 Swayam Prakash Srivastava
Swayam Prakash Srivastava Keizo Kanasaki
Keizo Kanasaki Julie E. Goodwin
Julie E. Goodwin- 1Department of Pediatrics, Yale University School of Medicine, New Haven, CT, United States
- 2Vascular Biology and Therapeutics Program, Yale University School of Medicine, New Haven, CT, United States
- 3Internal Medicine 1, Shimane University Faculty of Medicine, Izumo, Japan
Disruption of mitochondrial biosynthesis or dynamics, or loss of control over mitochondrial regulation leads to a significant alteration in fuel preference and metabolic shifts that potentially affect the health of kidney cells. Mitochondria regulate metabolic networks which affect multiple cellular processes. Indeed, mitochondria have established themselves as therapeutic targets in several diseases. The importance of mitochondria in regulating the pathogenesis of several diseases has been recognized, however, there is limited understanding of mitochondrial biology in the kidney. This review provides an overview of mitochondrial dysfunction in kidney diseases. We describe the importance of mitochondria and mitochondrial sirtuins in the regulation of renal metabolic shifts in diverse cells types, and review this loss of control leads to increased cell-to-cell transdifferentiation processes and myofibroblast-metabolic shifts, which affect the pathophysiology of several kidney diseases. In addition, we examine mitochondrial-targeted therapeutic agents that offer potential leads in combating kidney diseases.
Introduction
Chronic kidney disease (CKD), which affects 10–15% of people, is a leading cause of death worldwide (Levin et al., 2017). Almost 75% of CKD incidents are related to diabetic kidney disease (DKD) and linked-hypertensive kidney disease (HKD) (Levin et al., 2017). Angiotensin-converting enzyme inhibitors (ACEis) and angiotensin II receptor blockers (ARBs) are two classes of anti-hypertensive agents that can effectively reduce the incidence of end-stage kidney disease and are first-line drugs for therapy in diabetic kidney disease (Laverman et al., 2004; Palmer et al., 2015; Gu et al., 2016; Srivastava et al., 2020a; Srivastava et al., 2020b). In addition, the renal protective nature of SGLT-2 inhibitors, DPP-4 inhibitors and statins has been studied in the mouse models and controlled clinical trials (Kanasaki et al., 2014; Edwards, 2016; Wanner et al., 2016; Bae et al., 2019; Hanssen and Jandeleit-Dahm, 2019). However, there remains a lack of efficacious drugs that can retard CKD or DKD (Zelnick et al., 2017). This lack of progress is likely due to poor understanding of the mechanisms of kidney diseases (Breyer and Susztak, 2016). Renal fibrosis is the final consequence of all types of progressive kidney disease, including DKD, that results in end-stage renal disease (ESRD) (Allison, 2019; Cooper and Warren, 2019). Renal fibrosis results in damage to normal cellular functions and structures and is a result of severe inflammation and loss of control over wound healing mechanisms which ultimately lead to an excess accumulation of extracellular matrix (ECM) and fibrosis-associated proteins (Srivastava et al., 2019b). Renal fibroblasts accumulation play a crucial role during fibrogenic processes however, the genesis of fibroblasts is not clear and is a matter of ongoing discussion (Srivastava et al., 2019b).
The primary functions of the kidneys are to balance electrolytes, acid-base status and to maintain water homeostasis and remove toxic substance from the body, all of which are highly energetic processes. Since catabolism of free fatty acids produces more ATP than does catabolism of glucose, kidney tubule segments are mostly dependent on fatty acid oxidation (FAO) and have enormous numbers of mitochondria; they utilize mitochondrial oxidative phosphorylation (OXPHOS) to supply their energy demands (Kang et al., 2015). Mitochondrial synthesis needs the expression of both nuclear- and mitochondrial-coded proteins (Tanaka et al., 2020). The production of cellular energy, in the form of ATP, is the primary function of this organelle (O’Rourke and Blatter, 2009; Kuhlbrandt, 2015). However, the mitochondrion also participates in calcium homeostasis, heat production, cell-signaling and apoptosis (O’Rourke and Blatter, 2009). The metabolic enzymes in the mitochondria are highly regulated by cellular energy status and play an important role in metabolic control (O’Rourke and Blatter, 2009).Doleris et al. reported mitochondrial cytopathy cases from patients who had glomerulosclerosis (Doleris et al., 2000). M2-to-M1 macrophages conversion had been observed in the ESRD patients and which are associated associated with metabolic shifts from oxidative phosphorylation to glycolysis, indicates that suppression of mitochondrial oxidative phosphorylation is positively linked with inflammation and CKD (Ravi et al., 2014; Quadri et al., 2019). Loss of control over mitochondrial biogenesis, mitochondrial function or regulation affects fibrogenic phenotypes in kidney cells (Qin et al., 2018; Srivastava et al., 2018; Chung et al., 2019). The association among renal function, FAO, and bio-energetics suggests that alterations in tubule cell metabolism lead to CKD, DKD and activation of fibrogenic events (Kang et al., 2015). Over-expression of peroxisome proliferator-activated receptor alpha (PPARa) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1a) in epithelial cells, or pharmacological activation of PPARa by fenofibrate were reported to be renal protective in several mouse models of renal fibrosis (Kang et al., 2015).
The Unifying hypothesis suggests that defects in mitochondrial oxidative phosphorylation is a shared pathway in the pathogenesis of microvascular complications of diabetes, including diabetic nephropathy and CKD progression (Brownlee, 2005) however, the various aspects and validity of this theory have been challenged and remain to be carefully addressed (Galvan et al., 2017).
The present review will asses the association between the loss of control over mitochondrial bioengergetics in CKD development and, provide new insights into the role of mitochondrial sirtuins in the regulation of kidney diseases. A comprehensive analysis and its underlying mechanisms offer future therapeutic approaches in the management of kidney diseases.
Loss of Mitochondrial Control in Kidney Disease
Evidence suggests that excessive production of mitochondrial ROS is linked to cellular damage and progression of renal disease (Galvan et al., 2017; Forbes and Thorburn, 2018). Autophagy plays an essential role in the homeostasis of diverse cell types including kidney endothelial cells. Autophagy defects in endothelial cells lead to IL-6 (interleukin 6)-dependent endothelial-to-mesenchymal transition (EndMT) and organ fibrosis with metabolic defects in mice (Takagaki et al., 2020). Mitophagy is the removal of damaged mitochondria and recycling of useful components. Identification of the Parkin–phosphoubiquitin complex, PINK–ubiquitin complex and prohibitin 2, a mitophagy receptor contribute to mitophagy (Kumar et al., 2017; Schubert et al., 2017; Wei et al., 2017). Mitophagy regulatory mechanisms can be ubiquitin-dependent or independent (Zachari and Ktistakis, 2020). In addition, mitochondrial dynamics are regulated by the PINK1–Parkin pathway for proteasomal degradation by targeting mitofusins (MFN) and Miro (outer mitochondrial membrane protein) (Shirihai et al., 2015). In receptor-mediated mitophagy, PHB2 (prohibitin 2) and cardiolipin interactiom play a crucial role in LC3 impairment (Wei et al., 2017; Zhou et al., 2020b). In diabetic nephropathy, mitochondrial debris accumulation has been observed in the kidneys, suggesting that defective clearance of abnormal mitochondria is associated with disease (Sheng et al., 2018; Zhang et al., 2018). Calpain10, which is a mitochondrial cysteine protease, is suppressed in streptozotocin-induced diabetic rats which activate PINK1 (Smith et al., 2012). Calpain10 suppression causes reduction in mitochondrial fusion and induction of mitochondrial fission and autophagy, suggesting that calpain10 negatively regulates mitochondrial autophagy in early diabetics (Smith et al., 2012). However, researchers believe that in early diabetes, removal of defective mitochondria takes place and mitochondrial autophagy is compensatorially increased, which is correlated with progression of diabetic nephropathy (Smith et al., 2012; Yamahara et al., 2013).
NLRP3 inflammatory bodies regulate the secretion of IL-1β and IL-18, which are the critical for the inflammatory response (Kelley et al., 2019). Activated NLRP3 is linked to the activation of caspase-1 (Kelley et al., 2019). This activated caspase-1 enhances the formation of IL-1β and IL-18 by cleaving pro-IL-1β and pro-IL-18 (Kelley et al., 2019). These interleukins are involved in both the inflammatory response and the innate immune response in the kidney cells. Higher ROS or mtDNA release activate NLRP3 inflammasome formation, whereas mitochondrial autophagy inhibits NLRP3 inflammasomes (Zhuang et al., 2015).
ER stress is characterized by alteration in calcium homeostasis, redox imbalance, impaired protein glycosylation and causes misfolded proteins to gather in the ER lumen (Molino et al., 2017). ER-mitochondria crosstalk and contact sites are crucial in autophagosome formation (Molino et al., 2017). The ER-derived mitochondria-associated membranes (MAMs) form contact sites between the ER and mitochondria (Molino et al., 2017). MAMs are involved in lipid biosynthesis, mitochondrial dynamics and bioenergetics, and autophagy (Molino et al., 2017). MAMs transmit stress signals from the ER to mitochondria (Molino et al., 2017). In MCD patients, ER stress and higher release of mitochondrial ROS accelerate the interstitial fibrosis (Lindenmeyer et al., 2008). The elevated ROS level, ER dysregulation, and inflammasome are major factors in the development of renal fibrosis in diabetic nephropathy (Quadri et al., 2019).
Loss of Mitochondrial Functions in Glomerular Cells
Glomerular disease is often linked to mesangial cell proliferation and extracellular matrix deposition (Scindia et al., 2010). Increased mesangial proliferation leads to ECM accumulation and glomerular sclerosis (Scindia et al., 2010). Under high glucose conditions, excessive ROS level, is associated with a decrease in MnSOD activity, mtDNA copy number, mitochondrial membrane potential, and ATP production (Xu et al., 2012). Excessive ROS activate nuclear factor-κB signaling and activated protein-1 and induce TGFβ1 which is associated with inflammation, ECM synthesis, and glomerular sclerosis in diabetic kidneys (Jha et al., 2016). ROS promote mesangial cell proliferation and ECM synthesis by inducing ERK1/2) (Chen et al., 2018). Cyt bc1 complex inhibitor stigmatellin and the respiratory chain complex I inhibitor rotenone inhibit mesangial proliferation and its associated ECM synthesis by suppressing ROS production (Huang et al., 2009; Akool et al., 2012).
Podocytes, GBM and glomerular endothelial cells constitute the glomerular filtration barrier (Garg, 2018). Disruption in the permeability of the glomerular filtration barrier leads to proteinuria (Garg, 2018). The complex formed by nephrin, CD2AP, and podocin plays an important role in maintaining the homeostasis of the glomerular filtration barrier (Mallipattu and Kravets, 2020). Mutations in the mitochondrial gene A3243G cause podocyte injury such as abnormalities in podocyte mitochondria size and structure, aberrant podocyte cell bodies and foot process fusion (Hotta et al., 2001). The puromycin aminonucleoside-induced mouse model of glomerular sclerosis is associated with a defect in oxidative phosphorylation, suppressed mtDNA copy number, and downregulated expression of respiratory chain enzyme complex subunits (Hagiwara et al., 2006). Suppressed level of oxidative phosphorylation results in podocytes cell apoptosis (Zhou et al., 2019). Mitochondrial fission caused by high-glucose leads to effacement of podocyte foot processes, through Drp-1 phosphorylation by Rho-associated coiled-coil-containing protein kinase 1 (ROCK1) (Wang et al., 2012). In addition, podocytes have shown higher mTORC-associated autophagy levels while being unable to regenerate (Cinà et al., 2012) and, as a result of compensatory podocyte loss, parietal cells show fibrogenic responses (Hakroush et al., 2014).
Loss of Mitochondrial Function and Metabolism in Tubular Epithelial Cells
Fibrosis in renal tubules is a final common outcome in all kinds of chronic kidney disease which lead to ESRD (Efstratiadis et al., 2009; Liu et al., 2018).TECs are highly susceptible to damage (Liu et al., 2018). Proteinuria, lipid loading, aberrant levels of cytokines, ischemia, hypoxia, and hyperglycemia can damage tubular functions (Liu et al., 2018). Injured TECs can undergo phenotypic transitions into mesenchymal cell phenotypes via epithelial-to-mesenchymal transition (EMT) (Grande et al., 2015; Lovisa et al., 2015; Srivastava et al., 2019a). During EMT events, altered sets of inflammatory cytokines disrupt normal TECs structure and lead to fibrosis (Grande et al., 2015; Lovisa et al., 2015; Srivastava et al., 2019a). mtDNA depletion and loss of control over mitochondrial function induce EMT process and the recovery of mtDNA and mitochondrial function can reverse the EMT phenotype via gain of endogenous E-cadherin, downregulation of α-SMA expression, and restoration of an epithelial cell phenotype (Yuan et al., 2012). In folic acid-induced and urinary obstruction renal fibrosis models, deterioration in mitochondrial structure and function can cause mitophagy and apoptotic necrosis, which lead to defective fatty acid metabolism and accelerate interstitial fibrosis (Kang et al., 2015; Bhargava and Schnellmann, 2017). Induction of transforming growth factor (TGF)-β signaling is involved in the pathogenesis of renal fibrosis (Meng et al., 2015; Chung et al., 2018). The TGFβ/Smad3 pathway plays an important role in EMT and EndMT events (Srivastava et al., 2013). EMT and EndMT processes are key phenomena in the formation of cancer-associated fibroblasts in diabetes (Amar et al., 2020; Srivastava and Goodwin, 2020).TGFβ impairs antioxidant status by enhancing pro-oxidant NADPH oxidase (Wan et al., 2016).
In contrary to previous findings (Hickey et al., 2011), found higher expression of key mitochondrial proteins from renal biopsies from diabetic nephropathy patients, suggesting mitochondrial biogenesis in renal fibrosis (Hickey et al., 2011). The level of c-AMP is positively related with mitochondrial copy numbers and ATP levels in the tubules (Ding et al., 2018). Restoring cAMP levels by rolipram, a phosphodiesterase (PDE4) inhibitor, improves kidney fibrosis by inhibiting the mitochondrial biogenesis pathway regulator C/EBP-β /PGC1-α (Ding et al., 2018). It was observed that monoallelic mutations in the gene encoding glycine amidinotransferase (GATM), a renal proximal tubular enzyme in the creatine biosynthetic pathway, caused the abnormal aggregation of GATM (Reichold et al., 2018).
The mitochondrial transcription factor A (TFAM) is the crucial human mtDNA binding protein which is involved in the expression and maintenance of mtDNA (Campbell et al., 2012). TFAM regulates metabolic activities by directly targeting PGC1a and PPARa (Scarpulla, 2008). Whole-body knockout of TFAM in mice is lethal (Larsson et al., 1998); however, tubule-specific knock out mice are associated with metabolic defects and kidney fibrosis (Chung et al., 2019). TFAM regulates the mitochondrial copy number and concentration of ATP (Chung et al., 2019). Loss of TFAM in the tubules results in cytokine activation and immune cell infiltration. TFAM deficiency leads the mtDNA to move into the cytoplasm and activate the stimulator of interferon genes (STING) pathway, which, in turn, potentially lead to tubular cell apoptosis, interstitial fibrosis and, renal failure. These results suggest that tubule cell-specific loss of TFAM or mitochondrial damage leads to renal fibrosis not only by causing defective metabolism and energy deficits but also by leaking mtDNA into the cytoplasm, resulting in the activation of STING-dependent NF-kB pathways (Chung et al., 2019). Induction of STING-associated renal inflammation is an important downstream event in the development of kidney disease and inhibiting the STING pathway ameliorates disease development processes in the kidney. Figure 1 depicts a schematic diagram showing the functional importance of TFAM in tubular epithelial cells.
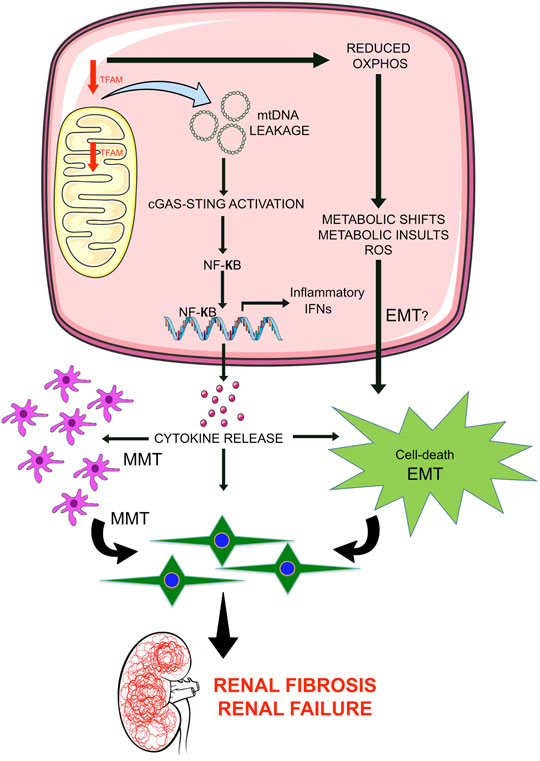
FIGURE 1. Mitochondrial damage leads to renal inflammation and fibrosis. Mitochondrial transcription factor A (TFAM) is critical in the regulation of mtDNA structure, replication, and stability. Suppression of TFAM is a key event in renal fibrosis in 2 ways: 1) By leading to the leakage of mtDNA in the cytosol which activates the STING pathway and results in transcription of NFkB-associated cytokines and release of cytokines outside the cells, induces pathological inflammation in tubular cells and neighboring cell types i.e. macrophages. Higher release of cytokine may influence macrophage-to-mesenchymal transition and epithelial-to-mesenchymal transition and contribute to the accumulation of mesenchymal-like cells in the extracellular matrix; 2) Deficiency of TFAM causes reduced oxidative phosphorylation that is involved in the alteration of metabolic shifts and metabolic insults, influences the reactive oxygen species level; cumulative effects may lead to epithelial-to-mesenchymal transition program and accumulations of EMT-derived myofibroblasts in the extracellular matrix and contributes to renal injury and renal fibrosis.
Loss of Mitochondrial Functions in Kidney Endothelial Cells
Glomerular endothelial cells (GECc) regulate hemodynamic homeostasis, ROS levels and metabolic homeostasis (Jourde-Chiche et al., 2019). GECs affect the integrity of the filtration barrier. Injuries to the endothelial cells lead to microvascular occlusion, glomerular capillary function loss and glomerular sclerosis (Jourde-Chiche et al., 2019). Loss of cristae membranes in the mitochondria of endothelial cells have been observed after ischemic injury in rats (Liu et al., 2014). Restoring mitochondrial structure effectively reduces the loss of peritubular capillaries and cortical arterioles (Liu et al., 2014). Endothelial cell dysfunction results in microalbuminuria in early diabetic nephropathy (Daehn, 2018). High glucose increases mitochondrial superoxide anion production with resultant decreased membrane potential and respiratory chain enzyme complex I deactivation (Sivitz and Yorek, 2010). In the early stages of the rat model of 5/6 nephrectomy, which is an established model of chronic progressive renal injury, with glomerular sclerosis and interstitial fibrosis are observed as well as increased GECs proliferation and apoptosis (Kang et al., 2002). Inflammation contributes to endothelial cell damage through EndMT (Galle et al., 2003; Bogdanova and Castellon, 2016; Zhou et al., 2020a). TNF-α stimulates mitochondrial membrane permeability, thereby inducing cytoplasmic entry of cytochrome c, induction of the proapoptotic protein Bak and suppression of the anti-apoptotic protein Bcl-xL (Meßmer et al., 2000). In addition, mitochondrial oxidative stress induces EndMT (Lin et al., 2018b; Thuan et al., 2018).
Interstitial endothelial cells play critical roles in health and disease processes of the kidneys (Kanasaki et al., 2014; Shi et al., 2015; Chung et al., 2019). Mitochondrial biogenesis and dynamics are important events in determining endothelial cell homeostasis (Wada and Nakatsuka, 2016; Hu et al., 2018). and are central for stress responses, that includes cell-differentiation and organ fibrosis (Stallons et al., 2014; Hu et al., 2018; Srivastava et al., 2018). Das et al. reported that microRNAs regulate mitochondrial function by regulating mitochondrial gene expression (Das et al., 2017). MiR-let-7a regulates glucose catabolism by generating ROS in cancinoma cells (Serguienko et al., 2015). Downregulation of miR-let-7 genesis has critical roles in aerobic glycolysis (Ma et al., 2014). Additionally, the clusters of miR-let-7 have diverse and critical roles in endothelial cell function and metabolism (Srivastava et al., 2013; Srivastava et al., 2014; Hu et al., 2018). Targeting the miR-let-7 biogenesis pathway can affect mitochondrial structure and function (Hu et al., 2018) and among all clusters of the miR-let-7 family, miR-let-7b is well-known to contribute to mitochondrial biogenesis (Kuppusamy et al., 2015). The fibroblast growth factor (FGF)/FGFR1 signaling pathway plays a crucial role in regulating both mitochondrial biogenesis and dynamics and endothelial cell homeostasis (Li et al., 2017a; Hu et al., 2018).
N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP), an endogenous tetrapeptide, plays a crucial role in kidney cell homeostasis (Srivastava et al., 2016; Srivastava et al., 2020b). AcSDKP induces the expression of FGFR1 and miR-let-7 in diabetic endothelium (Nagai et al., 2014; Nitta et al., 2016; Srivastava et al., 2016; Srivastava et al., 2019a). FGFR1 is required for the action of AcSDKP in regulating endothelial-mitochondrial dynamics by controlling miR-let-7b genesis (Hu et al., 2018). The FGF21/FGFR1 axis accelerates mitochondrial biogenesis in an AMP-activated-protein-kinase (AMPK)-dependent manner (Wang et al., 2016). miR-let-7a and miR-let-7b preserve endothelial cell-mitochondrial biogenesis and protect endothelial cells by mitigating ROS generation (Bao et al., 2014). In summary, mitochondrial dynamics, mitochondrial biogenesis, and mitophagy, play key roles in endothelial cell function and homeostasis (Sanchis-Gomar et al., 2014). Endothelial cell SIRT3 and endothelial cell glucocorticoid receptor deficiency is associated with endothelial-to-mesenchymal transition in the kidneys and endothelial FGFR1 deficiency results in severe organ fibrosis in both the kidney and heart via the induction of AcSDKP-resistant EndMT (Li et al., 2020a; Srivastava et al., 2020c). Figure 2 depicts the functional importance of FGFR1-miR-let-7s axis in the regulation of mitochondrial dynamics and suppression of FGFR1-miR-let-7s axis is associated with activation of endothelial-to-mesenchymal transition in the kidney.
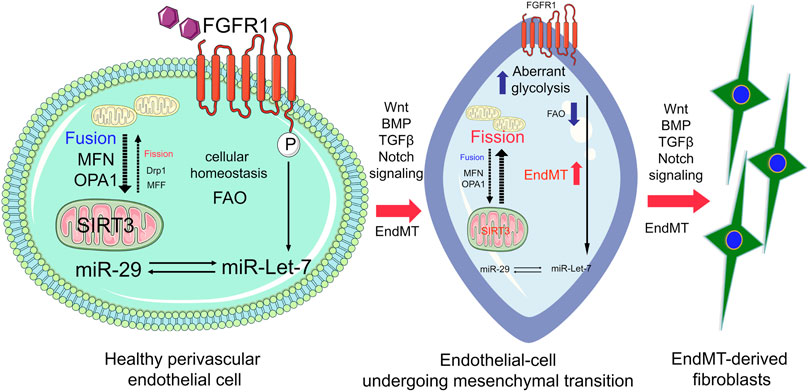
FIGURE 2. FGFR1-miR-let-7 axis maintains endothelial cell homeostasis by regulating mitochondrial dynamics and integrity through modulating SIRT3. In healthy endothelial cells, FGFR1-miR-let-7 maintains mitochondrial dynamics and influences SIRT3; however, suppression of the FGFR1-miR-let-7 axis disrupts the miR-29 level and SIRT3 level, and ultimately disrupts mitochondrial biogenesis and integrity. Cumulative effects of suppressed levels of FGFR1-miR-let-7 axis and SIRT3 lead to the activation of pro-mesenchymal signaling, Wnt signaling, BMP, Notch, and TGF-β signaling; resulting in EndMT events and accumulation of EndMT-derived myofibroblasts.
Mitochondria-Targetted Therapeutics in Kidney Diseases
In vitro and in vivo studies confirm the involvement of microRNAs in the pathogenesis of kidney diseases (Srivastava et al., 2019a; Metzinger-Le Meuth et al., 2019; Nascimento and Domingueti, 2019). Several microRNAs play a crucial role in mitochondria (Gomez et al., 2013; Jaquenod De Giusti et al., 2018; Bai et al., 2019). MiR-30e is suppressed in renal fibrosis, and its antagonism exerts an antifibrotic effect by targeting mitochondrial protein UCP2 (Jiang et al., 2013a). miR-21 contributes widely to organ fibrosis by acting on energy metabolism. miR-21 antagonism suppresses ROS production and significantly reduces glomerular sclerosis, interstitial fibrosis, and inflammatory responses (Chau et al., 2012; Kolling et al., 2017). miR-17 is capable of mitochondrial metabolism and promotes the growth of polycystic kidney cysts (Hajarnis et al., 2017).
The renal cortex of db/db mice have reduced levels of total and oxidized forms of Coenzyme Q10 and intervention with Q10 ameliorated mitochondrial functions and suppressed collagen deposition in these diabetic kidneys (Sourris et al., 2012). Similarly, supplementation of Q10 suppressed ROS levels and ameliorated renal function in nephrectomized rats (Ishikawa et al., 2010). Q10 mitigates nicotine-induced oxidative stress in tubular epithelial cells through activating the non-mitochondrial fork protein p66shc (Arany et al., 2016). A clinical randomized trial showed that hemodialysis patients benefitted from daily use of 1,200 mg of Q10 per day to control oxidative stress (Rivara et al., 2017).
MitoQ is used as a mitochondria-targeted antioxidant, exerts a protective effect on lipid peroxidation and oxidative stress (Kelso et al., 2001). MitoQ is known to reduce oxidative stress and protect renal function in the ischemia-reperfusion-induced renal injury (Rouschop et al., 2005) and has antifibrotic effects in Ins2Akita mouse model of type I diabetic nephropathy (Chacko et al., 2010) and in db/db mice (Ward et al., 2017). MitoQ suppresses oxidative stress through inducing autophagy, inhibiting mitochondrial membrane potential, suppressing fission protein Drp1, and restoring fusion protein Mfn2 expression in tubular epithelial cells (Xiao et al., 2017). MitoQ prevents hypertension, stimulates endothelial NO bioavailability and improves kidney structure in spontaneously-hypertensive rats (Graham et al., 2009). Oral MitoQ is in phase II clinical trials (Gane et al., 2010) (Snow et al., 2010).
SS-31 is a small peptide which suppresses excess ROS, stabilizes mitochondrial membrane potential, prevents cytochrome c translocation, and is associated with fibrogenesis in 5/6 nephrectomized rats (Zhao et al., 2017b). SS-31 significantly reduced tubular apoptosis, macrophage infiltration and maintained the integrity of mitochondrial function while inhibiting renal fibrosis in the UUO rat model (Mizuguchi et al., 2008) and in rat model of ischemia-reperfusion (Zhang et al., 2019). SS-31 restored mitochondrial function in podocytes (Sweetwyne et al., 2017) and parietal epithelial cells (Zhao et al., 2013), and reduced fibrosis in glomeruli and endothelial cells (Sweetwyne et al., 2017).
Rapamycin, an inhibitor of mTORC1, regulates mitochondrial autophagy (Bartolomé et al., 2017). Activation of the mTOR signaling pathway is the key pathogenic mechanism in diabetic kidney disease (Lloberas et al., 2006). Rapamycin inhibits kidney fibrosis, glomerulosclerosis, proteinuria and mesangial matrix deposition through mitigating the activation of mTOR (Lloberas et al., 2006; Li et al., 2019). Rapamycin can inhibit both mTORC1 and mTORC2 (Kawata et al., 2018). Rapamycin therapy is limited due to its side effects such as immunosuppression, and glucose intolerance in type II diabetic mice by reducing mTORC2 (Lamming et al., 2012) (Schreiber et al., 2019). mTORC2 regulates autophagy genes by FOXO3a phosphorylation and activation of the Akt pathway (Hung et al., 2012) (Chen et al., 2013). FOXO3a regulates mitochondrial autophagy through LC3, Bnip3, Nix, Atg4b, and Atg12l (Higgins and Coughlan, 2014). Further research is needed to establish rapamycin and its analogs as safe measures for treating fibrotic renal disease.
Metabolic Control by Posttranslational Modifications in Mitochondria
Metabolic control switches depend on the availability and scarcity of fuel. These regulatory mechanisms are highly conserved throughout evolution and affect many cellular signaling pathways linked to food intake and bioenergetics (Finkel et al., 2009; Morigi et al., 2018). Reversible acetylation is one important mechanism that regulates metabolic processes in mitochondria (Guan and Xiong, 2011). Reversible acetylation is regulated by the antagonistic activities of protein acetyltransferases (KATs) and deacetylases (HDACs) (Guan and Xiong, 2011). These proteins are encoded by multigene families, and are nuclearly-encoded (Guan and Xiong, 2011). In mammalian cells, thirty KATs and approximately eighteen HDACs are known (Guan and Xiong, 2011). The eighteen HDACs are classified into four types. Class I and II, which consist of ten members, are called “classical” HDACs; the enzyme activity of these classical HDACs can be repressed by trichostatin A, excluding HDAC11 that is unresponsive to trichostatin A. Class III HDACs, known as SIRTs (sirtuins), include 7 members and all are structurally different from HDACs. Most of the SIRTs need nicotinamide adenine dinucleotide (NAD+) as a co-substrate and are repressed by nicotinamide (NAM); however, these are unaffected by trichostatin A treatments (Guan and Xiong, 2011). Increasing evidence suggests metabolic function of these SIRTs whereas, the function of KATs and HDACs are less clear (Guan and Xiong, 2011). Out of the eleven HDACs, 4 (HDACs: 1, 2, 8, and 11) are localized in the nucleus and six (HDACs: 3, 4, 5, 7, 9, and 10) are either dispensed in or channeled between the nucleus and the cytosol (Seto and Yoshida, 2014). HDAC7 has been found to localize to the mitochondria (Seto and Yoshida, 2014).
Moreover, out of the seven SIRTs, SIRT3, 4, and 5 reside in mitochondria while SIRT2 is found in the cytosol and SIRT1 has been reported to be present in both the nucleus and the cytosol (Seto and Yoshida, 2014; Morigi et al., 2018). Figure 3 demonstrates the subcellular localization and biological functions of these sirtuins. The presence of many situins in mitochondria suggests a crucial role of metabolic control by the mitochondria (Wakino et al., 2015; Hershberger et al., 2017). Studies of several sirtuins in different model organisms have suggested that sirtuin genes play a role in life span, caloric-restriction, nutrient responses and bioenergetics (Lin et al., 2000; Finkel et al., 2009).
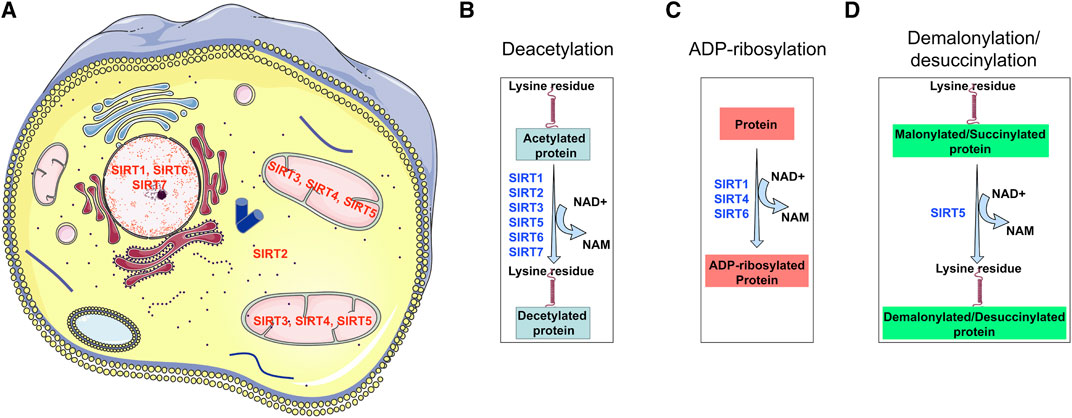
FIGURE 3. Sub-cellular localization of diverse types of sirtuins and their functional properties. SIRT1, SIRT6, and SIRT7 localize in the nucleus; SIRT3, SIRT4, and SIRT5 localize in mitochondrial; however only SIRT4 is known to reside in the cytosol. SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7 have deacetylation properties; SIRT1, SIRT4 and, SIRT6 have ADP-ribosylation activity, however, SIRT5 has two unique properties of demalonylation and desuccinylation. These functional differences make each sirtuin of diverse biological importance in renal health.
Deacetylase and Autosomal Dominant Polycystic Ribosylase Activity
In humans, the deacetylase domain of SIRTs is distinct from class I and class II HDACs, which are zinc dependent. SIRTs utilize one NAD+ to produce acetyl-ADP-ribose and NAM in the deacetylation process. Defective mitochondrial pathways can lead to metabolic diseases, oxidative damage, organ fibrosis and cancer (Pearce et al., 2009; Kang et al., 2015; Carrico et al., 2018; Srivastava et al., 2018). A proteomic approach revealed that twenty percent of mitochondrial proteins that are involved in life-span control and in metabolic control are present in the acetylated form (Kim et al., 2006). Reversible deacetylation of mitochondrial proteins is a crucial mechanism of metabolic control (Carrico et al., 2018). Acetyl-CoA and NAD+ are critical markers of energy levels in cells. Acetyl CoA is the substrate for histone acyl transferases while NAD+ is a co-substrate for SIRTs (Carrico et al., 2018). The deacetylation processes of mitochondrial SIRTs are known to have key roles in metabolic shifts in cancer cells (Carrico et al., 2018).
The deacetylation ability of the histone H4 peptide is different among the sirtuins. SIRT 1, 2, 3, 4, 5, 6, and 7 show increased tendencies toward histone H4 polypeptide whereas, SIRT 1,4,6 have mono-ADP-ribosylation activity (Lee et al., 2019). SIRT1 deacetylates histones, p53, Ku70 and FOXO. Among the sirtuins, only SIRT 2 deacetylates tubulin (Morigi et al., 2018). Similarly, the mono-ADP-ribosylating activity of sirtuins differs among different groups and is associated with SIRT4 and SIRT6. Mono-ADP-ribosylation is a process in which ADP-ribose from nicotinamide-adenine-dinucleotide is moved to the target acetylated protein (Carrico et al., 2018). This process is highly conserved from bacteria to humans (Saunders and Verdin, 2007). In addition to mono-ADP-ribosylation, SIRT4 has robust deacylase activity as well as substrate-dependent lipoamidase and deacetylase properties (Lee et al., 2019; Tomaselli et al., 2020). Beside deacetylase activity, SIRT5 has also been found to have demalonylase and desuccinylase activity (Lee et al., 2019).
Regulation of Mitochondrial Sirtuins
Among all mitochondrial SIRTs, SIRT3 mainly targets those proteins that are involved in metabolic homeostasis (Lombard et al., 2007; Houtkooper et al., 2012). For example, SIRT3 targets long-chain acyl CoA dehydrogenase, a key protein in FAO in prolonged fasting conditions (Hirschey et al., 2010). Deficiency of Sirt3 disrupts lipolysis, and lipid catabolism, and thus promotes diet-induced obesity (Hirschey et al., 2011). SIRT3 deacetylates 3-hydroxy-3-methylglutaryl-CoA-synthase 2, which controls the synthesis of ketone bodies, a crucial energy source for the brain under fasting conditions (Shimazu et al., 2010). During caloric restriction, SIRT3 induces the enzyme activitiy of isocitrate dehydrogenase (Someya et al., 2010), glutamate dehydrogenase and the enzymes of the TCA cycle (Lombard et al., 2007). SIRT3 deacetylates components of ETC such as complex I, complex II and complex III, and these are associated with oxidative-phosphorylation, the final stage of aerobic respiration (Ahn et al., 2008; Finley et al., 2011b; Jing et al., 2011). In addition, SIRT3 protects cells from oxidative stress by mitigating ROS levels (Someya et al., 2010; Jing et al., 2011) and activating the enzyme activity of superoxide-dismutase 2, a key antioxidant enzyme in mitochondria (Qiu et al., 2010). Caloric restriction increases SIRT3-associated deacetylation of IDH2, thereby increasing the reduced-to-oxidized glutathione ratio, and hence iinhibiting ROS (Someya et al., 2010).
SIRT4 primarily plays a role in metabolic control (Han et al., 2019). SIRT4 inhibits the GDH enzyme activity by ADP-ribosylation, and hence, blocks amino-acid-linked insulin secretion (Argmann and Auwerx, 2006). As a result, SIRT4 knock out mice have elevated levels of plasma insulin, in fed, as well as fasted,conditions (Argmann and Auwerx, 2006). In addition, SIRT4 controls FAO in cultured hepatocytes and myotubes, and knockdown of SIRT4 in the liver is associated with higher FAO (Nasrin et al., 2010). Interestingly, SIRT3 and SIRT4 have shown antagonistic properties in the regulation of GDH (Haigis et al., 2006; Lombard et al., 2007) and FAO (Hirschey et al., 2010; Nasrin et al., 2010). Further scientific advancement is required to analyze how SIRT3 and SIRT4 coordinate similar nutrient states to achieve opposite responses.
SIRT5 deacetylates CPS1 in a fasting state and promotes ammonia detoxification in the urea cycle (Nakagawa et al., 2009). SIRT5 may not primarily act as a deacetylase (Nakagawa et al., 2009) however, it acts as a demalonylase and desuccinylase (Peng et al., 2011), even for the described deacetylase target CPS1 (Du et al., 2011).
Sirtuin3 Regulates Mitochondrial Dynamics
In spite of the oval-shaped structure of mitochondria, these organelles can exist in an active, dynamic nexus and continuously go through fission and fusion phenomena (Lesnefsky et al., 2001; Otera and Mihara, 2011; Chan, 2012). Mitochondria divide by a simple binary fission process which requires only mtDNA for its function (Otera and Mihara, 2011; Chan, 2012). Both fission and fusion are highly linked to replication of mtDNA. Several proteins involved in the fission process have been implicated in mitochondrial diseases (Otera and Mihara, 2011; Chan, 2012).
Mitochondria maintain their active dynamic form by coordinating networks and sustaining a series of fusion (mitofusin-2, MFN-2; optic atrophy protein 1, OPA1) and fission cycles (dynamin-related protein-1, DRP1) (Dorn et al., 2015; Wada and Nakatsuka, 2016). The effector molecules differ in the outer and inner membranes of the mitochondria (Wada and Nakatsuka, 2016). Membrane-bound dynamins arbitrate fusion between outer-membranes and are identified as MFN1 and MFN2 proteins (Wada and Nakatsuka, 2016). However, a single-dynamin OPA1 coordinates fusion processes between inner-membranes (Wada and Nakatsuka, 2016). This protein is well-conserved among mammals, flies, and yeast. OPA1 is anchored to the inner membrane of mitochondria. OPA1 assists in preserving cristae structure and protectscells from apoptosis (Frezza et al., 2006). SIRT3 can directly target OPA1 (Samant et al., 2014). OPA1 is hyper-acetylated under stress conditions, and hyperacetylated OPA1 reduces its GTPase enzyme activity, thereby mitigating its biological functions (Frezza et al., 2006). SIRT3 activates OPA1 by deacetyation and consequently alters mitochondrial dynamics (Samant et al., 2014). Therefore, SIRT3 activates the function of mitochondria not only by affecting the enzyme activity level but by directly regulating mitochondrial dynamics by activating OPA1 (Samant et al., 2014). The DRP1 protein, a dynamin protein of large GTPases, positively regulates the mitochondrial fission process by pinching off the membrane stalk between two forming daughter mitochondria (Dorn et al., 2015; Wada and Nakatsuka, 2016). SIRT3-loss is associated with activation of mitochondrial-fission by moldulating DRP1 protein level.
Sirtuin3 in Renal Health and Metabolism
SIRT3 plays a major role in kidney health. Available evidence suggests that SIRT3 maintains mitochondrial energy homeostasis in proximal and distal tubule compartments (Morigi et al., 2018). A role for SIRT3 in the regulation of tubular-cell homeostasis has also been demonstrated and suggests that SIRT3 regulates microtubule-dependent transport of mitochondria among tubular epithelial cells, which is a process that conserves cell bioenergetic profiles and antioxidant mechanisms (Morigi et al., 2018).
However, our recent studies suggest that SIRT3 has a protective role in renal fibrosis and diabetic kidney disease (Srivastava et al., 2018). Renal fibrosis is the dominant cause of end-stage renal disease across the world (Roxburgh et al., 2009). It is characterized by deposition of collagen, myofibroblasts, and pro-inflammatory cells (Zeisberg et al., 2003; LeBleu et al., 2013). Renal fibroblasts have a crucial role in such fibrotic events, but, the genesis of these fibroblasts is not clear (Grande and Lopez-Novoa, 2009; Zeisberg and Neilson, 2010; Liu, 2011; Schrimpf and Duffield, 2011; Grgic et al., 2012; Srivastava et al., 2013; Srivastava et al., 2019b).
Available data suggest that activated myofibroblasts and fibroblast formation are caused by activated resident fibroblasts and/or activation of mesenchymal cell differentiation processes in neighboring cells such as epithelial cells, endothelial cells, pericytes and M2-derived macrophages. (Srivastava et al., 2013; Srivastava et al., 2019b). SIRT3 acts as a tumour-suppressor, maintains stability in the genome (Kim et al., 2010) and inhibits the features of organ fibrosis by mitigating TGF-β/Smad signaling (Sundaresan et al., 2009; Chen et al., 2015; Sundaresan et al., 2015; Bindu et al., 2017; Sosulski et al., 2017). However, during the cell-to-cell transition process, the metabolic switch is altered (Jiang et al., 2013b; DeNicola and Cantley, 2015; Liu et al., 2016). Thus, the fuel choice or energy sources of these injured cells is a matter of ongoing debate (Jiang et al., 2013b; DeNicola and Cantley, 2015; Liu et al., 2016). These reprogrammed metabolic shifts cause production of myofibroblast precursors and may assist in fibroblast growth and survival (Zeisberg et al., 2003; Kalluri and Weinberg, 2009; Jiang et al., 2013b).
Sirtuin3, in Association With Activated STAT3, Regulates Aberrant Glycolysis
In response to cytokines, signal transducer and activator of transcription (STAT)3 phosphorylation on its tyrosine 705 residue (Y-P) is mediated by receptor-associated JAK kinases (Schindler et al., 2007). Tyrosine705 phosphorylation causes dimerization and translocation from the cytosol to the nucleus where it binds to gene promoters and modulates the transcription (Yu et al., 2014). Besides regulation by phosphorylation on Tyr705, STAT3 is also regulated by phosphorylation on Ser727 by some members of the MAP kinases. Ser727 phosphorylation is a secondary step for enhancing transcriptional activity of STAT3 (Wen et al., 1995). STAT3 is mainly regulated by the cytokine IL-6 and other cytokines such as IL-11 and IL-27 which work through a gp130 signal transducer in their receptors (Zhong et al., 1994; Niemand et al., 2003). IL-10, IL-21 and leptin can also activate STAT3 which is independent of gp130 signal transduction (Niemand et al., 2003). STAT3 activation is also regulated by phosphatases and by the Suppressors of Cytokine Signaling (SOCS), which interfere with nuclear translocation or promote STAT3 degradation (Ward et al., 1994).
STAT3 affects energy metabolism by influencing the pathways both at nucleus and the mitochondrion, depending on specific post-transcriptional modifications (Y-P or S-P) triggered by diverse stimuli (Poli and Camporeale, 2015). Y-P nuclear STAT3 accumulation mediates transcriptional upregulation of HIF1α and the downregulation of mitochondrial genes (Poli and Camporeale, 2015). This leads to aberrant aerobic glycolysis, suppressed ETC activity, and decreased ROS generation, thus enhancing cell-proliferation and inhibiting apoptosis (Poli and Camporeale, 2015). S-P STAT3 mitochondrial activity also leads to increased cell-proliferation and limits apoptosis through retaining ETC activity, stimulating aerobic glycolysis, decreasing ROS generation, and inhibiting the opening of the mitochondrial permeability transition pore (Poli and Camporeale, 2015; Meier et al., 2017).
STAT3 promotes Complex I activity and mitochondrial respiration by binding on GRIM-19, which is a component of Complex I of the electron transport chain (ETC) (Lufei, 2003). In addition, STAT3 can bind to Complex II or Complex V (ATP synthase) and modulates ATP production (Gough et al., 2009). Mitochondrial STAT3 binds to cyclophilin D, and inhibits the opening of the mitochondrial permeability transition pore, hence reducing ROS generation (Meier et al., 2017). GRIM-19 functions as a chaperone which helps in recruiting STAT3 to the mitochondrial inner membrane complex and the mitochondrial importer Tom20 is involved in STAT3 recruitment into mitochondria (Boengler et al., 2010; Tammineni et al., 2013). Mitochondrial recruitment of Stat3 is enhanced by its acetylation, but the mechanism remains unclear; mitochondrial sirtuins may be involved in this mechanism (Xu et al., 2016b). The role of mitochondrial STAT3 has been demonstrated primarily in cancer, cardiology, neuroscience, organ fibrosis and in diabetic kidney disease (Yang and Rincon, 2016). JAK inhibitors and STAT3 inhibitors may have the potential to develop a new generation of therapeutics (Rincon and Pereira, 2018).
Recent studies suggest that SIRT3 deficiency in diabetic kidneys leads to the induction of aberrant glycolysis and linked fibrogenic programming through activation of the TGFβ/Smad3 signaling pathway in tubular epithelial cells that promotes an epithelial-to-mesenchymal transition program (Srivastava et al., 2018). SIRT3 deficiency-linked abnormal glycolysis is due to higher PKM2-dimer formation, HIF1α and activated STAT3 signaling (Srivastava et al., 2018; Li et al., 2020b). Disruption in glucose metabolism is associated with metabolic reprogramming in damaged cells and mesenchymal activation and gain of fibrogenic properties in diabetic kidneys (Srivastava et al., 2018). Suppression of SIRT3, HIF-1α accumulation and STAT3 phosphorylation are linked with EMT phenotype and defective glucose metabolism (Finley et al., 2011a; Palmirotta et al., 2016). Inhibition of glycolysis by either by 2-deoxy-glucose (2-DG) or with dichloroacetate (DCA) leads to reduction in EMT processes and cancer cell metastasis (Sottnik et al., 2011; Lu et al., 2015; Zhao et al., 2017a). HIF-1α and STAT3 phosphorylation in renal epithelial cells is linked to mesenchymal activation and renal fibrogenesis (Higgins et al., 2007; Sun et al., 2009). Proximal-tubular-cells (PTCs) are widely unprotected to excessive glucose uptake from the urine in severe-diabetes; it may possible that the urinary glucose can be utilized as a substrate for glycose catabolism by TECs (Hato et al., 2016). Hence, cumulative effects of SIRT3 deficiency and HIF-1α accumulation and defective central metabolism, triggered by higher glucose reabsorption, stimulate PTCs to transform into an intermediate-type mesenchymal- and complete-mesenchymal cell phenotype (Srivastava et al., 2018). In our study, it was demostated that loss of SIRT3-linked PKM2 tetramer-to-dimerization occurred in diabetic kidneys and in cultured TECs exposed to high-glucose-stimulated cell media (Srivastava et al., 2018). PKM2-dimerization causes transactivation of HIF1α and is a crucial mechanism for abnormal glucose metabolism in cancer cells by enhancing the Warburg effect (Greer et al., 2012; Soga, 2013; Palsson-McDermott et al., 2015).
The pathogenic role of defective glucose metabolism has been demonstrated in diabetic kidney disease (Qi et al., 2017). Tubular interstitial pathology was improved with increased enzyme activity of the PKM2 tetramer (Qi et al., 2017). TEPP-46 induced PKM2 tetramer formation, suppresseed accumulation of fibronectin and type-I-collagen, and mitigated TGFβ1 levels in the injured tubules. Conversely, TEPP-46 has less effect on glomerular collagen deposition since TGFβ1 signaling is higher in damaged tubules and not induced in the glomeruli of diabetic kidneys (Qi et al., 2017). This study suggests that aberrant glycolysis in TECs confers a renal disease phenotype in diabetes. Higher SIRT3 expression levels inhibit glucose-stimulated cell-senescence through FOXO1-mediated signaling mechanisms (Zhang et al., 2013) and enhance cellular resistance to oxidative stress damage (Morigi et al., 2018).
SGLT2 inhibitors and glycolysis inhibitors act primarily on kidney proximal tubular cells. One recent study suggests that SGLT2 inhibition and glycolysis inhibition in diabetic tubules impact central metabolism through restorating SIRT3 protein level, by causing a reduction in EMT events reducing abnormal glycolysis attributed to STAT3-phosphorylation, HIF1α-transactivation and PKM2-dimerization (Li et al., 2020b). SGLT2 inhibition suppresses EMT events in proximal tubular cells and linked EndMT in perivascular endothelial cells (Li et al., 2020b) and has been the focus of intensive discussion as a potential source of myofibroblasts (Srivastava et al., 2019a). TECs are injured in diabetic kidneys. They undergo phenotypic changes and acquire the features of matrix-generating mesenchymal cells, and fibrogenesis markers such as αSMA, fibronectin and FSP1. Renal fibrosis is influenced by intercommunication among several cell types in the kidney (Srivastava et al., 2019b). EMT influences the mesenchymal activation of perivascular endothelial cells, pericytes and macrophages through soluble-factors. Figure 4 represents the role of aberrant glucose metabolism in the induction of EMT events. EMT releases soluble factors that may accelerate EndMT events in diabetic kidneys.
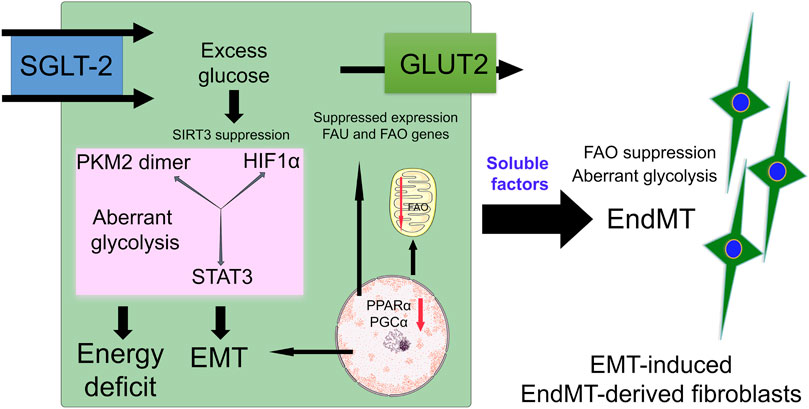
FIGURE 4. Defective central metabolism in diabetic tubular epithelial cells. Diabetic tubular epithelial cells have higher expression of SGLT-2 which transports urine glucose into tubular cells. Excess glucose accumulates in the cytosol which results in activation of SIRT3 deficiency-linked disruption in central metabolism. This defective central metabolism is characterized by PKM2-and STAT3-linked aberrant glycolysis and suppression of fatty acid oxidation. Defective central metabolism contributes to EMT events. These EMT events produce and release soluble factors which affect the homeostasis of endothelial cells and induce EndMT events in the kidneys.
Sirtuin3 Deficiency Disrupts Fatty Acid Oxidation
PTECs require excessive energy for proper function and have enormous quantities of functional mitochondria. Free fatty acids (FAs) are used as the most favored metabolic fuel for TECs, since catabolism of FAs synthesizes more ATP per molecule than does glucose catabolism (Kang et al., 2015). FA uptake is facilited by the transporter protein CD36, and fatty acids transporter proteins (Susztak et al., 2005). Catabolism of FFAs is dependent on transport into mitochondria that is catalyzed thorugh carnitine-palmitoyltransferase 1 (CPT1) (Schug and Li, 2011). The peroxisome-proliferator-activated-receptor-α (PPARα) and PPAR-γ-coactivator-1α (PGC1α) are crucial transcription factors that control the expression of genes/proteins in FA uptake and oxidation (Tran et al., 2011; Kang et al., 2015). In healthy TECs, FA uptake, FAO and FA biosynthesis are highly regulated to abstain from intra-cellular lipid deposition (Kang et al., 2015). The accumulation of lipids in TECs and their pathological role in acute and diabetic kidney disease is the subject of debate among researchers (Decleves et al., 2014; Srivastava et al., 2014). In addition, defective FA utilization and oxidations leads to mesenchymal activation and fibrogensis (Kang et al., 2015) since higher triglyceride accumulation in TECs catalyzes lipotoxicity, and augments the induction of mesenchymal activation and progression of kidney fibrosis (Decleves et al., 2014).
In diabetic kidneys with tubules undergoing mesenchymal cell formation, SIRT3 suppression is associated with defective FAO and concomitant induction of abnormal glycolysis (Srivastava et al., 2018; Srivastava et al., 2020b). Defective fatty acid oxidation has been found to play a critical role in humans and in mouse models of tubulointerstitial fibrosis (Kang et al., 2015; Srivastava et al., 2020b). Injured tubulointerstitial cells have been shown to have suppressed levels of regulatory enzymes of FAO and accumulation of intracellular lipids (Kang et al., 2015). FAO inhibition in TECs by the small molecule etomoxir causes ATP-deficits, cell-death, cell-differentiation, and lipid-accumulation, mimicking features of renal fibrosis (Kang et al., 2015). However, normalizing FAO by genetic or pharmacological means using small chemicals protects against renal fibrosis, suggesting that normalization of the renal-metabolic abnormality might be utilized for the treatment of chronic kidney disease (Kang et al., 2015).
Sirtuin4 in Renal Health and Metabolism
SIRT4 is a critical molecule in mitochondrial physiology. Researchers have investigated a pathological connection between SIRT4 and diabetic nephropathy in high-glucose-stimulated cultured podocytes (Shi et al., 2017). Glucose stimulation remarkably induces podocyte apoptosis which is associated with diminished protein levels of SIRT4, suggesting that SIRT4 suppression is critical in diabetic nephropathy (Shi et al., 2017). Over-expression of SIRT4 suppresses podocyte apoptosis, stimulates mitochondrial-membrane potential and decreases ROS generation (Shi et al., 2017). SIRT4-overexpression suppresses the level of apoptosis-linked effector molecules such as NOX1, Bax and p38 phosphorylation and increases the Bcl-2 protein expression in high-glucose-treated cultured podocyte cells (Shi et al., 2017). These results demonstrate that SIRT4 overexpression protects against hyperglycemia-associated podocyte cell death and ROS generation and that deficiency of podocyte SIRT4 represents a critical development in the understanding of diabetic nephropathy (Shi et al., 2017).
SIRT4 inhibits renal tumor metabolism, especially glutamine metabolism, and therefore functions as a tumor suppressor gene in the kidneys (Jeong et al., 2013). It is thought to be a gatekeeper of glutamine metabolism energetics (Mathias et al., 2014). Indeed, a metabolic shift is a hallmark of all types of tumors (Faubert et al., 2013). Tumor cells typically show Warburg metabolism and are dependent on enhanced glucose and glutamine uptake and catabolism to meet the large energy demand for tumor development (Daye and Wellen, 2012). In the future, it will be desirable to determine the corelation between SIRT4 levels and the prognosis of renal clear cell carcinoma, pending the existence of a suitable number of patients, which would allow further analysis of the influence of SIRT4 on the biological behavior of these cancer cells.
Sirtuin5 in Renal Health and Metabolism
SIRT5 has diverse, unique functional properties in its substrate choice for succinyllysine, malonyllysine, and glutaryllysine; it enhances fatty acid oxidation in hepatcytes and cardiac myocytes (Rardin et al., 2013; Du et al., 2018). Intriguingly, SIRT5 has been shown to localize to peroxisomes as well (Chiba et al., 2019). In contrast to its effect on mitochondrial FAO, SIRT5 suppresses peroxisomal FAO in vitro and in rodent liver (Chiba et al., 2019). SIRT5 knock out mice are protected against ischemic- and cisplatin-mediated AKI (Chiba et al., 2019). Although the mitochondrial function is moderately suppressed in SIRT5 KO kidneys, the peroxisome function is increased in kidneys from mice subjected to acute kidney injury (Chiba et al., 2019). These results suggest that SIRT5 controls the balance of mitochondrial versus peroxisomal FAO in PTECs and protects from acute kidney injury (Chiba et al., 2019).
The loss-of-function of SIRT5 is renoprotective (Chiba et al., 2019). The role of SIRT5 is antagonistic to that of SIRT1 and SIRT3, as their loss promotes acute kidney injury. Knock-down of SIRT3 in proximal tubular epithelial cells disrupts mitochondrial-fatty acid oxidation through hypersuccinylation and therefore, reduces the enzyme activity of key proteins involved in FAO (Chiba et al., 2019). Metabolic adaptation to blocked mitochondrial-FAO in proximal tubular epithelial cells reveals the compensatory FAO in the peroxisome, hence mitigating oxygen necessity, reducing reactive oxygen species, and protecting against kidney injury (Chiba et al., 2019).
Metabolic and Mitochondrial Reprogramming in Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common, monogenic disorders and is caused mostly by gene mutations in polycystic kidney disease 1 (PKD1) and 2 (PKD2), which encode polycystin 1 and polycystin 2, respectively (Harris and Torres, 2009). ADPKD is characterized by bilateral renal cyst development that impairs kidney function, leading to ESRD (Torres et al., 2007). Multiple pathways are dysregulated in the cystic epithelium including alterations in cell metabolism which have emerged as a hallmark of ADPKD (Rowe et al., 2013; Menezes et al., 2016; Podrini et al., 2020). Evidence suggests that the cystic epithelial lining shares neoplastic features (Rowe et al., 2013). Impaired mitochondrial structure and function play a role in ADPKD disease progression (Cassina et al., 2020). Metabolic reprogramming in PKD is similar to that reported in cancer (Rowe et al., 2013). Studies suggest that aerobic glycolysis is present in the disease, along with other metabolic defects such as augmentation of the pentose phosphate pathway, and increases in glutamine anaplerosis and fatty acid biosynthesis,; fatty acid oxidation and mitochondrial metabolism are suppressed (Podrini et al., 2020). ADPKD cells alter their energy dependency from oxidative phosphorylation to glycolysis (Podrini et al., 2020).
The precise origin of metabolic shifts has not been clearly demonstrated, however, two hypotheses has been postulated (Podrini et al., 2020). First, the polycystins have the ability to regulate mitochondrial function and structure either by regulating Ca++ uptake in mitochondria, or by a direct translocation of a small fragment protein into the mitochondrial matrix (Kuo et al., 2019). Second, loss of mitochondrial functions in ADPKD is driven by multiple signaling pathways, which include AMPK, PPARα, PGC1α, mTORC1, cAMP and cystic fibrosis transmembrane conductance regulator (CFTR)-mediated ion transport as well as the expression of crucial components of the mitochondrial energy production apparatus (Hajarnis et al., 2017). PKD1-deficient mouse embryonic fibroblasts were found to have increased glucose uptake and glycolysis as their primary source of energy, even in normoxic conditions (Rowe et al., 2013). Importantly, these effects are dependent on the upregulation of mTORC1 signaling, resulting in the inhibition of AMP-activated protein kinase (AMPK), which led to the hypothesis that, in ADPKD, cells preferentially use aerobic glycolysis for energy production (Rowe et al., 2013). Menezes et al. found reduced oxidative phosphorylation in Pkd-/- cells that had fatty acids as their main energy source, suggesting that FAO is reduced (Menezes et al., 2016), and is accompanied by a compensatory glycolysis and administration of 2-deoxyglucose slowed disease progression (Riwanto et al., 2016).
In adition, a renal transcriptomic analysis and urine metabolomic analysis in a mouse model of ADPKD revealed altered metabolic pathways that are associated with cyst formation (Menezes et al., 2012). Among these altered pathways were high levels of acetylcarnitine in the urine of ADPKD mice, suggesting the presence of defective fatty acid metabolism in mitochondria. Transcriptional profiling and metabolomic analysis of progressive ADPKD found alterations in lipid metabolism (Menezes et al., 2016). The defective FAO in mice with renal tubule-specific Pkd1 deletion occurs via signaling involving miR-17 and PPARα (Hajarnis et al., 2017). Inhibition of miR-17 restored defective FAO and suppressed cyst formation in PKD mouse models (Hajarnis et al., 2017). Moreover, mutations in genes encoding components of the OXPHOS and FAO pathways, as well as in PPARα target genes, result in clinical disorders that include cystic kidneys (Hackl et al., 2017). Renal cysts develop in patients with glutaric acidaemia type II (Whitfield et al., 1996), which is caused by gene mutations in electron transfer flavoprotein subunit-α (ETFα), ETFβ or ETF dehydrogenase (ETFDH), which are components of an OXPHOS enzyme, ETF complex. Polycystins affect the function and morphology of mitochondria (Padovano et al., 2017; Lin et al., 2018a). The clear mechanisms underlying these alterations identified in ADPKD will need further investigation.
Perspectives and Future Directions
TFAM is important for mitochondrial integrity and its loss causes metabolic insults in tubular cells and cytosolic mtDNA leak, resulting in activation of an inflammatory response and kidney damage (Chung et al., 2019). Loss of TFAM causes tubular cell inflammation which is key for the activation of mesenchymal programming in epithelial cells and in neighboring cells. However, it is hard to determine the severity of mitochondrial DNA leakage in CKD or DKD subjects, and it would be striking to unravel the contribution of the upstream-effectors of STING that could have a potential impact on disease progression. Mitochondrial sirtuins and their association with TFAM may be a key link in the pathogenesis of kidney disease.
Mitochondrial sirtuins are a prominent class of metabolic regulators that exert effects on energy metabolism by protein acetylation and are linked to several biological effects on kidney health. Restoring SIRT3 has shown to have renal protective effects in age-associated renal fibrosis, as well as several models of kidney injury and diabetic models as well (Srivastava et al., 2018). However, dissecting the diverse functions of SIRT3 in different cell types remains a challenge. How SIRT3 expression levels change in different compartments and what the impact of the distribution of subcellular SIRT3 is on cellular health has not yet been fully investigated.
As compared to SIRT3, little is known about the role of SIRT4 and SIRT5 in the regulation of central metabolism in diverse cell types in the kidney. ADP-ribosylation activity by SIRT4, and demalonylation and desuccinylation activity by SIRT5, distinguish them from SIRT3 (Morigi et al., 2018). Available clinical data describing the effects of SIRT4 and SIRT5 on patients is not yet sufficient enough to decipher the role of SIRT4 and SIRT5 in the kidney (Morigi et al., 2018). The understanding of different sirtuins in renal physiology is still in its early stages. However, scientific advancement in evaluating the broad-spectrum of sirtuins targets involved in protective and developmental mechanisms has been studied but still needs more attention in the development of suitable therapeutics for combating kidney diseases.
In mouse models of diabetic kidney disease, SGLT-2 inhibitors and glycolysis inhibitors have shown protective effects on renal tubules; however, an unbiased study is needed to test FAO modulators, DPP-4 inhibitors, Wnt signaling inhibitors, antifibrotic peptides, ACE inhibitors and ARBs on their ability to restore SIRT3 levels in injured kidneys (Kanasaki et al., 2014; Srivastava et al., 2020a; Li et al., 2020b).
Further studies are required to identify small molecules that activate sirtuins activity, known as sirtuin-activating-compounds (STAC). Mostly, these STAC are related to naturally-occuring polyphenols (Morigi et al., 2018). Resveratrol was a well-known sirtuin activator discovered from these naturally-occurring polyphenols (Morigi et al., 2018). Resveratrol induces SIRT1 and SIRT3 and can act as an allosteric modulator, leading to conformational changes in the substrate, which can then influence the binding affinity for sirtuins (Xu et al., 2016a; Morigi et al., 2018). These studies suggest that there is a need to search for compounds that influence sirtuin level and activity in diabetic kidneys, such as flavonoids, chalcones, polyhydroquinolines, propiophenone derivatives, deoxyandrographolides, 2-methoxy-estradiol (2-ME) and thiazolidin-4-one derivatives; all of these compounds have shown protective effects in mouse models of diabetes mellitus (Kumar et al., 2010; Srivastava et al., 2010; Srivsatava et al., 2010; Shukla et al., 2011; Jaiswal et al., 2012; Kumar et al., 2012; Verma et al., 2012; Jaiswal, 2013; Mishra et al., 2013; Raza et al., 2013; Balaramnavar et al., 2014; Arha et al., 2015; Kanasaki et al., 2017), and can be further tested and potentially could be used in the treatment of diabetic kidney disease. The non-coding microRNAs play a critical role in the pathogenesis of diabetes mellitus; in spite of certain limitations about their specificity, the tissue-specific expression of these microRNAs is needs to be to analyzed (Kaur et al., 2011; Pandey et al., 2011; Srivastava et al., 2019a).
In current literature, it is reported that curcumin, silybin and AICAR induce SIRT3 levels, improve renal function and improve mitochondrial physiology in cisplatin-induced AKI (Li et al., 2017b; Ortega-Dominguez et al., 2017). Stanniocalcin reduces renal damage by SIRT3-linked activation of AMPK and UCP2 in a mouse model of renal ischemia-reperfusion injury (Pan et al., 2015). Honokiol decreases renal damage by activating SIRT3 in sepsis-associated AKI and hypertensive nephropathy (Kume et al., 2010; Li et al., 2014). Of great interest for future development would be new pharmacological strategies to target the effector molecules that control NAD + syntheses such as NAPMT and pharmacological compounds that reduce the activity of NAD + -depleting enzymes such as ADP-ribosyl-cyclases (Morigi et al., 2015; Camacho-Pereira et al., 2016). Further research and large double-blind clinical trials will be required to advance our understanding in this field. In summary, mitochondrial SIRT3 could be a novel candidate for treating renal diseases.
Author Contributions
SS proposed the concept, designed the figures, wrote and edited the manuscript. KK provided intellectual input. JG edited the manuscript.
Funding
JG is supported by the National Institutes of Health (HL131952).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, B. H., Kim, H. S., Song, S., Lee, I. H., Liu, J., Vassilopoulos, A., et al. (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U. S. A. 105, 14447–14452. doi:10.1073/pnas.0803790105
Akool, E.-S., Gauer, S., Osman, B., Doller, A., Schulz, S., Geiger, H., et al. (2012). Cyclosporin A and tacrolimus induce renal Erk1/2 pathway via ROS-induced and metalloproteinase-dependent EGF-receptor signaling. Biochem. Pharmacol. 83, 286–295. doi:10.1016/j.bcp.2011.11.001
Allison, S. J. (2019). Ubiquitylation of PTEN drives fibrosis in diabetic kidney disease. Nat. Rev. Nephrol. 15, 254.
Amar, S. K., Srivastav, A. K., and Srivastava, S. P. (2020). “Advances of the current therapeutic approach for the management of breast cancer,” in Current advances in breast cancer research: a molecular approach, (Sharjah, UAE: Bentham Science Publishers), 328–345. doi:10.2174/9789811451447120010016
Arany, I., Carter, A., Hall, S., Fulop, T., and Dixit, M. (2016). Coenzyme Q10 protects renal proximal tubule cells against nicotine-induced apoptosis through induction of p66shc-dependent antioxidant responses. Apoptosis 22, 220–228. doi:10.1038/s41581-019-0130-y
Argmann, C., and Auwerx, J. (2006). Insulin secretion: SIRT4 gets in on the act. Cell 126, 837–839. doi:10.1016/j.cell.2006.08.031
Arha, D., Pandeti, S., Mishra, A., Srivastava, S. P., Srivastava, A. K., Narender, T., et al. (2015). Deoxyandrographolide promotes glucose uptake through glucose transporter-4 translocation to plasma membrane in L6 myotubes and exerts antihyperglycemic effect in vivo. Eur. J. Pharmacol. 768, 207–216. doi:10.1016/j.ejphar.2015.10.055
Bae, J. H., Kim, S., Park, E.-G., Kim, S. G., Hahn, S., and Kim, N. H. (2019). Effects of dipeptidyl peptidase-4 inhibitors on renal outcomes in patients with type 2 diabetes: a systematic review and meta-analysis. Endocrinol. Metabol. 34, 80–92. 10.3803/EnM.2019.34.1.80
Bai, M., Chen, H., Ding, D., Song, R., Lin, J., Zhang, Y., et al. (2019). MicroRNA-214 promotes chronic kidney disease by disrupting mitochondrial oxidative phosphorylation. Kidney Int. 95, 1389–1404. doi:10.1016/j.kint.2018.12.028
Balaramnavar, V. M., Srivastava, R., Rahuja, N., Gupta, S., Rawat, A. K., Varshney, S., et al. (2014). Identification of novel PTP1B inhibitors by pharmacophore based virtual screening, scaffold hopping and docking. Eur. J. Med. Chem. 87, 578–594. doi:10.1016/j.ejmech.2014.09.097
Bao, M. H., Zhang, Y. W., Lou, X. Y., Cheng, Y., and Zhou, H. H. (2014). Protective effects of let-7a and let-7b on oxidized low-density lipoprotein induced endothelial cell injuries. PLoS One 9, e106540. doi:10.1016/j.ejmech.2014.09.097
Bartolomé, A., García-Aguilar, A., Asahara, S.-I., Kido, Y., Guillén, C., Pajvani, U. B., et al. (2017). MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol. Cell Biol. 37, e00441-17. doi:10.1128/MCB.00441-17
Bhargava, P., and Schnellmann, R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646. doi:10.1038/nrneph.2017.107
Bindu, S., Pillai, V. B., Kanwal, A., Samant, S., Mutlu, G. M., Verdin, E., et al. (2017). SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L68–L78. doi:10.1152/ajplung.00188.201
Boengler, K., Hilfiker-Kleiner, D., Heusch, G., and Schulz, R. (2010). Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 105, 771–785. doi:10.1007/s00395-010-0124-1
Bogdanova, V., and Castellon, X. (2016). Chronic inflammatory diseases and endothelial dysfunction. Aging Dis. 7, 81–89. doi:10.14336/AD.2015.0803
Breyer, M. D., and Susztak, K. (2016). The next generation of therapeutics for chronic kidney disease. Nat. Rev. Drug Discov. 15, 568–588. doi:10.1038/nrd.2016.67
Brownlee, M. (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625. doi:10.2337/diabetes.54.6.1615
Camacho-Pereira, J., Tarrago, M. G., Chini, C. C. S., Nin, V., Escande, C., Warner, G. M., et al. (2016). CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23, 1127–1139. doi:10.1016/j.cmet.2016.05.006
Campbell, C. T., Kolesar, J. E., and Kaufman, B. A. (2012). Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 1819, 921–929. doi:10.1016/j.bbagrm.2012.03.002
Carrico, C., Meyer, J. G., He, W., Gibson, B. W., and Verdin, E. (2018). The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 27, 497–512. doi:10.1016/j.cmet.2018.01.016
Cassina, L., Chiaravalli, M., and Boletta, A. (2020). Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. Faseb. J. 34, 6493–6507. doi:10.1096/fj.201901739RR
Chacko, B. K., Reily, C., Srivastava, A., Johnson, M. S., Ye, Y., Ulasova, E., et al. (2010). Prevention of diabetic nephropathy in Ins2+/−AkitaJ mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 432, 9–19. doi:10.1042/BJ20100308
Chan, D. C. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265–287. doi:10.1146/annurev-genet-110410-132529
Chau, B. N., Xin, C., Hartner, J., Ren, S., Castano, A. P., Linn, G., et al. (2012). MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 4, 121ra118. doi:10.1126/scitranslmed.3003205
Chen, F., Ma, Y., Sun, Z., and Zhu, X. (2018). Tangeretin inhibits high glucose-induced extracellular matrix accumulation in human glomerular mesangial cells. Biomed. Pharmacother. 102, 1077–1083. doi:10.1016/j.biopha.2018.03.169
Chen, S., Han, Q., Wang, X., Yang, M., Zhang, Z., Li, P., et al. (2013). IBP-mediated suppression of autophagy promotes growth and metastasis of breast cancer cells via activating mTORC2/Akt/FOXO3a signaling pathway. Cell Death Dis. 4, e842. doi:10.1038/cddis.2013.380
Chen, T., Li, J., Liu, J., Li, N., Wang, S., Liu, H., et al. (2015). Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-beta/Smad3 pathway. Am. J. Physiol. Heart Circ. Physiol. 308, H424–H434. doi:10.1152/ajpheart.00454.2014
Chiba, T., Peasley, K. D., Cargill, K. R., Maringer, K. V., Bharathi, S. S., Mukherjee, E., et al. (2019). Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J. Am. Soc. Nephrol. 30, 2384–2398. doi:10.1681/ASN.2019020163
Chung, K. W., Dhillon, P., Huang, S., Sheng, X., Shrestha, R., Qiu, C., et al. (2019). Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 30, 784–799.e5. doi:10.1016/j.cmet.2019.08.003
Chung, S., Overstreet, J. M., Li, Y., Wang, Y., Niu, A., Wang, S., et al. (2018). TGF-beta promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 3, e123563. doi:10.1172/jci.insight.123563
Cinà, D. P., Onay, T., Paltoo, A., Li, C., Maezawa, Y., De Arteaga, J., et al. (2012). Inhibition of MTOR disrupts autophagic flux in podocytes. J. Am. Soc. Nephrol. 23, 412–420. doi:10.1681/ASN.2011070690
Cooper, M., and Warren, A. M. (2019). A promising outlook for diabetic kidney disease. Nat. Rev. Nephrol. 15, 68–70. doi:10.1038/s41581-018-0092-5
Daehn, I. S. (2018). Glomerular endothelial cells stress and cross-talk with podocytes in the development of diabetic kidney disease. Front. Med. 5, 76. doi:10.3389/fmed.2018.00076
Das, S., Kohr, M., Dunkerly-Eyring, B., Lee, D. I., Bedja, D., Kent, O. A., et al. (2017). Divergent effects of miR-181 family members on myocardial function through protective cytosolic and detrimental mitochondrial microRNA targets. J. Am. Heart Assoc. 6, e004694. doi:10.1161/JAHA.116.004694
Daye, D., and Wellen, K. E. (2012). Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin. Cell Dev. Biol. 23, 362–369. doi:10.1016/j.semcdb.2012.02.002
Decleves, A. E., Zolkipli, Z., Satriano, J., Wang, L., Nakayama, T., Rogac, M., et al. (2014). Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int. 85, 611–623. doi:10.1016/j.molcel.2015.10.018
Denicola, G. M., and Cantley, L. C. (2015). Cancer’s fuel choice: new flavors for a picky eater. Mol Cell 60, 514–523. doi:10.1016/j.molcel.2015.10.018
Ding, H., Bai, F., Cao, H., Xu, J., Fang, L., Wu, J., et al. (2018). PDE/cAMP/Epac/C/EBP-beta signaling cascade regulates mitochondria biogenesis of tubular epithelial cells in renal fibrosis. Antioxid. Redox Signal 29, 637–652. doi:10.1089/ars.2017.7041
Doleris, L. M., Hill, G. S., Chedin, P., Nochy, D., Bellanne-Chantelot, C., Hanslik, T., et al. (2000). Focal segmental glomerulosclerosis associated with mitochondrial cytopathy. Kidney Int. 58, 1851–1858. doi:10.1111/j.1523-17552000.00356.x
Dorn, G. W., Vega, R. B., and Kelly, D. P. (2015). Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29, 1981–1991. doi:10.1101/gad.269894.115
Du, J., Zhou, Y., Su, X., Yu, J. J., Khan, S., Jiang, H., et al. (2011). Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809. doi:10.1126/science.1207861
Du, Y., Hu, H., Qu, S., Wang, J., Hua, C., Zhang, J., et al. (2018). SIRT5 deacylates metabolism-related proteins and attenuates hepatic steatosis in ob/ob mice. EBioMedicine 36, 347–357. doi:10.1016/j.ebiom.2018.09.037
Edwards, J. L. (2016). Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 374, 1094. doi:10.1056/NEJMc1600827
Efstratiadis, G., Divani, M., Katsioulis, E., and Vergoulas, G. (2009). Renal fibrosis. Hippokratia 13, 224–229.
Faubert, B., Boily, G., Izreig, S., Griss, T., Samborska, B., Dong, Z., et al. (2013). AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17, 113–124. doi:10.1016/j.cmet.2012.12.001
Finkel, T., Deng, C. X., and Mostoslavsky, R. (2009). Recent progress in the biology and physiology of sirtuins. Nature 460, 587–591. doi:10.1038/nature08197
Finley, L. W., Carracedo, A., Lee, J., Souza, A., Egia, A., Zhang, J., et al. (2011a). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Canc. Cell 19, 416–428. doi:10.1016/j.ccr.2011.02.014
Finley, L. W., Haas, W., Desquiret-Dumas, V., Wallace, D. C., Procaccio, V., Gygi, S. P., et al. (2011b). Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PloS One 6, e23295. doi:10.1371/journal.pone.0023295
Forbes, J. M., and Thorburn, D. R. (2018). Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 14, 291–312. doi:10.1038/nr10.1038/nrneph.2018.9neph.2018.9
Frezza, C., Cipolat, S., Martins De Brito, O., Micaroni, M., Beznoussenko, G. V., Rudka, T., et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. doi:10.1016/j.cell.2006.06.025
Galle, J., Quaschning, T., Seibold, S., and Wanner, C. (2003). Endothelial dysfunction and inflammation: what is the link?. Kidney Int. 63, S45–S49. doi:10.1046/j.1523-1755.63.s84.12.x
Galvan, D. L., Green, N. H., and Danesh, F. R. (2017). The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 92, 1051–1057. doi:10.1016/j.kint.2017.05.034
Gane, E. J., Weilert, F., Orr, D. W., Keogh, G. F., Gibson, M., Lockhart, M. M., et al. (2010). The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 30, 1019–1026. doi:10.1111/j.1478-3231.2010.02250.x
Gomez, I. G., Grafals, M., Portilla, D., and Duffield, J. S. (2013). MicroRNAs as potential therapeutic targets in kidney disease. J. Formos. Med. Assoc. 112, 237–243. doi:10.1016/j.jfma.2012.12.011
Gough, D. J., Corlett, A., Schlessinger, K., Wegrzyn, J., Larner, A. C., and Levy, D. E. (2009). Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324, 1713–1716. doi:10.1126/science.1171721
Graham, D., Huynh, N. N., Hamilton, C. A., Beattie, E., Smith, R. A. J., Cochemé, H. M., et al. (2009). Mitochondria-targeted antioxidant MitoQ10 endothelial function and hypertrophy, improves endothelial function and attenuates cardiac hypertrophy. Hypertension 54, 322–328. doi:10.1161/HYPERTENSIONAHA
Grande, M. T., and Lopez-Novoa, J. M. (2009). Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 5, 319–328. doi:10.1038/nrneph.2009.74
Grande, M. T., Sanchez-Laorden, B., Lopez-Blau, C., De Frutos, C. A., Boutet, A., Arevalo, M., et al. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997. doi:10.1038/nm.3901
Greer, S. N., Metcalf, J. L., Wang, Y., and Ohh, M. (2012). The updated biology of hypoxia-inducible factor. EMBO J. 31, 2448–2460. doi:10.1038/emboj.2012.125
Grgic, I., Duffield, J. S., and Humphreys, B. D. (2012). The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 27, 183–193. doi:10.1007/s00467-011-1772-6
Gu, J., Yang, M., Qi, N., Mei, S., Chen, J., Song, S., et al. (2016). Olmesartan prevents microalbuminuria in db/db diabetic mice through inhibition of angiotensin II/p38/SIRT1-Induced podocyte apoptosis. Kidney Blood Press Res. 41, 848–864. doi:10.1159/000452588
Guan, K. L., and Xiong, Y. (2011). Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 36, 108–116. doi:10.1016/j.tibs.2010.09.003
Hackl, A., Mehler, K., Gottschalk, I., Vierzig, A., Eydam, M., Hauke, J., et al. (2017). Disorders of fatty acid oxidation and autosomal recessive polycystic kidney disease-different clinical entities and comparable perinatal renal abnormalities. Pediatr. Nephrol. 32, 791–800. doi:10.1007/s00467-016-3556-5
Hagiwara, M., Yamagata, K., Capaldi, R. A., and Koyama, A. (2006). Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis. Kidney Int. 69, 1146–1152. doi:10.1038/sj.ki.5000207
Haigis, M. C., Mostoslavsky, R., Haigis, K. M., Fahie, K., Christodoulou, D. C., Murphy, A. J., et al. (2006). SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954. doi:10.1016/j.cell.2006.06.057
Hajarnis, S., Lakhia, R., Yheskel, M., Williams, D., Sorourian, M., Liu, X., et al. (2017). microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat. Commun. 8, 14395. doi:10.1038/ncomms14395
Hakroush, S., Cebulla, A., Schaldecker, T., Behr, D., Mundel, P., and Weins, A. (2014). Extensive podocyte loss triggers a rapid parietal epithelial cell response. J. Am. Soc. Nephrol. 25, 927–938. doi:10.1681/ASN.2013070687
Han, Y., Zhou, S., Coetzee, S., and Chen, A. (2019). SIRT4 and its roles in energy and redox metabolism in health, disease and during exercise. Front. Physiol. 10, 1006. doi:10.3389/fphys.2019.01006
Hanssen, N. M. J., and Jandeleit-Dahm, K. a. M. (2019). Dipeptidyl peptidase-4 inhibitors and cardiovascular and renal disease in type 2 diabetes: what have we learned from the CARMELINA trial? Diabetes Vasc. Dis. Res. 16, 303–309. doi:10.1177/1479164119842339
Harris, P. C., and Torres, V. E. (2009). Polycystic kidney disease. Annu. Rev. Med. 60, 321–337. doi:10.1146/annurev.med.60.101707.125712
Hato, T., Friedman, A. N., Mang, H., Plotkin, Z., Dube, S., Hutchins, G. D., et al. (2016). Novel application of complementary imaging techniques to examine in vivo glucose metabolism in the kidney. Am. J. Physiol. Renal. Physiol. 310, F717–F725. doi:10.1152/ajprenal.00535.2015
Hershberger, K. A., Martin, A. S., and Hirschey, M. D. (2017). Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 13, 213–225. doi:10.1038/nrneph.2017.5
Hickey, F. B., Corcoran, J. B., Docherty, N. G., Griffin, B., Bhreathnach, U., Furlong, F., et al. (2011). IHG-1 promotes mitochondrial biogenesis by stabilizing PGC-1α. J. Am. Soc. Nephrol. 22, 1475–1485. doi:10.1681/ASN.2010111154
Higgins, G. C., and Coughlan, M. T. (2014). Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br. J. Pharmacol. 171, 1917–1942. doi:10.1111/bph.12503
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B., et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 117, 3810–3820. doi:10.1172/JCI30487
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. doi:10.1038/nature08778
Hirschey, M. D., Shimazu, T., Jing, E., Grueter, C. A., Collins, A. M., Aouizerat, B., et al. (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell. 44, 177–190. doi:10.1016/j.molcel.2011.07.019
Hotta, O., Inoue, C. N., Miyabayashi, S., Furuta, T., Takeuchi, A., and Taguma, Y. (2001). Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 59, 1236–1243. doi:10.1046/j.1523-1755.2001.0590041236.x
Houtkooper, R. H., Pirinen, E., and Auwerx, J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell. Biol. 13, 225–238. doi:10.1038/nrm3293
Hu, Q., Li, J., Nitta, K., Kitada, M., Nagai, T., Kanasaki, K., et al. (2018). FGFR1 is essential for N-acetyl-seryl-aspartyl-lysyl-proline regulation of mitochondrial dynamics by upregulating microRNA let-7b-5p. Biochem. Biophys. Res. Commun. 495, 2214–2220. doi:10.1016/j.bbrc.2017.12.089
Huang, S., Zhang, A., Ding, G., and Chen, R. (2009). Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am. J. Physiol. Ren. Physiol. 296, F1323–F1333. doi:10.1152/ajprenal.90428.2008
Hung, C. M., Garcia-Haro, L., Sparks, C. A., and Guertin, D. A. (2012). mTOR-dependent cell survival mechanisms. Cold Spring Harbor Perspect. Biol. 4, a008771. doi:10.1101/cshperspect.a008771
Ishikawa, A., Kawarazaki, H., Ando, K., Fujita, M., Fujita, T., and Homma, Y. (2010). Renal preservation effect of ubiquinol, the reduced form of coenzyme Q10. Clin. Exp. Nephrol. 15, 30–33. doi:10.1007/s10157-010-0350-8
Jaiswal, N. (2013). Inhibition of alpha-glucosidase by Acacia nilotica prevents hyperglycemia along with improvement of diabetic complications via aldose reductase inhibition. J. Diabetes Metabol. 1, doi:10.4172/2155-6156.s6-004
Jaiswal, N., Bhatia, V., Srivastava, S. P., Srivastava, A. K., and Tamrakar, A. K. (2012). Antidiabetic effect of Eclipta alba associated with the inhibition of alpha-glucosidase and aldose reductase. Nat. Prod. Res. 26, 2363–2367. doi:10.1080/14786419.2012.662648
Jaquenod De Giusti, C., Roman, B., and Das, S. (2018). The influence of MicroRNAs on mitochondrial calcium. Front. Physiol. 9, doi:10.3389/fphys.2018.01291
Jeong, S. M., Xiao, C., Finley, L. W., Lahusen, T., Souza, A. L., Pierce, K., et al. (2013). SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Canc. Cell 23, 450–463. doi:10.1016/j.ccr.2013.02.024
Jha, J. C., Banal, C., Chow, B. S. M., Cooper, M. E., and Jandeleit-Dahm, K. (2016). Diabetes and kidney disease: role of oxidative stress. Antioxid. Redox Signal. 25, 657–684. doi:10.1089/ars.2016.6664
Jiang, L., Qiu, W., Zhou, Y., Wen, P., Fang, L., Cao, H., et al. (2013a). A microRNA-30e/mitochondrial uncoupling protein 2 axis mediates TGF-beta1-induced tubular epithelial cell extracellular matrix production and kidney fibrosis. Kidney Int. 84, 285–296. doi:10.1038/ki.2013.80
Jiang, Y. S., Jiang, T., Huang, B., Chen, P. S., and Ouyang, J. (2013b). Epithelial-mesenchymal transition of renal tubules: divergent processes of repairing in acute or chronic injury? Med. Hypotheses 81, 73–75. doi:10.1016/j.mehy.2013.03.020
Jing, E., Emanuelli, B., Hirschey, M. D., Boucher, J., Lee, K. Y., Lombard, D., et al. (2011). Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. U. S. A. 108, 14608–14613. doi:10.1073/pnas.1111308108
Jourde-Chiche, N., Fakhouri, F., Dou, L., Bellien, J., Burtey, S., Frimat, M., et al. (2019). Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 15, 87–108. doi:10.1038/s41581-018-0098-z
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. The J. Clin. Invest. 119, 1420–1428. doi:10.1172/JCI39104
Kanasaki, K., Shi, S., Kanasaki, M., He, J., Nagai, T., Nakamura, Y., et al. (2014). Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 63, 2120–2131. doi:10.2337/db13-1029
Kanasaki, M., Srivastava, S. P., Yang, F., Xu, L., Kudoh, S., Kitada, M., et al. (2017). Deficiency in catechol-o-methyltransferase is linked to a disruption of glucose homeostasis in mice. Sci. Rep. 7, 7927. doi:10.1038/s41598-017-08513-w
Kang, D. H., Nakagawa, T., Feng, L., and Johnson, R. J. (2002). Nitric oxide modulates vascular disease in the remnant kidney model. Am. J. Pathol. 161, 239–248. doi:10.1016/S0002-9440(10)64175-2
Kang, H. M., Ahn, S. H., Choi, P., Ko, Y. A., Han, S. H., Chinga, F., et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46. doi:10.1038/nm.3762
Kaur, K., Pandey, A. K., Srivastava, S., Srivastava, A. K., and Datta, M. (2011). Comprehensive miRNome and in silico analyses identify the Wnt signaling pathway to be altered in the diabetic liver. Mol. Biosyst. 7, 3234–3244. doi:10.1039/c1mb05041a
Kawata, T., Tada, K., Kobayashi, M., Sakamoto, T., Takiuchi, Y., Iwai, F., et al. (2018). Dual inhibition of the mTORC1 and mTORC2 signaling pathways is a promising therapeutic target for adult T-cell leukemia. Canc. Sci. 109, 103–111. doi:10.1111/cas.13431
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20, 3328. doi:10.3390/ijms20133328
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., et al. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells. J. Biol. Chem. 276, 4588–4596. doi:10.1074/jbc.M009093200
Kim, H. S., Patel, K., Muldoon-Jacobs, K., Bisht, K. S., Aykin-Burns, N., Pennington, J. D., et al. (2010). SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Canc. Cell 17, 41–52. doi:10.1016/j.ccr.2009.11.023
Kim, S. C., Sprung, R., Chen, Y., Xu, Y., Ball, H., Pei, J., et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618. doi:10.1016/j.molcel.2006.06.026
Kolling, M., Kaucsar, T., Schauerte, C., Hubner, A., Dettling, A., Park, J. K., et al. (2017). Therapeutic miR-21 silencing ameliorates diabetic kidney disease in mice. Mol. Ther. 25, 165–180. doi:10.1016/j.ymthe.2016.08.001
Kuhlbrandt, W. (2015). Structure and function of mitochondrial membrane protein complexes. BMC Biol. 13, 89. doi:10.1186/s12915-015-0201-x
Kumar, A., Chaugule, V. K., Condos, T. E. C., Barber, K. R., Johnson, C., Toth, R., et al. (2017). Parkin–phosphoubiquitin complex reveals cryptic ubiquitin-binding site required for RBR ligase activity. Nat. Struct. Mol. Biol. 24, 475–483. doi:10.1038/nsmb.3400
Kumar, A., Sharma, S., Gupta, L. P., Ahmad, P., Srivastava, S. P., Rahuja, N., et al. (2012). Synthesis of propiophenone derivatives as new class of antidiabetic agents reducing body weight in db/db mice. Bioorg. Med. Chem. 20, 2172–2179. doi:10.1016/j.bmc.2011.12.027
Kumar, A., Sharma, S., Tripathi, V. D., Maurya, R. A., Srivastava, S. P., Bhatia, G., et al. (2010). Design and synthesis of 2,4-disubstituted polyhydroquinolines as prospective antihyperglycemic and lipid modulating agents. Bioorg. Med. Chem. 18, 4138–4148. doi:10.1016/j.bmc.2009.11.06
Kume, S., Uzu, T., Horiike, K., Chin-Kanasaki, M., Isshiki, K., Araki, S., et al. (2010). Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. 120, 1043–1055. doi:10.1172/JCI41376
Kuo, I. Y., Brill, A. L., Lemos, F. O., Jiang, J. Y., Falcone, J. L., Kimmerling, E. P., et al. (2019). Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 12, eaat7397.
Kuppusamy, K. T., Jones, D. C., Sperber, H., Madan, A., Fischer, K. A., Rodriguez, M. L., et al. (2015). Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 112, E2785–E2794. doi:10.1073/pnas.1424042112
Lamming, D. W., Ye, L., Katajisto, P., Goncalves, M. D., Saitoh, M., Stevens, D. M., et al. (2012). Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643. doi:10.1126/science.1215135
Larsson, N. G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., et al. (1998). Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236. doi:10.1038/ng0398-231
Laverman, G. D., Remuzzi, G., and Ruggenenti, P. (2004). ACE inhibition versus angiotensin receptor blockade: which is better for renal and cardiovascular protection?. J. Am. Soc. Nephrol. 1 (Suppl. 15), S64–S70. doi:10.1097/01.asn.0000093368.27046.3c
Lebleu, V. S., Taduri, G., O’connell, J., Teng, Y., Cooke, V. G., Woda, C., et al. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053. doi:10.1038/nm.3218
Lee, S. H., Lee, J. H., Lee, H. Y., and Min, K. J. (2019). Sirtuin signaling in cellular senescence and aging. BMB Rep. 52, 24–34. doi:10.5483/BMBRep.2019.52.1.290
Lesnefsky, E. J., Moghaddas, S., Tandler, B., Kerner, J., and Hoppel, C. L. (2001). Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 33, 1065–1089. doi:10.1006/jmcc.2001.1378
Levin, A., Tonelli, M., Bonventre, J., Coresh, J., Donner, J. A., Fogo, A. B., et al. (2017). Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 390, 1888–1917. doi:10.1016/S0140-6736(17)30788-2
Li, J., Liu, H., Prakash Srivastava, S., Hu, Q., Gao, R., Li, S., et al. (2020a). Endothelial FGFR1 (fibroblast growth factor receptor 1) deficiency contributes differential fibrogenic effects in kidney and heart of diabetic mice. Hypertension 76, 1935–1944. doi:10.1161/HYPERTENSIONAHA.120.15587
Li, J., Liu, H., Takagi, S., Nitta, K., Kitada, M., Srivastava, S. P., et al. (2020b). Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight 5, e129034.
Li, J., Shi, S., Srivastava, S. P., Kitada, M., Nagai, T., Nitta, K., et al. (2017a). FGFR1 is critical for the anti-endothelial mesenchymal transition effect of N-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway. Cell Death Dis. 8, e2965. doi:10.1038/cddis.2017.353
Li, N., Xie, H., Li, L., Wang, J., Fang, M., Yang, N., et al. (2014). Effects of honokiol on sepsis-induced acute kidney injury in an experimental model of sepsis in rats. Inflammation 37, 1191–1199. doi:10.1007/s10753-014-9845-x
Li, Q., Zeng, Y., Jiang, Q., Wu, C., and Zhou, J. (2019). Role of mTOR signaling in the regulation of high glucose-induced podocyte injury. Exp. Ther. Med. 17, 2495–2502. doi:10.3892/etm.2019.7236
Li, Y., Ye, Z., Lai, W., Rao, J., Huang, W., Zhang, X., et al. (2017b). Activation of sirtuin 3 by silybin attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Front. Pharmacol. 8, 178. doi:10.3389/fphar.2017.00178
Lin, C.-C., Kurashige, M., Liu, Y., Terabayashi, T., Ishimoto, Y., Wang, T., et al. (2018a). A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci. Rep. 8, 2743. doi:10.1038/s41598-018-20856-6
Lin, J.-R., Zheng, Y.-J., Zhang, Z.-B., Shen, W.-L., Li, X.-D., Wei, T., et al. (2018b). Suppression of endothelial-to-mesenchymal transition by SIRT (sirtuin) 3 alleviated the development of hypertensive renal injury. Hypertension 72, 350–360. doi:10.1161/HYPERTENSIONAHA.118.10482
Lin, S. J., Defossez, P. A., and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128. doi:10.1126/science.289.5487.2126
Lindenmeyer, M. T., Rastaldi, M. P., Ikehata, M., Neusser, M. A., Kretzler, M., Cohen, C. D., et al. (2008). Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J. Am. Soc. Nephrol. 19, 2225–2236. doi:10.1681/ASN.2007121313
Liu, Y. (2011). Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7, 684–696. doi:10.1038/nrneph.2011.149
Liu, B.-C., Tang, T.-T., Lv, L.-L., and Lan, H.-Y. (2018). Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 93, 568–579. doi:10.1016/j.kint.2017.09.033
Liu, M., Quek, L.-E., Sultani, G., and Turner, N. (2016). Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Canc. Metabol. 4, 19. doi:10.1186/s40170-016-0160-x
Liu, S., Soong, Y., Seshan, S. V., and Szeto, H. H. (2014). Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am. J. Physiol. Renal. Physiol. 306, F970–F980. doi:10.1152/ajprenal.00697.2013
Lloberas, N., Cruzado, J. M., Franquesa, M., Herrero-Fresneda, I., Torras, J., Alperovich, G., et al. (2006). Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J. Am. Soc. Nephrol. 17, 1395–1404. doi:10.1681/ASN.2005050549
Lombard, D. B., Alt, F. W., Cheng, H. L., Bunkenborg, J., Streeper, R. S., Mostoslavsky, R., et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814. doi:10.1128/MCB.01636-07
Lovisa, S., Lebleu, V. S., Tampe, B., Sugimoto, H., Vadnagara, K., Carstens, J. L., et al. (2015). Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 21, 998–1009. doi:10.1038/nm.3902
Lu, J., Tan, M., and Cai, Q. (2015). The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 356, 156–164. doi:10.1016/j.canlet.2014.04.001
Lufei, C. (2003). GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J. 22, 1325–1335. doi:10.1093/emboj/cdg135
Ma, X., Li, C., Sun, L., Huang, D., Li, T., He, X., et al. (2014). Lin28/let-7 axis regulates aerobic glycolysis and cancer progression via PDK1. Nat. Commun. 5, 5212. doi:10.1038/ncomms6212
Mallipattu, S. K., and Kravets, I. (2020). The role of podocytes and podocyte-associated biomarkers in diagnosis and treatment of diabetic kidney disease. J. Endocrine Soc. 4, bvaa029. doi:10.1210/jendso/bvaa029
Mathias, R. A., Greco, T. M., Oberstein, A., Budayeva, H. G., Chakrabarti, R., Rowland, E. A., et al. (2014). Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625. doi:10.1016/j.cell.2014.11.046
Meier, J. A., Hyun, M., Cantwell, M., Raza, A., Mertens, C., Raje, V., et al. (2017). Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 10, eaag2588. doi:10.1126/scisignal.aag2588
Menezes, L. F., Lin, C.-C., Zhou, F., and Germino, G. G. (2016). Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 5, 183–192. doi:10.1016/j.ebiom.2016.01.027
Menezes, L. F., Zhou, F., Patterson, A. D., Piontek, K. B., Krausz, K. W., Gonzalez, F. J., et al. (2012). Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4alpha as a disease modifier. PLoS Genet. 8, e1003053. doi:10.1371/journal.pgen.1003053
Meng, X. M., Tang, P. M., Li, J., and Lan, H. Y. (2015). TGF-beta/Smad signaling in renal fibrosis. Front. Physiol. 6, 82. doi:10.3389/fphys.2015.00082
Meßmer, U. K., Winkel, G., Briner, V. A., and Pfeilschifter, J. (2000). Suppression of apoptosis by glucocorticoids in glomerular endothelial cells: effects on proapoptotic pathways. Br. J. Pharmacol. 129, 1673–1683. doi:10.1038/sj.bjp.0703255
Metzinger-Le Meuth, V., Fourdinier, O., Charnaux, N., Massy, Z. A., and Metzinger, L. (2019). The expanding roles of microRNAs in kidney pathophysiology. Nephrol. Dial. Transplant. 34, 7–15. doi:10.1093/ndt/gfy140
Mishra, A., Srivastava, R., Srivastava, S. P., Gautam, S., Tamrakar, A. K., Maurya, R., et al. (2013). Antidiabetic activity of heart wood of Pterocarpus marsupium Roxb. and analysis of phytoconstituents. Indian J. Exp. Biol. 51, 363–374.
Mizuguchi, Y., Chen, J., Seshan, S. V., Poppas, D. P., Szeto, H. H., and Felsen, D. (2008). A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 295, F1545–F1553. doi:10.1152/ajprenal.00395.2007
Molino, D., Nascimbeni, A. C., Giordano, F., Codogno, P., and Morel, E. (2017). ER-driven membrane contact sites: evolutionary conserved machineries for stress response and autophagy regulation?. Commun. Integr. Biol. 10, e1401699. doi:10.1080/19420889.2017.1401699
Morigi, M., Perico, L., and Benigni, A. (2018). Sirtuins in renal health and disease. J. Am. Soc. Nephrol. 29, 1799–1809. doi:10.1681/ASN.2017111218
Morigi, M., Perico, L., Rota, C., Longaretti, L., Conti, S., Rottoli, D., et al. (2015). Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest. 125, 715–726. doi:10.1172/JCI77632
Nagai, T., Kanasaki, M., Srivastava, S., Nakamura, Y., Ishigaki, Y., Kitada, M., et al. (2014). N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. BioMed Res. Int. 2014, 696475. doi:10.1155/2014/696475
Nakagawa, T., Lomb, D. J., Haigis, M. C., and Guarente, L. (2009). SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570. doi:10.1016/j.cell.2009.02.026
Nascimento, L. R. D., and Domingueti, C. P. (2019). MicroRNAs: new biomarkers and promising therapeutic targets for diabetic kidney disease. J. Bras Nefrol. 41, 412–422. doi:10.1590/2175-8239-JBN-2018-0165
Nasrin, N., Wu, X., Fortier, E., Feng, Y., Bare, O. C., Chen, S., et al. (2010). SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 285, 31995–32002. doi:10.1074/jbc.M110.124164
Niemand, C., Nimmesgern, A., Haan, S., Fischer, P., Schaper, F., Rossaint, R., et al. (2003). Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J. Immunol. 170, 3263–3272. doi:10.4049/jimmunol.170.6.3263
Nitta, K., Shi, S., Nagai, T., Kanasaki, M., Kitada, M., Srivastava, S. P., et al. (2016). Oral administration of N-Acetyl-seryl-aspartyl-lysyl-proline ameliorates kidney disease in both type 1 and type 2 diabetic mice via a therapeutic regimen. BioMed Res. Int. 2016, 9172157. doi:10.1155/2016/9172157
O'rourke, B., and Blatter, L. A. (2009). Mitochondrial Ca2+ uptake: tortoise or hare?. J. Mol. Cell. Cardiol. 46, 767–774. doi:10.1016/j.yjmcc.2008.12.011
Ortega-Dominguez, B., Aparicio-Trejo, O. E., Garcia-Arroyo, F. E., Leon-Contreras, J. C., Tapia, E., Molina-Jijon, E., et al. (2017). Curcumin prevents cisplatin-induced renal alterations in mitochondrial bioenergetics and dynamic. Food Chem. Toxicol. 107, 373–385. doi:10.1016/j.fct.2017.07.018
Otera, H., and Mihara, K. (2011). Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2, 167–172. doi:10.4161/sgtp.2.3.16486
Padovano, V., Kuo, I. Y., Stavola, L. K., Aerni, H. R., Flaherty, B. J., Chapin, H. C., et al. (2017). The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol. Biol. Cell 28, 261–269. doi:10.1091/mbc.E16-08-0597
Palmer, S. C., Mavridis, D., Navarese, E., Craig, J. C., Tonelli, M., Salanti, G., et al. (2015). Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet 385, 2047–2056. doi:10.1016/S0140-6736(14)62459-4
Palmirotta, R., Cives, M., Della-Morte, D., Capuani, B., Lauro, D., Guadagni, F., et al. (2016). Sirtuins and cancer: role in the epithelial-mesenchymal transition. Oxid. Med. Cell Longev. 2016, 3031459. doi:10.1155/2016/3031459
Palsson-Mcdermott, E. M., Curtis, A. M., Goel, G., Lauterbach, M. A., Sheedy, F. J., Gleeson, L. E., et al. (2015). Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 21, 65–80. doi:10.1016/j.cmet.2014.12.005
Pan, J. S., Huang, L., Belousova, T., Lu, L., Yang, Y., Reddel, R., et al. (2015). Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an AMP-activated protein kinase-dependent pathway. J. Am. Soc. Nephrol. 26, 364–378. doi:10.1681/ASN.2013070703
Pandey, A. K., Verma, G., Vig, S., Srivastava, S., Srivastava, A. K., and Datta, M. (2011). miR-29a levels are elevated in the db/db mice liver and its overexpression leads to attenuation of insulin action on PEPCK gene expression in HepG2 cells. Mol. Cell. Endocrinol. 332, 125–133. doi:10.1016/j.mce.2010.10.004
Pearce, E. L., Walsh, M. C., Cejas, P. J., Harms, G. M., Shen, H., Wang, L. S., et al. (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. doi:10.1038/nature08097
Peng, C., Lu, Z., Xie, Z., Cheng, Z., Chen, Y., Tan, M., et al. (2011). The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteom. 10, M111.012658. doi:10.1074/mcp.M111.012658
Podrini, C., Cassina, L., and Boletta, A. (2020). Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell. Signal. 67, 109495. doi:10.1016/j.cellsig.2019.109495
Poli, V., and Camporeale, A. (2015). STAT3-Mediated metabolic reprograming in cellular transformation and implications for drug resistance. Front. Oncol. 5, 121. doi:10.3389/fonc.2015.00121
Qi, W., Keenan, H. A., Li, Q., Ishikado, A., Kannt, A., Sadowski, T., et al. (2017). Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 23, 753–762. doi:10.1038/nm.4328
Qin, J., Peng, Z. Z., Li, Q., Wen, R., and Tao, L. J. (2018). Renal fibrosis and mitochondrial damage. Chin. Med. J. (Engl) 131, 2769–2772. doi:10.4103/0366-6999.245272
Qiu, X., Brown, K., Hirschey, M. D., Verdin, E., and Chen, D. (2010). Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667. doi:10.1016/j.cmet.2010.11.015
Quadri, M. M., Fatima, S. S., Che, R. C., and Zhang, A. H. (2019). Mitochondria and renal fibrosis. Adv. Exp. Med. Biol. 1165, 501–524. doi:10.1007/978-981-13-8871-2_25
Rardin, M. J., He, W., Nishida, Y., Newman, J. C., Carrico, C., Danielson, S. R., et al. (2013). SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933. doi:10.1016/j.cmet.2013.11.013
Ravi, S., Mitchell, T., Kramer, P. A., Chacko, B., and Darley-Usmar, V. M. (2014). Mitochondria in monocytes and macrophages-implications for translational and basic research. Int. J. Biochem. Cell Biol. 53, 202–207. doi:10.1016/j.biocel.2014.05.019
Raza, S., Srivastava, S. P., Srivastava, D. S., Srivastava, A. K., Haq, W., and Katti, S. B. (2013). Thiazolidin-4-one and thiazinan-4-one derivatives analogous to rosiglitazone as potential antihyperglycemic and antidyslipidemic agents. Eur. J. Med. Chem. 63, 611–620. doi:10.1016/j.ejmech.2013.01.054
Reichold, M., Klootwijk, E. D., Reinders, J., Otto, E. A., Milani, M., Broeker, C., et al. (2018). Glycine amidinotransferase (GATM), renal fanconi syndrome, and kidney failure. J. Am. Soc. Nephrol. 29, 1849–1858. doi:10.1681/ASN.2017111179
Rincon, M., and Pereira, F. (2018). A new perspective: mitochondrial Stat3 as a regulator for lymphocyte function. Int. J. Mol. Sci. 19, 1656. doi:10.3390/ijms19061656
Rivara, M. B., Yeung, C. K., Robinson-Cohen, C., Phillips, B. R., Ruzinski, J., Rock, D., et al. (2017). Effect of coenzyme Q10 on biomarkers of oxidative stress and cardiac function in hemodialysis patients: the CoQ10 biomarker trial. Am. J. Kidney Dis. 69, 389–399. doi:10.1053/j.ajkd.2016.08.041
Riwanto, M., Kapoor, S., Rodriguez, D., Edenhofer, I., Segerer, S., and Wuthrich, R. P. (2016). Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS One 11, e0146654. doi:10.1371/journal.pone.0146654
Rouschop, K. M. A., Roelofs, J. J. T. H., Claessen, N., Martins, P. D. C., Zwaginga, J.-J., Pals, S. T., et al. (2005). Protection against renal ischemia reperfusion injury by CD44 disruption. J. Am. Soc. Nephrol. 16, 2034–2043. doi:10.1681/ASN.2005010054
Rowe, I., Chiaravalli, M., Mannella, V., Ulisse, V., Quilici, G., Pema, M., et al. (2013). Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 19, 488–493. doi:10.1038/nm.3092
Roxburgh, S. A., Kattla, J. J., Curran, S. P., O'meara, Y. M., Pollock, C. A., Goldschmeding, R., et al. (2009). Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes 58, 1641–1650. doi:10.2337/db08-1365
Samant, S. A., Zhang, H. J., Hong, Z., Pillai, V. B., Sundaresan, N. R., Wolfgeher, D., et al. (2014). SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol. 34, 807–819. doi:10.1128/MCB.01483-13
Sanchis-Gomar, F., Garcia-Gimenez, J. L., Gomez-Cabrera, M. C., and Pallardo, F. V. (2014). Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr. Pharm. Des. 20, 5619–5633. doi:10.2174/1381612820666140306095106
Saunders, L. R., and Verdin, E. (2007). Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 26, 5489–5504. doi:10.1038/sj.onc.1210616
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638. doi:10.1152/physrev.00025.2007
Schindler, C., Levy, D. E., and Decker, T. (2007). JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063. doi:10.1074/jbc.R700016200
Schreiber, K. H., Arriola Apelo, S. I., Yu, D., Brinkman, J. A., Velarde, M. C., Syed, F. A., et al. (2019). A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat. Commun. 10, 3194. doi:10.1038/s41467-019-11174-0
Schrimpf, C., and Duffield, J. S. (2011). Mechanisms of fibrosis: the role of the pericyte. Curr. Opin. Nephrol. Hypertens. 20, 297–305. doi:10.1097/MNH.0b013e328344c3d4
Schubert, A. F., Gladkova, C., Pardon, E., Wagstaff, J. L., Freund, S. M. V., Steyaert, J., et al. (2017). Structure of PINK1 in complex with its substrate ubiquitin. Nature 552, 51–56. doi:10.1038/nature24645
Schug, T. T., and Li, X. (2011). Sirtuin 1 in lipid metabolism and obesity. Ann. Med. 43, 198–211. doi:10.3109/07853890.2010.547211
Scindia, Y. M., Deshmukh, U. S., and Bagavant, H. (2010). Mesangial pathology in glomerular disease: targets for therapeutic intervention☆. Adv. Drug Deliv. Rev. 62, 1337–1343. doi:10.1016/j.addr.2010.08.011
Serguienko, A., Grad, I., Wennerstrom, A. B., Meza-Zepeda, L. A., Thiede, B., Stratford, E. W., et al. (2015). Metabolic reprogramming of metastatic breast cancer and melanoma by let-7a microRNA. Oncotarget 6, 2451–2465. doi:10.18632/oncotarget.3235
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713. doi:10.1101/cshperspect.a018713
Sheng, J., Li, H., Dai, Q., Lu, C., Xu, M., Zhang, J., et al. (2018). NR4A1 promotes diabetic nephropathy by activating mff-mediated mitochondrial fission and suppressing parkin-mediated mitophagy. Cell. Physiol. Biochem. 48, 1675–1693. doi:10.1159/000492292
Shi, J. X., Wang, Q. J., Li, H., and Huang, Q. (2017). SIRT4 overexpression protects against diabetic nephropathy by inhibiting podocyte apoptosis. Exp. Ther. Med. 13, 342–348. doi:10.3892/etm.2016.3938
Shi, S., Srivastava, S. P., Kanasaki, M., He, J., Kitada, M., Nagai, T., et al. (2015). Interactions of DPP-4 and integrin beta1 influences endothelial-to-mesenchymal transition. Kidney Int. 88, 479–489. doi:10.1038/ki.2015.103
Shimazu, T., Hirschey, M. D., Hua, L., Dittenhafer-Reed, K. E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661. doi:10.1016/j.cmet.2010.11.003
Shirihai, O. S., Song, M., and Dorn, G. W. (2015). How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 116, 1835–1849. doi:10.1161/CIRCRESAHA.116.306374
Shukla, P., Srivastava, S. P., Srivastava, R., Rawat, A. K., Srivastava, A. K., and Pratap, R. (2011). Synthesis and antidyslipidemic activity of chalcone fibrates. Bioorg. Med. Chem. Lett 21, 3475–3478. doi:10.1016/j.bmcl.2011.03.057
Sivitz, W. I., and Yorek, M. A. (2010). Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 12, 537–577. doi:10.1089/ars.2009.2531
Smith, M. A., Covington, M. D., and Schnellmann, R. G. (2012). Loss of calpain 10 causes mitochondrial dysfunction during chronic hyperglycemia. Arch. Biochem. Biophys. 523, 161–168. doi:10.1016/j.abb.2012.04.020
Snow, B. J., Rolfe, F. L., Lockhart, M. M., Frampton, C. M., O'sullivan, J. D., Fung, V., et al. (2010). A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 25, 1670–1674. doi:10.1002/mds.23148
Soga, T. (2013). Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 104, 275–281. doi:10.1111/cas.12085
Someya, S., Yu, W., Hallows, W. C., Xu, J., Vann, J. M., Leeuwenburgh, C., et al. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812. doi:10.1016/j.cell.2010.10.002
Sosulski, M. L., Gongora, R., Feghali-Bostwick, C., Lasky, J. A., and Sanchez, C. G. (2017). Sirtuin 3 deregulation promotes pulmonary fibrosis. J. Gerontol. A Biol. Sci. Med. Sci. 72, 595–602. doi:10.1093/gerona/glw151
Sottnik, J. L., Lori, J. C., Rose, B. J., and Thamm, D. H. (2011). Glycolysis inhibition by 2-deoxy-D-glucose reverts the metastatic phenotype in vitro and in vivo. Clin. Exp. Metastasis 28, 865–875. doi:10.1007/s10585-011-9417-5
Sourris, K. C., Harcourt, B. E., Tang, P. H., Morley, A. L., Huynh, K., Penfold, S. A., et al. (2012). Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic. Biol. Med. 52, 716–723. doi:10.1016/j.freeradbiomed.2011.11.017
Srivastava, S. P., and Goodwin, J. E. (2020). Cancer biology and prevention in diabetes. Cells 9, 1380. doi:10.3390/cells9061380
Srivastava, S. P., Goodwin, J. E., Kanasaki, K., and Koya, D. (2020a). Inhibition of angiotensin-converting enzyme ameliorates renal fibrosis by mitigating DPP-4 level and restoring antifibrotic MicroRNAs. Genes 11, 211. doi:10.3390/genes11020211
Srivastava, S. P., Goodwin, J. E., Kanasaki, K., and Koya, D. (2020b). Metabolic reprogramming by N-acetyl-seryl-aspartyl-lysyl-proline protects against diabetic kidney disease. Br. J. Pharmacol. 177, 3691–3711. doi:10.1111/bph.15087
Srivastava, S. P., Hedayat, A. F., Kanasaki, K., and Goodwin, J. E. (2019a). microRNA crosstalk influences epithelial-to-mesenchymal, endothelial-to-mesenchymal, and macrophage-to-mesenchymal transitions in the kidney. Front. Pharmacol. 10, 904. doi:10.3389/fphar.2019.00904
Srivastava, S. P., Hedayat, F. A., Kanasaki, K., and Goodwin, J. E. (2019b). microRNA crosstalk influences epithelial-to-mesenchymal, endothelial-to-mesenchymal, and macrophage-to-mesenchymal transitions in the kidney. Front. Pharmacol. 10, 904. doi:10.3389/fphar.2019.00904
Srivastava, S. P., Koya, D., and Kanasaki, K. (2013). MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. BioMed Res. Int. 2013, 125469. doi:10.1155/2013/125469
Srivastava, S. P., Li, J., Kitada, M., Fujita, H., Yamada, Y., Goodwin, J. E., et al. (2018). SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. 9, 997. doi:10.1038/s41419-018-1057-0
Srivastava, S. P., Mishra, A., Bhatia, V., Narender, T., and Srivastava, A. K. (2010). Acacia catechu hard wood: potential anti-diabetic cum anti-dyslipidemic. Med. Chem. Res. 20, 1732–1739. doi:10.1007/s00044-010-9479-y
Srivastava, S. P., Shi, S., Kanasaki, M., Nagai, T., Kitada, M., He, J., et al. (2016). Effect of antifibrotic MicroRNAs crosstalk on the action of N-acetyl-seryl-aspartyl-lysyl-proline in diabetes-related kidney fibrosis. Sci. Rep. 6, 29884. doi:10.1038/srep29884
Srivastava, S. P., Shi, S., Koya, D., and Kanasaki, K. (2014). Lipid mediators in diabetic nephropathy. Fibrogenesis Tissue Repair 7, 12. doi:10.1186/1755-1536-7-12
Srivastava, S. P., Zhou, H., Setia, O., Dardik, A., Fernandez-Hernando, C., and Goodwin, J. (2020c). Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. bioRxiv, doi:10.1101/2020.06.29.178228
Srivsatava, R., Srivastava, S. P., Jaiswal, N., Mishra, A., Maurya, R., and Srivastava, A. K. (2010). Antidiabetic and antidyslipidemic activities of Cuminum cyminum L. in validated animal models. Med. Chem. Res. 20, 1656–1666. doi:10.1007/s00044-010-9483-2
Stallons, L. J., Whitaker, R. M., and Schnellmann, R. G. (2014). Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 224, 326–332. doi:10.1016/j.toxlet.2013.11.014
Sun, S., Ning, X., Zhang, Y., Lu, Y., Nie, Y., Han, S., et al. (2009). Hypoxia-inducible factor-1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 75, 1278–1287. doi:10.1038/ki.2009.62
Sundaresan, N. R., Bindu, S., Pillai, V. B., Samant, S., Pan, Y., Huang, J. Y., et al. (2015). SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3beta. Mol. Cell Biol. 36, 678–692. doi:10.1128/MCB.00586-15
Sundaresan, N. R., Gupta, M., Kim, G., Rajamohan, S. B., Isbatan, A., and Gupta, M. P. (2009). Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771. doi:10.1172/JCI39162
Susztak, K., Ciccone, E., Mccue, P., Sharma, K., and Bottinger, E. P. (2005). Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2, e45. doi:10.1371/journal.pmed.0020045
Sweetwyne, M. T., Pippin, J. W., Eng, D. G., Hudkins, K. L., Chiao, Y. A., Campbell, M. D., et al. (2017). The mitochondrial-targeted peptide, SS-31, improves glomerular architecture in mice of advanced age. Kidney Int. 91, 1126–1145. doi:10.1016/j.kint.2016.10.036
Takagaki, Y., Lee, S. M., Dongqing, Z., Kitada, M., Kanasaki, K., and Koya, D. (2020). Endothelial autophagy deficiency induces IL6-dependent endothelial mesenchymal transition and organ fibrosis. Autophagy 16, 1905–1914. doi:10.1080/15548627.2020.1713641
Tammineni, P., Anugula, C., Mohammed, F., Anjaneyulu, M., Larner, A. C., and Sepuri, N. B. V. (2013). The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 288, 4723–4732. doi:10.1074/jbc.M112.378984
Tanaka, T., Nishimura, A., Nishiyama, K., Goto, T., Numaga-Tomita, T., and Nishida, M. (2020). Mitochondrial dynamics in exercise physiology. Pflugers Arch 472, 137–153. doi:10.1007/s00424-019-02258-3
Thuan, D. T. B., Zayed, H., Eid, A. H., Abou-Saleh, H., Nasrallah, G. K., Mangoni, A. A., et al. (2018). A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front. Immunol. 9, 1985. doi:10.3389/fimmu.2018.01985
Tomaselli, D., Steegborn, C., Mai, A., and Rotili, D. (2020). Sirt4: a multifaceted enzyme at the crossroads of mitochondrial metabolism and cancer. Front. Oncol. 10, 474. doi:10.3389/fonc.2020.00474
Torres, V. E., Harris, P. C., and Pirson, Y. (2007). Autosomal dominant polycystic kidney disease. Lancet 369, 1287–1301. doi:10.1016/S0140-6736(07)60601-1
Tran, M., Tam, D., Bardia, A., Bhasin, M., Rowe, G. C., Kher, A., et al. (2011). PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 121, 4003–4014. doi:10.1172/JCI58662
Verma, A. K., Singh, H., Satyanarayana, M., Srivastava, S. P., Tiwari, P., Singh, A. B., et al. (2012). Flavone-based novel antidiabetic and antidyslipidemic agents. J. Med. Chem. 55, 4551–4567. doi:10.1021/jm201107g
Wada, J., and Nakatsuka, A. (2016). Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med. Okayama 70, 151–158. doi:10.18926/AMO/54413
Wakino, S., Hasegawa, K., and Itoh, H. (2015). Sirtuin and metabolic kidney disease. Kidney Int. 88, 691–698. doi:10.1038/ki.2015.157
Wan, C., Su, H., and Zhang, C. (2016). Role of NADPH oxidase in metabolic disease-related renal injury: an update. Oxid. Med. Cell. Longevity 2016, 7813072. doi:10.1155/2016/7813072
Wang, W., Wang, Y., Long, J., Wang, J., Haudek, S. B., Overbeek, P., et al. (2012). Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metabol. 15, 186–200. doi:10.1016/j.cmet.2012.01.009
Wang, X. M., Xiao, H., Liu, L. L., Cheng, D., Li, X. J., and Si, L. Y. (2016). FGF21 represses cerebrovascular aging via improving mitochondrial biogenesis and inhibiting p53 signaling pathway in an AMPK-dependent manner. Exp. Cell Res. 346, 147–156. doi:10.1016/j.yexcr.2016.06.020
Wanner, C., Inzucchi, S. E., Lachin, J. M., Fitchett, D., Von Eynatten, M., Mattheus, M., et al. (2016). Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 375, 323–334. doi:10.1056/NEJMoa1515920
Ward, L. D., Howlett, G. J., Discolo, G., Yasukawa, K., Hammacher, A., Moritz, R. L., et al. (1994). High affinity interleukin-6 receptor is a hexameric complex consisting of two molecules each of interleukin-6, interleukin-6 receptor, and gp-130. J. Biol. Chem. 269, 23286–23289.
Ward, M. S., Flemming, N. B., Gallo, L. A., Fotheringham, A. K., Mccarthy, D. A., Zhuang, A., et al. (2017). Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci. Rep. 7, 15190. doi:10.1038/s41598-017-15589-x
Wei, Y., Chiang, W.-C., Sumpter, R., Mishra, P., and Levine, B. (2017). Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168, 224–238.e10. doi:10.1016/j.cell.2016.11.042
Wen, Z., Zhong, Z., and Darnell, J. E. (1995). Maximal activation of transcription by statl and stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250. doi:10.1016/0092-8674(95)90311-9
Whitfield, J., Hurst, D., Bennett, M. J., Sherwood, W. G., Hogg, R., and Gonsoulin, W. (1996). Fetal polycystic kidney disease associated with glutaric aciduria type II: an inborn error of energy metabolism. Am. J. Perinatol. 13, 131–134. doi:10.1055/s-2007-994309
Xiao, L., Xu, X., Zhang, F., Wang, M., Xu, Y., Tang, D., et al. (2017). The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11, 297–311. doi:10.1016/j.redox.2016.12.022
Xu, S., Gao, Y., Zhang, Q., Wei, S., Chen, Z., Dai, X., et al. (2016a). SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxid Med. Cell Longev. 2016, 7296092. doi:10.1155/2016/7296092
Xu, Y., Nie, L., Yin, Y.-G., Tang, J.-L., Zhou, J.-Y., Li, D.-D., et al. (2012). Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating SIRT1 in rat mesangial cells. Toxicol. Appl. Pharmacol. 259, 395–401. doi:10.1016/j.taap.2011.09.028
Xu, Y. S., Liang, J. J., Wang, Y., Zhao, X.-Z. J., Xu, L., Xu, Y.-Y., et al. (2016b). STAT3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci. Rep. 6, 395. doi:10.1038/srep39517
Yamahara, K., Yasuda, M., Kume, S., Koya, D., Maegawa, H., and Uzu, T. (2013). The role of autophagy in the pathogenesis of diabetic nephropathy. J. Diabetes Res. 2013, 193757. doi:10.1155/2013/193757
Yang, R., and Rincon, M. (2016). Mitochondrial Stat3, the need for design thinking. Int. J. Biol. Sci. 12, 532–544. doi:10.7150/ijbs.15153
Yu, H., Lee, H., Herrmann, A., Buettner, R., and Jove, R. (2014). Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Canc. 14, 736–746. doi:10.1038/nrc3818
Yuan, Y., Chen, Y., Zhang, P., Huang, S., Zhu, C., Ding, G., et al. (2012). Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic. Biol. Med. 53, 30–43. doi:10.1016/j.freeradbiomed.2012.03.015
Zachari, M., and Ktistakis, N. T. (2020). Mammalian mitophagosome formation: a focus on the early signals and steps. Front. Cell Develop. Biol. 8, 171. doi:10.3389/fcell.2020.00171
Zeisberg, M., and Neilson, E. G. (2010). Mechanisms of tubulointerstitial fibrosis. Journal of the American society of nephrology. JASN (J. Am. Soc. Nephrol.) 21, 1819–1834. doi:10.1681/ASN.2010080793
Zeisberg, M., Hanai, J., Sugimoto, H., Mammoto, T., Charytan, D., Strutz, F., et al. (2003). BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968. doi:10.1038/nm888
Zelnick, L. R., Weiss, N. S., Kestenbaum, B. R., Robinson-Cohen, C., Heagerty, P. J., Tuttle, K., et al. (2017). Diabetes and CKD in the United States population, 2009–2014. Clin. J. Am. Soc. Nephrol. 12, 1984–1990. doi:10.2215/CJN.03700417
Zhang, B., Cui, S., Bai, X., Zhuo, L., Sun, X., Hong, Q., et al. (2013). SIRT3 overexpression antagonizes high glucose accelerated cellular senescence in human diploid fibroblasts via the SIRT3-FOXO1 signaling pathway. Age (Dordr) 35, 2237–2253. doi:10.1007/s11357-013-9520-4
Zhang, C. X., Cheng, Y., Liu, D. Z., Liu, M., Cui, H., Zhang, B. L., et al. (2019). Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J Nanobiotechnol. 17, 18. doi:10.1186/s12951-019-0451-9
Zhang, Y., Feng, J., Wang, Q., Zhao, S., Yang, S., Tian, L., et al. (2018). Hyperglycaemia stress-induced renal injury is caused by extensive mitochondrial fragmentation, attenuated MKP1 signalling, and activated JNK-CaMKII-fis1 biological Axis. Cell. Physiol. Biochem. 51, 1778–1798. doi:10.1159/000495681
Zhao, H., Duan, Q., Zhang, Z., Li, H., Wu, H., Shen, Q., et al. (2017a). Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell Mol. Med. 21, 2055–2067. doi:10.1111/jcmm.13126
Zhao, H., Liu, Y.-J., Liu, Z.-R., Tang, D.-D., Chen, X.-W., Chen, Y.-H., et al. (2017b). Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. Eur. J. Pharmacol. 804, 57–67. doi:10.1016/j.ejphar.2017.03.037
Zhao, W.-Y., Han, S., Zhang, L., Zhu, Y.-H., Wang, L.-M., and Zeng, L. (2013). Mitochondria-targeted antioxidant peptide SS31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66Shc in renal tubular epithelial cells. Cell. Physiol. Biochem. 32, 591–600. doi:10.1159/000354463
Zhong, Z., Wen, Z., and Darnell, J. (1994). Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98. doi:10.1126/science.8140422
Zhou, D., Zhou, M., Wang, Z., Fu, Y., Jia, M., Wang, X., et al. (2019). PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 10, 524. doi:10.1038/s41419-019-1754-3
Zhou, H., Mehta, S., Srivastava, S. P., Grabinska, K., Zhang, X., Wong, C., et al. (2020a). Endothelial cell-glucocorticoid receptor interactions and regulation of Wnt signaling. JCI Insight 5, e131384. doi:10.1172/jci.insight.131384
Zhou, Y., Long, Q., and Liu, X. (2020b). A new sight: topology-dependent mitophagy. Cell Biol. Toxicol. 36, 199–204. doi:10.1007/s10565-020-09534-4
Keywords: mitochdrial damage, mitochondrial sirtuins, renal damage, kidney fibrosis, diabetic kidney disease, glycolysis, fatty acid oxidation, polycystic kidney disease
Citation: Srivastava SP, Kanasaki K and Goodwin JE (2020) Loss of Mitochondrial Control Impacts Renal Health. Front. Pharmacol. 11:543973. doi: 10.3389/fphar.2020.543973
Received: 19 March 2020; Accepted: 19 November 2020;
Published: 09 December 2020.
Edited by:
Norberto Perico, Mario Negri Pharmacological Research Institute (IRCCS), ItalyReviewed by:
Eric Stephen Goetzman, University of Pittsburgh, United StatesKrisztian Stadler, Pennington Biomedical Research Center, United States
Copyright © 2020 Srivastava, Kanasaki and Goodwin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swayam Prakash Srivastava, c3dheWFtLnNyaXZhc3RhdmFAeWFsZS5lZHU=; Julie E. Goodwin, anVsaWUuZ29vZHdpbkB5YWxlLmVkdQ==