Abstract
New drug discovery has been acknowledged as a complicated, expensive, time-consuming, and challenging project. It has been estimated that around 12 years and 2.7 billion USD, on average, are demanded for a new drug discovery via traditional drug development pipeline. How to reduce the research cost and speed up the development process of new drug discovery has become a challenging, urgent question for the pharmaceutical industry. Computer-aided drug discovery (CADD) has emerged as a powerful, and promising technology for faster, cheaper, and more effective drug design. Recently, the rapid growth of computational tools for drug discovery, including anticancer therapies, has exhibited a significant and outstanding impact on anticancer drug design, and has also provided fruitful insights into the area of cancer therapy. In this work, we discussed the different subareas of the computer-aided drug discovery process with a focus on anticancer drugs.
Introduction
Up to now, cancer remains a global and serious public health challenge. It is estimated that there are more than 200 different types of cancer, generally named according to the tissue where the cancer was recognized for the first time. Cancer is considered to be one of the significant causes for death in the 21st century and the most critical obstacle for the increase of global life expectancy. According to an analysis by the world health organization (WHO) in 2015, cancer is the second leading cause of death for patients younger than 70 years old in 91 countries and the third or fourth leading cause of death among 22 other countries (Yan et al., 2019). Moreover, a global increase of 18.1 million new cancer cases and 9.6 million cancer-related deaths have been reported in a previous study (Bray et al., 2018), especially 70% of the death caused by cancer occur in low-income and middle-income countries. The fast growth of the cancer incidence and mortality has turned out to be global health challenges. How to reduce the cancer-related death rate has attracted significant attention from the government, society, medical industry, as well as scientific communities, expecting the rapid development of effective and safe drugs for cancer treatment.
Despite of the impressive progress in biotechnologies and further understandings of the disease biology, the development of new, practical and innovative small molecule drugs remains an arduous, time-consuming, and expensive project, which requires collaborations from many expertise in multidisciplinary fields, including medicinal chemistry, computational chemistry, biology, drug metabolism, clinical research, etc. Furthermore, it has been illustrated that the successful discovery and development of a new drug costs 12 years, and expensive investment (Kapetanovic, 2008). Thus, novel drug development strategies with a reduced cost of time and money, as well as an enhanced efficiency are in high demand, which would contribute to a significant improvement in global health and life expectancy. Since the successful development of HIV protease inhibitor Viracept in the USA in 1997, which was the first drug design fully driven by its target structure (Kaldor et al., 1997), computational methods have served as an essential tool in drug discovery projects and have been a cornerstone for new drug development approaches. This makes the drug developmental process faster and cheaper. Recently, the fast growth in computational power, including massively parallel computing on graphical processing units (GPUs), the continuous advances in artificial intelligence (AI) tools (Chan et al., 2019; Yang et al., 2019), have translated fundamental research into practical applications (Zhavoronkov et al., 2019) in the drug discovery field. This attracted considerable attention for their outstanding performance on providing new promising perspectives and solutions to overcome life-threatening diseases.
In this review, we aim at providing an overview of different subjects of the computational-method-aided new drug discovery processes in general, and anti-cancer therapy discovery in particular. We reviewed some of the most representative examples and clarified fundamental principles by exploring studies on anticancer drug designs with the help of computational methods. A workflow of computational drug discovery is explained in Figure 1.
Figure 1
Anti-Cancer Drug Target Prediction
Human contains approximately 30,000 genes, among which around 6,000 to 8,000 sites are estimated as potential pharmacological targets. However, less than 400 encoded proteins have been proved to be effective for drug development until now (Drews, 2000; Chen et al., 2016). Cancer, compared to many other human diseases, now has a plethora of potential molecular targets for therapeutic development (Lazo and Sharlow, 2016). Traditional drug discovery mainly follows the paradigm of “one molecule - one target - one disease”, without considering the interactions between drugs and proteins. However, an important fact that many complex diseases are relevant to a variety of target proteins (Hopkins, 2008; Yamanishi et al., 2008; Chen et al., 2012) has been overlooked. Furthermore, unexpected drug functions derived from off-targets are an accidental and uncontrollable activities because of the “poly-pharmacological” properties of certain drugs, which might result in undesirable side effects. Those are particularly pronounced for cancer drugs. On the other hand, there are some positive examples that benefit from the different pathways targeted by one given molecule. For example, sildenafil (viagra) was developed to treat angina, but now it is used for erectile dysfunction therapy (Ghofrani et al., 2006). There are several drugs, including anticancer drugs, whose corresponding target proteins (both primary and non-target) remain yet unidentified or unknown (Takarabe et al., 2012). Furthermore, some attractive and potentially effective cancer targets remain outside of the scope of pharmacological regulation. Some of these targets such as phosphatases, transcription factors, and RAS family members have been described as undruggable, as they lack effective enzymatic active sites (Lazo and Sharlow, 2016). To make the full use of known drugs to treat new indications, the characterization of all potential new ligand binding sites has been illustrated as a key point in drug repositioning and repurposing. Therefore, new and highly qualitative bioinformatic target prediction methods are required for the accurate prediction of drug targets.
Up to now, a wide range of drug target interactive web servers has been established, providing a series of drug-target databases and prediction tools (see Tables 1 and 2). Moreover, various computational approaches have been used to study potential interactions between proteins and drugs. In particular, network-based models and ML-based models have emerged as important tools. A review by Chen et al. summarizes several available computational models for this application (Chen et al., 2016). Interestingly, a method proposed by Campillos et al. that uses the similarity of drug side effects to determine whether multiple drugs could interact with the same target proteins attracted our attention (Campillos et al., 2008). Based on this research, Takarabe et al. took advantage of the US FDA's adverse event reporting system (AERS) to define the pharmacological similarity of all potential medicines and developed a novel system to predict large-scale interactions between unknown drug-targets (Takarabe et al., 2012). Notably, AERS was employed to predict interactions between drugs and targets for the first time. In 2010, Klipp et al. summarized several available computational models for network-based drug-target prediction (Klipp et al., 2010). Moreover, various biological data settings, including structures of bioactive compounds, sequences of target proteins, and information of ligand-target interactions, have been combined. A series of machine learning-based approaches have been demonstrated as efficient tools in detecting relationships among drug structures and corresponding target proteins from a large amount of data, such as supervised learning method (Srivastava et al., 2014), bipartite graph learning method (Li and Chen, 2013), bipartite local model (Yildirim et al., 2007), and so on. A recent review by Mayr et al. compared the predictive performance of deep learning with other prediction approaches for multiple drug targets in the comparative studies of composite target prediction methods. As a result, feed-forward neural networks were identified with better performance in drug target prediction than other methods (Mayr et al., 2018).
Table 1
| Databases | Websites |
|---|---|
| DrugBank | https://www.drugbank.ca/ |
| TTD | http://bidd.nus.edu.sg/group/ttd/ttd.asp |
| MATADOR | http://matador.embl.de/ |
| SuperTarget | http://insilico.charite.de/supertarget/ |
| TDR targets | http://tdrtargets.org/ |
| PDTD | http://www.dddc.ac.cn/pdtd/ |
| ChEMBL | https://www.ebi.ac.uk/chembldb |
| STITCH | http://stitch.embl.de/ |
| BindingDB | http://www.bindingdb.org/ |
| CancerDR | http://crdd.osdd.net/raghava/cancerdr/ |
| DCDB | http://www.cls.zju.edu.cn/dcdb/ |
Drug-target database.
Table 2
| Computational tools | Websites |
|---|---|
| SEA | https://omictools.com/sea-2-tool |
| Pharmmapper | http://www.lilab-ecust.cn/pharmmapper/ |
| Chemmapper | https://omictools.com/chemmapper-tool |
| Tide | http://sysbio.molgen.mpg.de/tide |
| DINIES | http://www.genome.jp/tools/dinies/ |
| SuperPred | http://prediction.charite.de/ |
| SwissTarget Prediction | http://www.swisstargetprediction.ch/ |
Computational tools for target prediction.
As above, since a large number of compounds and vigorous efforts are abandoned and wasted due to the off-target effects during the classical drug development procedure, a greatly enhanced development of target prediction in new drug exploration exhibited attractive advantages and further expansion in this area are still highly desirable
Structure-Based Drug Discovery
Structure-based strategy relies on the known structural information to define the interaction effect between bioactive compounds and the corresponding receptors. (Wang et al., 2000). With the development of biomolecular spectroscopic technologies such as X-ray crystallography and nuclear magnetic resonance (NMR), remarkable progress has been made in this field, leading to considerable improvements in our structural understanding of the drug target. Taking advantages of the three-dimensional structure of the proteins, new ligands could be rationally designed to trigger therapeutic effects. Hence, structure-based design (SBD) could provide critical insights into new drug design and development via discovering and optimizing the initial lead compounds (Prada-Gracia et al., 2016; Lu et al., 2018a). The high affinity ligand regulates validated drug targets selectively to influence specific cellular activities, ultimately achieving the desired pharmacological, and therapeutic effects (Urwyler, 2011). Capoten (captopril), the first ACE (angiotensin-converting enzyme) inhibitor, was one of the first successful examples of using structural information to optimize drug designs in the 1980s (Anthony et al., 2012). Since this study, structure-based drug development started to serve as a novel and powerful algorithm and technique to promote faster, cheaper, and more effective drug development. In the past decade, extensive efforts have been made to promote the strategy of SBD, more and more successful applications played important roles in new medical research (Debnath et al., 2019; Hong et al., 2019; Mendoza et al., 2019; Itoh, 2020; Tondo et al., 2020).
Molecular Docking
Molecular docking is a typical structure-based protocol in rational drug design by studying and predicting the binding patterns and interaction affinities among the ligand and receptor biomolecules (Ferreira et al., 2015). It could be categorized as rigid docking and flexible docking according to the flexibility of the ligands involved in the computational process (Halperin et al., 2002; Dias and De Azevedo, 2008). The rigid docking method is a binding model which only considers the static geometrical, physical, and chemical complementarity between the ligand and the target proteins, while ignores the flexibility and the induced-fit theory (Salmaso and Moro, 2018). In general, the rigid docking, which is fast and highly effective, is applied to the high throughput virtual screening with a large number of small-molecule databases to be time-efficient. While the flexible docking method considers more detailed and accurate information. With the rapid improvement of computing resources and efficiency, flexible docking methods developed continuously and became more easily accessible. There are different types of software available for docking, such as Glide, FlexX, DOCK, AutoDock, Discovery Studio, Sybyl, etc.
The molecular docking process is mainly composed of three steps. First, the structures of small molecules and target proteins should be prepared in advance. In this step, abundant experimentally solved structures are available in the open access PDB database (http://www.rcsb.org), which can be used to understand many physiological processes based on the crystal structures, and also for homologous template models if docking structures are of interest. Second, it can act as an engine for predicting conformations, orientations, and positional spaces in the ligand binding site (Mathi et al., 2018). Conformational search algorithms carry out this task to predict the conformations of binary complexes by applying the methods of systematic and stochastic search. Systematic search techniques include: (i) Exhaustive search; (ii) Fragmentation; (iii) Conformational Ensemble. On the other hand, stochastic methods include: (i) Monte Carlo (MC) methods; (ii) Tabu search methods; (iii) Evolutionary Algorithms (EA); (IV) Swarm optimization (SO) methods (Ferreira et al., 2015). Finally, these programs evaluate the putative binding-free energy, which associates the scoring function to determine which compounds are more likely to bind to targets during the molecular docking (Huang et al., 2010). There are four essential types of scoring functions, including: (i) Consensus scoring functions (ii) Empirical scoring functions; (iii) Knowledge-based scoring functions; (iv) Force-field based scoring functions (Kortagere and Ekins, 2010). Furthermore, new scoring capabilities have been developed, for example (i) machine learning technologies; (ii) interactive fingerprints; (iii) quantum mechanical scores (Yuriev et al., 2015).
Structure-Based Pharmacophore Mapping
With the development in the past decades, the pharmacophore mapping method has been considered as one of the most useful technology during the process of drug discovery. All kinds of structure-based approaches have been conducted to improve pharmacophore modeling, which has been widely used for virtual screening, de novo design as well as lead optimization (Yang, 2010; Lu et al., 2018a). The structure-based pharmacophore (SBP) is another useful method. Based on the availability of ligand structures, SBP modeling methods can be cataloged into two types: target-ligand complex-based methods and target-binding site-based (without ligand) methods (Pirhadi et al., 2013). The approach based on the target-ligand complex can conveniently locate the ligand-binding pocket of the protein and assess the main ligand-protein interactions. This is exampled by LigandScout (Wolber et al., 2006), Pocket v.2 (Chen and Lai, 2006), and GBPM (Ortuso et al., 2006). It is worth noting that they cannot be used to the situations where ligands are unknown. The macromolecule (without ligand)-based method implemented in Discovery Studio (Lu et al., 2018b) is an obvious example which is not dependent on the ligands and the receptor-ligand interactions. The LUDI program (Bohm, 1992) defines the interactions within the binding site as pharmacological characteristics. Although this purely SBP method has the advantage of describing the entire interaction capability of a binding pocket, the main limitation of this method is that the derived interaction maps typically involve many unprioritized interaction features.
Ligand-Based Drug Discovery
Similarity Searching
The main principle and motivation behind the ligand-based approaches in drug discovery is a concept known as molecular similarity; based on this principle, molecules tend to perform similar biological effects due to the high structural similarity (Zhavoronkov et al., 2019). In other words, ligand-based drug discovery methods rely on the structural information of the active ligand that interacts with the target protein, and such a compound with interesting biological properties can be used as a query template in identifying and predicting new chemical entities with similar properties. Since only the structure of the known active small molecules are required, this methodology is considered as an indirective protocol for drug discovery. It offers an option when the 3D target protein structure is unknown or cannot be predicted. Hence, this approach is commonly applied to screen novel ligands with interesting biological activities in silico and to optimize the biological activities of ligands to improve drug pharmacokinetics including Adsorption, Distribution, Metabolism, Excretion, Toxicity (ADMET) properties.
This simple and most widely used technique is based on molecular descriptors. Physicochemical properties (e.g., molecular weight, logP, Energy of high occupied molecular orbital (EHOMO), Energy of lowest unoccupied orbital (ELUMO), charges), as well as 2D fingerprint and 3D shape-similarity searches can be introduced as coordinates to represent the reference compounds. The 2D fingerprint (Molprint2D and Unity 2D) and 3D shape similarity methods (MACCS), extended-connectivity fingerprints (ECFP), rapid overlay of chemical structures (ROCS), and Phase Shape, are more often used for molecular representation in virtual screening (Rush et al., 2005). For example, Bologa et al. (2006) applied 2D fingerprint and 3D shape-similarity methods to identify novel agonists of the estradiol receptor family receptor GPR30 (Bologa et al.). Furthermore, both methods have been successfully applied in virtual screenings, and both technology have exhibited better performance against a number of targets than docking methods in terms of the scalability and computational time. However, the main problem of the similar methods is their preference for input molecules and the difficulty in deciding which input structures to be used (Hu et al., 2012).
Ligand-Based Pharmacophore Mapping
Another more precise approach in comparison with the molecular descriptors is the pharmacophore-based approach, in which a pharmacophore model (PH4) is developed based on a group of active compounds. The IUPAC (International Union of Pure and Applied Chemistry) pointed out that a pharmacophore is “a collection of spatial and electronic characteristics necessary to ensure optimal supramolecular interactions with specific biological targets and to trigger (or block) their biological reactions” (Buckle et al., 2013). Thus, structural overlap of key molecular features derived from active compounds or a binding site in space are used as a pattern to represent the most probable chemical characteristics. The newly identified molecules that match and show a high complementation to the developed pharmacophore are likely to be active against the target protein of interests. Therefore, they can be selected as candidates for more further investigations. This approach has become a key computational strategy to promote and guide drug discoveries in the absence of macromolecular structures (Chao et al., 2007).
The process of pharmacophore modelling can be summarized as following: (i) Selection of a training set of ligands (active and inactive compounds). (ii) Molecular preparation (low energy conformations). (iii) Ligand alignment/superimposition and pharmacophore model generation. (iv) Validation of pharmacophore models (Chiang et al., 2009). Ligand-based pharmacophore modeling highly depends on the availability of a good training set of compounds manifesting the same binding mode.
QSAR Modeling
QSAR (Quantitative Structure Activity Relationship) is another ligand-based approach that relies on analyzing the biological activities of drugs using various molecular descriptors (MDs) or fingerprints (FPs). These models mathematically describe how the activities response to the targets according to the ligand's structural characteristics. QSAR was obtained by calculating the correlations between the properties of the ligand binding agent and the biological activity measured by experiments. Different ML and deep learning (DL) approaches have also been applied to develop QSAR models (Mendenhall and Meiler, 2016): including Support Vector Machine (SVM), Random Forest (RF), Polynomial Regression (PR), Multi Linear Regression (MLR), Artificial Neural Network (ANN). Unlike the pharmacophore models, QSAR models can measure biological activities quantitatively and can even find positive or negative effects according to certain characteristics of the molecule on its activity.
QSAR has been applied to many other molecular design purposes, such as predicting the new molecule analog activity, optimizing lead, and predicting new structural leads in drug discovery. In the classical 2D-QSAR approaches, the biological activity is related to physical and chemical features consisting of steric, electronic, and hydrophobic characters of drugs, and the relationships are represented as mathematical equations (Hansch and Fujita, 1964). More advanced 3D-QSAR approaches, such as comparative molecular field analysis (Cramer et al., 1988) and molecular similarity indexes in a comparative analysis (Klebe et al., 1994), are based on the force field calculations. The structural information of molecules is needed, and developed models are represented in 3D contour maps facilitating the visualization and interpretation.
Using MD simulation to Find New Drug Binding Sites
Many important biological events rely on the information of protein-ligand complex interactions. The recognition and characterization of LBP is the key to understand the function of endogenous ligands and synthetic drug molecules. GPCRs perform an important role in a variety of physiological processes. GPCRs are a class of commonly used targets in drug discovery (Conn et al., 2009). Recent discovery indicated that beside binding to orthosteric sites, ligands could bind to different allosteric sites that are far away from the targeted binding pockets (Tautermann, 2014; Flock et al., 2015; Devree et al., 2016). Unfortunately, the position of such allosteric pocket is unclear without the information of experimental structures, and predicting the existence of such sites could facilitate the discovery of new drugs (Tautermann, 2014). A recent overview described the progresses in important computational tools for the prediction of functional sites, such as 3DLigandStie (http://www.sbg.bio.ic.ac.uk/~3dligandsite/), COACH-D (http://yanglab.nankai.edu.cn/COACH-D/), or SiteMap (https://www.schrodinger.com/sitemap), and many others. However, these reported tools often create multiple possible ligand binding sites, and sometimes it is not easy for the user to confirm which active pocket is real one for the compound binding. To overcome this limitation, methods based on molecular dynamics (MD) have been developed in recent years. For example, the supervised MD is an efficient approach for precise sampling and the identification of ligand-binding sites (Sabbadin and Moro, 2014; Deganutti et al., 2015; Cuzzolin et al., 2016). The conventional long-timescale MD has also been successfully applied for new drug binding sites (Chan et al., 2018). Similarly, a study by Chan et al. (2020) reported that an additional sodium ion, which located in the vicinity of the orthosteric binding site, by MD simulations (Chan et al., 2020). MD could also be applied for the recognition of the allosteric sites involved in protein kinases (Tong and Seeliger, 2015), Ras proteins (Hancock, 2003), and Staphylococcus aureus Sortase A (Mazmanian et al., 1999). As above, information obtained from MD predictions provides new opportunities of drug discovery.
Artificial Intelligence in Anti-Cancer Drug Discovery
Computational drug design has successfully promoted the discovery of several new anticancer drugs, which has become a milestone in this area. Gefitinib (Muhsin et al., 2003), Erlotinib (Grunwald and Hidalgo, 2003), Sorafenib (Wilhelm et al., 2006), Lapatinib (Wood et al., 2004), Abiraterone (Jarman et al., 1998), Crizotinib (Butrynski et al., 2010) are all approved drugs that have been discovered based on computational drug methods. Until now, the anticancer drug research is rapidly progressing: computational, and AI methods are generating new promising results. As an example, SR13668 is optimized from indole-3-carbinol (I3C) using PH4 design. SR13668 has shown a strong effect on different cancers in phase I (Chao et al., 2007). Recently, Rodrigues et al. have successfully identified a potent inhibitor for 5-lipoxygenase by using machine learning (ML)-based method which was developed from physicochemical and pharmacophore characteristics (Reker et al., 2014; Rodrigues et al., 2018). With the arrival of AI, the design of anticancer drugs in silico has undergone unprecedented changes, and state-of-the-art deep learning approaches have the potential to produce the excellent chemical properties needed for new molecules (Gomez-Bombarelli et al., 2018). Similarly, Jann et al. have developed the first ML-based anti-cancer compound generator using variational autoencoders (VAEs) and have demonstrated that the compound production may be selective toward molecules with high predicted inhibition to a specific cancer (Born et al., 2019). This implied that models could be developed to yield drug candidates with highly desired efficacy (IC50) against a target of interest. This breakthrough could transform the design of anticancer drugs in silico by taking advantage of the bimolecular features of the disease to improve the success rate of lead compound discovery.
Successful Stories of Computational Drug Discovery
Computational methods have proved to play an essential role in modern drug discovery. Since computational methods could cover almost all stages of the drug discovery pipeline, the applications of computational methods in anticancer drug discoveries have shown great advantages in terms of the required investment, resources, and time. More recently, computational methods have become a potent and powerful tool in several successful cases of anticancer drug development. Herein, we list several successful applications of computational methods for small molecule drugs, which have been applied to cancer treatment or are at later stages in the clinical trial.
The development of Crizotinib is a successful example of applying structure-based design techniques (Cui et al., 2011; Kung et al., 2015). Crizotinib has been considered as a selective and potent cMet/ALK dual inhibitor, which was approved by FDA in 2011 (Cui et al., 2013). c-Met, also known as HGFR (hepatocyte growth factor receptor), and its corresponding natural ligand HGF (hepatocyte growth factor) play a critical role in different cell activities (Christensen et al., 2005). The over-expression of c-Met protein has been often detected in human cancers (including SCLC and NSCLC) (Bottaro et al., 1991; Liu et al., 2008), and abnormal function of c-Met signaling was observed in various solid and blood tumor cancers. Thus, c-MET is an attractive and promising oncology target.
The investigation started with evaluating a series of 3-substituted indolin-2-ones, a potent class of kinase inhibitors, indolin-2-one derivatives for c-MET inhibition. Among the derivatives, compound 1 (PHA-665752, Scheme 1) showed strong activity against the c-MET autophosphorylation process and the corresponding biological activations both in vitro and in vivo. However, the bad drug-like characteristics of compound 1 (PHA-665752) limited its further study. The co-crystal structure analysis of compound 1 with the kinase domain of c-MET elucidated the key inhibitor binding site, presenting opportunities for more efficient drug designs. In combination with re-designing the central rings of compound 1 (PHA-665752), a new set of 5-substituted 3-benzyloxy-2-aminopyridine series has been developed. Among these newly designed derivatives, compound 2 displayed promising inhibition against c-MET. It is noted that lipophilic efficiency (LipE) was employed as the parameter for the binding effectiveness to monitor the progress of optimization. To further improve the c-Met inhibitory potency, a docked structure of compound 2 with the c-Met kinase domain was carried out to guide the application of structure-based design techniques. Followed by optimization of 3-benzyloxy group, the functional group at 5-position of the 2-aminopyridine, and examination of the chiral center, crizotinib (PF-02341066) with effective tumor growth inhibition and good drug performance has been achieved (see Scheme 1A). Moreover, Crizotinib has demonstrated remarkable clinical efficacy on c-MET gene amplification against lung cancer, lymphoma, and esophageal cancers (Cui et al., 2011; Lennerz et al., 2011; Schwab et al., 2014).
Scheme 1
In 2012, Axitinib (AG-013736) was approved by the FDA as as a new therapy for advanced renal cell carcinoma (Meadows and Hurwitz, 2012) to treat VEHG. Axitinib was developed with a structure-based drug design strategy and served as an inhibitor by binding to the VEGF kinase domain in the DFG-out conformation (Kania, 2009; Kania et al., 2016). The VEGF (vascular endothelial growth factor) family functions as important regulators of many signaling networks which involves in angiogenesis. VEGF signaling was identified in tumor cells, and the VEGF signaling plays a crucial role in the development of malignant diseases. As the key receptors of VEGF, VEGFRs serve as ligands in the VEGF signaling network. The VEGF receptors are known as a class of the tyrosine kinases (RTKs), including VEGFR-1 (also called FLT1), VEGFR-2 (also called FLK1 and KDR) and VEGFR-3 (also called FLT4). Blocking the action of VEGFRs with a pan kinase inhibitor against VEGFR-1, VEGFR-2, and VEGFR-3 has been proved to be an efficient way of anti-angiogenic drug development.
During the developmental process, the crystal structure of phosphorylated construct (p-VEGFR2Δ50), the resolved structures of inhibitor–VEGFR2Δ50 (unphosphorylated kinase) complexes, and robust SAR provided important guidance to the rational drug design (Kania, 2009). Combining with the complex structure information, a collection of compounds has been evaluated, generating pyrazoles 3 and benzamide 4 as the starting point for the drug design. Further efforts have been made by the modeling of pyrazole 3 into the ligand-free p-VEGFR2Δ50 structure to modify the conformation of pyrazole 3 further, leading to the generation of indazole compounds as novel kinase inhibitors. Among these derivatives, compound 5 with a styryl functional group at the 3-position of the indazole ring was identified to exhibit potent inhibitory effect (Ki of 0.3 nM), with a high level of LipE and LE. The crystal structure of VEGFR2Δ50 with compound 5 revealed the detailed enzyme-ligand mode, showing the indazole core binding to the “open” DFG-in conformation of VEGFR2Δ50. Superimposing the other two VEGFR2Δ50–inhibitor co-crystal complex structures demonstrated a more precise 3D structure of the key binding sites for the induction of the DFG-out conformation. Inspired by the superposition result, a chimera design protocol was applied for the subsequent design to capture the above described inhibitor interactions, giving access to 6–sulfur linked indazole compound 6 and the corresponding amide analogs. Further studies on the overlay of VEGFR2Δ50 bound co-crystal structures of benzamide 4 and indazole 6 demonstrated that an additional amide group on the orthosteric site of S-phenyl group would help to make the two important hydrogen bonds with the hydrogen bonding groups from Glu885 and Asp1046 of VEGFR2Δ50 and provide highly potent inhibitors. Further applying the truncation strategy generated axitinib (AG-013736) (see Scheme 1B), which exhibited a remarkable improvement on cellular potency, desirable physiochemical, and PK properties. Very recently, axitinib (Inlyta®), in combination with pembrolizumab (KEYTRUDA®), was approved as the first-line anticancer drug against renal cell carcinoma (RCC)(Atkins et al., 2018).
Heat shock protein 90 (HSP90) has direct and essential effects on the correct performance of different proteins with their activation, conformation, stabilization, and localization functions, whose alterations are associated with cancer development. Thus, HSP90 has become a promising target for cancer treatment (Whitesell and Lindquist, 2005; Pearl and Prodromou, 2006; Sharp and Workman, 2006; Workman et al., 2007). The biological functions of HSP90 have been identified. Its crystal structures indicated that HSP90 has four functional domains: a middle domain, an N-term domain, ATP/ADP-binding domain, and a C-term dimerization domain (Pearl and Prodromou, 2006). Based on the structural information of HSP90, a high-throughput screening was conducted which generated the active drug inhibitor: compound 7 (CCT018159) (Cheung et al., 2005; Smith et al., 2006; Sharp et al., 2007). The subsequently obtained co-crystal structure of HSP90-compound 7 (CCT018159) complex revealed that further modification of compound 7 (CCT018159) by replacing or adding certain functional groups could improve the pharmacokinetic properties. Moreover, replacing the methyl group to an amide group (VER-49009), changing pyrazolyl ring to isoxazole aromatic ring (VER-50589), and modifying some other chemical groups (see Scheme 1C) led to a potent effect in animal cancer models. Followed by toxicology and safety evaluation, Luminespib (NVP-AUY922) has been proved to be a strong HSP90 inhibitor which is now in clinical trials. More recently, Luminespib, a drug in phase one clinical trials, exhibited positive results for patients with ALK rearrangements (Felip et al., 2018). Luminespib (NVP-AUY922) also exhibited potent anti-tumor activity in lung adenocarcinomas targeting EGFR exon 20 insertion mutations and cellular models in a confirmatory clinical trial (Jorge et al., 2018; Piotrowska et al., 2018). Moreover, Luminespib (NVP-AUY922) serves as one of the components in anticancer combination therapies, which are now at different stages of clinical trials (Garcia-Carbonero et al., 2013; Rong and Yang, 2018). To depict how computational drug discovery facilitates to the development of anticancer drugs, we listed the FDA-approved anticancer drug in recent 3 years which was obtained from National Cancer Institute database (Heller, 1951) in Table 3.
Table 3
| Name | Chemical Structure | Therapeutic area | Target and functiuon | Year of Approval |
|---|---|---|---|---|
| Alpelisib | 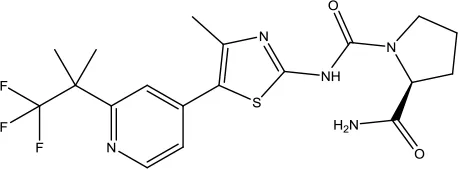 | Breast cancer | PI3K inhibitor | 2019 (Markham, 2019a) |
| Cladribine | 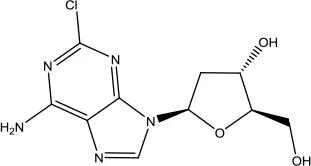 | Hairy cell leukemia | Adenosine deaminase inhibitor | 2019 (Bryson and Sorkin, 1993) |
| Darolutamide | 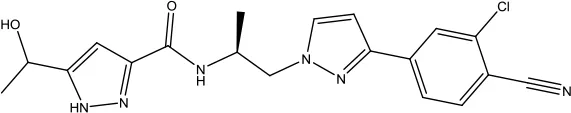 | Prostate cancer | Androgen receptor inhibitor | 2019 (Markham and Duggan, 2019) |
| Entrectinib | 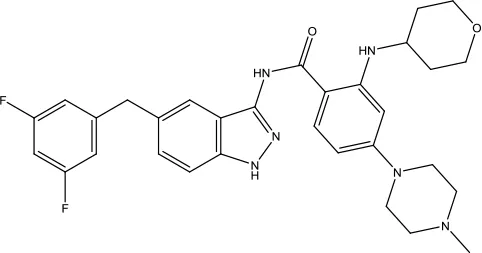 | Non-small cell lung cancer and Solid tumors | Tyrosine kinase inhibitor | 2019 (Al-Salama and Keam, 2019) |
| Erdafitinib | 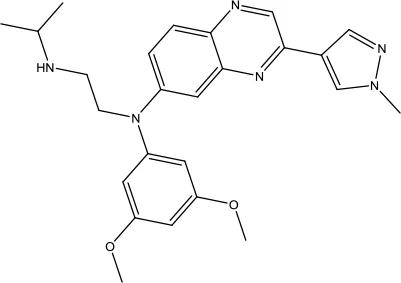 | Urothelial carcinoma | FGFR tyrosine inhibitor | 2019 (Markham, 2019b) |
| Fedratinib Hydrochloride | 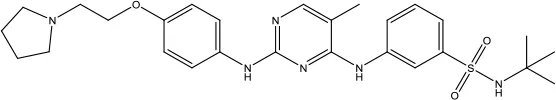 | Myelofibrosis | Tyrosine kinase inhibitor | 2019 (Zhang et al., 2014) |
| Selinexor | 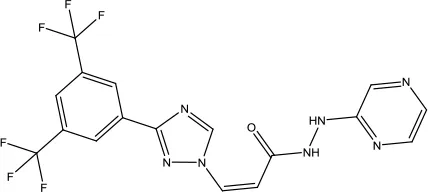 | Multiple myeloma | Nuclear export inhibitor | 2019 (Syed, 2019) |
| Zanubrutinib | 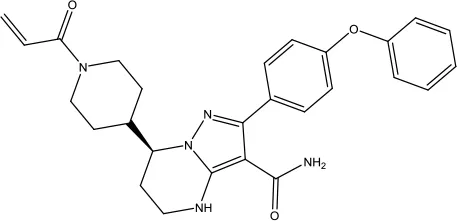 | Mantle cell lymphoma | Bruton's tyrosine kinase inhibitor | 2019 (Syed, 2020) |
| Abemaciclib | 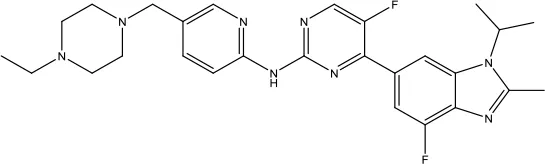 | Breast cancer | Cyclin-dependent kinase inhibitor | 2018 (Kim, 2017b) |
| Apalutamide | 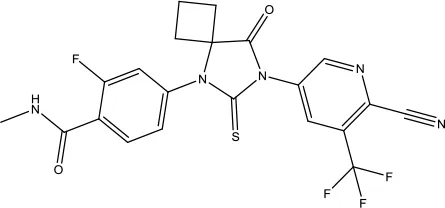 | Prostate cancer | Androgen receptor inhibitor | 2018 (Al-Salama, 2019) |
| Binimetinib | 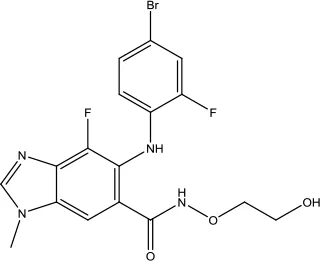 | Melanoma | MEk1 and MEK2 inhibitor | 2018 (Shirley, 2018) |
| Dacomitinib | 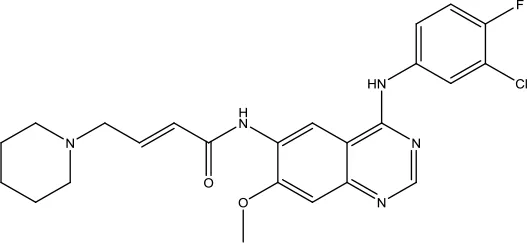 | Non-small cell lung cancer | Oral kinase inhibitor | 2018 (Sidaway, 2018) |
| Duvelisib | 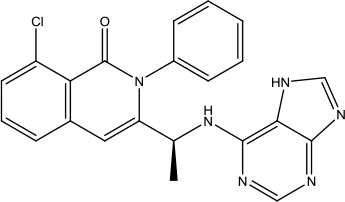 | Chronic lymphocytic leukemia (CLL) and follicular lymphoma (FL) | PI3K Kinase inhibitor | 2018 (Blair, 2018) |
| Encorafenib | 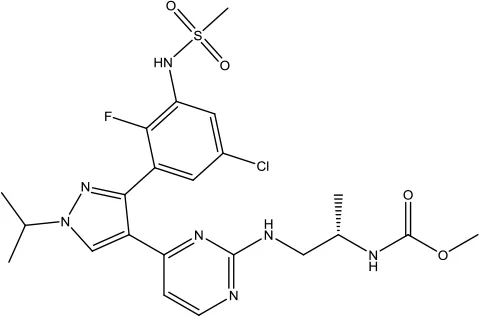 | Colorectal cancer and Melanoma | BRAF Kinase inhibitor | 2018 (Shirley, 2018) |
| Gilteritinib Fumarate | 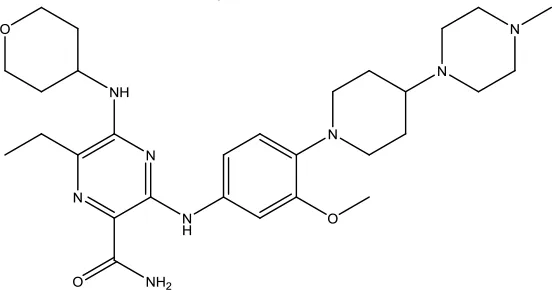 | Acute myeloid leukemia | Tyrosine kinase inhibitor | 2018 (Dhillon, 2019) |
| Glasdegib Maleate | 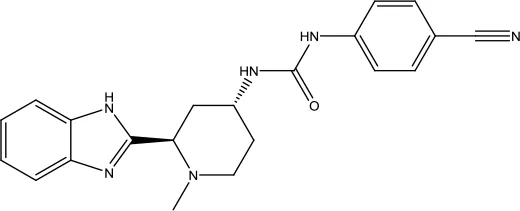 | Acute myeloid leukemia | Hedgehog pathway inhibitor | 2018 (Shaik et al., 2019) |
| Iobenguane I 131 |  | Pheochromocytoma | Radioactive therapeutic agent | 2018 (Giammarile et al., 2008) |
| Ivosidenib |  | Acute myeloid leukemia | Isocitrate dehydrogenase-1 (IDH1) inhibitor | 2018 (Dhillon, 2018) |
| Larotrectinib Sulfate |  | Solid tumors | Tropomyosin-related kinase (Trk) inhibitor | 2018 (Gajdosik, 2017) |
| Lorlatinib |  | Non-small cell lung cancer | Tyrosine kinase inhibitor | 2018 (Su et al., 2019) |
| Talazoparib Tosylate | 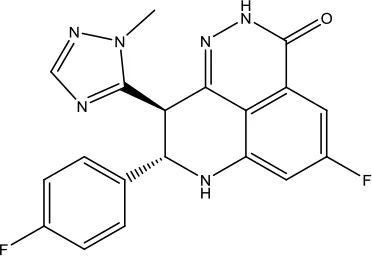 | Breast cancer | Poly (ADP-ribose) polymerase (PARP) inhibitor | 2018 (Eskiler, 2019) |
| Acalabrutinib | 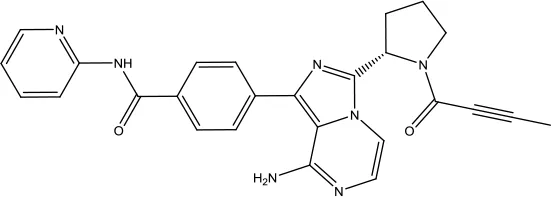 | Chronic lymphocytic leukemia, small lymphocytic lymphoma, and mantle cell lymphoma | Bruton's tyrosine kinase inhibitor | 2017 (Markham and Dhillon, 2018) |
| Brigatinib | 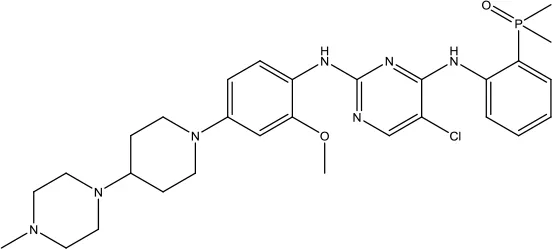 | Non-small cell lung cancer | Anaplastic lymphoma kinase (ALK) and epidermal growth factor receptor (EGFR) kinase inhibitor | 2017 (Markham, 2017a) |
| Copanlisib Hydrochloride | 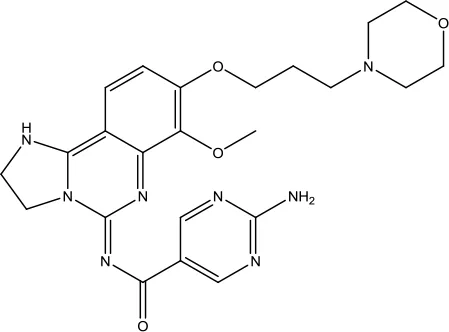 | Follicular lymphoma | Phosphoinositide 3-kinase (PI3K) inhibitor | 2017 (Markham, 2017b) |
| Enasidenib Mesylate | 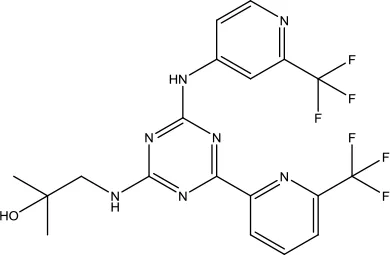 | Acute myeloid leukemia | Isocitrate dehydrogenase-2 inhibitor | 2017 (Gras, 2017) |
| Midostaurin | 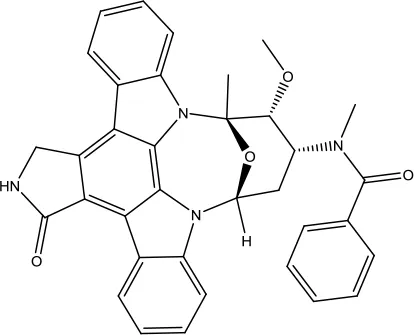 | Acute myeloid leukemia | Synthetic indolocarbazole multikinase inhibitor | 2017 (Kim, 2017a) |
| Neratinib Maleate | 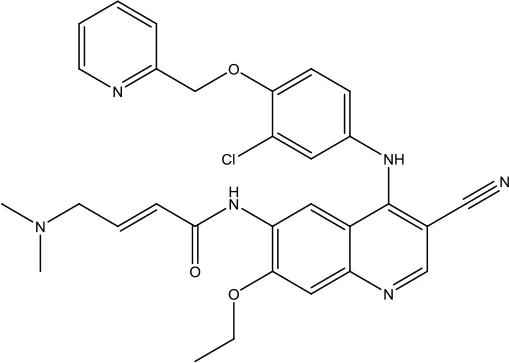 | Breast cancer | Receptor tyrosine kinases (RTKs), Human epidermal growth factor receptor 2 (HER2; ERBB2), and Human epidermal growth factor receptor (EGFR) inhibitor | 2017 (Kotecki et al., 2019) |
| Niraparib Tosylate Monohydrate | 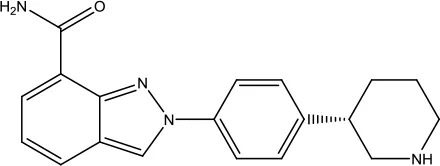 | Recurrent epithelial ovarian, fallopian tube and primary peritoneal cancer | Poly (ADP-ribose) polymerase (PARP) inhibitor | 2017 (Mittica et al., 2018) |
| Ribociclib | 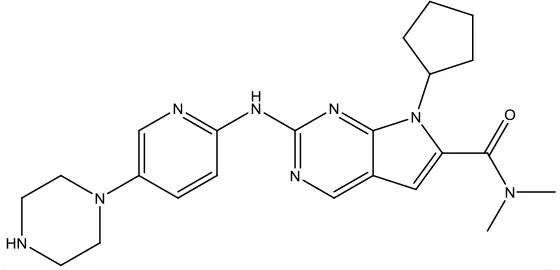 | Breast cancer | Cyclin-dependent kinase (CDK) inhibitor | 2017 (Syed, 2017) |
The list of FDA-approved anticancer drugs in recent 3 years from the National Cancer Institute database.
We further manually screened the database to remove drugs that do not directly target cancer. Drugs for ameliorating conditions related to cancer or limiting side effects of cancer therapies are not listed in this short list. We then identified the FDA label of the drugs in the shortlist by searching in the U.S. National Library of Medicine database “DailyMed”. The FDA approval date, drug function, and therapeutic area are retrieved from DailyMed database.
Conclusion and Perspective
Cancer has become a tangible threat to human health. About 9.6 million people are estimated to die from the various forms of cancer each year, according to a statistic report (Collaborators, 2019). Cancer has become the second-largest disease that causes human death (Reimann et al., 2020). However, developing a new drug molecule costs 12 years and 2.7 billion USD on average (Hauser et al., 2017). The drug development for cancer even becomes more complicated, especially considering the molecular pharmacology is still not well understood. Hence, the discovery and development of new drugs is considered very expensive and time-consuming. In this respect, computational methods could be constructive for performing different tasks including protein-interaction network analysis, drug-target prediction, binding site prediction, virtual screening, and many others. All these innovative methods could considerably facilitate the anti-cancer drug discovery. In recent years, with the advance of AI, more sophisticated methods, such as retro-synthetic routine plan, drug scaffold generation, drug binding affinity predictions, were developed. The useful predictions generated by computational models combined with experimental validations could further speed up the anti-cancer drug development.
Funding
This work was supported by the internal funding of Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences.
Statements
Author contributions
SY designed the whole review. WC directed the completion of the review. AA, SW, QY, and YL were supportive during the review.
Conflict of interest
SY is the cofounder of AlphaMol Science Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Al-SalamaZ. T.KeamS. J. (2019). Entrectinib: first global approval. Drugs79, 1477–1483. doi: 10.1007/s40265-019-01177-y
2
Al-SalamaZ. T. (2019). Apalutamide: A Review in Non-Metastatic Castration-Resistant Prostate Cancer. Drugs79, 1591–1598. doi: 10.1007/s40265-019-01194-x
3
AnthonyC. S.MasuyerG.SturrockE. D.AcharyaK. R. (2012). Structure Based Drug Design of Angiotensin-I Converting Enzyme Inhibitors. Curr. Med. Chem.19, 845–855. doi: 10.2174/092986712799034950
4
AtkinsM. B.PlimackE. R.PuzanovI.FishmanM. N.McdermottD. F.ChoD. C.et al. (2018). Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol.19, 405–415. doi: 10.1016/S1470-2045(18)30081-0
5
BlairH. A. (2018). Duvelisib: First global approval. Drugs78, 1847–1853. doi: 10.1007/s40265-018-1013-4
6
BohmH. J. (1992). The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J. Comp. Aided Mol. Design6, 61–78. doi: 10.1007/BF00124387
7
BologaC. G.RevankarC. M.YoungS. M.EdwardsB. S.ArterburnJ. B.KiselyovA. S.et al. (2006). Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol.2, 207–212. doi: 10.1038/nchembio775
8
BornJ.ManicaM.OskooeiA.Rodriguez MartínezM. (2019). PaccMannRL: Designing anticancer drugs from transcriptomic data via reinforcement learning.New York: Cornell University Press.
9
BottaroD. P.RubinJ. S.FalettoD. L.ChanA. M.KmiecikT. E.Vande WoudeG. F.et al. (1991). Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Sci. (New York N.Y.)251, 802–804. doi: 10.1126/science.1846706
10
BrayF.FerlayJ.SoerjomataramI.SiegelR. L.TorreL. A.JemalA. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-a Cancer J. Clin.68, 394–424. doi: 10.3322/caac.21492
11
BrysonH. M.SorkinE. M. (1993). Cladribine—A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in hematological malignancies. Drugs46, 872–894. doi: 10.2165/00003495-199346050-00007
12
BuckleD. R.ErhardtP. W.GanellinC. R.KobayashiT.PerunT. J.ProudfootJ.et al. (2013). Glossary of terms used in medicinal chemistry. Part II (IUPAC Recommendations 2013). Pure Appl. Chem.85, 1725–1758. doi: 10.1351/PAC-REC-12-11-23
13
ButrynskiJ. E.D'adamoD. R.HornickJ. L.Dal CinP.AntonescuC. R.JhanwarS. C.et al. (2010). Crizotinib in ALK-Rearranged Inflammatory Myofibroblastic Tumor. New Engl. J. Med.363, 1727–1733. doi: 10.1056/NEJMoa1007056
14
CampillosM.KuhnM.GavinA.-C.JensenL. J.BorkP. (2008). Drug target identification using side-effect similarity. Science321, 263–266. doi: 10.1126/science.1158140
15
ChanH. C. S.WangJ.PalczewskiK.FilipekS.VogelH.LiuZ.-J.et al. (2018). Exploring a new ligand binding site of G proteincoupled receptors. Chem. Sci.9, 11. doi: 10.1039/C8SC01680A
16
ChanH. C. S.ShanH.DahounT.VogelH.YuanS. (2019). Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci.40, 592–604. doi: 10.1016/j.tips.2019.06.004
17
ChanH. C. S.XuY.TanL.VogelH.ChengJ.WuD.et al. (2020). Enhancing the Signaling of GPCRs via Orthosteric Ions. ACS Cent. Sci.6, 274–282. doi: 10.1021/acscentsci.9b01247
18
ChaoW.-R.YeanD.AminK.GreenC.JongL. (2007). Computer-aided rational drug design: A novel agent (SR13668) designed to mimic the unique anticancer mechanisms of dietary indole-3-carbinol to block akt signaling. J. Med. Chem.50, 3412–3415. doi: 10.1021/jm070040e
19
ChenJ.LaiL. (2006). Pocket v.2: Further developments on receptor-based pharmacophore modeling. J. Chem. Inf. Model.46, 2684–2691. doi: 10.1021/ci600246s
20
ChenX.LiuM.-X.YanG.-Y. (2012). Drug-target interaction prediction by random walk on the heterogeneous network. Mol. Biosyst.8, 1970–1978. doi: 10.1039/c2mb00002d
21
ChenX.YanC. C.ZhangX.ZhangX.DaiF.YinJ.et al. (2016). Drug-target interaction prediction: databases, web servers and computational models. Briefings Bioinf.17, 696–712. doi: 10.1093/bib/bbv066
22
CheungK. M. J.MatthewsT. P.JamesK.RowlandsM. G.BoxallK. J.SharpS. Y.et al. (2005). The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett.15, 3338–3343. doi: 10.1016/j.bmcl.2005.05.046
23
ChiangY. K.KuoC. C.WuY. S.ChenC. T.CoumarM. S.WuJ. S.et al. (2009). Generation of Ligand-Based Pharmacophore Model and Virtual Screening for Identification of Novel Tubulin Inhibitors with Potent Anticancer Activity. J. Med. Chem.52, 4221–4233. doi: 10.1021/jm801649y
24
ChristensenJ. G.BurrowsJ.SalgiaR. (2005). c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett.225, 1–26. doi: 10.1016/j.canlet.2004.09.044
25
Collaborators, G.B.D.S (2019). Global, regional, and national burden of stroke 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol.18, 439–458. doi: 10.1016/s1474-4422(19)30034-1
26
ConnP. J.ChristopoulosA.LindsleyC. W. (2009). Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery8, 41–54. doi: 10.1038/nrd2760
27
CramerR. D.PattersonD. E.BunceJ. D. (1988). Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc.110, 5959–5967. doi: 10.1021/ja00226a005
28
CuiJ. J.Tran-DubeM.ShenH.NambuM.KungP.-P.PairishM.et al. (2011). Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal-Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem.54, 6342–6363. doi: 10.1021/jm2007613
29
CuiJ. J.MctigueM.KaniaR.EdwardsM. (2013). Case History: XalkoriTM (Crizotinib), a Potent and Selective Dual Inhibitor of Mesenchymal Epithelial Transition (MET) and Anaplastic Lymphoma Kinase (ALK) for Cancer Treatment. Annu. Rep. Med. Chem.48, 421–434. doi: 10.1016/b978-0-12-417150-3.00025-9
30
CuzzolinA.SturleseM.DeganuttiG.SalmasoV.SabbadinD.CiancettaA.et al. (2016). Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model.56, 687–705. doi: 10.1021/acs.jcim.5b00702
31
DebnathS.KanakarajuM.IslamM.YeeravalliR.SenD.DasA. (2019). In silico design, synthesis and activity of potential drug-like chrysin scaffoldd-erived selective EGFR inhibitors as anticancer agents. Comput. Biol. Chem.83. doi: 10.1016/j.compbiolchem.2019.107156
32
DeganuttiG.CuzzolinA.CiancettaA.MoroS. (2015). Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A(3) adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem.23, 4065–4071. doi: 10.1016/j.bmc.2015.03.039
33
DevreeB. T.MahoneyJ. P.Velez-RuizG. A.RasmussenS. G. F.KuszakA. J.EdwaldE.et al. (2016). Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature535, 182–18+. doi: 10.1038/nature18324
34
DhillonS. (2018). Ivosidenib: First Global Approval. Drugs78, 1509–1516. doi: 10.1007/s40265-018-0978-3
35
DhillonS. (2019). Gilteritinib: First Global Approval. Drugs79, 331–339. doi: 10.1007/s40265-019-1062-3
36
DiasR.De AzevedoW. F.Jr. (2008). Molecular Docking Algorithms. Curr. Drug Targets9, 1040–1047. doi: 10.2174/138945008786949432
37
DrewsJ. (2000). Drug discovery: a historical perspective. Sci. (New York N.Y.)287, 1960–1964. doi: 10.1126/science.287.5460.1960
38
EskilerG. G. (2019). Talazoparib to treat BRCA-positive breast cancer. Drugs Today55, 459–467. doi: 10.1358/dot.2019.55.7.3015642
39
FelipE.BarlesiF.BesseB.ChuQ.GandhiL.KimS.-W.et al. (2018). Phase 2 Study of the HSP-90 Inhibitor AUY922 in Previously Treated and Molecularly Defined Patients with Advanced Non-Small Cell Lung Cancer. J. Thoracic Oncol.13, 576–584. doi: 10.1016/j.jtho.2017.11.131
40
FerreiraL. G.Dos SantosR. N.OlivaG.AndricopuloA. D. (2015). Molecular Docking and Structure-Based Drug Design Strategies. Molecules20, 13384–13421. doi: 10.3390/molecules200713384
41
FlockT.RavaraniC. N. J.SunD.VenkatakrishnanA. J.KayikciM.TateC. G.et al. (2015). Universal allosteric mechanism for G alpha activation by GPCRs. Nature524, 173–17+. doi: 10.1038/nature14663
42
GajdosikZ. (2017). Larotrectinib sulfate. Drugs Future42, 275–280. doi: 10.1358/dof.2017.042.05.2623108
43
Garcia-CarboneroR.CarneroA.Paz-AresL. (2013). Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol.14, E358–E369. doi: 10.1016/S1470-2045(13)70169-4
44
GhofraniH. A.OsterlohI. H.GrimmingerF. (2006). Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discovery5, 689–702. doi: 10.1038/nrd2030
45
GiammarileF.ChitiA.LassmannM.BransB.FluxG. (2008). EANM procedure guidelines for I-131-meta-iodobenzylguanidine (I-131-mIBG) therapy. Eur. J. Nuclear Med. Mol. Imaging35, 1039–1047. doi: 10.1007/s00259-008-0715-3
46
Gomez-BombarelliR.WeiJ. N.DuvenaudD.Hernandez-LobatoJ. M.Sanchez-LengelingB.SheberlaD.et al. (2018). Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci.4, 268–276. doi: 10.1021/acscentsci.7b00572
47
GrasJ. (2017). Enasidenib mesylate. Drugs Future42, 15–20. doi: 10.1358/dof.2017.042.01.2579894
48
GrunwaldV.HidalgoM. (2003). Development of the epidermal growth factor receptor inhibitor Tarceva (TM) (OSI-774). New Trends in Cancer for the 21st Century.235–246. doi: 10.1007/978-1-4615-0081-0_19
49
HalperinI.MaB.WolfsonH.NussinovR. (2002). Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins47, 409–443. doi: 10.1002/prot.10115
50
HancockJ. F. (2003). Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol.4, 373–384. doi: 10.1038/nrm1105
51
HanschC.FujitaT. (1964). Additions and Corrections-ρ-σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc.86, 5710–5710. doi: 10.1021/ja01078a623
52
HauserA. S.AttwoodM. M.Rask-AndersenM.SchiothH. B.GloriamD. E. (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery16, 829–842. doi: 10.1038/nrd.2017.178
53
HellerJ.R. (1951). National Canaer Institute. JAMA-J. Am. Med. Assoc.146, 1248–1248. doi: 10.1001/jama.1951.03670130070024
54
HongJ. Y.PriceI. R.BaiJ. J.LieH. (2019). A Glycoconjugated SIRT2 Inhibitor with Aqueous Solubility Allows Structure-Based Design of SIRT2 Inhibitors. ACS Chem. Biol.14, 1802–1810. doi: 10.1021/acschembio.9b00384
55
HopkinsA. L. (2008). Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol.4, 682–690. doi: 10.1038/nchembio.118
56
HuG.KuangG.XiaoW.LiW.LiuG.TangY. (2012). Performance Evaluation of 2D Fingerprint and 3D Shape Similarity Methods in Virtual Screening. J. Chem. Inf. Model.52, 1103–1113. doi: 10.1021/ci300030u
57
HuangS.-Y.GrinterS. Z.ZouX. (2010). Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys. Chem. Chem. Phys.12, 12899–12908. doi: 10.1039/c0cp00151a
58
ItohY. (2020). Drug Discovery Researches on Modulators of Lysine-Modifying Enzymes Based on Strategic Chemistry Approaches. Chem. Pharmaceut. Bull.68, 34–45. doi: 10.1248/cpb.c19-00741
59
JarmanM.BarrieS. E.LleraJ. M. (1998). The 16,17-double bond is needed for irreversible inhibition of human cytochrome P450(17 alpha) by abiraterone (17-(3-pyridyl)androsta-5,16-dien-3 beta-ol) and related steroidal inhibitors. J. Med. Chem.41, 5375–5381. doi: 10.1021/jm981017j
60
JorgeS. E.Lucena-AraujoA. R.YasudaH.PiotrowskaZ.OxnardG. R.RangachariD.et al. (2018). EGFR Exon 20 Insertion Mutations Display Sensitivity to Hsp90 Inhibition in Preclinical Models and Lung Adenocarcinomas. Clin. Cancer Res.24, 6548–6555. doi: 10.1158/1078-0432.CCR-18-1541
61
KaldorS. W.KalishV. J.DaviesJ. F.ShettyB. V.FritzJ. E.AppeltK.et al. (1997). Viracept (nelfinavir mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem.40, 3979–3985. doi: 10.1021/jm9704098
62
KaniaR. S.BenderS. L.BorchardtA. J.CrippsS. J.HuaY.JohnsonM. D.et al. (2016) Indazole compounds and pharmaceutical compositions for Inhibiting protein kinases, and methods for their use [Accessed November 28, 2006].
63
KaniaR. (2009). “Structure-Based Design and Characterization of Axitinib. Kinase Inhibitor Drugs, Drug Discovery and Development. Eds. LiR.StaffordJ. A. (New York: Wiley).
64
KapetanovicI. M. (2008). Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact.171, 165–176. doi: 10.1016/j.cbi.2006.12.006
65
KimE. S. (2017a). Midostaurin: First Global Approval. Drugs77, 1251–1259. doi: 10.1007/s40265-017-0779-0
66
KimE. S. (2017b). Abemaciclib: first global approval. Drugs77, 2063–2070. doi: 10.1007/s40265-017-0840-z
67
KlebeG.AbrahamU.MietznerT. (1994). Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem.37, 4130–4146. doi: 10.1021/jm00050a010
68
KlippE.WadeR. C.KummerU. (2010). Biochemical network-based drug-target prediction. Curr. Opin. Biotechnol.21, 511–516. doi: 10.1016/j.copbio.2010.05.004
69
KortagereS.EkinsS. (2010). Troubleshooting computational methods in drug discovery. J. Pharmacol. Toxicol. Methods61, 67–75. doi: 10.1016/j.vascn.2010.02.005
70
KoteckiN.GombosA.AwadaA. (2019). Adjuvant therapeutic approaches of HER2-positive breast cancer with a focus on neratinib maleate. Expert Rev. Anticancer Ther.19, 447–454. doi: 10.1080/14737140.2019.1613892
71
KungP. P.JonesR. A.RichardsonP. (2015). Crizotinib (Xalkori): The First-in-Class ALK/ROS Inhibitor for Non-small Cell Lung Cancer (Inc: John Wiley & Sons).
72
LazoJ. S.SharlowE. R. (2016). Drugging Undruggable Molecular Cancer Targets.. Annu. Rev. Phar. Toxicol.56, 23–40. doi: 10.1146/annurev-pharmtox-010715-103440
73
LennerzJ. K.KwakE. L.AckermanA.MichaelM.FoxS. B.BergethonK.et al. (2011). MET Amplification Identifies a Small and Aggressive Subgroup of Esophagogastric Adenocarcinoma With Evidence of Responsiveness to Crizotinib. J. Clin. Oncol.29, 4803–4810. doi: 10.1200/JCO.2011.35.4928
74
LiX.ChenH. C. (2013). Recommendation as link prediction in bipartite graphs: A graph kernel-based machine learning approach. Decision Support Syst.54, 880–890. doi: 10.1016/j.dss.2012.09.019
75
LiuX.YaoW.NewtonR. C.ScherleP. A. (2008). Targeting the c-MET signaling pathway for cancer therapy. Expert Opin. Investigational Drugs17, 997–1011. doi: 10.1517/13543784.17.7.997
76
LuP.BevanD. R.LeberA.HontecillasR.Tubau-JuniN.Bassaganya-RieraJ. (2018a). “Computer-aided drug discovery,” in Accelerated Path to Cures (Springer), 7–24. doi: 10.1007/978-3-319-73238-1_2
77
LuX.YangH.ChenY.LiQ.HeS.-Y.JiangX.et al. (2018b). The Development of Pharmacophore Modeling: Generation and Recent Applications in Drug Discovery. Curr. Pharmaceut. Design24, 3424–3439. doi: 10.2174/1381612824666180810162944
78
MarkhamA.DhillonS. (2018). Acalabrutinib: First Global Approval. Drugs78, 139–145. doi: 10.1007/s40265-017-0852-8
79
MarkhamA.DugganS. (2019). Darolutamide: First Approval. Drugs79, 1813–1818. doi: 10.1007/s40265-019-01212-y
80
MarkhamA. (2017a). Brigatinib: First Global Approval. Drugs77, 1131–1135. doi: 10.1007/s40265-017-0776-3
81
MarkhamA. (2017b). Copanlisib: First Global Approval. Drugs77, 2057–2062. doi: 10.1007/s40265-017-0838-6
82
MarkhamA. (2019a). Alpelisib: first global approval. Drugs79, 1249–1253. doi: 10.1007/s40265-019-01161-6
83
MarkhamA. (2019b). Erdafitinib: First Global Approval79, 1017–1021. doi: 10.1007/s40265-019-01142-9
84
MathiP.PrasadM. V.BotlaguntaM.RaviM.RamachandranD. (2018). De novo design of selective Sortase-A inhibitors: Synthesis, structural and in vitro characterization. Chem. Data Collect.15, 126–133. doi: 10.1016/j.cdc.2018.04.007
85
MayrA.KlambauerG.UnterthinerT.SteijaertM.WegnerJ. K.CeulemansH.et al. (2018). Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci.9, 5441–5451. doi: 10.1039/C8SC00148K
86
MazmanianS. K.LiuG.Ton-ThatH.SchneewindO. (1999). Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Sci. (New York N.Y.)285, 760–763. doi: 10.1126/science.285.5428.760
87
MeadowsK. L.HurwitzH. I. (2012). Anti-VEGF Therapies in the Clinic. Cold Spring Harbor Perspect. Med.2, 27. doi: 10.1101/cshperspect.a006577
88
MendenhallJ.MeilerJ. (2016). Improving quantitative structure-activity relationship models using Artificial Neural Networks trained with dropout. J. Comp. Aided Mol. Design30, 177–189. doi: 10.1007/s10822-016-9895-2
89
MendozaJ. L.EscalanteN. K.JudeK. M.BellonJ. S.SuL.HortonT. M.et al. (2019). Structure of the IFN gamma receptor complex guides design of biased agonists. Nature567, 56–5+. doi: 10.1038/s41586-019-0988-7
90
MitticaG.GhisoniE.GiannoneG.GentaS.AgliettaM.SapinoA.et al (2018). PARP Inhibitors in Ovarian Cancer. Recent Pat. Anticancer Drug Discov.13, 392–410. doi: 10.2174/1574892813666180305165256
91
MuhsinM.GrahamJ.KirkpatrickP. (2003). Fresh from the pipeline - Gefitinib. Nat. Rev. Drug Discovery2, 515–516. doi: 10.1038/nrd1136
92
OrtusoF.LangerT.AlcaroS. (2006). GBPM: GRID-based pharmacophore model: concept and application studies to protein-protein recognition. Bioinformatics22, 1449–1455. doi: 10.1093/bioinformatics/btl115
93
PearlL. H.ProdromouC. (2006). Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem.75, 271–294. doi: 10.1146/annurev.biochem.75.103004.142738
94
PiotrowskaZ.CostaD. B.OxnardG. R.HubermanM.GainorJ. F.LennesI. T.et al. (2018). Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions. Ann. Oncol.29, 2092–2097. doi: 10.1093/annonc/mdy336
95
PirhadiS.ShiriF.GhasemiJ. B. (2013). Methods and applications of structure based pharmacophores in drug discovery. Curr. Top. Med. Chem.13, 1036–1047. doi: 10.2174/1568026611313090006
96
Prada-GraciaD.Huerta-YepezS.Moreno-VargasL. M. (2016). Application of computational methods for anticancer drug discovery, design, and optimization. Boletin Med. Del Hosp. Infantil. Mexico73, 411–423. doi: 10.1016/j.bmhimx.2016.10.006
97
ReimannZ.MillerJ. R.DahleK. M.HooperA. P.YoungA. M.GoatesM. C.et al. (2020). Executive functions and health behaviors associated with the leading causes of death in the United States: A systematic review. J. Health Psychol.25, 186–196. doi: 10.1177/1359105318800829
98
RekerD.RodriguesT.SchneiderP.SchneiderG. (2014). Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc. Natl. Acad. Sci. United States America111, 4067–4072. doi: 10.1073/pnas.1320001111
99
RodriguesT.WernerM.RothJ.Da CruzE. H. G.MarquesM. C.AkkapeddiP.et al. (2018). Machine intelligence decrypts -lapachone as an allosteric 5-lipoxygenase inhibitor. Chem. Sci.9, 6. doi: 10.1039/c8sc02634c.
100
RongB.YangS. (2018). Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: New discoveries and developments (Review). Int. J. Oncol.52, 321–336. doi: 10.3892/ijo.2017.4214
101
RushT. S.GrantJ. A.MosyakL.NichollsA. (2005). A Shape-Based 3-D Scaffold Hopping Method and Its Application to a Bacterial Protein?Protein Interaction. J. Med. Chem.48, 1489–1495. doi: 10.1021/jm040163o
102
SabbadinD.MoroS. (2014). Supervised Molecular Dynamics (SuMD) as a Helpful Tool To Depict GPCR-Ligand Recognition Pathway in a Nanosecond Time Scale. J. Chem. Inf. Model.54, 372–376. doi: 10.1021/ci400766b
103
SalmasoV.MoroS. (2018). Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol.9. doi: 10.3389/fphar.2018.00923
104
SchwabR.PetakI.KollarM.PinterF.VarkondiE.KohankaA.et al. (2014). Major partial response to crizotinib, a dual MET/ALK inhibitor, in a squamous cell lung (SCC) carcinoma patient with de novo c-MET amplification in the absence of ALK rearrangement. Lung Cancer83, 109–111. doi: 10.1016/j.lungcan.2013.10.006
105
ShaikN.HeeB.WeiH.LabadieR. R. (2019). Evaluation of the effects of formulation, food, or a proton-pump inhibitor on the pharmacokinetics of glasdegib (PF-04449913) in healthy volunteers: a randomized phase I study. Cancer Chemother. Pharmacol.83, 463–472. doi: 10.1007/s00280-018-3748-8
106
SharpS.WorkmanP. (2006). Inhibitors of the HSP90 molecular chaperone: Current status. Adv. Cancer Res.95. doi: 10.1016/s0065-230x(06)95009-x
107
SharpS. Y.BoxallK.RowlandsM.ProdromouC.RoeS. M.MaloneyA.et al. (2007). In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res.67, 2206–2216. doi: 10.1158/0008-5472.CAN-06-3473
108
ShirleyM. (2018). Encorafenib and Binimetinib: First Global Approvals. Drugs78, 1277–1284. doi: 10.1007/s40265-018-0963-x
109
SidawayP. (2018). Cemiplimab effective in cutaneous SCC. Nat. Rev. Clin. Oncol.15, 472–472. doi: 10.1038/s41571-018-0056-5
110
SmithN. F.HayesA.JamesK.NutleyB. P.McdonaldE.HenleyA.et al. (2006). Preclinical pharmacokinetics and metabolism of a novel diaryl pyrazole resorcinol series of heat shock protein 90 inhibitors. Mol. Cancer Ther.5, 1628–1637. doi: 10.1158/1535-7163.MCT-06-0041
111
SrivastavaN.HintonG.KrizhevskyA.SutskeverI.SalakhutdinovR. (2014). Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res.15, 1929–1958. doi: 10.5555/2627435.2670313
112
SuY.LongX.SongY.ChenP.LiS.YangH.et al. (2019). Distribution of ALK Fusion Variants and Correlation with Clinical Outcomes in Chinese Patients with Non-Small Cell Lung Cancer Treated with Crizotinib. Targeted Oncol.14, 159–168. doi: 10.1007/s11523-019-00631-x
113
SyedY. Y. (2017). Ribociclib: First Global Approval. Drugs77, 799–807. doi: 10.1007/s40265-017-0742-0
114
SyedY. Y. (2019). Selinexor: First Global Approval. Drugs79, 1485–1494. doi: 10.1007/s40265-019-01188-9
115
SyedY. Y. (2020). Zanubrutinib: First Approval. Drugs.80, 91–97. 1–7. doi: 10.1007/s40265-019-01252-4
116
TakarabeM.KoteraM.NishimuraY.GotoS.YamanishiY. (2012). Drug target prediction using adverse event report systems: a pharmacogenomic approach. Bioinformatics28, I611–I618. doi: 10.1093/bioinformatics/bts413
117
TautermannC. S. (2014). GPCR structures in drug design, emerging opportunities with new structures. Bioorg. Med. Chem. Lett.24, 4073–4079. doi: 10.1016/j.bmcl.2014.07.009
118
TondoA. R.CaputoL.MangiatordiG. F.MonaciL.LentiniG.LogriecoA. F.et al. (2020). Structure-Based Identification and Design of Angiotensin Converting Enzyme-Inhibitory Peptides from Whey Proteins. J. Agric. Food Chem.68, 541–548. doi: 10.1021/acs.jafc.9b06237
119
TongM.SeeligerM. A. (2015). Targeting Conformational Plasticity of Protein Kinases. ACS Chem. Biol.10, 190–200. doi: 10.1021/cb500870a
120
UrwylerS. (2011). Allosteric Modulation of Family C G-Protein-Coupled Receptors: from Molecular Insights to Therapeutic Perspectives. Pharmacol. Rev.63, 59–126. doi: 10.1124/pr.109.002501
121
WangJ. L.LiuD.ZhangZ. J.ShanS.HanX.SrinivasulaS. M.et al. (2000). Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. United States America97, 7124–7129. doi: 10.1073/pnas.97.13.7124
122
WhitesellL.LindquistS. L. (2005). HSP90 and the chaperoning of cancer. Nat. Rev. Cancer5, 761–772. doi: 10.1038/nrc1716
123
WilhelmS.CarterC.LynchM.LowingerT.DumasJ.SmithR. A.et al. (2006). Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discovery5, 835–844. doi: 10.1038/nrd2130
124
WolberG.DornhoferA. A.LangerT. (2006). Efficient overlay of small organic molecules using 3D pharmacophores. J. Comp. Aided Mol. Design20, 773–788. doi: 10.1007/s10822-006-9078-7
125
WoodE. R.TruesdaleA. T.McdonaldO. B.YuanD.HassellA.DickersonS. H.et al. (2004). A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res.64, 6652–6659. doi: 10.1158/0008-5472.CAN-04-1168
126
WorkmanP.BurrowsF.NeckersL.RosenN. (2007). ““Drugging the cancer chaperone HSP90 combinatorial therapeutic exploitation of oncogene addiction and tumor stress,” in Stress Responses in Biology and Medicine: Stress of Life in Molecules, Cells, Organisms, and Psychosocial Communities. Eds. CsermelyP.KorcsmarosT.SulyokK.. 1113, 202–216. doi: 10.1196/annals.1391.012
127
YamanishiY.ArakiM.GutteridgeA.HondaW.KanehisaM. (2008). Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics24, I232–I240. doi: 10.1093/bioinformatics/btn162
128
YanB.YangW. J.HanX. Y.HanL. H. (2019). Crystal structures and antitumor activity evaluation against gastric carcinoma of two novel coordination polymers. Main Group Chem.18, 239–246. doi: 10.3233/MGC-180748
129
YangX.WangY.ByrneR.SchneiderG.YangS. (2019). Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev.119, 10520–10594. doi: 10.1021/acs.chemrev.8b00728
130
YangS.-Y. (2010). Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discovery Today15, 444–450. doi: 10.1016/j.drudis.2010.03.013
131
YildirimM. A.GohK. I.CusickM. E.BarabasiA. L.VidalM. (2007). Drug-target network. Nat. Biotechnol.25, 1119–1126. doi: 10.1038/nbt1338
132
YurievE.HolienJ.RamslandP. A. (2015). Improvements, trends, and new ideas in molecular docking: 2012-2013 in review. J. Mol. Recogn.28, 581–604. doi: 10.1002/jmr.2471
133
ZhangQ.ZhangY.DiamondS.BoerJ.HarrisJ. J.LiY.et al. (2014). The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke's encephalopathy. Drug Metab. Dispos.42, 1656–1662. doi: 10.1124/dmd.114.058883
134
ZhavoronkovA.IvanenkovY. A.AliperA.VeselovM. S.AladinskiyV. A.AladinskayaA. V.et al. (2019). Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol.37, 1038–103+. doi: 10.1038/s41587-019-0224-x
Summary
Keywords
anti-cancer, CADD, drug discovery, AI, computational methods
Citation
Cui W, Aouidate A, Wang S, Yu Q, Li Y and Yuan S (2020) Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 11:733. doi: 10.3389/fphar.2020.00733
Received
12 March 2020
Accepted
01 May 2020
Published
20 May 2020
Volume
11 - 2020
Edited by
Weiwei Xue, Chongqing University, China
Reviewed by
Zhiwei Feng, University of Pittsburgh, United States; Shiliang Li, East China University of Science and Technology, China
Updates
Copyright
© 2020 Cui, Aouidate, Wang, Yu, Li and Yuan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuguang Yuan, shuguang.yuan@siat.ac.cn
This article was submitted to Pharmacology of Anti-Cancer Drugs, a section of the journal Frontiers in Pharmacology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.