 Mathias Hallberg
Mathias Hallberg Mats Larhed
Mats Larhed- 1The Beijer Laboratory, Division of Biological Research on Drug Dependence, Department of Pharmaceutical Biosciences, BMC, Uppsala University, Uppsala, Sweden
- 2Department of Medicinal Chemistry, Science for Life Laboratory, BMC, Uppsala University, Uppsala, Sweden
It was reported three decades ago that intracerebroventricular injection of angiotensin IV (Ang IV, Val-Tyr-Ile-His-Pro-Phe) improved memory and learning in the rat. There are several explanations for these positive effects of the hexapeptide and related analogues on cognition available in the literature. In 2001, it was proposed that the insulin-regulated aminopeptidase (IRAP) is a main target for Ang IV and that Ang IV serves as an inhibitor of the enzyme. The focus of this review is the efforts to stepwise transform the hexapeptide into more drug-like Ang IV peptidemimetics serving as IRAP inhibitors. Moreover, the discovery of IRAP inhibitors by virtual and substance library screening and direct design applying knowledge of the structure of IRAP and of related enzymes is briefly presented.
Introduction
Angiotensin IV (Ang IV) is a small bioactive peptide in the renin-angiotensin system (RAS) formed after proteolytic degradation of angiotensin II (Ang II). In 1988, Braszko et al. (1988), at the Medical University of Bialystok, Poland demonstrated that intracerebroventricular injection of Ang IV (Val-Tyr-Ile-His-Pro-Phe), 1 in rat (1 nmol dose) improved memory and learning (Figure 1). Furthermore, it was reported that Ang IV affects both passive and conditioned avoidance response as well as motor activity. The impact of the hexapeptide in various experimental models, e.g., for Barnes maze, swim mazes as well as radial arm mazes were subsequently explored (Wright et al., 1993; Wright et al., 1996; Wright et al., 1999; Lee et al., 2003; Braszko et al., 2008; De Bundel et al., 2009). Related Ang IV analogues were studied as well, such as Nle-Ang IV 2 (Nle-Tyr-Ile-His-Pro-Phe) and the endogenous LVV-hemorphin-7 3 (Leu-Val-Val-Tyr-Pro-Trp-Thr-Gln-Arg-Phe), both with structural similarities to Ang IV at the N-terminal part of the peptides (Figure 1). Nle-Ang IV 2 and LVV-hemorphin-7 3, the latter formed after degradation of β-globin, were demonstrated to be strong promoters of memory retention and retrieval in rats (Lee et al., 2003; Lee et al., 2004; De Bundel et al., 2009). In 1992, Wright and Harding identified binding sites for the hexapeptide Ang IV in several brain areas associated with cognition, motor and sensory physiological functions (Swanson et al., 1992), e.g., in hippocampus (Harding et al., 1992). The encouraging data obtained after administration of Ang IV in the experimental models and the impact of Ang IV on parameters anticipated to be linked to cognition promoted an interest in more detailed studies of the hexapeptide.
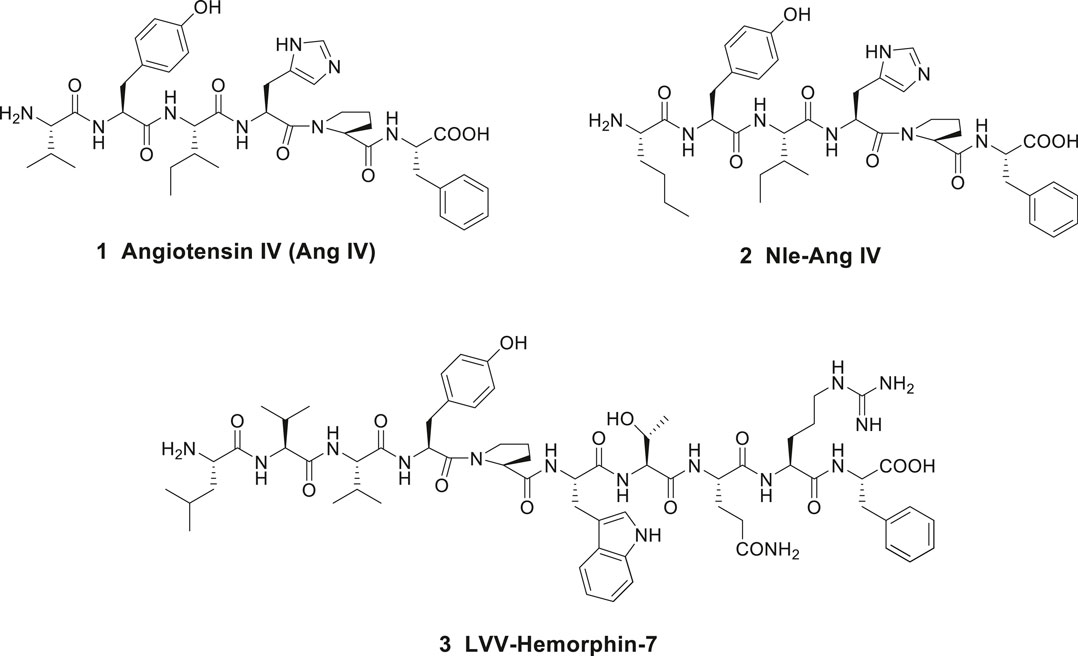
Figure 1. Peptides with high affinity to the angiotensin IV (AT4) receptor.
Excellent reviews on Ang IV and its contribution to cognition are available (Gard, 2008; Albiston et al., 2011; Ho and Nation, 2018; Jackson et al., 2018; Wright and Harding, 2019). Herein we summarize the efforts the last three decades to transform the hexapeptide into more drug-like peptidemimetics acting at its receptor(s). We discuss from a medicinal chemistry perspective, starting points in the design processes and the very different approaches applied. The incorporation of various unnatural amino acid residues into peptides was proven to be a very productive methodology. The iterative stepwise modifications of the parent ligand Ang IV is first presented.
Linear Angiotensin IV Analogues and Incorporation of Unnatural Amino Acids
After systematic structure activity studies (SAR) of Ang IV analogues, involving glycine and D-amino acid scans in combination with displacement and incorporation of various alternative amino acid residues it became clear that the N-terminal Val-Tyr-Ile residues of the peptide ligands were important for high affinity to the specific binding site identified (Sardinia et al., 1993), named the AT4 receptor (de Gasparo et al., 1995). Furthermore, amino acid residues with hydrophobic side chains in the 1-position of the Ang IV analogues, e.g., a norleucine residue (cf. 2) combined with an unsubstituted amino group in the N-terminal seemed optimal. On the contrary, alterations in the C-terminal seemed less critical for activity and for instance a deletion of the C-terminal phenylalanine residue of 1 had only a minor impact and the binding affinity was essentially maintained (Sardinia et al., 1993; Sardinia et al., 1994; Krishnan et al., 1999).
Ang IV and related analogues are susceptible to proteolytic cleavage of the peptide bond between the Val1 and Tyr2 amino acid residues. Thus, a reduction of this N-terminal peptide bond rendered peptides significantly less prone to hydrolysis. Notably, most of the affinity to the binding site was still maintained. The Ang IV analogue peptide 4 and the Nle-Ang IV analogue 5 provide examples comprising two basic amino groups accessible for protonation in the N-terminal of the peptides and flanking hydrophobic 2-isopropyl and n-butyl side chains, respectively (Figure 2) (Sardinia et al., 1994). A reduced peptide bond between the amino acid residues Ile3 (or Val3) and His4 is the characteristic feature of divalinal-Ang IV 6 and norleual 7, two pseudopeptides that both for a long time served as important research tools (Krebs et al., 1996; Kramár et al., 2001).
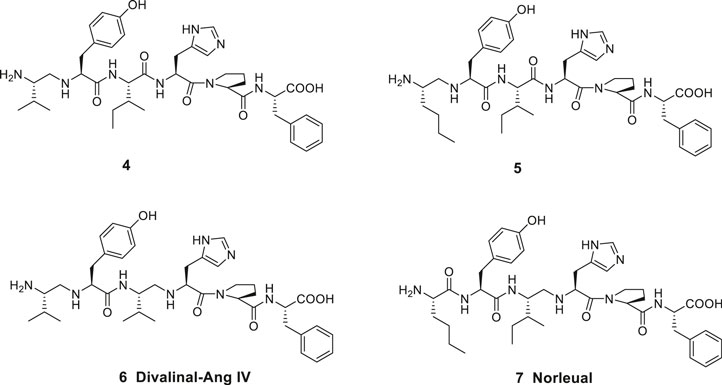
Figure 2. Linear peptides with high affinity to the angiotensin IV (AT4) receptor and that are less prone to proteolytic degradation.
In the late 1990s Taisho Pharmaceuticals filed patent applications covering compounds that according to competitive experiments with radiolabeled [125I]Ang IV exhibited high affinity to hippocampus membranes from guinea pig (Kobori et al., 1997; Kobori et al., 1998). Kobori et al. (1997), Kobori et al. (1998) characterized the compounds as Ang IV receptor agonists and some of them, e.g., 8 with the straight four-carbon chain of the Nle residue at position one, exhibited very high affinity to the binding site (IC50 values < 1 nM) (Figure 3). The drug-like 8 encompasses a styrene moiety replacing His-Pro-Phe residues in the C-terminal of Ang IV. Furthermore, it was demonstrated that saturation of the trans styrene double bond of 8 was deleterious for activity. Reduction of the remaining peptide bond of 8 furnished 9, a compound that still bind to the binding site although with 40 times lower affinity. From the series of compounds that were disclosed it appears that the binding site favors two amine functions in the N-terminal part of the ligands in order to achieve an efficient binding to the receptor (cf. 4, 5, 6). Hence, a compound with an intact peptide bond between the Nle1 and Tyr2 residues but with a reduced peptide bond between the Tyr2 and Ile3 residues was essentially inactive (Kobori et al., 1997; Kobori et al., 1998).
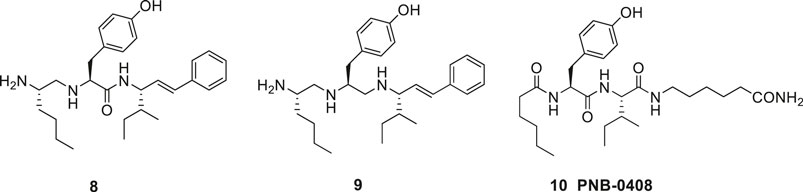
Figure 3. The low molecular weight high-affinity angiotensin IV (AT4) receptor binding analogues 8 and 9 incorporate one or two reduced peptide bonds. PNB-0408 (10), devoid of an N-terminal amino group crosses the blood-brain barrier, enhances cognition and is interfering with the hepatocyte growth factor/c-Met receptor system.
In 2001, Albiston and Chai suggested that Ang IV exerts its procognitive actions by inhibition of a peptidase, insulin-regulated aminopeptidase (IRAP) (Albiston et al., 2001) and not through binding to a G-protein-coupled receptor as is the case of its major precursor angiotensin II (Ang II, Asp-Arg-Val-Tyr-Ile-His-Pro-Phe), that mediates its effects mainly through the Ang II receptor type 1 (AT1R) and the angiotensin receptor type 2 (AT2R). Thus, while the octapeptide Ang II exerts a powerful hypertensive effect and acts as a receptor agonist, it is degraded by proteolysis to a bioactive hexapeptide metabolite with a very different pharmacological profile that acts as an enzyme inhibitor. Notably, there are several other examples known demonstrating that small neuropeptides can be converted into fragments with significantly different biological effects, e.g., the nociceptive substance P is degraded to the antinociceptive substance P (1–7) (Fransson et al., 2008; Hallberg, 2015). Ang IV binds to both AT1R and AT2R but only at micromolar concentrations (Bosnyak et al., 2011; Hallberg et al., 2017). Furthermore, the IRAP inhibitors Ang IV (1) and LVV-hemorphin-7 (3) (Lee et al., 2003; Lew et al., 2003) inhibit also aminopeptidase N (AP-N, EC 3.4.11.2) activity (Garreau et al., 1998; Fruitier-Arnaudin et al., 2002) and LVV-hemorphin-7 (3), in addition, binds to the μ-opioid receptor (Nyberg et al., 1996).
IRAP (EC 3.4.11.3) is a single-spanning transmembrane zinc-metallopeptidase that belongs to the M1 family of aminopeptidases. The enzyme was cloned and characterized in adipocytes in vesicles containing the insulin-regulated glucose transporter GLUT4 (Keller et al., 1995; Keller, 2003), was identified as placental leucine aminopeptidase, oxytocinase, gp160 and vp165 and is a cystinyl aminopeptidase (Tsujimoto et al., 1992; Kandror and Pilch, 1994; Rogi et al., 1996; Rasmussen et al., 2000; De Bundel et al., 2008). IRAP is able to cleave the peptide bond between the N-terminal amino acid residues from several bioactive peptides in vitro, e.g., met-enkephalin and leu-enkephalin, dynorphin A, lysine-bradykinin, neurokinin A1, cholecystokinin-8, somatostatin, oxytocin and vasopressin (Lew et al., 2003) although the key substrates seem to be the macrocyclic oxytocin and arginine-vasopresin (Albiston et al., 2010a). Studies on the IRAP knockout (KO) mice suggest that vasopressin, oxytocin and somatostatin are physiologically important substrates (Wallis et al., 2007; De Bundel et al., 2015). Hence, the N-terminal peptide bonds of macrocyclic peptidic regulators of cognition, as vasopressin and oxytocin can be cleaved (Alescio-Lautier et al., 2000; Matsumoto et al., 2000; Matsumoto et al., 2001; Wallis et al., 2007). The aminopeptidase is expressed in areas of the brain associated with cognition (Fernando et al., 2005; Albiston et al., 2007; Albiston et al., 2011), and the observation that Ang IV improves memory and learning has attracted attention to the aminopeptidase IRAP as a potential macromolecular target for drugs for treatment of cognitive disorders (Wolfe, 2002; Gard, 2008; Wright and Harding, 2008; Hallberg, 2009). Moreover, the enzyme mediates a series of other important physiological activities that are related to immunology (Stratikos, 2014), e.g., antigen processing and trafficking of T-cells receptors and of glucose transporters (Waters et al., 1997; Bryant et al., 2002). The catalytic site of IRAP is located in the extracellular region and is highly homologous to two other important members of the M1 aminopeptidase family, endoplasmic reticulum aminopeptidase 1 (ERAP1) and 2 (ERAP2) (Evnouchidou et al., 2009). IRAP, ERAP1, and ERAP2 are the three so-called antigen processing aminopeptidases responsible for the processing of the N-terminus of antigenic peptides and the activity of the three enzymes constitutes an important regulatory node in antigen processing, vide infra (Stratikos, 2014).
It should be emphasized that macromolecular targets other than IRAP have been proposed to account for the positive physiological effects on cognition observed in vivo after administration of Ang IV and related analogues (Vanderheyden, 2009; Kawas et al., 2012; Benoist et al., 2014). The tyrosine kinase receptor c-Met that binds hepatocyte growth factor (HGF) and that is associated with memory and learning consolidation is one candidate (Wright et al., 2008; Yamamoto et al., 2010) being implicated in Alzheimer’s disease (AD) (Wright and Harding, 2011). Consequently, it is suggested that Ang IV exerts its action through the c-Met signaling pathway (Wright and Harding, 2019) and an overexpression HGF in the nervous system enhances learning and memory performance in mice (Kato et al., 2012). The Ang IV analogue Norleual 7 inhibits HGF-mediated effects at picomolar concentrations and blocks [125I]HGF binding to c-Met (Yamamoto et al., 2010). The drug-like PNB-0408 (N-hexanoyl-Tyr-Ile-N′-(5-carbamoylpentyl)amide 10, synthesized in the laboratories of Wright and Harding is of particular interest and penetrates the blood-brain barrier and improves cognitive activity. (Figure 3) (Wright and Harding, 2008; Wright and Harding, 2009; McCoy, 2010). PNB-0408 (dihexa) with a primary amid function in the C-terminal attached via a lipophilic tether to the Ile residue is devoid of the N-terminal amino group found in most Ang IV analogues, vide supra. The compound 10, derived from Nle-Ang IV 2 facilitates formation of new functional synaptic connections, facilitates LTP in hippocampal slices and amplifies memory consolidation in animal models of AD (Wright and Harding, 2015; Wright and Harding, 2019). Even though the brain hepatocyte growth factor/c-Met receptor system has attracted considerable attention (Wright and Harding, 2019), with regard to synthesis of small molecules and medicinal chemistry, it seems that most efforts so far have been devoted to identify new selective inhibitors of IRAP. To the best of our knowledge, no data on inhibition of IRAP by compounds 8 and 9 and related ligands from the Kobori laboratory nor by PNB-0408 (10) are yet available in literature.
After the pioneering work by Wright and Harding at Washington State University and by Kobori et al. (1997), Kobori et al. (1998). at Taisho Pharmaceuticals in Japan who identified low molecule strong binders to the Ang IV receptor in the 1990s, several groups have been engaged in the search for improved compounds interacting with the Ang IV receptor. The fundamental discovery by Albiston and Chai that IRAP is a molecular target for Ang IV and that IRAP seems to play an important physiological role and is associated with cognition encouraged focused research aimed at identifying new efficient IRAP inhibitors that could serve as pharmaceutical agents. Several complementary approaches were applied; 1) iterative modifications of the endogenous Ang IV, 2) virtual and substance library screening, and 3) direct design applying knowledge of the structure of IRAP. To make orally bioavailable IRAP inhibitors that reach the brain and that are metabolically relatively stable is a tremendous challenge.
Previously metal chelators (e.g., EDTA and phenantroline) were normally added in the experimental settings for determination of binding affinity. Although many of the ligands that in the 1990 ties had been identified as high-affinity binders inhibit IRAP (Lee et al., 2003; Lew et al., 2003), the ligands often demonstrated differences in potencies and rank orders in the IRAP assay in experiments where chelators were absent. It was postulated that the differences observed were attributed to the absence of zinc in the active site in presence of metal chelators (Lew et al., 2003; Demaegdt et al., 2004; Laeremans et al., 2005; Demaegdt et al., 2006). It became apparent that chelators must be omitted to obtain physiologically relevant data (Demaegdt et al., 2009).
Introduction of unnatural amino acids into peptide structures was demonstrated to provide a successful concept in the search for potent and metabolically stable inhibitors of IRAP. Hence, a β-homoamino acid scan of Ang IV performed at Vrije University in Brussels resulted in the identification of the metabolically stable derivative 11 (AL-11) with five-fold higher affinity to IRAP than Ang IV (Ki = 7.6 vs. 62 nM for Ang IV) and exhibiting a high selectivity for IRAP over AP-N (Figure 4) (Lukaszuk et al., 2008). Thus, insertion of methylene groups in both in the N- and C-terminals of Ang IV improved the inhibitors. The His4 and Pro5 residues of Ang IV were subsequently replaced by conformationally constrained residues and the highly selective and stable IRAP inhibitor 12 (AL-40, IVDE77) was identified, with >30-fold higher affinity (Ki = 1.7 nM) than Ang IV and improved both selectivity and metabolic stability (Lukaszuk et al., 2008; Lukaszuk et al., 2009; Nikolaou et al., 2013). In addition, the Belgium group assessed the roles of Tyr2, Pro5, and Phe6 in Ang IV by systematic incorporation of constrained residues. The replacements of Tyr2 by residues with restricted flexibility were deleterious for activity. For example, the compound 13 (racemic) is a very week IRAP inhibitor (Lukaszuk et al., 2011) and compound 14 with a 4-hydroxydiphenylmethane scaffold as a substitute for Tyr2 is essentially inactive (Figure 4) (Andersson et al., 2008). Thus, in summery the lipophilic character of the side chain in position 1 is essential and the proper orientation of the Tyr2 side chain in space is critical for activity and IRAP inhibition while various alternations and topography at the C-terminal are more acceptable.
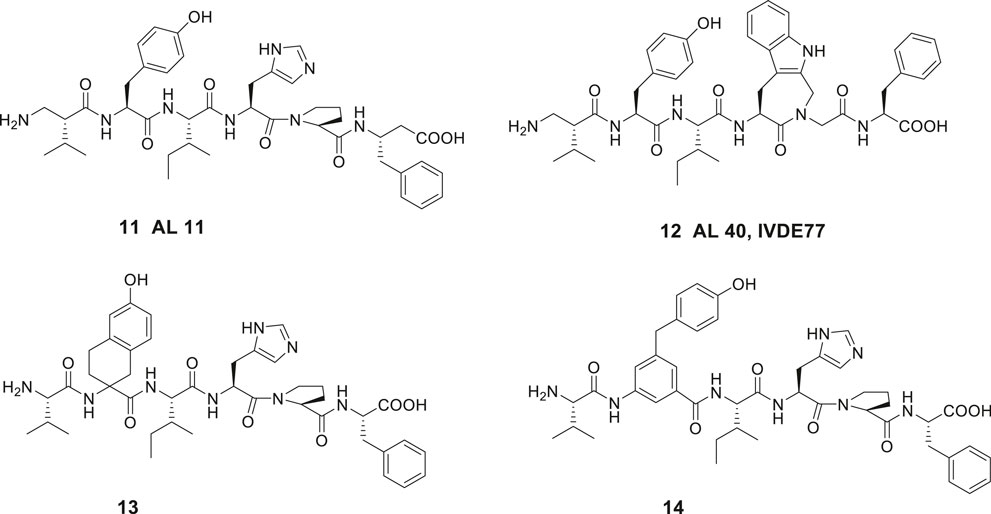
Figure 4. The high affinity and metabolically stable IRAP inhibitors 11 (AL-11) and 12 (AL-40) both comprising β amino acid residues. The compounds 13 and 14 where the Tyr2 residue is replaced are inactive as IRAP inhibitors.
Macrocyclic Angiotensin IV Analogues
The capability of IRAP to degrade physiologically important cyclic peptides has inspired the development of macrocyclic analogues of Ang IV. Our group synthesized a series of macrocyclic peptides with the aim to determine bioactive conformations and better understand the mode of binding and structural requirements for efficient binding of Ang IV to IRAP (Axén et al., 2006; Axén et al., 2007; Andersson et al., 2010; Andersson et al., 2011). Steric constrains were introduced. Attempts to obtain high affinity binding inhibitors by cyclization in the C-terminal was productive as expected from previously obtained data. Hence, the macrocyclic disulphide 15 encompassing an 11-membered macrocycle was more potent as an IRAP inhibitor than the native Ang IV and exhibited a Ki value of 26 nM (Figure 5). Since the macrocyclic system of 15, as deduced from modeling tends to adopt a γ-turn (Schmidt et al., 1997; Lindman et al., 2001), the entire C-terminal tripeptide fragment His-Pro-Phe was replaced by a 2-(aminomethyl)phenylacetic acid moiety anticipated to serve as a proper γ-turn mimic. The structurally simplified Ang IV peptidemimetic 16 was almost as active as Ang IV as an IRAP inhibitor while 17 was five-fold and 18 and the open chain 19 were 20-fold less active (Figure 5). Furthermore, compound 16 is degraded considerably more slowly in membrane preparations than Ang IV and stimulates proliferation of mouse neural stem cells at low concentrations, supporting a role of IRAP in neurogenesis (Axén et al., 2007).
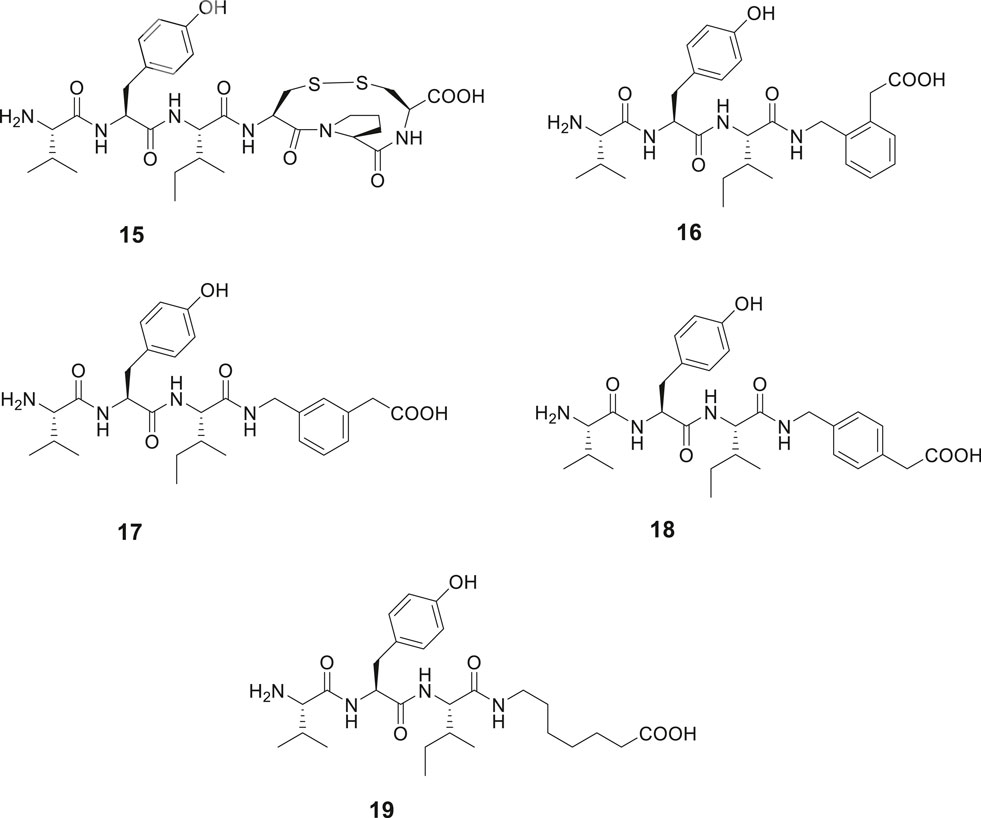
Figure 5. Modifications in the C-terminal. Compound 15 with an 11-membered macrocyclic ring adopting a γ-turn in the C-terminal binds better than Ang IV to IRAP and 16 with a scaffold serving as a γ-turn mimetic incorporated in the C-terminal is essentially as active as Ang IV. Compounds 17, 18, and 19 bind to IRAP but are more than five-fold less potent.
Contrary to the results obtained after modification in the C-terminal of Ang IV where essentially all compounds examined exhibited some activity, oxidative cyclization of the side chains of Cys1 and Cys3 and consequently creation of an 11-membered disulfide macrocycle in the N-terminal rendered the inactive compound 20 (Figure 6). However, a widening of the ring system was productive and the Ang IV analogue 21 with a 13-membered macrocycle exhibited a moderate IRAP inhibitory capacity. Compound 22, a hybrid with the N-terminal from 21 and the C-terminal from 16 demonstrated a further enhanced potency (Ki = 23 nM) and a high selectivity for IRAP over AP-N (Andersson et al., 2010). Notably 16, devoid of a macrocyclic ring system in the N-terminal is not selective and inhibits both IRAP and AP-N activity, suggesting that introduction of proper conformational constrains in the N-terminal by macrocyclization improves selectivity (Axén et al., 2007). As deduced from molecular modeling, 15 and Ang IV seem to adopt a γ-turn at the C-terminal when binding to IRAP, while in the case of 21, a less well-defined turn conformation is adopted at the N-terminal. Further structural optimization delivered a large number of macrocyclic disulfides and among them 23 (HA08) encompassing a β3-homotyrosine residue in a 13-membered ring system and exhibiting the lowest Ki value in the series (Ki = 3.3 nM). HA08 is formed after an Hcy1/Cys3 side chain cyclization and is a competitive inhibitor and 20 times more potent than Ang IV and less prone to proteolytic degradation (Figure 6). The compound was designed to provide both high IRAP inhibitory capacity and an improved metabolic stability attributed to the introduction of an β-amino acid residue adjacent to the predicted scissile peptide bond. Enlargement of the ring system of 23, by oxidatative cyclization of Hcy1/Hcy3 side chains rather than the Hcy1/Cys3 side chains of 23 resulted in a compound comprising a 14-membered macrocycle but with a somewhat lower affinity to IRAP (Ki = 5.1 nM), vide infra. Furthermore, one important characteristic of 23 (HA08) is its high 2,000-fold selectivity for IRAP over AP-N and high selectivity vs. the homologous enzymes ER aminopeptidase 1 (ERAP1) and ER aminopeptidase 2 (ERAP2). Replacement of the L-amino acids of 23 with D-amino acids were not productive (Andersson et al., 2010). Deletion of the N-terminal amino group of 23 had a dramatic effect. Hence, 24 is inactive as an IRAP inhibitor, cf. the potent cognitive enhancer 10 (PNB-0408, dihexa), derived from Ang IV, vide supra. Furthermore, removal of the methylene carboxy group at the C-terminal of 23 resulted in a less efficient IRAP inhibitor. Introduction of a primary carboxamide in the C-terminal (cf. PNB-0408, 10) to replace the carboxy group of 23 provided 25 that inhibits IRAP but exhibits a two-fold lower activity (Figure 6). A reduction of the N-terminal peptide bond of 25, with the ambition to achieve a compound even more resistant to proteolytic cleavage, provided 26 (Barlow et al., 2020). This maneuver was not fruitful and compound 26 displays a Ki value in the micromolar range in sharp contrast to what could be expected considering the results obtained after reduction of Ang IV or Nle-Ang IV, cf. 1 vs. 4 and 2 vs. 5 where the bioactivities were essentially retained after reduction of the carbonyl group of the peptide bond, vide supra. Moreover, in attempts to replace the hydroxyl group of the tyrosine moiety to avoid potential problems with phase II metabolism compound 27 was prepared (Barlow et al., 2020). This fluoro compound (27) is 10-fold less potent than 25 demonstrating that interactions with the hydroxyl group is important, although not critical for activity.
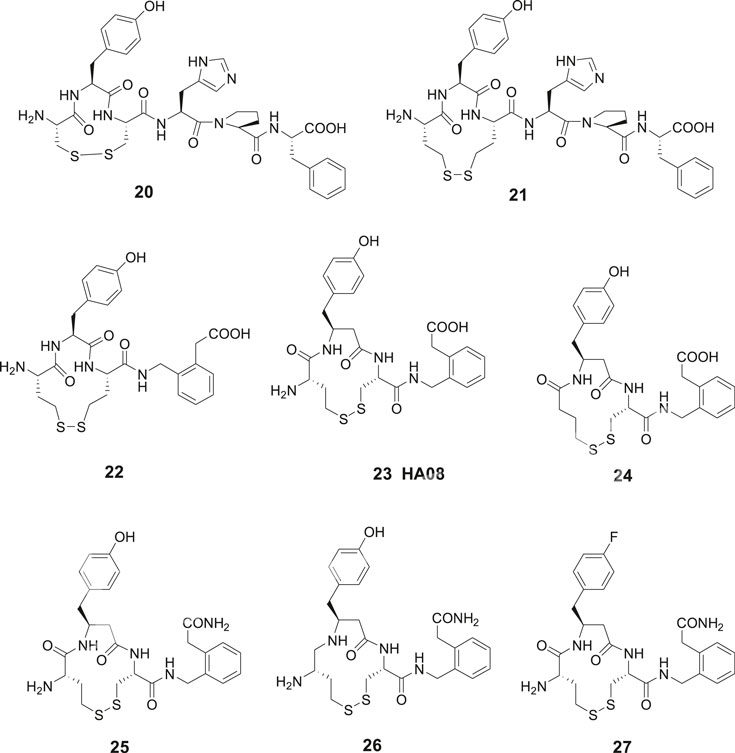
Figure 6. Macrocyclizations in the N-terminal. The Ang IV analogue 20 encompassing an 11-membered macrocycle is inactive while 21 with a 13-membered ring system inhibits IRAP efficiently. The selective compound 23 (HA08) is the most potent IRAP inhibitor in the series and deletion of the N-terminal amino group of 23 makes the ligand (24) inactive. The carboxamide 25 is two-fold and the fluoro compound 27 is 10-fold less potent than 23 as IRAP inhibitors. Compound 26 comprising a reduced peptide bond in the N-terminal is a weak IRAP inhibitor.
Crystal structures of IRAP were recently solved (Hermans et al., 2015; Mpakali et al., 2015). Based on analyses of available crystal structures a tentative model for 23 (HA08) binding to IRAP was created in 2016 by applying MD simulations and calculating associated binding free energies by the linear interaction energy (LIE) method. According to the model, the carbonyl group of the N-terminal peptide bond of 23 is coordinated to the Zn2+ ion, whereas the terminal amine is fixed by three glutamate carboxylates (Glu431, Glu487, and Glu295) (Diwakarla et al., 2016b). Regarding the affinity of the fluoro compound 27, it was anticipated that the hydroxyl group of the tyrosine side chain of 25 makes a hydrogen-bond with Glu494 of IRAP, an interaction that is lost in the case of 27 according to the free energy perturbation (FEP) analysis (Jespers et al., 2019), explaining the reduction in binding affinity of compound 27 as compared to the analogous compound 25. Moreover, it was postulated that the aromatic ring of the β3-homotyrosine side chain and the C-terminal phenyl ring interact and Phe550 in IRAP further stabilizes aromatic/hydrophobic packing (Barlow et al., 2020).
More recently, a crystal structure of IRAP with the macrocyclic peptide inhibitor 23 (HA08) was reported (Mpakali et al., 2020). After a comparison with the known IRAP structures and a combination with small angle X-ray scattering experiments it was proposed that IRAP is an open dimer in solution and assumes a more compact conformation upon HA08 binding. Thus, 23 (HA08) stabilizes the closed conformation of IRAP. Compound 23 is combining the structural elements from Ang IV and the physiological substrates vasopressin and oxytocin. The cleavage of the scissile bond of substrates as oxytocin and vasopressin is anticipated to take place after a nucleophilic attack by water assisted by the carboxylate function of Glu465 in the active site of IRAP. Importantly, the corresponding cleavage of the scissile bond of the 13-membered 23 (HA08) in the closed conformation of IRAP is less prone to occur since there is not enough space for motion of water molecules to interact with the carboxylate group of Glu465. In fact, no ordered water molecule is found in the crystal structure at that location. The structure reveals that the side-chain of the β-tyrosine amino acid residue is situated in the S1 pocket of the enzyme and that the disulfide bond abuts to the S1′ pocket. The phenylacetic acid group of HA08 was found in two different orientations; a) the carboxyl group makes electrostatic interactions with Arg439 and Arg929 of IRAP and b) phenyl ring of the phenylacetic acid group makes pi-stacking interactions with Tyr961. Moreover, it was suggested from inhibition data that 23 (HA08) acting through a competitive mechanism and Ang IV may occupy the same binding site and operate by the same mode of action. The structure of the IRAP (Mpakali et al., 2020) was found very similar to that reported previously with a phosphinic pseudotripeptide transition-state analogue binding to the enzyme, vide infra (Mpakali et al., 2017). It was concluded that HA08 binds to IRAP in a very similar conformation to what was proposed by the MD simulations (Diwakarla et al., 2016b; Barlow et al., 2020).
The metathesis reaction was applied to obtain a series of macrocyclic compounds, devoid of the reactive disulfide function and with the potential to become oral bioavailable and more metabolically stable (Driggers et al., 2008; Marsault and Peterson, 2011). Several carba analogues comprising a 13- or 14-membered heterocycle were synthesized and assessed (Andersson et al., 2011). Among those, the tyrosine derivative 28 exhibited a Ki value of 4.1 nM and the corresponding analogue with a saturated double bond, compound 29, a Ki value of 25 nM, demonstrating that a proper constrain of flexibility of the ring system, alternatively an electron density in the center of the lipophilic bridge between amino acid residues in positions 1 and 3 is favorable for affinity to IRAP (Figure 7). Compound 30 encompassing a homo tyrosine in a 14-membered macrocycle and with a trans double bond provided the lowest Ki value in the series, 1.8 nM, while the corresponding tyrosine analogues with the 13-membered ring system exhibited a Ki value of 50 nM. Compound 31, the cis isomer of 30, demonstrated a lower affinity to the enzyme (Ki = 30.4 nM). The high activity of 30 prompted a synthesis of the corresponding disulfide 32 with a 14-membered ring system anticipated be able to adopt a similar “trans conformation” as 30. A high activity was encountered, Ki = 5.1 nM, to be compared with 23 (HA08) with a Ki of 3.3 nM, the latter comprising a 13-membered macrocyclic system. Insertion of amide bonds at various locations in the carbon chain was not productive. The lactams 33 and 34 comprising 14-membered ring systems were most active in the series but nonetheless 100-fold less potent than the best disulfide and carba analogues (Barlow et al., 2020).
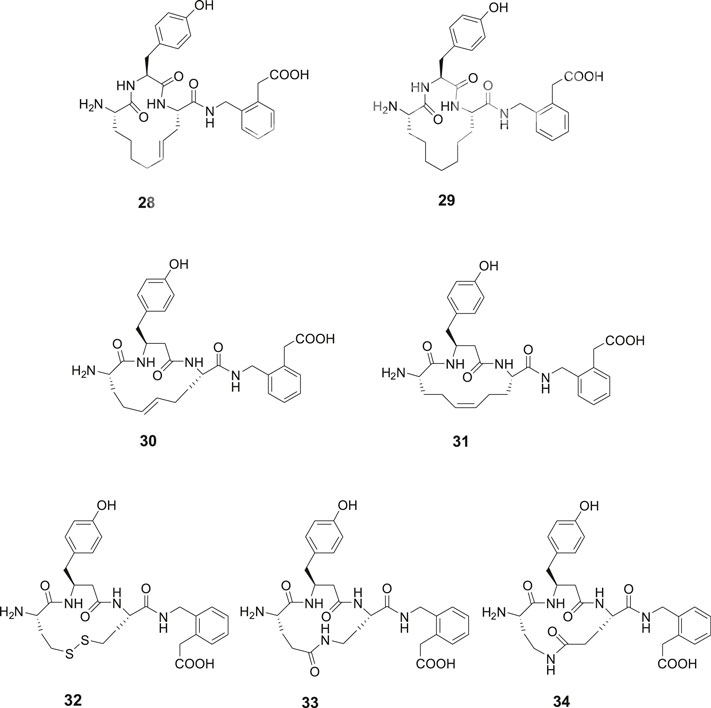
Figure 7. Macrocyclizations in the N-terminal. A comparison of the activity of a series of cyclized Ang IV analogues encompassing 14-membered ring systems. The macrocyclic 28 and 30, made by metathesis reactions are the most potent IRAP inhibitors in the series and somewhat more potent than the disulfide analogue 32. Compound 29 with a saturated carbon bridge and the cis isomer 31 are 10-fold less potent and the amides 33 and 34 more than 100-fold less potent than the most effective IRAP inhibitor compound 30.
The disulfide and carba analogues examined demonstrated a relative high stability against proteolysis by metallopeptidases, despite the similarities in the N-terminal to oxytocin 35 and vasopressin 36 that both serve as substrates to IRAP (Alescio-Lautier et al., 2000; Matsumoto et al., 2000; Matsumoto et al., 2001; Wallis et al., 2007; Hermans et al., 2015). This is most likely attributed to the inability of the constrained 13- and 14-membered macrocycles to adopt the proper transition states required for cleavage of the N-terminal peptide bond (Andersson et al., 2011) (Figure 8).
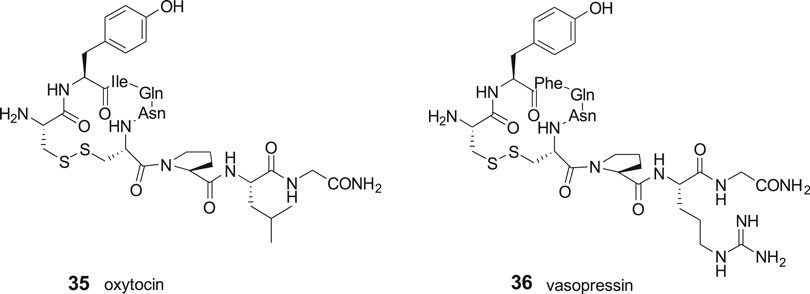
Figure 8. The macrocyclic disulfides oxytocin (35) and vasopressin (36) are IRAP substrates.
To conclude, macrocyclizations by oxidative disulfide formations in the N-terminal of Ang IV analogues or alternatively displacement of the disulfide unite with a trans double bond can provide very potent inhibitors of IRAP, as exemplified with 23 (HA08) comprising a 13-membered ring system and 28, 30, and 32 comprising 14-membered ring systems. Macrocyclization can convey drug-like properties of larger molecules (Driggers et al., 2008; Brandt et al., 2010; Marsault and Peterson, 2011; Mallinson and Collins, 2012; Giordanetto and Kihlberg, 2014) that includes improved membrane permeability compared to acyclic matched pairs and oral bioavailability but also often improved affinity for the target, improved selectivity and reduced metabolism (Hess et al., 2008; Bogdan et al., 2011; Yap et al., 2016).
The macrocycle 23 was selected as a proper research tool for more detailed studies since the molecule is convenient to prepare and due to its structural similarity to the IRAP substrates oxytocin and vasopressin in its N-terminal. A decline in the dendritic spine density (DSD) is an early characteristic feature of many neurodegenerative diseases (Fiala et al., 2002; Bourne and Harris, 2008; van Spronsen and Hoogenraad, 2010; Penzes et al., 2011) and drugs able to enhance DSD are suggested as potential candidates for future treatment of memory disorders (Lynch et al., 2008). Dendritic spines, small protrusions from the dendrites that acts as contacts with neighboring axons and contain all of the molecular machinery required for synaptic plasticity and storage of memories. Hence, the procognitive activity of a molecule in vivo correlates well with its in vitro ability to alter dendritic spine structure and DSD (Moser et al., 1994; O’Malley et al., 1998; O’Malley et al., 2000; Benoist et al., 2011; Fu et al., 2012; Lai et al., 2012; McCoy et al., 2013). The specific loss of stubby/mushroom spines, which morphologically are characterized by a larger spine head and functionally have stronger synapses (Nusser et al., 1998; Matsuzaki et al., 2001; Murthy et al., 2001) is assumed to have a more pronounced impact on cognitive decline (Kasai et al., 2003). The competitive IRAP inhibitor 23 (HA08) enhances the numbers of dendritic spines in hippocampal cell cultures and HA08 treatment resulted in an increasing number of stubby and mushroom-like spines, a morphology typically associated with mature spines, which are believed to have strengthened synaptic connectivity. The dendritic spines were also vesicular glutamate transporter 1 (vGLUT1) positive, indicating that spines were receptive to glutamatergic signaling. Notably, in contrast to 23 (HA08), a structurally very similar epimer (HA09) which differs only with regard to one stereogenic center and exhibiting a 100-fold lower affinity to IRAP did not affect spine morphology, indicating a correlation between the capacity to inhibit IRAP and a positive impact on DSD and spine morphology. HA09 comprises a Cys3 residue with R rather than S-configuration. The effect of 23 (HA08) was similar to that of brain-derived neurotrophic factor (BDNF) that is a known inducer of spine development (Diwakarla et al., 2016b).
Small Molecule Insulin-Regulated Aminopeptidase Inhibitors From In Silico Screening
In 2008, Siew Chai and her group in Australia disclosed the first generation of drug-like small molecular weight inhibitors of IRAP (Albiston et al., 2008). An in silico screening of a homology model of IRAP based on the crystal structure of LTA4H (Leukotriene-A4 hydrolase) was applied (Thunnissen et al., 2001). The virtual screening of a library of approximately two million compounds resulted in a lead series of inhibitors encompassing a benzopyran scaffold. Among the more potent compounds were the racemic pyridine derivative 37 (HFI-419), and the quinoline derivatives 38 (HFI-435) and 39 (HFI-437) exhibiting Ki values of 420, 360, and 20 nM, respectively (Figure 9). The benzopyran-based IRAP inhibitor 37, with high selectivity vs. aminopeptidases such APN, ERAP1, ERAP2, and LTA4H, exerted a cognitive-enhancing effect in rodents after icv administration similar to that of Ang IV 1 and LVV-H7 3 (Albiston et al., 2008; De Bundel et al., 2009).
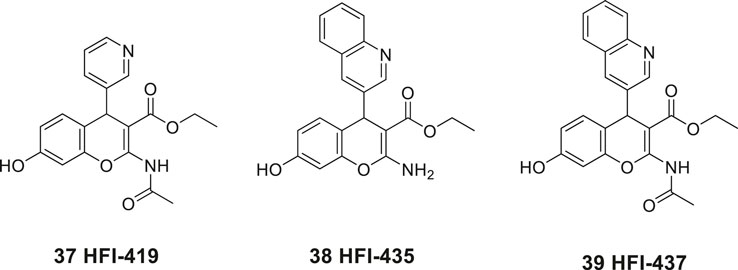
Figure 9. The first drug-like small molecule IRAP inhibitors reported. The benzopyran derivatives 37, 38, and 39 were discovered after in silico screening.
As deduced from computational docking the S-isomer is the preferred binding mode of the inhibitors although alternative binding conformations were suggested (Albiston et al., 2010b) However, after determination of the crystal structure of IRAP, computational docking of, e.g., 37, 38, and 39 into the IRAP structure demonstrated that these inhibitors all bind in the same orientation relative to the active site (Hermans et al., 2015), in contrast to conclusions drawn in previous modeling studies. Moreover, a rationale for the unique specificity of IRAP to process endogenous macrocyclic peptides such as oxytocin (35) and vasopressin (36) was presented (Hermans et al., 2015). Structure–activity relationship of a large series of benzopyran analogues has been established and the structural elements most important for binding have been determined. The lead candidate 37 (HFI-419) exhibits brain exposure following intravenous administration in rats but was found to be rapidly degraded to the corresponding deacetylated and less active IRAP inhibitor (Mountford et al., 2014).
An increase in the activity of matrix-metalloproteases (MMPs) or an increase in neuronal glucose uptake are two likely mechanisms by which inhibition of IRAP can provide an enhancement of memory. Modulation of either of the two systems is known to improve memory and learning as well as affect DSD. The benzopyran 37 (HFI-419) enhances spatial working memory in rats and the inhibition of IRAP by 37 increased DSD prior to peak dendritic growth in hippocampal neurons (Seyer et al., 2020). Moreover, it was concluded that this enhancement was likely to be driven by GLUT4-mediated changes to DSD. In particular, the inhibition of IRAP led to an enhancement of the proportion of mushroom/stubby-like spines. Furthermore, the spines were estimated to be functional based on their expression of the pre-synaptic markers vesicular glutamate transporter 1 and synapsin. The spine formation was inhibited in the case when the GLUT4-mediated glucose uptake was blocked. Thus, these results strongly suggest that IRAP inhibitors may facilitate memory by increasing hippocampal DSD via a GLUT4-mediated mechanism (Seyer et al., 2020).
Small Molecule Insulin-Regulated Aminopeptidase Inhibitors From Substance Library Screening
Our laboratory screened a substance library screen of 10 500 low-molecular-weight compounds in an enzyme inhibition assay with IRAP originating from Chinese hamster ovary (CHO). Three structurally different classes of compounds considered to be of particular interest as starting points for the development of small-molecule IRAP inhibitors were identified. The arylsulfonamide 40 with no structural similarities to Ang IV was one of them (Engen et al., 2016) (Figure 10). Compound 40 comprises a tetrazole ring in the meta position of an aromatic ring of an arylsulfonamide which is the characteristic feature of the sulfonamide class of compounds. Subsequently a large series of tetrazole derivatives were prepared and examined (Borhade et al., 2014) and a few of them were studied in detail. For example 41, that is a competitive inhibitor of IRAP and exhibits an IC50 of 540 nM in a recombinant human IRAP assay. This sulfonamide (41) demonstrates a high metabolic stability and alters dendritic spine morphology and increases spine density in primary cultures of hippocampal neurons (Diwakarla et al., 2016a; Vanga et al., 2018). Molecular dynamics simulations and binding affinity estimations with the linear interaction energy method were performed for a large series of the arylsulfonamides. The significant agreement with experimental affinities suggested one of several tentatively proposed binding modes. Thus, the side chain of Arg439 of IRAP was estimated to interact with the tetrazole ring of the inhibitors while one of the oxygens of the sulfonamide function binds to the zinc ion of the enzyme. The NH of the sulfonamide binds to Glu 431 och Glu 295 via a water molecule bridge. This proposal was supported by the essentially perfect correlation for binding affinity differences between the selected pair of compounds obtained by rigorous free energy perturbation calculations by Gutierrez-de-Teran’s group (Vanga et al., 2018).
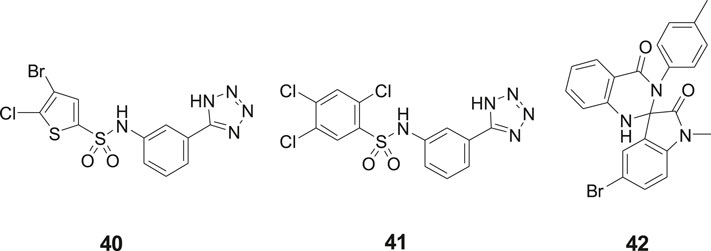
Figure 10. The sulfonamides 40 and 41 and the spiro-oxindole dihydroquinazolinone 42 were identified after screening a substance library of 10,500 low-molecular-weight compounds.
In addition to the sulfonamides, e.g., 40 and 41, the spiro-oxindole dihydroquinazolinone derivative 42 was identified in the screening campaign and subsequently a large series of related analogues were made, e.g., by applying rapid MW-assisted reactions and thereafter examined as IRAP inhibitors in bioassays (Engen et al., 2020) (Figure 10). Compounds that were selective toward the closely related APN and that exhibited sub-μM affinity were identified. Enantiomers were separated and according to computational modeling the inhibitory capacity of the compounds were attributed to the S-configuration of the spiro-oxindole dihydroquinazolinones. The derivatives with corresponding R-configuration were postulated to be essentially inactive in all cases. Notably, the proposed binding mode is compatible with the simultaneous binding of the substrate L-Leu-pNA, in agreement with the uncompetitive IRAP inhibition determined for a representative compound. Unfortunately, the compounds suffer from poor in vitro metabolic stability (Engen et al., 2020).
Direct Design From the Structure of Insulin-Regulated Aminopeptidase
In 2013, laboratories in Greece and Stratikos’ and Georgiadis’ groups reported that the phosphinic pseudotripeptide 43 (DG013A) is a potent inhibitor of IRAP with an IC50 of 57 nM (Figure 11). In contrast to 23 (HA08), the pseudotripeptide 43 was not selective and the related ERAP1 and ERAP2 were inhibited as well showing IC50 values of 48 and 80 nM, respectively (Zervoudi et al., 2013). As deduced from an X-ray crystallographic analysis of 43 in complex with ERAP2 it was concluded that the three first specificity pockets of the enzyme was of particular importance for binding to the enzyme (Zervoudi et al., 2013). The two oxygen atoms of the hydroxyphosphinyl group of 43 are interacting with the zinc ion whilst hydrogen bonds with Glu371 and Tyr455 essential for catalysis are created. Phosphinic peptides often share structural similarities with the transition-state of peptide substrate upon hydrolysis (Georgiadis and Dive, 2015).
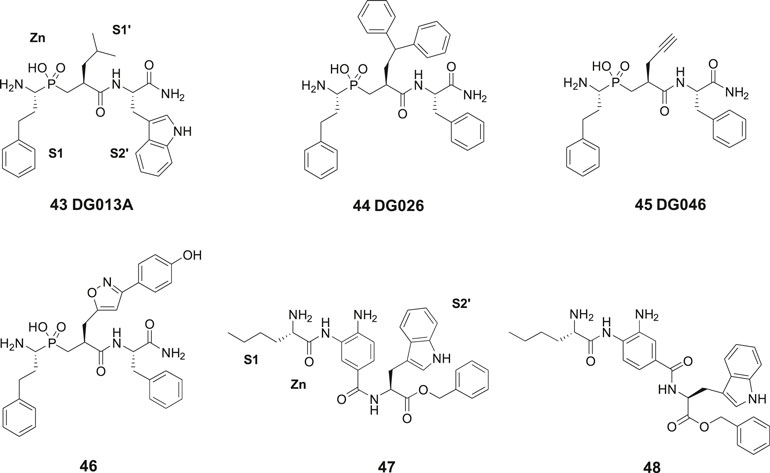
Figure 11. The phosphinic pseudotripeptide 43 (DG013A) is a potent IRAP inhibitor but is not selective and is equally effective inhibiting both ERAP1 and ERAP2. Compound 44 (DG026) comprising a bulky P1ʹ substituent is more IRAP selective than 43. Compounds 45 (DG046) and 46 that are further modified in the P1ʹ position are the most potent IRAP inhibitors in the series and approximately 10-fold selective vs. ERAP1 and ERAP2. The IRAP inhibitors 47 and 48 encompassing a 3,4-diaminobenzoic acid (DABA) scaffold are significantly less efficient than 43–46 as IRAP inhibitors.
The phosphinic group is, as compared to, e.g., thiols and hydroxamic acids more weakly binding to zinc ions in metalloproteases and a significantly improved selectivity could be achieved by systematic variations of the side chains at P1′ and P2′ positions of 43. Thus, the affinity to IRAP was improved after an enlargement of the P1ʹ as exemplified by 44 (DG026) exhibiting an IC50 of 32 nM while the affinity to ERAP1 dropped 100-fold. The crystal structure of IRAP in complex with 44 reveals that the enzyme undergoes structural reconfiguration that allows the accommodation of bulky side chains of the inhibitor. A closed conformational state of IRAP is created that is believed to be induced upon ligand binding. A hollow structure is formed excluding access of external solvent to the catalytic center (Mpakali et al., 2017). Notably, a propargyl group at P1′ or alternatively an extended P1′ side chain resulted both in very potent and fairly selective IRAP inhibitors, 45 (DG046) with IC50 values of 2 nM and 46 with a IC50 value of 4 nM (Figure 11). Thus, the potent IRAP inhibitor 45 demonstrated for ERAP1 an IC50 of 43 nM and for ERAP2 an IC50 value of 37 nM. The corresponding IC50 values for the oxazole derivative 46 with the extended P1ʹ side chain were 35 and 57 nM, respectively (Kokkala et al., 2016). Recently, a high-resolution crystal structure of phosphinic pseudopeptide inhibitor 45 (DG046) in the closed-conformation of ERAP1 was disclosed (Giastas et al., 2019b) and a mechanism for antigen peptide selection by ERAP1 presented (Giastas et al., 2019a). A review on inhibitors ERAP1 and ERAP2 was recently published (Georgiadis et al., 2019).
In 2015, Papakyriakou et al. reported a large series of inhibitors of ERAP1, ERAP2, and IRAP. This set of compounds comprises a 3,4-diaminobenzoic acid (DABA) scaffold (Papakyriakou et al., 2015). Compound 47 exhibited the best IRAP inhibitory capacity (IC50 = 105 nM) and demonstrated an almost 10-fold selectivity vs. ERAP1 (IC50 = 900 nM) and 15-fold improved selectivity vs. ERAP2 (IC50 = 1,600 nM) (Figure 11). L-Nle was preferred in the N-terminal for optimal inhibition and zinc coordination to the oxygen of the benzamide function is estimated to allow the lipophilic P1 substituent to be easily accommodated in the S1 pocket. In such conformation, the terminal amine group can form salt bridge interactions with two conserved glutamates, Glu295 and Glu431. Furthermore, the free aniline group could form a hydrogen bond with the catalytic E465 residue of IRAP. Deprotection of 47 to furnish an inhibitor with a carboxylate in the C-terminal exhibited a somewhat lower binding affinity for IRAP (IC50 = 296 nM). L-Val-OBn rather than L-Trp-OBn in the C-terminal led to a ten times less potent inhibitor of IRAP. The IRAP inhibitor 48 was the best inhibitor in a second series of bioisosteric diaminobenzoic acid inhibitors but was both less potent and selective as compared 47 (Papakyriakou et al., 2015).
Most small molecule binders of the oxytocinase subfamily of M1 aminopeptidases, e.g., the phosphinic pseudotripeptides 43–46 address the active site and establish strong interactions with the catalytic zinc ion. Highly potent enzyme inhibitors can be achieved but the selectivity obtained is often insufficient. Vourloumis’ group explored a large series of weaker zinc binding groups as potential alternatives to, e.g., the zinc binding amide function found in the 3,4-diaminobenzoic acid derivatives 47 and 48 (Tsoukalidou et al., 2019). Functionalized pyridinone- and pyridinethione-scaffolds, nicotinic-, isonicotinic-, aminobenzoic- and hydrazinobenzoic acids were prepared and examined as bioisosters, but no significant improvement of affinity was encountered by this maneuver. It was concluded that the potency of the compounds in the oxytocinase subfamily is mainly attributed to the occupation of the active site specificity pockets and their orientation in the enzymes (Tsoukalidou et al., 2019).
Conclusion
A series linear Ang IV analogues demonstrating improved metabolic stability and very high affinity to IRAP have been reported, e.g., the potent hexapeptides 11 (AL-11) and 12 (IVDE77) with Ki values of 7.6 and 1.7 nM, respectively, data to be compared with the Ki value of Ang IV of 62 nM in the same binding assay. These pseudopeptides that have been studied in some detail are encompassing β-amino acid residues and 12, in addition a conformationally constrained residue close to the C-terminal of the peptide. Furthermore, various macrocyclizations of Ang IV and subsequent simplifications of the structures delivered a series of selective high affinity ligands mimicking the substrates oxytocin and vasopressin in the N-terminal. For example, the disulfide 23 (HA08) that comprises a 13-membered ring system, a γ-turn mimetic in the C-terminal and a β-amino acid residue in the N-terminal ring system exhibits a Ki of 3.3 nM. The density of dendritic spines is closely associated with memory enhancement and 23 was found to increase the density of spines in the hippocampus significantly. To summarize, although very potent IRAP inhibitors have been discovered after systematic alterations starting from Ang IV none of the compounds are expected to reach the brain, at least not after oral administration.
Furthermore, ligands derived from the hexapeptide Ang IV but that are not proven to act as IRAP inhibitors have been reported. Thus, according to patent literature the drug-like 8 and 9 exhibit a very high affinity to the Ang IV binding site named the AT4 receptor. Moreover 10 (PNB-0408), a low-molecular weight molecule lacking the N-terminal amino group found in Ang IV analogues enhances cognition and is reported to crosses the blood-brain barrier and subsequently exert its action by interacting with the hepatocyte growth factor/c-Met receptor system. The compound has been studied in detail and promising pharmacological data have been reported. We believe that the positive effects of 10 on cognition are not attributed to inhibition of IRAP.
The HFI series of compounds, e.g., 37 (HFI 419) identified in 2008 after the virtual screening are much more drug-like than the hexapeptides 11 (AL-11), 12 (IVDE77), and the potent macrocyclic Ang IV analogue 23 (HA08) and can hopefully after further systematic optimization be converted into bioactive small molecules with capacity to cross the blood-brain barrier. However, while HFI 419 exerts a proven cognitive-enhancing effect in rodents after icv administration similar to that of Ang IV 1 and LVV-H7 3, the Ki value of 420 nM seems not optimal. The high throughput screening campaign of the substance library provided several hit compounds one of those the drug-like aryl sulfonamide 40 that was subsequently optimized into 41 that is a metabolically stable and reversible IRAP inhibitor. The sulfonamide 41, like 23 (HA08) and 37 (HFI 419) increases spine density in primary cultures of hippocampal neurons but is unfortunately a too poor inhibitor of IRAP and is demonstrating a Ki value of 540 nM. Similarly, compounds from the spiro-oxindole dihydroquinazolinone series identified in the screening, e.g., 42 are weak inhibitors and besides that not stable in in vitro assays.
The phosphinic pseudotripeptide series of compounds designed from structural and mechanistic knowledge of IRAP and the related ERAP1 and ERAP2 are promising and some of them exert powerful IRAP inhibitory effects. Thus, pseudotripeptides 45 and 46 exhibit Ki values of 2 and 4 nM, respectively and an approximately 10-fold selectivity vs. ERAP1 and ERAP2. This class of inhibitors holds promise for the future. The 3,4-diaminobenzoic acid series of compounds, e.g., 47 with a Ki value of 105 nM are less efficient IRAP inhibitors.
In summary, both direct design relying on mechanistic knowledge and available structural data from the oxytocinase subfamily of M1 aminopeptidases as well as iterative manipulations of the parent peptide Ang IV, involving insertion of unnatural amino acids as β-amino acids, macrocyclizations and various structural simplifications have provided highly potent selective Ang IV peptidemimetics inhibiting IRAP. Nevertheless, no inhibitor has yet reached phase I clinical trials and further efforts are needed to circumvent all obstacles related to absorption, metabolism and various issues on pharmacokinetics in order to eventually achieve orally bioavailable drug candidates acting as cognitive enhancers in vivo.
Author Contributions
MH and ML contributed equally in the preparation of the manuscript.
Funding
We thank the Kjell and Märta Beijer Foundation, the Swedish Brain Foundation and King Gustaf V and Queen Victoria’s Foundation of Freemasons for economic support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.The handling editor declared a past co-authorship with the authors.
Acknowledgments
We are very grateful for valuable discussions and constructive comments on the manuscript from Professor Anders Hallberg.
References
Albiston, A. L., Diwakarla, S., Fernando, R. N., Mountford, S. J., Yeatman, H. R., Morgan, B., et al. (2011). Identification and development of specific inhibitors for insulin-regulated aminopeptidase as a new class of cognitive enhancers. Br. J. Pharmacol. 164, 37–47. doi:10.1111/j.1476-5381.2011.01402.x
Albiston, A. L., Fernando, R. N., Yeatman, H. R., Burns, P., Ng, L., Daswani, D., et al. (2010a). Gene knockout of insulin-regulated aminopeptidase: loss of the specific binding site for angiotensin IV and age-related deficit in spatial memory. Neurobiol. Learn. Mem. 93, 19–30. doi:10.1016/j.nlm.2009.07.011
Albiston, A. L., Pham, V., Ye, S., Ng, L., Lew, R. A., Thompson, P. E., et al. (2010b). Phenylalanine-544 plays a key role in substrate and inhibitor binding by providing a hydrophobic packing point at the active site of insulin-regulated aminopeptidase. Mol. Pharmacol. 78, 600–607. doi:10.1124/mol.110.065458
Albiston, A. L., Mcdowall, S. G., Matsacos, D., Sim, P., Clune, E., Mustafa, T., et al. (2001). Evidence that the Angiotensin IV (AT4) receptor is the enzyme insulin-regulated aminopeptidase. J. Biol. Chem. 276, 48623–48626. doi:10.1074/jbc.c100512200
Albiston, A. L., Morton, C. J., Ng, H. L., Pham, V., Yeatman, H. R., Ye, S., et al. (2008). Identification and characterization of a new cognitive enhancer based on inhibition of insulin‐regulated aminopeptidase. Faseb. J. 22, 4209–4217. doi:10.1096/fj.08-112227
Albiston, A. L., Peck, G. R., Yeatman, H. R., Fernando, R., Ye, S., and Chai, S. Y. (2007). Therapeutic targeting of insulin-regulated aminopeptidase: heads and tails? Pharmacol. Ther. 116, 417–427. doi:10.1016/j.pharmthera.2007.07.006
Alescio-Lautier, B., Paban, V., and Soumireu-Mourat, B. (2000). Neuromodulation of memory in the hippocampus by vasopressin. Eur. J. Pharmacol. 405, 63–72. doi:10.1016/s0014-2999(00)00542-2
Andersson, H., Demaegdt, H., Johnsson, A., Vauquelin, G., Lindeberg, G., Hallberg, M., et al. (2011). Potent macrocyclic inhibitors of insulin-regulated aminopeptidase (IRAP) by olefin ring-closing metathesis. J. Med. Chem. 54, 3779–3792. doi:10.1021/jm200036n
Andersson, H., Demaegdt, H., Vauquelin, G., Lindeberg, G., Karlén, A., and Hallberg, M. (2008). Ligands to the (IRAP)/AT4 receptor encompassing a 4-hydroxydiphenylmethane scaffold replacing Tyr2. Bioorg. Med. Chem. 16, 6924–6935. doi:10.1016/j.bmc.2008.05.046
Andersson, H., Demaegdt, H., Vauquelin, G., Lindeberg, G., Karlén, A., Hallberg, M., et al. (2010). Disulfide cyclized tripeptide analogues of angiotensin IV as potent and selective inhibitors of insulin-regulated aminopeptidase (IRAP). J. Med. Chem. 53, 8059–8071. doi:10.1021/jm100793t
Axén, A., Andersson, H., Lindeberg, G., Rönnholm, H., Kortesmaa, J., Demaegdt, H., et al. (2007). Small potent ligands to the insulin-regulated aminopeptidase (IRAP)/AT4 receptor. J. Pept. Sci. 13, 434–444. doi:10.1002/psc.859
Axén, A., Lindeberg, G., Demaegdt, H., Vauquelin, G., Karlén, A., and Hallberg, M. (2006). Cyclic insulin-regulated aminopeptidase (IRAP)/AT4 receptor ligands. J. Pept. Sci. 12, 705–713. doi:10.1002/psc.782
Barlow, N., Vanga, S. R., Sävmarker, J., Sandström, A., Burns, P., Hallberg, A., et al. (2020). Macrocyclic peptidomimetics as inhibitors of insulin-regulated aminopeptidase (IRAP). RSC Med. Chem. 11, 234–244. doi:10.1039/c9md00485h
Benoist, C. C., Kawas, L. H., Zhu, M., Tyson, K. A., Stillmaker, L., Appleyard, S. M., et al. (2014). The procognitive and synaptogenic effects of angiotensin IV-derived peptides are dependent on activation of the hepatocyte growth factor/c-met system. J. Pharmacol. Exp. Therapeut. 351, 390–402. doi:10.1124/jpet.114.218735
Benoist, C. C., Wright, J. W., Zhu, M., Appleyard, S. M., Wayman, G. A., and Harding, J. W. (2011). Facilitation of hippocampal synaptogenesis and spatial memory by C-terminal truncated Nle1-angiotensin IV analogs. J. Pharmacol. Exp. Therapeut. 339, 35–44. doi:10.1124/jpet.111.182220
Bogdan, A. R., Davies, N. L., and James, K. (2011). Comparison of diffusion coefficients for matched pairs of macrocyclic and linear molecules over a drug-like molecular weight range. Org. Biomol. Chem. 9, 7727–7733. doi:10.1039/c1ob05996c
Borhade, S. R., Rosenström, U., Sävmarker, J., Lundbäck, T., Jenmalm-Jensen, A., Sigmundsson, K., et al. (2014). Inhibition of insulin-regulated aminopeptidase (IRAP) by arylsulfonamides. ChemistryOpen 3, 256–263. doi:10.1002/open.201402027
Bosnyak, S., Jones, E. S., Christopoulos, A., Aguilar, M.-I., Thomas, W. G., and Widdop, R. E. (2011). Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin. Sci. (Lond.) 121, 297–303. doi:10.1042/cs20110036
Bourne, J. N., and Harris, K. M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47–67. doi:10.1146/annurev.neuro.31.060407.125646
Brandt, W., Joachim Haupt, V., and Wessjohann, L. A. (2010). Chemoinformatic analysis of biologically active macrocycles. Curr. Top. Med. Chem. 10, 1361–1379. doi:10.2174/156802610792232060
Braszko, J. J., Kupryszewski, G., Witczuk, B., and Wiśniewski, K. (1988). Angiotensin II-(3-8)-hexapeptide affects motor activity, performance of passive avoidance and a conditioned avoidance response in rats. Neuroscience 27, 777–783. doi:10.1016/0306-4522(88)90182-0
Braszko, J. J., Wielgat, P., and Walesiuk, A. (2008). Effect of D3 dopamine receptors blockade on the cognitive effects of angiotensin IV in rats. Neuropeptides 42, 301–309. doi:10.1016/j.npep.2008.02.001
Bryant, N. J., Govers, R., and James, D. E. (2002). Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 3, 267–277. doi:10.1038/nrm782
De Bundel, D., Fafouri, A., Csaba, Z., Loyens, E., Lebon, S., El Ghouzzi, V., et al. (2015). Trans-modulation of the somatostatin type 2A receptor trafficking by insulin-regulated aminopeptidase decreases limbic seizures. J. Neurosci. 35, 11960–11975. doi:10.1523/jneurosci.0476-15.2015
De Bundel, D., Smolders, I., Vanderheyden, P., and Michotte, Y. (2008). Ang II and Ang IV: unraveling the mechanism of action on synaptic plasticity, memory, and epilepsy. CNS Neurosci. Ther. 14, 315–339. doi:10.1111/j.1755-5949.2008.00057.x
De Bundel, D., Smolders, I., Yang, R., Albiston, A. L., Michotte, Y., and Chai, S. Y. (2009). Angiotensin IV and LVV-haemorphin 7 enhance spatial working memory in rats: effects on hippocampal glucose levels and blood flow. Neurobiol. Learn. Mem. 92, 19–26. doi:10.1016/j.nlm.2009.02.004
De Gasparo, M., Husain, A., Alexander, W., Catt, K. J., Chiu, A. T., Drew, M., et al. (1995). Proposed update of angiotensin receptor nomenclature. Hypertension 25, 924–927. doi:10.1161/01.hyp.25.5.924
Demaegdt, H., Laeremans, H., De Backer, J.-P., Mosselmans, S., Le, M. T., Kersemans, V., et al. (2004). Synergistic modulation of cystinyl aminopeptidase by divalent cation chelators. Biochem. Pharmacol. 68, 893–900. doi:10.1016/j.bcp.2004.05.046
Demaegdt, H., Lenaerts, P.-J., Swales, J., De Backer, J.-P., Laeremans, H., Le, M. T., et al. (2006). Angiotensin AT4 receptor ligand interaction with cystinyl aminopeptidase and aminopeptidase N: [125I]Angiotensin IV only binds to the cystinyl aminopeptidase apo-enzyme. Eur. J. Pharmacol. 546, 19–27. doi:10.1016/j.ejphar.2006.07.005
Demaegdt, H., Lukaszuk, A., De Buyser, E., De Backer, J.-P., Szemenyei, E., Tóth, G., et al. (2009). Selective labeling of IRAP by the tritiated AT4 receptor ligand [3H]Angiotensin IV and its stable analog [3H]AL-11. Mol. Cell. Endocrinol. 311, 77–86. doi:10.1016/j.mce.2009.07.020
Diwakarla, S., Nylander, E., Grönbladh, A., Vanga, S. R., Khan, Y. S., Gutiérrez-de-Terán, H., et al. (2016a). Aryl sulfonamide inhibitors of insulin-regulated aminopeptidase enhance spine density in primary hippocampal neuron cultures. ACS Chem. Neurosci. 7, 1383–1392. doi:10.1021/acschemneuro.6b00146
Diwakarla, S., Nylander, E., Grönbladh, A., Vanga, S. R., Khan, Y. S., Gutiérrez-de-Terán, H., et al. (2016b). Binding to and inhibition of insulin-regulated aminopeptidase by macrocyclic disulfides enhances spine density. Mol. Pharmacol. 89, 413–424. doi:10.1124/mol.115.102533
Driggers, E. M., Hale, S. P., Lee, J., and Terrett, N. K. (2008). The exploration of macrocycles for drug discovery—an underexploited structural class. Nat. Rev. Drug Discov. 7, 608–624. doi:10.1038/nrd2590
Engen, K., Rosenström, U., Axelsson, H., Konda, V., Dahllund, L., Otrocka, M., et al. (2016). Identification of drug-like inhibitors of insulin-regulated aminopeptidase through small-molecule screening. Assay Drug Dev. Technol. 14, 180–193. doi:10.1089/adt.2016.708
Engen, K., Vanga, S. R., Lundbäck, T., Agalo, F., Konda, V., Jensen, A. J., et al. (2020). Synthesis, evaluation and proposed binding pose of substituted spiro‐oxindole dihydroquinazolinones as IRAP inhibitors. ChemistryOpen 9, 325–337. doi:10.1002/open.201900344
Evnouchidou, I., Papakyriakou, A., and Stratikos, E. (2009). A new role for Zn(II) aminopeptidases: antigenic peptide generation and destruction. Curr. Pharmaceut. Des. 15, 3656–3670. doi:10.2174/138161209789271816
Fernando, R. N., Larm, J., Albiston, A. L., and Chai, S. Y. (2005). Distribution and cellular localization of insulin-regulated aminopeptidase in the rat central nervous system. J. Comp. Neurol. 487, 372–390. doi:10.1002/cne.20585
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54. doi:10.1016/s0165-0173(02)00158-3
Fransson, R., Botros, M., Nyberg, F., Lindeberg, G., Sandström, A., and Hallberg, M. (2008). Small peptides mimicking substance P (1-7) and encompassing a C-terminal amide functionality. Neuropeptides 42, 31–37. doi:10.1016/j.npep.2007.11.002
Fruitier-Arnaudin, I., Cohen, M., Bordenave, S., Sannier, F., and Piot, J.-M. (2002). Comparative effects of angiotensin IV and two hemorphins on angiotensin-converting enzyme activity. Peptides 23, 1465–1470. doi:10.1016/s0196-9781(02)00083-9
Fu, M., Yu, X., Lu, J., and Zuo, Y. (2012). Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature 483, 92–95. doi:10.1038/nature10844
Gard, P. R. (2008). Cognitive-enhancing effects of angiotensin IV. BMC Neurosci. 9 (Suppl. 2), S15. doi:10.1186/1471-2202-9-s2-s15
Garreau, I., Chansel, D., Vandermeersch, S., Fruitier, I., Piot, J. M., and Ardaillou, R. (1998). Hemorphins inhibit angiotensin IV binding and interact with aminopeptidase N. Peptides 19, 1339–1348. doi:10.1016/s0196-9781(98)00075-8
Georgiadis, D., and Dive, V. (2015). Phosphinic peptides as potent inhibitors of zinc-metalloproteases. Top. Curr. Chem. 360, 1–38. doi:10.1007/128_2014_571
Georgiadis, D., Mpakali, A., Koumantou, D., and Stratikos, E. (2019). Inhibitors of ER aminopeptidase 1 and 2: from design to clinical application. Curr. Med. Chem. 26, 2715–2729. doi:10.2174/0929867325666180214111849
Giastas, P., Mpakali, A., Papakyriakou, A., Lelis, A., Kokkala, P., Neu, M., et al. (2019a). Mechanism for antigenic peptide selection by endoplasmic reticulum aminopeptidase 1. Proc. Natl. Acad. Sci. U.S.A. 116, 26709–26716. doi:10.1073/pnas.1912070116
Giastas, P., Neu, M., Rowland, P., and Stratikos, E. (2019b). High-resolution crystal structure of endoplasmic reticulum aminopeptidase 1 with bound phosphinic transition-state analogue inhibitor. ACS Med. Chem. Lett. 10, 708–713. doi:10.1021/acsmedchemlett.9b00002
Giordanetto, F., and Kihlberg, J. (2014). Macrocyclic drugs and clinical candidates: what can medicinal chemists learn from their properties? J. Med. Chem. 57, 278–295. doi:10.1021/jm400887j
Hallberg, M. (2009). Targeting the insulin-regulated aminopeptidase/AT4 receptor for cognitive disorders. Drug News Perspect. 22, 133–139. doi:10.1358/dnp.2009.22.3.1325032
Hallberg, M. (2015). Neuropeptides: metabolism to bioactive fragments and the pharmacology of their receptors. Med. Res. Rev. 35, 464–519. doi:10.1002/med.21323
Hallberg, M., Savmarker, J., and Hallberg, A. (2017). Angiotensin peptides as AT2 receptor agonists. Curr. Protein Pept. Sci. 18, 809–818. doi:10.2174/1389203718666170203150344
Harding, J. W., Cook, V. I., Miller-Wing, A. V., Hanesworth, J. M., Sardinia, M. F., Hall, K. L., et al. (1992). Identification of an AII(3-8) [AIV] binding site in guinea pig hippocampus. Brain Res. 583, 340–343. doi:10.1016/s0006-8993(10)80047-2
Hermans, S. J., Ascher, D. B., Hancock, N. C., Holien, J. K., Michell, B. J., Chai, S. Y., et al. (2015). Crystal structure of human insulin-regulated aminopeptidase with specificity for cyclic peptides. Protein Sci. 24, 190–199. doi:10.1002/pro.2604
Hess, S., Linde, Y., Ovadia, O., Safrai, E., Shalev, D. E., Swed, A., et al. (2008). Backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administrated drug lead for treating obesity. J. Med. Chem. 51, 1026–1034. doi:10.1021/jm701093y
Ho, J. K., and Nation, D. A. (2018). Cognitive benefits of angiotensin IV and angiotensin-(1-7): a systematic review of experimental studies. Neurosci. Biobehav. Rev. 92, 209–225. doi:10.1016/j.neubiorev.2018.05.005
Jackson, L., Eldahshan, W., Fagan, S. C., and Ergul, A. (2018). Within the brain: the renin angiotensin system. Int. J. Mol. Sci. 19, 876. doi:10.3390/ijms19030876
Jespers, W., Esguerra, M., Aqvist, J., and Gutierrez-De-Teran, H. (2019). QligFEP: an automated workflow for small molecule free energy calculations in Q. J. Cheminf. 11, 26. doi:10.1186/s13321-019-0348-5
Kandror, K. V., and Pilch, P. F. (1994). gp160, a tissue-specific marker for insulin-activated glucose transport. Proc. Natl. Acad. Sci. U.S.A. 91, 8017–8021. doi:10.1073/pnas.91.17.8017
Kasai, H., Matsuzaki, M., Noguchi, J., Yasumatsu, N., and Nakahara, H. (2003). Structure-stability-function relationships of dendritic spines. Trends Neurosci. 26, 360–368. doi:10.1016/s0166-2236(03)00162-0
Kato, T., Funakoshi, H., Kadoyama, K., Noma, S., Kanai, M., Ohya-Shimada, W., et al. (2012). Hepatocyte growth factor overexpression in the nervous system enhances learning and memory performance in mice. J. Neurosci. Res. 90, 1743–1755. doi:10.1002/jnr.23065
Kawas, L. H., Mccoy, A. T., Yamamoto, B. J., Wright, J. W., and Harding, J. W. (2012). Development of angiotensin IV analogs as hepatocyte growth factor/Met modifiers. J. Pharmacol. Exp. Therapeut. 340, 539–548. doi:10.1124/jpet.111.188136
Keller, S. R. (2003). The insulin-regulated aminopeptidase a companion and regulator of GLUT4. Front. Biosci. 8, s410–s420. doi:10.2741/1078
Keller, S. R., Scott, H. M., Mastick, C. C., Aebersold, R., and Lienhard, G. E. (1995). Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J. Biol. Chem. 270, 23612–23618. doi:10.1074/jbc.270.40.23612
Kobori, T., Goda, K., Sugimoto, K., Ota, T., and Tomisawa, K. (1997). Preparation of peptide derivatives as angiotensin IV receptor agonists. WO 97/03093 A1.
Kobori, T., Goda, K., Sugimoto, K., Ota, T., and Tomisawa, K. (1998). Preparation of amino acid derivatives as angiotensin IV receptor agonists. WO 98/05624 A1.
Kokkala, P., Mpakali, A., Mauvais, F.-X., Papakyriakou, A., Daskalaki, I., Petropoulou, I., et al. (2016). Optimization and structure-activity relationships of phosphinic pseudotripeptide inhibitors of aminopeptidases that generate antigenic peptides. J. Med. Chem. 59, 9107–9123. doi:10.1021/acs.jmedchem.6b01031
Kramár, E. A., Armstrong, D. L., Ikeda, S., Wayner, M. J., Harding, J. W., and Wright, J. W. (2001). The effects of angiotensin IV analogs on long-term potentiation within the CA1 region of the hippocampus in vitro. Brain Res. 897, 114–121. doi:10.1016/s0006-8993(01)02100-x
Krebs, L. T., Kramár, E., Hanesworth, J. M., Sardinia, M. F., Ball, A. E., Wright, J. W., et al. (1996). Characterization of the binding properties and physiological action of divalinal-angiotensin IV, a putative AT4 receptor antagonist. Regul. Pept. 67, 123–130. doi:10.1016/s0167-0115(96)00121-8
Krishnan, R., Hanesworth, J. M., Wright, J. W., and Harding, J. W. (1999). Structure-binding studies of the adrenal AT4 receptor: analysis of position two- and three-modified angiotensin IV analogs. Peptides 20, 915–920. doi:10.1016/s0196-9781(99)00081-9
Laeremans, H., Demaegdt, H., De Backer, J.-P., Le, M. T., Kersemans, V., Michotte, Y., et al. (2005). Metal ion modulation of cystinyl aminopeptidase. Biochem. J. 390, 351–357. doi:10.1042/bj20050349
Lai, C. S. W., Franke, T. F., and Gan, W.-B. (2012). Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature 483, 87–91. doi:10.1038/nature10792
Lee, J., Albiston, A. L., Allen, A. M., Mendelsohn, F. A. O., Ping, S. E., Barrett, G. L., et al. (2004). Effect of I.C.V. injection of AT4 receptor ligands, NLE1-angiotensin IV and LVV-hemorphin 7, on spatial learning in rats. Neuroscience 124, 341–349. doi:10.1016/j.neuroscience.2003.12.006
Lee, J., Mustafa, T., Mcdowall, S. G., Mendelsohn, F. A. O., Brennan, M., Lew, R. A., et al. (2003). Structure-activity study of LVV-hemorphin-7: angiotensin AT4 receptor ligand and inhibitor of insulin-regulated aminopeptidase. J. Pharmacol. Exp. Therapeut. 305, 205–211. doi:10.1124/jpet.102.045492
Lew, R. A., Mustafa, T., Ye, S., Mcdowall, S. G., Chai, S. Y., and Albiston, A. L. (2003). Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J. Neurochem. 86, 344–350. doi:10.1046/j.1471-4159.2003.01852.x
Lindman, S., Lindeberg, G., Gogoll, A., Nyberg, F., Karlén, A., and Hallberg, A. (2001). Synthesis, receptor binding affinities and conformational properties of cyclic methylenedithioether analogues of angiotensin II. Bioorg. Med. Chem. 9, 763–772. doi:10.1016/s0968-0896(00)00294-7
Lukaszuk, A., Demaegdt, H., Feytens, D., Vanderheyden, P., Vauquelin, G., and Tourwé, D. (2009). The replacement of His(4) in angiotensin IV by conformationally constrained residues provides highly potent and selective analogues. J. Med. Chem. 52, 5612–5618. doi:10.1021/jm900651p
Lukaszuk, A., Demaegdt, H., Szemenyei, E., Tóth, G., Tymecka, D., Misicka, A., et al. (2008). β-Homo-amino acid scan of angiotensin IV. J. Med. Chem. 51, 2291–2296. doi:10.1021/jm701490g
Lukaszuk, A., Demaegdt, H., Van Den Eynde, I., Vanderheyden, P., Vauquelin, G., and Tourwé, D. (2011). Conformational constraints in angiotensin IV to probe the role of Tyr2, Pro5 and Phe6. J. Pept. Sci. 17, 545–553. doi:10.1002/psc.1365
Lynch, G., Rex, C. S., Chen, L. Y., and Gall, C. M. (2008). The substrates of memory: defects, treatments, and enhancement. Eur. J. Pharmacol. 585, 2–13. doi:10.1016/j.ejphar.2007.11.082
Mallinson, J., and Collins, I. (2012). Macrocycles in new drug discovery. Future Med. Chem. 4, 1409–1438. doi:10.4155/fmc.12.93
Marsault, E., and Peterson, M. L. (2011). Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 54, 1961–2004. doi:10.1021/jm1012374
Matsumoto, H., Nagasaka, T., Hattori, A., Rogi, T., Tsuruoka, N., Mizutani, S., et al. (2001). Expression of placental leucine aminopeptidase/oxytocinase in neuronal cells and its action on neuronal peptides. Eur. J. Biochem. 268, 3259–3266. doi:10.1046/j.1432-1327.2001.02221.x
Matsumoto, H., Rogi, T., Yamashiro, K., Kodama, S., Tsuruoka, N., Hattori, A., et al. (2000). Characterization of a recombinant soluble form of human placental leucine aminopeptidase/oxytocinase expressed in Chinese hamster ovary cells. Eur. J. Biochem. 267, 46–52. doi:10.1046/j.1432-1327.2000.00949.x
Matsuzaki, M., Ellis-Davies, G. C. R., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092. doi:10.1038/nn736
McCoy, A. (2010). Pharmacokinetic characterization of angiotensin IV analogs with therapeutic potential for cancer and dementia. PhD thesis. Pullman, WA: Washington State University.
McCoy, A. T., Benoist, C. C., Wright, J. W., Kawas, L. H., Bule-Ghogare, J. M., Zhu, M., et al. (2013). Evaluation of metabolically stabilized angiotensin IV analogs as procognitive/antidementia agents. J. Pharmacol. Exp. Therapeut. 344, 141–154. doi:10.1124/jpet.112.199497
Moser, M. B., Trommald, M., and Andersen, P. (1994). An increase in dendritic spine density on hippocampal CA1 pyramidal cells following spatial learning in adult rats suggests the formation of new synapses. Proc. Natl. Acad. Sci. U.S.A. 91, 12673–12675. doi:10.1073/pnas.91.26.12673
Mountford, S. J., Albiston, A. L., Charman, W. N., Ng, L., Holien, J. K., Parker, M. W., et al. (2014). Synthesis, structure-activity relationships and brain uptake of a novel series of benzopyran inhibitors of insulin-regulated aminopeptidase. J. Med. Chem. 57, 1368–1377. doi:10.1021/jm401540f
Mpakali, A., Saridakis, E., Giastas, P., Maben, Z., Stern, L. J., Larhed, M., et al. (2020). Structural basis of inhibition of insulin-regulated aminopeptidase by a macrocyclic peptidic inhibitor. ACS Med. Chem. Lett. 11, 1429–1434. doi:10.1021/acsmedchemlett.0c00172
Mpakali, A., Saridakis, E., Harlos, K., Zhao, Y., Kokkala, P., Georgiadis, D., et al. (2017). Ligand-induced conformational change of insulin-regulated aminopeptidase: insights on catalytic mechanism and active site plasticity. J. Med. Chem. 60, 2963–2972. doi:10.1021/acs.jmedchem.6b01890
Mpakali, A., Saridakis, E., Harlos, K., Zhao, Y., Papakyriakou, A., Kokkala, P., et al. (2015). Crystal structure of insulin-regulated aminopeptidase with bound substrate analogue provides insight on antigenic epitope precursor recognition and processing. J. Immunol. 195, 2842–2851. doi:10.4049/jimmunol.1501103
Murthy, V. N., Schikorski, T., Stevens, C. F., and Zhu, Y. (2001). Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32, 673–682. doi:10.1016/s0896-6273(01)00500-1
Nikolaou, A., Eynde, I. V. D., Tourwé, D., Vauquelin, G., Tóth, G., Mallareddy, J. R., et al. (2013). [3H]IVDE77, a novel radioligand with high affinity and selectivity for the insulin-regulated aminopeptidase. Eur. J. Pharmacol. 702, 93–102. doi:10.1016/j.ejphar.2013.01.026
Nusser, Z., Lujan, R., Laube, G., Roberts, J. D. B., Molnar, E., and Somogyi, P. (1998). Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21, 545–559. doi:10.1016/s0896-6273(00)80565-6
Nyberg, G., Sanderson, K., Andrén, P., Thörnwall, M., Einarsson, M., Danielson, B., et al. (1996). Isolation of haemorphin-related peptides from filter membranes collected in connection with haemofiltration of human subjects. J. Chromatogr. A 723, 43–49. doi:10.1016/0021-9673(95)00811-x
O’Malley, A., O’Connell, C., Murphy, K. J., and Regan, C. M. (2000). Transient spine density increases in the mid-molecular layer of hippocampal dentate gyrus accompany consolidation of a spatial learning task in the rodent. Neuroscience 99, 229–232. doi:10.1016/s0306-4522(00)00182-2
O’Malley, A., O’Connell, C., and Regan, C. M. (1998). Ultrastructural analysis reveals avoidance conditioning to induce a transient increase in hippocampal dentate spine density in the 6 hour post-training period of consolidation. Neuroscience 87, 607–613. doi:10.1016/s0306-4522(98)00178-x
Papakyriakou, A., Zervoudi, E., Tsoukalidou, S., Mauvais, F.-X., Sfyroera, G., Mastellos, D. C., et al. (2015). 3,4-diaminobenzoic acid derivatives as inhibitors of the oxytocinase subfamily of M1 aminopeptidases with immune-regulating properties. J. Med. Chem. 58, 1524–1543. doi:10.1021/jm501867s
Penzes, P., Cahill, M. E., Jones, K. A., Vanleeuwen, J.-E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi:10.1038/nn.2741
Rasmussen, T. E., Pedraza-Díaz, S., Hardré, R., Laustsen, P. G., Carríon, A. G., and Kristensen, T. (2000). Structure of the human oxytocinase/insulin-regulated aminopeptidase gene and localization to chromosome 5q21. Eur. J. Biochem. 267, 2297–2306. doi:10.1046/j.1432-1327.2000.01234.x
Rogi, T., Tsujimoto, M., Nakazato, H., Mizutani, S., and Tomoda, Y. (1996). Human placental leucine aminopeptidase/oxytocinase. J. Biol. Chem. 271, 56–61. doi:10.1074/jbc.271.1.56
Sardinia, M. F., Hanesworth, J. M., Krebs, L. T., and Harding, J. W. (1993). AT4 receptor binding characteristics: D-amino acid- and glycine-substituted peptides. Peptides 14, 949–954. doi:10.1016/0196-9781(93)90071-n
Sardinia, M. F., Hanesworth, J. M., Krishnan, F., and Harding, J. W. (1994). AT4 receptor structure-binding relationship: angiotensin IV analogues. Peptides 15, 1399–1406. doi:10.1016/0196-9781(94)90115-5
Schmidt, B., Lindman, S., Tong, W., Lindeberg, G., Gogoll, A., Lai, Z., et al. (1997). Design, synthesis, and biological activities of four angiotensin II receptor ligands with γ-turn mimetics replacing amino acid residues 3−5. J. Med. Chem. 40, 903–919. doi:10.1021/jm960553d
Seyer, B., Diwakarla, S., Burns, P., Hallberg, A., Grӧnbladh, A., Hallberg, M., et al. (2020). Insulin‐regulated aminopeptidase inhibitor‐mediated increases in dendritic spine density are facilitated by glucose uptake. J. Neurochem. 153, 485–494. doi:10.1111/jnc.14880
Stratikos, E. (2014). Regulating adaptive immune responses using small molecule modulators of aminopeptidases that process antigenic peptides. Curr. Opin. Chem. Biol. 23, 1–7. doi:10.1016/j.cbpa.2014.08.007
Swanson, G. N., Hanesworth, J. M., Sardinia, M. F., Coleman, J. K. M., Wright, J. W., Hall, K. L., et al. (1992). Discovery of a distinct binding site for angiotensin II (3-8), a putative angiotensin IV receptor. Regul. Pept. 40, 409–419. doi:10.1016/0167-0115(92)90527-2
Thunnissen, M. M., Nordlund, P., and Haeggström, J. Z. (2001). Crystal structure of human leukotriene A(4) hydrolase, a bifunctional enzyme in inflammation. Nat. Struct. Biol. 8, 131–135. doi:10.1038/84117
Tsoukalidou, S., Kakou, M., Mavridis, I., Koumantou, D., Calderone, V., Fragai, M., et al. (2019). Exploration of zinc-binding groups for the design of inhibitors for the oxytocinase subfamily of M1 aminopeptidases. Bioorg. Med. Chem. 27, 115177. doi:10.1016/j.bmc.2019.115177
Tsujimoto, M., Mizutani, S., Adachi, H., Kimura, M., Nakazato, H., and Tomoda, Y. (1992). Identification of human placental leucine aminopeptidase as oxytocinase. Arch. Biochem. Biophys. 292, 388–392. doi:10.1016/0003-9861(92)90007-j
Vanderheyden, P. M. L. (2009). From angiotensin IV binding site to AT4 receptor. Mol. Cell. Endocrinol. 302, 159–166. doi:10.1016/j.mce.2008.11.015
Vanga, S. R., Sävmarker, J., Ng, L., Larhed, M., Hallberg, M., Åqvist, J., et al. (2018). Structural basis of inhibition of human Insulin-Regulated Aminopeptidase (IRAP) by aryl sulfonamides. ACS Omega. 3, 4509–4521. doi:10.1021/acsomega.8b00595
Van Spronsen, M., and Hoogenraad, C. C. (2010). Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 10, 207–214. doi:10.1007/s11910-010-0104-8
Wallis, M. G., Lankford, M. F., and Keller, S. R. (2007). Vasopressin is a physiological substrate for the insulin-regulated aminopeptidase IRAP. Am. J. Physiol. Endocrinol. Metab. 293, E1092–E1102. doi:10.1152/ajpendo.00440.2007
Waters, S. B., D’Auria, M., Martin, S. S., Nguyen, C., Kozma, L. M., and Luskey, K. L. (1997). The amino terminus of insulin-responsive aminopeptidase causes Glut4 translocation in 3T3-L1 adipocytes. J. Biol. Chem. 272, 23323–23327. doi:10.1074/jbc.272.37.23323
Wolfe, M. S. (2002). Therapeutic strategies for Alzheimer's disease. Nat. Rev. Drug Discov. 1, 859–866. doi:10.1038/nrd938
Wright, J. W., Clemens, J. A., Panetta, J. A., Smalstig, E. B., Weatherly, L. S., Kramár, E., et al. (1996). Effects of LY231617 and angiotensin IV on ischemia-induced deficits in circular water maze and passive avoidance performance in rats. Brain Res. 717, 1–11. doi:10.1016/0006-8993(95)01454-3
Wright, J. W., and Harding, J. W. (2008). The angiotensin AT4 receptor subtype as a target for the treatment of memory dysfunction associated with Alzheimer’s disease. J. Renin Angiotensin Aldosterone Syst. 9, 226–237. doi:10.1177/1470320308099084
Wright, J. W., and Harding, J. W. (2009). The brain angiotensin IV/AT4receptor system as a new target for the treatment of Alzheimer’s disease. Drug Dev. Res. 70, 472–480. doi:10.1002/ddr.20328
Wright, J. W., and Harding, J. W. (2011). Brain renin-angiotensin-A new look at an old system. Prog. Neurobiol. 95, 49–67. doi:10.1016/j.pneurobio.2011.07.001
Wright, J. W., and Harding, J. W. (2015). The brain hepatocyte growth factor/c-met receptor system: a new target for the treatment of Alzheimer’s disease. J. Alzheimers Dis. 45, 985–1000. doi:10.3233/jad-142814
Wright, J. W., and Harding, J. W. (2019). Contributions by the brain renin-angiotensin system to memory, cognition, and alzheimer’s disease. J. Alzheimers Dis. 67, 469–480. doi:10.3233/jad-181035
Wright, J. W., Miller-Wing, A. V., Shaffer, M. J., Higginson, C., Wright, D. E., Hanesworth, J. M., et al. (1993). Angiotensin II(3-8) (ANG IV) hippocampal binding: potential role in the facilitation of memory. Brain Res. Bull. 32, 497–502. doi:10.1016/0361-9230(93)90297-o
Wright, J. W., Stubley, L., Pederson, E. S., Kramár, E. A., Hanesworth, J. M., and Harding, J. W. (1999). Contributions of the brain angiotensin IV-AT4Receptor subtype system to spatial learning. J. Neurosci. 19, 3952–3961. doi:10.1523/jneurosci.19-10-03952.1999
Wright, J., Yamamoto, B., and Harding, J. (2008). Angiotensin receptor subtype mediated physiologies and behaviors: new discoveries and clinical targets. Prog. Neurobiol. 84, 157–181. doi:10.1016/j.pneurobio.2007.10.009
Yamamoto, B. J., Elias, P. D., Masino, J. A., Hudson, B. D., Mccoy, A. T., Anderson, Z. J., et al. (2010). The angiotensin IV analog Nle-Tyr-Leu-ψ-(CH2-NH2)3-4-His-Pro-Phe (Norleual) can act as a hepatocyte growth factor/c-Met inhibitor. J. Pharmacol. Exp. Therapeut. 333, 161–173. doi:10.1124/jpet.109.161711
Yap, B. K., Harjani, J. R., Leung, E. W. W., Nicholson, S. E., Scanlon, M. J., Chalmers, D. K., et al. (2016). Redox-stable cyclic peptide inhibitors of the SPSB2-iNOS interaction. FEBS Lett. 590, 696–704. doi:10.1002/1873-3468.12115
Zervoudi, E., Saridakis, E., Birtley, J. R., Seregin, S. S., Reeves, E., Kokkala, P., et al. (2013). Rationally designed inhibitor targeting antigen-trimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc. Natl. Acad. Sci. U.S.A. 110, 19890–19895. doi:10.1073/pnas.1309781110
Keywords: angiotensin IV, peptidemimetics, aminopeptidase N, cystinyl aminopeptidase, insulin-regulated aminopeptidase
Citation: Hallberg M and Larhed M (2020) From Angiotensin IV to Small Peptidemimetics Inhibiting Insulin-Regulated Aminopeptidase. Front. Pharmacol. 11:590855. doi:10.3389/fphar.2020.590855
Received: 03 August 2020; Accepted: 18 September 2020;
Published: 15 October 2020.
Edited by:
Efstratios Stratikos, National Centre of Scientific Research Demokritos, GreeceReviewed by:
Marcin Poreba, Wrocław University of Science and Technology, PolandAkira Hattori, Kyoto University, Japan
Copyright © 2020 Hallberg and Larhed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathias Hallberg, TWF0aGlhcy5IYWxsYmVyZ0BmYXJtYmlvLnV1LnNl