 Li-Sha Wang1
Li-Sha Wang1 Qi Chang
Qi Chang- 1Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2National Key Laboratory of Human Factors Engineering and the State Key Laboratory of Space Medicine Fundamentals and Application, China Astronaut Research and Training Center, Beijing, China
Amyloid-β1–42 (Aβ1–42) oligomers play an important role at the early stage of Alzheimer’s disease (AD) and have been a vital target in the development of therapeutic drugs for AD. Cajaninstilbene acid (CSA), a major bioactive stilbene isolated from pigeon pea (Cajanus cajan) leaves, exerted the neuroprotective property in our previous studies. The present study utilized a validated mouse model of early-stage AD induced by bilateral injection of Aβ1–42 oligomers into hippocampal CA1 regions (100 pmol/mouse) to investigate the cognitive enhancing effects of CSA and the underlying mechanism, by a combination of animal behavioral tests, immunohistochemistry, liquid chromatography-tandem mass spectrometry analysis, and Western blot methods. Intragastric administration of CSA (7.5, 15, and 30 mg/kg) attenuated the impairment of learning and memory induced by Aβ1–42 oligomers. CSA stimulated Aβ clearance and prevented microglial activation and astrocyte reactivity in the hippocampus of model mice. It also decreased the high levels of Glu but increased the low levels of GABA. In addition, CSA inhibited excessive expression of GluN2B-containing NMDARs and upregulated the downstream PKA/CREB/BDNF/TrkB signaling pathway. These results suggest that CSA could be a potential therapeutic agent at the early stage of AD.
Introduction
Alzheimer’s disease (AD) is a main form of dementia in the elderly and characterized by the accumulation of senile plaques which are composed of fibrillar amyloid-β peptide (Aβ) (Cohen and Calkins, 1959). Recent experiments suggest that Aβ oligomers are more toxic than fibrils (Dahlgren et al., 2002; Balducci et al., 2010; DaRocha-Souto et al., 2011) and have already been detected in AD-affected human brains, 7PA2 cells and Tg2576 mice(Walsh et al., 2002; Gong et al., 2003; Lesne et al., 2006), inducing AD-type pathology (Walsh et al., 2002; De Felice et al., 2008; Jin et al., 2011). Based on the important role of Aβ oligomers in Alzheimer’s dementia, the Aβ oligomers hypothesis was proposed (Klein, 2002). Aβ oligomers form readily at low concentrations, whereas significant higher levels of Aβ monomer are required to foster protofibrils formation, suggesting that Aβ oligomers would be first to appear and involve in early-stage AD(Klein, 2002). Aβ1–42 aggregates more rapidly than Aβ1–40 (Snyder et al., 1994) and is more toxic than Aβ1–40 (Sultana et al., 2009). Mutations in amyloid precursor protein, presenilin-1 or presenilin-2 have been shown to increase the production of Aβ1–42 and lead to early onset of AD (Selkoe, 2001). Therefore, Aβ1–42 oligomers play an important role at the early stage of AD.
Currently available treatments for AD are symptomatic and do not decelerate or prevent AD progression (Herrmann et al., 2011). In light of past failures of anti-Aβ antibodies (solanezumab, bapineuzumab, and gantenerumab) in phase III trials, the AD community has increasingly embraced the idea that disease modification depends on earlier initiation of treatment (Mullard, 2016). Animal models with excess Aβ burden in the brain are vital tools for modeling AD states. Pathogen-induced AD models produced by direct injection of Aβ into the brain can not only show AD-like behavioral abnormalities but also exhibit Aβ pathology to some extent (Kim et al., 2016). Compared with transgenic models, the Aβ-injected animal models are economical, time-saving, and allow researchers to control different conditions to focus on the downstream pathology of Aβ abnormalities. Accumulating studies have adopted the Aβ1–42 oligomers-injected animal models to screen therapeutic agents for early-stage AD.
After intracerebral injection, Aβ1–42 oligomers have been shown to impair animal recognition memory in the object recognition task (Figueiredo et al., 2013), spatial learning, and memory in Morris water maze (MWM) (Zhang et al., 2014), working memory in Y maze (Moon et al., 2011), and fear-aggravated memory in passive avoidance test (Choi et al., 2011). The underlying mechanism involves the abnormal status of neurons, astrocytes and microglia (Choi et al., 2011; Ledo et al., 2016; Kasza et al., 2017; Chun et al., 2018). Activated microglia release proinflammatory mediators which influence the efficiency of astrocytic glutamate (Glu) uptake (Zhao et al., 2004). Aβ also downregulates Glu uptake capacity of astrocytes (Matos et al., 2008) and induces the release of astrocytic and microglial Glu (Noda et al., 1999; Talantova et al., 2013). The inhibitory amino acid neurotransmitter γ-aminobutyric acid (GABA) synthesized from Glu contributes to the progression of AD (Bai et al., 2015). Dyshomeostasis of Glu and GABA cause neuronal damage through their downstream receptors.
Among Glu receptors, N-methyl-D-aspartate receptors (NMDARs) are the most important channels mediating Glu excitotoxicity due to their widespread distributions in the central nervous system. The majority of NMDARs are tetrameric complexes containing two obligatory GluN1 subunits and two regulatory GluN2A-D subunits (Cull-Candy et al., 2001). Recent evidence shows that alternation of NMDARs activity plays a vital role in Aβ1–42 oligomers-induced neurotoxicity. Low molecular weight fractions of Aβ1–42 oligomers induce internalization of GluN1 in cultured hippocampal neurons (Figueiredo et al., 2013). Aβ1–42 oligomers significantly reduce synaptic GluN1, GluN2A and GluN2B expression in primary hippocampal neurons (Geng et al., 2013), but activate extrasynaptic GluN2B (Li et al., 2011; Talantova et al., 2013). The activation of extrasynaptic NMDARs contributes to dephosphorylation of cAMP response element binding protein (CREB) (Hardingham et al., 2002), a transcription factor required for long-term memory and neuronal survival and activated by cAMP-dependent protein kinase (PKA). CREB shut-off suppresses the expression of the gene encoding brain-derived neurotrophic factor (BDNF) and leads to less BDNF binding to tropomyosin receptor kinase B (TrkB).
Cajaninstilbene acid (CSA), a main bioactive stilbene from pigeon pea [Cajanus cajan (L.) Millsp.] leaves, possesses many bioactivities, including anti-bacterial (Kong et al., 2010), anti-tumor (Fu et al., 2015), anti-osteoporosis (Sun et al., 2019), anti-oxidative (Wu et al., 2011), anti-inflammatory (Patel and Bhutani, 2014; Huang et al., 2016; Schuster et al., 2016), and neuroprotective (Jiang et al., 2014; Liu et al., 2014a) effects. It is reported that stilbenes containing extract-fraction from Cajanus cajan (L.) leaves (sECC) ameliorated Aβ25–35-induced cognitive impairment (Ruan et al., 2009). As a component in sECC, CSA protected corticosterone-induced impairment in PC12 cells through the inhibition of oxidative stress, endoplasmic reticulum stress-mediated and mitochondrial apoptotic pathways in our previous studies (Jiang et al., 2014; Liu et al., 2014a). In addition, some stilbenes, including resveratrol (Wang et al., 2017), trans-2,4-dimethoxystibene (Ruan et al., 2010a; Ruan et al., 2010b), rhapontigenin (Misiti et al., 2006) and piceatannol (Kim et al., 2007), with similar chemical structures of CSA were reported to have protective effects against Aβ neurotoxicity. Therefore, it is hypothesized that CSA could ameliorate cognitive impairment and be a potential therapeutic agent at the early stage of AD.
To our knowledge, there has been no in vivo publication reported the cognitive effects of CSA so far. The present study was conducted to evaluate the cognitive enhancing effects of CSA using a validated mouse model of early-stage AD induced by intrahippocampal injection of Aβ1–42 oligomers. The underlying mechanism was explored by monitoring the status of neurons, astrocytes and microglia, detecting the intrahippocampal amount of Aβ, measuring the homeostasis of Glu and GABA, and determining the expression of GluN1 and GluN2B subunits and downstream PKA/CREB/BDNF/TrkB signaling pathway.
Materials and Methods
Preparation of Aβ1–42 Oligomers
Aβ1–42 oligomers were generated as the previously described method (Stine et al., 2003; Fa et al., 2010). Briefly, Aβ1–42 (A9810, Sigma, USA) was initially dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, 105228, Sigma, USA) to a final concentration of 1 mM and incubated at room temperature for 2 h. The peptide solution was aliquoted and dried in the fume hood. Traces of HFIP was removed under vacuum in a speedvac (Labconco, USA), and the thin clear peptide film was stored over desiccant at −80ºC. For aggregation, the aliquoted peptide film was dissolved in dimethyl sulfoxide (DMSO) to 5 mM. The peptide in DMSO was diluted directly into phosphate buffered saline (PBS, 1×) at 100 μM and incubated at 4 ºC for 16 h to make the oligomeric form of Aβ1–42. Following incubation, the solution was centrifuged at 14,000 × g for 10 min at 4 ºC. Then the supernatant was transferred to a new tube and diluted with the same volume of PBS to a final concentration of 50 μM before stereotactic injections. The Aβ1–42 oligomers preparations were routinely characterized by atomic force microscopy (AFM)(Stine et al., 1996) and Western blot using anti-Aβ antibody 6E10 (Lambert et al., 1998) (Table 1).
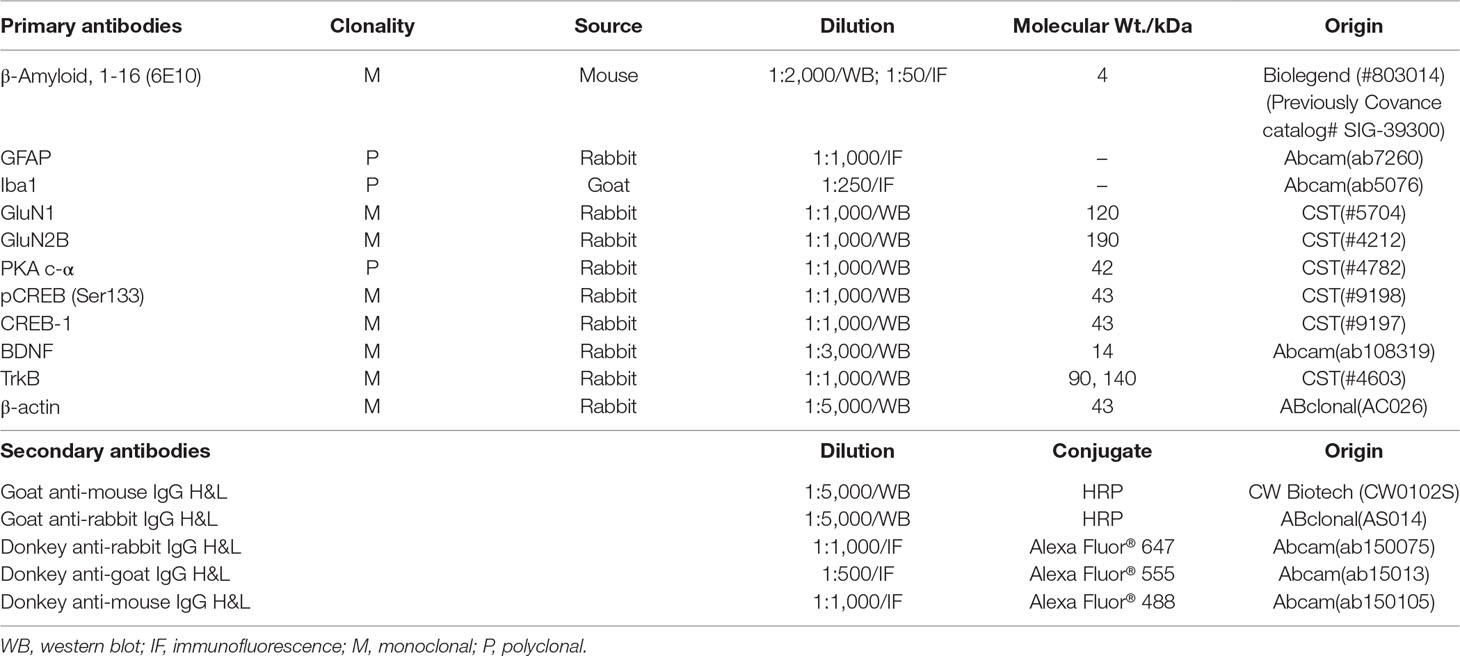
Table 1 Antibodies used in this study.
Animals and Stereotactic Injections
Male ICR mice (25–30 g, 6 w) were purchased from Vital River Experimental Animal CO. LTD (Beijing, China). Animals were housed with free access to standard food and water under a 12 h light/dark cycle with controlled room temperature and humidity. Animals were habituated for 1 week to the new environment before initiating surgery. All procedures were conducted in accordance with the protocols approved by the Animal Ethical Committee at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences.
Mice were anesthetized with 2% isoflurane and placed in a stereotaxic apparatus (Benchmark, USA). The Aβ1–42 oligomers preparations (50 μM) or an equivalent volume of vehicle (1% DMSO in PBS) were injected into the bilateral CA1 molecular layer of hippocampus (1 μL in each side) using a Hamilton syringe (10 μL, 701N, ga26s/51 mm/pst3; Hamilton, Bonaduz, GR, Switzerland) and mounted on a nano-pump syringe (Benchmark, USA) with a speed of 0.4 μL/min. The coordinates of the injection sites were adapted from the Paxinos and Franklin atlas (Paxinos and Franklin, 2001): −2.0 mm posterior to bregma, ± 1.5 mm lateral to the midline, and 1.5 mm deep from dura. After injection, the needle was left in place for another 5 min and withdrawn slowly.
Groups and Drug Administration
The mice were numbered according to their body weight and then randomly divided into six experimental groups using a random number table: (1) a control group (without stereotactic injections); (2) a sham-operated group; (3) Aβ1–42 oligomers group; (4) Aβ1–42 oligomers + CSA 7.5 mg/kg/d group; (5) Aβ1–42 oligomers + CSA 15 mg/kg/d; (6) Aβ1–42 oligomers + CSA 30 mg/kg/d. CSA (98.0% purity by HPLC) was separated from the leaves of Pigeon pea in our laboratory (Liu et al., 2014b), suspended in 0.5% carboxymethylcellulose sodium (CMC-Na) water solution and intragastrically administered after surgery. Meanwhile, the control, sham and Aβ1–42 oligomers groups were received the same volume of 0.5% CMC-Na water solution intragastrically. Learning and memory capacity was assessed using object location recognition (OLR), open filed, MWM, and passive avoidance tests. The experimental schedule is shown in Figure 1. Another same experiment was operated without behavioral tests, and mice were sacrificed for tissue collection seven days after stereotactic injections.
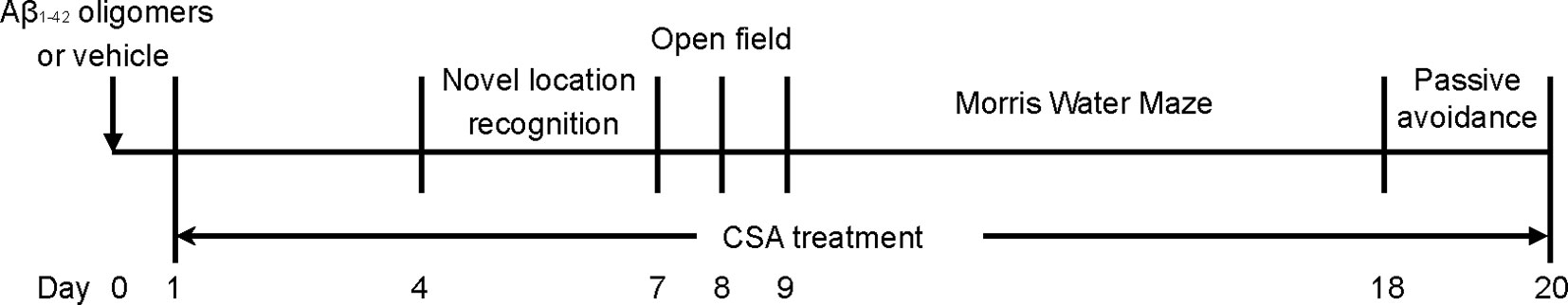
Figure 1 Experimental schedule of behavioral performance follow-up.
Behavioral Tests
Object Location Recognition (OLR) Test
Object location memory was measured using published methods (Dong et al., 2019). The test was performed in an open field arena, to which animals were first habituated for three consecutive days. The next day, two identical objects were introduced to the arena. In familiar trials, mice were allowed to freely explore the arena with the two objects for 5 min. In test trials 20 min thereafter, mice again encountered the two objects for 5 min, except that one of them had been moved to a new location. The trials were recorded using a camera mounted above the arena and scored for the amount of time spent sniffing or touching the objects. The object-location discrimination index is calculated as dividing the time spent exploring the object in the novel location over the total exploring time.
Open Field Test
The locomotor activity of mice was assessed using an open-field computer-aided control system (Lu et al., 2018). After the OLR test, mice were placed in a square box and allowed to freely move for 10 min while being recorded by an overhead camera. The total distance of moving was recorded automatically.
Morris Water Maze (MWM) Test
The spatial memory was evaluated according to the protocol of MWM (Vorhees and Williams, 2006). A black stainless-steel circular tank (100 cm diameter, 40 cm height) was filled with water (23 ± 1°C) being opaque by adding nontoxic black ink and divided into four equal quadrants. A black circular escape platform (6 cm diameter) was submerged 1.5 cm below the water surface. Some visual cues surrounding this tank kept permanent place throughout all MWM tests. Mice were monitored by a video camera tracking system directly above the water tank as they swim. Parameters were measured using the software system developed by the China Astronaut Center and the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, and Peking Union Medical College.
In the spatial acquisition test, mice were trained three trials per day for five consecutive days. The platform was placed in the target quadrant, and one of the other three quadrants was chosen in each trial as the start location in a semi-random set. Before each trial, mice were allowed to stay in the platform for 10 s. After that, mice were released into the water and were given a maximum of 90 s to find the platform. If mice did not find the platform within 90 s, they were guided to the platform and assigned an escape latency of 90 s for this trial. Once the mice reached the platform, they were allowed to stay on the platform for 10 s and then dried and returned to their home cage.
Twenty-four hours after the last training trial, a probe test was performed in which the platform was removed. The start location was opposite to the target quadrant and each mouse completed a single, 90-s probe test. The number of platform crossings was recorded to assess reference memory.
After the probe test, the working memory test was performed in which the platform was relocated every day for three consecutive days (Zhou et al., 2019). Mice were given two trials per day. In the first trial (acquisition), mice had to learn the new location of the platform by trial-and-error in 90 s before staying in the platform for 15 s. After the 15-s inter-trial interval, the second trial (retrieval) was performed. Mice started from a different point with the same platform position. If the mouse recalled the first trial, it would swim to the goal faster on the second trial. As the platform was moved daily, no learning of platform position from the previous day could be transferred to the next day’s task. Therefore, recall on each day during the second trial was dependent on that day’s first trial and measured only temporary or working memory. The average escape latency of each trial for three days was recorded to assess working or trial-dependent learning and memory.
Passive Avoidance Task
The passive avoidance test was carried out in a trough-shaped apparatus consisted of a white illuminated chamber and a dark chamber (20 cm × 12 cm × 60 cm, respectively) as previously reported (Xue et al., 2010; Bu et al., 2011; Reid et al., 2018). On the first day, mice were allowed to explore both chambers for 4 min twice to acclimatize. Next day the training trial was performed for 5 min after a 3-min adaption. When mice entered the dark chamber, an electric foot shock (40 V, 2 mA) was delivered. After 24 h, the consolidation trial was performed for 5 min in the same way as training and the tracking system was started once mice were placed into the light chamber. The step-through latency (up to a maximum of 300 s) and error times to enter the dark chamber were recorded.
Nissl Staining and Immunofluorescence
Three mice in each group were perfused transcardially with PBS, followed by cold 4% paraformaldehyde (PFA) in PBS. Brains were removed and postfixed in 4% PFA in PBS at 4ºC overnight and then immersed in a freshly made 15% sucrose solution in PBS. Next day, brains were transferred to 30% sucrose solution in PBS for cryoprotection. After 24 h, brains were embedded in optimal cutting temperature compound (Sakura Finetek USA, Inc., Torrance, CA, USA) and frozen quickly onto specimen discs with quick-freeze shelf in the freezing microtome (Leica CM3050 S, Nussloch, Germany). Serial 10 μm-thick coronal sections were cut and thaw mounted on positively charge adhesion slides which were stored at −20°C until use.
Nissl staining was carried out as described before (Lindroos and Leinonen, 1983). Brain sections were dehydrated in ascending grades of ethanol, immersed in xylene, rehydrated in descending grades of ethanol, and hydrated in distilled water. Subsequently, the sections were stained with toluidine blue, dehydrated in ethanol and cleared in xylene before coverslipping.
For immunofluorescence analysis, brain sections were briefly rinsed in PBS three times for 5 min and incubated in PBS containing 0.2% Triton X-100 for 30 min. After rinsing in PBS three times, the sections were incubated with 2% BSA in PBST (0.2% Tween 20 in PBS) for 1 h to block unspecific binding of the antibodies. Primary antibodies (Table 1) were diluted in blocking solution and incubated with sections overnight at 4°C, followed by incubation with Alexa Fluor 488, Alexa Fluor 555, and Alexa Fluor 647-conjugated secondary antibodies (Table 1) for 1 h at room temperature in the dark. After rinsing in PBS three times, coverslips were mounted with mounting medium containing the nuclear counterstain DAPI (4’,6-diamidino-2-phenylindole, SouthernBiotech) and sealed with nail polish to prevent drying and movement.
Image Analysis
All sections were digitalized at 20× magnification utilizing a TissueFaxs System coupled with a Zeiss Axio Imager Z2 Microscope (Jena, Germany). Hippocampal regions were acquired using the TissueFaxs (TissueGnostics, Vienna, Austria) under the same exposure time and acquisition parameters. With a threshold that best discriminated staining from the background, the integrated optical density (IOD, for Nissl or Aβ analysis) and area (for GFAP or Iba1 analysis) above the threshold were calculated for each image with equal size by ImageJ software (NIH version). For each hippocampal region (CA1, CA3, and DG), the obtained value was first normalized with respect to control, and the values from the different levels were then averaged.
Hippocampus Levels of Glu and GABA
Glu and GABA levels in mouse hippocampus were measured by a liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis method as published before (Wang et al., 2019). Briefly, mice were sacrificed by decapitation and hippocampus was rapidly separated, weighed and homogenized with water in an ice bath. An aliquot of 50 μL hippocampus homogenate was spiked with 20 μL internal standard solution (500 ng/mL), followed by mixing with 10 μL trifluoroacetic acid to precipitate protein. The mixture was vortexed and centrifuged at 4°C, 20,000 × g for 15 min. The supernatant was collected and a 1 µL aliquot was injected into the LC-MS/MS system. Gradient elution was performed on a Restek Ultra Aqueous C18 column (100 mm × 2.1 mm, 3 µm, Bellefonte, PA, USA) using acetonitrile and water containing 0.1% FA as the mobile phase. The mass detection was performed by conducting multiple reaction monitoring (MRM) analysis with an electrospray ionization source in positive mode. The MRM transitions are as follows: m/z 148.2→84.0 (Glu), 104.1→87.0 (GABA), and 152.0→109.9 (4-acetamidophenol, internal standard).
Western Blot
Total proteins from hippocampus tissues were using lysis buffer containing 1% protease inhibitor cocktail and 1% phosphatase inhibitor cocktail (CW Biotech, Beijing, China). Protein concentrations were determined by BCA assay. Protein samples were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with 7.5% nonfat dried milk for 2 h and incubated with primary antibodies (Table 1) at 4°C overnight. Next day, membranes were subsequently washed with TBST (0.05% Tween 20 in Tris-buffered saline) three times and incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (Table 1) at room temperature for 2 h. After washing three times with TBST, the protein bands were visualized using an ECL system (CW Biotech, Beijing, China), imaged in a Bio-Rad imaging system (Bio-Rad, Hercules, CA, USA) and analyzed by ImageJ.
Statistical Analysis
Data were analyzed with SPSS version 16.0 and expressed as mean ± SEM. The escape latency during spatial acquisition in the MWM test was examined by repeated measures analysis of variance (ANOVA) followed by Fisher’s least significant difference (LSD) post hoc test. Other data were analyzed using one-way ANOVA followed by LSD post hoc test (only for normality parameters with equal variances) or using the Mann-Whitney U test. Statistical significance was considered when p < 0.05.
Results
Aβ1–42 Oligomers Characterization
Before in vivo experiments, oligomeric Aβ1–42 structures were characterized by AFM (Figure 2A) and Western blot analysis (Figure 2B). As shown in Figure 2A, Aβ1–42 assembled into predominantly 1–6-nm globular structures after incubation, and no fibril formation was detected under this condition. Western blot analysis of SDS-PAGE (Figure 2B) revealed the preparations contained a mixture of Aβ1–42 monomers, trimers, and tetramers, hexamers and large oligomers. Taken together, these data indicate that the predominant compositions of the present soluble Aβ1–42 preparations were Aβ1–42 monomers and oligomers rather than fibrils.
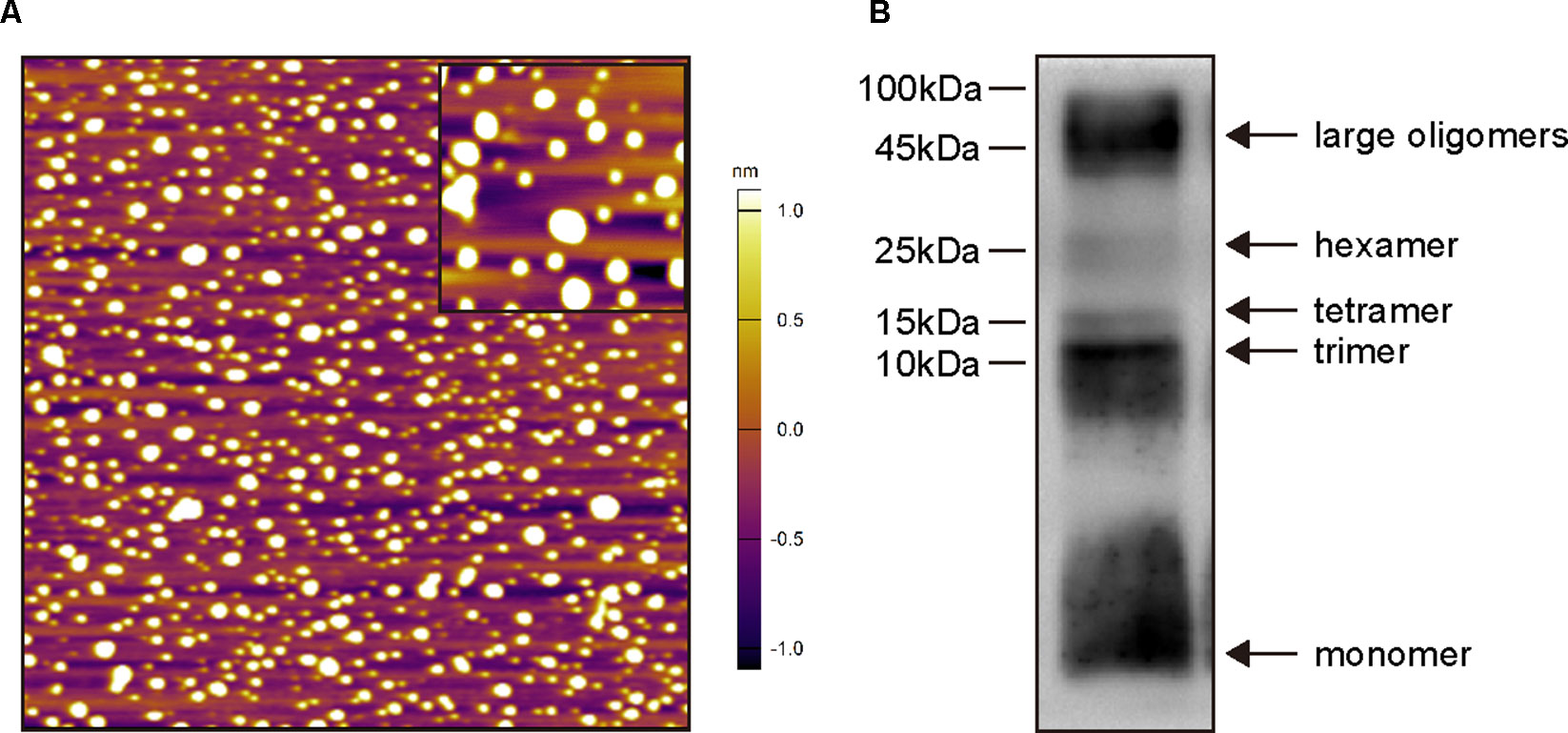
Figure 2 Biophysical characterization of Aβ1–42 oligomers preparations. (A) Atomic force microscopy images of Aβ1–42 oligomers preparations. Only the globular structure of oligomers was observed in the representative 2 × 2-μm x–y image (inset image, 350 × 350-nm x–y). (B) Western blot analysis of the Aβ1–42 oligomers preparations which were separated by SDS-PAGE and probed with monoclonal antibody 6E10 (recognizing residues 1–16 of Aβ). Monomers, trimers, tetramers, hexamers and large oligomers are visible.
CSA Treatment Rescued Aβ1–42 Oligomers-Induced Impairment of Mice Learning and Memory
Open field test was used to determine the spontaneous motor activity. Mice traveled a similar distance without significant changes among all groups (Figure 3A). The results indicate that CSA had no effects on the locomotor activity of mice.
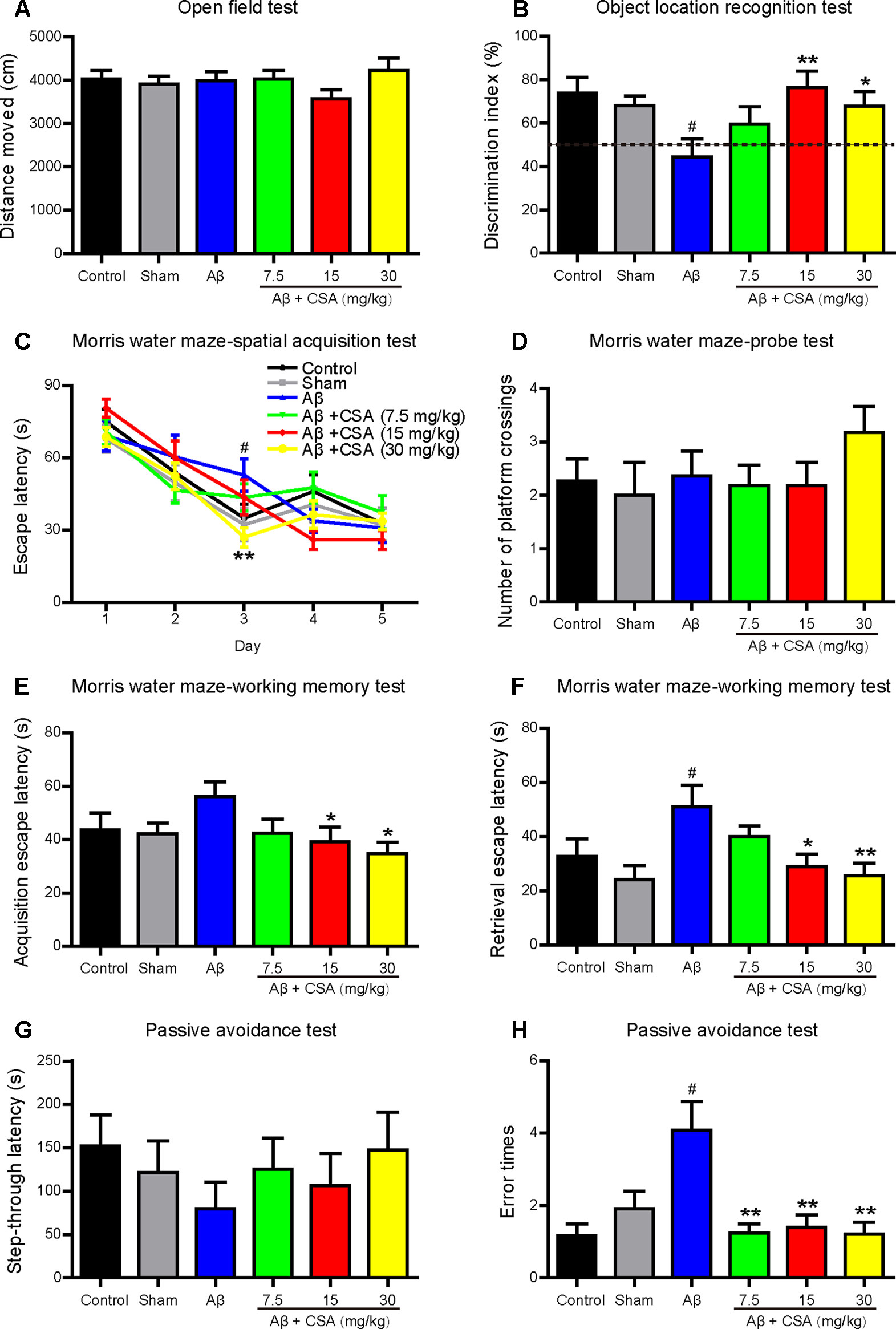
Figure 3 Effects of CSA on cognitive impairment of mice induced by intrahippocampal injection of Aβ1–42 oligomers. (A) Distance traveled in the open field test during 10 min (Mann-Whitney U test, n = 11–12 mice per group). (B) The discrimination index in test trials of object location recognition test (one-way ANOVA followed by LSD post hoc test, n = 10–12 mice per group). (C) The mean daily escape latency calculated from three trials per day in spatial acquisition test of Morris water maze (MWM) (repeated measures ANOVA followed by LSD post hoc test, n = 11 mice per group). (D) The number of times that the mice crossed the original platform location in the probe test of MWM (Mann-Whitney U test, n = 11 mice per group). Acquisition (E) and retrieval (F) escape latency in working memory test of MWM (Mann-Whitney U test, n = 9–11 mice per group). The step-through latency (G) and error times (H) into the dark chamber in the consolidation trial of passive avoidance test (Mann-Whitney U test, n = 9–12 mice per group). All values are mean ± SEM. #P < 0.05 as compared to the sham-operated group. *P < 0.05 and **P < 0.01 as compared to the Aβ1–42 oligomers-injected group.
To investigate the effects of CSA on cognitive performance, mice were tested in OLR, MWM, and passive avoidance tests. The OLR test is based on the spontaneous tendency for rodents to spend more time exploring a novel location rather than a familiar location to assess spatial memory and discrimination. Mice showed no preference for the two locations during familiar trials. In test trials (Figure 3B), like in control group, mice in the sham-operated group spent more time for exploring the novel location than the familiar location with a discrimination index nearly 70%. In contrast, the discrimination index of Aβ1–42 oligomers-injected group was decreased significantly to around 50% compared with that of the sham-operated group. Treatments with CSA (15 and 30 mg/kg) elevated the discrimination index with significance (P = 0.003, P = 0.026), suggesting that CSA (15 and 30 mg/kg) treatments could reverse Aβ1–42 oligomers-induced cognitive deficits.
In spatial acquisition test of MWM, the escape latency gradually decreased during the first three training days in all groups (Figure 3C). The repeated measurement of ANOVA showed the interaction effect between group and day on escape latency was significant (P = 0.018), indicating that the effect of group varied with day. Then the group effect on each day was measured by LSD post hoc test to localize which day showed group differences. Longer escape latency was observed in Aβ1–42 oligomers-injected group on day 3 in contrast to that in the sham-operated group (P = 0.020), indicating that the spatial learning ability of mice was significantly affected by Aβ1–42 oligomers. On the same training day, CSA (30 mg/kg) treatment significantly shortened the escape latency prolongation caused by Aβ1–42 oligomers (P = 0.004). In probe test, the number of platform crossings of mice in CSA treatment (30 mg/kg) group was the highest among all groups even without statistical significance compared with that in Aβ1–42 oligomers-injected group (Figure 3D). These results suggest that intrahippocampal injection of Aβ1–42 oligomers impaired mice spatial learning and memory and CSA treatment rescued the impairment. In working memory test, the acquisition escape latency was decreased in Aβ1–42 oligomers-injected group (P = 0.060) while CSA (15 and 30 mg/kg) treatments reversed the decrease significantly (P = 0.034, P = 0.011, Figure 3E). In the retrieval trial, the latency was significantly decreased in Aβ1–42 oligomers-injected group (P = 0.018), and CSA (15 and 30 mg/kg) treatments reversed the decrease significantly (P = 0.022, P = 0.007, Figure 3F) as well. These findings indicate that CSA treatment could enhance both acquisition and retrieval of Aβ1–42 oligomers-impaired working memory.
The passive avoidance task is a fear-aggravated test used to evaluate non-spatial learning and memory. In the consolidation trial (Figures 3G, H), Aβ1–42 oligomers-induced cognitive deficits were observed as shorter latency and more error times (P = 0.035) into the dark chamber than those of sham-operated mice. Compared with Aβ1–42 oligomers-injected group, CSA (7.5, 15, and 30 mg/kg) treatments reversed the increase of error times significantly (P = 0.002, P = 0.009, P = 0.005), and prolonged the latency even without statistical significance.
Aβ1–42 Oligomers Injection and CSA Treatment Did Not Cause Hippocampal Neuronal Loss
The protective effects of CSA were investigated using Nissl staining in mice hippocampus. Neurons from all groups had a distinct profile and a spherical nucleus neatly arranged with no difference in neuronal morphology (Figure 4A). The IOD of Nissl bodies was quantified to show neuronal status. It was found that there were no significant differences in the hippocampal CA1, CA2, or DG cell layers between control and any other groups (Figure 4B). Therefore, no detectable hippocampal neuronal loss was observed after the injection of Aβ1–42 oligomers and CSA treatment, suggesting that the protective effects of CSA did not involve in the modulation of neuron death.
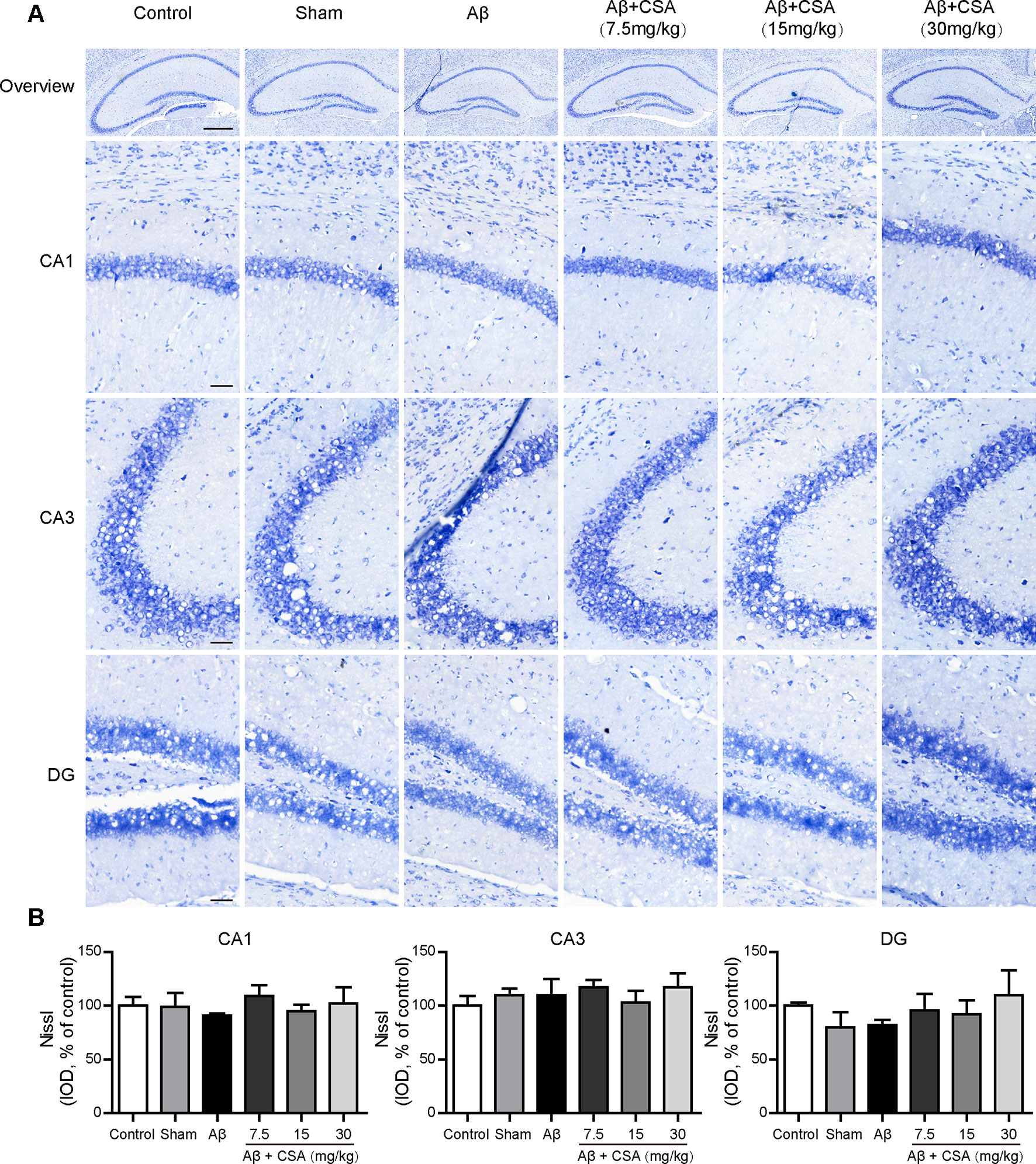
Figure 4 Effects of CSA on the status of the hippocampal neuron of mice induced by intrahippocampal injection of Aβ1–42 oligomers. (A) Representative micrographs of Nissl staining in the hippocampus. The scale bar represents 500 μm in the overview image of hippocampus or 50 μm in the detail image of hippocampal subregions (CA1, CA3, and DG). (B) Quantification of Nissl integrated optical density (IOD) in the CA1, CA3, and DG regions of the hippocampus (one-way ANOVA followed by LSD post hoc test, n = 3 mice per group). Values represent mean ± SEM.
CSA Treatment Attenuated Aβ1–42 Oligomers-Induced Activation of Microglia and Reactivity of Astrocytes in the Hippocampus
Sections from the hippocampus were further immunostained for the presence of microglia (anti-Iba1 antibody) and astrocytes (anti-GFAP antibody). Compared with the sham-operated group, Aβ1–42 oligomers-injected group showed markedly increased immunoreactivities for both Iba1 and GFAP in the hippocampus (Figures 5 and 6). The activation of microglia and reactivity of astrocytes by Aβ1–42 oligomers were attenuated by CSA treatment significantly (Figures 5 and 6).
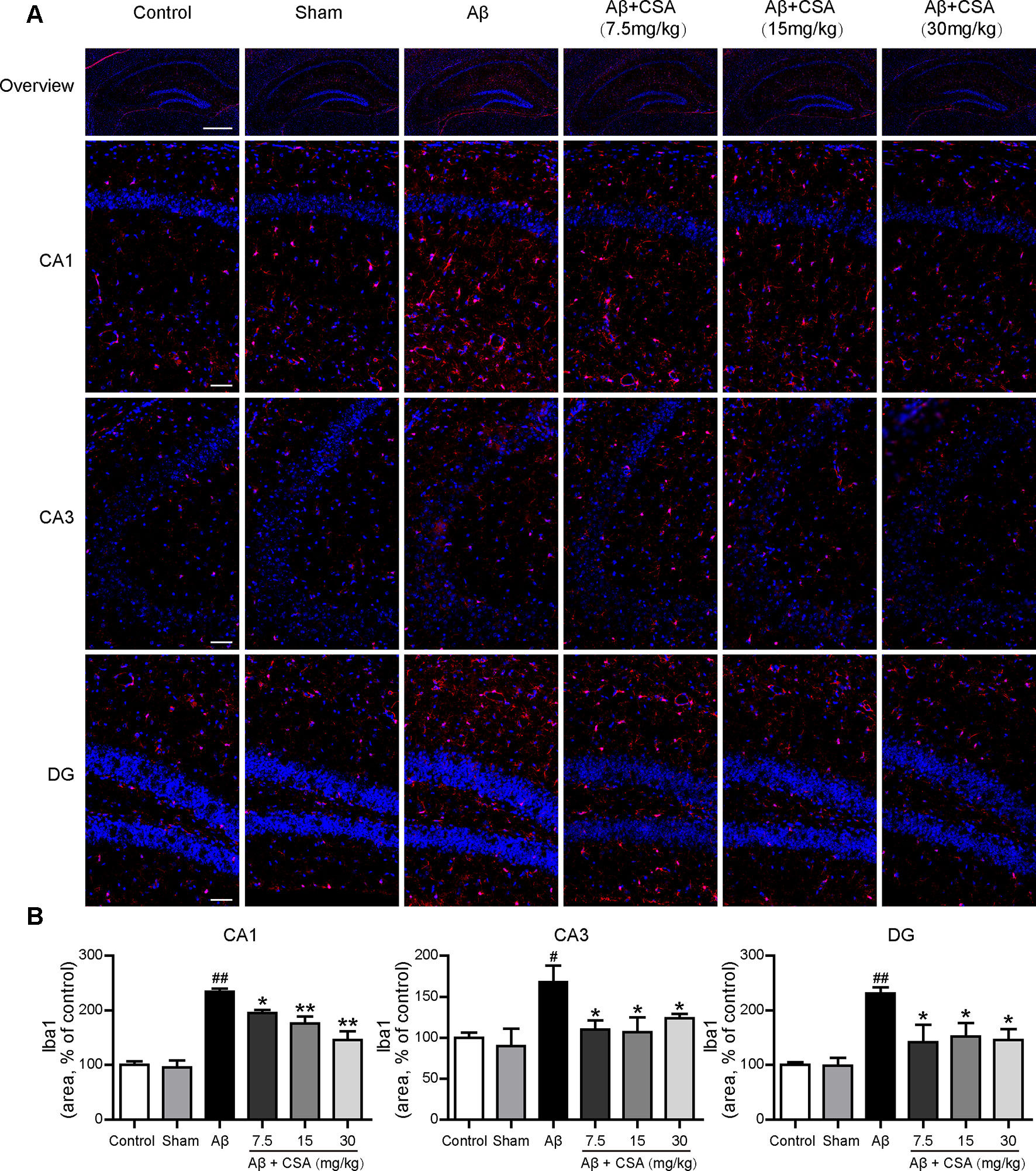
Figure 5 Effects of CSA on microglial activation in the hippocampus of mice induced by intrahippocampal injection of Aβ1–42 oligomers. (A) Representative micrographs showing Iba1 (red) immunofluorescence in the hippocampus. Nuclei are in blue (DAPI). The scale bar represents 500 μm in the overview image of hippocampus or 50 μm in the detail image of hippocampal subregions (CA1, CA3, and DG). (B) Quantification of Iba1 area in the CA1, CA3 and DG regions of the hippocampus (one-way ANOVA followed by LSD post hoc test or Mann-Whitney U test, n = 3 mice per group). Values represent mean ± SEM. #P < 0.05 and ##P < 0.01 as compared to the sham-operated group. *P < 0.05 and **P < 0.01 as compared to the Aβ1–42 oligomers-injected group.
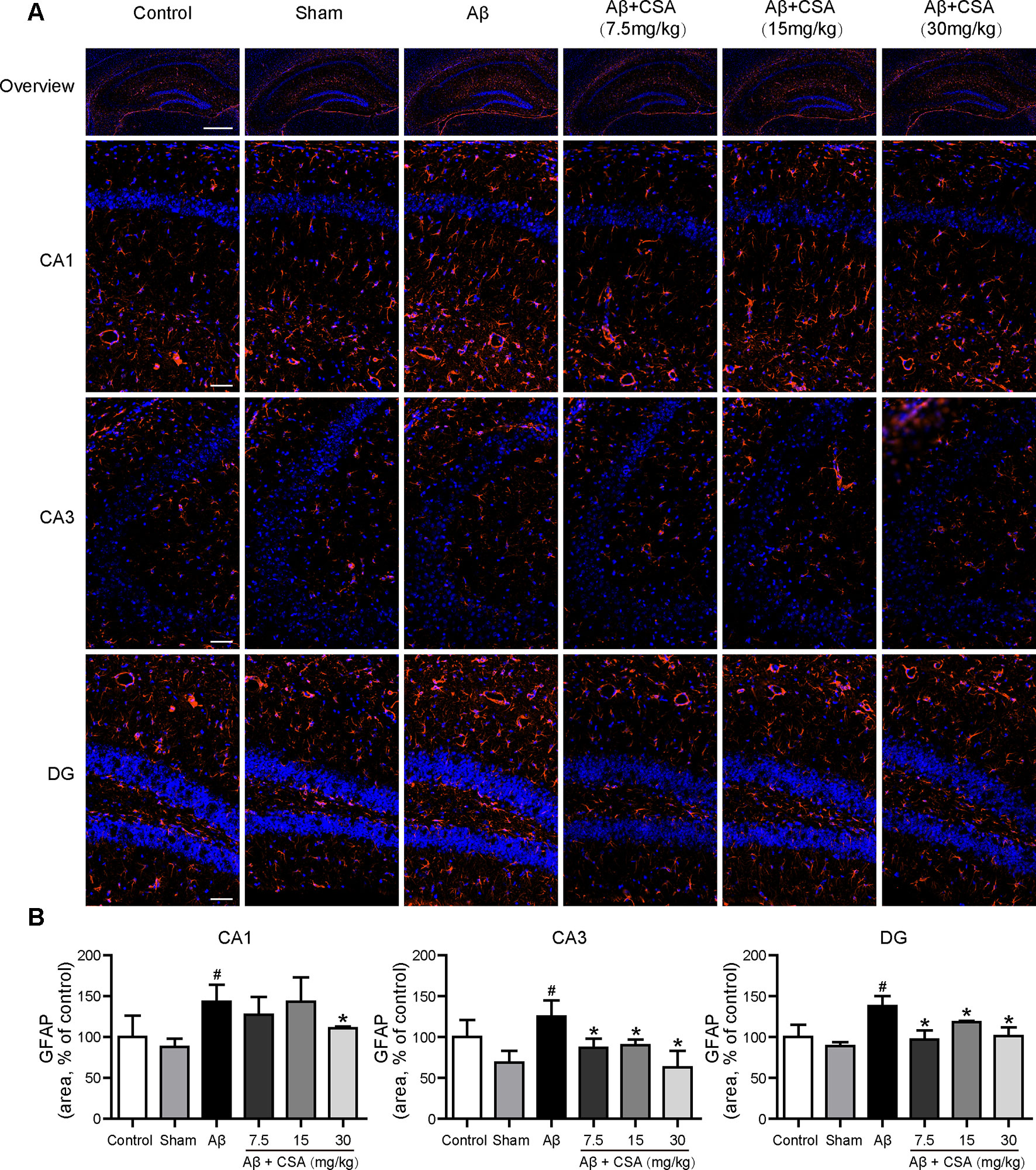
Figure 6 Effects of CSA on astrocyte reactivity in the hippocampus of mice induced by intrahippocampal injection of Aβ1–42 oligomers. (A) Representative micrographs showing GFAP (orange) immunofluorescence in the hippocampus. Nuclei are in blue (DAPI). The scale bar represents 500 μm in the overview image of hippocampus or 50 μm in the detail image of hippocampal subregions (CA1, CA3, and DG). (B) Quantification of GFAP area in the CA1, CA3, and DG regions of the hippocampus (Mann-Whitney U test, n = 3 mice per group). Values represent mean ± SEM. #P < 0.05 as compared to the sham-operated group. *P < 0.05 as compared to the Aβ1–42 oligomers-injected group.
CSA Treatment Reduced Aβ1–42 Levels in the Hippocampus
Immunostaining with anti-Aβ (6E10) confirmed the widespread diffusion of Aβ oligomers after intracerebral injection. All hippocampal regions (CA1, CA3, and DG) of Aβ1–42 oligomers-injected mice presented significantly abundant Aβ immunoreactivity compared with those of sham-operated mice (P = 0.004, P = 0.049, P = 0.002). In the CA1 region, CSA (15 and 30 mg/kg) treatments, except the lowest dose (7.5 mg/kg), reduced Aβ1–42 levels significantly compared with Aβ1–42 oligomers-injected group (P = 0.009, P = 0.020). In the CA3 and DG regions, all doses of CSA (7.5, 15 and 30 mg/kg) could reduce Aβ1–42 levels significantly (Figure 7).
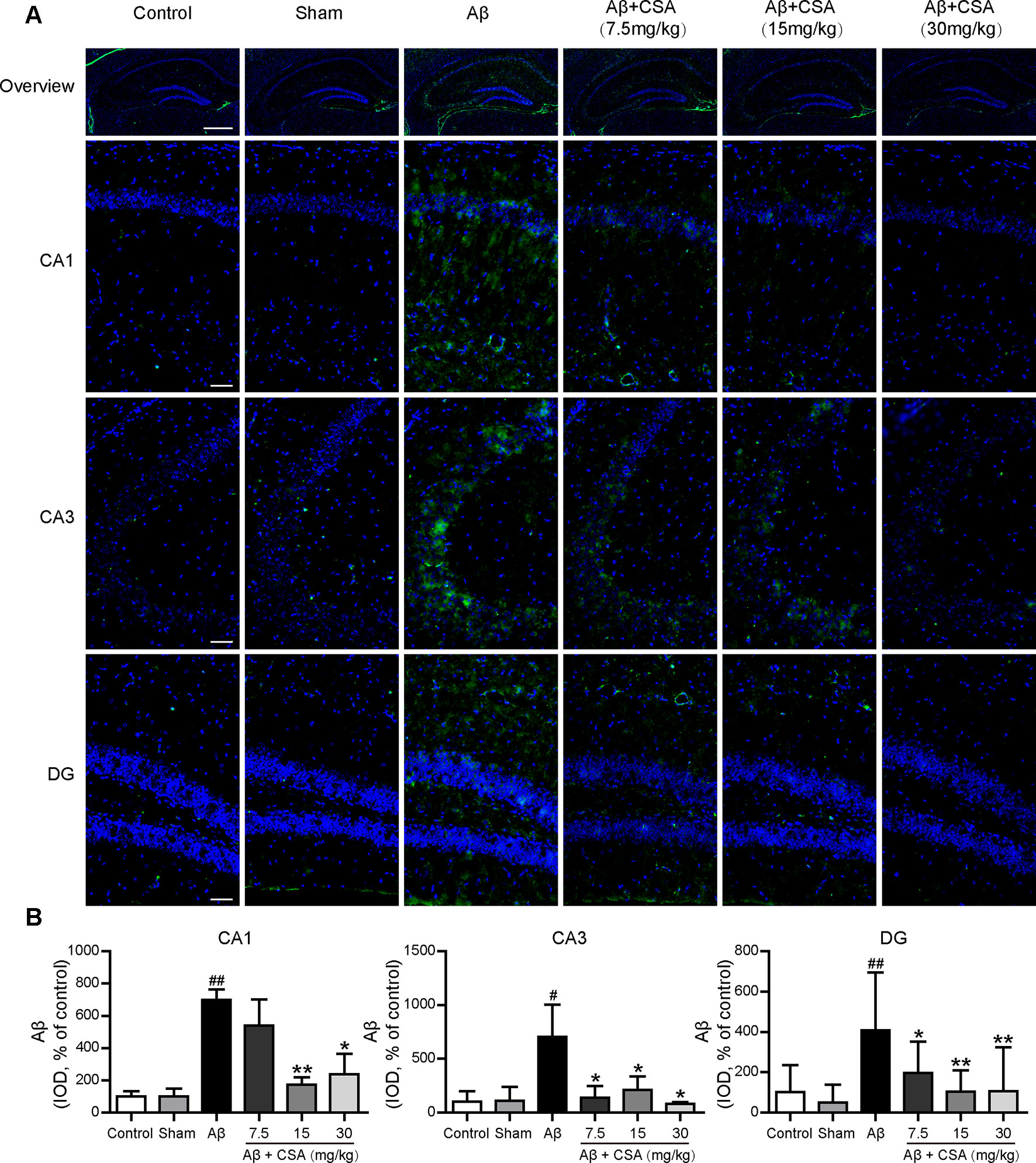
Figure 7 Effects of CSA on hippocampal Aβ1–42 levels of mice induced by intrahippocampal injection of Aβ1–42 oligomers. (A) Representative micrographs showing Aβ (green) immunofluorescence in the hippocampus. Nuclei are in blue (DAPI). The scale bar represents 500 μm in the overview image of hippocampus or 50 μm in the detail image of hippocampal subregions (CA1, CA3, and DG). (B) Quantification of Aβ integrated optical density (IOD) in the CA1, CA3, and DG regions of the hippocampus (one-way ANOVA followed by LSD post hoc test or Mann-Whitney U test, n = 3 mice per group). Values represent mean ± SEM. #P < 0.05 and ##P < 0.01 as compared to the sham-operated group. *P < 0.05 and **P < 0.01 as compared to the Aβ1–42 oligomers-injected group.
CSA Treatment Reversed Aβ1–42 Oligomers-Induced Dyshomeostasis of Glu and GABA in the Hippocampus
The concentrations of Glu and GABA in the hippocampus are shown in Figure 8. Compared with the sham-operated group, Glu levels were significantly increased and GABA levels were significantly decreased in the hippocampus of Aβ1–42 oligomers-injected group (P = 0.035, P = 0.049). CSA treatment significantly decreased Glu levels at the dose of 15 mg/kg (P = 0.035) and significantly increased GABA levels at the dose of 30 mg/kg (P = 0.037) compared with Aβ1–42 oligomers-injected group.
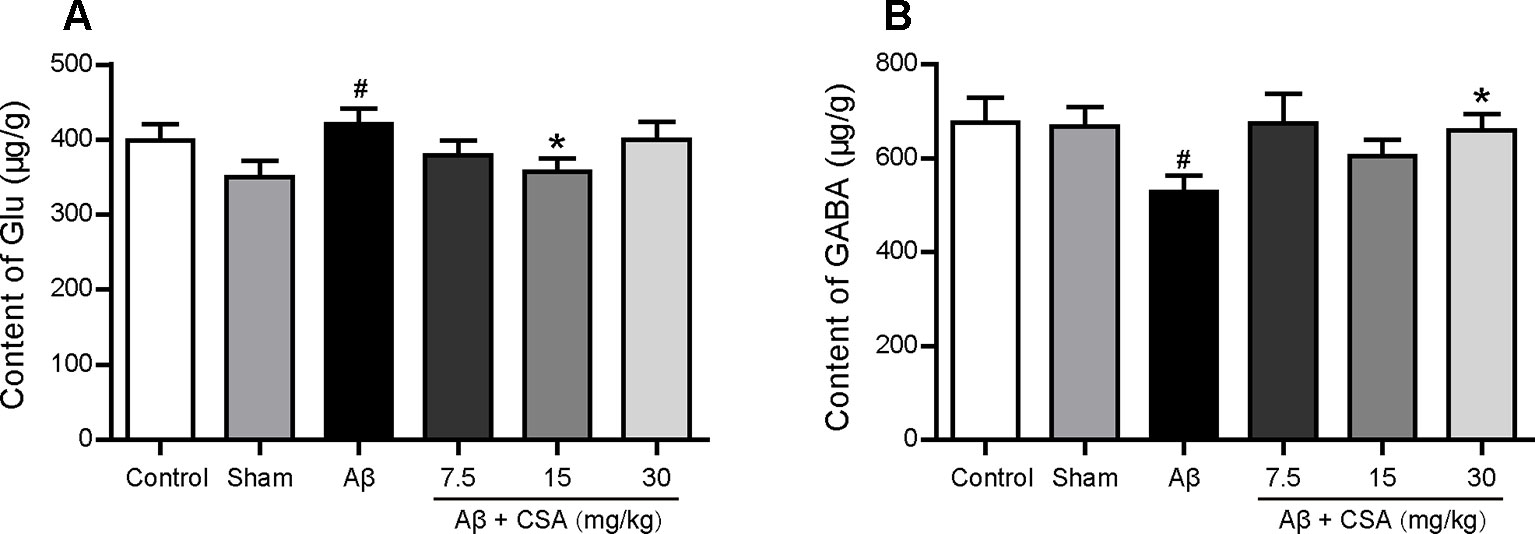
Figure 8 Effects of CSA on hippocampal dyshomeostasis of glutamate (Glu) and γ-aminobutyric acid (GABA) of mice induced by intrahippocampal injection of Aβ1–42 oligomers. (A) The content of Glu in the hippocampus (Mann-Whitney U test, n = 7–9 mice per group). (B) The content of GABA in the hippocampus (Mann-Whitney U test, n = 7-9 mice per group). All values are mean ± SEM. #P < 0.05 as compared to the sham-operated group. *P < 0.05 as compared to the Aβ1–42 oligomers-injected group
CSA Treatment Prevented Aβ1–42 Oligomers-Induced Increase of GluN2B and Decrease of Downstream PKA/CREB/BDNF/TrkB Signaling Pathway
To investigate the protective effects of CSA, Western blot analysis was used to measure the expression levels of GluN2B, GluN1, PKA c-α, pCREB/CREB, BDNF, and TrkB. As shown in Figure 9A, GluN2B expression was markedly increased after Aβ1–42 oligomers injection (P = 0.004) but reduced significantly when treated with CSA (P = 0.035, P = 0.045, P = 0.013). However, there was no significant difference of GluN1 levels among all groups (Figure 9B). In the subsequent signaling pathway, the expression of PKA c-α, pCREB/CREB, BDNF or TrkB was downregulated in Aβ1–42 oligomers-injected group compared with those in the sham-operated group (P = 0.016, P = 0.00002, P = 0.016, and P = 0.016) but upregulated by CSA treatments (Figures 9C–F).
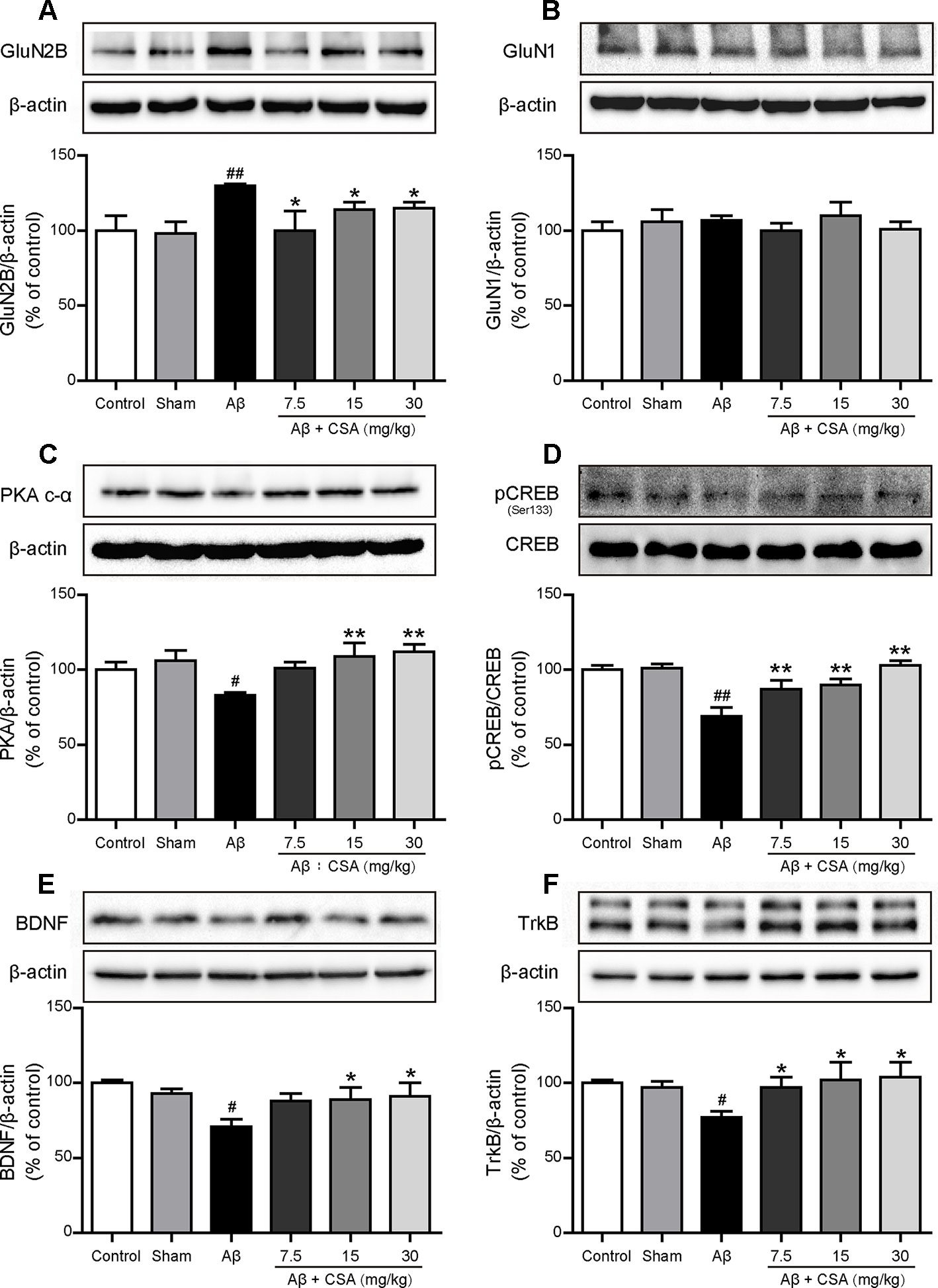
Figure 9 Effects of CSA on hippocampal expression of GluN2B, GluN1 and PKA/CREB/BDNF/TrkB signaling pathway of mice induced by intrahippocampal injection of Aβ1–42 oligomers. (A) The expression levels of GluN2B normalized to β-actin (Mann-Whitney U test, n = 6 mice per group). (B) The expression levels of GluN1 normalized to β-actin (one-way ANOVA followed by LSD post hoc test, n = 7 mice per group). (C) The expression levels of PKA normalized to β-actin (one-way ANOVA followed by LSD post hoc test, n = 5 mice per group). (D) The ratio of pCREB/CREB expression levels normalized to β-actin (one-way ANOVA followed by LSD post hoc test, n = 5 mice per group). (E) The expression levels of BDNF normalized to β-actin (one-way ANOVA followed by LSD post hoc test, n = 5 mice per group). (F) The expression levels of TrkB normalized to β-actin (Mann-Whitney U test, n = 5 mice per group). Values represent mean ± SEM. #P < 0.05 and ##P < 0.01 as compared to the sham-operated group. *P < 0.05 and **P < 0.01 as compared to the Aβ1–42 oligomers-injected group.
Discussion
The Aβ injection model has been used widely, but there are still missing operation standards. Numerous factors can affect the outcome of this model, such as the preparation methods of Aβ, injection doses, injection sites, and the duration of modeling. Therefore, we validated these factors before using this model to evaluate the effects of CSA on learning and memory. Distinct Aβ aggregates are formed by depending on the initial structure in lyophilized stocks of commercial Aβ, Aβ species and the incubation conditions, including concentration, time, temperature, pH, and ionic strength. Many studies (Choi et al., 2012; Yang et al., 2012; Choi et al., 2013; Chen et al., 2015) adopted the incubation method of Aβ peptides in Maurice’s report (Maurice et al., 1996). Aβ peptides were directly dissolved in sterile double-distilled water (or saline or PBS) and incubated at 37 ºC for 4 days. Light microscopic observation indicated that two types of insoluble precipitates birefringent fibril-like structures and globular aggregates were formed after incubation (Maurice et al., 1996). Different preexisting structures in lyophilized stocks of commercial Aβ lead to different types of Aβ aggregates after the same incubation condition, which might result in distinct outcomes of in vivo or in vitro experiments. Therefore, the removal of any preexisting structures in lyophilized stocks of Aβ peptide by HFIP is required for controlling aggregation studies (Stine et al., 2003). After a standard incubation method, we characterized the structures of Aβ1–42 aggregates by AFM and Western blot analysis. The results indicate that the predominant compositions of the present soluble Aβ1–42 preparations were Aβ1–42 monomers and oligomers rather than fibrils (Figure 2). The clarified structures of Aβ1–42 preparations laid a critical foundation for exploring the in vivo mechanism of soluble Aβ1–42 oligomers neurotoxicity and CSA treatment.
The amount of injected Aβ was calculated to reach a concentration of the same order of magnitude as the actual level of soluble Aβ observed in AD brains. The mean concentration of soluble Aβ in the brains of AD patients was 0.29 μg/g in the study of McLean et al. (1999) or 1.5 μg/g in the study of Tabaton and Piccini (2005), while that of normal subjects was at a quite low level (ng/g). The amount of intrahippocampal injection of Aβ1–42 oligomers in the study of Epelbaum et al. (2015) was 50 pmol/mouse which could induce memory impairment. Other amounts, such as 10, 30, 222, and 500 pmol/mouse, were used as well (Youssef et al., 2008; Dineley et al., 2010; Choi et al., 2011; Brito-Moreira et al., 2017). After taking these references into account, we injected an aliquot of 2 μL Aβ1–42 oligomers preparations (50 μM), equivalent to 100 pmol of Aβ1–42 monomer, into the mouse brain (about 500 mg). If the Aβ1–42 preparations freely diffused in the whole brain, the final concentration would be 0.9 μg/g, reaching to a similar concentration in the brains of AD patients. Therefore, our study chose 100 pmol/mouse as the amount of Aβ injection.
Before the in vivo experiments to assess the effects of CSA on learning and memory, we operated a preliminary experiment to choose a suitable injection site to build the animal model. Unilateral injection into the ventricle (−0.2 mm AP, 1.0 mm ML, 2.5 mm DV), bilateral injection into the ventricle (−0.2 mm AP, ± 1.0 mm ML, 2.5 mm DV) and bilateral injection into the CA1 region of hippocampus (−2.0 mm AP, ± 1.5 mm ML, 1.5 mm DV) were compared under the same volume of Aβ1–42 oligomers solution (100 pmol/mouse). Learning and memory capacity was assessed using OLR, open filed and MWM tests following nearly the same experimental schedule (Figure 1). Mice in all groups had similar locomotor activity without significant difference. Compared with the mice in the corresponding sham-operated group, mice in Aβ1–42 oligomers-injected group performed differently in the OLR test and working memory test in MWM (Figure 10). Only into the bilateral CA1 region of hippocampus could Aβ1–42 oligomers cause a significant decrease of discrimination index in recognition test (P = 0.021) and a marked increase of retrieval escape latency in working memory test (P = 0.003). The results of intracerebroventricular injection are inconsistent with some outcomes published before (Youssef et al., 2008; Figueiredo et al., 2013; Brito-Moreira et al., 2017). These inconsistent results might be due to different amounts of injected Aβ oligomers or mice species which were summarized in a review we published previously (Wang et al., 2018). In our experimental environment, Aβ1–42 oligomers (100 pmol/mouse) injected bilaterally into the CA1 region of hippocampus could impair cognition successfully and be used for evaluating the role of CSA in learning and memory.
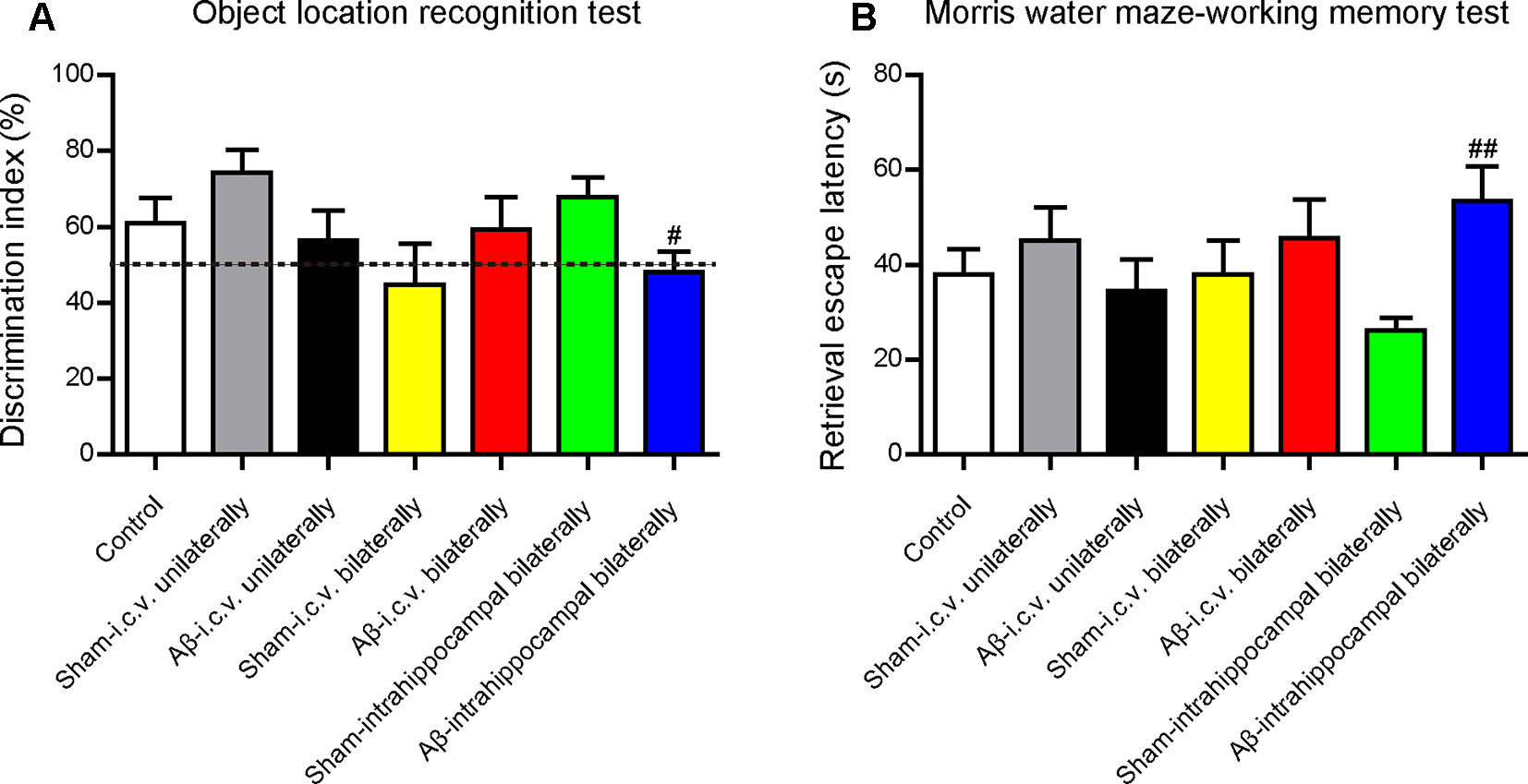
Figure 10 Effects of injection sites of Aβ1–42 oligomers on mice cognition detected by behavioral tests. (A) The discrimination index in test trials of object location recognition test (Mann-Whitney U test, n = 11–12 mice per group). (B) Retrieval escape latency in working memory test of Morris water maze (Mann-Whitney U test, n = 10–12 mice per group). Values represent mean ± SEM. #P < 0.05 and ##P < 0.01 as compared to the corresponding sham-operated group. Intracerebroventricular, i.c.v.
Accumulating evidence shows that soluble Aβ oligomers are more toxic than insoluble fibrils and responsible for cognitive impairment in AD pathogenesis (Dahlgren et al., 2002; Gong et al., 2003; Balducci et al., 2010; DaRocha-Souto et al., 2011). In our study, bilateral intrahippocampal injection of Aβ1–42 oligomers impaired the short-term spatial memory and discrimination in the OLR test, slowed down the pace of spatial learning in acquisition test of MWM, damaged working memory, and impaired the non-spatial fear-aggravated memory in passive avoidance task. These results provide additional evidence for the neurotoxic effects of Aβ1–42 oligomers. Compared with Aβ1–42 oligomers-injected group, CSA treatment significantly elevated discrimination index in the OLR test, shortened escape latency and enhanced working memory in MWM, and reduced error times in passive avoidance task, suggesting that CSA could rescue Aβ1–42 oligomers-induced impairment of mice learning and memory.
The Aβ oligomers hypothesis proposed by Klein (2002) suggests that early stage memory impairment in AD stems not from neuron death but rather from malfunctions in memory-specific signal transduction required for synaptic plasticity. Consistent with the hypothesis, significant neuron death was not found in the hippocampus after bilateral intrahippocampal injection of Aβ1–42 oligomers in our study. This result is in accordance with the report of Kasza et al. (2017). Ágnes Kasza compared the neurotoxic effects of different kinds of Aβ1–42 oligomers at three different aggregation concentrations (25, 75, and 200 μM) under two different aggregation times (24 and 168 h). The Aβ1–42 oligomers used in our study was incubated at 100 μM for 16 h, which is similar to the Aβ1–42 oligomers (75 μM/24 h) investigated by Ágnes Kasza. Their Aβ1–42 oligomers (75 μM/24 h) significantly impaired rat spatial memory in MWM and reduced long-term potentiation (LTP) levels but did not decrease the number of viable neurons significantly by Nissl staining in the hippocampus. The consistent result can also be found in the report of Yang et al. (2018). Aβ1–42 oligomers (100 μM/24 h) not only impaired spatial learning and memory of rats in MWM but also suppressed LTP in the DG region. However, there was no obvious difference in the number of NeuN positive cells in DG region between the Aβ1–42 oligomers-injected group and the control group. Therefore, bilateral intrahippocampal injection of Aβ1–42 oligomers (100 μM/16 h) in our study impaired learning and memory without neuron death and could successfully simulate the early stage of AD. CSA treatment had no impact on neuron death as well, suggesting that the protective effects of CSA might not be involved in the modulation of neuron death.
It has been recognized that brain inflammation involving glial cells is a prominent feature of AD (Chun et al., 2018). The activated microglia and reactive astrocytes have been observed in the brains of AD patients and animal models (Nagele et al., 2003; Heneka et al., 2005; Olabarria et al., 2010; Fan et al., 2017). Consistent with a previous study (Ledo et al., 2013), we also demonstrated that Aβ1–42 oligomers could induce the activation of microglia and reactivity of astrocytes in the hippocampus of mice. On one hand, the activated microglia and reactive astrocytes in Aβ1–42 oligomers-injected group might, in turn, stimulate the clearance of Aβ and delay the pathological progression (Wyss-Coray et al., 2003; Mandrekar et al., 2009). On the other hand, the microglial activation and astrocyte reactivity and associated inflammation could become deleterious, contributing to neurotoxicity (Hickman et al., 2008; Verkhratsky et al., 2010). In the present study, CSA significantly reduced the level of Aβ and attenuated Aβ1–42 oligomers-induced activation of microglia and reactivity of astrocytes in the hippocampus. These results suggest that CSA could stimulate the clearance of Aβ and the underlying mechanism needs to be explored in further studies. Downregulation of the microglial activation and astrocyte reactivity by CSA might be due to the decreasing level of Aβ or the anti-inflammatory effects of CSA (Patel and Bhutani, 2014; Huang et al., 2016; Schuster et al., 2016).
Glu and GABA dysregulation also contribute to the progression of AD. Attenuated Glu transport and decreased GABA levels were observed previously in the brains of AD patients (Masliah et al., 1996; Bai et al., 2015). We found elevated Glu levels and decreased GABA levels in the hippocampus of Aβ1–42 oligomers-injected group. Many factors contribute to this aberrant rise in Glu levels. Proinflammatory mediators potentially released from activated microglia in pathological conditions have been shown to influence the efficiency of astrocytic Glu uptake (Zhao et al., 2004). Aβ downregulates Glu uptake capacity of astrocytes and thereby induces a dysfunctional extracellular Glu clearance (Matos et al., 2008). The Glu released from neurons cannot be cleared from the synapse by astrocytes which increases the spillover of Glu from synapses to extrasynaptic space (Pal, 2018). Aβ also induces the release of microglial and astrocytic Glu, which in turn activates extrasynaptic NMDA receptors (Noda et al., 1999; Talantova et al., 2013). CSA treatment decreased the high levels of Glu but increased the low levels of GABA in the hippocampus. The prohibition against Aβ1–42 oligomers-induced high levels of Glu by CSA might be owing to its clearance of Aβ or prevention of microglial activation and astrocyte reactivity.
Synapse loss, a structural correlate involved in cognitive decline, is an early event in the progression of AD (Scheff et al., 2006). NMDARs play an essential role in synaptic function and plasticity (Collingridge and Singer, 1990) but can also be harmful (Waxman and Lynch, 2005). An explanation for the ‘NMDARs paradox’ revealed recently is the location of NMDARs: the activation of synaptic NMDARs initiates plasticity and stimulates cell survival, whereas the activation of extrasynaptic NMDARs promotes cell death and thus contributes to the etiology of AD (Wang and Reddy, 2017). Numerous studies have demonstrated that Aβ oligomers decrease synaptic NMDARs expression (Geng et al., 2013) but activate extrasynaptic GluN2B (Li et al., 2011; Talantova et al., 2013). In our study, Aβ1–42 oligomers increased the expression of GluN2B but decreased the expression of pCREB/CREB and BDNF in the hippocampus. Because synaptic NMDARs induce CREB activity and BDNF gene expression whereas extrasynaptic NMDARs lead to dephosphorylation of CREB that blocks induction of BDNF expression (Hardingham et al., 2002), it can be concluded that the increasing levels of GluN2B induced by Aβ1–42 oligomers might mainly occur at the extrasynaptic membrane. Based on the fact that PKA activity is necessary and sufficient for synaptic clustering of NMDARs (Crump et al., 2001), the decreasing levels of PKA c-α, a kind of catalytic subunit of PKA, in Aβ oligomers-injected group indicate that Aβ1–42 oligomers might inhibit PKA activity and thus downregulate synaptic NMDARs. Taken together, extrasynaptic GluN2B might be upregulated while synaptic NMDARs might be downregulated after bilateral intrahippocampal injection of Aβ1–42 oligomers in the present study. CSA treatment reversed the Aβ1–42 oligomers-induced excessive expression of GluN2B, decreased the high level of Glu, enhanced the downstream PKA/CREB/BDNF/TrkB signaling pathway and might decrease extrasynaptic NMDARs but increase synaptic NMDARs activity. More direct evidence of CSA effects on extrasynaptic or synaptic NMDARs needs to be provided by an electrophysiological method using MK-801 to block active synaptic NMDARs in the further experiment (Li et al., 2011). No significant difference of GluN1 levels was observed among all groups, suggesting that the effects of Aβ1–42 oligomers or CSA did not involve in the modulation of GluN1 total expression.
Conclusions
In summary, our study provides a validated animal model to simulate the early stage of AD by bilateral injection of Aβ1–42 oligomers into the CA1 region of the hippocampus. CSA effectively improved the cognitive impairment induced by Aβ1–42 oligomers. Its cognitive enhancing effects might be mediated by clearing Aβ, preventing microglial activation and astrocyte reactivity, reversing Glu and GABA dyshomeostasis, inhibiting excessive expression of GluN2B and enhancing the downstream PKA/CREB/BDNF/TrkB signaling pathway in the hippocampus. Our study provides the possibility for CSA to be a potential therapeutic agent at the early stage of AD.
Data Availability
All datasets generated for this study are included in the manuscript/Supplementary Files.
Ethics Statement
The animal study was reviewed and approved by the Animal Ethical Committee at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences.
Author Contributions
QC, R-LP, X-ML, Y-HL and L-SW participated in research design. L-SW, XT, Y-FZ and M-DZ conducted experiments and analyzed data. L-SW and QC wrote the manuscript. All authors reviewed the manuscript.
Funding
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-1-012), the National S&T Major Projects for New Drug Innovation and Development (2017ZX09301029) and the National Key R&D Program of China (2018YFC1602105).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CSA, cajaninstilbene acid; AD, Alzheimer’s disease; Aβ, amyloid β peptide; MWM, Morris water maze; Glu, glutamate; GABA, γ-aminobutyric acid; NMDARs, N-methyl-D-aspartate receptors; CREB, cAMP response element binding protein; PKA, cAMP-dependent protein kinase; BDNF, brain-derived neurotrophic factor; TrkB, tropomyosin receptor kinase B; sECC, stilbenes containing extract-fraction from Cajanus cajan (L.) leaves; HFIP, 1,1,1,3,3,3-hexafluoro-2-propanol; DMSO, dimethyl sulfoxide; PBS, phosphate buffered saline; AFM, atomic force microscopy; CMC-Na, carboxymethylcellulose sodium; OLR, object location recognition; PFA, paraformaldehyde; IOD, integrated optical density; MRM, multiple reactions monitoring; ANOVA, analysis of variance; LSD, least significant difference; LTP, long-term potentiation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.01084/full#supplementary-material
References
Bai, X., Edden, R. A., Gao, F., Wang, G., Wu, L., Zhao, B., et al. (2015). Decreased gamma-aminobutyric acid levels in the parietal region of patients with Alzheimer’s disease. J. Magn. Reson. Imaging 41 (5), 1326–1331. doi: 10.1002/jmri.24665
Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., et al. (2010). Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U. S. A. 107 (5), 2295–2300. doi: 10.1073/pnas.0911829107
Brito-Moreira, J., Lourenco, M. V., Oliveira, M. M., Ribeiro, F. C., Ledo, J. H., Diniz, L. P., et al. (2017). Interaction of amyloid-beta (Abeta) oligomers with neurexin 2alpha and neuroligin 1 mediates synapse damage and memory loss in mice. J. Biol. Chem. 292 (18), 7327–7337. doi: 10.1074/jbc.M116.761189
Bu, L., Shi, Z., Sun, X., Liu, X., Zhao, M., Gao, J. (2011). An animal model for evaluating the effect of memory improvement assisted by functional foods. Chin. J. Food Hyg. 23 (5), 402–405. doi: 10.13590/j.cjfh.2011.05.004
Chen, C., Li, X., Gao, P., Tu, Y., Zhao, M., Li, J., et al. (2015). Baicalin attenuates alzheimer-like pathological changes and memory deficits induced by amyloid beta1–42 protein. Metab. Brain Dis. 30 (2), 537–544. doi: 10.1007/s11011-014-9601-9
Choi, D. Y., Lee, J. W., Peng, J., Lee, Y. J., Han, J. Y., Lee, Y. H., et al. (2012). Obovatol improves cognitive functions in animal models for Alzheimer’s disease. J. Neurochem. 120 (6), 1048–1059. doi: 10.1111/j.1471-4159.2011.07642.x
Choi, J. G., Moon, M., Kim, H. G., Mook-Jung, I., Chung, S. Y., Kang, T. H., et al. (2011). Gami-Chunghyuldan ameliorates memory impairment and neurodegeneration induced by intrahippocampal Abeta 1–42 oligomer injection. Neurobiol. Learn. Mem. 96 (2), 306–314. doi: 10.1016/j.nlm.2011.06.004
Choi, J. Y., Cho, E. J., Lee, H. S., Lee, J. M., Yoon, Y. H., Lee, S. (2013). Tartary buckwheat improves cognition and memory function in an in vivo amyloid-beta-induced Alzheimer model. Food Chem. Toxicol. 53, 105–111. doi: 10.1016/j.fct.2012.11.002
Chun, H., Marriott, I., Lee, C. J., Cho, H. (2018). Elucidating the interactive roles of glia in alzheimer’s disease using established and newly developed experimental models. Front. Neurol. 9, 797. doi: 10.3389/fneur.2018.00797
Cohen, A. S., Calkins, E. (1959). Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 183 (4669), 1202–1203. doi: 10.1038/1831202a0
Collingridge, G. L., Singer, W. (1990). Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol. Sci. 11 (7), 290–296. doi: 10.1016/0165-6147(90)90011-V
Crump, F. T., Dillman, K. S., Craig, A. M. (2001). cAMP-dependent protein kinase mediates activity-regulated synaptic targeting of NMDA receptors. J. Neurosci. 21 (14), 5079–5088. doi: 10.1523/JNEUROSCI.21-14-05079.2001
Cull-Candy, S., Brickley, S., Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11 (3), 327–335. doi: 10.1016/S0959-4388(00)00215-4
Dahlgren, K. N., Manelli, A. M., Stine, W. B., Baker, L. K., Krafft, G. A., LaDu, M. J. (2002). Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277 (35), 32046–32053. doi: 10.1074/jbc.M201750200
DaRocha-Souto, B., Scotton, T. C., Coma, M., Serrano-Pozo, A., Hashimoto, T., Sereno, L., et al. (2011). Brain oligomeric beta-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J. Neuropathol. Exp. Neurol. 70 (5), 360–376. doi: 10.1097/NEN.0b013e318217a118
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 29 (9), 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
Dineley, K. T., Kayed, R., Neugebauer, V., Fu, Y., Zhang, W. R., Reese, L. C., et al. (2010). Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J. Neurosci. Res. 88 (13), 2923–2932. doi: 10.1002/jnr.22445
Dong, L., Wang, Y., Lv, J., Zhang, H., Jiang, N., Lu, C., et al. (2019). Memory enhancement of fresh ginseng on deficits induced by chronic restraint stress in mice. Nutr. Neurosci. 22 (4), 235–242. doi: 10.1080/1028415X.2017.1373928
Epelbaum, S., Youssef, I., Lacor, P. N., Chaurand, P., Duplus, E., Brugg, B., et al. (2015). Acute amnestic encephalopathy in amyloid-beta oligomer-injected mice is due to their widespread diffusion in vivo. Neurobiol. Aging 36 (6), 2043–2052. doi: 10.1016/j.neurobiolaging.2015.03.005
Fa, M., Orozco, I. J., Francis, Y. I., Saeed, F., Gong, Y., Arancio, O. (2010). Preparation of oligomeric beta-amyloid 1–42 and induction of synaptic plasticity impairment on hippocampal slices. J. Visualized Exp. JoVE (41), 1884. doi: 10.3791/1884
Fan, Z., Brooks, D. J., Okello, A., Edison, P. (2017). An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 140 (3), 792–803. doi: 10.1093/brain/aww349
Figueiredo, C. P., Clarke, J. R., Ledo, J. H., Ribeiro, F. C., Costa, C. V., Melo, H. M., et al. (2013). Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 33 (23), 9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013
Fu, Y., Kadioglu, O., Wiench, B., Wei, Z., Gao, C., Luo, M., et al. (2015). Cell cycle arrest and induction of apoptosis by cajanin stilbene acid from Cajanus cajan in breast cancer cells. Phytomedicine 22 (4), 462–468. doi: 10.1016/j.phymed.2015.02.005
Geng, D., Kang, L., Su, Y., Jia, J., Ma, J., Li, S., et al. (2013). Protective effects of EphB2 on Abeta1–42 oligomer-induced neurotoxicity and synaptic NMDA receptor signaling in hippocampal neurons. Neurochem. Int. 63 (4), 283–290. doi: 10.1016/j.neuint.2013.06.016
Gong, Y., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., et al. (2003). Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U. S. A. 100 (18), 10417–10422. doi: 10.1073/pnas.1834302100
Hardingham, G. E., Fukunaga, Y., Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5 (5), 405–414. doi: 10.1038/nn835
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Dewachter, I., Walter, J., Klockgether, T., et al. (2005). Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J. Neuroinflam. 2, 22. doi: 10.1186/1742-2094-2-22
Herrmann, N., Chau, S. A., Kircanski, I., Lanctot, K. L. (2011). Current and emerging drug treatment options for Alzheimer’s disease: a systematic review. Drugs 71 (15), 2031–2065. doi: 10.2165/11595870-000000000-00000
Hickman, S. E., Allison, E. K., El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28 (33), 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Huang, M. Y., Lin, J., Lu, K., Xu, H. G., Geng, Z. Z., Sun, P. H., et al. (2016). Anti-inflammatory effects of cajaninstilbene acid and its derivatives. J. Agric. Food Chem. 64 (14), 2893–2900. doi: 10.1021/acs.jafc.6b00227
Jiang, B. P., Liu, Y. M., Le, L., Li, Z. Y., Si, J. Y., Liu, X. M., et al. (2014). Cajaninstilbene acid prevents corticosterone-induced apoptosis in PC12 cells by inhibiting the mitochondrial apoptotic pathway. Cell Physiol. Biochem. 34 (3), 1015–1026. doi: 10.1159/000366317
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., Selkoe, D. J. (2011). Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U. S. A. 108 (14), 5819–5824. doi: 10.1073/pnas.1017033108
Kasza, A., Penke, B., Frank, Z., Bozso, Z., Szegedi, V., Hunya, A., et al. (2017). Studies for improving a rat model of Alzheimer’s disease: ICV administration of well-characterized beta-amyloid 1–42 oligomers induce dysfunction in spatial memory. Molecules 22 (11), 2007. doi: 10.3390/molecules22112007
Kim, H. J., Lee, K. W., Lee, H. J. (2007). Protective effects of piceatannol against beta-amyloid-induced neuronal cell death. Ann. N. Y. Acad. Sci. 1095, 473–482. doi: 10.1196/annals.1397.051
Kim, H. Y., Lee, D. K., Chung, B. R., Kim, H. V., Kim, Y. (2016). Intracerebroventricular injection of amyloid-beta peptides in normal mice to acutely induce alzheimer-like cognitive deficits. J. Visualized Exp. JoVE (109), 53308. doi: 10.3791/53308
Klein, W. L. (2002). Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 41 (5), 345–352. doi: 10.1016/S0197-0186(02)00050-5
Kong, Y., Fu, Y. J., Zu, Y. G., Chang, F. R., Chen, Y. H., Liu, X. L., et al. (2010). Cajanuslactone, a new coumarin with anti-bacterial activity from pigeon pea [Cajanus cajan (L.) Millsp.] leaves. Food Chem. 121 (4), 1150–1155. doi: 10.1016/j.foodchem.2010.01.062
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from a beta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U. S. A. 95 (11), 6448–6453. doi: 10.1073/pnas.95.11.6448
Ledo, J. H., Azevedo, E. P., Beckman, D., Ribeiro, F. C., Santos, L. E., Razolli, D. S., et al. (2016). Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer’s amyloid-beta oligomers in mice. J. Neurosci. 36 (48), 12106–12116. doi: 10.1523/JNEUROSCI.1269-16.2016
Ledo, J. H., Azevedo, E. P., Clarke, J. R., Ribeiro, F. C., Figueiredo, C. P., Foguel, D., et al. (2013). Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol. Psychiatry 18 (10), 1053–1054. doi: 10.1038/mp.2012.168
Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440 (7082), 352–357. doi: 10.1038/nature04533
Li, S., Jin, M., Koeglsperger, T., Shepardson, N. E., Shankar, G. M., Selkoe, D. J. (2011). Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31 (18), 6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011
Lindroos, O. F., Leinonen, L. M. (1983). Rapid Nissl staining for frozen sections of fresh brain. Stain Technol. 58 (4), 240–242. doi: 10.3109/10520298309066795
Liu, Y., Shen, S., Li, Z., Jiang, Y., Si, J., Chang, Q., et al. (2014a). Cajaninstilbene acid protects corticosterone-induced injury in PC12 cells by inhibiting oxidative and endoplasmic reticulum stress-mediated apoptosis. Neurochem. Int. 78, 43–52. doi: 10.1016/j.neuint.2014.08.007
Liu, Y. M., Jiang, B. P., Shen, S. N., Guo, Z., Li, Z. Y., Si, J. Y., et al. (2014b). Chemical constituents from leaves of Cajanus cajan. Chin. Tradit. Herbal Drugs 45 (4), 466–470. doi: 10.1002/chin.201423216
Lu, C., Wang, Y., Lv, J., Jiang, N., Fan, B., Qu, L., et al. (2018). Ginsenoside Rh2 reverses sleep deprivation induced cognitive deficit in mice. Behav. Brain Res. 349, 109–115. doi: 10.1016/j.bbr.2018.03.005
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D. M., Landreth, G. E. (2009). Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 29 (13), 4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009
Masliah, E., Alford, M., DeTeresa, R., Mallory, M., Hansen, L. (1996). Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 40 (5), 759–766. doi: 10.1002/ana.410400512
Matos, M., Augusto, E., Oliveira, C. R., Agostinho, P. (2008). Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 156 (4), 898–910. doi: 10.1016/j.neuroscience.2008.08.022
Maurice, T., Lockhart, B. P., Privat, A. (1996). Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 706 (2), 181–193. doi: 10.1016/0006-8993(95)01032-7
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Beyreuther, K., et al. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46 (6), 860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M
Misiti, F., Sampaolese, B., Mezzogori, D., Orsini, F., Pezzotti, M., Giardina, B., et al. (2006). Protective effect of rhubarb derivatives on amyloid beta (1–42) peptide-induced apoptosis in IMR-32 cells: a case of nutrigenomic. Brain Res. Bull. 71 (1–3), 29–36. doi: 10.1016/j.brainresbull.2006.07.012
Moon, M., Choi, J. G., Nam, D. W., Hong, H. S., Choi, Y. J., Oh, M. S., et al. (2011). Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-beta1–42 oligomer-injected mice. J. Alzheimers Dis. 23 (1), 147–159. doi: 10.3233/JAD-2010-101263
Mullard, A. (2016). Alzheimer amyloid hypothesis lives on. Nat. Rev. Drug Discov. 16 (1), 3–5. doi: 10.1038/nrd.2016.281
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V., Wang, H. Y. (2003). Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971 (2), 197–209. doi: 10.1016/S0006-8993(03)02361-8
Noda, M., Nakanishi, H., Akaike, N. (1999). Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience 92 (4), 1465–1474. doi: 10.1016/S0306-4522(99)00036-6
Olabarria, M., Noristani, H. N., Verkhratsky, A., Rodriguez, J. J. (2010). Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58 (7), 831–838. doi: 10.1002/glia.20967
Pal, B. (2018). Involvement of extrasynaptic glutamate in physiological and pathophysiological changes of neuronal excitability. Cell Mol. Life Sci. 75 (16), 2917–2949. doi: 10.1007/s00018-018-2837-5
Patel, N. K., Bhutani, K. K. (2014). Pinostrobin and Cajanus lactone isolated from Cajanus cajan (L.) leaves inhibits TNF-alpha and IL-1beta production: in vitro and in vivo experimentation. Phytomedicine 21 (7), 946–953. doi: 10.1016/j.phymed.2014.02.011
Paxinos, G., Franklin, K. B. J. (2001). The mouse brain in stereotaxic coordinates. 2nd ed. Vol. 28. Psychoneuroendocrinology. doi: 10.1016/S0306-4530(03)00088-X
Reid, S. N. S., Ryu, J. K., Kim, Y., Jeon, B. H. (2018). GABA-enriched fermented Laminaria japonica improves cognitive impairment and neuroplasticity in scopolamine- and ethanol-induced dementia model mice. Nutr. Res. Pract. 12 (3), 199–207. doi: 10.4162/nrp.2018.12.3.199
Ruan, C. J., Li, Z., Zhang, L., Chen, D. H., Du, G. H., Sun, L. (2010a). Protective effects of trans-2, 4-dimethoxystibene on cognitive, impairments induced by Abeta(25–35) in, hypercholesterolemic rats. Brain 4, 016. doi: 10.1016/j.brainresbull.2010.04.016
Ruan, C. J., Si, J. Y., Zhang, L., Chen, D. H., Du, G. H., Su, L. (2009). Protective effect of stilbenes containing extract-fraction from Cajanus cajan L. Neurosci. Lett. 467 (2), 159–163. doi: 10.1016/j.neulet.2009.10.029
Ruan, C. J., Zhang, L., Chen, D. H., Li, Z., Du, G. H., Sun, L. (2010b). Effects of trans-2,4-dimethoxystibene against the neurotoxicity induced by Abeta(25–35) both in vitro and in vivo. Neurosci. Res. 67 (3), 209–214. doi: 10.1016/j.neures.2010.03.009
Scheff, S. W., Price, D. A., Schmitt, F. A., Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27 (10), 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Schuster, R., Holzer, W., Doerfler, H., Weckwerth, W., Viernstein, H., Okonogi, S., et al. (2016). Cajanus cajan- a source of PPARgamma activators leading to anti-inflammatory and cytotoxic effects. Food Funct. 7 (9), 3798–3806. doi: 10.1039/C6FO00689B
Selkoe, D. J. (2001). Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81 (2), 741–766. doi: 10.1152/physrev.2001.81.2.741
Snyder, S. W., Ladror, U. S., Wade, W. S., Wang, G. T., Barrett, L. W., Matayoshi, E. D., et al. (1994). Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys. J. 67 (3), 1216–1228. doi: 10.1016/S0006-3495(94)80591-0
Stine, W. B., Jr., Dahlgren, K. N., Krafft, G. A., LaDu, M. J. (2003). In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278 (13), 11612–11622. doi: 10.1074/jbc.M210207200
Stine, W. B., Jr., Snyder, S. W., Ladror, U. S., Wade, W. S., Miller, M. F., Perun, T. J., et al. (1996). The nanometer-scale structure of amyloid-beta visualized by atomic force microscopy. J. Protein Chem. 15 (2), 193–203. doi: 10.1007/BF01887400
Sultana, R., Perluigi, M., Butterfield, D. A. (2009). Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol. 118 (1), 131–150. doi: 10.1007/s00401-009-0517-0
Sun, Y., Liu, Y., He, W., Wang, C., Tickner, J., Kuek, V., et al. (2019). Cajaninstilbene acid inhibits osteoporosis through suppressing osteoclast formation and RANKL-induced signaling pathways. J. Cell. Physiol. 234 (7), 11792–11804. doi: 10.1002/jcp.27868
Tabaton, M., Piccini, A. (2005). Role of water-soluble amyloid-beta in the pathogenesis of Alzheimer’s disease. Int. J. Exp. Pathol. 86 (3), 139–145. doi: 10.1111/j.0959-9673.2005.00428.x
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S., et al. (2013). Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U. S. A. 110 (27), E2518–E2527. doi: 10.1073/pnas.1306832110
Verkhratsky, A., Olabarria, M., Noristani, H. N., Yeh, C. Y., Rodriguez, J. J. (2010). Astrocytes in Alzheimer’s disease. Neurotherapeutics 7 (4), 399–412. doi: 10.1016/j.nurt.2010.05.017
Vorhees, C. V., Williams, M. T. (2006). Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 1 (2), 848–858. doi: 10.1038/nprot.2006.116
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416 (6880), 535–539. doi: 10.1038/416535a
Wang, L. S., Liu, X. M., Tao, X., Zhang, M. D., Zhou, Y. F., Chang, Q. (2018). Application of the animal model of intracerebral injection of amyloid-β oligomers to the study of Alzheimer’s disease. Acta Pharm. Sin. 53 (7), 1060–1067. doi: 10.16438/j.0513-4870.2018-0088
Wang, L. S., Zhang, M. D., Tao, X., Zhou, Y. F., Liu, X. M., Pan, R. L., et al. (2019). LC-MS/MS-based quantification of tryptophan metabolites and neurotransmitters in the serum and brain of mice. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1112, 24–32. doi: 10.1016/j.jchromb.2019.02.021
Wang, R., Reddy, P. H. (2017). Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 57 (4), 1041–1048. doi: 10.3233/JAD-160763
Wang, R., Zhang, Y., Li, J., Zhang, C. (2017). Resveratrol ameliorates spatial learning memory impairment induced by Abeta1–42 in rats. Neuroscience 344, 39–47. doi: 10.1016/j.neuroscience.2016.08.051
Waxman, E. A., Lynch, D. R. (2005). N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist 11 (1), 37–49. doi: 10.1177/1073858404269012
Wu, N., Kong, Y., Fu, Y., Zu, Y., Yang, Z., Yang, M., et al. (2011). In vitro antioxidant properties, DNA damage protective activity, and xanthine oxidase inhibitory effect of cajaninstilbene acid, a stilbene compound derived from pigeon pea [Cajanus cajan (L.) Millsp.] leaves. J. Agric. Food Chem. 59 (1), 437–443. doi: 10.1021/jf103970b
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anankov, R., Yan, F., et al. (2003). Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 9 (4), 453–457. doi: 10.1038/nm838
Xue, D., Chen, S. G., Xu, S. P., Zhe, S., Li, H. Q., Yin, T. H., et al. (2010). Establishment of a highly automated and intelligent experimental system of passive avoidance for mice. J. Clin. Rehabil. Tissue Eng. Res. 14 (15), 2778–2782.
Yang, R., Chen, L., Wang, H., Xu, B., Tomimoto, H., Chen, L. (2012). Anti-amnesic effect of neurosteroid PREGS in Abeta25-35-injected mice through sigma1 receptor- and alpha7nAChR-mediated neuroprotection. Neuropharmacology 63 (6), 1042–1050. doi: 10.1016/j.neuropharm.2012.07.035
Yang, Y., Ji, W. G., Zhu, Z. R., Wu, Y. L., Zhang, Z. Y., Qu, S. C. (2018). Rhynchophylline suppresses soluble Abeta1–42-induced impairment of spatial cognition function via inhibiting excessive activation of extrasynaptic NR2B-containing NMDA receptors. Neuropharmacology 135, 100–112. doi: 10.1016/j.neuropharm.2018.03.007
Youssef, I., Florent-Bechard, S., Malaplate-Armand, C., Koziel, V., Bihain, B., Olivier, J. L., et al. (2008). N-truncated amyloid-beta oligomers induce learning impairment and neuronal apoptosis. Neurobiol. Aging 29 (9), 1319–1333. doi: 10.1016/j.neurobiolaging.2007.03.005
Zhang, L. L., Sui, H. J., Liang, B., Wang, H. M., Qu, W. H., Yu, S. X., et al. (2014). Atorvastatin prevents amyloid-beta peptide oligomer-induced synaptotoxicity and memory dysfunction in rats through a p38 MAPK-dependent pathway. Acta Pharmacol. Sin. 35 (6), 716–726. doi: 10.1038/aps.2013.203
Zhao, W., Xie, W., Le, W., Beers, D. R., He, Y., Henkel, J. S., et al. (2004). Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 63 (9), 964–977. doi: 10.1093/jnen/63.9.964
Keywords: Alzheimer’s disease, amyloid-β oligomer, cajaninstilbene acid, cognition, microglia, astrocyte, glutamate, N-methyl-D-aspartate receptor
Citation: Wang L-S, Tao X, Liu X-M, Zhou Y-F, Zhang M-D, Liao Y-H, Pan R-L and Chang Q (2019) Cajaninstilbene Acid Ameliorates Cognitive Impairment Induced by Intrahippocampal Injection of Amyloid-β1–42 Oligomers. Front. Pharmacol. 10:1084. doi: 10.3389/fphar.2019.01084
Received: 10 June 2019; Accepted: 26 August 2019;
Published: 24 September 2019.
Edited by:
Ashok Kumar, University of Florida, United StatesReviewed by:
Luca Ferraro, University of Ferrara, ItalyLucio Tremolizzo, University of Milano Bicocca, Italy
Yona Levites, University of Florida, United States
Copyright © 2019 Wang, Tao, Liu, Zhou, Zhang, Liao, Pan and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui-Le Pan, cmxwYW5AaW1wbGFkLmFjLmNu; Qi Chang, cWNoYW5nQGltcGxhZC5hYy5jbg==