 Colleen S. Curran
Colleen S. Curran Donna R. Rivera2
Donna R. Rivera2 Jeffrey B. Kopp
Jeffrey B. Kopp- 1Critical Care Medicine Department, Clinical Center, National Institutes of Health, Bethesda, MD, United States
- 2Surveillance Research Program, Division of Cancer Control and Population Sciences, National Cancer Institute, National Institutes of Health, Rockville, MD, United States
- 3Kidney Disease Section, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, United States
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection causes coronavirus disease 2019 (COVID-19). SARS-CoV-2 binds the angiotensin-converting enzyme 2 (ACE2) on the cell surface and this complex is internalized. ACE2 serves as an endogenous inhibitor of inflammatory signals associated with four major regulator systems: the renin-angiotensin-aldosterone system (RAAS), the complement system, the coagulation cascade, and the kallikrein-kinin system (KKS). Understanding the pathophysiological effects of SARS-CoV-2 on these pathways is needed, particularly given the current lack of proven, effective treatments. The vasoconstrictive, prothrombotic and pro-inflammatory conditions induced by SARS-CoV-2 can be ascribed, at least in part, to the activation of these intersecting physiological networks. Moreover, patients with immune deficiencies, hypertension, diabetes, coronary heart disease, and kidney disease often have altered activation of these pathways, either due to underlying disease or to medications, and may be more susceptible to SARS-CoV-2 infection. Certain characteristic COVID-associated skin, sensory, and central nervous system manifestations may also be linked to viral activation of the RAAS, complement, coagulation, and KKS pathways. Pharmacological interventions that target molecules along these pathways may be useful in mitigating symptoms and preventing organ or tissue damage. While effective anti-viral therapies are critically needed, further study of these pathways may identify effective adjunctive treatments and patients most likely to benefit.
Introduction
The first cases of coronavirus disease 2019 (COVID-19), the infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), appeared in China in late 2019 (Chen Z. L. et al., 2020). Although SARS-CoV-2 likely originated from an animal host in China (Zhang T. et al., 2020), regular and seasonal travel patterns of individuals near the source allowed the virus to spread globally, prompting a pandemic declaration by the World Health Organization in March 2020 (Chen Z. L. et al., 2020; Sohrabi et al., 2020).
Of the four genera of coronaviruses (Alphacoronavirus, Betacoronavirus, Gammacoronavirus, Deltacoronavirus), three affect mammals (Alphacoronavirus, Betacoronavirus, Deltacoronavirus), and within the first two genera, seven known human coronaviruses (HCoV) are identified (Alphacoronavirus: HCoV-NL63, HCoV-229E; Betacoronavirus: SARS-CoV, SARS-CoV-2, MERS-CoV, HCoV-OC43, HCoV-HKU1) (Dominguez et al., 2014). Coronavirus genomes are continuous, single-stranded, non-segmented, positive-sense RNA molecules that contain a 5’ cap and a 3’ poly-A tail for efficient translation by eukaryotic host ribosomes. The four main coronavirus structural proteins are spike (S)-protein, membrane (M)-protein, envelope (E)-protein, and nucleocapsid-(N)-protein, while a fifth protein called hemagglutinin-esterase (HE)-protein is present in a subset of Betacoronaviruses (Fehr and Perlman, 2015) (Figure 1).
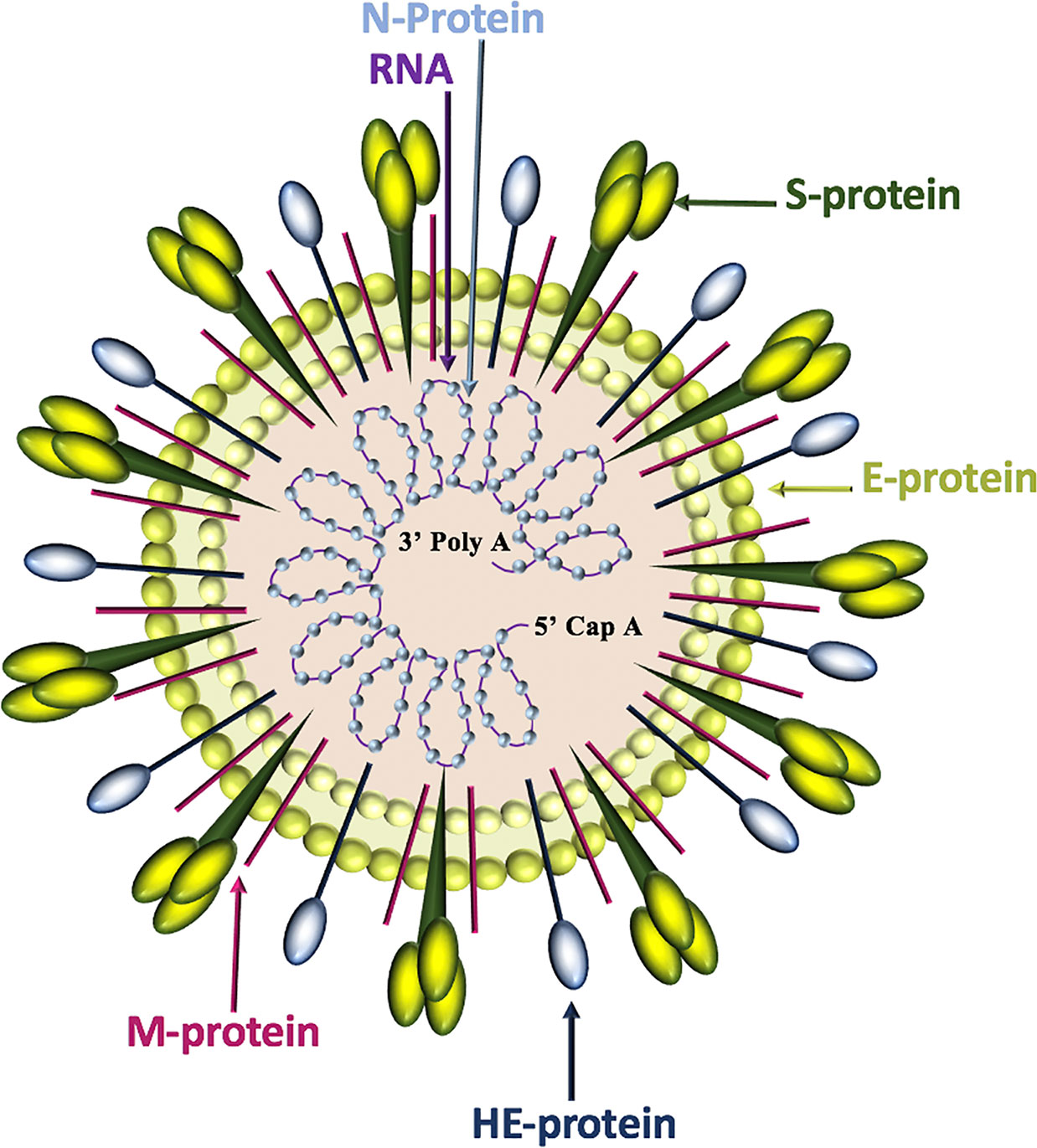
Figure 1 Coronavirus structure. Coronaviruses contain a trimeric spike (S)-protein that mediates attachment to the host receptor, abundant membrane (M)-proteins, envelope (E)-proteins that facilitate assembly and release of the virus, nucleocapsid (N)-proteins that bind positive-strand RNA, and hemagglutinin-esterase (HE)-proteins that bind sialic acids on surface glycoproteins and manifest acetyl-esterase activity.
Similar to the human coronaviruses HCoV-NL63 and SARS-CoV, SARS-CoV-2 binds angiotensin-converting enzyme 2 (ACE2) on host cell plasma membranes (Wu et al., 2009; Hoffmann et al., 2020). SARS-CoV-2, and all other human coronaviruses, can also use transmembrane protease, serine 2 (TMPRSS2, epitheliasin) (Milewska et al., 2018; Shirato et al., 2018; Widagdo et al., 2019; Hoffmann et al., 2020), which cleaves ACE2 and activates the S-protein for membrane fusion and viral internalization (Glowacka et al., 2011; Heurich et al., 2014) (Figure 2). The extensive sequence homology between SARS-CoV-2 and SARS-CoV (Grifoni et al., 2020) allows for informative comparisons in evaluating SARS-Cov-2 pathogenesis and possible therapeutic treatment targets for COVID-19 patients.
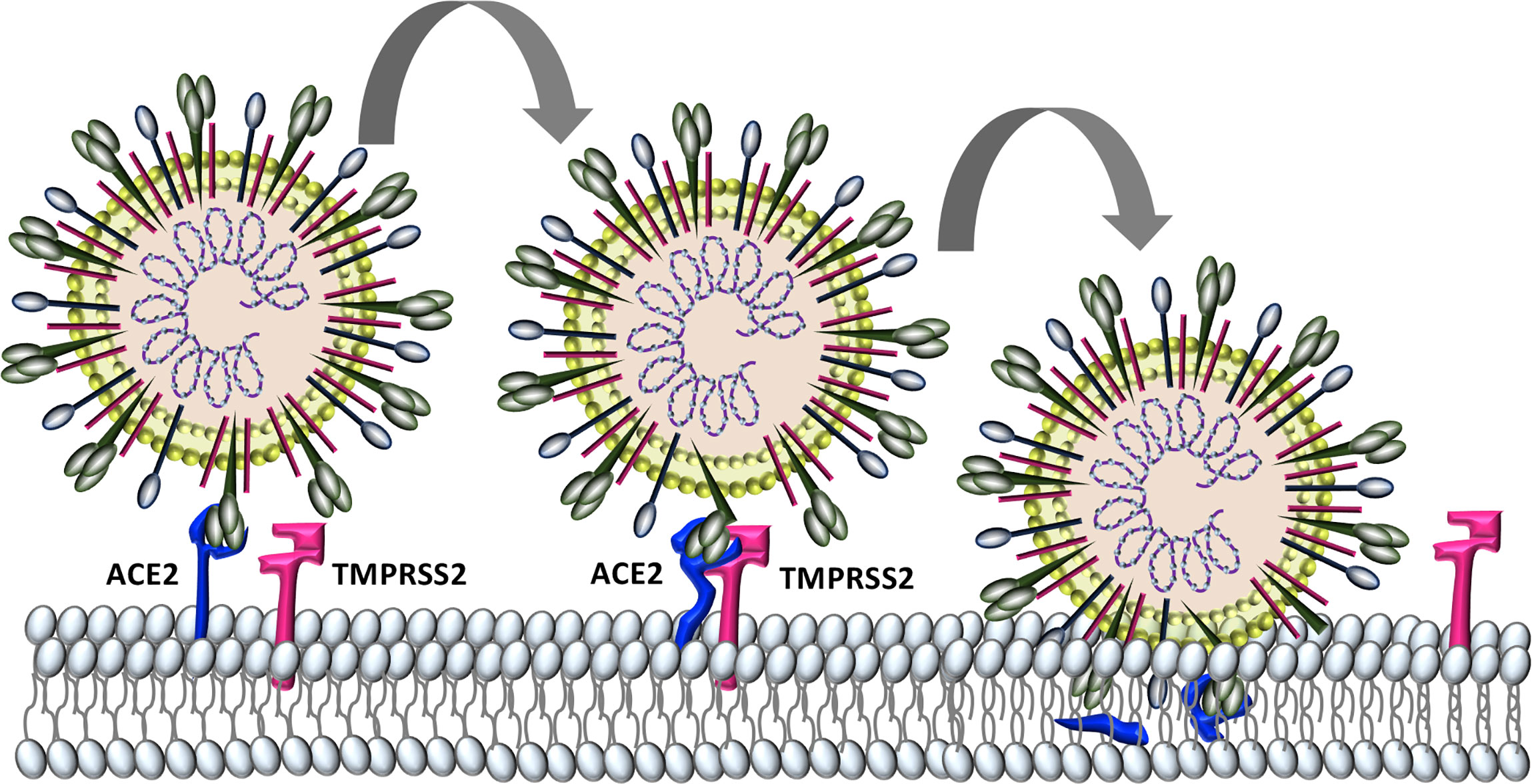
Figure 2 Characterized SARS-CoV-2 entry. SARS-CoV-2 binds to host cell angiotensin converting enzyme-2 (ACE2). The protease TMPRSS2 is recruited and cleaves ACE2 and activates the S protein for membrane fusion and viral entry.
COVID-19 patients may be asymptomatic, have mild symptoms, or present with a febrile pneumonia. Three stages have been described: early infection, pulmonary involvement, and systemic hyperinflammation, sometimes with sepsis (Guan et al., 2020; Huang et al., 2020; Siddiqi and Mehra, 2020). The early phases are characterized by variable combinations of fever, fatigue, dry cough, shortness of breath, headache, pharyngitis, rhinorrhea, hemoptysis, nausea, vomiting, abdominal pain, and diarrhea (Guan et al., 2020; Wang D. et al., 2020). Some patients exhibit cutaneous manifestations (Galvan Casas et al., 2020) or a disruption in smell or taste (Sedaghat et al., 2020). Common clinical findings at admission include ground-glass opacity and bilateral patchy shadowing on chest imaging, lymphocytopenia, neutrophilia, and elevated levels of C-reactive protein (CRP) (Chen G. et al., 2020; Chen N. et al., 2020; Guan et al., 2020). Disease progression is most common in patients with hypertension, diabetes, coronary heart disease and immune deficiencies and these populations are also at increased risk of multiple organ failure and death (Kuderer et al., 2020; Zhou et al., 2020). Greater disease severity is associated with higher levels of alanine aminotransferase, interleukin (IL)-6, CRP, tumor necrosis factor (TNF), and fibrin degradation products (D-dimers) as well as markedly lower levels of blood lymphocytes (Chen G. et al., 2020). Multi-system involvement can include cardiovascular, pulmonary, hepatic, and renal effects, as well as COVID-19 manifesting coagulopathies, shock and multiple organ failure.
Thus, COVID-19 pathogenesis is complex and while intensive investigations have been made, there is undoubtedly much still to be learned in this is rapidly developing field. Here we focus on SARS-CoV-2 pathophysiology in the renin-angiotensin-aldosterone system (RAAS), the complement pathway, the coagulation pathway, and the kallikrein-kinin system that are each impacted by the primary SARS-CoV-2 receptor, ACE2. Dysfunction in these pathways is linked to three systemic manifestations that occur in many patients with COVID-19. First, patients are at an increased risk for thromboembolism and systemic vasculitis. Second, patients often have low platelet counts, elevated D-dimer levels, prolonged pro-thrombin time and may develop disseminated intravascular coagulation. Third, COVID-19 patients may develop localized pulmonary angioedema, manifesting as fever, dry cough, dyspnea, and in some cases respiratory failure and/or a systemic cytokine storm (Figure 3).
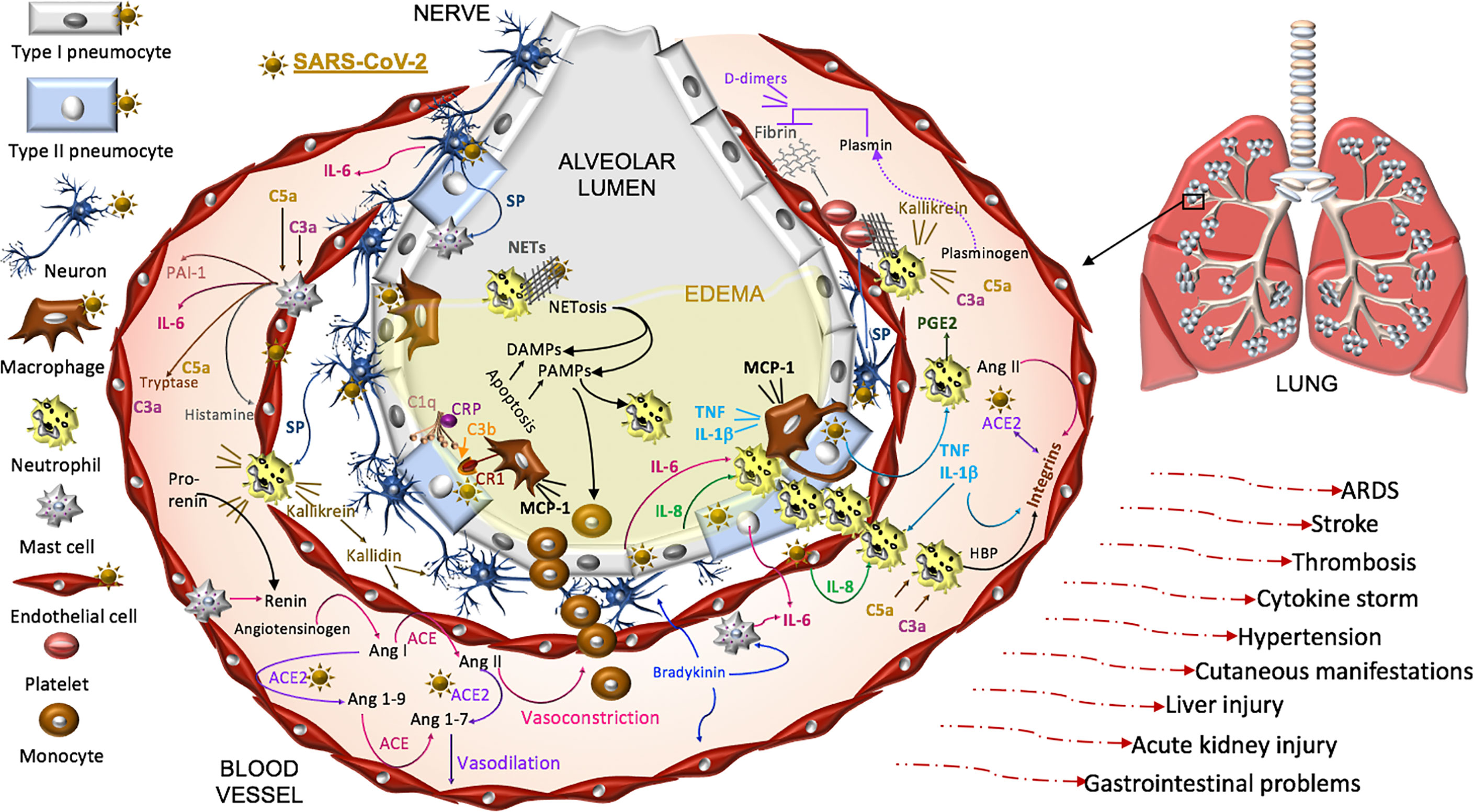
Figure 3 Proposed model of SARS-CoV-2 pathogenesis. SARS-CoV-2 binds ACE2 on alveolar type I and type II cells, macrophages, neurons, and arterial and venous endothelial cells. Complement, damage-and pathogen-associated molecular pattern ligands (DAMPs, PAMPs), and cytokines prime circulating neutrophils that are recruited in response to IL-8 and IL-6 released from infected cells and mast cells. SARS-CoV-2-induces epithelial apoptosis. Alveolar macrophages remove apoptotic cells through complement independent and dependent mechanisms. Apoptotic cells generate antigens that bind C1q and the interaction may be enhanced by C reactive protein (CRP). The activation of C1q induces C3b deposition for macrophage complement receptor 1 (CR1) binding and phagocytosis. Activated neutrophils produce heparin binding protein (HBP), prostaglandins (PGE2), and extracellular traps (NETs) to capture and kill the virus. NET activity induces a form of cell death called NETosis. Both NETosis and apoptosis generate DAMPs and PAMPs that bind and activate toll-like receptors in promoting inflammation. Mast cells and neutrophils are activated by complement factors and the neuropeptide, substance P (SP), which promotes their degranulation. Mast cells also produce histamine that promotes vasodilation, tryptase involved in complement factor production, and renin in the RAAS. Excessive inflammation, associated with MCP-1-recruited monocytes, promotes the accumulation of fluid, leading to alveolar edema. Platelets are activated by SP and exhibit cross-talk with neutrophil NETs in promoting coagulation. The kallikrein-kinin system is activated by damaged tissue and cells, such as neutrophils. Kallikrein functions as a precursor to bradykinin, activates pro-renin, and cleaves complement C3 and C5 as well as plasminogen. The latter generates plasmin involved in the degradation of fibrin and the formation of D-dimers identified in COVID-19 patient serum. SARS-CoV-2-induced degradation of ACE2 promotes RAAS activity, vasoconstriction, and hypertension. Angiotensin II, various cytokines, and HBP induce the expression of endothelial integrins. In the absence of ACE2, which also binds integrins, the functions of integrins may be dysregulated, promoting inflammation, hypertension, and thromboses. The infection can proceed to acute respiratory distress syndrome (ARDS) and culminates in additional tissues and organs in response to systemic infection.
Highlighted Cell Types in SARS-CoV-2 Pathophysiology
Epithelial Cells
Cell surface receptors on oral (Xu et al., 2020) and nasal epithelial cells (Sungnak et al., 2020) are points of entry for SARS-CoV-2. Local viral proliferation in the airway disrupts the ciliated epithelium, induces ciliary dyskinesia and viral movement down the lower respiratory tract epithelium (Chilvers et al., 2001) through the activity of phosphodiesterase, which is a potential therapeutic target (Joskova et al., 2020). Infected bronchial epithelial cells may produce complement (Peters-Hall et al., 2015). In the alveolar epithelium, type I pneumocytes cover approximately 95% of the gas exchange surface area, and the type II pneumocytes cover the remaining area and secrete surfactant in maintaining lung compliance (McElroy and Kasper, 2004). Receptors for SARS-CoV-2 on these cells (Hamming et al., 2004) facilitate infection, increase the production of surfactant and decrease gas exchange in the alveoli (McElroy and Kasper, 2004). SARS-CoV-activated epithelial cells produce cytokines such as IL-6 and IL-8, involved in neutrophil recruitment and reduced lymphocyte activity (Yoshikawa et al., 2009). These cells also undergo apoptosis in COVID-19 (Li et al., 2020). Cell signaling through the pneumocyte surface receptor Mas, associated with the RAAS system (Figure 4), prevents angiotensin (Ang) II-induced apoptosis (Gopallawa and Uhal, 2016).
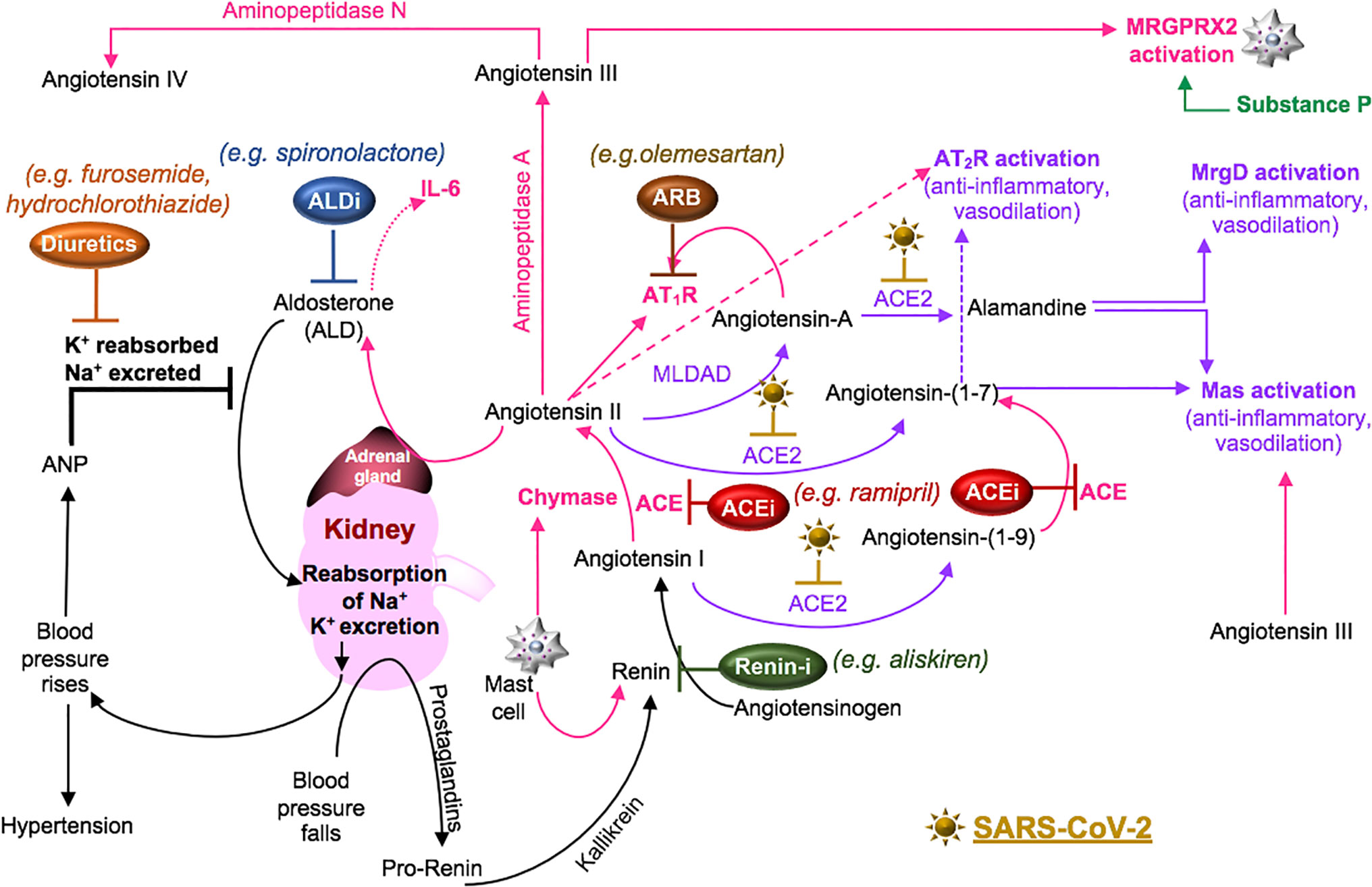
Figure 4 SARS-CoV-2 in the RAAS pathway. Prostaglandins stimulate the release of pro-renin from juxtaglomerular cells. Pro-renin is cleaved to renin by kallikrein. Renin is also produced by activated mast cells. Renin transforms angiotensinogen into angiotensin I. ACE or mast cell chymase converts angiotensin I into angiotensin II, which binds AT1R, and stimulates the production of aldosterone and subsequently IL-6. Aldosterone promotes renal distal tubular reabsorption of sodium, increasing blood pressure. In response to intravascular volume expansion, cells in the atrial wall release atrial natriuretic peptide (ANP), which down-regulates angiotensin II activity. Mononuclear leukocyte-derived aspartate decarboxylase (MLDAD) converts the octapeptide angiotensin II to another octapeptide, angiotensin A, which promotes the activation or AT1R, or may generate the anti-inflammatory heptapeptide (seven amino acid) ligand, alamandine, via ACE2 activity. ACE2 also converts angiotensin II to angiotensin-(1-7), which is a ligand for both the Mas receptor and AT2R involved in vasodilation and anti-inflammatory responses. Additional aminopeptidases convert angiotensin II into angiotensin III and IV. Angiotensin III can bind Mas and both angiotensin III and substance P activate MGRPRX2 on mast cells. A lack of ACE2 in RAAS due to SARS-CoV-2-induced degradation may suggest a benefit for intervention along the ACE/angiotensin II/AT1R/aldosterone pathway. These may include renin inhibitors (Renin-i), AT1R blockers (ARB), ACE inhibitors (ACEi), aldosterone blockers (ALDi), or diuretics.
Macrophages
In the alveolar lumen, macrophages regularly clear debris, surfactant, and microorganisms to maintain lung compliance and protect the epithelium (Curran et al., 2018). The exact interactions of SARS-CoV-2 with macrophages are not clearly known. Macrophages may phagocytose the virus through opsonic mechanisms (e.g. complement, antibodies or surfactant protein-A) or become infected through ACE2 receptor binding interactions (Figure 2) (Keidar et al., 2005; Curran et al., 2018). Pathogen- and damage-associated molecular pattern ligands (PAMPs and DAMPs) bind and activate macrophage Toll-like receptors (TLRs) (Zhao and Zhao, 2020). In response to inflammation, macrophages produce the neuropeptide substance P (SP) and the chemokine, monocyte chemoattractant-1 (MCP-1/CCL2), and express the Mas receptor, which attenuates the production of pro-inflammatory cytokines (e.g. IL-6, TNF) upon ligation (Germonpre et al., 1999; Simoes e Silva et al., 2013). Macrophages produce complement factors (e.g. C1q, C1r, C1s, C2, C3, C4, C5) and respond to complement deposited on apoptotic cells or bound to receptors for C3a and C5a (Lubbers et al., 2017). The differentiation of macrophages is regulated, in part, by components of the complement cascade. Specifically, C3a and C5a activate inflammasomes in macrophages, promoting a pro-inflammatory M1-type macrophage whereas the opsonic molecules [C1q, mannose binding lectin (MBL)] promote phagocytosis and reduced inflammation, forming an M2-type macrophage (Bohlson et al., 2014). The neuropeptide SP also promotes the formation of an anti-inflammatory, tissue-reparative M2-type macrophage (Lim et al., 2017). Macrophage polarization states at various stages of infection may play role in viral evasion (Atri et al., 2018).
Mast Cells
Some viruses are able to activate and infect mast cells, which induces the release of antimicrobial peptides (e.g. cathelicidins) that protect the lung, as well as enzymes (tryptase, chymase) and mediators (e.g. TNF, prostaglandins, histamine), that promote vasodilation and inflammation associated with highly pathogenic infections (Graham et al., 2015; Mollerherm et al., 2016). A functional role of mast cell extracellular traps in viral host defense or inflammation has not been significantly explored (Mollerherm et al., 2016). In the absence of direct viral interaction, mast cells are activated by immunoglobulins (IgE), TLR ligands, complement (C3a, C5a), integrin binding interactions, and thrombin (Theoharides et al., 2011; Graham et al., 2015). Mast cells are additionally activated by tachykinins (SP, neurokinin A, neurokinin B), which bind neurokinin receptors and members of the Mas family of receptors (Kleij and Bienenstock, 2005; Karnik et al., 2017; Green et al., 2019). Mast cells and nerves are in constant contact with each other in both physiologic and pathologic situations and exhibit cross-talk in the brain, lung, and gut (Kleij and Bienenstock, 2005; Traina, 2019). Activated mast cells generate tryptase and serotonin, which induce the release of tachykinins from sensory nerve endings that in turn, activate mast cells (Kleij and Bienenstock, 2005). Mast cells are the major source of heparin throughout the body (Humphries et al., 1999) and produce renin (Theoharides et al., 2011) and angiotensin-converting enzyme (ACE) (Chao et al., 2011) in response to oxidative stress. Mast cells are also a target of approved therapeutics.
Neurons
The process of respiratory gas exchange sends signals from vagal afferent nerves to respiratory neurons in the brain, which activate muscles in the thorax and abdomen to contract and relax, altering pressure in the lung (Cherniack and Cherniack, 2007). The activation of certain vagal afferent nerves in the respiratory tract can lead to dyspnea and an urge to cough (Mazzone and Undem, 2016). Dry cough is a common feature of COVID-19 (Qin et al., 2020), which could be a result of SARS-CoV-2 binding to neuronal ACE2 (Xiao et al., 2013). Pulmonary unmyelinated sensory neurons (C-fibers) can also be activated by tachykinins or bradykinin to induce cough and the response is enhanced by prostaglandins and histamine (Canning, 2009). Moreover, activated sensory neurons release tachykinins that act on bronchial smooth muscles, the mucosal vasculature, and submucosal glands to promote bronchoconstriction, vascular permeability, edema, and inflammatory cell chemotaxis (Solway and Leff, 1991). The release of SP by intra-adrenal nerve fibers stimulates aldosterone secretion via SP binding to neurokinin type 1 receptors (NK1) expressed by aldosterone-producing adrenocortical cells (Wils et al., 2020). The additional finding of SARS-CoV-2 in COVID-19 patient cerebral spinal fluid and the development encephalitis and Guillain-Barré syndrome in some COVID-19 patients identifies a distinct role for neurons in the disease (Ellul et al., 2020).
Endothelial Cells
The pulmonary capillaries that surround the alveoli consist of a thin layer of endothelial cells to allow rapid exchange of respiratory gases between capillary blood and type I pneumocytes (Gonzales et al., 2015). In examining deceased COVID-19 patient lungs, diffuse alveolar damage and severe endothelial injury associated with intracellular SARS-CoV-2, thrombosis, and signs of intussusceptive angiogenesis were identified (Ackermann et al., 2020). This latter regenerative process of blood vessel splitting in lung injury is a poorly understood process that may contribute to wound repair and restoration of the endothelium (Niethamer et al., 2020). Intussusceptive angiogenesis is also an identified contributor to the progression of cancers and occlusive vascular diseases (Weddell and Imoukhuede, 2018). This process may involve the complement system. Complement factor C1q consists of collagen-like [cC1q] and globular head [gC1q] regions that bind receptors cC1qR and gC1qR, respectively. The activation of both of these receptors on endothelial cells contributes to angiogenesis (Ghebrehiwet et al., 2019). gC1qR also binds factor XII and high molecular weight kininogen in initiating the kallikrein-kinin system and the secondary hemostasis intrinsic pathway (Kaira et al., 2020). The latter pathway is additionally triggered by endothelial damage, which produces endothelial collagen that binds platelet glycoprotein VI (GPVI) (Gale, 2011). Endothelial cells can express RAAS pathway enzymes (ACE, ACE2) as wells as receptors that promote [Ang II type 1 receptor (AT1R)] and inhibit (Mas, AT1R) vasoconstriction, fibrosis, and inflammation (Nehme et al., 2019).
Platelets
In response to injury, platelets adhere to the endothelium, are activated, and aggregate to form a platelet plug (Reyes Gil, 2019). Platelets are activated by extracellular matrix molecules that bind integrins (Bennett, 2005), complement (Ghebrehiwet et al., 2019), thrombin (Major et al., 2003), and TLR ligands, (Aslam et al., 2006; Koupenova et al., 2014). Influenza ssRNA activates TLR7 in platelets and induces platelet granule release of complement C3, which augments the release of DNA from neutrophils and promotes the formation of platelet–neutrophil aggregates associated with thrombosis (Koupenova et al., 2019). Platelets can also be activated by Ang II and their adhesiveness is down-regulated by ACE2 activity, possibly through Mas activation or through mediators released from Mas-activated endothelial cells (Fraga-Silva et al., 2010). Activated platelets release granules from dense organelles (containing serotonin, ATP, ADP, histamine, thromboxane A2) and alpha organelles (containing factors involved in both promoting and inhibiting fibrinolysis and angiogenesis) (Bhagavan and Ha, 2015; Reyes Gil, 2019).
Neutrophils
Circulating neutrophils are primed by cytokines (Wright et al., 2010), complement (Fung et al., 2001), and TLR ligands (Wang et al., 2008), which alters their cytoskeletal structure for effective migration across the endothelium, through the pulmonary interstitium, across the epithelium and into alveolar spaces (Potey et al., 2019). In response to thrombin or histamine, endothelial cells rapidly release the most potent neutrophil chemokine, IL-8 (Utgaard et al., 1998). Endothelial cells also produce IL-8, as well as MCP-1, in response to IL-6 complexed to the soluble receptor (sIL-6Ra), which binds the cell surface receptor, gp130 (Romano et al., 1997). In mice, neutrophil trafficking is promoted by IL-6 signaling (JAK/STAT3) through gp130 (Fielding et al., 2008). Neutrophils release neutrophil extracellular traps (NETs), neutrophil elastase, reactive oxygen species (ROS) and proteases to eliminate an invading pathogen, which can also produce additional tissue injury (Potey et al., 2019). Neutrophils shed sIL-6Ra from their cell surface and the IL-6:sIL-6Ra complex stimulates fibroblasts and macrophages to produce MCP-1 involved in the recruitment of monocytes (Kaplanski et al., 2003).
Monocytes
Increased production of MCP-1 promotes the recruitment of monocytes to the alveoli, where they differentiate into macrophages in response to cytokines, PAMPs and DAMPs in the microenvironment (Huang et al., 2018). A sustained monocyte influx is correlated with the severity of respiratory failure (Rosseau et al., 2000) and may be linked to IL-17 producing T helper cells (Th17) (Jiang et al., 2017).
Lymphocytes
Circulating lymphocyte numbers decline in severe COVID-19 (Chen G. et al., 2020; Qin et al., 2020). Both CD8+ T cells and CD4+ T cells have elevated levels of the programmed death (PD)-1 receptor in COVID-19 patients, a sign of T cell functional exhaustion (Diao et al., 2020). Compared to healthy controls, COVID-19 patient CD8+ T cells and CD4+ T cells activated in vitro produce more IL-17, indicating skewing toward a Th17 phenotype (De Biasi et al., 2020). Because the levels of IL-17 tend to increase with disease severity, an underlying function of Th17 cells has been suggested (Wang J. et al., 2020), despite the lower lymphocyte numbers identified in COVID-19 patients (Chen G. et al., 2020; Qin et al., 2020). Because mast cells and neutrophils are also known to release IL-17 from their extracellular traps (Lin et al., 2011), additional research into the source of IL-17 in COVID-19 patients is needed.
The RAAS Pathway
The RAAS pathway is a cascade of enzymatic reactions that function in the homeostatic control of extracellular volume, arterial pressure, tissue perfusion, electrolyte balance, and wound healing (Atlas, 2007). A prominent component of the RAAS pathway is located within the juxtaglomerular apparatus. There, the macula densa cells are strategically located in the distal tubule, in a region adjacent to the afferent arteriole, at a point where these elements meet the glomerulus. Sensors in the macula densa cells respond to a low sodium chloride concentration within the lumen of the distal tubule. In response, these cells produce adenosine and ATP, that constrict the afferent arteriole and thereby reduce glomerular perfusion. These cells also produce cyclooxygenase-2 (COX-2), which generates prostaglandins. Juxtaglomerular cells in the afferent glomerular arteriole release pro-renin in response to stimulation by prostaglandins (Kriz, 2004; Atlas, 2007; Peti-Peterdi and Harris, 2010).
Pro-renin is transformed into active renin by proteases, including kallikrein (Biswas et al., 2016). Renin may also be released from mast cells activated by oxidative injury (Theoharides et al., 2011). Liver-produced angiotensinogen is cleaved by renin into Ang I, which is further cleaved by a ubiquitous membrane and soluble ectoprotein, ACE, into Ang II (Igic and Behnia, 2003; Jiang et al., 2014). The binding of Ang II to AT1R promotes vasoconstriction; induces the production of aldosterone, which promotes renal tubular sodium reabsorption; has a biphasic effect on sodium reabsorption in the gut (promotes at low concentrations, inhibits at high concentrations) and increases blood pressure (Nehme et al., 2019), which is associated with increased neuronal and immune cell production of the vasodilator, SP (Calvillo et al., 2019). Ang II also binds the type 2 receptor (AT2R), which exerts inhibitory actions on AT1R cell signals by promoting vasodilation and natriuresis (Atlas, 2007; Nehme et al., 2019). Transformation of Ang II to III and III to IV by aminopeptidases A and N, respectively, activates AT4R, which is also known as insulin-regulated aminopeptidase (Goldstein et al., 2017).
The ACE homolog, ACE2, cleaves Ang I into Ang-(1-9) and processes Ang II and Ang-(1-9) into Ang-(1-7), thereby inactivating Ang II. Ang II-mediated signaling is involved in apoptosis, reactive oxygen species production, epithelial-to-mesenchymal transformation (EMT), and alveolar fluid retention (Sriram and Insel, 2020). Thus, ACE2 functions as an endogenous inhibitor of the ACE/Ang II/AT1R pathway and opposes the vasoconstrictive, inflammatory, prothrombotic, and fibrotic effects associated with ACE/Ang II/AT1R activity (Fraga-Silva et al., 2010; Santos et al., 2018). Consequently, ACE2 and drugs that oppose ACE/Ang II/AT1R activity improve the tissue response to injury (Sriram and Insel, 2020). Ang II can also be cleaved by mononuclear leukocyte-derived aspartate decarboxylase (MLDAD), generating angiotensin A (differing from Ang II by one amino acid) that is processed into a newly recognized peptide, alamandine [a decarboxylated form of Ang-(1-7)], by ACE2 (Hrenak et al., 2016; Tetzner et al., 2018). Ang-(1-7) binds Mas and AT2R (Karnik et al., 2017). Both Ang-(1-7) and alamandine bind the Mas-related G-protein coupled (MrgD) receptor. These cell signals antagonize inflammation and AT1R responses (Hrenak et al., 2016; Karnik et al., 2017; de Carvalho Santuchi et al., 2019) (Figure 4).
Moreover, Ang II induces the expression of intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (Alvarez et al., 2004), P-selectin (Piqueras et al., 2000), and integrins (Kawano et al., 2000; Li et al., 2006) in experimental models. These adhesive interactions promote the recruitment of neutrophils (Piqueras et al., 2000; Alvarez et al., 2004). In in vitro assays, cellular ACE2 binds integrin β1 (ITGB1) and integrin α5 (ITGA5), which enhances adhesion and focal adhesion kinase (FAK) signaling, whereas soluble ACE2 inhibits FAK cell signals (Clarke et al., 2012). This may indicate that ectodomain shedding of ACE2 is important in regulating Ang II-induced leukocyte adhesion and recruitment.
In summary, the RAAS pathway is an intricate system that coordinates the activities of many cell types, organ systems and the vasculature to regulate electrolyte balance, blood pressure and cardiovascular function. Dysregulation in this system over an extended period of time promotes renal and cardiovascular diseases (Munoz-Durango et al., 2016). In acute pulmonary infections, the RAAS pathway contributes to the development of acute respiratory distress syndrome (ARDS) and subsequent pulmonary fibrosis (Kuba et al., 2006), which has an estimated incidence rate of 19.5% in COVID-19 patients (Zhu et al., 2020).
SARS-CoV-2 and RAAS Pathway
ACE2 is expressed in type I and type II pneumocytes, oral and nasal epithelial cells, neurons, and arterial and venous endothelial cells (Hamming et al., 2004; Xiao et al., 2013). In single cell transcriptome data from healthy human lung tissues, ACE2 and TMPRSS2 expression levels were highest in the subsegmental bronchial branches (Lukassen et al., 2020), highlighting likely SARS-CoV-2 binding interactions within the lung. In a murine model, SARS-CoV infection reduced lung function and ACE2 protein levels. A subsequent study by this group assessed a mouse model of acid aspiration-induced lung injury followed by intraperitoneal injection of recombinant SARS-CoV spike-Fc protein compared to a control Fc protein. These data revealed that the spike protein worsened lung injury compared to the control and this response was attenuated by an AT1R blocker (ARB) (Kuba et al., 2005). In another model, ACE2 knockout mice exhibited reduced lung function that was improved by the administration of either recombinant ACE2 or an ARB (Imai et al., 2005).
These models demonstrate the potential for interventions that target components of the RAAS pathway to ameliorate acute lung injury. Previous concerns regarding a potential risk of increased SARS-CoV-2 infection in response to these drugs has been alleviated by two large retrospective studies in Italy (Mancia et al., 2020) and the USA (Reynolds et al., 2020). Moreover, in a retrospective review of 42 hospitalized COVID-19 patients with hypertension (median age 64), 17 subjects received ACE inhibitors (ACEi) or ARB therapy and 25 subjects received other antihypertensive drugs. Those treated with ACEi or ARBs had a trend toward less severe disease and tended to have increased T cell counts, reduced viral loads, and lower levels of circulating IL-6 compared to subjects receiving other antihypertensive drugs (Meng et al., 2020). These findings suggest that blocking the RAAS pathway with ACEi or ARBs improves immune responses in COVID-19 patients and indicates a need for randomized controlled trials for further evaluation. In the interim, current clinical guidelines recommend continued use of these therapies in COVID-19 patients with pre-existing conditions such as hypertension (Mehta et al., 2020).
Predicting the possible benefits of a RAAS pathway intervention is complicated by possible ectodomain shedding of ACE2 (Xiao et al., 2014). In vitro, ligation of the SARS-CoV S-protein to ACE2 induced the activity of TNF-converting enzyme (TACE) and TACE-dependent shedding of the catalytically active ectodomain of ACE2 (Haga et al., 2008). The functions of soluble ACE2 are not fully known. In patients with heart failure, increased levels of plasma soluble ACE2 correlate with greater disease severity (Epelman et al., 2008). In another study involving patients with acute decompensated heart failure, increased serum soluble ACE2 levels during intensive medical therapy predicted improved outcomes (Shao et al., 2013). These apparently discordant findings indicate that additional study of soluble ACE2 is warranted, particularly prior to using soluble ACE2 as a therapy to block SARS-CoV-2 binding interactions with host cells (Monteil et al., 2020; Zhang H. et al., 2020).
While the effects of ACEi/ARBs and soluble ACE2 in COVID-19 are not fully known, ACE2 is an endogenous inhibitor of the ACE/Ang II/AT1R pathway and the downstream vasoconstrictive, inflammatory, prothrombotic, and fibrotic responses (Fraga-Silva et al., 2010; Santos et al., 2018). Possibly, ACE2 dampens Ang II-induced production of interleukin (IL)-6 (via aldosterone) (Luther et al., 2006) and reduces the downstream formation of Th17 cells (Madhur et al., 2010). In COVID-19 patients, plasma IL-6 levels are elevated (Ranucci et al., 2020), prompting the clinical trial investigation of therapeutics that target IL-6 (siltuximab, NCT04329650), its receptor (tocilizumab, NCT04377659), its cell signals [e.g. Janus kinases, JAKs (baricitinib, NCT04373044) and downstream production of IL-17 (secukinumab, NCT04403243)] (Schett et al., 2020). Initial studies in COVID-19 patients with tocilizumab (Klopfenstein et al., 2020; Luo P. et al., 2020; Toniati et al., 2020) and baricitinib (Cantini et al., 2020) have shown promising results. However, tocilizumab use requires caution in patients with hepatic impairment and can induce liver injury in some COVID-19 patients (Muhovic et al., 2020). The potential for additional opportunistic pulmonary infection is also associated with monoclonal antibodies such as tocilizumab. An increased risk of pulmonary infections may also arise in autoimmune patients treated with baricitinib (Khoo et al., 2020) or anti-IL-17 antibodies (ixekizumab) (Mease et al., 2019), suggesting that the use of these drugs requires careful evaluation.
Further exploration of these networks may require targeting Mas, MrgD, or AT2R receptors or the use of other pharmacologic interventions, such as direct renin inhibitors or diuretics, which act on several elements of the RAAS (Table 1) (Guichard et al., 2013; Byrd et al., 2019). The use of a highly selective NK1 antagonist (aprepitant), which blocks SP binding to NK1 on adrenal zona glomerulosa cells, may also be beneficial in reducing aldosterone levels (Wils et al., 2020) that in turn, promote IL-6 production (Luther et al., 2006). The continued investigation of these pathways will be essential to defining COVID-19 pathophysiology and devising adjunctive therapies to current anti-viral therapies (e.g. remdesivir, ClinicalTrials.gov Identifier: NCT04280705) and immunogenic approaches (Figures 2 and 3).
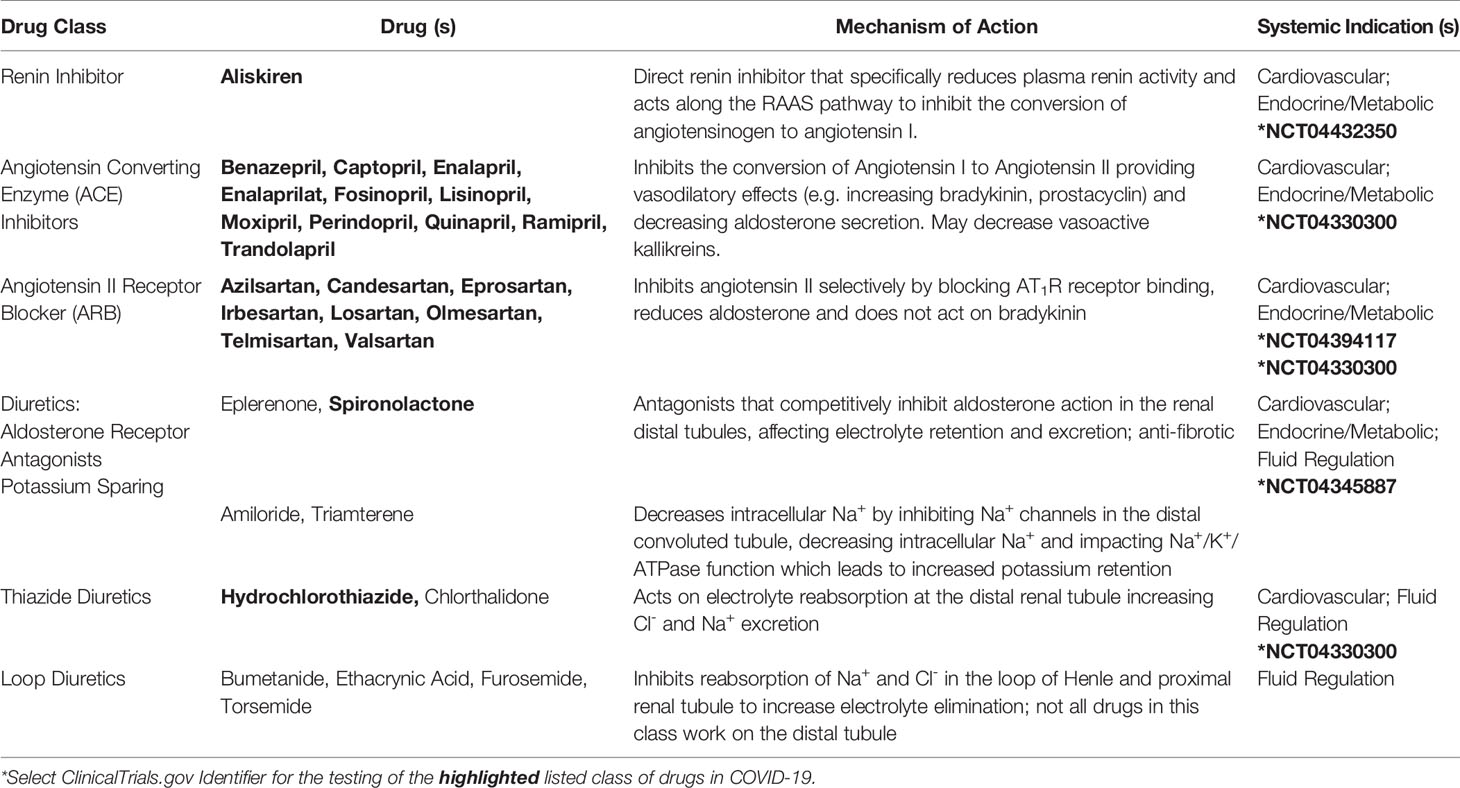
Table 1 Medications that Target the RAAS Pathway.
The Complement System
The complement system is composed of soluble and cell membrane proteins that regulate the activity of the classical, lectin and alternative pathways (Figure 5). The molecules in these pathways act as sensors to tissue damage and pathogens and as effectors to kill microbes and to clear damaged cells (Reis et al., 2019). In the classical pathway, C1q recognizes pathogens or apoptotic cells directly or indirectly through antibody complexes, or through associations with pentraxins, such as CRP (Sproston and Ashworth, 2018). In the lectin pathway, complement activation is initiated by an interaction involving mannose binding lectin (MBL). Serine proteases [C1r/C1s and MBL-associated serine protease (MASP)] complex with C1q (C1r/C1s) and MBL (MASP). This leads to the cleavage of C4 to its fragments (C4b and C4a) and the formation of a C3 and C5 convertase. In the alternative pathway, C3 is spontaneously hydrolyzed and through the activity of factor D, a C3 convertase is formed, which leads to the formation of a C5 convertase (Ricklin and Lambris, 2007). Additionally, C3 and C5 may also be cleaved by mast cell tryptase, thrombin or kallikrein (Ricklin and Lambris, 2007; Ali, 2010).
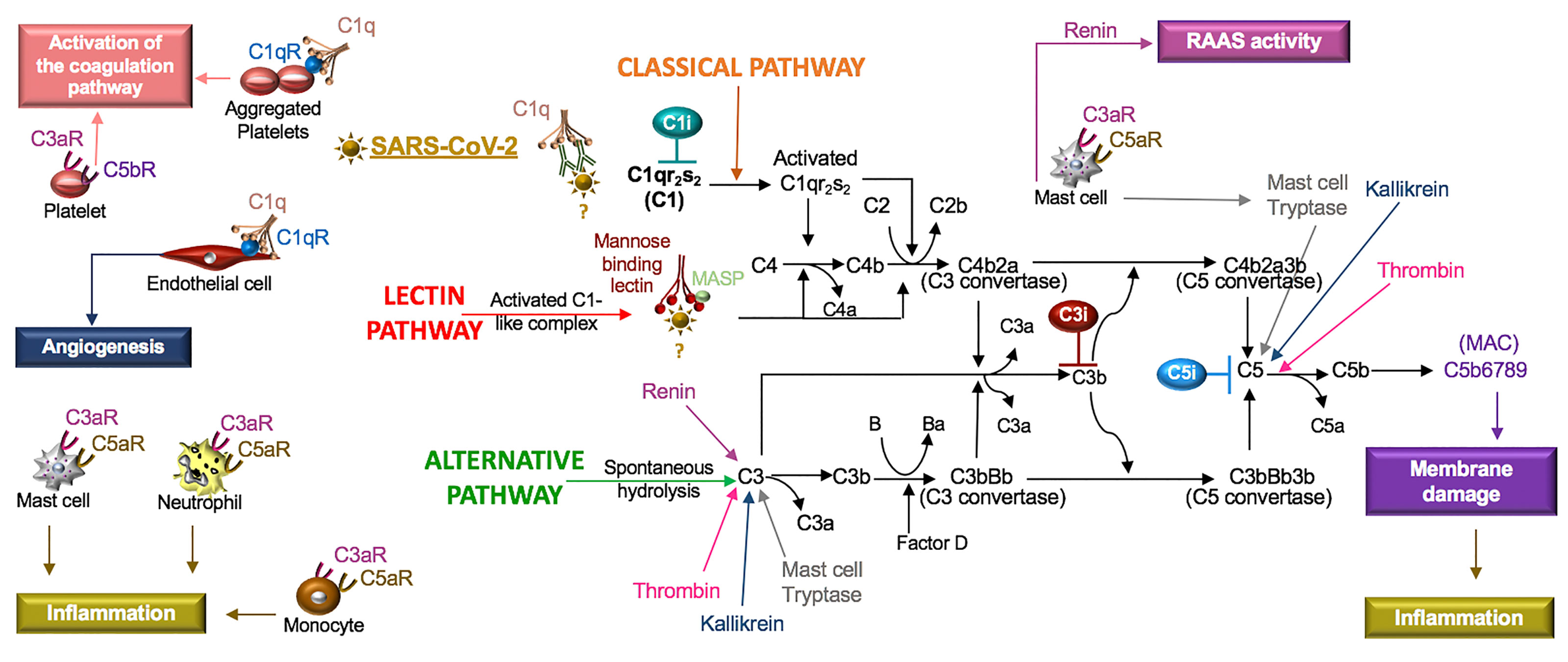
Figure 5 SARS-CoV-2 in the complement system. Classical, lectin, and alternative are the three pathways in the complement system. Complement components C1, C2, C3, and C4 are present in plasma in inactive forms. In the classical pathway, the C1 component, C1q, recognizes apoptotic cells directly or pathogens indirectly through antibody complexes or associations with pentraxins. In the lectin pathway, mannose-binding lectin (MBL) binds the surface of the pathogen. Serine proteases complex with C1q (C1r/C1s) and MBL (MASP: MBL-associated serine protease), which leads to cleavage of C4 to its fragments (C4b and C4a) and the formation of a C3 and C5 convertase. In the alternative pathway, C3 is spontaneously hydrolyzed and through the activity of factor D forms a C3 convertase and subsequently a C5 convertase. Mast cell tryptase, thrombin or kallikrein can also cleave C3 and C5 whereas renin cleaves only C3. Cleavage fragments from these pathways (e.g. C3a, C5a, C5b) activate immune cell subsets to produce inflammation or coagulation. The terminal product of these pathways, C5b6789, is a membrane attack complex (MAC), that creates a pore in cell membranes by displacing phospholipids. The resulting cell lysis induces inflammatory responses. C1q also acts independent of the complement system and binds its receptor (C1qR) on aggregated platelets and endothelial cells in the promotion of coagulation and angiogenesis, respectively. Tissue and organ damage and excessive inflammation in some COVID-19 patients may indicate that SARS-CoV-2 activates the complement cascade. C1, C3, and C5 inhibitors (i) block factor formation in the complement cascade.
Activation of any of these pathways results in the insertion of the membrane attack complex (MAC, composed of C5b–9) into targeted cells and generation of active complement fragments such as C3a, C3b, C4a, C4b, and C5a, which bind complement receptors on a various cell types (Reis et al., 2019) (Figure 5). C3a and C5a are anaphylatoxins and potent stimulators of neutrophils, monocytes, mast cells and platelets, resulting in the release of mediators and the expression of adhesion receptors (Fung et al., 2001; Ali, 2010). Mast cells at rest produce tissue-type plasminogen activator (t-PA) but in the presence of C5a, mast cells generate plasminogen activator inhibitor (PAI)-1 (Wojta et al., 2002). Moreover, C1q also binds a cell surface receptor, C1qR, on aggregated platelets and endothelial cells, resulting in the activation of the coagulation pathway and angiogenesis, respectively (Ghebrehiwet et al., 2019). In viral infections, such as SARS-CoV, PAMPs and DAMPs bind and activate TLRs (Zhao and Zhao, 2020), which regulate the production and function of complement (Hajishengallis and Lambris, 2016). Thus, the innate immune responses involving the complement system are highly implicated in SARS-CoV-2.
SARS-CoV-2 and the Complement System
In COVID-19 patients, elevated plasma CRP levels are a prognostic indicator of adverse outcomes. Threshold values of an adverse outcome have been reported as 27 mg/L (Wang G. et al., 2020) and as 41 mg/L (Luo X. et al., 2020). CRP is primarily synthesized by IL-6-dependent hepatic biosynthesis (Sproston and Ashworth, 2018). In a small study of COVID-19 patients, a combination of IL-6 levels > 80 pg/mL and CRP levels > 97 mg/L were highly predictive of the need for mechanical ventilation (Herold et al., 2020). In human skin fibroblasts, IL-6 induces the production of complement factor B and C3 involved in the activation of the alternative pathway (Katz et al., 1989). Because complement participates in various inflammatory skin diseases (Giang et al., 2018), complement may be produced in response to IL-6 in COVID-19 patients, promoting the cutaneous skin disorders characteristic of COVID-19 (Galvan Casas et al., 2020).
Research involving intranasal infection with recombinant mouse-adapted SARS-CoV (MA15) identified C3 fragments in the lungs of mice one day after infection. Additionally, C3-/- mice exposed to this virus manifested reduced neutrophil and monocyte recruitment and less respiratory dysfunction compared to control mice (Gralinski et al., 2018), demonstrating an active role for the alternative pathway in SARS-CoV. The effects of C3 inhibitors (AMY-101 and APL-9) in COVID-19 subjects with ARDS (ClinicalTrials.gov Identifier: NCT04395456 and NCT04402060) are being tested in clinical trials. Mixed reports involving the binding of the SARS-CoV spike protein to MBL (Leth-Larsen et al., 2007; Zhou et al., 2010), indicate that the function of the lectin pathway in SARS-CoV and SARS-CoV-2 requires further study. Lastly, despite increased production of CRP in SARS-CoV-2 (Wang G. et al., 2020), the functions of the classical pathway have not been systematically explored in SARS-CoV or SARS-CoV-2. Clinical investigations with a C1 inhibitor (Conestat alfa, NCT04414631) and a C5 inhibitor (Zilucoplan, NCT04382755) in severe COVID-19 patients are in progress. Monoclonal antibodies against C5 (e.g. eculizumab, ravulizumab) are also available therapeutics that block excessive complement activation.
In cryoinjured mice, ARB treatment was associated with lower systemic and local levels of C1q, decreased fibrosis and increased myofiber regeneration compared to the controls. The response was reversed by topical C1q and the mechanisms were linked to changes in macrophage C1q production (Yabumoto et al., 2015). Excessive activation of macrophages is associated with the pathophysiology of COVID-19 (Verdoni et al., 2020) and may therefore include macrophage C1q production. Blocking Bruton tyrosine kinase (BTK) is a proposed mechanism to suppress macrophage activation (Roschewski et al., 2020) and is currently being tested in clinical trials of COVID-19 subjects (Acalabrutinib, NCT04380688). Blocking BTK may therefore also affect C1q levels. Moreover, in a rat model of Ang II-induced renal damage, increased circulating levels of complement (C1q, C3, C3c, and C5b-9), CRP, and renal TNF were reduced by a direct renin inhibitor (aliskiren) and also by an ARB (losartan) (Shagdarsuren et al., 2005). The exact roles of Ang II and the SARS-CoV-2 receptor, ACE2, in the complement system require further study.
The increased numbers of apoptotic type I and II pneumocytes and endothelial cells in COVID-19 patient lung tissue (Li et al., 2020) are suggestive of a dysregulated host response in the clearance of these cells. This may involve changes in phagocyte cell surface receptors, the activation state of phagocytes, and/or the response of these cells to components of the complement system (Gordon and Pluddemann, 2018). Neutrophils also undergo apoptosis in inflamed tissue but in ARDS, this process is impaired. In assessing the function of peripheral blood neutrophils from ARDS patients, neutrophils were activated in vitro and produced more NETs and exhibited increased viability compared to healthy control neutrophils. In addition, human monocyte-derived macrophages from the ARDS patients were unable to effectively phagocytose apoptotic neutrophils. However, in the presence of metformin, a 5’ AMP-activated protein kinase (AMPK) activator, the response was improved (Gregoire et al., 2018). Complement component C1q also induces AMPK activation in macrophages (Galvan et al., 2014) and similar to ARDS patients, COVID-19 patient serum exhibits increased NET activity (Zuo et al., 2020). Thus, SARS-CoV-2 may alter the activity or production of C1q, its receptor, the C1 proteases (C1r, C1S), and/or the regulatory crosstalk known to occur between complement and the various TLR ligands (e.g. dsDNA and ssRNA) released from NETs and apoptotic cells (Hajishengallis and Lambris, 2016).
Mast cell-produced tryptase contributes to the cleavage of C3 and C5 (Ali, 2010). Mast cell-produced renin cleaves C3 but not C5 (Bekassy et al., 2018). Because ligands from C3 and C5 cleavage (C3a, C5a) activate mast cells (Ali, 2010) and mast cells may additionally produce ACE (Chao et al., 2011), persistent cross-talk between the complement system and mast cells likely maintains homeostasis in the RAAS and complement pathways. Mast cells also express Mas-related G protein coupled receptor X2 (MRGPRX2) (Ali, 2016). The Ang-(1-7) receptors (Mas, MrgD) and MRGPRX2 are members of a family of ~40 orphan receptors that exhibit ligand promiscuity with AT1R and AT2R in the regulation of the RAAS (Karnik et al., 2017). Moreover, Ang III activates Mas and MRGPRX2 (Gembardt et al., 2008), indicating that Ang III may compete with the ACE2 product, Ang-(1-7), in binding to its receptors (Mas, MrgD, and AT2R) (Figure 4).
The mast cell receptor MRGPRX2 also binds the neuropeptide SP and in response, mast cells produce chemokines (Green et al., 2019). SP, a tachykinin, binds NK1, is released from immune cells and neurons, and enhances inflammatory processes in the lung, gut, and skin (Johnson et al., 2016), which are common sites of inflammation and sources of symptoms in COVID-19 patients (Galvan Casas et al., 2020; Wong et al., 2020). SP and the complement fragments (C3a, C5a) similarly activate mast cells via distinct pathways (el-Lati et al., 1994). SP also acts synergistically with C5a in the recruitment and activation of neutrophils (Perianin et al., 1989). Regulatory cell signals generated by complement fragments and the ligands to Mas-related receptors in mast cells, neutrophils and additional immune subsets may be important to the recruitment of neutrophils and the pathogenesis of various COVID-19 inflammatory disorders.
The Coagulation Pathway
The hemostatic system is divided into three phasic processes. In primary hemostasis, activated platelets aggregate to form a platelet plug. In secondary hemostasis, activated coagulation factors on the surface of the endothelium and platelets form a fibrin mesh that stabilizes the plug, forming a fibrin clot. These processes are balanced by tertiary hemostasis, which activates fibrinolysis for the dissolution of the clot. During primary hemostasis, platelets adhere to von Willebrand factor released by the injured endothelium. Their activation is induced by various PAMPs and DAMPs, which encourages platelet aggregation and the formation of a necessary physical platform for the activation of the coagulation cascade (Reyes Gil, 2019).
Secondary hemostasis involves a cascade of serine proteases that are subdivided into an intrinsic pathway (surface-contact factors) and an extrinsic pathway (tissue factor initiated). The intrinsic pathway commences after endothelial damage with the release of endothelial collagen. This activates factor XII to factor XIIa, which acts as a catalyst to activate factor XI to factor XIa. Factor XIa converts factor IX to factor IXa and factor IXa converts factor X to factor Xa, which is a point of convergence in the two pathways. In the extrinsic pathway, local tissue injury releases tissue factor into the blood. Tissue factor activates factor VII to factor VIIa and the complex of tissue factor and factor VIIa activates factor X and factor IX. The resulting factor Xa is incorporated into a prothrombinase complex (composed of factor Xa: factor Va: Ca2+: platelet phospholipid) that converts factor II (pro-thrombin) to factor IIa (thrombin) (Gale, 2011). This initiates the coagulation cascade and the accumulation of thrombin, which exhibits proteolytic functions, acting upon substrates such as fibrinogen and factor XIII (fibrin stabilizing factor), resulting in the production of fibrinopeptides and active factor XIII (FXIIIa), respectively (Stassen et al., 2004; Huntington, 2005). Concomitantly, thrombin induces the activation of mast cells that produce molecules that promote (IL-6) and inhibit (heparin) coagulation (Theoharides et al., 2011). The fibrinopeptides spontaneously polymerize to form fibrin, which is covalently cross-linked by FXIIIa to form a stable nascent fibrin clot (Huntington, 2005).
Tertiary hemostasis involves clot dissolution and the initiation of wound remodeling (Reyes Gil, 2019). Plasmin is generated by plasminogen activators, such as urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA), which cleave plasminogen into the proteolytically active plasmin enzyme (Stassen et al., 2004). This process of fibrinolysis is controlled by plasminogen activator inhibitors (PAI-1 and PAI-2) and plasmin inhibitors (α2-antiplasmin and α2-macroglobulin) (Stassen et al., 2004) (Figure 6).
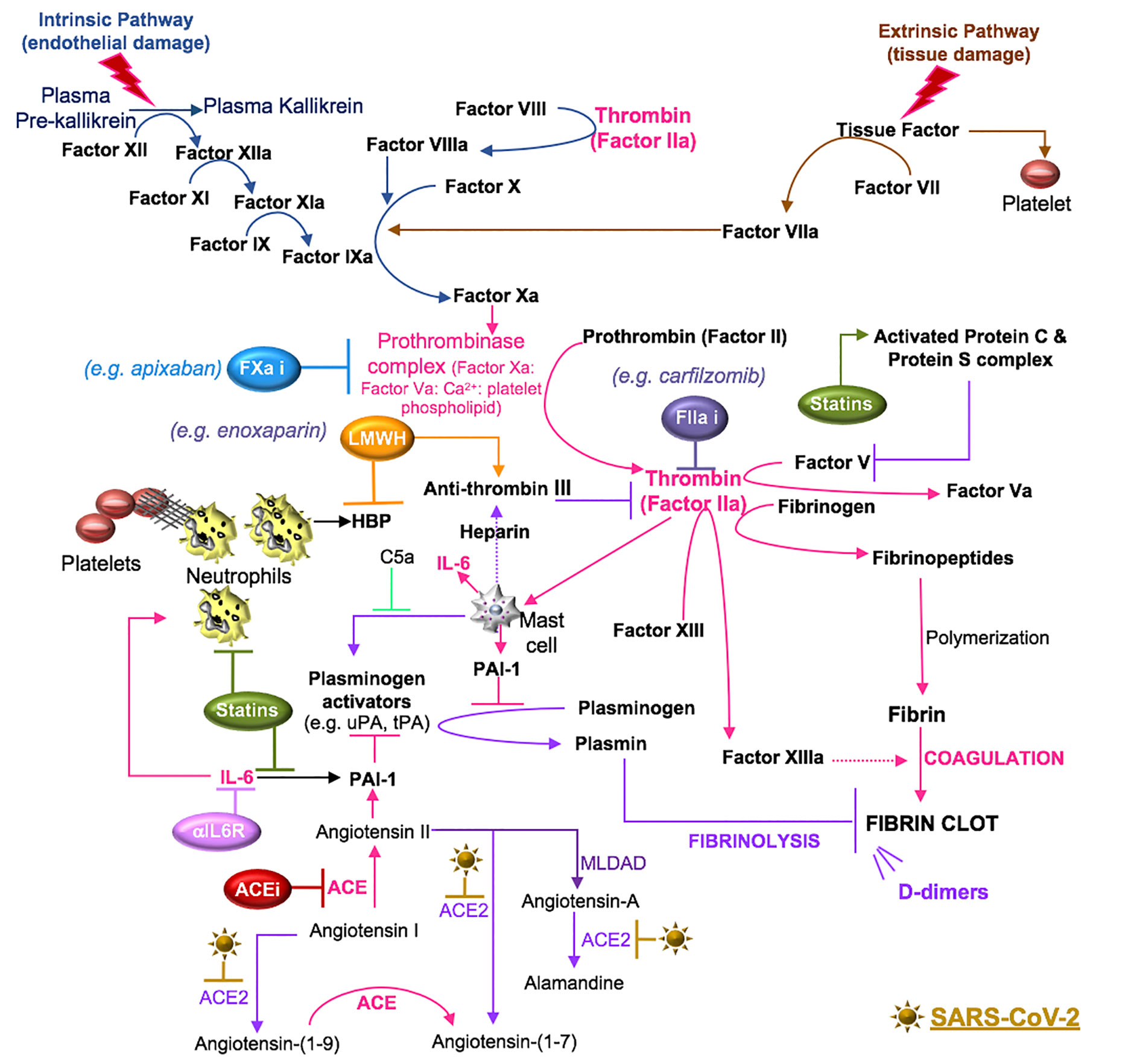
Figure 6 SARS-CoV-2 in coagulation. Primary hemostasis controls platelet aggregation and secondary hemostasis promotes fibrin formation through the clotting cascade, the latter involving intrinsic and extrinsic pathways. In the intrinsic pathway, damage-induced release of endothelial collagen activates factor XII (factor XIIa), which is a reaction that is also involved in the initiation of plasma kallikrein. Factor XIIa acts as a catalyst to activate factor XI to factor XIa. Factor XIa activates factor IX to factor IXa and the latter, acting with factor VIIIa as a cofactor, activates factor X to factor Xa. Tissue injury releases tissue factor into the blood, which activates platelets to induce neutrophil extracellular trap (NET) formation. The components of NETs reciprocally activate platelets and their aggregation. In the extrinsic pathway, tissue factor activates factor VII (factor VIIa), which activates factor X (factor Xa). The common coagulation pathway commences at factor X, with factor Xa, factor Va, calcium and platelet phospholipids forming the prothrombinase complex, which activates prothrombin (factor II) to thrombin (factor IIa). Thrombin cleaves factor V, VIII, factor XIII and fibrinogen. Polymerization of the formed fibrinopeptides produces fibrin, which is covalently cross-linked by FXIIIa to form a stable nascent fibrin clot. These processes can be antagonized by the heparin-dependent activity of antithrombin and plasmin degradation of fibrin into soluble fibrin degradation products (e.g. D-dimer). Plasmin formation is reduced by angiotensin II- or IL-6-induced plasminogen activator inhibitor (PAI)-1, which antagonizes the activity of urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA). SARS-CoV-2-induced degradation of ACE2 reduces ACE2 cleavage of angiotensin I and II and the anti-inflammatory effects of the ACE2 fragment [Ang-(1-7)], suggesting that inhibitors of RAAS [e.g. ACE inhibitor (ACEi)] may counter the effects of SARS-CoV-2. Mast cells activated by thrombin or complement (C5a) also contribute to coagulation through the preferential production of IL-6 and PAI-1. Drugs that inhibit IL-6 signals (e.g. anti-IL-6 receptor antibodies) may inhibit IL-6-induced PAI-1 production and IL-6 recruitment of neutrophils. Statins also inhibit IL-6 production, the activity of neutrophils and the functions of activated protein C. The use of inhibitors (i) to the clotting cascade such as factor Xa (e.g. apixaban) and factor IIa (e.g. carfilzomib) may impede SARS-CoV-2-induced coagulation responses. Lastly, low molecular weight heparin (LMWH) therapy may promote the effects of anti-thrombin and inhibit the functions of the alarmin, heparin binding protein (HBP), which binds glycosaminoglycan moieties of cell surface proteoglycans and promotes endothelial permeability.
SARS-CoV-2 and Coagulation
Elevated circulating plasminogen levels may contribute to SARS-CoV-2 susceptibility and worse clinical outcomes in patients with hypertension, diabetes, coronary heart disease, and chronic kidney disease (Ji et al., 2020). Because Ang II induces the production of PAI-1 in endothelial cells (Vaughan et al., 1995), SARS-CoV-2 may indirectly regulate PAI-1 via ACE2 down-regulation. In a rat model of abdominal vena cava thrombosis, decreased ACE2 activity is associated with thrombus formation whereas pharmacological (e.g. xanthenone) activation of ACE2 attenuates platelet vessel attachment and thrombus formation (Fraga-Silva et al., 2010), highlighting a potential regulatory role of ACE2 in coagulation.
A retrospective study of hospitalized COVID-19 patients indicates a potential benefit of anticoagulation therapy, particularly in mechanically-ventilated patients (N = 395, 29% mortality), compared to patients that did not receive anticoagulation therapy (63% mortality). Consequently, algorithms are evolving regarding diagnostic markers (IL-6, D-dimer levels, pro-thrombin time, fibrinogen, chest vessel enlargement on computerized tomography) to identify and implement best practice approaches to prophylactic and therapeutic anticoagulation therapy (Oudkerk et al., 2020). Elevated D-dimer levels in COVID-19 patients are common and indicative of coagulation, secondary hyperfibrinolysis, and a possible increased risk of venous thromboembolism (Cui et al., 2020; Tang et al., 2020b; Yin S. et al., 2020). In COVID-19 patients with D-dimer levels more than six-fold over the upper limit of normal, low molecular weight heparin (LMWH) therapy may be associated with better prognosis (Tang et al., 2020a). In another study of COVID-19 patients with elevated D-dimer levels, increased fibrinogen levels were also identified and associated with increased IL-6 levels (Ranucci et al., 2020). Because IL-6 also induces PAI-1 production (Dong et al., 2005; Rega et al., 2005), which promotes a prothrombotic state, this inflammatory cytokine is a therapeutic target in the coagulation pathway (Figure 6).
Moreover, circulating levels of IL-6 induce the recruitment of neutrophils to the lung (Suwa et al., 2001). Activated neutrophils release the alarmin, heparin binding protein (HBP), which binds glycosaminoglycan moieties of cell surface proteoglycans on the endothelium, promoting endothelial permeability; this pro-inflammatory response can be attenuated by LMWH (Fisher and Linder, 2017). The interactions between activated neutrophils and platelets also promote coagulation (Zucoloto and Jenne, 2019). HMG CoA reductase inhibitors (statins) reduce the activation of neutrophils (Fisher and Linder, 2017), promote fibrinolysis through increased activated protein C (Undas et al., 2014) and inhibit IL-6-induced PAI-1 production (Dong et al., 2005). For all these reasons, statins may have a therapeutic effect in COVID-19-induced thrombotic responses.
C5a activation of mast cells stimulates production of plasminogen activator inhibitor (PAI)-1 and antagonizes t-PA production (Wojta et al., 2002), suggesting that additional mast cell activators, such as thrombin (Theoharides et al., 2011) and SP (Hermans et al., 2018), may have similar effects. SP also induces monocyte tissue factor expression (Khan et al., 2012) and promotes platelet clot formation (Azma et al., 2009). Understanding SP binding interactions with NK1 (Johnson et al., 2016) and Mas-related receptors (Green et al., 2019) on monocytes, mast cells and platelets may therefore be important hemostatic regulators in COVID-19 patients.
Additional approaches to modulate the coagulation cascade may include direct thrombin inhibitors, factor Xa inhibitors, and heparin, and these are all being assessed in clinical trials (Table 2) (Stassen et al., 2004; Padmanabhan, 2014; Solari and Varacallo, 2020). Clarifying the roles of these agents in the context of COVID-19 pathophysiology is needed to better understand the associated thrombotic complications that arise in the clinical course of infection. This research may also shed light on complement-mediated microvascular injury and thrombosis in COVID-19 patients (Magro et al., 2020).
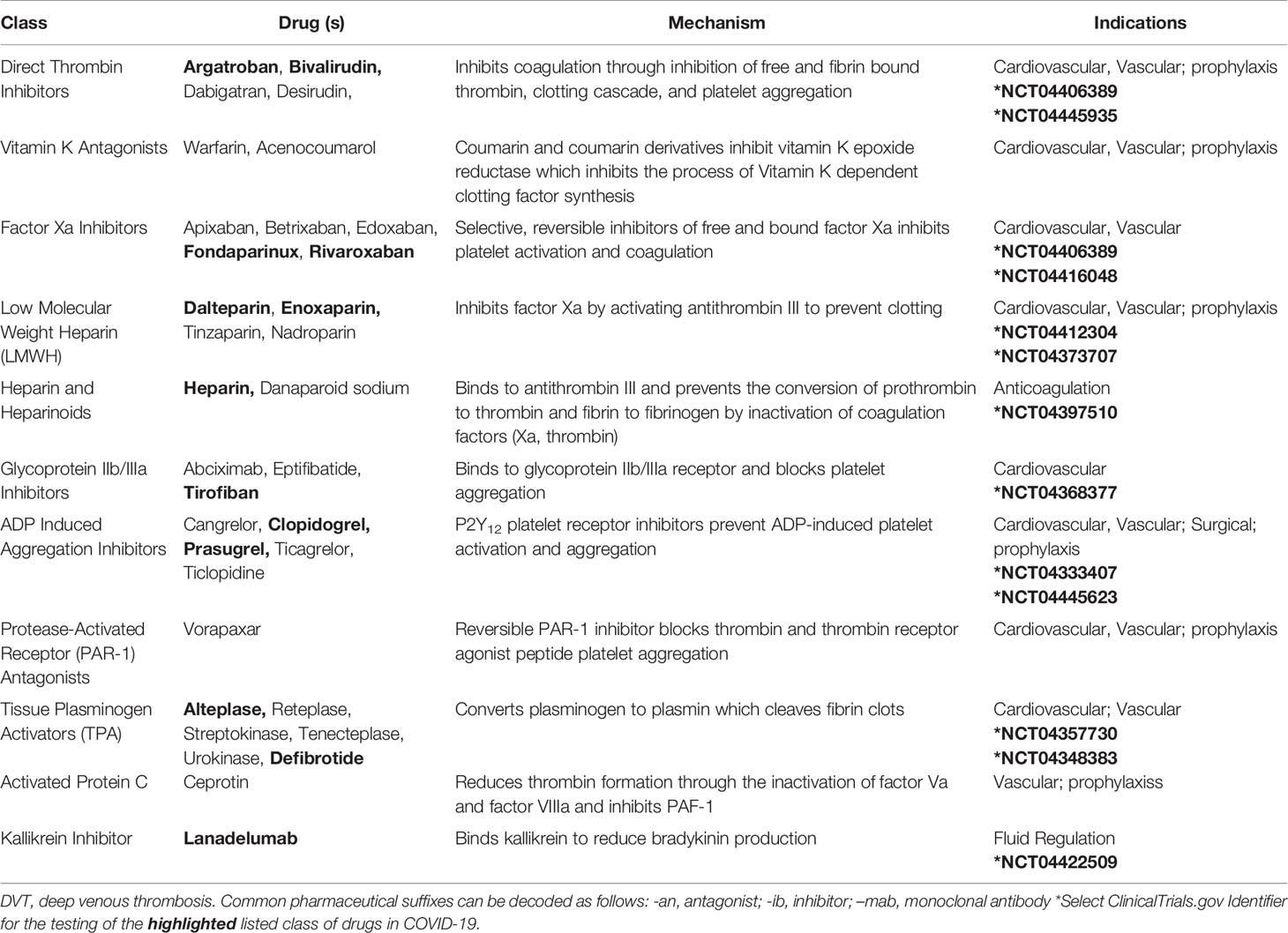
Table 2 Medications that Target the Clotting Cascade.
The Kallikrein-Kinin System
The kallikrein-kinin system is an enzymatic cascade of molecules whose functions are interlaced with the activation of the coagulation and RAAS pathways and associated with vascular permeability and inflammation (Nokkari et al., 2018). The process begins with prekallikrein sourced from tissue and plasma. Tissue prekallikrein (KLK1, true tissue kallikrein) is found in arteries and veins; various organs including heart, pituitary and adrenal glands; and immune cell subsets including neutrophils (Rhaleb et al., 2011; Lizama et al., 2015). Release of proteolytic enzymes in damaged tissue activates tissue prekallikrein to form kallikrein, which catalyzes the formation of kallidin from low molecular weight kininogen (Nokkari et al., 2018). The enzymatic interaction of kallidin with an aminopeptidase generates bradykinin, which is also a derivative of plasma kallikrein (KLKB1, Fletcher factor) (Rhaleb et al., 2011; Nokkari et al., 2018). Plasma prekallikrein is mainly produced in the liver, activated by Hageman factor (factor XII), and preferentially releases bradykinin from high molecular weight kininogen (Nokkari et al., 2018). Kallidin and bradykinin can be further processed by carboxypeptidase-M/kininase I into des-Arg10-kallidin and des-Arg9-bradykinin, respectively, and both of these peptides activate the bradykinin 1 receptor (B1R) (Kakoki and Smithies, 2009). Kallidin and bradykinin activate bradykinin 2 receptor (B2R) and both can be degraded by angiotensin-converting enzyme (ACE, kininase II) into inactive peptides (Kakoki and Smithies, 2009; Nokkari et al., 2018).
The activation of B1R and B2R receptors induces a calcium flux that promotes nitric oxide (NO) production and the activation of phospholipase A2 (PLA2) enzymes, which liberates arachidonic acid from cell membrane phospholipids (Park et al., 2006; Kakoki and Smithies, 2009). Constitutive cyclooxygenases-1 (COX-1) and inducible cyclooxygenases-2 (COX-2), that may be induced by B2R NF-κB activation (Chen et al., 2004), oxidize arachidonic acid to form prostaglandins (Park et al., 2006). Prostaglandins promote vasodilation and can be released from activated neutrophils (e.g. TNF or IL-1ß activation) as a compensatory mediator to down-regulate inflammation (Wright et al., 2010).
Moreover, activation of B1R and B2R on neurons induces a calcium flux that releases SP (Thornton et al., 2010). Tachykinins (SP, neurokinin- A and B, neuropeptide- K and γ) bind neurokinin receptors (NK1, NK2, NK3) where SP exhibits a high affinity for NK1 on nerve cells localized in bronchial vessels, and on epithelial cells, submucosal glands and the vascular endothelium (O’Connor et al., 2004). Consequently, SP contributes to the regulation of cardiovascular and respiratory function, emetic reflux, and the recruitment and activation of leukocytes in response to pathogens, allergens, and self-antigens (O’Connor et al., 2004; Thornton et al., 2010; Mashaghi et al., 2016) (Figure 7).
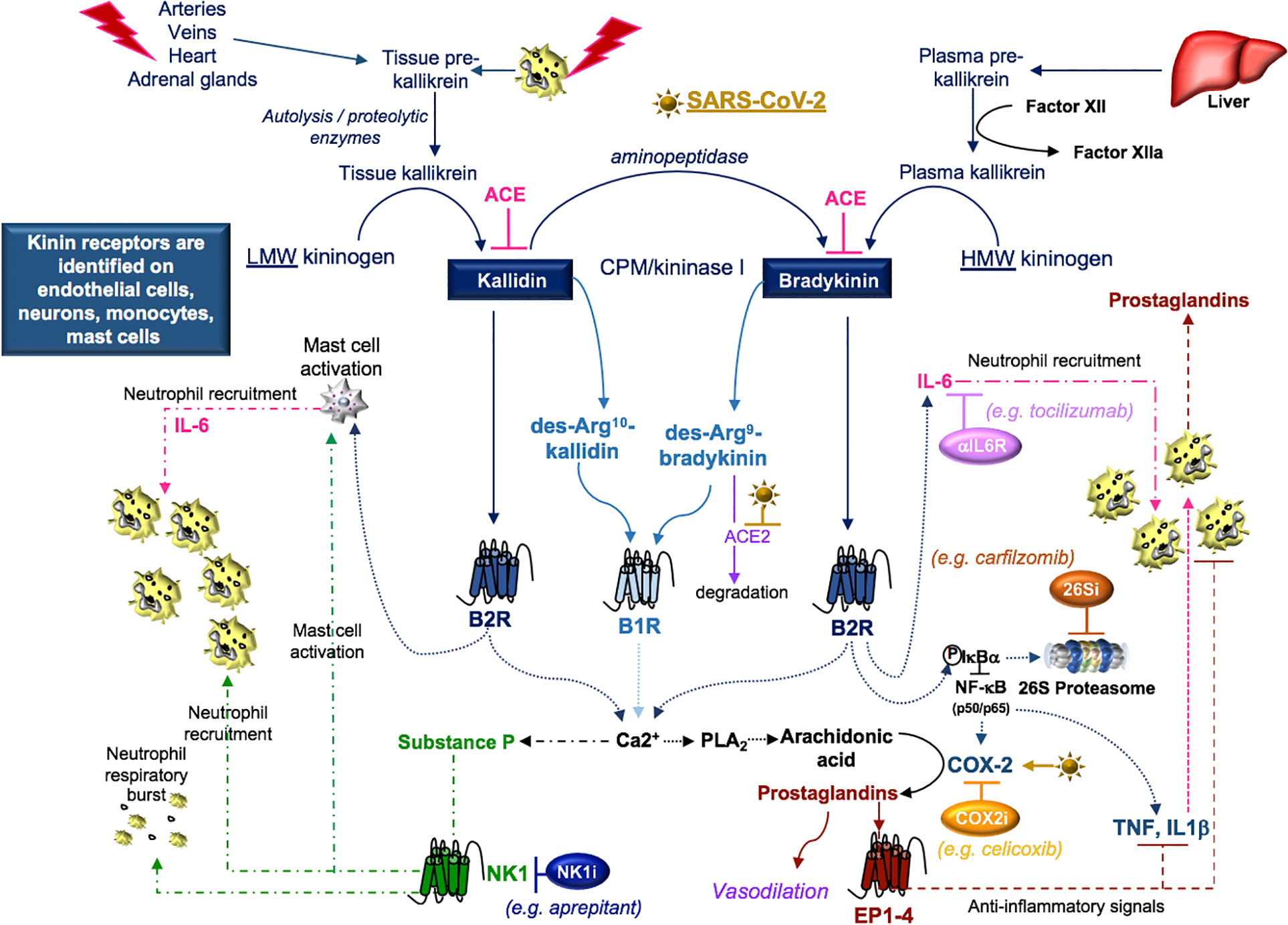
Figure 7 SARS-CoV-2 and kinins. Tissue prekallikrein released from neutrophils and additional cell types is cleaved by cell surface proteases into tissue kallikrein, which promotes the transformation of low molecular weight (LMW) kininogen into kallidin. An aminopeptidase cleaves kallidin and generates bradykinin, which is a product of factor IIa-activated plasma kallikrein and high molecular weight (HMW) kininogen. Kallidin and bradykinin can be cleaved by ACE (kininase II) to form inactive peptides or carboxypeptidase-M (CPM/kininase I), which forms bradykinin 1 receptor (B1R) ligands des-Arg10-kallidin and des-Arg9-bradykinin. ACE2 is the SARS-CoV-2 receptor, suggesting that ACE2 degradation of des-Arg9-bradykinin may be impaired in COVID-19 patients. Both kallidin and bradykinin activate the bradykinin receptor, B2R, which induces the activation of mast cells, the phosphorylation/degradation of IkBα through the 26S proteasome and the release of NF-kB transcription factors involved in the production of cytokines and the prostaglandin-generating enzyme COX-2. Proteasome inhibitors (26Si) antagonize NF-kB activation. B1R and B2R induce a calcium flux that promotes substance P production and the activation of phospholipase A2 (PLA2) enzymes. PLA2 liberates arachidonic acid for COX-2-induced production of prostaglandins which can be blocked with COX-2 inhibitors (COX2i). Prostaglandins induce anti-inflammatory signals. Substance P and bradykinin-induced IL-6 recruit neutrophils into tissues. Substance P also promotes the degranulation of mast cells and neutrophils. Neurokinin-1 (NK1) receptor antagonist (NKi) may block neutrophil recruitment and respiratory burst activity.
SARS-CoV-2 and the Kallikrein-Kinin System
Disruption of the kallikrein-kinin system is identified in cardiovascular diseases, chronic kidney diseases, and Alzheimer disease (Kayashima et al., 2012; Ji et al., 2019) and each of these conditions can increase the susceptibility to SARS-CoV-2 (Ji et al., 2020; Kuo et al., 2020). SARS-CoV-induced loss of ACE2 (Kuba et al., 2005) reduces ACE2-dependent degradation of des-Arg9-bradykinin (Sodhi et al., 2018) and activates this pathway (Figure 7). Further, in a mouse model of acute lung infection, neutrophil influx in response to endotoxin was associated with reduced ACE2 lung protein production. This response was attenuated by a B1R antagonist (Sodhi et al., 2018), suggesting that a loss of ACE2 in the lung enhances neutrophil recruitment through the production of bradykinin and the des-Arg9-bradykinin peptide. In additional studies, neutrophil influx was amplified by B1R- and B2R-induced production of prostaglandins and SP, which promote vasodilation and neutrophil recruitment (O’Connor et al., 2004; Thornton et al., 2010; Nokkari et al., 2018). In COVID-19 patients, the neutrophil-to-lymphocyte ratio increases with disease severity (Chen G. et al., 2020; Qin et al., 2020). This may indicate that blocking kallikrein (ClinicalTrials.gov Identifier: NCT04422509) or B2R (van de Veerdonk et al., 2020) may reduce neutrophilia in COVID-19 patients.
Cellular responses to B1R and B2R activation are implicated in several autoimmune disorders that can manifest in the skin and central nervous system (Dutra, 2017). Increasingly, various atypical skin lesions (Jimenez-Cauhe et al., 2020; Sachdeva et al., 2020) and central nervous disorders (Yin R. et al., 2020), including demyelinating lesions (Zanin et al., 2020), are identified in COVID-19 patients. Whether SAR-CoV-2 contributes to the promotion of autoimmunity in COVID-19 patients through kallikrein-kinin system activation is not known.
An important target of B2R cell signaling is NF-κB (Chen et al., 2004), which is activated by the phosphorylation and targeted proteasomal degradation of inhibitors of NF-κB (e.g. IκBα) (Liu et al., 2017). In two different murine models involving the murine hepatitis virus strain (MHV), conflicting results with the proteasome inhibitor (PS-341/bortezomib/Velcade) revealed improved survival in one model involving the MHV-1 strain (Ma et al., 2010) and increased infection and mortality in the other model involving the MHV-A59 strain (Raaben et al., 2010a). These differences may, in part, be due to differences in the virus (MHV-1 (pneumotropic) versus MHV-A59 (neurotropic)), mouse strains (A/J versus C57BL/6), drug concentrations (0.25 mg/kg versus 1 mg/kg), and the route of administration (subcutaneous versus intraperitoneal), respectively. In both studies, drug-induced inhibition of viral replication in vitro was noted and has been further supported in studies identifying the importance of the ubiquitin proteasome system in viral entry, RNA synthesis and mRNA translation (Raaben et al., 2010b). Bortezomib is commonly used in the treatment of multiple myeloma and in particular patient subsets, severe pulmonary toxicity has been reported (Li et al., 2016). The effects of proteasome inhibition in COVID-19 are not clear; however, the extensive pro-inflammatory response in severe COVID-19 patients (Chen G. et al., 2020; Qin et al., 2020) suggests that drugs targeting NF-κB (Table 3) (Baeuerle and Baichwal, 1997; Miller et al., 2010) could be of benefit in dampening excessive cytokine responses in some COVID-19 patients.
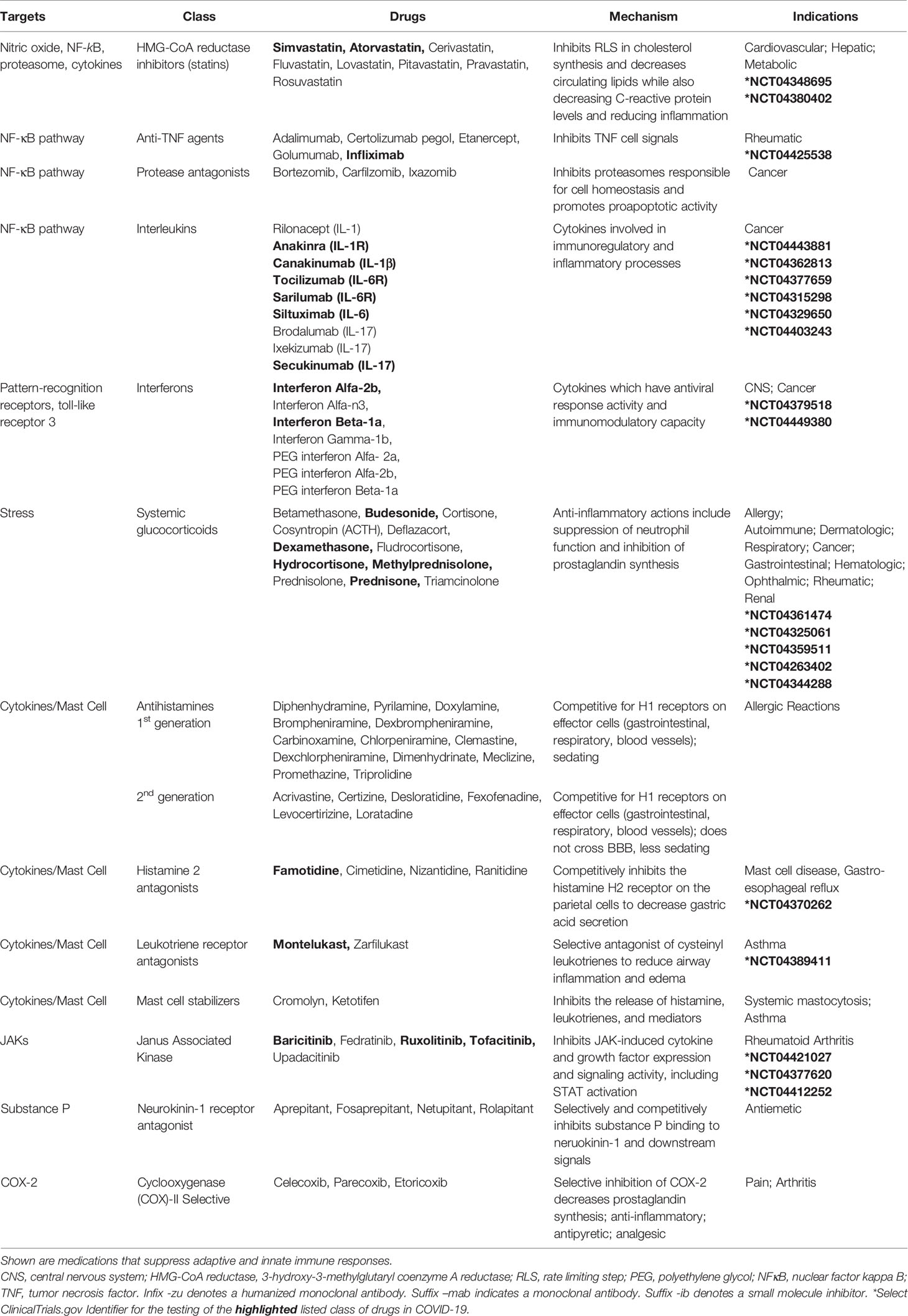
Table 3 Medications that Target the Immune Response.
B2R also signals through NF-κB to induce cytokine production and to activate COX-2, promoting prostaglandin production (Chen et al., 2004). The SARS-CoV components, S-protein (Liu et al., 2007) and N-protein (Yan et al., 2006), are implicated in the activation of COX-2 via calcium dependent and independent signaling. Inhibition of COX-2 via siRNA or the NS-398 COX-2 inhibitor revealed that COX-2 is required for replication of the MHV-A59 strain in vitro (Raaben et al., 2007). In a murine model of influenza A/H5N1, intraperitoneal treatment with the antiviral neuraminidase zanamivir improved viability that was further enhanced with the addition of the COX-2 inhibitor celecoxib (Zheng et al., 2008). In COX-2 deficient mice infected with influenza A/Hong Kong/8/68 (H3N2), the COX-2-deficient mice exhibited enhanced survival compared to wild-type controls (Carey et al., 2005), highlighting the potential benefits of COX-2 inhibition in viral infection.
Non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, antagonize COX enzymes. In a rat model of streptozotocin-induced diabetes, oral gavage with ibuprofen reduced the production of Ang II and ACE but induced the production of ACE2 in cardiac tissue (Qiao et al., 2015). In murine bone marrow-derived dendritic cells and human peripheral blood monocytes, B1R and B2R agonists or an ACE inhibitor (captopril) blocked recombinant interferon-α- or TLR-induced type I interferon responses. In the additional presence of an NSAID (indomethacin), the type I interferon response was restored (Seliga et al., 2018). Because SARS-CoV is able to delay TLR-induced type I interferon responses (Kindler et al., 2016) and activate COX-2 (Liu et al., 2007), a role for kallikrein-kinin signals that stimulate prostaglandin production may exist in the pathogenesis of SARS-CoV and SARS-CoV-2. The potential for NSAIDs to induce ACE2 production and possibly increase opportunities for SARS-CoV-2 to bind host cell ACE2 has created controversy regarding the use these drugs (Russell et al., 2020). NSAIDs are predominantly non-selective inhibitors of COX-1 activity (Warner and Mitchell, 2004). Classes of NSAIDS include salicylates (e.g. aspirin), salicylic acid derivatives (e.g. 5-aminosalicyclic acid), acetic acid derivatives (e.g. sulindac), oxicams (e.g. piroxicam), propionic acid derivatives (e.g. ibuprofen), and COX-2 inhibitors (e.g. celecoxib). Whether selective COX-2 inhibition has an effect on ACE2 production, SARS-COV-2 replication, or COVID-19 inflammation requires further investigation (Figure 7, Table 3). Other classes of NSAIDS, aside from aspirin, have beneficial anti-inflammatory effects in general; however, potential cardiovascular effects may limit use in clinical treatment, and are therefore not included in the associated figure and table.
Lastly, bradykinin-induced B2R signals generate IL-6 (Hayashi et al., 2000; Huang et al., 2003) and the activation of B1R or B2R can result in SP production (O’Connor et al., 2004). Both IL-6 and SP participate in the recruitment of neutrophils (Suwa et al., 2001; O’Connor et al., 2004) and the activation of JAK2 cell signals (Hermans et al., 2018; Schett et al., 2020). SP-initiated intracellular signals induce mast cell degranulation (Hermans et al., 2018) and neutrophil respiratory burst activity (O’Connor et al., 2004), suggesting that blocking SP may dampen inflammation. SP also regulates the lung response to inhaled antigens and promotes the pathogenesis of viruses (Munoz and Covenas, 2014). Moreover, cutaneous skin disorders (Vena et al., 2018), altered taste sensitivity (Huang and Wu, 2018), and gastrointestinal disorders (Koon and Pothoulakis, 2006) are also linked to SP as well as COVID-19 (Galvan Casas et al., 2020; Sedaghat et al., 2020; Wong et al., 2020), suggesting that SP may play a role in COVID-19 symptomology. Antagonists to the SP receptor, NK1, are in development for use as antitussive therapy (Smith et al., 2020), which may indicate that SP-activation is involved in the dry cough that is a common in the clinical presentation of COVID-19. The use of inhibitors to bradykinin, SP, IL-6, their receptors, or the downstream signal JAK2 in the kallikrein-kinin system may provide supportive therapy and modulate the COVID-19 inflammatory response in various clinical trials currently in progress (Table 3).
Summary and Implications
SARS-CoV-2 binding to ACE2 is critical to COVID-19 pathophysiological manifestations that develop through the RAAS pathway, the complement system, the coagulation cascade, and the kallikrein-kinin system. The functional disruption of ACE2 by SARS-CoV-2 tends to promote the activation of these pathways, which may already be at a heightened activation state by underlying diseases in patients most susceptible to the virus. The reduced production and activity of ACE2 promotes the formation of Ang II, which in association with SARS-CoV-2-induced DAMPs and PAMPs, promotes the production of complement. This pro-inflammatory response is enhanced by the reduced levels of the ACE2 cleavage product, Ang-(1-7), which is an anti-inflammatory ligand that signals through Mas, MrgD, and AT2R (Karnik et al., 2017). Understanding the binding interactions between Ang III and Ang-(1-7) for Mas may be important to the pathophysiology of COVID-19. In addition, both Ang III and SP bind MRGPRX2 (Gembardt et al., 2008; Green et al., 2019). Mast cells are activated by MGRPRX2 ligands and complement fragments (C3a, C5a) (el-Lati et al., 1994; Green et al., 2019). The activity of mast cells is identified in each of these pathways. Specifically, mast cells produce renin and chymase in RAAS, activate complement with renin and tryptase, release heparin and plasminogen factors in the hemostatic system, and produce the pro-inflammatory cytokine IL-6 in response to bradykinin (Theoharides et al., 2011). Mast cells are additionally identified in ARDS and associated with fibroproliferation (Liebler et al., 1998), suggesting that these cells, as well as the more common neutrophils, contribute to ARDS that is a severe and sometimes terminal manifestation in COVID-19 patients (Zhu et al., 2020). Mast cells and their downstream pro-inflammatory mediators may therefore be additional intervention targets in COVID-19.
The activation of COX-2 by SARS-CoV suggests that similar responses may occur with SARS-CoV-2. In addition to cell signals from kallikrein-kinin activation and potential SARS-CoV-2 signals, COX-2 and prostaglandin production can be induced by SP activated NF-kB (Sio et al., 2010) and JAK/STAT (Koon et al., 2006) cell signaling pathways. Prostaglandins promote RAAS activity by stimulating the release of renin, which in turn generates Ang I (Atlas, 2007; Peti-Peterdi and Harris, 2010). SARS-CoV-2 inhibition of ACE2 promotes ACE/Ang II/AT1R activity, leading to hypertension-induced SP production (Calvillo et al., 2019). This positive-feedback loop may be supported by the additional release of prostaglandins from neutrophils (Wright et al., 2010) that can also be activated by SP (O’Connor et al., 2004). Thus, because SP is implicated in the pathophysiology of COVID-19 and is a potential factor in nearly all COVID-19 disorders, investigation of interventions that affect SP production and/or functions in COVID-19 patients appears warranted.
Anti-viral therapies will almost certainly be the cornerstone of effective treatments against COVID-19 associated disease. In critically ill patients, additional therapeutic interventions are needed to prevent systemic inflammatory responses that induce organ damage and failure. The physiological manifestations that arise from dysfunction in the RAAS pathway, the complement system, the coagulation cascade, and the kallikrein-kinin system suggest that existing therapeutics along these pathways may be effective in the treatment or mitigation of symptoms in COVID-19 patients.
Author Contributions
Wrote or contributed to the writing of the manuscript: CC, DR, JK. Illustrations: CC. Developed tables: DR. Edited manuscript: CC, DR, JK.
Funding
This work was supported by the Intramural Research Program,NIH, Bethesda, MD, including NIDD (ZO1 DK 043308).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Michael A. Solomon, MD, NIH for insightful comments and the careful review of the manuscript.
References
Ackermann, M., Verleden, S. E., Kuehnel, M., Haverich, A., Welte, T., Laenger, F., et al. (2020). Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 383 (2), 120–128. doi: 10.1056/NEJMoa2015432
Ali, H. (2010). Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol. Lett. 128 (1), 36–45. doi: 10.1016/j.imlet.2009.10.007
Ali, H. (2016). Mas-related G protein coupled receptor-X2: A potential new target for modulating mast cell-mediated allergic and inflammatory diseases. J. Immunobiol. 1 (4). doi: 10.4172/2476-1966.1000115
Alvarez, A., Cerda-Nicolas, M., Naim Abu Nabah, Y., Mata, M., Issekutz, A. C., Panes, J., et al. (2004). Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood 104 (2), 402–408. doi: 10.1182/blood-2003-08-2974
Aslam, R., Speck, E. R., Kim, M., Crow, A. R., Bang, K. W., Nestel, F. P., et al. (2006). Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 107 (2), 637–641. doi: 10.1182/blood-2005-06-2202
Atlas, S. A. (2007). The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 13 (8 Suppl B), 9–20. doi: 10.18553/jmcp.2007.13.s8-b.9
Atri, C., Guerfali, F. Z., Laouini, D. (2018). Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 19 (6), 1801. doi: 10.3390/ijms19061801
Azma, T., Matsubara, Y., Kinoshita, H., Hidaka, I., Shiraishi, S., Nakao, M., et al. (2009). Prothrombotic roles of substance-P, neurokinin-1 receptors and leukocytes in the platelet-dependent clot formation in whole blood. J. Thromb. Thrombolysis 27 (3), 280–286. doi: 10.1007/s11239-008-0215-0
Baeuerle, P. A., Baichwal, V. R. (1997). NF-kappa B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv. Immunol. 65, 111–137. doi: 10.1016/S0065-2776(08)60742-7
Bekassy, Z. D., Kristoffersson, A. C., Rebetz, J., Tati, R., Olin, A. I., Karpman, D. (2018). Aliskiren inhibits renin-mediated complement activation. Kidney Int. 94 (4), 689–700. doi: 10.1016/j.kint.2018.04.004
Bennett, J. S. (2005). Structure and function of the platelet integrin alphaIIbbeta3. J. Clin. Invest. 115 (12), 3363–3369. doi: 10.1172/JCI26989
Bhagavan, N. V., Ha, C.-E. (2015). “Chapter 34 - Biochemistry of Hemostasis,” in Essentials of Medical Biochemistry, 2nd ed. Eds. Bhagavan, N. V., Ha, C.-E. (San Diego: Academic Press), 637–660.
Biswas, N., Maihofer, A. X., Mir, S. A., Rao, F., Zhang, K., Khandrika, S., et al. (2016). Polymorphisms at the F12 and KLKB1 loci have significant trait association with activation of the renin-angiotensin system. BMC Med. Genet. 17, 21. doi: 10.1186/s12881-016-0283-5
Bohlson, S. S., O’Conner, S. D., Hulsebus, H. J., Ho, M. M., Fraser, D. A. (2014). Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front. Immunol. 5, 402. doi: 10.3389/fimmu.2014.00402
Byrd, J. B. V. C., Ram, S., Lerma, E. V. (2019). Nephrology Secrets (FOURTH EDITION). Science Direct CHAPTER 69 - Pharmacologic treatment of hypertension. 477–482.
Calvillo, L., Gironacci, M. M., Crotti, L., Meroni, P. L., Parati, G. (2019). Neuroimmune crosstalk in the pathophysiology of hypertension. Nat. Rev. Cardiol. 16 (8), 476–490. doi: 10.1038/s41569-019-0178-1
Canning, B. J. (2009). Central regulation of the cough reflex: therapeutic implications. Pulm. Pharmacol. Ther. 22 (2), 75–81. doi: 10.1016/j.pupt.2009.01.003
Cantini, F., Niccoli, L., Matarrese, D., Nicastri, E., Stobbione, P., Goletti, D. (2020). Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 81 (2), 318–356. doi: 10.1016/j.jinf.2020.04.017
Carey, M. A., Bradbury, J. A., Seubert, J. M., Langenbach, R., Zeldin, D. C., Germolec, D. R. (2005). Contrasting effects of cyclooxygenase-1 (COX-1) and COX-2 deficiency on the host response to influenza A viral infection. J. Immunol. 175 (10), 6878–6884. doi: 10.4049/jimmunol.175.10.6878
Chao, J., Blanco, G., Wood, J. G., Gonzalez, N. C. (2011). Renin released from mast cells activated by circulating MCP-1 initiates the microvascular phase of the systemic inflammation of alveolar hypoxia. Am. J. Physiol. Heart Circ. Physiol. 301 (6), H2264–H2270. doi: 10.1152/ajpheart.00461.2011
Chen, B. C., Yu, C. C., Lei, H. C., Chang, M. S., Hsu, M. J., Huang, C. L., et al. (2004). Bradykinin B2 receptor mediates NF-kappaB activation and cyclooxygenase-2 expression via the Ras/Raf-1/ERK pathway in human airway epithelial cells. J. Immunol. 173 (8), 5219–5228. doi: 10.4049/jimmunol.173.8.5219
Chen, G., Wu, D., Guo, W., Cao, Y., Huang, D., Wang, H., et al. (2020). Clinical and immunologic features in severe and moderate Coronavirus Disease 2019. J. Clin. Invest. 130 (5), 2620–2629. doi: 10.1172/JCI137244
Chen, N., Zhou, M., Dong, X., Qu, J., Gong, F., Han, Y., et al. (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395 (10223), 507–513. doi: 10.1016/S0140-6736(20)30211-7
Chen, Z. L., Zhang, Q., Lu, Y., Guo, Z. M., Zhang, X., Zhang, W. J., et al. (2020). Distribution of the COVID-19 epidemic and correlation with population emigration from wuhan, China. Chin. Med. J. (Engl.) 133 (9), 1044–1050. doi: 10.1097/CM9.0000000000000782
Cherniack, N. S., Cherniack, E. P. (2007). “Respiratory System,” in Encyclopedia of Gerontology, 2nd ed. Ed. Birren, J. E. (New York: Elsevier), 442–449.
Chilvers, M. A., McKean, M., Rutman, A., Myint, B. S., Silverman, M., O’Callaghan, C. (2001). The effects of coronavirus on human nasal ciliated respiratory epithelium. Eur. Respir. J. 18 (6), 965–970. doi: 10.1183/09031936.01.00093001
Clarke, N. E., Fisher, M. J., Porter, K. E., Lambert, D. W., Turner, A. J. (2012). Angiotensin converting enzyme (ACE) and ACE2 bind integrins and ACE2 regulates integrin signalling. PLoS One 7 (4), e34747. doi: 10.1371/journal.pone.0034747
Cui, S., Chen, S., Li, X., Liu, S., Wang, F. (2020). Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 18 (6), 1421–1424. doi: 10.1111/jth.14830
Curran, C. S., Bolig, T., Torabi-Parizi, P. (2018). Mechanisms and Targeted Therapies for Pseudomonas aeruginosa Lung Infection. Am. J. Respir. Crit. Care Med. 197 (6), 708–727. doi: 10.1164/rccm.201705-1043SO
De Biasi, S., Meschiari, M., Gibellini, L., Bellinazzi, C., Borella, R., Fidanza, L., et al. (2020). Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 11 (1), 3434. doi: 10.1038/s41467-020-17292-4
de Carvalho Santuchi, M., Dutra, M. F., Vago, J. P., Lima, K. M., Galvao, I., de Souza-Neto, F. P., et al. (2019). Angiotensin-(1-7) and Alamandine Promote Anti-inflammatory Response in Macrophages In Vitro and In Vivo. Mediators Inflammation 2019, 2401081. doi: 10.1155/2019/2401081
Diao, B., Wang, C., Tan, Y., Chen, X., Liu, Y., Ning, L., et al. (2020). Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 11, 827. doi: 10.3389/fimmu.2020.00827
Dominguez, S. R., Shrivastava, S., Berglund, A., Qian, Z., Goes, L. G. B., Halpin, R. A., et al. (2014). Isolation, propagation, genome analysis and epidemiology of HKU1 betacoronaviruses. J. Gen. Virol. 95 (Pt 4), 836–848. doi: 10.1099/vir.0.059832-0
Dong, J., Fujii, S., Li, H., Nakabayashi, H., Sakai, M., Nishi, S., et al. (2005). Interleukin-6 and mevastatin regulate plasminogen activator inhibitor-1 through CCAAT/enhancer-binding protein-delta. Arterioscler. Thromb. Vasc. Biol. 25 (5), 1078–1084. doi: 10.1161/01.ATV.0000159701.24372.49
Dutra, R. C. (2017). Kinin receptors: Key regulators of autoimmunity. Autoimmun. Rev. 16 (2), 192–207. doi: 10.1016/j.autrev.2016.12.011
el-Lati, S. G., Dahinden, C. A., Church, M. K. (1994). Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J. Invest. Dermatol. 102 (5), 803–806. doi: 10.1111/1523-1747.ep12378589
Ellul, M. A., Benjamin, L., Singh, B., Lant, S., Michael, B. D., Easton, A., et al. (2020). Neurological associations of COVID-19. Lancet Neurol. 1–17. doi: 10.1016/S1474-4422(20)30221-0
Epelman, S., Tang, W. H., Chen, S. Y., Van Lente, F., Francis, G. S., Sen, S. (2008). Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J. Am. Coll. Cardiol. 52 (9), 750–754. doi: 10.1016/j.jacc.2008.02.088
Fehr, A. R., Perlman, S. (2015). Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 1282, 1–23. doi: 10.1007/978-1-4939-2438-7_1
Fielding, C. A., McLoughlin, R. M., McLeod, L., Colmont, C. S., Najdovska, M., Grail, D., et al. (2008). IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 181 (3), 2189–2195. doi: 10.4049/jimmunol.181.3.2189
Fisher, J., Linder, A. (2017). Heparin-binding protein: a key player in the pathophysiology of organ dysfunction in sepsis. J. Intern. Med. 281 (6), 562–574. doi: 10.1111/joim.12604
Fraga-Silva, R. A., Sorg, B. S., Wankhede, M., Dedeugd, C., Jun, J. Y., Baker, M. B., et al. (2010). ACE2 activation promotes antithrombotic activity. Mol. Med. 16 (5-6), 210–215. doi: 10.2119/molmed.2009.00160
Fung, M., Loubser, P. G., Undar, A., Mueller, M., Sun, C., Sun, W. N., et al. (2001). Inhibition of complement, neutrophil, and platelet activation by an anti-factor D monoclonal antibody in simulated cardiopulmonary bypass circuits. J. Thorac. Cardiovasc. Surg. 122 (1), 113–122. doi: 10.1067/mtc.2001.114777
Gale, A. J. (2011). Continuing education course #2: current understanding of hemostasis. Toxicol. Pathol. 39 (1), 273–280. doi: 10.1177/0192623310389474
Galvan Casas, C., Catala, A., Carretero Hernandez, G., Rodriguez-Jimenez, P., Fernandez Nieto, D., Rodriguez-Villa Lario, A., et al. (2020). Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br. J. Dermatol. 183 (1), 71–77. doi: 10.1111/bjd.19163
Galvan, M. D., Hulsebus, H., Heitker, T., Zeng, E., Bohlson, S. S. (2014). Complement protein C1q and adiponectin stimulate Mer tyrosine kinase-dependent engulfment of apoptotic cells through a shared pathway. J. Innate Immun. 6 (6), 780–792. doi: 10.1159/000363295
Gembardt, F., Grajewski, S., Vahl, M., Schultheiss, H. P., Walther, T. (2008). Angiotensin metabolites can stimulate receptors of the Mas-related genes family. Mol. Cell Biochem. 319 (1-2), 115–123. doi: 10.1007/s11010-008-9884-4
Germonpre, P. R., Bullock, G. R., Lambrecht, B. N., Van De Velde, V., Luyten, W. H., Joos, G. F., et al. (1999). Presence of substance P and neurokinin 1 receptors in human sputum macrophages and U-937 cells. Eur. Respir. J. 14 (4), 776–782. doi: 10.1034/j.1399-3003.1999.14d08.x
Ghebrehiwet, B., Geisbrecht, B. V., Xu, X., Savitt, A. G., Peerschke, E. I. B. (2019). The C1q Receptors: Focus on gC1qR/p33 (C1qBP, p32, HABP-1)(1). Semin. Immunol. 45, 101338. doi: 10.1016/j.smim.2019.101338
Giang, J., Seelen, M. A. J., van Doorn, M. B. A., Rissmann, R., Prens, E. P., Damman, J. (2018). Complement Activation in Inflammatory Skin Diseases. Front. Immunol. 9, 639. doi: 10.3389/fimmu.2018.00639
Glowacka, I., Bertram, S., Muller, M. A., Allen, P., Soilleux, E., Pfefferle, S., et al. (2011). Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 85 (9), 4122–4134. doi: 10.1128/JVI.02232-10
Goldstein, B., Trivedi, M., Speth, R. C. (2017). Alterations in Gene Expression of Components of the Renin-Angiotensin System and Its Related Enzymes in Lung Cancer. Lung Cancer Int. 2017, 6914976. doi: 10.1155/2017/6914976
Gonzales, J. N., Lucas, R., Verin, A. D. (2015). The Acute Respiratory Distress Syndrome: Mechanisms and Perspective Therapeutic Approaches. Austin J. Vasc. Med. 2 (1), 1009.
Gopallawa, I., Uhal, B. D. (2016). Angiotensin-(1-7)/mas inhibits apoptosis in alveolar epithelial cells through upregulation of MAP kinase phosphatase-2. Am. J. Physiol. Lung Cell Mol. Physiol. 310 (3), L240–L248. doi: 10.1152/ajplung.00187.2015
Gordon, S., Pluddemann, A. (2018). Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front. Immunol. 9, 127. doi: 10.3389/fimmu.2018.00127
Graham, A. C., Temple, R. M., Obar, J. J. (2015). Mast cells and influenza a virus: association with allergic responses and beyond. Front. Immunol. 6, 238. doi: 10.3389/fimmu.2015.00238
Gralinski, L. E., Sheahan, T. P., Morrison, T. E., Menachery, V. D., Jensen, K., Leist, S. R., et al. (2018). Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 9 (5), e01753–18. doi: 10.1128/mBio.01753-18
Green, D. P., Limjunyawong, N., Gour, N., Pundir, P., Dong, X. (2019). A Mast-Cell-Specific Receptor Mediates Neurogenic Inflammation and Pain. Neuron 101 (3), 412–420.e413. doi: 10.1016/j.neuron.2019.01.012
Gregoire, M., Uhel, F., Lesouhaitier, M., Gacouin, A., Guirriec, M., Mourcin, F., et al. (2018). Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 52 (2), 1702590. doi: 10.1183/13993003.02590-2017
Grifoni, A., Sidney, J., Zhang, Y., Scheuermann, R. H., Peters, B., Sette, A. (2020). A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe. 27 (4), 671–680. doi: 10.1016/j.chom.2020.03.002
Guan, W. J., Ni, Z. Y., Hu, Y., Liang, W. H., Ou, C. Q., He, J. X., et al. (2020). Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 382 (18), 1708–1720. doi: 10.1056/NEJMoa2002032
Guichard, J. L., Clark, D, 3., Calhoun, D. A., Ahmed, M. I. (2013). Aldosterone receptor antagonists: current perspectives and therapies. Vasc. Health Risk Manag. 9, 321–331. doi: 10.2147/VHRM.S33759
Haga, S., Yamamoto, N., Nakai-Murakami, C., Osawa, Y., Tokunaga, K., Sata, T., et al. (2008). Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. U.S.A. 105 (22), 7809–7814. doi: 10.1073/pnas.0711241105
Hajishengallis, G., Lambris, J. D. (2016). More than complementing Tolls: complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol. Rev. 274 (1), 233–244. doi: 10.1111/imr.12467
Hamming, I., Timens, W., Bulthuis, M. L., Lely, A. T., Navis, G., van Goor, H. (2004). Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203 (2), 631–637. doi: 10.1002/path.1570
Hayashi, R., Yamashita, N., Matsui, S., Fujita, T., Araya, J., Sassa, K., et al. (2000). Bradykinin stimulates IL-6 and IL-8 production by human lung fibroblasts through ERK- and p38 MAPK-dependent mechanisms. Eur. Respir. J. 16 (3), 452–458. doi: 10.1034/j.1399-3003.2000.016003452.x
Hermans, M. A. W., Schrijver, B., van Holten-Neelen, C., Gerth van Wijk, R., van Hagen, P. M., van Daele, P. L. A., et al. (2018). The JAK1/JAK2- inhibitor ruxolitinib inhibits mast cell degranulation and cytokine release. Clin. Exp. Allergy 48 (11), 1412–1420. doi: 10.1111/cea.13217
Herold, T., Jurinovic, V., Arnreich, C., Lipworth, B. J., Hellmuth, J. C., Bergwelt-Baildon, M. V., et al. (2020). Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J. Allergy Clin. Immunol. 146 (1), 128–136. doi: 10.1016/j.jaci.2020.05.008
Heurich, A., Hofmann-Winkler, H., Gierer, S., Liepold, T., Jahn, O., Pohlmann, S. (2014). TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 88 (2), 1293–1307. doi: 10.1128/JVI.02202-13
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Kruger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181 (2), 271–280. doi: 10.1016/j.cell.2020.02.052
Hrenak, J., Paulis, L., Simko, F. (2016). Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. Int. J. Mol. Sci. 17 (7), 1098. doi: 10.3390/ijms17071098
Huang, A. Y., Wu, S. Y. (2018). Substance P as a putative efferent transmitter mediates GABAergic inhibition in mouse taste buds. Br. J. Pharmacol. 175 (7), 1039–1053. doi: 10.1111/bph.14142
Huang, C. D., Tliba, O., Panettieri, R. A., Jr., Amrani, Y. (2003). Bradykinin induces interleukin-6 production in human airway smooth muscle cells: modulation by Th2 cytokines and dexamethasone. Am. J. Respir. Cell Mol. Biol. 28 (3), 330–338. doi: 10.1165/rcmb.2002-0040OC
Huang, X., Xiu, H., Zhang, S., Zhang, G. (2018). The Role of Macrophages in the Pathogenesis of ALI/ARDS. Mediators Inflammation 2018, 1264913. doi: 10.1155/2018/1264913
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395 (10223), 497–506. doi: 10.1016/S0140-6736(20)30183-5
Humphries, D. E., Wong, G. W., Friend, D. S., Gurish, M. F., Qiu, W. T., Huang, C., et al. (1999). Heparin is essential for the storage of specific granule proteases in mast cells. Nature 400 (6746), 769–772. doi: 10.1038/23481
Huntington, J. A. (2005). Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 3 (8), 1861–1872. doi: 10.1111/j.1538-7836.2005.01363.x
Igic, R., Behnia, R. (2003). Properties and distribution of angiotensin I converting enzyme. Curr. Pharm. Des. 9 (9), 697–706. doi: 10.2174/1381612033455459
Imai, Y., Kuba, K., Rao, S., Huan, Y., Guo, F., Guan, B., et al. (2005). Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436 (7047), 112–116. doi: 10.1038/nature03712
Ji, B., Wang, Q., Xue, Q., Li, W., Li, X., Wu, Y. (2019). The Dual Role of Kinin/Kinin Receptors System in Alzheimer’s Disease. Front. Mol. Neurosci. 12, 234. doi: 10.3389/fnmol.2019.00234
Ji, H. L., Zhao, R., Matalon, S., Matthay, M. A. (2020). Elevated Plasmin(ogen) as a Common Risk Factor for COVID-19 Susceptibility. Physiol. Rev. 100 (3), 1065–1075. doi: 10.1152/physrev.00013.2020
Jiang, F., Yang, J., Zhang, Y., Dong, M., Wang, S., Zhang, Q., et al. (2014). Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat. Rev. Cardiol. 11 (7), 413–426. doi: 10.1038/nrcardio.2014.59
Jiang, Z., Zhou, Q., Gu, C., Li, D., Zhu, L. (2017). Depletion of circulating monocytes suppresses IL-17 and HMGB1 expression in mice with LPS-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 312 (2), L231–L242. doi: 10.1152/ajplung.00389.2016
Jimenez-Cauhe, J., Ortega-Quijano, D., Carretero-Barrio, I., Suarez-Valle, A., Saceda-Corralo, D., Moreno-Garcia Del Real, C., et al. (2020). Erythema multiforme-like eruption in patients with COVID-19 infection: clinical and histological findings. Clin. Exp. Dermatol. doi: 10.1111/ced.14281
Johnson, M. B., Young, A. D., Marriott, I. (2016). The Therapeutic Potential of Targeting Substance P/NK-1R Interactions in Inflammatory CNS Disorders. Front. Cell Neurosci. 10, 296. doi: 10.3389/fncel.2016.00296
Joskova, M., Mokry, J., Franova, S. (2020). Respiratory Cilia as a Therapeutic Target of Phosphodiesterase Inhibitors. Front. Pharmacol. 11, 609. doi: 10.3389/fphar.2020.00609
Kaira, B. G., Slater, A., McCrae, K. R., Dreveny, I., Sumya, U. M., Mutch, N. J., et al. (2020). Factor XII and kininogen asymmetric assembly with gC1qR/C1QBP/P32 is governed by allostery. Blood. doi: 10.1182/blood.2020004818
Kakoki, M., Smithies, O. (2009). The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int. 75 (10), 1019–1030. doi: 10.1038/ki.2008.647
Kaplanski, G., Marin, V., Montero-Julian, F., Mantovani, A., Farnarier, C. (2003). IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 24 (1), 25–29. doi: 10.1016/s1471-4906(02)00013-3
Karnik, S. S., Singh, K. D., Tirupula, K., Unal, H. (2017). Significance of angiotensin 1-7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. Br. J. Pharmacol. 174 (9), 737–753. doi: 10.1111/bph.13742
Katz, Y., Revel, M., Strunk, R. C. (1989). Interleukin 6 stimulates synthesis of complement proteins factor B and C3 in human skin fibroblasts. Eur. J. Immunol. 19 (6), 983–988. doi: 10.1002/eji.1830190605
Kawano, H., Cody, R. J., Graf, K., Goetze, S., Kawano, Y., Schnee, J., et al. (2000). Angiotensin II enhances integrin and alpha-actinin expression in adult rat cardiac fibroblasts. Hypertension 35 (1 Pt 2), 273–279. doi: 10.1161/01.hyp.35.1.273
Kayashima, Y., Smithies, O., Kakoki, M. (2012). The kallikrein-kinin system and oxidative stress. Curr. Opin. Nephrol. Hypertens. 21 (1), 92–96. doi: 10.1097/MNH.0b013e32834d54b1
Keidar, S., Gamliel-Lazarovich, A., Kaplan, M., Pavlotzky, E., Hamoud, S., Hayek, T., et al. (2005). Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ. Res. 97 (9), 946–953. doi: 10.1161/01.RES.0000187500.24964.7A
Khan, M. M., Douglas, S. D., Benton, T. D. (2012). Substance P-neurokinin-1 receptor interaction upregulates monocyte tissue factor. J. Neuroimmunol. 242 (1-2), 1–8. doi: 10.1016/j.jneuroim.2011.10.012
Khoo, J. K., Barnes, H., Key, S., Glaspole, I. N., Ostor, A. J. (2020). Pulmonary adverse events of small molecule JAK inhibitors in autoimmune disease: systematic review and meta-analysis. Rheumatol. (Oxford). doi: 10.1093/rheumatology/keaa117
Kindler, E., Thiel, V., Weber, F. (2016). Interaction of SARS and MERS Coronaviruses with the Antiviral Interferon Response. Adv. Virus Res. 96, 219–243. doi: 10.1016/bs.aivir.2016.08.006
Kleij, H. P., Bienenstock, J. (2005). Significance of Conversation between Mast Cells and Nerves. Allergy Asthma Clin. Immunol. 1 (2), 65–80. doi: 10.1186/1710-1492-1-2-65
Klopfenstein, T., Zayet, S., Lohse, A., Balblanc, J. C., Badie, J., Royer, P. Y., et al. (2020). Tocilizumab therapy reduced intensive care unit admissions and/or mortality in COVID-19 patients. Med. Mal. Infect. 50 (5), 397–400. doi: 10.1016/j.medmal.2020.05.001
Koon, H. W., Pothoulakis, C. (2006). Immunomodulatory properties of substance P: the gastrointestinal system as a model. Ann. N. Y. Acad. Sci. 1088, 23–40. doi: 10.1196/annals.1366.024
Koon, H. W., Zhao, D., Zhan, Y., Rhee, S. H., Moyer, M. P., Pothoulakis, C. (2006). Substance P stimulates cyclooxygenase-2 and prostaglandin E2 expression through JAK-STAT activation in human colonic epithelial cells. J. Immunol. 176 (8), 5050–5059. doi: 10.4049/jimmunol.176.8.5050
Koupenova, M., Vitseva, O., MacKay, C. R., Beaulieu, L. M., Benjamin, E. J., Mick, E., et al. (2014). Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 124 (5), 791–802. doi: 10.1182/blood-2013-11-536003
Koupenova, M., Corkrey, H. A., Vitseva, O., Manni, G., Pang, C. J., Clancy, L., et al. (2019). The role of platelets in mediating a response to human influenza infection. Nat. Commun. 10 (1), 1780. doi: 10.1038/s41467-019-09607-x
Kriz, W. (2004). Adenosine and ATP: traffic regulators in the kidney. J. Clin. Invest. 114 (5), 611–613. doi: 10.1172/JCI22669
Kuba, K., Imai, Y., Rao, S., Gao, H., Guo, F., Guan, B., et al. (2005). A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11 (8), 875–879. doi: 10.1038/nm1267
Kuba, K., Imai, Y., Penninger, J. M. (2006). Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 6 (3), 271–276. doi: 10.1016/j.coph.2006.03.001
Kuderer, N. M., Choueiri, T. K., Shah, D. P., Shyr, Y., Rubinstein, S. M., Rivera, D. R., et al. (2020). Clinical impact of COVID-19 on patients with cancer (CCC19): a cohort study. Lancet 395 (10241), 1907–1918. doi: 10.1016/S0140-6736(20)31187-9
Kuo, C. L., Pilling, L. C., Atkins, J. L., Masoli, J. A. H., Delgado, J., Kuchel, G. A., et al. (2020). APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. J. Gerontol. A. Biol. Sci. Med. Sci. doi: 10.1093/gerona/glaa131
Leth-Larsen, R., Zhong, F., Chow, V. T., Holmskov, U., Lu, J. (2007). The SARS coronavirus spike glycoprotein is selectively recognized by lung surfactant protein D and activates macrophages. Immunobiology 212 (3), 201–211. doi: 10.1016/j.imbio.2006.12.001
Li, S., Wang, X., Qiu, J., Si, Q., Wang, H., Guo, H., et al. (2006). Angiotensin II stimulates endothelial integrin beta3 expression via nuclear factor-kappaB activation. Exp. Aging Res. 32 (1), 47–60. doi: 10.1080/01902140500325049
Li, J., Chen, S., Hu, Y., Cai, J. (2016). Bortezomib-induced severe pulmonary complications in multiple myeloma: A case report and literature review. Oncol. Lett. 11 (3), 2255–2260. doi: 10.3892/ol.2016.4204
Li, S., Jiang, L., Li, X., Lin, F., Wang, Y., Li, B., et al. (2020). Clinical and pathological investigation of patients with severe COVID-19. JCI Insight 5 (12), e138070. doi: 10.1172/jci.insight.138070
Liebler, J. M., Qu, Z., Buckner, B., Powers, M. R., Rosenbaum, J. T. (1998). Fibroproliferation and mast cells in the acute respiratory distress syndrome. Thorax 53 (10), 823–829. doi: 10.1136/thx.53.10.823
Lim, J. E., Chung, E., Son, Y. (2017). A neuropeptide, Substance-P, directly induces tissue-repairing M2 like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNgamma. Sci. Rep. 7 (1), 9417. doi: 10.1038/s41598-017-09639-7
Lin, A. M., Rubin, C. J., Khandpur, R., Wang, J. Y., Riblett, M., Yalavarthi, S., et al. (2011). Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 187 (1), 490–500. doi: 10.4049/jimmunol.1100123
Liu, M., Yang, Y., Gu, C., Yue, Y., Wu, K. K., Wu, J., et al. (2007). Spike protein of SARS-CoV stimulates cyclooxygenase-2 expression via both calcium-dependent and calcium-independent protein kinase C pathways. FASEB J. 21 (7), 1586–1596. doi: 10.1096/fj.06-6589com
Liu, T., Zhang, L., Joo, D., Sun, S. C. (2017). NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2, 17023. doi: 10.1038/sigtrans.2017.23
Lizama, A. J., Andrade, Y., Colivoro, P., Sarmiento, J., Matus, C. E., Gonzalez, C. B., et al. (2015). Expression and bioregulation of the kallikrein-related peptidases family in the human neutrophil. Innate Immun. 21 (6), 575–586. doi: 10.1177/1753425914566083
Lubbers, R., van Essen, M. F., van Kooten, C., Trouw, L. A. (2017). Production of complement components by cells of the immune system. Clin. Exp. Immunol. 188 (2), 183–194. doi: 10.1111/cei.12952
Lukassen, S., Lorenz Chua, R., Trefzer, T., Kahn, N. C., Schneider, M. A., Muley, T., et al. (2020). SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 39, 1–15. doi: 10.15252/embj.20105114
Luo, P., Liu, Y., Qiu, L., Liu, X., Liu, D., Li, J. (2020). Tocilizumab treatment in COVID-19: A single center experience. J. Med. Virol. 92 (7), 814–818. doi: 10.1002/jmv.25801
Luo, X., Zhou, W., Yan, X., Guo, T., Wang, B., Xia, H., et al. (2020). Prognostic value of C-reactive protein in patients with COVID-19. Clin. Infect. Dis. doi: 10.1093/cid/ciaa641
Luther, J. M., Gainer, J. V., Murphey, L. J., Yu, C., Vaughan, D. E., Morrow, J. D., et al. (2006). Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 48 (6), 1050–1057. doi: 10.1161/01.HYP.0000248135.97380.76
Ma, X. Z., Bartczak, A., Zhang, J., Khattar, R., Chen, L., Liu, M. F., et al. (2010). Proteasome inhibition in vivo promotes survival in a lethal murine model of severe acute respiratory syndrome. J. Virol. 84 (23), 12419–12428. doi: 10.1128/JVI.01219-10
Madhur, M. S., Lob, H. E., McCann, L. A., Iwakura, Y., Blinder, Y., Guzik, T. J., et al. (2010). Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55 (2), 500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094
Magro, C., Mulvey, J. J., Berlin, D., Nuovo, G., Salvatore, S., Harp, J., et al. (2020). Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 220, 1–13. doi: 10.1016/j.trsl.2020.04.007
Major, C. D., Santulli, R. J., Derian, C. K., Andrade-Gordon, P. (2003). Extracellular mediators in atherosclerosis and thrombosis: lessons from thrombin receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 23 (6), 931–939. doi: 10.1161/01.ATV.0000070100.47907.26
Mancia, G., Rea, F., Ludergnani, M., Apolone, G., Corrao, G. (2020). Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 382 (25), 2431–2440. doi: 10.1056/NEJMoa2006923
Mashaghi, A., Marmalidou, A., Tehrani, M., Grace, P. M., Pothoulakis, C., Dana, R. (2016). Neuropeptide substance P and the immune response. Cell Mol. Life Sci. 73 (22), 4249–4264. doi: 10.1007/s00018-016-2293-z
Mazzone, S. B., Undem, B. J. (2016). Vagal Afferent Innervation of the Airways in Health and Disease. Physiol. Rev. 96 (3), 975–1024. doi: 10.1152/physrev.00039.2015
McElroy, M. C., Kasper, M. (2004). The use of alveolar epithelial type I cell-selective markers to investigate lung injury and repair. Eur. Respir. J. 24 (4), 664–673. doi: 10.1183/09031936.04.00096003
Mease, P., Roussou, E., Burmester, G. R., Goupille, P., Gottlieb, A., Moriarty, S. R., et al. (2019). Safety of Ixekizumab in Patients With Psoriatic Arthritis: Results From a Pooled Analysis of Three Clinical Trials. Arthritis Care Res. (Hoboken) 71 (3), 367–378. doi: 10.1002/acr.23738
Mehta, N., Kalra, A., Nowacki, A. S., Anjewierden, S., Han, Z., Bhat, P., et al. (2020). Association of Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. doi: 10.1001/jamacardio.2020.1855
Meng, J., Xiao, G., Zhang, J., He, X., Ou, M., Bi, J., et al. (2020). Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 9 (1), 757–760. doi: 10.1080/22221751.2020.1746200
Milewska, A., Nowak, P., Owczarek, K., Szczepanski, A., Zarebski, M., Hoang, A., et al. (2018). Entry of Human Coronavirus NL63 into the Cell. J. Virol. 92 (3), e01933–17. doi: 10.1128/JVI.01933-17
Miller, S. C., Huang, R., Sakamuru, S., Shukla, S. J., Attene-Ramos, M. S., Shinn, P., et al. (2010). Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem. Pharmacol. 79 (9), 1272–1280. doi: 10.1016/j.bcp.2009.12.021
Mollerherm, H., von Kockritz-Blickwede, M., Branitzki-Heinemann, K. (2016). Antimicrobial Activity of Mast Cells: Role and Relevance of Extracellular DNA Traps. Front. Immunol. 7, 265. doi: 10.3389/fimmu.2016.00265
Monteil, V., Kwon, H., Prado, P., Hagelkruys, A., Wimmer, R. A., Stahl, M., et al. (2020). Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 181 (4), 905–913. doi: 10.1016/j.cell.2020.04.004
Muhovic, D., Bojovic, J., Bulatovic, A., Vukcevic, B., Ratkovic, M., Lazovic, R., et al. (2020). First case of drug-induced liver injury associated with the use of tocilizumab in a patient with COVID-19. Liver Int. doi: 10.1111/liv.14516
Munoz, M., Covenas, R. (2014). Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 46 (7), 1727–1750. doi: 10.1007/s00726-014-1736-9
Munoz-Durango, N., Fuentes, C. A., Castillo, A. E., Gonzalez-Gomez, L. M., Vecchiola, A., Fardella, C. E., et al. (2016). Role of the Renin-Angiotensin-Aldosterone System beyond Blood Pressure Regulation: Molecular and Cellular Mechanisms Involved in End-Organ Damage during Arterial Hypertension. Int. J. Mol. Sci. 17 (7), 797. doi: 10.3390/ijms17070797
Nehme, A., Zouein, F. A., Zayeri, Z. D., Zibara, K. (2019). An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 6 (2), 14. doi: 10.3390/jcdd6020014
Niethamer, T. K., Stabler, C. T., Leach, J. P., Zepp, J. A., Morley, M. P., Babu, A., et al. (2020). Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. Elife 9, e53072. doi: 10.7554/eLife.53072
Nokkari, A., Abou-El-Hassan, H., Mechref, Y., Mondello, S., Kindy, M. S., Jaffa, A. A., et al. (2018). Implication of the Kallikrein-Kinin system in neurological disorders: Quest for potential biomarkers and mechanisms. Prog. Neurobiol. 165-167, 26–50. doi: 10.1016/j.pneurobio.2018.01.003
O’Connor, T. M., O’Connell, J., O’Brien, D. I., Goode, T., Bredin, C. P., Shanahan, F. (2004). The role of substance P in inflammatory disease. J. Cell Physiol. 201 (2), 167–180. doi: 10.1002/jcp.20061
Oudkerk, M., Buller, H. R., Kuijpers, D., van Es, N., Oudkerk, S. F., McLoud, T. C., et al. (2020). Diagnosis, Prevention, and Treatment of Thromboembolic Complications in COVID-19: Report of the National Institute for Public Health of the Netherlands. Radiology, 201629. doi: 10.1148/radiol.2020201629
Padmanabhan, S. (Ed.) (2014). “Handbook of Pharmacogenomics and Stratified Medicine,” in Handbook of Pharmacogenomics and Stratified Medicine (San Diego: Academic Press), iii.
Park, J. Y., Pillinger, M. H., Abramson, S. B. (2006). Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin. Immunol. 119 (3), 229–240. doi: 10.1016/j.clim.2006.01.016
Perianin, A., Snyderman, R., Malfroy, B. (1989). Substance P primes human neutrophil activation: a mechanism for neurological regulation of inflammation. Biochem. Biophys. Res. Commun. 161 (2), 520–524. doi: 10.1016/0006-291x(89)92630-2
Peters-Hall, J. R., Brown, K. J., Pillai, D. K., Tomney, A., Garvin, L. M., Wu, X., et al. (2015). Quantitative proteomics reveals an altered cystic fibrosis in vitro bronchial epithelial secretome. Am. J. Respir. Cell Mol. Biol. 53 (1), 22–32. doi: 10.1165/rcmb.2014-0256RC
Peti-Peterdi, J., Harris, R. C. (2010). Macula densa sensing and signaling mechanisms of renin release. J. Am. Soc. Nephrol. 21 (7), 1093–1096. doi: 10.1681/ASN.2009070759
Piqueras, L., Kubes, P., Alvarez, A., O’Connor, E., Issekutz, A. C., Esplugues, J. V., et al. (2000). Angiotensin II induces leukocyte-endothelial cell interactions in vivo via AT(1) and AT(2) receptor-mediated P-selectin upregulation. Circulation 102 (17), 2118–2123. doi: 10.1161/01.cir.102.17.2118
Potey, P. M., Rossi, A. G., Lucas, C. D., Dorward, D. A. (2019). Neutrophils in the initiation and resolution of acute pulmonary inflammation: understanding biological function and therapeutic potential. J. Pathol. 247 (5), 672–685. doi: 10.1002/path.5221
Qiao, W., Wang, C., Chen, B., Zhang, F., Liu, Y., Lu, Q., et al. (2015). Ibuprofen attenuates cardiac fibrosis in streptozotocin-induced diabetic rats. Cardiology 131 (2), 97–106. doi: 10.1159/000375362
Qin, C., Zhou, L., Hu, Z., Zhang, S., Yang, S., Tao, Y., et al. (2020). Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 71 (15), 762–768. doi: 10.1093/cid/ciaa248
Raaben, M., Einerhand, A. W., Taminiau, L. J., van Houdt, M., Bouma, J., Raatgeep, R. H., et al. (2007). Cyclooxygenase activity is important for efficient replication of mouse hepatitis virus at an early stage of infection. Virol. J. 4, 55. doi: 10.1186/1743-422X-4-55
Raaben, M., Grinwis, G. C., Rottier, P. J., de Haan, C. A. (2010a). The proteasome inhibitor Velcade enhances rather than reduces disease in mouse hepatitis coronavirus-infected mice. J. Virol. 84 (15), 7880–7885. doi: 10.1128/JVI.00486-10
Raaben, M., Posthuma, C. C., Verheije, M. H., te Lintelo, E. G., Kikkert, M., Drijfhout, J. W., et al. (2010b). The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 84 (15), 7869–7879. doi: 10.1128/JVI.00485-10
Ranucci, M., Ballotta, A., Di Dedda, U., Bayshnikova, E., Dei Poli, M., Resta, M., et al. (2020). The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 18 (7), 1747–1751. doi: 10.1111/jth.14854
Rega, G., Kaun, C., Weiss, T. W., Demyanets, S., Zorn, G., Kastl, S. P., et al. (2005). Inflammatory cytokines interleukin-6 and oncostatin m induce plasminogen activator inhibitor-1 in human adipose tissue. Circulation 111 (15), 1938–1945. doi: 10.1161/01.CIR.0000161823.55935.BE
Reis, E. S., Mastellos, D. C., Hajishengallis, G., Lambris, J. D. (2019). New insights into the immune functions of complement. Nat. Rev. Immunol. 19 (8), 503–516. doi: 10.1038/s41577-019-0168-x
Reyes Gil, M. (2019). “Chapter 91 - Overview of the Coagulation System,” in Transfusion Medicine and Hemostasis, 3rd ed. Eds. Shaz, B. H., Hillyer, C. D., Reyes Gil, M. (Elsevier), 559–564.
Reynolds, H. R., Adhikari, S., Pulgarin, C., Troxel, A. B., Iturrate, E., Johnson, S. B., et al. (2020). Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N. Engl. J. Med. 382 (25), 2441–2448. doi: 10.1056/NEJMoa2008975
Rhaleb, N. E., Yang, X. P., Carretero, O. A. (2011). The kallikrein-kinin system as a regulator of cardiovascular and renal function. Compr. Physiol. 1 (2), 971–993. doi: 10.1002/cphy.c100053
Ricklin, D., Lambris, J. D. (2007). Complement-targeted therapeutics. Nat. Biotechnol. 25 (11), 1265–1275. doi: 10.1038/nbt1342
Romano, M., Sironi, M., Toniatti, C., Polentarutti, N., Fruscella, P., Ghezzi, P., et al. (1997). Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 6 (3), 315–325. doi: 10.1016/s1074-7613(00)80334-9
Roschewski, M., Lionakis, M. S., Sharman, J. P., Roswarski, J., Goy, A., Monticelli, M. A., et al. (2020). Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 5 (48), eabd0110. doi: 10.1126/sciimmunol.abd0110
Rosseau, S., Hammerl, P., Maus, U., Walmrath, H. D., Schutte, H., Grimminger, F., et al. (2000). Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 279 (1), L25–L35. doi: 10.1152/ajplung.2000.279.1.L25
Russell, B., Moss, C., Rigg, A., Van Hemelrijck, M. (2020). COVID-19 and treatment with NSAIDs and corticosteroids: should we be limiting their use in the clinical setting? Ecancermedicalscience 14, 1023. doi: 10.3332/ecancer.2020.1023
Sachdeva, M., Gianotti, R., Shah, M., Lucia, B., Tosi, D., Veraldi, S., et al. (2020). Cutaneous manifestations of COVID-19: Report of three cases and a review of literature. J. Dermatol. Sci. 98 (2), 75–81. doi: 10.1016/j.jdermsci.2020.04.011
Santos, R. A. S., Sampaio, W. O., Alzamora, A. C., Motta-Santos, D., Alenina, N., Bader, M., et al. (2018). The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 98 (1), 505–553. doi: 10.1152/physrev.00023.2016
Schett, G., Sticherling, M., Neurath, M. F. (2020). COVID-19: risk for cytokine targeting in chronic inflammatory diseases? Nat. Rev. Immunol. 20 (5), 271–272. doi: 10.1038/s41577-020-0312-7
Sedaghat, A. R., Gengler, I., Speth, M. M. (2020). Olfactory Dysfunction: A Highly Prevalent Symptom of COVID-19 With Public Health Significance. Otolaryngol. Head Neck Surg. 163 (1), 12–15. doi: 10.1177/0194599820926464
Seliga, A., Lee, M. H., Fernandes, N. C., Zuluaga-Ramirez, V., Didukh, M., Persidsky, Y., et al. (2018). Kallikrein-Kinin System Suppresses Type I Interferon Responses: A Novel Pathway of Interferon Regulation. Front. Immunol. 9, 156. doi: 10.3389/fimmu.2018.00156
Shagdarsuren, E., Wellner, M., Braesen, J. H., Park, J. K., Fiebeler, A., Henke, N., et al. (2005). Complement activation in angiotensin II-induced organ damage. Circ. Res. 97 (7), 716–724. doi: 10.1161/01.RES.0000182677.89816.38
Shao, Z., Shrestha, K., Borowski, A. G., Kennedy, D. J., Epelman, S., Thomas, J. D., et al. (2013). Increasing serum soluble angiotensin-converting enzyme 2 activity after intensive medical therapy is associated with better prognosis in acute decompensated heart failure. J. Card. Fail 19 (9), 605–610. doi: 10.1016/j.cardfail.2013.06.296
Shirato, K., Kawase, M., Matsuyama, S. (2018). Wild-type human coronaviruses prefer cell-surface TMPRSS2 to endosomal cathepsins for cell entry. Virology 517, 9–15. doi: 10.1016/j.virol.2017.11.012
Siddiqi, H. K., Mehra, M. R. (2020). COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J. Heart Lung Transplant. 39 (5), 405–407. doi: 10.1016/j.healun.2020.03.012
Simoes e Silva, A. C., Silveira, K. D., Ferreira, A. J., Teixeira, M. M. (2013). ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 169 (3), 477–492. doi: 10.1111/bph.12159
Sio, S. W., Ang, S. F., Lu, J., Moochhala, S., Bhatia, M. (2010). Substance P upregulates cyclooxygenase-2 and prostaglandin E metabolite by activating ERK1/2 and NF-kappaB in a mouse model of burn-induced remote acute lung injury. J. Immunol. 185 (10), 6265–6276. doi: 10.4049/jimmunol.1001739
Smith, J., Allman, D., Badri, H., Miller, R., Morris, J., Satia, I., et al. (2020). The Neurokinin-1 Receptor Antagonist Orvepitant Is a Novel Antitussive Therapy for Chronic Refractory Cough: Results From a Phase 2 Pilot Study (VOLCANO-1). Chest 157 (1), 111–118. doi: 10.1016/j.chest.2019.08.001
Sodhi, C. P., Wohlford-Lenane, C., Yamaguchi, Y., Prindle, T., Fulton, W. B., Wang, S., et al. (2018). Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 314 (1), L17–L31. doi: 10.1152/ajplung.00498.2016
Sohrabi, C., Alsafi, Z., O’Neill, N., Khan, M., Kerwan, A., Al-Jabir, A., et al. (2020). World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 76, 71–76. doi: 10.1016/j.ijsu.2020.02.034
Solari, F., Varacallo, M. (2020). “Low Molecular Weight Heparin (LMWH),” in StatPearls. (Treasure Island (FL): StatPearls Publishing LLC). Available from: https://www.ncbi.nlm.nih.gov/books/NBK525957/
Solway, J., Leff, A. R. (1991). Sensory neuropeptides and airway function. J. Appl. Physiol. (1985) 71 (6), 2077–2087. doi: 10.1152/jappl.1991.71.6.2077
Sproston, N. R., Ashworth, J. J. (2018). Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 9, 754. doi: 10.3389/fimmu.2018.00754
Sriram, K., Insel, P. A. (2020). A hypothesis for pathobiology and treatment of COVID-19: The centrality of ACE1/ACE2 imbalance. Br. J. Pharmacol. doi: 10.1111/bph.15082
Stassen, J. M., Arnout, J., Deckmyn, H. (2004). The hemostatic system. Curr. Med. Chem. 11 (17), 2245–2260. doi: 10.2174/0929867043364603
Sungnak, W., Huang, N., Becavin, C., Berg, M., Queen, R., Litvinukova, M., et al. (2020). SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26 (5), 681–687. doi: 10.1038/s41591-020-0868-6
Suwa, T., Hogg, J. C., Klut, M. E., Hards, J., van Eeden, S. F. (2001). Interleukin-6 changes deformability of neutrophils and induces their sequestration in the lung. Am. J. Respir. Crit. Care Med. 163 (4), 970–976. doi: 10.1164/ajrccm.163.4.2005132
Tang, N., Bai, H., Chen, X., Gong, J., Li, D., Sun, Z. (2020a). Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 18 (5), 1094–1099. doi: 10.1111/jth.14817
Tang, N., Li, D., Wang, X., Sun, Z. (2020b). Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 18 (4), 844–847. doi: 10.1111/jth.14768
Tetzner, A., Naughton, M., Gebolys, K., Eichhorst, J., Sala, E., Villacanas, O., et al. (2018). Decarboxylation of Ang-(1-7) to Ala(1)-Ang-(1-7) leads to significant changes in pharmacodynamics. Eur. J. Pharmacol. 833, 116–123. doi: 10.1016/j.ejphar.2018.05.031
Theoharides, T. C., Sismanopoulos, N., Delivanis, D. A., Zhang, B., Hatziagelaki, E. E., Kalogeromitros, D. (2011). Mast cells squeeze the heart and stretch the gird: their role in atherosclerosis and obesity. Trends Pharmacol. Sci. 32 (9), 534–542. doi: 10.1016/j.tips.2011.05.005
Thornton, E., Ziebell, J. M., Leonard, A. V., Vink, R. (2010). Kinin receptor antagonists as potential neuroprotective agents in central nervous system injury. Molecules 15 (9), 6598–6618. doi: 10.3390/molecules15096598
Toniati, P., Piva, S., Cattalini, M., Garrafa, E., Regola, F., Castelli, F., et al. (2020). Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun. Rev. 19 (7), 102568. doi: 10.1016/j.autrev.2020.102568
Traina, G. (2019). Mast Cells in Gut and Brain and Their Potential Role as an Emerging Therapeutic Target for Neural Diseases. Front. Cell Neurosci. 13, 345. doi: 10.3389/fncel.2019.00345
Undas, A., Brummel-Ziedins, K. E., Mann, K. G. (2014). Anticoagulant effects of statins and their clinical implications. Thromb. Haemost. 111 (3), 392–400. doi: 10.1160/TH13-08-0720
Utgaard, J. O., Jahnsen, F. L., Bakka, A., Brandtzaeg, P., Haraldsen, G. (1998). Rapid secretion of prestored interleukin 8 from Weibel-Palade bodies of microvascular endothelial cells. J. Exp. Med. 188 (9), 1751–1756. doi: 10.1084/jem.188.9.1751
van de Veerdonk, F. L., Netea, M. G., van Deuren, M., van der Meer, J. W., de Mast, Q., Bruggemann, R. J., et al. (2020). Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife 9, e57555. doi: 10.7554/eLife.57555
Vaughan, D. E., Lazos, S. A., Tong, K. (1995). Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J. Clin. Invest. 95 (3), 995–1001. doi: 10.1172/JCI117809
Vena, G. A., Cassano, N., Di Leo, E., Calogiuri, G. F., Nettis, E. (2018). Focus on the role of substance P in chronic urticaria. Clin. Mol. Allergy 16, 24. doi: 10.1186/s12948-018-0101-z
Verdoni, L., Mazza, A., Gervasoni, A., Martelli, L., Ruggeri, M., Ciuffreda, M., et al. (2020). An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 395 (10239), 1771–1778. doi: 10.1016/S0140-6736(20)31103-X
Wang, J. P., Bowen, G. N., Padden, C., Cerny, A., Finberg, R. W., Newburger, P. E., et al. (2008). Toll-like receptor-mediated activation of neutrophils by influenza A virus. Blood 112 (5), 2028–2034. doi: 10.1182/blood-2008-01-132860
Wang, D., Hu, B., Hu, C., Zhu, F., Liu, X., Zhang, J., et al. (2020). Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 323 (11), 1061–1069. doi: 10.1001/jama.2020.1585
Wang, G., Wu, C., Zhang, Q., Wu, F., Yu, B., Lv, J., et al. (2020). C-Reactive Protein Level May Predict the Risk of COVID-19 Aggravation. Open Forum Infect. Dis. 7 (5), ofaa153. doi: 10.1093/ofid/ofaa153
Wang, J., Jiang, M., Chen, X., Montaner, L. J. (2020). Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 108 (1), 17–41. doi: 10.1002/JLB.3COVR0520-272R
Warner, T. D., Mitchell, J. A. (2004). Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J. 18 (7), 790–804. doi: 10.1096/fj.03-0645rev
Weddell, J. C., Imoukhuede, P. I. (2018). “Computational Systems Biology for the VEGF Family in Angiogenesis,” in Encyclopedia of Cardiovascular Research and Medicine. Eds. Vasan, R. S., Sawyer, D. B. (Oxford: Elsevier), 659–676.
Widagdo, W., Sooksawasdi Na Ayudhya, S., Hundie, G. B., Haagmans, B. L. (2019). Host Determinants of MERS-CoV Transmission and Pathogenesis. Viruses 11 (3), 280. doi: 10.3390/v11030280
Wils, J., Duparc, C., Cailleux, A. F., Lopez, A. G., Guiheneuf, C., Boutelet, I., et al. (2020). The neuropeptide substance P regulates aldosterone secretion in human adrenals. Nat. Commun. 11 (1), 2673. doi: 10.1038/s41467-020-16470-8
Wojta, J., Kaun, C., Zorn, G., Ghannadan, M., Hauswirth, A. W., Sperr, W. R., et al. (2002). C5a stimulates production of plasminogen activator inhibitor-1 in human mast cells and basophils. Blood 100 (2), 517–523. doi: 10.1182/blood.v100.2.517
Wong, S. H., Lui, R. N., Sung, J. J. (2020). Covid-19 and the digestive system. J. Gastroenterol. Hepatol. 35 (5), 744–748. doi: 10.1111/jgh.15047
Wright, H. L., Moots, R. J., Bucknall, R. C., Edwards, S. W. (2010). Neutrophil function in inflammation and inflammatory diseases. Rheumatol. (Oxford) 49 (9), 1618–1631. doi: 10.1093/rheumatology/keq045
Wu, K., Li, W., Peng, G., Li, F. (2009). Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U.S.A. 106 (47), 19970–19974. doi: 10.1073/pnas.0908837106
Xiao, L., Haack, K. K., Zucker, I. H. (2013). Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am. J. Physiol. Cell Physiol. 304 (11), C1073–C1079. doi: 10.1152/ajpcell.00364.2012
Xiao, F., Zimpelmann, J., Agaybi, S., Gurley, S. B., Puente, L., Burns, K. D. (2014). Characterization of angiotensin-converting enzyme 2 ectodomain shedding from mouse proximal tubular cells. PLoS One 9 (1), e85958. doi: 10.1371/journal.pone.0085958
Xu, H., Zhong, L., Deng, J., Peng, J., Dan, H., Zeng, X., et al. (2020). High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral. Sci. 12 (1), 8. doi: 10.1038/s41368-020-0074-x
Yabumoto, C., Akazawa, H., Yamamoto, R., Yano, M., Kudo-Sakamoto, Y., Sumida, T., et al. (2015). Angiotensin II receptor blockade promotes repair of skeletal muscle through down-regulation of aging-promoting C1q expression. Sci. Rep. 5, 14453. doi: 10.1038/srep14453
Yan, X., Hao, Q., Mu, Y., Timani, K. A., Ye, L., Zhu, Y., et al. (2006). Nucleocapsid protein of SARS-CoV activates the expression of cyclooxygenase-2 by binding directly to regulatory elements for nuclear factor-kappa B and CCAAT/enhancer binding protein. Int. J. Biochem. Cell Biol. 38 (8), 1417–1428. doi: 10.1016/j.biocel.2006.02.003
Yin, R., Feng, W., Wang, T., Chen, G., Wu, T., Chen, D., et al. (2020). Concomitant neurological symptoms observed in a patient diagnosed with coronavirus disease 2019. J. Med. Virol. doi: 10.1002/jmv.25888
Yin, S., Huang, M., Li, D., Tang, N. (2020). Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis. 1–4. doi: 10.1007/s11239-020-02105-8
Yoshikawa, T., Hill, T., Li, K., Peters, C. J., Tseng, C. T. (2009). Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J. Virol. 83 (7), 3039–3048. doi: 10.1128/JVI.01792-08
Zanin, L., Saraceno, G., Panciani, P. P., Renisi, G., Signorini, L., Migliorati, K., et al. (2020). SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir. (Wien). 162 (7), 1491–1494. doi: 10.1007/s00701-020-04374-x
Zhang, H., Penninger, J. M., Li, Y., Zhong, N., Slutsky, A. S. (2020). Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 46 (4), 586–590. doi: 10.1007/s00134-020-05985-9
Zhang, T., Wu, Q., Zhang, Z. (2020). Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 301346-1351 (7), e1342. doi: 10.1016/j.cub.2020.03.022
Zhao, C., Zhao, W. (2020). NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front. Immunol. 11, 211. doi: 10.3389/fimmu.2020.00211
Zheng, B. J., Chan, K. W., Lin, Y. P., Zhao, G. Y., Chan, C., Zhang, H. J., et al. (2008). Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc. Natl. Acad. Sci. U.S.A. 105 (23), 8091–8096. doi: 10.1073/pnas.0711942105
Zhou, Y., Lu, K., Pfefferle, S., Bertram, S., Glowacka, I., Drosten, C., et al. (2010). A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 84 (17), 8753–8764. doi: 10.1128/JVI.00554-10
Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., et al. (2020). Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395 (10229), 1054–1062. doi: 10.1016/S0140-6736(20)30566-3
Zhu, J., Ji, P., Pang, J., Zhong, Z., Li, H., He, C., et al. (2020). Clinical characteristics of 3,062 COVID-19 patients: a meta-analysis. J. Med. Virol. 10. doi: 10.1002/jmv.25884
Zucoloto, A. Z., Jenne, C. N. (2019). Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 6, 85. doi: 10.3389/fcvm.2019.00085
Keywords: SARS-CoV-2, renin-angiotensin-aldosterone system, COVID-19, pharmacotherapy, angiotensin II, bradykinin, coagulation, substance P
Citation: Curran CS, Rivera DR and Kopp JB (2020) COVID-19 Usurps Host Regulatory Networks. Front. Pharmacol. 11:1278. doi: 10.3389/fphar.2020.01278
Received: 01 June 2020; Accepted: 03 August 2020;
Published: 14 August 2020.
Edited by:
Xian-Tao Zeng, Wuhan University, ChinaReviewed by:
Christina Lamers, University of Basel, SwitzerlandAnthony Ashton, The University of Sydney, Australia
Copyright © 2020 Curran, Rivera and Kopp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Colleen S. Curran, Y29sbGVlbi5jdXJyYW5AbmloLmdvdg==