Abstract
Cannabis sativa, commonly known as marijuana, contains a pool of secondary plant metabolites with therapeutic effects. Besides Δ9-tetrahydrocannabinol that is the principal psychoactive constituent of Cannabis, cannabidiol (CBD) is the most abundant nonpsychoactive phytocannabinoid and may represent a prototype for anti-inflammatory drug development for human pathologies where both the inflammation and oxidative stress (OS) play an important role to their etiology and progression. To this regard, Alzheimer's disease (AD), Parkinson's disease (PD), the most common neurodegenerative disorders, are characterized by extensive oxidative damage to different biological substrates that can cause cell death by different pathways. Most cases of neurodegenerative diseases have a complex etiology with a variety of factors contributing to the progression of the neurodegenerative processes; therefore, promising treatment strategies should simultaneously target multiple substrates in order to stop and/or slow down the neurodegeneration. In this context, CBD, which interacts with the eCB system, but has also cannabinoid receptor-independent mechanism, might be a good candidate as a prototype for anti-oxidant drug development for the major neurodegenerative disorders, such as PD and AD. This review summarizes the multiple molecular pathways that underlie the positive effects of CBD, which may have a considerable impact on the progression of the major neurodegenerative disorders.
Introduction
Oxidative stress (OS) plays a crucial role in aging and occurs manly when the activity of the anti-oxidants enzymes is not sufficient to counterbalance the generation of reactive oxygen species (ROS). In the latter condition, high production of ROS can alter the structure of proteins, lipids, nucleic acids, and matrix components leading to programmed cell death (Cassano et al., 2016). Different tissues present different susceptibility to OS. The central nervous system (CNS) is extremely sensitive to this type of damage for several reasons. To this regard, the CNS has a low level of antioxidant enzymes, a high content of oxidizable substrates, and a large amount of ROS produced during neurochemical reactions (Trabace et al., 2004; Uttara et al., 2009). In addition to several other environmental or genetic factors, OS contributes to neurodegeneration since free radicals attack neural cells. Therefore, neurons suffer a functional or sensory loss during the neurodegenerative process. Even if oxygen is indispensable for life, an unbalanced metabolism and an excess production of ROS ends up in a series of pathological conditions, such as Alzheimer's disease (AD), Parkinson's disease (PD), and many other neural disorders. Free radicals cause lesions to protein and DNA, activate inflammatory process and subsequent cell apoptosis (Cassano et al., 2012).
In the last years, there is an urgent need to discover new drug targets that can effectively combat cell alteration caused by the stress of cell membranes. In this perspective, the endocannabinoid (eCB) system has attracted considerable interest due to the current interplay between eCB and different redox-dependent signaling pathways. The two well-characterized eCBs are N-arachidonoyl-ethanolamine or anandamide (AEA) and 2-arachidonoyl-glycerol (2-AG), which are synthesized on demand in response to elevations of intracellular calcium (Howlett et al., 2002; Di Marzo et al., 2005) and respectively metabolized by fatty acid amide hydrolase (FAAH) and monoglyceride lipase (MAGL) (Piomelli, 2003; Di Marzo, 2008; Kunos et al., 2009). Cannabinoid (CB) receptors exist in two different subtypes: type 1 (CB1) and type 2 (CB2) (Matsuda et al., 1990; Munro et al., 1993; Howlett et al., 2002). The CB1 receptors, first cloned in 1990, are widely distributed in the body and in the CNS are distributed at the level of basal ganglia, cerebellum, hippocampus, caudate nucleus, putamen, hypothalamus, amygdala, and spinal cord (Matsuda et al., 1990). The CB2 receptors, cloned in 1993, are mainly located in cells of the immune system with high density in the spleen, T lymphocytes, and macrophages (Munro et al., 1993). Their anatomical distribution correlates them to the actions for which they are responsible: the activation of the CB1 receptors has euphoric effects and an antioxidant, antiemetic, analgesic, antispasmodic, and appetite stimulating actions. As for CB2 receptors, their stimulation is attributable to the anti-inflammatory and immunomodulatory actions of CB (Cassano et al., 2017).
Converging evidence strongly suggests that eCBs act as retrograde synaptic messengers (Kano et al., 2002; Freund et al., 2003). This phenomenon is initiated postsynaptically by an elevation of cytoplasmic calcium concentration that induces the production and release into the synaptic space of eCBs. Thereafter, eCBs activate CB1 receptors at presynaptic levels and block the release from the terminals of neurons of different transmitters, such as gamma-aminobutyric acid (GABA), glutamate, dopamine (DA), noradrenaline, serotonin, and acetylcholine (Howlett et al., 2002; Pertwee and Ross, 2002; Szabo and Schlicker, 2005). These mechanisms mediated by the activation of presynaptic CB1 receptors are termed depolarization-induced suppression of inhibition (DSI) and excitation (DSE), respectively when are involved the inhibitory (GABA) or excitatory (glutamate) synaptic transmissions (Kano et al., 2002; Freund et al., 2003). Likewise, CB2 receptors can modulate the production and function of certain inflammatory cytokines at multiple levels by activating the immune cells and modulating their migration both within and outside the CNS (Freund et al., 2003; Walter and Stella, 2004). Antioxidant enzymes can be modulated by eCBs, not only acting on the CB1 and CB2 receptors, but also through the transient receptor potential vanilloid-1 (TRPV1), the peroxisome proliferator-activated receptor alpha (PPAR-alpha), and the orphan receptors N-arachidonyl glycine receptor or G-protein-coupled receptors 18 (GPR18) GPR19 and GPR55 (Piomelli, 2003; McHugh et al., 2010; Howlett et al., 2011; McHugh, 2012). Therefore, the direct and/or indirect modulation of pathways through which the eCBs damper the OS may represent a promising strategy for reducing the damage caused by a redox imbalance (Gallelli et al., 2018). Moreover, antioxidants are now seen as a convincing therapy against severe neurodegeneration, as they have the ability to fight it by blocking the OS. Diet and medicinal herbs are an important source of antioxidants. The recognition of antioxidant therapy upstream and downstream of OS has proven to be an effective tool to improve any neuronal damage as well as to eliminate free radicals. Antioxidants have a wide field of application and can prevent OS interacting with the metal ions, which play an important role in the build-up of neuronal plaque (Uttara et al., 2009).
In the last decade there are increasing evidences that secondary plant metabolites, extracted from medicinal herbs, may represent lead compounds for the production of medications against inflammation and OS, protecting from neuronal cell loss (Giudetti et al., 2018). Among these medicinal herbs, Cannabis sativa, commonly known as marijuana, contains a pool of secondary plant metabolites with therapeutic effects (Gugliandolo et al., 2018). In this context, cannabidiol (CBD) the nonpsychotropic CB extract from Cannabis sativa may represent a prototype for anti-inflammatory drug development for those human pathologies where both the inflammation and OS play a key role to their etiology and progression (Izzo et al., 2009). To this regard, therapies that effectively combat disease progression are still lacking in the field of neurodegenerative disorders, and mostly with AD. CBD, which modulates the eCB system, but has also CB receptor-independent mechanism, seems to be a prototype for anti-inflammatory drug development.
Therefore, the present review summarizes the main molecular mechanisms through which CBD exerts its beneficial effects that may have a considerable impact on the progression of the major neurodegenerative disorders.
Cannabis sativa
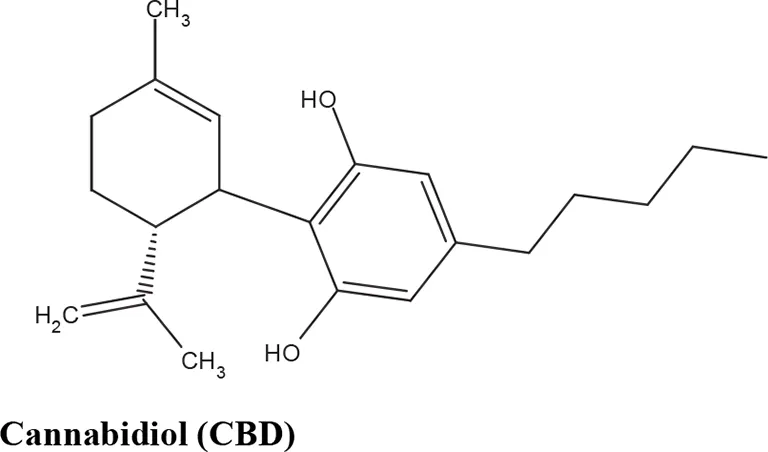
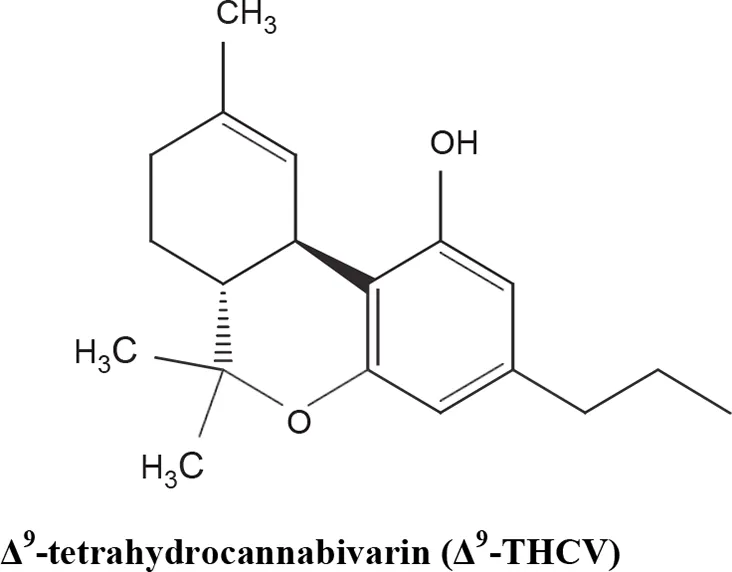
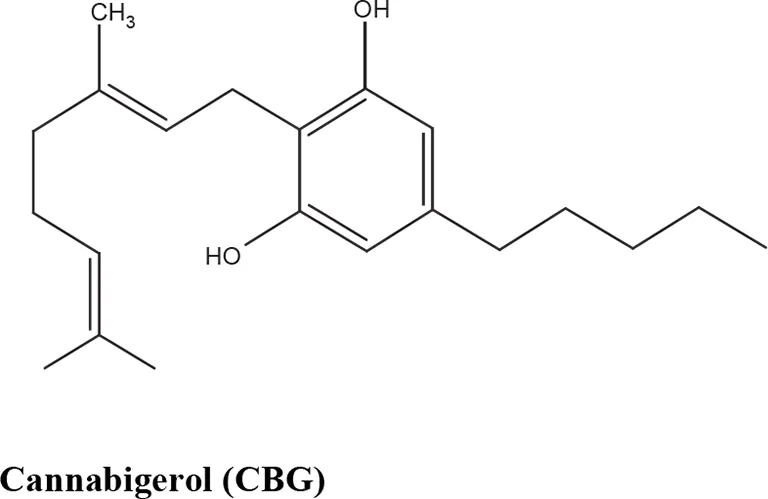
The medical and psychotropic effects of Cannabis sativa have been well known since long time. A multitude of secondary metabolites was extracted from this plant and most of them were used for therapeutic purpose by many cultures. So far more than 400 chemical compounds have been isolated from Cannabis sativa and among them more than 100 terpeno-phenol compounds named phytocannabinoids have been detected (Mechoulam and Hanus, 2000; Mechoulam et al., 2007). As such Cannabis sativa can be regarded as a natural library of unique compounds. The most abundant phytocannabinoid is the Δ9-tetrahydrocannabinol (delta-9-THC), responsible for the psychotropic effect associated with Cannabis consumption, and then the nonpsychoactive constituent CBD and cannabigerol (CBG) (Mechoulam and Hanus, 2000; Mechoulam et al., 2007; Gugliandolo et al., 2018). Table 1 shows the list of the most abundant nonpsychoactive phytocannabinoids isolated from Cannabis sativa. Phytocannabinoids mimic the effects of eCBs that regulate the transmission of nerve impulses in some synapses of the nerve pathways, causing in particular a reduction in the release of signals between the cells (Piomelli, 2003).
Table 1
| Phytocannabinoids | Mechanisms | Effects | References |
|---|---|---|---|
| CB2 inverse agonist | Anti-inflammatory effects | Thomas et al., 2005 | |
| CB1, CB2 antagonist | Antispasmodic effect | ||
| FAAH inhibition | Reduces FAAH expression in the inflamed intestine | Ligresti et al., 2006 | |
| TRPA1 agonist | Analgesic effects | De Petrocellis et al., 2008 | |
 | TRPM8 antagonist | Analgesic effects. Potential role in prostate carcinoma | |
| TRPV1 agonist | Antipsychotic and analgesic effects | ||
| Adenosine uptake competitive inhibitor | Anti-inflammatory effects | Carrier et al., 2006 | |
| PPARγ agonist | Vasorelaxation and stimulation of fibroblasts into adipocytes | O'Sullivan et al., 2009 | |
| 5HT1A agonist | Anti-ischemic and anxiolytic properties | Campos and Guimarães, 2008 Resstel et al., 2009 | |
| Ca2+ channel | Neuroprotective and antiepileptic properties | Drysdale et al., 2006 Ryan et al., 2009 | |
| Suppressor of tryptophan degradation | Potential role in pain, inflammation and depression | Jenny et al., 2009 | |
 | CB1 antagonist | Increases central inhibitory neurotransmission | Thomas et al., 2005 Dennis et al., 2008 Ma et al., 2008 |
| CB2 partial agonist | Stimulates mesenchymal stem cells | Scutt and Williamson, 2007 | |
 | |||
| TRPV1 agonist | Potential role in analgesia | Ligresti et al., 2006 | |
| TRPA1 agonist | De Petrocellis et al., 2008 | ||
| TRPM8 antagonist | |||
 | TRPA1 partial agonist | Potential role in analgesia | De Petrocellis et al., 2008 |
| TRPM8 antagonist | |||
 | TRPA1 partial agonist | De Petrocellis et al., 2008 | |
| TRPM8 antagonist | Potential role in analgesia | ||
| TRPV1 agonist | Ligresti, A., et al., 2006 | ||
| COX-2 inhibitor | Potential role in inflammation | Takeda et al., 2008 |
Most abundant nonpsychoactive phytocannabinoids isolated from Cannabis sativa: chemical structures and pharmacological actions.
Due to its high lipophilicity and its affinity for lipid membranes, delta-9-THC was supposed to bind non-specifically variety of cell membranes modifying their fluidity rather than to activate a specific receptor (Hillard et al., 1985). Later this first hypothesis was completely discarded and was demonstrated that delta-9-THC exerts its effects by combining with a selective receptor (Devane et al., 1988; Howlett et al., 1990; Matsuda et al., 1990). In fact, many authors have demonstrated that delta-9-THC exerts its psychoactive effects acting on CB1 receptors, whereas CDB and CBG, two nonpsychoactive CBs, have low affinity for both CB1 and CB2 receptors and inhibit FAAH, resulting in increased levels of eCBs, which in turn further activate the CB1 receptor (Devane et al., 1988; Howlett et al., 1990; Matsuda et al., 1990; Appendino et al., 2011). Among the nonpsychoactive phytocannabinoids, most of the evidences have focused on CBD, which possesses a high antioxidant and anti-inflammatory activity, together with neuroprotective, anxiolytic and anticonvulsant properties (Pellati et al., 2018).
Mechanisms of CBD Action
After delta-9-THC, CBD is the second most abundant phytocannabinoids and is one of the major nonpsychoactive CB constituents in the plant of Cannabis sativa representing up to 40% of Cannabis extract. Adams and colleagues first isolated the CBD, while Mechoulam and colleagues analyzed its structure and stereochemistry (reviewed in Pertwee, 2006). Therapeutically CBD is already available alone and in formulation with delta-9-THC (Booz, 2011). In particular, a drug containing only CBD (Epidiolex) is used for children affected by epilepsy resistant to other treatments, as well as in combination (1:1 ratio) with delta-9-THC (CBD/delta-9-THC, Sativex/Nabiximols) is currently used to treat the spasticity observed in patients affected by multiple sclerosis (Pertwee, 2008; Devinsky et al., 2016). Compared to delta-9-THC, CBD possess a better safety profile and it is well tolerated when administered at animals and patients even at high doses (up to 1,500 mg/day) (Bergamaschi et al., 2011). In fact, authors demonstrated that CBD did not alter cardiovascular parameters, body temperature, psychomotor, and psychological functions, as well as did not induce catalepsy like delta-9-THC (Bergamaschi et al., 2011). Unlike delta-9-THC, CBD does not target directly the CB receptors and this characteristic may justify its better safety profile compared to delta-9-THC (Pertwee, 2006; Thomas et al., 2007).
Although the pharmacodynamics of CBD is not fully clarified, different evidences have been accumulated showing that CBD seems to act throughout different pathways. To this regard, although CBD shows much lower affinity than delta-9-THC for CB1 and CB2 receptors, it is able to antagonize CB1/CB2 receptor agonists in vitro at reasonably low concentrations (nanomolar range) (Thomas et al., 2007). In particular, it has been shown by in vitro studies that CBD is able to act as CB1/CB2 receptors inverse agonist an action that underlies its antagonism of CP55940 and R-(+)-WIN55212 at the CB1/CB2 receptor (Thomas et al., 2007). It has been hypothesized that the anti-inflammatory actions of CBD might be due to its ability to act as a CB2 receptor inverse agonist (Pertwee, 2006). Besides CB receptors, CBD has been profiled also towards other pharmacological substrates. To this regard, CBD showed also affinity to the peroxisome proliferation-activated receptors (PPARs), which are a family of ligand-inducible transcription factors that belong to the nuclear hormone receptor superfamily. In humans, there are three PPAR isoforms PPARα, PPARβ/δ, and PPARγ that are encoded by separate genes and are differently expressed in organs and tissues (Michalik et al., 2006). CBD seems to activate the transcriptional activity of PPARγ, which play a primary role in the regulation of adipocyte formation, insulin sensitivity and activation of inflammatory response (O'Sullivan, 2007; O'Sullivan and Kendall, 2010; Hind et al., 2016; O'Sullivan, 2016). To this regard, CBD activates PPARγ receptors leading to a lower expression of proinflammatory genes, which were inhibited by PPARγ antagonists (Esposito et al., 2007; Esposito et al., 2011; O'Sullivan, 2016).
Moreover, CBD exerts a more potent antioxidant effects than other antioxidants, such as ascorbate or α-tocopherol, in in vitro study where cortical neurons were treated with toxic concentrations of glutamate (Hampson et al., 1998; Campos et al., 2016).
The neuroprotective effect was present regardless of whether the insult was due to the activation of N-methyl-D-aspartate (NMDA) receptor, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, or kainate receptors and, more interestingly, it was not mediated by CB receptors since the CB antagonist was unaffected (Hampson et al., 1998). The latter result suggests that CBD may be a potent antioxidant without psychotropic side effects, which are mediated by the direct action on CB receptors.
The anti-inflammatory effect of CBD is also mediated by the adenosine A2A (A2A) receptor whose activation dampers the immune system, leading to a reduction of the antigen presentation, immune cell trafficking, immune cell proliferation, production of the proinflammatory cytokine, and cytotoxicity (Magen et al., 2009). In fact, it has been shown that CBD enhances A2A receptor signaling by the inhibition of cellular update of an adenosine transporter leading to anti-inflammatory and antioxidant effects (Carrier et al., 2006). Likewise, CGS-21680, which is an agonist of the A2A receptor, mimics the actions of CBD that were suppressed by an A2A antagonist (i.e. ZM241,385) (Martín-Moreno et al., 2011).
The CBD neuroprotective property seems to be due also to the activation of 5-hydroxytryptamine subtypes 1A (5-HT1A) receptors, which are located in pre- and post-synaptic membranes in several brain regions (Hoyer et al., 1986). Russo and colleagues first demonstrated that CBD is able to activate the 5-HT1A receptors (Russo et al., 2005). Further support to this first observation was given by a recent study where the authors found that the effect of CBD was blocked by WAY-100135, a selective 5-TH1A receptor antagonist (Galaj et al., 2019).
Finally, it has been demonstrated that CBD has a direct effect on mitochondria (da Silva et al., 2018). To this regard, it has been widely accepted that mitochondrial dysfunction can contribute to neurodegeneration due to the overproduction of ROS and iron accumulation (Mills et al., 2010; Serviddio et al., 2011; Cassano et al., 2012; Cassano et al., 2016; Romano et al., 2017). In particular, iron overload induces several mitochondrial alterations, such as increased mitochondrial DNA (mtDNA) deletions and reduction of epigenetic mtDNA modulation, mitochondrial ferritin levels, and succinate dehydrogenase activity, which may altogether alter cellular viability leading to neurodegenerative process (da Silva et al., 2018). Interestingly, all these iron-induced mitochondrial alterations were completely reversed by CBD, which promotes neural cell survival (da Silva et al., 2018). Moreover, doxorubicin, a broad-spectrum chemotherapeutic drug, induces a dose-dependent cardiotoxicity through the dysregulation of various metabolic signaling pathways, including mitochondrial dysfunction (Hao et al., 2015). In particular, doxorubicin reduces the activity of myocardial mitochondrial complexes (I and II) and glutathione peroxidase leading to an increase of ROS generation (Hao et al., 2015). Interestingly, CBD significantly attenuated doxorubicin-induced cardiotoxicity and cardiac dysfunction by improving mitochondrial complex I activity and enhancing mitochondrial biogenesis (Hao et al., 2015).
Since CBD targets multiple substrates, it may be a good candidate as a multimodal drug for the major neurodegenerative disorders, such as PD and AD. Figure 1 shows the effects of CBD in PD and AD.
Figure 1
CBD and PD
PD is a progressive neurodegenerative disorder characterized manly by motor alterations, such as akinesia, bradykinesia, tremors, postural instability, and rigidity. Although the etiology of PD is still largely elusive, its pathophysiology is characterized by loss of midbrain substantia nigra DA neurons and overwhelming evidence indicates that OS is a central factor in PD pathophysiology (Hirsch et al., 1988; Branchi et al., 2010; Aureli et al., 2014). It has been demonstrated in animal model of PD that CBD exerts a neuroprotective effect as antioxidant compound acting through a mechanism that is CB receptor-independent (Fernández-Ruiz et al., 2013). In fact, in 6-hydroxydopamine-lesioned mice CBD was able to significantly reduce the DA depletion and to attenuate the OS increasing the expression of Cu,Zn-superoxide dismutase (SOD), which is an important endogenous mechanism that defences cell against OS (Fernández-Ruiz et al., 2013; Martinez et al., 2015). The latter evidence indicates that CBs having antioxidant CB receptor-independent properties attenuate the neurodegeneration of nigrostriatal dopaminergic fibers occurring in PD (García-Arencibia et al., 2007). This thesis is reinforced by the observation that CBD reduces the neuronal cell death in the striatum occurring after the administration of 3-nitropropionic acid (3NP), an inhibitor of mitochondrial complex II. In particular, the authors demonstrated that 3NP administration causes a reduction of both GABA levels and striatal atrophy of the GABAergic neurons as indicated by a depletion of mRNA levels of proenkephalin (PENK), substance P (SP), and neuronal-specific enolase (NSE). Moreover, the inhibition of mitochondrial complex II induced by 3NP reduces the mRNA expression of superoxide dismutase-1 (SOD-1) and -2 (SOD-2), which are endogenous defences against the OS. Interestingly, after 3NP administration CBD can completely abolish the atrophy of the GABAergic neurons and significantly increase the mRNA levels of SOD-2, as well as attenuate the reduction of mRNA levels of SOD-1 and PENK. Differently, after 3NP administration the administration of arachidonyl-2-chloroethylamide (ACEA) or HU-308, respectively agonist of CB1 and CB2 receptor, did not revert the striatal atrophy of the GABAergic neurons, as well as did not restore the endogenous defences against the OS induced by 3NP (Sagredo et al., 2007). Taken together, these results suggest that CBD exerts a neuroprotective role on the GABAergic neurons that project from the striatum to the substantia nigra and further confirm that its mechanism is CB receptor-independent (Sagredo et al., 2007). Furthermore, in another study authors explored whether CBD was able to attenuate the pathological symptoms of PD modulating the GPR55. In particular, mice were treated for 5 weeks with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid (MPTPp), which induced motor function impairment and loss of tyrosine hydroxylase-positive neurons and DA levels in the brain. This chronic mouse model of PD was treated with abnormal-CBD (Abn-CBD), a synthetic CBD isomer and GPR55 agonist. Authors found that the key features of PD induced by MPTPp were prevented by the pharmacological treatment, suggesting that the activation of GPR55 may be a good strategy for the treatment of PD (Celorrio et al., 2017).
CBD and AD
AD is the most common form of dementia affecting elderly people and its pathology is characterized by the accumulation of amyloid-β (Aβ) plaques and tau neurofibrillary tangles (NFTs) in the brain (Querfurth and LaFerla, 2010).
Although the etiology of AD appears to be linked to a multitude of mechanisms, inflammation seems to play a crucial role in its pathogenesis (Bronzuoli et al., 2018; Scuderi et al., 2018). Expected benefits of current therapies are limited (Sabbagh, 2009; Neugroschl and Sano, 2010), so that there is pressing demand for discovering new treatments able to slow disease progression or prevent its onset.
In this contest, the anti-inflammatory properties of CBD were evaluated by both in vitro and in vivo studies in an animal model of Aβ-induced neuroinflammation (Iuvone et al., 2004; Esposito et al., 2006; Esposito et al., 2007; Esposito et al., 2011). In particular, authors demonstrated that CBD reduces the tau protein hyperphosphorylation through the inhibition of Wingless-type MMTV integration site family member (Wnt) pathways and significantly attenuates all the markers of the Aβ-induced neuroinflammation, including the glial fibrillary acidic protein (GFAP) and inducible nitric oxide synthase (iNOS) protein expression, nitrite production, and interleukin 1 β (IL-1β) (Iuvone et al., 2004; Esposito et al., 2006; Esposito et al., 2007; Iuvone et al., 2009; Esposito et al., 2011). CBD pre-treatment induces a reduction of ROS production, lipid peroxidation, caspase-3 levels, and DNA fragmentation in PC12 cells stimulated by Aβ, an in vitro model of AD (Iuvone et al., 2004; Bedse et al., 2014; Gallelli et al., 2018).
The beneficial effects of CBD were further confirmed by another study where mice were chronically treated (for 3 weeks) with CBD after been injected intracerebroventricularly with fibrillar Aβ (Martín-Moreno et al., 2011). CBD counteracts the Aβ-induced microglial activation, the production of proinflammatory cytokine tumor necrosis factor α (TNF-α) and ameliorates the memory alterations observed in a spatial memory task (Martín-Moreno et al., 2011).
Moreover, Aβ can gradually accumulate in mitochondria, where it can cause reduction of both activity of the respiratory chain complexes and the rate of oxygen consumption leading to a free radical generation and oxidative damage (Caspersen et al., 2005; Lin and Beal, 2006; Manczak et al., 2006; Cassano et al., 2012). To this regard, CBD is able to counteract mitochondrial alterations by the reduction of ROS production induced by both the Aβ and nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase (NOX) (Hao et al., 2015; Vallée et al., 2017).
It is well know that tau hyperphosphorylation, mostly at serine (Ser) or threonine (Thr) residues, plays a crucial role in the pathogenesis of AD, thereby molecules that reduce phospho-tau aggregates may represent a good candidate for the AD treatment. To this regard, it has been demonstrated that CBD reduces the expression of genes, which encode kinases (GSK-3β, CMK, and MAPK) responsible for aberrant tau phosphorylation, leading to a reduction of tau hyperphosphorylation and subsequent NFT formation (Libro et al., 2016). Likewise, CBD activates the PI3K/Akt signaling through the TRPV1, which is able to inhibit the kinase GSK-3β, thereby decreasing tau phosphorylation (Libro et al., 2016). Finally, CBD downregulates β- and γ-secretase genes leading to a reduction of Aβ production (Libro et al., 2016).
Conclusion
The present review provided evidence that the nonpsychoactive phytocannabinoids CBD could be a potential pharmacological tool for the treatment of neurodegenerative disorders; its excellent safety and tolerability profile in clinical studies renders it a promising therapeutic agent.
The molecular mechanisms associated with CBD's improvement in PD and AD are likely multifaceted, and although CBD may act on different molecular targets all the beneficial effects are in some extent linked to its antioxidant and anti-inflammatory profile, as observed in in vitro and in vivo studies. Therefore, this review describes evidence to prove the therapeutical efficacy of CBD in patients affected by neurodegenerative disorders and promotes further research in order to better elucidate the molecular pathways involved in the therapeutic potential of CBD.
Funding
This article was published with a contribution from the University of Foggia.
Statements
Author contributions
All authors have contributed to the writing, design, and preparation of figures. The senior authors TC and GS have carried out coordination of efforts.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GB declared a past co-authorship with one of the authors TC to the handling editor.
References
1
AppendinoG.ChianeseG.Taglialatela-ScafatiO. (2011). Cannabinoids: occurrence and medicinal chemistry. Curr. Med. Chem.18, 1085–1099. doi: 10.2174/092986711794940888
2
AureliC.CassanoT.MasciA.FranciosoA.MartireS.CoccioloA.et al. (2014). 5-S-cysteinyldopamine neurotoxicity: Influence on the expression of α-synuclein and ERp57 in cellular and animal models of Parkinson's disease. J. Neurosci. Res.92, 347–358. doi: 10.1002/jnr.23318
3
BedseG.RomanoA.CianciS.LavecchiaA. M.LorenzoP.ElphickM. R.et al. (2014). Altered expression of the CB1 cannabinoid receptor in the triple transgenic mouse model of Alzheimer's disease. J. Alzheimers Dis.40, 701–712. doi: 10.3233/JAD-131910
4
BergamaschiM. M.QueirozR. H.ZuardiA. W.CrippaJ. A. (2011). Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr. Drug Saf6, 237–249. doi: 10.2174/157488611798280924
5
BoozG. W. (2011). Cannabidiol as an emergent therapeutic strategy for lessening the impact of inflammation on oxidative stress. Free Radic. Biol. Med.51, 1054–1061. doi: 10.1016/j.freeradbiomed.2011.01.007
6
BranchiI.D'AndreaI.ArmidaM.CarnevaleD.Ajmone-CatM. A.PèzzolaA.et al. (2010). Striatal 6-OHDA lesion in mice: investigating early neurochemical changes underlying Parkinson's disease. Behav. Brain Res.208, 137–143. doi: 10.1016/j.bbr.2009.11.020
7
BronzuoliM. R.FacchinettiR.SteardoL.Jr.RomanoA.SteccaC.PassarellaS.et al. (2018). Palmitoylethanolamide dampens reactive astrogliosis and improves neuronal trophic support in a triple transgenic model of Alzheimer's disease: in vitro and in vivo evidence. Oxid. Med. Cell Longev2018, 4720532. doi: 10.1155/2018/4720532
8
CamposA. C.GuimarãesF. S. (2008). Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology (Berl). 199, 223–230. doi: 10.1007/s00213-008-1168-x
9
CamposA. C.FogaçaM. V.SonegoA. B.GuimarãesF. S. (2016). Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res.112, 119–127. doi: 10.1016/j.phrs.2016.01.033
10
CarrierE. J.AuchampachJ. A.HillardC. J. (2006). Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc. Natl. Acad. Sci. U S A.103, 7895–7900. doi: 10.1073/pnas.0511232103
11
CaspersenC.WangN.YaoJ.SosunovA.ChenX.LustbaderJ. W.et al. (2005). Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J.19, 2040–2041. doi: 10.1096/fj.05-3735fje
12
CassanoT.ServiddioG.GaetaniS.RomanoA.DipasqualeP.CianciS.et al. (2012). Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging33, 1121.e1–12. doi: 10.1016/j.neurobiolaging.2011.09.021
13
CassanoT.PaceL.BedseG.LavecchiaA. M.De MarcoF.GaetaniS.et al. (2016). Glutamate and mitochondria: two prominent players in the oxidative stress-induced neurodegeneration. Curr. Alzheimer Res.13, 185–197. doi: 10.2174/1567205013666151218132725
14
CassanoT.CalcagniniS.PaceL.De MarcoF.RomanoA.GaetaniS. (2017). Cannabinoid receptor 2 signaling in neurodegenerative disorders: from pathogenesis to a promising therapeutic target. Front. Neurosci.11, 30. doi: 10.3389/fnins.2017.00030
15
CelorrioM.Rojo-BustamanteE.Fernández-SuárezD.SáezE.Estella-Hermoso de MendozaA.MüllerC. E.et al. (2017). GPR55: A therapeutic target for Parkinson's disease? Neuropharmacology125, 319–332. doi: 10.1016/j.neuropharm.2017.08.017
16
da SilvaV. K.de FreitasB. S.DornellesV. C.KistL. W.BogoM. R.SilvaM. C.et al. (2018). Novel insights into mitochondrial molecular targets of iron-induced neurodegeneration: Reversal by cannabidiol. Brain Res. Bull.139, 1–8. doi: 10.1016/j.brainresbull.2018.01.014
17
DennisI.WhalleyB. J.StephensG. J. (2008). Effects of Delta9-tetrahydrocannabivarin on [35S]GTPgammaS binding in mouse brain cerebellum and piriform cortex membranes. Br. J. Pharmacol.154, 1349-1358. doi: 10.1038/bjp.2008.190
18
De PetrocellisL.VellaniV.Schiano-MorielloA.MariniP.MagheriniP. C.OrlandoP.et al. (2008). Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J. Pharmacol. Exp. Ther.325, 1007–1015. doi: 10.1124/jpet.107.134809
19
DevaneW. A.Dysarz,. F. A.3rdJohnsonM. R.MelvinL. S.HowlettA. C. (1988). Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol.34, 605–613.
20
DevinskyO.MarshE.FriedmanD.ThieleE.LauxL.SullivanJ.et al. (2016). Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol.15, 270–278. doi: 10.1016/S1474-4422(15)00379-8
21
Di MarzoV.De PetrocellisL.BisognoT. (2005). The biosynthesis, fate and pharmacological properties of endocannabinoids. Handb. Exp. Pharmacol.168, 147–185. doi: 10.1007/3-540-26573-2_5
22
Di MarzoV. (2008). Targeting the endocannabinoid system: to enhance or reduce? Nat. Rev. Drug Discovery7, 438–455. doi: 10.1038/nrd2553
23
DrysdaleA. J.RyanD.PertweeR. G.PlattB. (2006). Cannabidiol-induced intracellular Ca2+ elevations in hippocampal cells. Neuropharmacology.50, 621–631. doi: 10.1016/j.neuropharm.2005.11.008
24
EspositoG.De FilippisD.MaiuriM. C.De StefanoD.CarnuccioR.IuvoneT. (2006). Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett.399, 91–95. doi: 10.1016/j.neulet.2006.01.047
25
EspositoG.ScuderiC.SavaniC.SteardoL.Jr.De FilippisD.CottoneP.et al. (2007). Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br. J. Pharmacol.151, 1272–1279. doi: 10.1038/sj.bjp.0707337
26
EspositoG.ScuderiC.ValenzaM.TognaG. I.LatinaV.De FilippisD.et al. (2011). Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PloS One6, e28668. doi: 10.1371/journal.pone.0028668
27
Fernández-RuizJ.SagredoO.PazosM. R.GarcíaC.PertweeR.MechoulamR. (2013). Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharmacol.75, 323–333. doi: 10.1111/j.1365-2125.2012.04341.x
28
FreundT. F.KatonaI.PiomelliD. (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev.83, 1017–1066. doi: 10.1152/physrev.00004.2003
29
GalajE.BiG. H.YangH. J.XiZ. X. (2019). Cannabidiol attenuates the rewarding effects of cocaine in rats by CB2, 5-TH(1A) and TRPV1 receptor mechanisms. Neuropharmacology19, 107740. doi: 10.1016/j.neuropharm.2019.107740
30
GallelliC. A.CalcagniniS.RomanoA.KoczwaraJ. B.de CegliaM.DanteD.et al. (2018). Modulation of the oxidative stress and lipid peroxidation by endocannabinoids and their lipid Analogues. Antioxid (Basel)18, 7. doi: 10.3390/antiox7070093
31
García-ArencibiaM.GonzálezS.de LagoE.RamosJ. A.MechoulamR.Fernández-RuizJ. (2007). Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson's disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res.1134, 162–170. doi: 10.1016/j.brainres.2006.11.063
32
GiudettiA. M.SalzetM.CassanoT. (2018). Oxidative stress in aging brain: nutritional and pharmacological interventions for neurodegenerative disorders. Oxid. Med. Cell Longev.2018, 3416028. doi: 10.1155/2018/3416028
33
GugliandoloA.PollastroF.GrassiG.BramantiP.MazzonE. (2018). In vitro model of neuroinflammation: efficacy of cannabigerol, a non-psychoactive cannabinoid. Int. J. Mol. Sci.19, 7. doi: 10.3390/ijms19071992
34
HampsonA. J.GrimaldiM.AxelrodJ.WinkD. (1998). Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. U. S. A.95, 8268–8273. doi: 10.1073/pnas.95.14.8268
35
HaoE.MukhopadhyayP.CaoZ.ErdélyiK.HolovacE.LiaudetL.et al. (2015). Cannabidiol Protects against Doxorubicin-Induced Cardiomyopathy by Modulating Mitochondrial Function and Biogenesis. Mol. Med.21, 38–45. doi: 10.2119/molmed.2014.00261
36
HillardC. J.HarrisR. A.BloomA. S. (1985). Effects of the cannabinoids on physical properties of brain membranes and phospholipid vesicles: fluorescence studies. J. Pharmacol. Exp. Ther.232, 579–588.
37
HindW. H.EnglandT. J.O'SullivanS. E. (2016). Cannabidiol protects an in vitro model of the blood-brain barrier from oxygen-glucose deprivation via PPARγ and 5-HT1A receptors. Br. J. Pharmacol.173, 815–825. doi: 10.1111/bph.13368
38
HirschE.GraybielA. M.AgidY. A. (1988). Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature334, 345–348. doi: 10.1038/334345a0
39
HowlettA. C.Bidaut-RussellM.DevaneW. A.MelvinL. S.JohnsonM. R.HerkenhamM. (1990). The cannabinoid receptor: biochemical, anatomical and behavioral characterization. Trends Neurosci.13, 420–423. doi: 10.1016/0166-2236(90)90124-s
40
HowlettA. C.BarthF.BonnerT. I.CabralG.CasellasP.DevaneW. A.et al. (2002). International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev.54, 161–202. doi: 10.1124/pr.54.2.161
41
HowlettA. C.ReggioP. H.ChildersS. R.HampsonR. E.UlloaN. M.DeutschD. G. (2011). Endocannabinoid tone versus constitutive activity of cannabinoid receptors. Br. J. Pharmacol.163, 1329–1343. doi: 10.1111/j.1476-5381.2011.01364.x
42
HoyerD.PazosA.ProbstA.PalaciosJ. M. (1986). Serotonin receptors in the human brain. I. Characterization and autoradiographic localization of 5-HT1A recognition sites. Apparent absence of 5-HT1B recognition sites. Brain Res.376, 85–96. doi: 10.1016/0006-8993(86)90902-9
43
IuvoneT.EspositoG.EspositoR.SantamariaR.Di RosaM.IzzoA. A. (2004). Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem.89, 134–141. doi: 10.1111/j.1471-4159.2003.02327.x
44
IuvoneT.EspositoG.De FilippisD.ScuderiC.SteardoL. (2009). Cannabidiol: a promising drug for neurodegenerative disorders? CNS Neurosci. Ther.15, 65–75. doi: 10.1111/j.1755-5949.2008.00065.x
45
IzzoA. A.BorrelliF.CapassoR.Di MarzoV.MechoulamR. (2009). Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci.30, 515–527. doi: 10.1016/j.tips.2009.07.006
46
JennyM.SanterE.PirichE.SchennachH.FuchsD. (2009). Delta9-tetrahydrocannabinol and cannabidiol modulate mitogen-induced tryptophan degradation and neopterin formation in peripheral blood mononuclear cells in vitro. J. Neuroimmunol.207, 75–82. doi: 10.1016/j.jneuroim.2008.12.004
47
KanoM.Ohno-ShosakuT.MaejimaT. (2002). Retrograde signaling at central synapses via endogenous cannabinoids. Mol. Psychiatry7, 234–235. doi: 10.1038/sj.mp.4000999
48
KunosG.Osei-HyiamanD.BátkaiS.SharkeyK. A.MakriyannisA. (2009). Should peripheral CB(1) cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol. Sci.30, 1–7. doi: 10.1016/j.tips.2008.10.001
49
LibroR.DiomedeF.SciontiD.PiattelliA.GrassiG.PollastroF.et al. (2016). Cannabidiol Modulates the Expression of Alzheimer's Disease-Related Genes in Mesenchymal Stem Cells. Int. J. Mol. Sci.18, E26. doi: 10.3390/ijms18010026
50
LinM. T.BealM. F. (2006). Alzheimer's APP mangles mitochondria. Nat. Med.12, 1241–1243. doi: 10.1038/nm1106-1241
51
LigrestiA.CascioM. G.PryceG.KulasegramS.BeletskayaI.De PetrocellisL.et al. (2006). New potent and selective inhibitors of anandamide reuptake with antispastic activity in a mouse model of multiple sclerosis. Br. J. Pharmacol.147, 83–91. doi: 10.1038/sj.bjp.0706418
52
MaY. L.WestonS. E.WhalleyB. J.StephensG. J. (2008). The phytocannabinoid Delta(9)-tetrahydrocannabivarin modulates inhibitory neurotransmission in the cerebellum. Br. J. Pharmacol.154, 204–215. doi: 10.1038/bjp.2008.57
53
MagenI.AvrahamY.AckermanZ.VorobievL.MechoulamR.BerryE. M. (2009). Cannabidiol ameliorates cognitive and motor impairments in mice with bile duct ligation. J. Hepatol.51, 528–534. doi: 10.1016/j.jhep.2009.04.021
54
ManczakM.AnekondaT. S.HensonE.ParkB. S.QuinnJ.ReddyP. H. (2006). Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet.15, 1437–1449. doi: 10.1093/hmg/ddl066
55
Martín-MorenoA. M.ReigadaD.RamírezB. G.MechoulamR.InnamoratoN.CuadradoA.et al. (2011). Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer's disease. Mol. Pharmacol.79, 964–973. doi: 10.1124/mol.111.071290
56
MartinezA. A.MorgeseM. G.PisanuA.MachedaT.PaquetteM. A.SeillierA.et al. (2015). Activation of PPAR gamma receptors reduces levodopa-induced dyskinesias in 6-OHDA-lesioned rats. Neurobiol. Dis.74, 295–304. doi: 10.1016/j.nbd.2014.11.024
57
MatsudaL. A.LolaitS. J.BrownsteinM. J.YoungA. C.BonnerT. I. (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature346, 561–564. doi: 10.1038/346561a0
58
McHughD.HuS. S.RimmermanN.JuknatA.VogelZ.WalkerJ. M.et al. (2010). N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci.11, 44. doi: 10.1186/1471-2202-11-44
59
McHughD. (2012). GPR18 in microglia: implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol.167, 1575–1582. doi: 10.1111/j.1476-5381.2012.02019.x
60
MechoulamR.HanusL. (2000). A historical overview of chemical research on cannabinoids. Chem. Phys. Lipids108, 1–13. doi: 10.1016/s0009-3084(00)00184-5
61
MechoulamR.PetersM.Murillo-RodriguezE.HanusL. O. (2007). Cannabidiol–recent advances. Chem. Biodivers.4, 1678–1692. doi: 10.1002/cbdv.200790147
62
MichalikL.AuwerxJ.BergerJ. P.ChatterjeeV. K.GlassC. K.GonzalezF. J.et al. (2006). International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev.58, 726–741. doi: 10.1124/pr.58.4.5
63
MillsE.DongX. P.WangF.XuH. (2010). Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Future Med. Chem.2, 51–64. doi: 10.4155/fmc.09.140
64
MunroS.ThomasK. L.Abu-ShaarM. (1993). Molecular characterization of a peripheral receptor for cannabinoids. Nature365, 61–65. doi: 10.1038/365061a0
65
NeugroschlJ.SanoM. (2010). Current treatment and recent clinical research in Alzheimer's disease. Mt Sinai J. Med.77, 3–16. doi: 10.1002/msj.20165
66
O’SullivanS. E.SunY.BennettA. J.RandallM. D.KendallD. A. (2009). Time-dependent vascular actions of cannabidiol in the rat aorta. Eur J. Pharmacol.612, 61–68. doi: 10.1016/j.ejphar.2009.03.010
67
O'SullivanS. E.KendallD. A. (2010). Cannabinoid activation of peroxisome proliferator-activated receptors: potential for modulation of inflammatory disease. Immunobiology215, 611–616. doi: 10.1016/j.imbio.2009.09.007
68
O'SullivanS. E. (2007). Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol.152, 576–582. doi: 10.1038/sj.bjp.0707423
69
O'SullivanS. E. (2016). An update on PPAR activation by cannabinoids. Br. J. Pharmacol.173, 1899–1910. doi: 10.1111/bph.13497
70
PellatiF.BorgonettiV.BrighentiV.BiagiM.BenvenutiS.CorsiL. (2018). Cannabis sativa L. and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. BioMed. Res. Int.2018, 1691428. doi: 10.1155/2018/1691428
71
PertweeR. G.RossR. A. (2002). Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids66, 101–121. doi: 10.1054/plef.2001.0341
72
PertweeR. G. (2006). Cannabinoid pharmacology: the first 66 years. Br. J. Pharmacol.147, 63–71. doi: 10.1038/sj.bjp.0706406
73
PertweeR. G. (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol.153, 199–215. doi: 10.1038/sj.bjp.0707442
74
PiomelliD. (2003). The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci.4, 873–884. doi: 10.1038/nrn1247
75
QuerfurthH. W.LaFerlaF. M. (2010). Alzheimer's disease. N Engl. J. Med.362, 329–344. doi: 10.1056/NEJMra0909142
76
ResstelL. B.TavaresR. F.LisboaS. F.JocaS. R.CorrêaF. M.GuimarãesF. S. (2009). 5-HT1A receptors are involved in the cannabidiol-induced attenuation of behavioural and cardiovascular responses to acute restraint stress in rats. Br. J. Pharmacol.156, 181–188. doi: 10.1111/j.1476-5381.2008.00046.x
77
RomanoA.ServiddioG.CalcagniniS.VillaniR.GiudettiA. M.CassanoT.et al. (2017). Linking lipid peroxidation and neuropsychiatric disorders: focus on 4-hydroxy-2-nonenal. Free Radic. Biol. Med.111, 281–293. doi: 10.1016/j.freeradbiomed.2016.12.046
78
RussoE. B.BurnettA.HallB.ParkerK. K. (2005). Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res.30, 1037–1043. doi: 10.1007/s11064-005-6978-1
79
RyanD.DrysdaleA. J.LafourcadeC.PertweeR. G.PlattB. (2009). Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci.29, 2053–63. doi: 10.1523/JNEUROSCI.4212-08.2009
80
SabbaghM. N. (2009). Drug development for Alzheimer's disease: where are we now and where are we headed? Am. J. Geriatr. Pharmacother7, 167–185. doi: 10.1016/j.amjopharm.2009.06.003
81
SagredoO.RamosJ. A.DecioA.MechoulamR.Fernández-RuizJ. (2007). Cannabidiol reduced the striatal atrophy caused 3-nitropropionic acid in vivo by mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors. Eur. J. Neurosci.26, 843–851. doi: 10.1111/j.1460-9568.2007.05717.x
82
ScuderiC.BronzuoliM. R.FacchinettiR.PaceL.FerraroL.BroadK. D.et al. (2018). Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer's disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry8, 32. doi: 10.1038/s41398-017-0076-4
83
ServiddioG.RomanoA. D.CassanoT.BellantiF.AltomareE.VendemialeG. (2011). Principles and therapeutic relevance for targeting mitochondria in aging and neurodegenerative diseases. Curr. Pharm. Des.17, 2036–2055. doi: 10.2174/138161211796904740
84
ScuttA.WilliamsonE. M. (2007). Cannabinoids stimulate fibroblastic colony formation by bone marrow cells indirectly via CB2 receptors. Calcif. Tissue Int.80, 50–59.
85
SzaboB.SchlickerE. (2005). Effects of cannabinoids on neurotransmission. Handb. Exp. Pharmacol.168, 327–365. doi: 10.1007/3-540-26573-2_11
86
TakedaS.MisawaK.YamamotoI.WatanabeK. (2008). Cannabidiolic acid as a selective cyclooxygenase-2 inhibitory component in cannabis. Drug Metab. Dispos.36, 1917–1921. doi: 10.1124/dmd.108.020909
87
ThomasA.StevensonL. A.WeaseK. N.PriceM. R.BaillieG.RossR. A.et al. (2005). Evidence that the plant cannabinoid Delta9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br. J. Pharmacol.146, 917-926. doi: 10.1038/sj.bjp.0706414
88
ThomasA.BaillieG. L.PhillipsA. M.RazdanR. K.RossR. A.PertweeR. G. (2007). Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol.150, 613–623. doi: 10.1038/sj.bjp.0707133
89
TrabaceL.CassanoT.TucciP.SteardoL.KendrickK. M.CuomoV. (2004). The effects of nitric oxide on striatal serotoninergic transmission involve multiple targets: an in vivo microdialysis study in the awake rat. Brain Res.1008, 293–298. doi: 10.1016/j.brainres.2004.01.090
90
UttaraB.SinghA. V.ZamboniP.MahajanR. T. (2009). Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol7, 65–74. doi: 10.2174/157015909787602823
91
ValléeA.LecarpentierY.GuillevinR.ValléeJ. N. (2017). Effects of cannabidiol interactions with Wnt/β-catenin pathway and PPARγ on oxidative stress and neuroinflammation in Alzheimer's disease. Acta Biochim. Biophys. Sin. (Shanghai)49, 853–866. doi: 10.1093/abbs/gmx073
92
WalterL.StellaN. (2004). Cannabinoids and neuroinflammation. Br. J. Pharmacol.141, 775–785. doi: 10.1038/sj.bjp.0705667
Summary
Keywords
Cannabis sativa, oxidative stress, phytocannabinoids, cannabidiol, Alzheimer's disease, Parkinson's disease
Citation
Cassano T, Villani R, Pace L, Carbone A, Bukke VN, Orkisz S, Avolio C and Serviddio G (2020) From Cannabis sativa to Cannabidiol: Promising Therapeutic Candidate for the Treatment of Neurodegenerative Diseases. Front. Pharmacol. 11:124. doi: 10.3389/fphar.2020.00124
Received
26 November 2019
Accepted
29 January 2020
Published
06 March 2020
Volume
11 - 2020
Edited by
Andrea Tarozzi, University of Bologna, Italy
Reviewed by
Gaurav Bedse, Vanderbilt University Medical Center, United States; Pasqualina Castaldo, Marche Polytechnic University, Italy
Updates
Copyright
© 2020 Cassano, Villani, Pace, Carbone, Bukke, Orkisz, Avolio and Serviddio.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tommaso Cassano, tommaso.cassano@unifg.it
This article was submitted to Neuropharmacology, a section of the journal Frontiers in Pharmacology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.