and acute effects of different antihypertensive drugs on femoral artery relaxation of l-NAME hypertensive rats. Physiol. Res. 56 Suppl 2, S85–S91.
Soleimani, M., Barone, S., Xu, J., Shull, G. E., Siddiqui, F., Zahedi, K., et al. (2012). Double knockout of pendrin and Na–Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc. Natl. Acad. Sci. U.S.A. 109, 13368–13373. doi: 10.1073/pnas.1202671109
Stanton, H. C., White, J. B., Jr. (1965). Hypotensive actions of drugs on unanesthetized normotensive and “metacorticoid” hypertensive rats determined by a direct recording technique. Arch. Int. Pharmacodyn. Ther. 154, 351–363.
Svendsen, U. G., Ibsen, H., Rasmussen, S., Leth, A., Nielsen, M. D., Dige-Petersen, H., et al. (1983). Effects of amiloride on plasma and total body potassium, blood pressure, and the renin–angiotensin–aldosterone system in thiazide-treated hypertensive patients. Clin. Pharmacol. Ther. 34, 448–453. doi: 10.1038/clpt.1983.196
Swenson, E. R. (2014). New insights into carbonic anhydrase inhibition, vasodilation, and treatment of hypertensive-related diseases. Curr. Hypertens. Rep. 16, 467. doi: 10.1007/s11906-014-0467-3
Talso, P. J., Carballo, A. J. (1960). Effects of benzothiadiazines on serum and total body electrolytes. Ann. N. Y. Acad. Sci. 88, 822–840. doi: 10.1111/j.1749-6632.1960.tb20075.x
Tamargo, J., Segura, J., Ruilope, L. M. (2014). Diuretics in the treatment of hypertension. Part 1: thiazide and thiazide-like diuretics. Expert. Opin. Pharmacother. 15, 527–547. doi: 10.1517/14656566.2014.879118
Tapia, F. A., Dustan, H. P., Schenckloth, R. A., Corcoran, A. C., Page, I. H. (1957). Enhanced effectiveness of ganglion-blocking agents in hypertensive patients during administration of a saluretic agent (chlorothiazide). Lancet 273, 831–833. doi: 10.1016/S0140-6736(57)91494-0
Tarazi, R. C., Dustan, H. P., Frohlich, E. D. (1970). Long-term thiazide therapy in essential hypertension. Evidence for persistent alteration in plasma volume and renin activity. Circulation 41, 709–717. doi: 10.1161/01.CIR.41.4.709
Tobian, L. (1967). Why do thiazide diuretics lower blood pressure in essential hypertension? Annu. Rev. Pharmacol. 7, 399–408. doi: 10.1146/annurev.pa.07.040167.002151
Tobian, L., Coffee, K. (1964). Effect of thiazide drugs on renovascular hypertension in contrast to their effect on essential hypertension. Proc. Soc. Exp. Biol. Med. 115, 196–198. doi: 10.3181/00379727-115-28868
Tobian, L., Janecek, J., Foker, J., Ferreira, D. (1962). Effect of chlorothiazide on renal juxtaglomerular cells and tissue electrolytes. Am. J. Physiol. 202, 905–908. doi: 10.1152/ajplegacy.1962.202.5.905
Tran, J. M., Farrell, M. A., Fanestil, D. D. (1990). Effect of ions on binding of the thiazide-type diuretic metolazone to kidney membrane. Am. J. Physiol. 258, F908–F915. doi: 10.1152/ajprenal.1990.258.4.F908
Tsukamoto, T., Kobayashi, T., Kawamoto, K., Fukase, M., Chihara, K. (1995). Possible discrimination of Gitelman’s syndrome from Bartter’s syndrome by renal clearance study: report of two cases. Am. J. Kidney Dis 25, 637–641. doi: 10.1016/0272-6386(95)90137-X
Turner, S. T., Boerwinkle, E., O’Connell, J. R., Bailey, K. R., Gong, Y., Chapman, A. B., et al. (2013). Genomic association analysis of common variants influencing antihypertensive response to hydrochlorothiazide. Hypertension 62, 391–397. doi: 10.1161/HYPERTENSIONAHA.111.00436
Tutakhel, O. A., Jeleń, S., Valdez-Flores, M., Dimke, H., Piersma, S. R., Jimenez, C. R., et al. (2016). Alternative splice variant of the thiazide-sensitive NaCl cotransporter: a novel player in renal salt handling. Am. J. Physiol. 310, F204–F216. doi: 10.1152/ajprenal.00429.2015
van Brummelen, P., Man in ‘t Veld, A. J., Schalekamp, M. A. (1980). Hemodynamic changes during long-term thiazide treatment of essential hypertension in responders and nonresponders. Clin. Pharmacol. Ther. 27, 328–336. doi: 10.1038/clpt.1980.44
Veterans Administration Cooperative Study on Antihypertensive Agents (1962). Double blind control study of antihypertensive agents: III. Chlorothiazide alone and in combination with other agents; preliminary results. Arch. Intern. Med. 110, 230–236. doi: 10.1001/archinte.1962.03620200090016
Villarreal, H., Exaire, J. E., Revollo, A., Soni, J. (1962). Effects of chlorothiazide on systemic hemodynamics in essential hypertension. Circulation 26, 405–408. doi: 10.1161/01.CIR.26.3.405
Vogt, L., Waanders, F., Boomsma, F., de Zeeuw, D., Navis, G. (2008). Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J. Am. Soc. Nephrol. 19, 999–1007. doi: 10.1681/ASN.2007060693
Vongpatanasin, W. (2014). Resistant hypertension: a review of diagnosis and management. JAMA 311, 2216–2224. doi: 10.1001/jama.2014.5180
Wang, L., Dong, C., Xi, Y. G., Su, X. (2015a). Thiazide-sensitive Na+-Cl– cotransporter: genetic polymorphisms and human diseases. Acta Biochim. Biophys. Sin. (Shanghai). 47, 325–334. doi: 10.1093/abbs/gmv020
Wang, J., Qiu, B., Du, J. L., Deng, S. B., Liu, Y. J., She, Q. (2015b). The effects of a low-salt diet on the efficacy of different antihypertensive drug regimens. J. Clin. Pharmacol. 55, 1362–1368. doi: 10.1002/jcph.559
Ware, J. S., Wain, L. V., Channavajjhala, S. K., Jackson, V. E., Edwards, E., Lu, R., et al. (2017). Phenotypic and pharmacogenetic evaluation of patients with thiazide-induced hyponatremia. J. Clin. Invest 127, 3367–3374. doi: 10.1172/JCI89812
Wilkins, R. W., Hollander, W., Chobanian, A. V. (1958). Chlorothiazide in hypertension: studies on its mode of action. Ann. N. Y. Acad. Sci. 71, 465–472. doi: 10.1111/j.1749-6632.1958.tb46775.x
Wilson, I. M., Freis, E. D. (1959). Relationship between plasma and extracellular fluid volume depletion and the antihypertensive effect of chlorothiazide. Circulation 20, 1028–1036. doi: 10.1161/01.CIR.20.6.1028
Winer, B. M. (1961). The antihypertensive actions of benzothiadiazines. Circulation 23, 211–218. doi: 10.1161/01.CIR.23.2.211
Xiao, X., Du, H. J., Hu, W. J., Shaw, P. X. (2013). The influence of long term hydrochlorothiazide administration on the relationship between renin–angiotensin–aldosterone system activity and plasma glucose in patients with hypertension. Oxid. Med. Cell. Longev. doi: 10.1155/2013/434618
Xu, J., Barone, S., Zahedi, K., Brooks, M., Soleimani, M. (2018). SLC4A8 in the kidney: expression, subcellular localization and role in salt reabsorption. Cell. Physiol. Biochem. 50, 1361–1375. doi: 10.1159/000494596
Yang, S. S., Fang, Y. W., Tseng, M. H., Chu, P. Y., Yu, I. S., Wu, H. C., et al. (2013). Phosphorylation regulates NCC stability and transporter activity in vivo. J. Am. Soc. Nephrol. 24, 1587–1597. doi: 10.1681/ASN.2012070742
Yu, D., Chen, Y., Hao, K. (2015). The pharmacokinetic–pharmacodynamic model of telmisartan and hydrochlorothiazide on blood pressure and plasma potassium after long-term administration in spontaneously hypertensive rats. Fundam. Clin. Pharmacol. 29, 543–552. doi: 10.1111/fcp.12152
Zhu, Z., Zhu, S., Liu, D., Cao, T., Wang, L., Tepel, M. (2005). Thiazide-like diuretics attenuate agonist-induced vasoconstriction by calcium desensitization linked to Rho kinase. Hypertension 45, 233–239. doi: 10.1161/01.HYP.0000152701.97426.5f
Keywords: hypertension, thiazide diuretics, diuresis, plasma volume, vasoconstriction, arterial blood pressure, renal, extrarenal
Citation: Rapoport RM and Soleimani M (2019) Mechanism of Thiazide Diuretic Arterial Pressure Reduction: The Search Continues. Front. Pharmacol. 10:815. doi: 10.3389/fphar.2019.00815
Received: 12 March 2019; Accepted: 24 June 2019;
Published: 27 August 2019.
Edited by:
Issy Laher, University of British Columbia, CanadaReviewed by:
Chris Rembold, University of Virginia, United StatesJosé Ramón López-López, University of Valladolid, Spain
Copyright © 2019 Rapoport and Soleimani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert M. Rapoport, cm9iZXJ0LnJhcG9wb3J0QHVjLmVkdQ==