 Yuanying Fang1†Daoyong Hu2†Huilan Li1,3Jianguo Hu4Yanhua Liu4Zhifeng Li1Guoliang Xu3*Lanying Chen1Yi Jin1Shilin Yang1
Yuanying Fang1†Daoyong Hu2†Huilan Li1,3Jianguo Hu4Yanhua Liu4Zhifeng Li1Guoliang Xu3*Lanying Chen1Yi Jin1Shilin Yang1 Zunhua Yang4*
Zunhua Yang4*- 1National Engineering Research Center for Manufacturing Technology of TCM Solid Preparation, Jiangxi University of Traditional Chinese Medicine, Nanchang, China
- 2The Key Laboratory of Oral Biomedicine, The Affiliated Stomatological Hospital of Nanchang University, Nanchang, China
- 3Research Center for Differentiation and Development of Basic Theory of Traditional Chinese Medicine, Jiangxi University of Traditional Chinese Medicine, Nanchang, China
- 4College of Pharmacy, Jiangxi University of Traditional Chinese Medicine, Nanchang, China
A series of ester and amide derivatives of triterpenoid saponin Pulsatilla saponin D (PSD) were designed, synthesized, and evaluated for their antiproliferative activity. Compounds 1 and 6 displayed 1.7–8.3 times more potent cytotoxicity (IC50 = 1.2–4.7 and 1.7–4.5 μM, respectively) against five human tumor cell lines (SMMC-7721, MCF-7, NCI-H460, A549, and HCT-116) in vitro and lower acute toxicity to mice in vivo than did PSD. Furthermore, compound 6 was observed to show potent tumor growth inhibition against mice H22 hepatocellular cells (49.8% at 20 mg/kg) and induce cell cycle at G1 phase and apoptosis in HCT-116 cells.
Introduction
The Pulsatilla chinensis (Bunge) Regel, a traditional Chinese herb, is used for “blood cooling,” stopping dysentery, and detoxification (Chen et al., 2015; Liu et al., 2013; Jiang et al., 2009; Fang et al., 2016). A series of triterpenoid saponins were isolated from the plant and reported to possess various biological activities such as anticancer, anti-inflammatory, antiviral and antibacterial, and so on (Liang et al., 2011; Ye et al., 2014; Guan et al., 2015; Acebey-Castellon et al., 2011). In our efforts to discover anticancer agents, we focus on the modification of natural product as an effective and powerful strategy, which is also a promising approach to develop new lead compounds (Che et al., 2014).
Among these triterpenoid saponins, several abundant components including hederacolchiside A1, Pulsatilla saponin D (PSD), and α-hederin (Figure 1) exhibited potent antitumor activities in vitro and in vivo (Chen et al., 2018; Zhang et al., 2015; Shu et al., 2013; Xu et al., 2012; Yan et al., 2008; Kim et al., 2013). However, due to the hemolytic toxicity of triterpenoid saponins, it is impossible to develop them as clinical medicines directly. We have previously investigated hederacolchiside A1 derivatives bearing furoxan fragment as a nitric oxide donor against solid tumor in animal models (Fang et al., 2017). Nevertheless, since the isolation of hederacolchiside A1 was in a lower yield than PSD, it was difficult to obtain the material, which limited its modification.
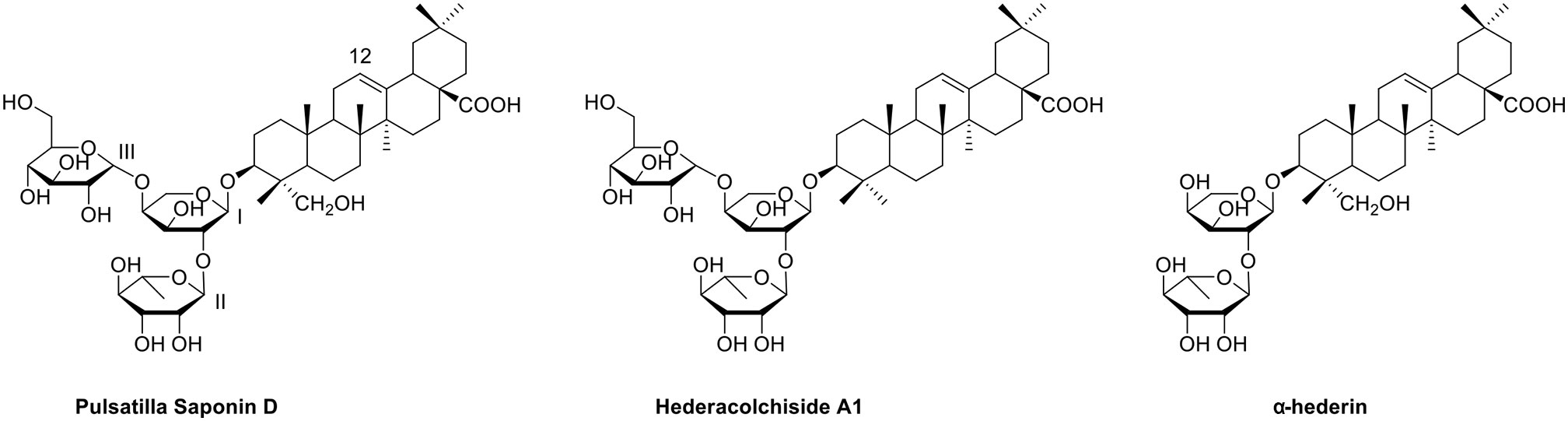
Figure 1 The structures of three triterpenoid saponins.
In our attempts to discover the valuable anticancer agents, PSD was chosen as the lead compound. PSD (hederagenin 3-O-a-L-rhamnopyranosyl-(1/2)-[b-D-glucopyranosyl-(1/4)]-a-L-arabinopyranoside), also named SB365 in the literature, was isolated from P. chinensis (Bunge) Regel in our department and showed excellent bioactivity (Kim et al., 2004; Bang et al., 2005). A structure–activity relationship study indicated that the 28-COOH is the moiety causing hemolytic toxicity, so modification of PSD was conducted by converting it to ester or amide bearing various functional groups such as alkyl, amino, halogen, hydroxyl, and carboxyl groups. We would see whether these moieties were beneficial for enhancement of antitumor activity and lowering of toxicity. In addition, we envisaged that the shelter of carboxyl group would significantly improve antiproliferative activities and hemolysis issue. Herein we presented the synthesis, biological evaluation, and mode of action of PSD derivatives as antitumor agents.
Results and Discussion
Chemistry
PSD derivatives were generated from the natural product PSD, following the procedures and conditions as shown in Schemes 1 and 2. Esterification of PSD with iodomethane, iodoethane, or 1-iodopropane in the presence of K2CO3/DMF at room temperature afforded esters 1, 2, and 3, respectively, in good yields. Reaction of PSD with dichloroethane in the presence of K2CO3/NaI/DMF at 50°C provided chlorinated ester 5 in 52%. Condensation of PSD with bromo-substituted materials in the presence of K2CO3/DMF at 50°C yielded substituted esters 4 and 6–10, respectively. The amides 11–13 were obtained via condensation of PSD with amines in the presence of EDCI/HOBt/DIPEA/DMF at room temperature.
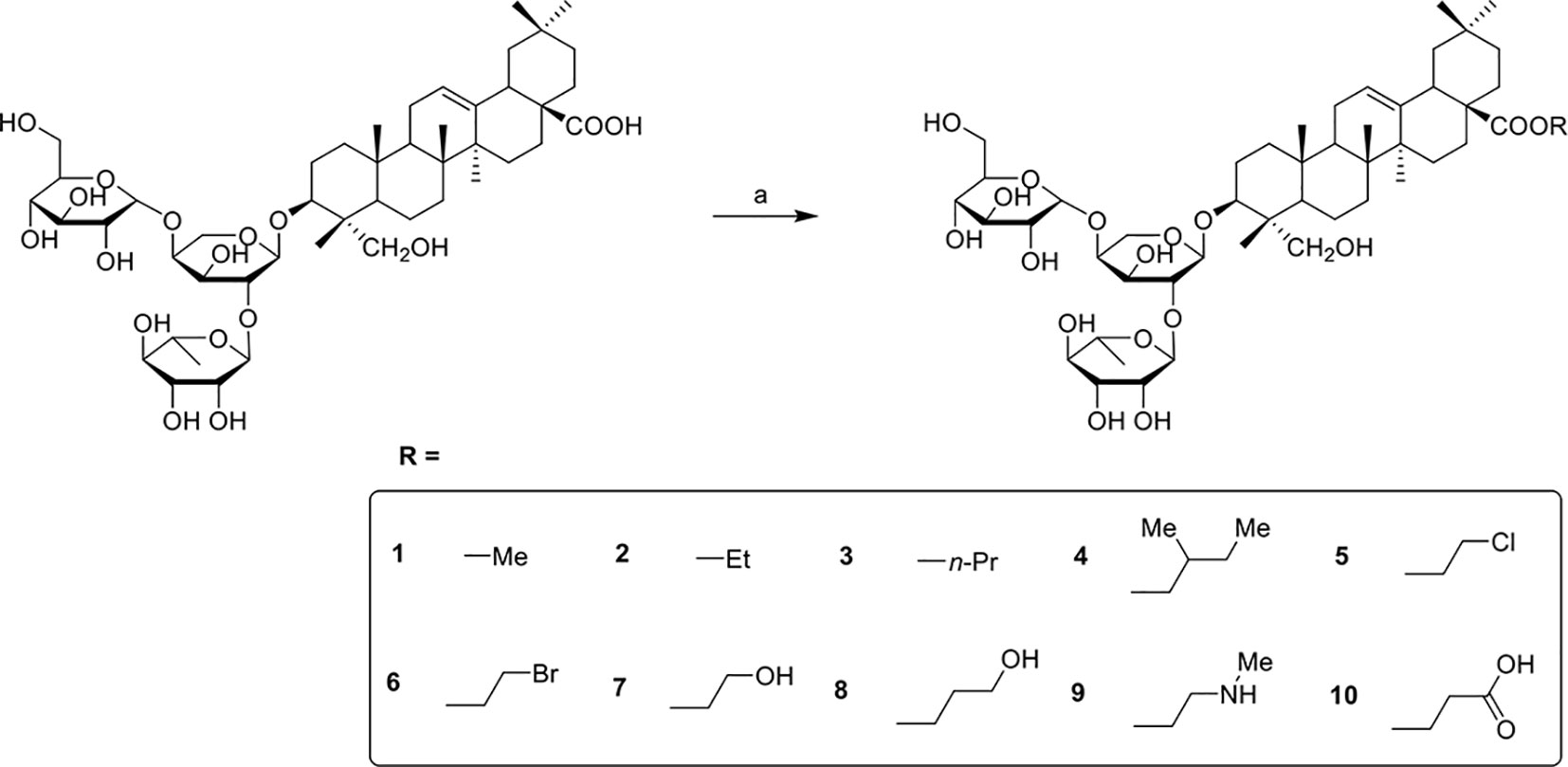
Scheme 1 Reagents and conditions: (a) R-I, K2CO3, DMF, R.T., 12h or R-Br, K2CO3, DMF, 50°C, 12h or R-Cl, K2CO3, DMF, NaI, 50°C, 12h.
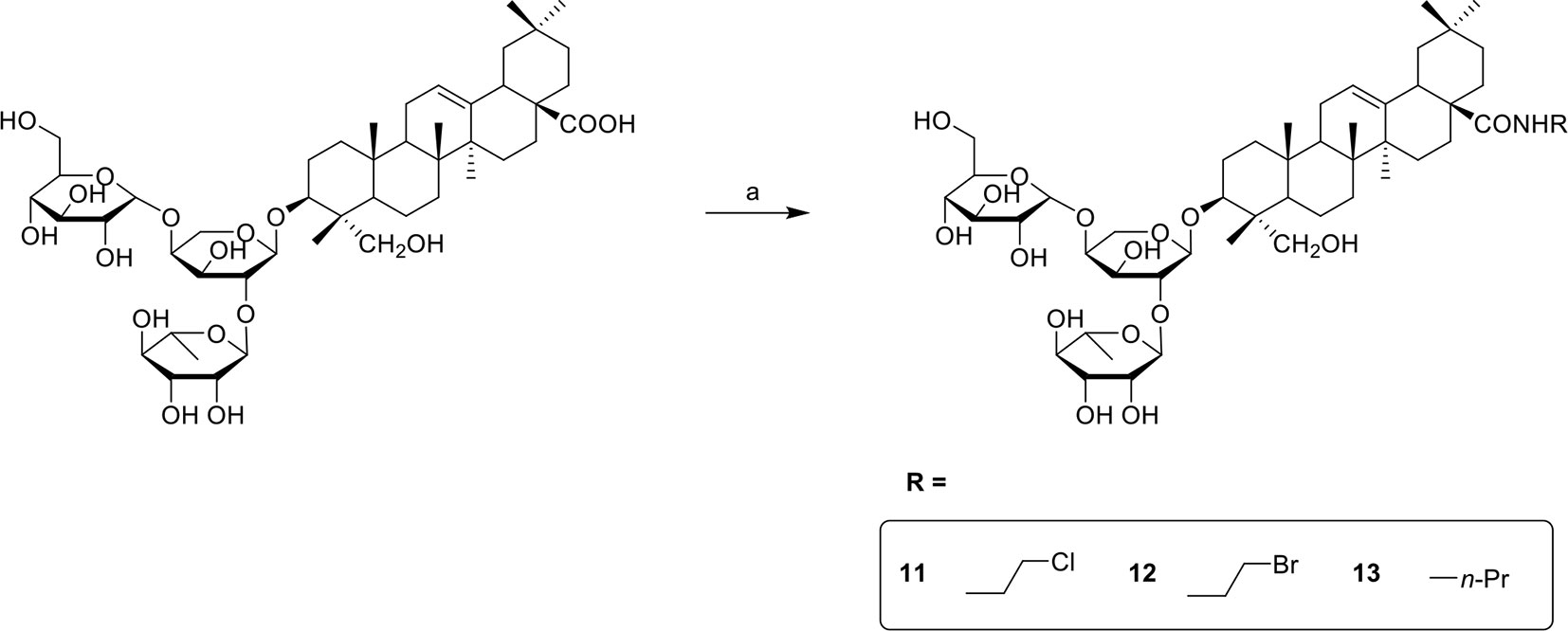
Scheme 2 Reagents and conditions: (a) NH2-R, EDCI, HOBt, DIPEA, DMF, R.T., 12h.
Antiproliferative Activity
Compounds 1–13 were evaluated for their antiproliferative activities against SMMC-7721, MCF-7, NCI-H460, A549, and HCT-116 human cancer cell lines using the MTT assay. PSD was selected as a positive control, and the cytotoxicity assay results are illustrated in Table 1. Compounds 1 and 6, methyl ester and β-bromoethyl ester of PSD, exhibited 1.7–8.3 times more potent antiproliferative activities than did PSD against all five cancer cell lines. The ethyl ester 2 and β-chloroethyl ester 5 displayed stronger cytotoxicity than did PSD against the four cell lines MCF-7, NCI-H460, A549, and HCT-116 with IC50 less than 10 μM, and similar activities to those of PSD against SMMC-7721. In general, the halogen substitution and ester derivatives were favorable to the activities compared with hydroxyl and amide, respectively. However, the β-carboxylic ethyl ester 10 also showed superior cytotoxicity to PSD against the three cell lines MCF-7, NCI-H460, and HCT-116.
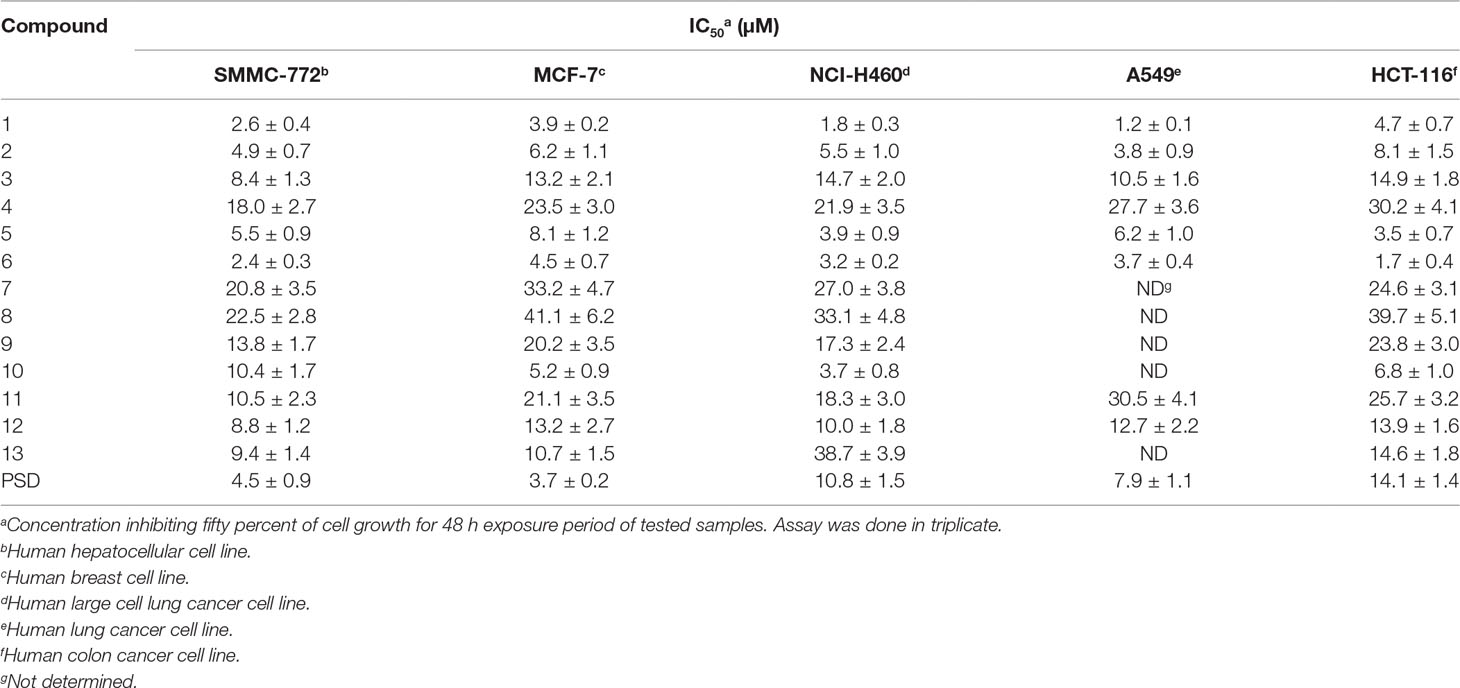
Table 1 Cytotoxicity of PSD derivatives against 5 human cancer cell lines after 48 h incubation.
Mice Acute Toxicity
PSD derivatives 1 and 6 exhibited potent antitumor activities on five human cancer cell lines (IC50 = 1.2–4.7 and 1.7–4.5 μM, respectively) and were selected to identify the mice acute toxicities compared with PSD. Due to the poor solubility, these three compounds were made in micelle formation with methoxyl poly(ethylene glycol)-poly(lactide) copolymer (mPEG-PLA; molecular weight, 4000) at 1.0-mg/ml concentration of PSD and at 2.4-mg/ml concentration of 1 and 6. The mice were injected with the solutions via the caudal vein at a dose of 0.1 ml/10 g/d for 7 days and continued to be under observation for another 7 days without injection. As shown in Table 2, the tolerance dose of natural product PSD was less than 10 mg/kg, and 12 mice were all dead in 1 h after injection. The tolerance doses of 1 and 6 were both higher than 168 mg/kg, and no mouse was dead in 14 days. The results indicated that 1 and 6 declined the acute toxicity greatly and supplied an evidence to design the suitable dose for tumor inhibition rate trial.
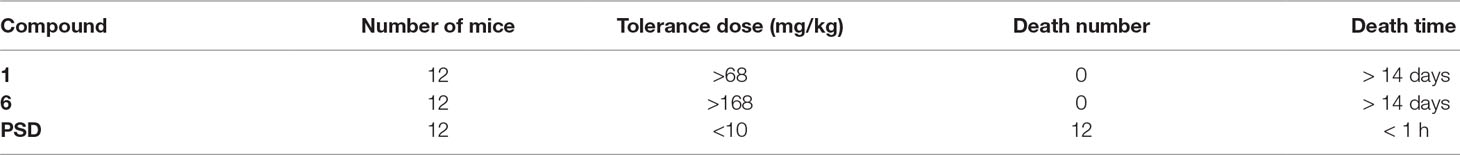
Table 2 Mice acute toxicity of compound 1 and 6.
Tumor Growth Inhibition Study In Vivo
Compound 6 was observed to exhibit significant and consistent cytotoxicity in five human cancer cell lines without apparent mice acute toxicity. The effect of compound 6 on tumor growth inhibition was evaluated using H22 hepatocellular carcinoma model in male mice with PSD as a positive control. Compounds 6 and PSD were made in micelle with mPEG-PLA at concentrations of 2.4 and 1.0 mg/ml, respectively. Then compound 6 was injected via the caudal vein at doses of 10 and 20 mg/kg/d for 14 days, respectively; PSD was injected at a dose of 6 mg/kg/3 d five times. The results are illustrated in Table 3. Treatment with 6 at 10 mg/kg significantly inhibited the growth of H22 tumor in 42.8%, but treatment with 6 at 20 mg/kg enhanced the inhibitory effect on the growth of H22 tumor (49.8%), which was almost equal to the treatment of PSD (50.2%). Remarkably, no mice were dead after treatment with 6 in 14 days, which indicated that derivative 6 could reduce the toxicity greatly.

Table 3 In vivo H22 xenograft studies of compound 6.
Cell Cycle Study
To elucidate the relationship between the mechanism of cell growth suppression and cell cycle arrest, cell cycle distribution on HCT-116 cells by treating with compound 6 was investigated. As shown in Figure 2, the cells of the control group in G1, S, and G2 phases accounted for 51.8%, 4.9%, and 33.3%, respectively. After cells were treated with 6 at concentrations of 2.5, 5 and 10 μg/ml, the cell percentages of G1 phase increased to 68.3%, 70.6%, and 74.7%, respectively. These results indicated that compounds 6 could inhibit tumor cell proliferation by blocking the cell cycle at G1 phase in a concentration-dependent manner.
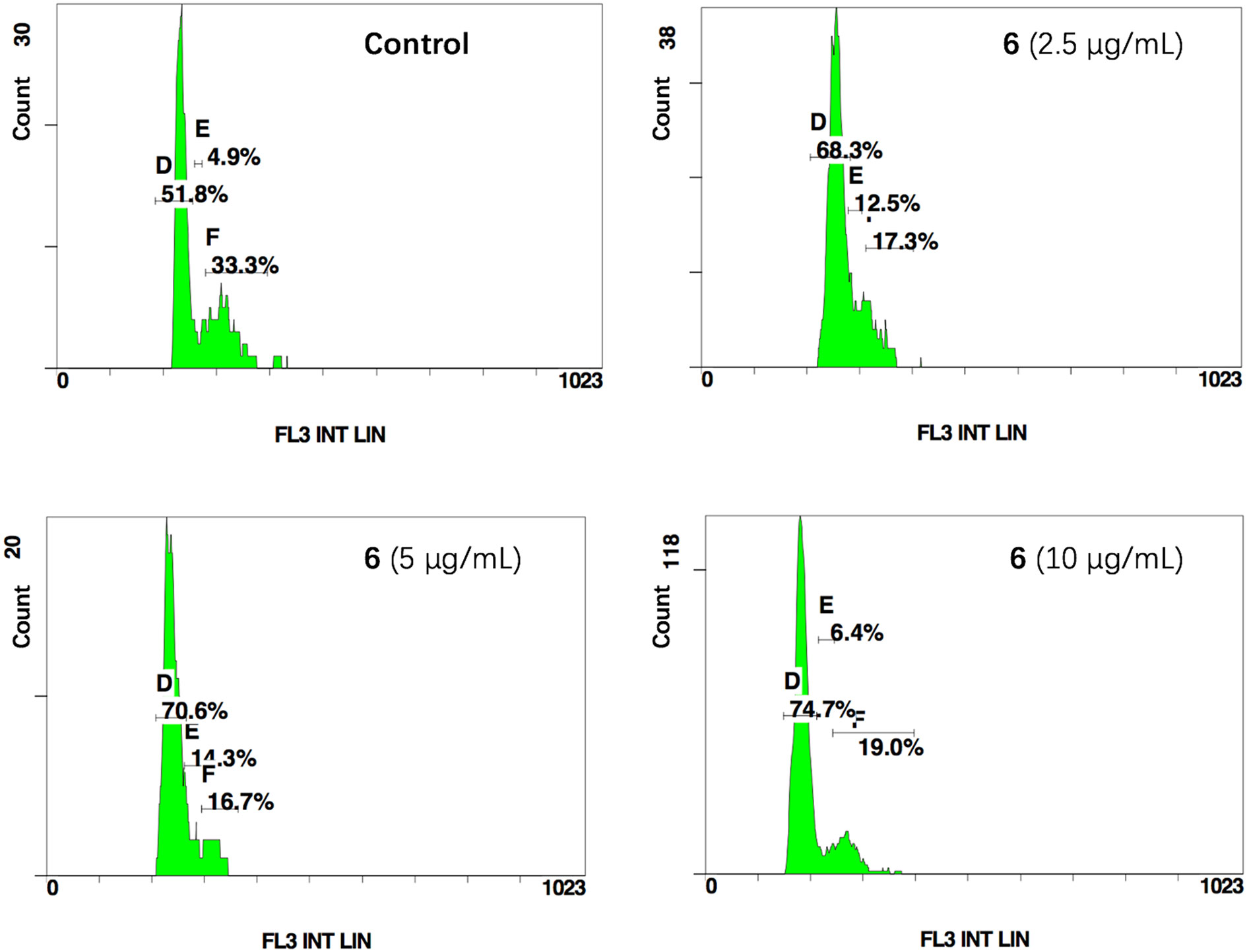
Figure 2 Compound 6 induced G1 cycle arrest in HCT-116. D, E and F represent G1, S and G2 phase.
Apoptosis Induction
Activation of apoptosis is always involved in the mechanism of action of antiproliferative agents. An Annexin V–FITC and propidium iodide (PI) double-staining assay was used to evaluate the effect of compound 6 on the induction of apoptosis. The HCT-116 cells were treated with three concentrations of 6 (5, 10, and 20 μg/ml) for 48 h, and the apoptotic cells were determined. As shown in Figure 3, the ratios of early and late apoptotic cells were 27.6%, 85.1%, and 87.0% at the respective concentrations, which indicated that compound 6 induced apoptosis in a concentration-dependent manner also.
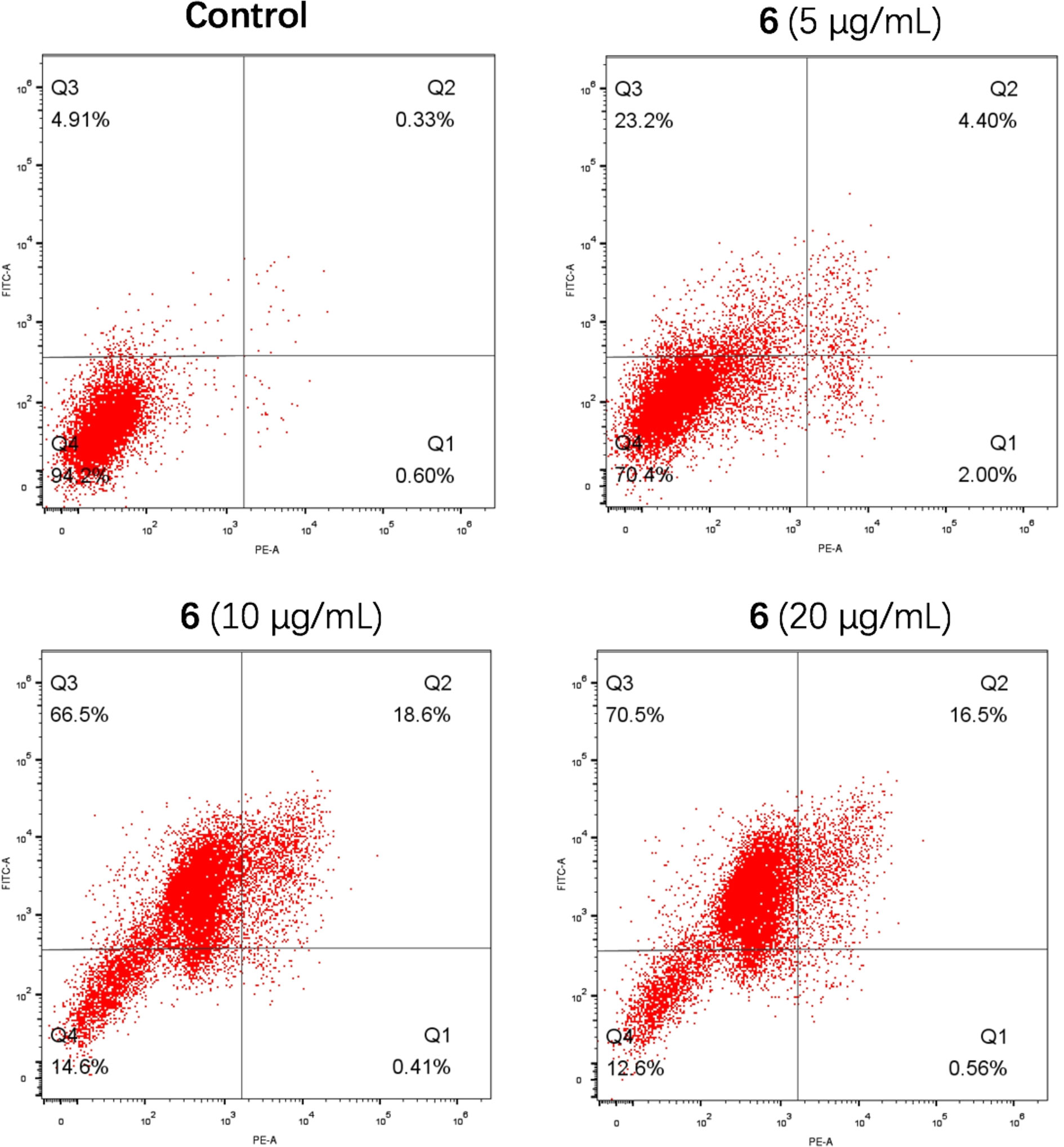
Figure 3 Compound 6 induced apoptosis in HCT-116. Q2 and Q3 represent late and early apoptosis.
Statistical Methods
T-test was used to evaluate the statistical differences between samples. Differences were considered to be significant when p < 0.05.
Conclusion
In summary, 13 PSD derivatives were designed, synthesized, and evaluated for their antiproliferative activities. Among them, compounds 1 and 6 were revealed to exhibit the most potent cytotoxicity against all five cancer cell lines with greatly reduced mice acute toxicity. Remarkably, compound 6 displayed 8.3 times more potent cytotoxicity against HCT-116 (IC50 = 1.7 μM) than did PSD and equal potent tumor inhibition rate in vivo (49.8%). The mode of action study revealed that compound 6 caused cell cycle arrest of G1 phase and induced apoptosis in HCT-116 cells. These results encourage us to develop a preclinical candidate as an anticancer agent from natural products. The follow-up studies on pharmacokinetics of active compounds are in progress, and the results will be reported in due course.
Materials and Methods
Chemistry
All starting materials were obtained from commercial suppliers and used without further purification. 1H-NMR and 13C-NMR spectra were recorded on a Bruker AVANCE III HD 600 (600 Hz) spectrometer. Chemical shifts are reported in parts per million downfield relative to tetramethylsilane as an internal standard. Peak splitting patterns are abbreviated as s (singlet), br s (broad singlet), d (doublet), t (triplet), dd (doublet of doublet), and m (multiplet). Mass spectra were recorded on a Thermo Fisher (LCQ Fleet). High-resolution mass spectrometry (HRMS) spectra were recorded on an AB SCIEX (Triple TOF 5600+). Thin-layer chromatography was performed on silica F254 purchased from the Branch of Qingdao Haiyang Chemical Co. and detected by UV light at 254 and 365 nm or by charring with sulfuric acid. Column chromatography was performed on silica gel column (200–300 mesh; Branch of Qingdao Haiyang Chemical Co.).
General Procedure to Synthesize Compounds 1-3
To a solution of PSD (100 mg, 0.11 mmol) in DMF (3 ml) was added K2CO3 (45 mg, 0.33 mmol) and alkyl iodide (0.33 mmol), respectively. The reaction was stirred at room temperature overnight. Then the mixture was poured into ice water and extracted with ethyl acetate (25 ml × 2). The organic phase was combined, washed with saturated brine (15 ml × 2), dried over anhydrous MgSO4, and filtered. The filtrate was evaporated to give the crude product, which was purified by column chromatography (dichloromethane methanol mixture as eluent) to afford the target compounds.
Methyl Ester of PSD (1)
White solid, 75% yield. 1H-NMR (600 Hz, MeOD): δ 5.27 (t, J = 3.4 Hz, 1H, H-12), 5.23 (s, 1H, H-1II), 4.51 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.6 Hz, 1H, H-1I), 3.64 (s, 3H, COOCH3), 1.26 (d, J = 6.2 Hz, 3H, H-6II), 1.19 (s, 3H), 0.99 (s, 3H), 0.96 (s, 3H), 0.93 (s, 3H), 0.77 (s, 3H), 0.73 (s, 3H). HRMS–time-of-flight mass spectrometry (TOF; m/z) calcd for C48H78O17 [M + H]+: 927.5317, found, 927.5329.
Ethyl Ester of PSD (2)
White solid, 71% yield. 1H-NMR (600 MHz, MeOD) δ 5.25 (t, J = 3.3 Hz, 1H, H-12), 5.22 (s, 1H, H-1II), 4.49 (d, J = 6.1 Hz, 1H, H-1III), 4.44 (d, J = 7.6 Hz, 1H, H-1I), 4.11–4.02 (m, 2H, COOCH2CH3), 1.23 (t, J = 6.9 Hz, 6H, COOCH2CH3), 1.18 (s, 3H), 0.98 (s, 3H), 0.94 (s, 3H), 0.91 (s, 3H), 0.77 (s, 3H), 0.71 (s, 3H). HRMS-TOF (m/z) calcd for C49H80O17 [M + H]+: 941.5474, found 941.5463.
n-Propyl Ester of PSD (3)
White solid, 64% yield. 1H-NMR (600 MHz, MeOD) δ 5.27 (t, J = 3.5 Hz, 1H, H-12), 5.23 (d, J = 1.4 Hz, 1H, H-1II), 4.52 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.6 Hz, 1H, H-1I), 4.04–3.96 (m, 2H, COOCH2CH2CH3), 1.70 (m, 2H, COOCH2CH2CH3), 1.26 (d, J = 6.2 Hz, 3H, H-6II), 1.18 (s, 3H), 0.99 (m, 3H, COOCH2CH2CH3), 0.98 (s, 3H), 0.95 (s, 3H), 0.93 (s, 3H), 0.78 (s, 3H), 0.73 (s, 3H). HRMS-TOF (m/z) calcd for C50H82O17 [M + H]+: 955.5630, found 933.5644.
General Procedure to Synthesize Compounds 4-10
To a solution of PSD (100 mg, 0.11 mmol) in DMF (3 ml) was added K2CO3 (45 mg, 0.33 mmol) and bromide (0.33 mmol; or 0.33 mmol of dichloroethane plus catalytic amount of NaI for 5), respectively. The reaction was stirred at 50°C overnight. Then the mixture was poured into ice water and extracted with ethyl acetate (25 ml × 2). The organic phase was combined, washed with saturated brine (15 ml × 2), dried over anhydrous MgSO4, and filtered. The filtrate was evaporated to give the crude product, which was purified by column chromatography (dichloromethane methanol mixture as eluent) to afford the target compounds.
β-Methyl Butyl Ester of PSD (4)
White solid, 72% yield. 1H-NMR (600 MHz, MeOD) δ 5.27 (t, J = 3.5 Hz, 1H, H-12), 5.23 (d, J = 1.3 Hz, 1H, H-1II), 4.51 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.6 Hz, 1H, H-1I), 3.92–3.89 (m, 2H, COOCH2CH(CH3)CH2CH3), 1.52–1.47 (m, 2H, COOCH2CH(CH3)CH2CH3), 1.26 (d, J = 6.2, 3H, H-6II), 1.20 (s, 3H), 1.03 (m, 1H, COOCH2CH(CH3)CH2CH3), 0.99 (s, 3H), 0.98 (d, J = 6.8 Hz, 3H, COOCH2CH(CH3)CH2CH3), 0.96 (s, 3H), 0.94 (m, 6H, COOCH2CH(CH3)CH2CH3), 0.77 (s, 3H), 0.73 (s, 3H). HRMS-TOF (m/z) calcd for C52H87O17 [M + H]+: 983.5943, found 983.5935.
β-Chloroethyl Ester of PSD (5)
White solid, 58% yield. 1H-NMR (600 MHz, MeOD) δ 5.29 (t, J = 3.5 Hz, 1H, H-12), 5.23 (d, J = 1.4 Hz, 1H, H-1II), 4.51 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.6 Hz, 1H, H-1I), 4.35–4.29 (m, 1H, COOCH2CH2Cl), 4.23 (m, 1H, COOCH2CH2Cl), 3.74 (m, 2H, COOCH2CH2Cl), 1.26 (d, J = 6.2 Hz, 3H), 1.20 (s, 3H), 0.99 (s, 3H), 0.97 (s, 3H), 0.94 (s, 3H), 0.79 (s, 3H), 0.73 (s, 3H). 13C-NMR (150Hz, MeOD) δ 177.6, 143.4, 122.6, 104.7, 103.2, 100.5, 80.8, 78.4, 76.7, 76.5, 75.5, 73.9, 72.6, 72.5, 70.7, 70.6, 69.9, 65.4, 64.1, 63.7, 63.1, 61.2, 46.8, 46.7, 45.6, 42.5, 41.6, 41.5, 41.4, 39.2, 38.3, 36.2, 35.5, 33.4, 32.3, 32.1, 32.0, 30.2, 27.4, 25.1, 25.0, 23.1, 22.6, 22.5, 17.4, 16.6, 16.4, 15.0, 12.4. HRMS-TOF (m/z) calcd for C49H79ClO17 [M + H]+: 975.5084, found 975.5106.
β-Bromoethyl Ester of PSD (6)
White solid, 73% yield. 1H-NMR (600 MHz, MeOD) δ 5.29 (t, J = 3.6 Hz, 1H, H-12), 5.23 (s, 1H, H-1II), 4.51 (d, J = 6.1 Hz, 1H, H-1III), 4.45 (t, J = 6.9 Hz, 1H, H-1I), 4.40–4.35 (m, 1H, COOCH2CH2Br), 4.32–4.27 (m, 1H, COOCH2CH2Br), 3.60-3.58 (m, 2H, COOCH2CH2Br), 1.25 (d, J = 6.1 Hz, 3H), 1.19 (s, 3H), 0.99 (s, 3H), 0.96 (s, 3H), 0.93 (s, 3H), 0.79 (s, 3H), 0.73 (s, 3H). 13C-NMR (150Hz, MeOD) δ 177.5, 143.4, 122.7, 104.7, 103.2, 100.5, 80.8, 78.4, 76.7, 76.5, 75.5, 73.9, 72.6, 72.5, 70.7, 70.6, 69.9, 68.7, 64.0, 63.7, 63.2, 61.2, 46.8, 46.7, 45.7, 42.6, 41.5, 41.3, 39.3, 38.3, 36.2, 35.6, 33.4, 32.3, 32.1, 32.0, 30.2, 28.9, 27.4, 25.1, 25.0, 23.1, 22.6, 22.5, 17.4, 16.6, 16.5, 15.0, 12.4. HRMS-TOF (m/z) calcd for C49H79BrO17 [M + H]+: 1019.4579, found 1019.4585.
β-Hydroxyethyl Ester of PSD (7)
White solid, 70% yield. 1H-NMR (600 MHz, MeOD) δ 5.27 (t, J = 3.5 Hz, 1H, H-12), 5.21 (d, J = 1.4 Hz, 1H, H-1II), 4.49 (d, J = 6.1 Hz, 1H, H-1III), 4.44 (d, J = 7.6 Hz, 1H, H-1I), 4.13 (m, 2H, COOCH2CH2OH), 3.71 (m, 2H, COOCH2CH2OH), 1.24 (d, J = 6.2 Hz, 3H), 1.18 (s, 3H), 0.97 (s, 3H), 0.94 (s, 3H), 0.91 (s, 3H), 0.77 (s, 3H), 0.71 (s, 3H). HRMS-TOF (m/z) calcd for C49H80O18 [M + H]+: 957.5423, found 957.5433.
γ-Hydroxypropyl Ester of PSD (8)
White solid, 55% yield. 1H-NMR (600 MHz, MeOD) δ 5.28 (t, J = 3.6 Hz, 1H, H-12), 5.20 (d, J = 1.3 Hz, 1H, H-1II), 4.48 (d, J = 6.2 Hz, 1H, H-1III), 4.42 (d, J = 7.6 Hz, 1H, H-1I), 4.18–4.14 (m, 2H, COOCH2CH2CH2OH), 3.83–3.69 (m, 2H, COOCH2CH2CH2OH), 1.88–1.83 (m, 2H, COOCH2CH2CH2OH), 1.25 (d, J = 6.2 Hz, 3H), 1.15 (s, 3H), 0.98 (s, 3H), 0.95 (s, 3H), 0.91 (s, 3H), 0.76 (s, 3H), 0.71 (s, 3H). HRMS-TOF (m/z) calcd for C50H82O18 [M + H]+: 971.5579, found 971.5595.
β-Methylaminoethyl Ester of PSD (9)
White solid, 51% yield. 1H-NMR (600 MHz, MeOD) δ 5.26 (s, 1H, H-12), 5.23 (s, 1H, H-1II), 4.52 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.6 Hz, 1H, H-1I), 4.36–4.31 (m, 2H, COOCH2CH2NHCH3), 3.38 (m, 2H, COOCH2CH2NHCH3), 3.37 (m, 3H, COOCH2CH2NHCH3), 1.26 (d, J = 6.2 Hz, 3H), 1.20 (s, 3H), 1.00 (s, 3H), 0.96 (s, 3H), 0.93 (s, 3H), 0.84 (s, 3H), 0.73 (s, 3H). HRMS-TOF (m/z) calcd for C50H83NO17 [M + H]+: 970.5739, found 970.5753.
β-Carboxyethyl Ester of PSD (10)
White solid, 42% yield. 1H-NMR (600 MHz, MeOD) δ 5.26 (s, 1H, H-12), 5.24 (d, J = 1.3 Hz, 1H, H-1II), 4.52 (d, J = 6.1 Hz, 1H, H-1III), 4.47 (d, J = 7.7 Hz, 1H, H-1I), 4.39–4.34 (m, 2H, COOCH2CH2COOH), 2.67–2.57 (m, 2H, COOCH2CH2COOH), 1.26 (d, J = 6.2 Hz, 3H), 1.19 (s, 3H), 0.99 (s, 3H), 0.96 (s, 3H), 0.93 (s, 3H), 0.84 (s, 3H), 0.73 (s, 3H). HRMS-TOF (m/z) calcd for C50H80O19 [M + H]+: 985.5372, found 985.5386.
General Procedure to Synthesize Compounds 11-13
To a solution of PSD (100 mg, 0.11 mmol) in DMF (3 ml) was added EDCI (42 mg, 0.22 mmol), HOBt (30 mg, 0.22 mmol), DIPEA (43 mg, 0.33 mmol), and aliphatic amine (0.33 mmol), respectively. The reaction was stirred at room temperature overnight. Then the mixture was poured into ice water and extracted with ethyl acetate (25 ml × 2). The organic phase was combined, washed with saturated brine (15 ml × 2), dried over anhydrous MgSO4, and filtered. The filtrate was evaporated to give the crude product, which was purified by column chromatography (dichloromethane methanol mixture as eluent) to afford the target compounds.
β-Chloroethyl Amide of PSD (11)
White solid, 67% yield. 1H-NMR (600 MHz, MeOD) δ 5.38 (t, J = 3.3 Hz, 1H, H-12), 5.23 (s, 1H, H-1II), 4.72–4.66 (m, 1H, CONHCH2CH2Cl), 4.66–4.60 (m, 1H, CONHCH2CH2Cl), 4.51 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.7 Hz, 1H, H-1I), 3.65–3.58 (m, 2H, CONHCH2CH2Cl), 1.26 (d, J = 6.2 Hz, 3H), 1.20 (s, 3H), 0.97 (s, 3H), 0.94–0.93 (m, 3H), 0.93 (s, 3H), 0.74 (s, 3H), 0.71 (s, 3H). HRMS-TOF (m/z) calcd for C49H80ClNO16 [M + H]+: 974.5244, found 974.5230.
β-Bromoethyl Amide of PSD (12)
White solid, 58% yield. 1H-NMR (600 MHz, MeOD) δ 5.37 (t, J = 3.4 Hz, 1H, H-12), 5.23 (d, J = 1.3 Hz, 1H, H-1II), 4.73–4.66 (m, 1H, CONHCH2CH2Br), 4.63 (m, 1H, CONHCH2CH2Br), 4.51 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.7 Hz, 1H, H-1I), 3.61 (m, 2H, CONHCH2CH2Br), 1.26 (d, J = 6.2 Hz, 3H), 1.20 (s, 3H), 0.97 (s, 3H), 0.94 (s, 3H), 0.93 (s, 3H), 0.74 (s, 3H), 0.71 (s, 3H). HRMS-TOF (m/z) calcd for C49H80BrNO16 [M + H]+: 1018.4739, found 1018.4755.
n-Propyl Amide of PSD (13)
White solid, 70% yield. 1H-NMR (600 MHz, MeOD) δ 5.37 (t, J = 3.3 Hz, 1H, H-12), 5.23 (d, J = 1.3 Hz, 1H, H-1II), 4.52 (d, J = 6.1 Hz, 1H, H-1III), 4.46 (d, J = 7.6 Hz, 1H, H-1I), 3.19 (m, 1H, CONHCH2CH2CH3), 3.04 (m, 1H, CONHCH2CH2CH3), 1.52 (m, 2H, CONHCH2CH2CH3), 1.26 (d, J = 6.2 Hz, 3H), 1.20 (s, 3H), 0.99 (s, 3H), 0.97 (s, 3H), 0.93 (m, 6H, CONHCH2CH2CH3), 0.81 (s, 3H), 0.73 (s, 3H). 13C-NMR (150 MHz, MeOD) δ 178.9, 144.1, 122.6, 104.7, 103.2, 100.5, 80.8, 78.3, 76.7, 76.5, 75.5, 73.9, 72.6, 72.5, 70.7, 70.6, 69.9, 68.7, 63.7, 63.9, 1, 61.3, 46.8, 46.3, 46.1, 42.6, 41.7, 41.2, 41.1, 39.3, 38.3, 36.2, 33.7, 33.0, 32.2, 31.9, 30.2, 29.4, 27.1, 25.1, 25.0, 23.2, 22.6, 22.5, 22.2, 17.4, 16.6, 16.5, 15.0, 12.4, 10.5. HRMS-TOF (m/z) calcd for C50H83NO16 [M + H]+: 954.5790, found 954.5814.
Mtt Assay
Cell culture and cytotoxicity test: The human cancer cell lines were purchased from Shanghai Institutes for Biological Sciences and maintained in a humidified atmosphere at 37°C in 5% CO2. The cells were grown in RPMI-1640 (GIBCO) media containing 10% heat-inactivated fetal bovine serum. Cytotoxicity was determined by the MTT assay according to the manufacturer’s protocol. Briefly, cells were seeded in 96-well microtiter plates at a density of 8 × 103 cells per well. After a 24-h incubation, cells were treated with various concentrations of PSD derivatives, cultured for 48 h. At the end of the treatment period, 20 μl of the MTT (5 mg/ml) reagent was added to each well. After a 4-h incubation at 37°C, the supernatant was aspirated, and formazan crystals were dissolved in 150 μl DMSO for 10 min with gentle agitation. The absorbance per well was measured at 490 nm with a SpectraMax i3 (Molecular Devices Corp.). The assay was done in triplicate. The IC50 values were then determined for each compound from a plot of log (drug concentration) versus percentage of loss of viability
Tumor Growth Inhibition In Vivo
Male KM mice with a weight of 18–22 g were inoculated subcutaneously with 5 × 106 H22 cells (purchased from the Chinese Academy of Medical Sciences). After 72 h, the tumor-bearing mice were randomized into four groups. The three test groups were treated via caudal vein injection with 6 mg/kg/3 d of PSD five times, or 10 or 20 mg/kg/d of compound 6 for 14 days, respectively. After 24 h of the last administration, the mice were sacrificed, and their tumors were dissected out and weighted.
Cell Cycle Analysis
HCT-116 cells were plated in 6-well plates and incubated at 37°C for 24 h, then incubated with or without 2.5, 5, and 10 μg/ml of 6, respectively. After 48 h, the cells were centrifuged and fixed in 70% ethanol at 4°C overnight, washed with PBS, treated with 20 μl RNase A (1 mg/ml), and stained with 20 μl PI (1 mg/ml) for 30 min. Cellular DNA content was determined using a flow cytometer (Beckman–Coulter Gallios).
Apoptosis Analysis
Apoptosis-mediated cell death of tumor cells was examined using an FITC–Annexin V/PI apoptosis detection kit (BD Pharmingen™) according to the manufacturer’s instructions. In brief, 1 × 106 cells were harvested and washed with PBS. They were resuspended in 500 μl of binding buffer, and then 5 μl of FITC–Annexin V and 5 μl of PI were added. Flow cytometric analysis was performed immediately after supravital staining. Data acquisition and analysis were performed with BD FACSCalibur™ flow cytometer. The cells in early stages of apoptosis were Annexin V positive and PI negative, whereas the cells in the late stage of apoptosis were both Annexin V and PI positive.
Data Availability Statement
All datasets generated for this study are included in the manuscript/supplementary files.
Ethics Statement
The animal study was reviewed and approved by the experimental animal ethics committee, Jiangxi University of Traditional Chinese Medicine.
Author Contributions
YF, GX, and ZY designed the project. YF, JH, and YL performed the chemistry experiments. DH, HL, and ZL completed the biology experiments. YF, DH, and ZY analyzed the data and wrote the manuscript. LC, YJ, and SY provided instructions for the project.
Funding
This work was financially supported by the Jiangxi Provincial Department of Science and Technology (20171BAB205103, 20181BAB215043, 20192ACB21012), Education Department of Jiangxi Province on Science and Technology Project Foundation (GJJ180667), Health and Family Planning Commission of Jiangxi Province (2017A297, 20185523), Jiangxi University of Traditional Chinese Medicine (JXSYLXK-ZHYAO013), and Nanchang Innovative Talent Team (No. [2018]274).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acebey-Castellon, I. L., Voutquenne-Nazabadioko, L., Mai, H. D. T., Roseau, N., Bouthagane, N., Muhammad, D., et al. (2011). Triterpenoid saponins from Symplocos lancifolia. J. Nat. Prod. 74, 163–168. doi: 10.1021/np100502y
Bang, S., Lee, J., Song, G., Kim, D., Yoon, M., Ahn, B. (2005). Antitumor activity of Pulsatilla koreana saponins and their structure–activity relationship. Chem. Pharm. Bull. 53, 1451–1454. doi: 10.1248/cpb.53.1451
Che, H., Fang, Y., Gurung, S. K., Luo, J., Yoon, D., Sung, G., et al. (2014). Synthesis of new 4-(tert-octyl)phenol derivatives and their anticancer activity against human prostate and lung cancer cell lines. Bull. Korean Chem. Soc. 35, 2038–2042. doi: 10.5012/bkcs.2014.35.7.2038
Chen, Z., Duan, H., Wang, M., Han, L., Liu, Y., Zhu, Y., et al. (2015). Synthesis, cytotoxicity and haemolytic activity of Pulsatilla saponin A, D derivatives, Bioorg. Med. Chem. Lett. 25, 2550–2554. doi: 10.1016/j.bmcl.2015.04.049
Chen, Z., Duan, H., Tong, X., Hsu, P., Han, L., Morris-Natschke, S. L., et al. (2018). Cytotoxicity, hemolytic toxicity, and mechanism of action of Pulsatilla saponin D and its synthetic derivatives. J. Nat. Prod. 81, 465–474. doi: 10.1021/acs.jnatprod.7b00578
Fang, Y., Yang, Z., Ouyang, H., Wang, R., Li, J., Huang, H., et al. (2016). Synthesis and biological evaluation of hederacolchiside A1 derivatives as anticancer agents, Bioorg. Med. Chem. Lett. 26, 4576–4579. doi: 10.1016/j.bmcl.2016.08.077
Fang, Y., Wang, R., Li, J., Huang, H., Yang, Z., Wang, Q., et al. (2017). Nitric oxide-donating derivatives of hederacolchiside A1: synthesis and biological evaluation in vitro and in vivo as anticancer agents. Bioorg. Med. Chem. Lett. 27, 98–101. doi: 10.1016/j.bmcl.2016.11.021
Guan, Y., Liu, H., Luan, X., Xu, J., Lu, Q., Liu, Y., et al. (2015). a triterpenoid saponin isolated from Anemone raddeana, suppresses the angiogenesis and growth of human colorectal tumor by inhibiting VEGFR2 signaling. Phytomedicine 22, 103–110. doi: 10.1016/j.phymed.2014.11.008
Jiang, F., Chen, F., Sun, S. (2009). Experiment research and clinical application of Pulsatilla. Medical Recapitulate 15, 3785–3787.
Kim, Y., Bang, S., Lee, J., Ahn, B. (2004). Pulsatilla saponin D: The antitumor principle from Pulsatilla koreana. Arch. Pharmacal Res. 27, 915–918. doi: 10.1007/BF02975843
Kim, M., Lim, E., Jung, M. (2013). First total synthesis of natural Pulsatilla saponin D via highly stereospecific glycosylation. Tetrahedron 69, 5481–5486. doi: 10.1016/j.tet.2013.04.095
Liang, D., Hao, Z., Zhang, G., Zhang, Q., Chen, R., Yu, D. (2011). Cytotoxic triterpenoid saponins from Lysimachia clethroides. J. Nat. Prod. 74, 2128–2136. doi: 10.1021/np2004038
Liu, Y., Song, Y., Xu, Q., Su, D., Feng, Y., Li, X., et al. (2013). Validated rapid resolution LC-ESI–MS/MS method for simultaneous determination of five pulchinenosides from Pulsatilla chinensis (Bunge) Regel in rat plasma: application to pharmacokinetics and bioavailability studies. J. Chromatogr. B. 942–943, 141–150. doi: 10.1016/j.jchromb.2013.10.036
Shu, Z., Chen, Z., Liu, Y., Zhu, W., Feng, Y., Xu, Q., et al. (2013). Nat. Prod. Res. 27, 2196–2201. doi: 10.1080/14786419.2013.814052
Xu, Q., Shu, Z., He, W., Chen, L., Yang, S., Yang, G., et al. (2012). Antitumor activity of Pulsatilla chinensis (Bunge) Regel saponins in human liver tumor 7402 cells in vitro and in vivo. Phytomedicine 19, 293–300. doi: 10.1016/j.phymed.2011.08.066
Yan, M., Liu, Y., Lu, W., Wang, H., Sha, Y., Cheng, M. (2008). Facile synthesis and cytotoxicity of triterpenoid saponins bearing a unique disaccharide moiety: hederacolchiside A1 and its analogues. Carbohyd. Res. 343, 780–784. doi: 10.1016/j.carres.2007.12.014
Ye, X., Yu, S., Liang, Y., Huang, H., Lian, X., Zhang, Z. (2014). Bioactive triterpenoid saponins and phenolic compounds against glioma cells, Bioorg. Med. Chem. Lett. 24, 5157–5163. doi: 10.1016/j.bmcl.2014.09.087
Keywords: Pulsatilla saponin D, synthesis, antiproliferative activity, acute toxicity, apoptosis
Citation: Fang Y, Hu D, Li H, Hu J, Liu Y, Li Z, Xu G, Chen L, Jin Y, Yang S and Yang Z (2019) Synthesis, Biological Evaluation, and Mode of Action of Pulsatilla Saponin D Derivatives as Promising Anticancer Agents. Front. Pharmacol. 10:1208. doi: 10.3389/fphar.2019.01208
Received: 19 July 2019; Accepted: 20 September 2019;
Published: 15 October 2019.
Edited by:
Jiangjiang Qin, Zhejiang Chinese Medical University, ChinaReviewed by:
Hongwei Gao, Guangxi University of Chinese Medicine, ChinaWen Zhou, Guangzhou University of Chinese Medicine, China
Ying Wang, University of Macau, China
Copyright © 2019 Fang, Hu, Li, Hu, Liu, Li, Xu, Chen, Jin, Yang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoliang Xu, eHVndW9saWFuZzY2MDZAMTI2LmNvbQ==; Zunhua Yang, am9zaHl5eUAxMjYuY29t
†These authors contributed equally to this work