Abstract
Non-alcoholic steatohepatitis (NASH) develops from non-alcoholic fatty liver disease (NAFLD). Currently, around 25% of the population is estimated to have NAFLD, and 25% of NAFLD patients are estimated to have NASH. NASH is typically characterized by liver steatosis inflammation, and fibrosis driven by metabolic disruptions such as obesity, diabetes, and dyslipidemia. NASH patients with significant fibrosis have increased risk of developing cirrhosis and liver failure. Currently, NASH is the second leading cause for liver transplant in the United States. More importantly, the risk of developing hepatocellular carcinoma from NASH has also been highlighted in recent studies. Patients may have NAFLD for years before progressing into NASH. Although the pathogenesis of NASH is not completely understood, the current “multiple-hits” hypothesis suggests that in addition to fat accumulation, elevated oxidative and ER stress may also drive liver inflammation and fibrosis. The development of clinically relevant animal models and pharmacological treatments for NASH have been hampered by the limited understanding of the disease mechanism and a lack of sensitive, non-invasive diagnostic tools. Currently, most pre-clinical animal models are divided into three main groups which includes: genetic models, diet-induced, and toxin + diet-induced animal models. Although dietary models mimic the natural course of NASH in humans, the models often only induce mild liver injury. Many genetic and toxin + diet-induced models rapidly induce the development of metabolic disruption and serious liver injury, but not without their own shortcomings. This review provides an overview of the “multiple-hits” hypothesis and an evaluation of the currently existing animal models of NASH. This review also provides an update on the available interventions for managing NASH as well as pharmacological agents that are currently undergoing clinical trials for the treatment of NASH.
Introduction
First discovered in 1980, non-alcoholic steatohepatitis (NASH) is a type of fatty liver disease characterized by excessive liver fat accumulation, hepatic inflammation and fibrosis (Ludwig et al., 1980; Kleiner et al., 2005; Diehl and Day, 2017). NASH is falls within the large, overarching theme of non-alcoholic fatty liver disease (NAFLD) which encompasses varying degrees of liver injury (Friedman et al., 2018a). NASH is histologically distinct from a simple fatty liver, where there is only an accumulation of fat without the presence of inflammation and fibrosis (Brunt et al., 2011).
Epidemiology of Non-Alcoholic Steatohepatitis
The growing epidemics of obesity, dyslipidemia and insulin resistance serve as major risk factors for the development of NASH (Saklayen, 2018). Epidemiological studies show that roughly 82% of NASH patients are obese, 83% exhibit hyperlipidemia and 48% are diagnosed with type 2 diabetes (Younossi et al., 2016b). NAFLD tends to be more prevalent in middle-aged to elderly patients as older patients exhibit more characteristics of metabolic syndrome (Frith et al., 2009; Williams et al., 2011). Nevertheless, NAFLD can also be diagnosed in children/adolescences who are as young as 13 years old (Goyal and Schwimmer, 2016). According to the survey conducted by the National Health and Nutrition Examination in the United States, the incidence of NAFLD in adolescent and young adults (aged 19–35) have risen by approximately 2.5 times in the last 20 years (Welsh et al., 2013). More importantly, longitudinal follow-up studies suggest that adolescent diagnosed with NAFLD/NASH have increased risk of cirrhosis and mortality compared to age-matched average population (Feldstein et al., 2009; Goyal and Schwimmer, 2016; Doycheva et al., 2017). Currently, it is estimated that approximately 25% of the world population has NAFLD, and further, 20–25% of this NAFLD patient population will go on to develop NASH (Younossi et al., 2016a; Estes et al., 2018) (Figure 1). If left untreated, the risk of developing cirrhosis, and subsequently liver failure and hepatocellular carcinoma will increase and eventually causing death (Alexander et al., 2019). NASH-induced cirrhosis has been recognized as one of the fastest-growing liver diseases, and is the second greatest contributor to an indication for liver transplantation in the United States (Wong et al., 2015). Based on current trends, there will be an estimated global NAFLD incidence of 101 million by 2030, and the number of NASH cases is projected to increase to 27 million by 2030 (Estes et al., 2018). In parallel with the development and progression of obesity and type 2 diabetes a recent study highlighted that the annual health care cost associated with NAFLD in the United States was approximately US$103 billion, and €35 billion in four European countries combined (Germany, Italy, United Kingdom., and France) (Younossi et al., 2016a). These costs are estimated to rise to US$908 and €302 billion in the United States and in these European countries, respectively within 10 years (Younossi et al., 2016a). Thus, early detection, diagnosis and treatment of fatty liver disease are of paramount importance in controlling the impact of this disease.
FIGURE 1
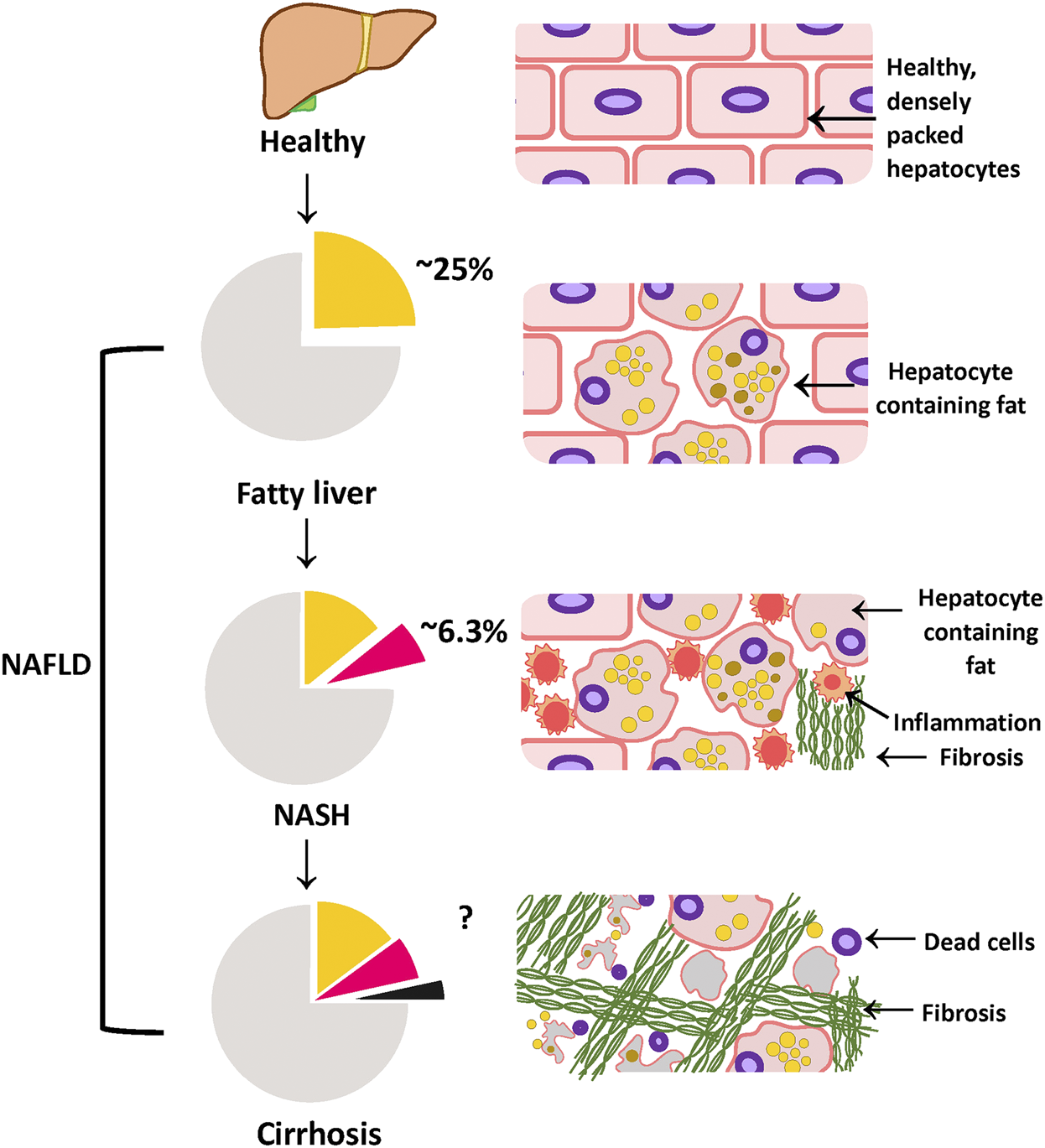
Different phases of NAFLD: progressing from healthy to cirrhosis NAFLD represents a spectrum of fatty liver diseases ranging from fatty liver to cirrhosis. Approximately 25% of the population worldwide is estimated to have fatty liver, characterized by more than 5% fat accumulating in the liver. If left untreated, fatty liver can progress onto the more severe form; NASH, defined by severe liver injury and inflammation in addition to fat. Currently, a further 25% of the NAFLD population is estimated to have NASH (which is roughly calculated to be 6.3% of the population). NASH patients are estimated to have a higher risk of developing cirrhosis, which is the extensive liver tissue scarring. Figure is designed and drawn using Inkscape (http://www.inkscape.org/.).
Diagnosis and Detection Methods
NASH itself can often be asymptomatic, although patients with a high body mass index (>25 kg/m2) and T2DM features such as hyperglycemia and insulin resistance are encouraged to be screened for the presence of fatty liver disease (Chalasani et al., 2012; Friedman et al., 2018a). Nevertheless, a recent population study has highlighted that NASH patients have a higher incidence of fatigue and abdominal discomfort which are shown to be correlated with hepatic lobular inflammation (Huber et al., 2019). This may be because that hepatic inflammation is associated with elevated plasma inflammatory cytokines (Ajmera et al., 2017) which creates a metabolically inflamed milieu that can negatively affect the mood (Rethorst et al., 2014; Huber et al., 2019).
Patients consuming less than the excessive alcohol intake threshold of >20–30 g/day are classified as having NAFLD, and patients who consumed above that threshold would be diagnosed as having alcoholic fatty liver disease (AFLD), typically treated by alcohol abstinence (Scaglioni et al., 2011). Although NAFLD/NASH is not a result of excessive alcohol intake, it shares many histological similarities with AFLD, such as liver steatosis and inflammation (Williams et al., 2011). Nevertheless, it might not be possible to determine whether low alcohol use contributes to the development of NAFLD/NASH (Scaglioni et al., 2011).
Elevation in the plasma of the liver enzymes alanine transaminase (ALT) and aspartate aminotransferase (AST) in a routine blood test is generally the first line of diagnosis (Kim et al., 2008; Siddiqui et al., 2018). ALT and AST are highly expressed in hepatocytes. In the event of hepatocyte necrosis, ALT and AST leak into the circulation, and are thus biomarkers for liver injury (Kew, 2000). Nevertheless, NASH patients may have normal plasma ALT/AST levels and the presence of other diseases, such as viral hepatitis, may also induce ALT and AST elevation (Dyson et al., 2014). ALT and AST are thus insufficiently specific and sensitive enough to determine the presence or severity of NASH (Friedman et al., 2018a). To confirm the presence of fatty liver, computed tomography (CT) scan or magnetic resonance imaging (MRI) can potentially be used as a non-invasive diagnostic tool to assess the percentage of fat in the liver (Friedman et al., 2018a). However, using MRI as a diagnostic tool in the clinic may not be practical due to the high cost and limited availability. Patients in rural/regional areas and/or from low socioeconomic areas would be unlikely to be able to access it (Friedman et al., 2018a). More importantly, the percentage of hepatic fat alone does not indicate the level of liver inflammation, hepatocyte damage and tissue fibrosis. Thus pathologist scoring of liver biopsy histological features remains the gold standard for determining the presence and severity of NASH (Siddiqui et al., 2018). Liver hematoxylin and eosin staining are assigned an ordinal score on a scale of 0–3 for steatosis, 0–3 for inflammation and 0–2 for ballooning hepatocytes by a panel of pathologists (Table 1). Ordinal scores of these 3 parameters are combined to give a total NAFLD activity score (NAS). In both clinical and preclinical studies, there is a general consensus that a total NAS ≥5 is classified as definitive NASH rather than a simple fatty liver disease (Kleiner et al., 2005). Nevertheless, the invasive nature of liver biopsy has made the histological diagnosis method less favored. Further studies in elucidating the molecular mechanisms of NASH to discover sensitive and highly NASH-specific biomarkers are warranted.
TABLE 1
| Features | Score | Description |
|---|---|---|
Healthy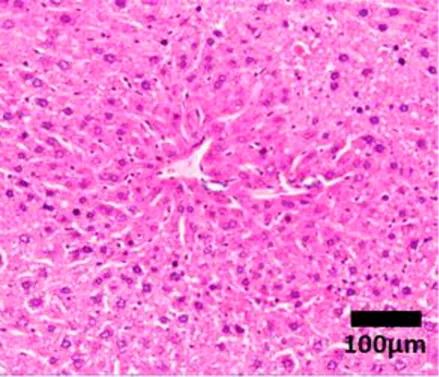 | None | Healthy liver |
| Hepatocytes are nicely arranged and densely packed | ||
Steatosis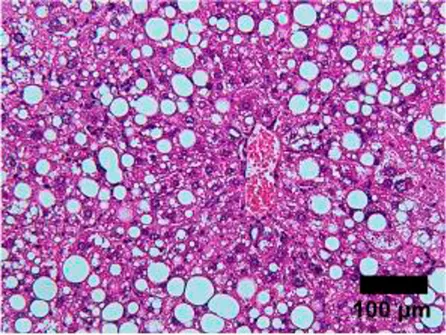 | 0 | <5% of liver tissue (per field of view) |
| 1 | <33% of liver tissue (per field of view) | |
| 2 | 34–66% of liver tissue (per field of view) | |
| 3 | >66% of liver tissue (per field of view) | |
Inflammation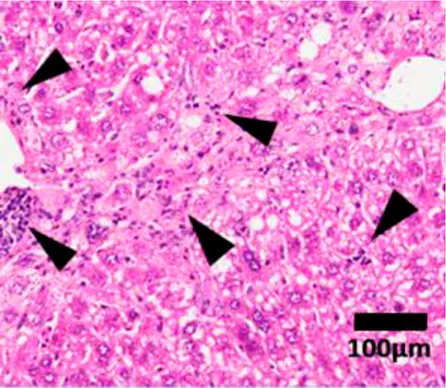 | 0 | None |
| 1 | 1–2 foci | |
| 2 | 3–4 foci | |
| 3 | >4 foci | |
Ballooning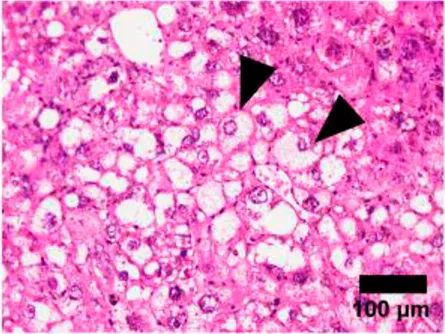 | 0 | None |
| 1 | Few | |
| 2 | Many |
NAFLD Activity Score (NAS) system with representative H&E images. The NAS system is an internationally recognized method of determining the severity of fatty liver disease (see text for references). Steatosis score represents the percentage of lipid droplets present in each field of view, inflammation score represents the number of inflammatory cell clusters (1 cluster = 1 foci) and the ballooning score is indicative of the number of hepatocytes that have altered cell structure due to excess lipid accumulation. The pathologist can give a score between 0 and 3 for each of steatosis and inflammation, and 0–2 for ballooning, based on the characteristics of the samples. Combining the scores from each of the parameters give rise to the total NAS. H&E-stained representative images are provided by our laboratory. All images were taken under ×200 magnification. Black arrows are marking the specific location of the histological features.
Pathogenesis of Non-Alcoholic Steatohepatitis
Day and James (1998) hypothesized the “two-hit” NASH model, i.e., that obesity, as an external stressor, can increase the accumulation of fat in the liver, but is normally not sufficient to cause inflammation and fibrosis. Hence a “second-hit” is required to further exaggerate liver injury. Recent findings on NASH shed a new light on the disease pathogenesis, shifting from the traditional “two-hit” model to a model where multiple parallel pathogenic influences are present that may act synergistically to drive the development of NASH, as indicated in Figure 2 (Buzzetti et al., 2016). Although the exact mechanism of disease pathogenesis remains to be elucidated, epidemiological studies have highlighted common metabolic comorbidities of NAFLD/NASH patients including obesity, insulin resistance and hyperlipidemia (Younossi et al., 2016a). In support of this concept, several clinical studies have suggested that NAFLD/NASH may have a role in the development of other metabolic diseases including cardiovascular diseases (Targher et al., 2010; Labenz et al., 2020) and chronic kidney disease (Kaps et al., 2020).
FIGURE 2
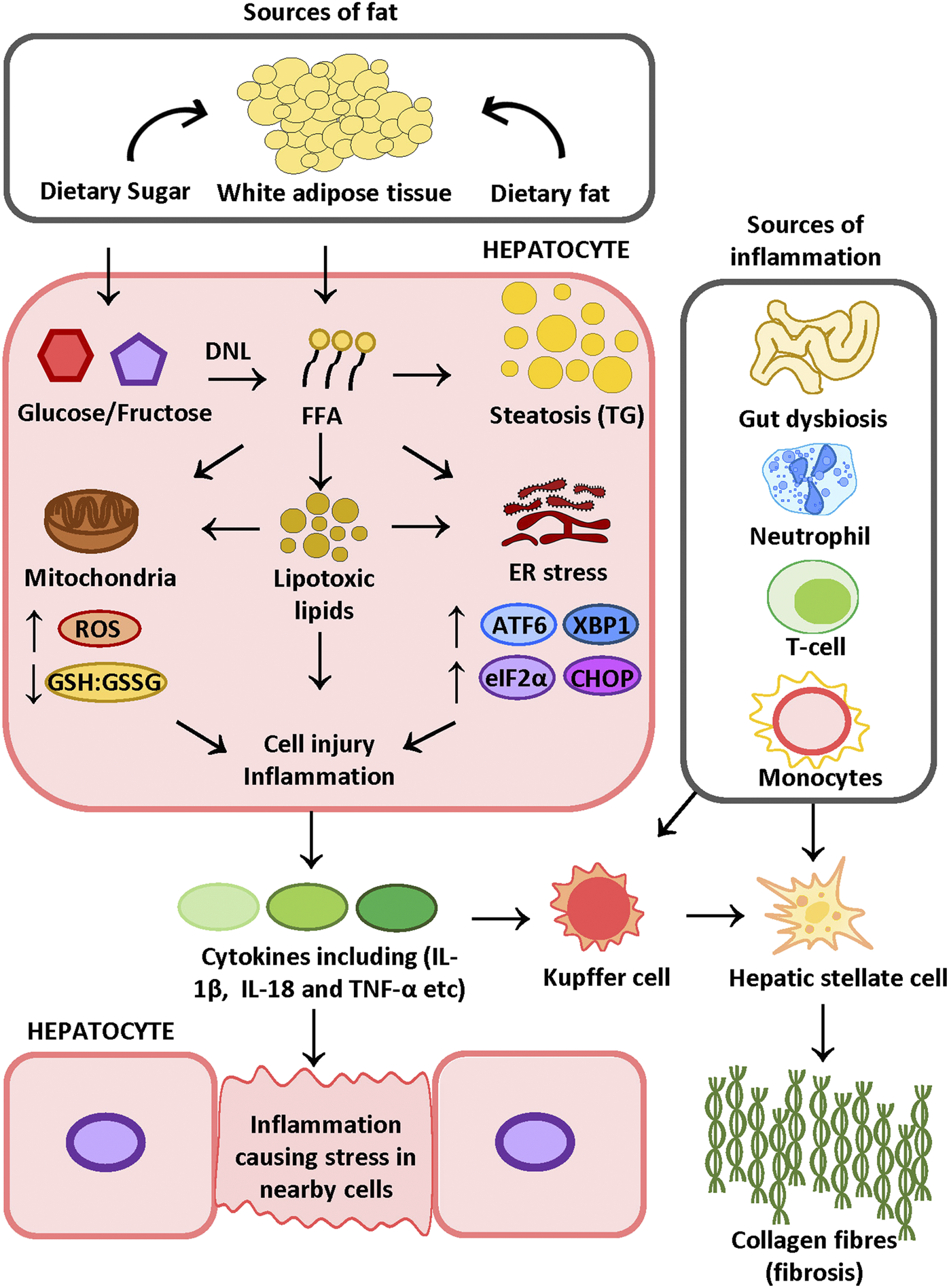
Proposed mechanism of NASH FFAs released from adipose tissue due to insulin resistance and dietary-sugar-induced DNL increase the FFA pool in the liver. FFAs can be stored as TG in the hepatocyte or be metabolized into lipotoxic lipids. Lipid mediators may induce oxidative stress and ER stress, which ultimately results in cell injury and inflammation. Cell injury induces inflammatory cell recruitment and activation. A leaky gut due to gut dysbiosis can further contribute to liver inflammation. The combination of inflammation and tissue damage triggers HSC activation and collagen deposition. FFA: free fatty acid, KC: Kupffer cell, HSC: hepatic stellate cell, TG: triglyceride, DNL: de novo lipogenesis, IL-18: interleukin 18, IL-1β: interleukin one beta, TNF-α: tumor necrosis factor α, ATF6: activating transcription factor 6; TXBP-1: total X-box protein-1; CHOP: C/EBP Homologous Protein, eIF2α: eukaryotic translation initiation factor 2α. Figure is designed and drawn using Inkscape (http://www.inkscape.org/.).
Recently, there has been increasing interest in the role of metabolic inflammation and the crosstalk network between liver and other organs in driving metabolic diseases (Gehrke and Schattenberg, 2020; Wang et al., 2020). The elevated pro-inflammatory gut microbes influencing the liver during gut dysbiosis may initiate and/or exacerbate hepatic inflammation (Hildebrandt et al., 2009; Ogawa et al., 2018). Moreover, the presence of disrupted metabolism such as insulin resistance in the adipose tissue is known to be associated with increased hepatic steatosis as well as hepatic macrophage activation (Rosso et al., 2019; Gehrke and Schattenberg, 2020). In addition to extrahepatic stressors, intrahepatic cellular stressors such as liver oxidative stress and endoplasmic reticulum (ER) stress are also known to be part of the multiple parallel influences mechanism which may lead to the development of NAFLD/NASH (Sanyal et al., 2001; Friedman et al., 2018a; Lebeaupin et al., 2018a). Adding to the complexity of the disease, the likelihood of NASH development also appears closely associated with genetic factors (Eslam et al., 2018).
The Role of Genetics and Ethnicity
Familial studies indicate that children from parents with higher hepatic fat contents are more likely to develop NAFLD and cirrhosis (Schwimmer et al., 2009). Twin studies demonstrated a significantly higher intra-pair correlation between the level of liver fat and plasma ALT in monozygotic twin than dizygotic twins (Makkonen et al., 2009). In addition, genome-wide association studies have recently identified numerous genetic factors that associate strongly with the development of NAFLD. Genetic variants in genes such as transmembrane six superfamily member 2 (TM6SF2) (Mahdessian et al., 2014), glucokinase regulatory protein (GCKR) (Petta et al., 2014) and patatin-like phospholipase domain-containing-3 (PNPLA3) are found to associate with NAFLD and NASH, with PNPLA3 classified as one of the most common genetic variations (Eslam et al., 2018). Patients who have the PNPLA3 genetic polymorphism produce a truncated lipase enzyme which impedes triglyceride breakdown and subsequently reduces liver triglyceride (TG) secretion in the form of very-low-density lipoproteins (VLDL) (Dongiovanni, 2013). Interestingly, a population study conducted in the United States revealed differences in susceptibility to triglyceride accumulation, and the development of NAFLD, in different ethnic groups. The PNPLA3 I148M polymorphism is more frequently present in the Hispanic population, less in those of European descent, and lowest in African-American populations (Romeo et al., 2008). Investigators suggested that the lower frequency of the PNPLA3 I148M polymorphism offers a potential explanation for the lower prevalence of NAFLD observed in the African-American population, despite a higher rate of obesity and diabetes (Romeo et al., 2008; Chinchilla-lópez et al., 2019). Notably, gene polymorphisms alone do not completely confer full NAFLD/NASH development (Diehl and Day, 2017). Rather, genetic predisposition combined with environmental factors such as obesity, increased abdominal fat mass and excessive carbohydrate and sugar consumption may confer a higher risk of developing NASH (Davis et al., 2010).
The Role of Obesity and Systemic Insulin Resistance
Epidemiological studies have revealed a strong link between obesity and type 2 diabetes with the development of NASH, suggesting that an insulin-resistant milieu may be an important initial driving force for the development of NASH (Lomonaco et al., 2012; Younossi et al., 2016b). Nevertheless, accumulating evidence suggests that lean subjects, particularly from the Asian ethnic group, may also develop NAFLD, commonly referred to as having “lean NAFLD” (Das and Chowdhury, 2013; Feng et al., 2014; Chen et al., 2020). Lean NAFLD subjects often exhibit excess visceral white adipose tissue despite a normal BMI (25 kg/m2 in Caucasian and 23 kg/m2 in Asian populations) (Ruderman et al., 1998; Feng et al., 2014; Chen et al., 2020). Excess white adipose tissue associated with increased plasma and adipose tissue pro-inflammatory cytokines such as TNF-α and IL-6 has been reported in both patients with NAFLD and in animal models of NAFLD (Hotamisligil et al., 1993; Hotamisligil et al., 1995; Weisberg et al., 2003). In addition to contributing to chronic low-grade systemic inflammation, adipose-tissue-derived cytokines are also reported to induce systemic insulin resistance, by impeding downstream insulin signaling (Peraldi et al., 1996; Xu et al., 2003). Upon insulin binding, insulin receptor activation initiates tyrosine phosphorylation of the downstream cytosolic insulin receptor substrate (IRS) (White, 1997). This signaling cascade is transduced by IRS as it phosphorylates phosphoinositide 3-kinase (PI3K) and Akt to further elicit insulin-mediated effects (White, 1997). TNF-α inhibits insulin downstream signaling by activating c-Jun NH2-terminal kinase (JNK) which phosphorylates IRS-1 at Ser307 (Aguirre et al., 2000). As a result of insulin resistance, adipose-tissue-released free fatty acids (FFA) accumulate in the plasma (Morigny et al., 2016). Clinical studies of NAFLD patients have revealed a positive correlation between insulin resistance and elevated hepatic TGs, suggesting that the adipose-tissue-released FFAs may ultimately be taken up by the liver and metabolized into TG (Lomonaco et al., 2012). In addition to adipose-tissue-derived FFAs, increased dietary fat and carbohydrate uptake (especially fructose) can also contribute to steatosis in the liver (Faeh et al., 2005; Lomonaco et al., 2012; Lambert et al., 2014).
The Role of Dietary Fat Intake and de novo Lipogenesis
Steatosis is defined as excess TG deposition in the liver, which gives rise to lipid droplets scattered through the liver tissue (Figure 2). Notably, of all TGs found in the liver of NAFLD patients, 59% are derived from plasma FFAs, whereas 15% and 26% are derived from dietary fat and de novo lipogenesis, respectively (Donnelly et al., 2005). This is consistent with the role of dietary fat intake and de novo lipogenesis in triggering liver steatosis, in addition to adipose-tissue-derived FFAs (Lomonaco et al., 2012; Lambert et al., 2014; Luukkonen et al., 2018). In support of this, a dietary study using a stabilized isotope tracer demonstrated that human subjects on a diet rich in saturated-fat exhibited increased adipose tissue triglyceride storage and increased intrahepatic TG levels (Luukkonen et al., 2018). Moreover, long-term consumption of diets with 45–68% energy derived from fats has been reported to elevate intrahepatic TG in rodents (Wang et al., 2006; Koppe et al., 2009). Apart from direct fat uptake, TGs derived from de novo lipogenesis are reportedly elevated in subjects who were on a high carbohydrate diet (Faeh et al., 2005; Luukkonen et al., 2018). Dietary studies in rodents fed with a high fructose diet showed activation of the lipogenic transcription factor, sterol regulatory element-binding protein 1c (SCREBP1c), which is responsible for inducing the transcription of lipogenic enzymes for catalyzing TG synthesis (Aragno et al., 2009; Softic et al., 2016). Overall, hepatic FFA accumulation contributes to the development of fatty liver disease. It is, however, suggested that the ensuing hepatic lipotoxicity is potentially driving the development of liver injury and inflammation that is characteristic of NASH (Neuschwander-Tetri, 2010).
The Role of Hepatic Lipotoxicity
Many patients with fatty liver disease show only steatosis for many years without additional characteristics of NASH (Calzadilla Bertot and Adams, 2016). Although the exact mechanism that drives the development of NASH from simple steatosis is unclear, lipotoxic lipid-inflicted cell injury is proposed to be a major contributor (Neuschwander-Tetri, 2010). In agreement with this hypothesis, common lipotoxic lipids such as cholesterol, TG and DAG are reported to be significantly elevated in the liver of NASH patients when compared to control subjects (Puri et al., 2007; Magkos et al., 2012). Animal studies, where rodents were fed a high fat or high cholesterol diet revealed that liver resident macrophages can be activated by engulfing cholesterol crystals resulting in liver inflammation (Van Rooyen et al., 2011; Mridha et al., 2017). DAG is known for its ability to exacerbate hepatic insulin resistance by interfering with insulin signaling via PKC activation (Samuel et al., 2004; Mota et al., 2016). Moreover, the accumulation of TG in the liver leads to steatosis, which is a hallmark of fatty liver disease (Thomas et al., 2005). The role of other FFA-derived lipid species such as ceramide is inconclusive. NAFLD animal models showed elevated levels of ceramide and inhibition of ceramide synthesis attenuated liver inflammation (Jiang et al., 2019; Montandon et al., 2019). Nevertheless, clinical observations from Magkos et al. (2012) reported that the severity of NAFLD/NASH is not correlated with hepatic ceramide content, although it is worth noting that this clinical study only included a small patient population of 16 subjects (Magkos et al., 2012). Interestingly, another study reported an elevated ceramide in the adipose tissue of obese and insulin resistant human subjects (Turpin et al., 2014). Clinical studies involving a wider patient cohort is warranted to confirm the findings from preclinical studies.
The Role of Hepatic Oxidative Stress
Oxidative stress-induced hepatocyte damage and apoptosis have been reported as one of the main drivers of tissue injury in NASH patients (Masarone et al., 2018). As an adaptation to minimize hepatic steatosis, the rate of the disposal of fatty acid via mitochondrial ß-oxidation was reported to be significantly upregulated in NAFLD and NASH patients (Sanyal et al., 2001). However, studies comparing mitochondrial function in NAFLD and NASH patients have highlighted that this adaptation is lost in later stage-NASH patients, due to excessive ROS-induced mitochondrial dysfunction (Kojima et al., 2007; Koliaki et al., 2015). Apart from mitochondria-derived ROS, NADPH oxidase 2 (NOX2) activation in liver-infiltrating macrophages is also reported to contribute to oxidative stress-induced liver damage in NAFLD (Kim et al., 2017). More importantly, Yesilova and colleagues (2005) documented that NAFLD/NASH patients exhibit the reduced activity of antioxidative mechanisms such as coenzyme Q10 and superoxide dismutase. In addition, the reduced glutathione: oxidized glutathione (GSH:GSSG) ratio in animals with diet-induced with NASH also highlights an imbalance between ROS and antioxidants (Iruarrizaga-Lejarreta et al., 2017). Compromised antioxidant capacity enables the generation of reactive oxygen/nitrogen species such as hydroxyl radical (•OH), superoxide anion (O2−•) and peroxynitrite (ONOO−) to accumulate and readily react with intracellular biomolecules, such as FFAs and DNA (Fujita et al., 2010; Mello et al., 2016). As a result, by-products of reactive oxygen species (ROS)-induced damage such as 4-hydroxynonenal and 3-nitrotyrosine was significantly enhanced in the plasma and liver, respectively in NAFLD/NASH patients (Loguercio et al., 2001; Kojima et al., 2007).
The Role of Hepatic ER Stress
Similar to oxidative stress, upregulated hepatic ER stress is closely associated with NASH (Lake et al., 2014). Kuo et al. (2012) provided evidence that an ER stress response is provoked in response to an increase in FFA accumulation in hepa. Consistent with this observation, Xiao et al. (2013) demonstrated that mice deficient in activating transcription factor 4 (ATF4), a major ER-stress mediator, were protected from high fructose diet-induced hepatic steatosis, highlighting the necessity of the ER stress response in driving the accumulation of fat in the liver. In general, when the concentration of intracellular unfolded proteins reaches a critical threshold, the ER initiates the unfolded protein response (UPR) in an attempt to maintain normal cell function (Lebeaupin et al., 2018). The UPR encompasses three main pathways: reduced protein translation by activating protein kinase RNA-like endoplasmic reticulum kinase-eukaryotic initiation factor 2 alpha (PERK-eIF2α) signaling (Harding et al., 1999); enhancing protein folding via the inositol-requiring enzyme 1 (IRE1) and X-box binding protein 1 (XBP1) signaling cascade (Ning et al., 2011), and inducing apoptosis and ER-associated degradation by activating transcription factor 6 (ATF6) associated pathway (Lebeaupin et al., 2018). However, prolonged unresolved ER stress is thought to induce the expression of the pro-apoptotic transcription factor C/EBP Homologous Protein (CHOP) (Zinszner et al., 1998). In an ER-stress induced NASH model induced by major urinary protein urokinase-type plasminogen activator (MUP-uPA, discussed in detail later), animals exhibit high levels of XBP1s as well as CHOP (Nakagawa et al., 2014). Although apoptosis was elevated together with increased CHOP expression, liver injury was not ameliorated in mice with CHOP ablation (Soon et al., 2010; Nakagawa et al., 2014). It is possible that CHOP is a downstream product of ER stress but not an active driver of liver injury in NAFLD/NASH. Clinical studies displayed varying degrees of ER stress gene and protein expression in NASH patients (Puri et al., 2008; Lake et al., 2014). It is noteworthy that both studies showed high variability within the NASH patient cohort, with Puri et al. (2008) having 21 NASH patients and Lake et al. (2014) having 13 NASH patients. Given the complexity of NAFLD/NASH pathology, the expression levels of ER stress mediators may be influenced by many different factors. The different results observed in these studies may be attributed to patient variability. Recruitment of a larger patient cohort and effective patient stratification may provide a better understanding of the drivers underpinning ER-stress-driven liver injury.
The Role of c-Jun N-Terminal Kinase Signaling
There are numerous in vitro and in vivo studies highlighting the pleiotropic role of intracellular signaling pathways such as JNK in the development of NASH (Gehrke and Schattenberg, 2020). In particular, JNK activation by TNF-α has been implicated in mediating insulin resistance by interfering with the IRS signaling pathway (Aguirre et al., 2000). In addition, FFA-induced JNK activation resulted in cell apoptosis in both cell line cells and primary mouse hepatocytes (Malhi et al., 2006). In contrast, hepatocytes that are isolated from JNK1 deficient mice had reduced apoptosis compared to cells from wild type animals when exposed to FFAs (Malhi et al., 2006). In support of this finding, mice genetically deficient in JNK1 exhibit attenuated hepatic steatosis compared to their wild type counterparts in a dietary model of NASH (Schattenberg et al., 2006). Interestingly, it has been reported that JNK1 deficiency in the adipose tissue indeed protects animals against hepatic steatosis (Sabio et al., 2009). However, JNK1 deficiency in the liver gave rise to glucose intolerance and insulin resistance in these animals with diet-induced NASH (Sabio et al., 2009). Therefore, future therapeutic targeting of the JNK pathway may need to take into consideration the differential effects that JNK1 blockade might have at different target organs.
The Role of Hepatic Inflammation
Inflammation is one of the features that distinguishes NASH from fatty liver disease (Kleiner et al., 2005). Although the exact mechanism that triggers inflammation in NASH patients is not well characterized, several key contributing factors have been suggested (Kleiner et al., 2005; Younossi et al., 2011). Adipose tissue-derived cytokines, such as TNF-α, are suggested to contribute to hepatic inflammation (Hotamisligil et al., 1993; Tilg and Moschen, 2006). In addition, gut dysbiosis caused by long-term HFD consumption can result in a leaky gut, enabling endotoxins, such as lipopolysaccharide to travel to the liver, triggering/enhancing liver inflammation during NASH (Hildebrandt et al., 2009; Ogawa et al., 2018). Moreover, metabolism associated molecular patterns (MAMPS) including FFAs and cholesterol have been reported to initiate inflammasome-induced inflammatory cell death in hepatocytes (Csak et al., 2011; Mridha et al., 2017; Wang et al., 2020). The resulting danger-associated molecular patterns (DAMPs) from inflammatory cell death can stimulate the activation of liver resident macrophages known as Kupffer cells (Seki and Brenner, 2008; Baffy, 2009). Activated Kupffer cells secrete TNF-α (Tosello-Trampont et al., 2012), a pro-inflammatory cytokine that mediates pleiotropic actions including amplifying insulin resistance and regulating NF-κB activation (Schütze et al., 1995; Peraldi et al., 1996). NF-κB has been suggested as a key player in exacerbating liver inflammation, as phosphorylated NF-κB levels are also elevated in preclinical models of NASH (dela Peña et al., 2005; Nakagawa et al., 2014). Moreover, pharmacological inhibition of NF-κB activation significantly reduced the expression of NF-κB downstream inflammatory genes in animal models of NASH (Leclercq et al., 2004). In addition to NF-κB activation, TNF-α also induces the expression of monocyte chemoattractant protein-1 (MCP-1) which is reported to be elevated in NASH patients (Haukeland et al., 2006; Greco et al., 2008; Tosello-Trampont et al., 2012). MCP-1 and its corresponding receptor, C-C chemokines receptor type 2 (CCR2), are important for the hepatic recruitment of Ly6C+ monocytes, which can amplify inflammation as they mature into macrophages (Baeck et al., 2012; Miura et al., 2012). In addition to monocytes and Kupffer cells, neutrophil-secreted myeloperoxidase has been proposed to exacerbate liver inflammation by generating oxidative stress (Rensen et al., 2009). In agreement with these findings, Zang and colleagues (2015) discovered that neutrophils are responsible for contributing to liver inflammation during early stages of NASH. Animals with Ly6G+-neutrophil depletion in the early stages of NASH displayed significantly reduced serum ALT, as well as reduced pro-inflammatory gene expression compared to diseased mice (Zang et al., 2015). Recent studies demonstrate that patients who progress to NASH exhibit a high level of natural killer T-cells and CD8+-T-cells (Tajiri et al., 2009; Gadd et al., 2014). A potential role for T-helper cells in mediating NASH progression and initiation of fibrogenesis has indeed been proposed (Rolla et al., 2016).
The Role of Hepatic Fibrosis
Hepatic fibrosis one of the hallmarks of NASH, is characterized by extensive accumulation of connective tissue which following extensive tissue damage (Kleiner et al., 2005). The process of fibrogenesis in the liver is thought to be mainly regulated by hepatic stellate cells (HSCs), a type of liver progenitor cell that is quiescent under physiological conditions (Tsuchida and Friedman, 2017). HSC can be activated to produce collagen I in response to elevated ER stress by overexpressing XBP1 (Kim et al., 2016). Apart from ER stress, liver-specific overexpression of NACHT, Leucine-rich-repeat and pyrin domain-containing protein 3 (NLRP3) induced marked HSC activation and fibrosis, indicating a possible role of inflammatory cell death in inducing HSC activation (Wree et al., 2014). Moreover, it has been elucidated that engulfment of apoptotic cell bodies induces HSC activation (Canbay et al., 2003). The activated HSC transforms from a dormant cell into an active myofibroblast which is characterized by increased production of collagen I, collagen III and transforming growth factor-β (TGF-β) (Dooley et al., 2000; Inagaki et al., 2001; Zhan et al., 2006). More importantly, TGFβ acts in a paracrine/autocrine fashion to activate quiescent HSCs, while also amplifying collagen deposition from activated HSCs (Hellerbrand et al., 1999). The increased collagen I and III gradually alters the composition of liver extracellular matrix and gave rise to tissue scarring (Maher and McGuire, 1990; Mak and Mei, 2017). If liver injury is not resolved, the continuous supply of inflammatory cytokines and apoptotic cell bodies will perpetuate the fibrogenic actions of HSC and promote further tissue remodeling (Bachem et al., 1992). When the collagen deposition is evident in most of the liver tissue, the disease has officially progressed beyond NASH to cirrhosis (Kleiner et al., 2005). Moreover, results from longitudinal studies suggested that NAFLD/NASH patients with severe liver fibrosis have increased risk of HCC and mortality compared to those with mild fibrosis (Ekstedt et al., 2015; Alexander et al., 2019).
Preclinical Models of Non-Alcoholic Steatohepatitis
NASH is a disease that encompasses a broad array of systemic metabolic disruptions as well as liver-specific abnormalities induced by a multitude of processes (Buzzetti et al., 2016). The complex nature of the disease has made it challenging to recapitulate the full spectrum of the disease phenotype in animal models (Friedman et al., 2018a). The currently established models are broadly categorized into three main areas: dietary-induced, diet-toxin-induced and diet-genetically mutated models (Table 2).
TABLE 2
Summary table of commonly used preclinical models of NASH.
Summarized key characteristics of NASH. ↑: modest increase; ↑↑: marked increase; ✕: did not exhibit this feature; *: shortcomings of this model mentioned in the text; HFD: High fat diet; WD: Western diet; MCD: methionine choline deficient diet; MD: methionine deficient diet; CD: choline deficient diet; AMLN (Amylin NASH diet): HFD containing cholesterol supplemented by high fructose and sucrose; STAM: streptozotocin + HFD; DEN: diethylnitrosamine; ob/ob: leptin-deficient mice; db/db: leptin-receptor-deficient mice; foz/foz: Alms1 genetic mutation mice; ApoE−/−: Apolipoprotein E Knock out; hApoE-KI: human Apolipoprotein E Knock in; PNPLA3I148M KI: 148 Isoleucine to Methionine protein variant (I148M) of patatin-like phospholipase domain-containing three knock-in mice; MUP-uPA, mice overexpressing urokinase plasminogen activator introduced into hepatocytes via adeno-associated virus.
Genetically Induced Non-Alcoholic Steatohepatitis Models
Genetically-induced obese mouse models of diabetes and pre-diabetes, such as ob/ob, db/db and foz/foz exhibit are also being used as models of NASH/NAFLD as they exhibit obesity, insulin resistance and hyperglycaemia (Bleisch et al., 1952; Marshall et al., 2011).
ob/ob and db/db Models
The ob gene transcribes leptin, an adipocyte hormone involved in the regulation of food intake and insulin sensitivity (Friedman et al., 1991). In ob/ob mice, there is a deficiency in the production of functional leptin (Friedman et al., 1991; Zhang et al., 1994). Therefore, animals with this genetic alteration develop hyperphagia and insulin resistance (Friedman and Halaas, 1998). Sutter and colleagues (2015) demonstrated that ob/ob mice fed with an HFD rapidly gained weight and developed insulin resistance and most of the liver NASH features except fibrosis. Complementing this finding, another study by Leclercq et al. (2002) reported that leptin is essential to promote liver fibrosis. Thus, the ob/ob model is deemed unsuitable for studying NASH due to this paradoxical shortcoming. Having a similar metabolic phenotype to the ob/ob animals, the db/db model exhibits leptin resistance caused by premature termination of leptin receptor transcription (Chen et al., 1996). Disruption in transcription gave rise to faulty leptin receptors which precluded normal leptin signaling (Chen et al., 1996). The db/db model gave rise to severe obesity, glucose intolerance and liver steatosis. Nonetheless, liver inflammation and fibrosis in this model were reported to be mild (Trak-Smayra et al., 2011). Several studies use db/db mice coupled with a methionine-choline-deficient (MCD) diet feeding to induce more severe liver injury (Sahai et al., 2004; Rinella et al., 2008; Staels et al., 2013). More importantly, it has been proposed that whilst ob/ob and db/db mice can be good models for studying obesity and insulin resistance, both ob and db mutations are rare in humans, therefore, these mice may be less clinically-relevant as animal models of NASH (Carlsson et al., 1997; Wang et al., 2014).
foz/foz Model
The foz/foz mice have also been used as an obese and diabetic NASH model (Van Rooyen et al., 2011; Haczeyni et al., 2017; Mridha et al., 2017). The foz/foz mice were generated from a recessive mutation on the AlstrÖm syndrome 1 (Alms1) gene which encodes proteins involved in ciliary function (Marshall et al., 2011). Mice that have genetic mutation typically develop hyperinsulinemia, hyperglycaemia, and hypercholesterolemia together with liver inflammation (Van Rooyen et al., 2011). Nevertheless, obeticholic acid (OCA), an FDA-approved drug for NASH (discussed in detail below), did not improve liver histology of foz/foz mice, like it did in NASH patients (Haczeyni et al., 2017; Younossi et al., 2019). This finding raises a question regarding the use of animal models that are merely a “phenocopy” of human NASH, as humans do not normally develop NASH due to the rare autosomal recessive Alms1 mutation (Marshall et al., 2011).
Apolipoprotein E Knock-Out and Knock-In Models
ApoE is a multifunctional protein that plays an important role in lipid transport, abnormality in the type 2 ApoE results in type III hyperlipoproteinemia (Huang and Mahley, 2014). Mice that are ApoE deficient (ApoE−/−) are commonly used as an animal model for atherosclerosis (Song et al., 2011). Although weight gain and abnormal glucose tolerance can be achieved in ApoE−/− animals, the model by itself only gave rise to negligible hepatic steatosis, inflammation and fibrosis (Schierwagen et al., 2015). Only when ApoE deficiency is combined with a high caloric diet, or a MCD diet does it then induce extensive liver injury (Schierwagen et al., 2015; Zang et al., 2015). Furthermore, hApoE2-knock-in mice, where the human ApoE2 gene replaced the murine gene, is used as another model for NASH studies (Shiri-Sverdlov et al., 2006; Staels et al., 2013). Interestingly, despite inflammatory and fibrotic genes both being upregulated in the liver, only mild steatosis was observed in the hApoE2 mice (Shiri-Sverdlov et al., 2006). Noteworthily, there are few clinical studies examining the association between ApoE2 polymorphism and NAFLD. In one clinical study 57 NAFLD patients from a Turkish ethnic background showed no significant association between ApoE2 and NAFLD (Sazci et al., 2008). Results from this study are consistent with a case controlled study by Demirag et al. (2007) involving 237 NAFLD patients, where subjects with ApoE2 polymorphism showed a significant association with dyslipidaemia but not with NAFLD. The ability to generate a model with dyslipidaemia was what made ApoE−/− and hApoE2 KI mice a potential model for the study of NAFLD/NASH (Sazci et al., 2008). However, Severson et al. (2016) in their systematic clinical review concluded that ApoE polymorphism may not play as important a role as other genetic polymorphisms such as PNPLA3.
Phospholipase Domain-Containing-3 Variant-Knock-In Model
Recently, there have been attempts to develop a fatty liver disease mouse model by introducing the human PNPLA3 polymorphism in mice to mimic human genetic mutant-induced NASH (Smagris et al., 2015). However, mice with human PNPLA3 variant knock-in (KI) only showed elevated hepatic fat when fed a HFD, whereas the extent of liver inflammation and fibrosis in PNPLA3 variant KI mice was comparable to wildtype animals (Smagris et al., 2015). It has been suggested that this model may be suitable for the study of fatty liver disease and hepatic insulin resistance (Kumashiro et al., 2013). Further studies of the PNPLA3 polymorphism in the context of NASH in humans and its mechanism of action are required, to confirm whether murine models with this genetic mutation are good preclinical models of NASH.
Major Urinary Protein Urokinase-Type Plasminogen Activator Model
A relatively new NASH model has been developed by transiently upregulating ER stress in the liver by delivering major urinary protein urokinase plasminogen activator (MUP-uPA) into the hepatocytes via adeno associated virus coupled with a HFD (Nakagawa et al., 2014; Febbraio et al., 2019). The method of generating MUP-uPA-transgenic mice was first described by Weglarz et al. (2000). The MUP-uPA transgenic mice is generated by delivering adeno-associated virus containing the uPA protein specifically to hepatocytes. This results in an accumulation of uPA protein in the ER of hepatocytes and thus transiently upregulates ER stress in the hepatocytes (Nakagawa et al., 2014). The MUP-uPA mice placed on a HFD exhibited significantly upregulated liver injury markers of NASH, including ER-stress, fibrosis and inflammation at 24 weeks (Nakagawa et al., 2014; Lebeaupin et al., 2018). Furthermore, MUP-uPA mice spontaneously progress from NASH to HCC by 32 weeks of age, exhibiting markers frequently observed in humans HCC tissues such as alpha fetoprotein and p62 (Nakagawa et al., 2014). In support of this finding, Shalapour et al. (2017) observed an elevation of immunosuppressive IgA+ cells, interleukin 10 and programmed cell death ligand-1 in both NASH-derived HCC patients and the MUP-uPA mice that were placed on HFD. More importantly, some degree of transcriptomic alignments were observed between human NASH/HCC subjects and the MUP-uPA model, highlighting the clinical relevance of this model (Febbraio et al., 2019).
Diet-Induced Models
Other than genetic predisposition, a diet high in fat and sugar is one of the major factors that is strongly associated with the development of NASH in humans (Faeh et al., 2005; Luukkonen et al., 2018). Diet-induced NASH models include, but are not limited to, MCD diet (Rinella et al., 2008), HFD (HFD) (Kohli et al., 2010), western diet (WD) (Bruckbauer et al., 2016) and Amylin diet (AMLN) (Clapper et al., 2013; Asgharpour et al., 2016) with only the most widely used models summarized here. MCD is a dietary model used for inducing NASH-like liver features with 40% of sucrose and 10% energy derived from lipids but is deficient in methionine and choline (Anstee and Goldin, 2006). Methionine and choline are essential nutrients for growth and development in humans (Zeisel and Da Costa, 2009). Feeding a diet which is deficient of these two nutrients can lead to the rapid development of hepatic lesions such as hepatic steatosis, inflammation and fibrosis (Oz et al., 2008). Nevertheless, Rinella et al. (2004) showed significantly lower body weight and unaltered plasma insulin in MCD diet-fed mice, highlighting the absence of key metabolic characteristics of NASH such as insulin resistance and obesity in this model. Overall, the field has reached a consensus that the MCD can exhibit histological features that are not only similar to, but are equally severe, as those in human NASH, though key metabolic features are missing (Leclercq et al., 2000; Rinella and Green, 2004; Rinella et al., 2008; Wortham et al., 2008). There have been attempts to use only methionine-deficient (MD) or choline deficient (CD) diets to induce NASH (Caballero et al., 2010). Despite animals on MD and CD had reduced weight loss, metabolic features that are present in NASH in human was not observed (Caballero et al., 2010; Febbraio et al., 2019). To develop a NASH model that mimics both systemic and hepatic pathology, many attempts have been made using varying degrees of fat (∼40–70% energy derived from fat) and 0.1–2% cholesterol in the diet (Anstee and Goldin, 2006). The use of high-fat content alone is normally referred to as the HFD model (Kohli et al., 2010), whereas WD represents a type of HFD with the addition of cholesterol (Machado et al., 2015; Bruckbauer et al., 2016). Models that received HFD or WD feeding develop weight gain, insulin resistance and hepatic steatosis which are concordant with the insulin resistance and hyperglycaemia of humans who have NASH (Zheng et al., 2008; Kohli et al., 2010). However, in some cases, HFD and WD models are reported to have minimal fibrosis (Febbraio et al., 2019). In recent years, the ALMN diet-induced NASH model developed by Amylin Pharmaceuticals (hence ALMN model), composed of 40% lipids, 2% cholesterol and water supplemented with fructose, has been reported to display both systemic and liver-specific characteristics of human NASH at 28–30 weeks of AMLN diet feeding (Clapper et al., 2013; Boland et al., 2019). Overall, NASH animal models induced by dietary interventions alone require a long time to achieve a mild to moderate NASH phenotype. Characteristics such as moderate to severe liver injury and fibrosis may take up to 20–30 weeks of dietary feeding (Charlton et al., 2011; Clapper et al., 2013). However, the longer the study period of animal experiments, the higher the chance of animals dying due to aging and age-related complications. In addition, such models are resource-draining and less time-effective. Thus, other alternative models are being explored in the attempt to induce severe liver injury in a shorter period.
Diet and Toxin-Induced Models
To increase the severity of liver injury in rodent NASH models, toxins such as streptozotocin (STZ) (Fujii et al., 2013), diethylnitrosamine (DEN) (Park et al., 2010) and carbon tetrachloride (CCL4) (Tsuchida et al., 2018) have been added to the modified diet.
STAM Model
In the STAM model, a single dose (200 μg) of the pancreatic ß-cell toxin STZ is administered subcutaneously to 2 day old neonatal C57BL/6 mice followed by 4–6 weeks of HFD feeding (Fujii et al., 2013; Saito et al., 2017; Middleton et al., 2018). By destroying pancreatic ß-cells, the STZ-induced hyperglycaemia is coupled with HFD to drive liver damage feeding (Fujii et al., 2013; Saito et al., 2017; Middleton et al., 2018). Although the STAM model gives rise to liver steatosis, inflammation and fibrosis, these animals develop conditions that resemble type 1 rather than type 2 diabetes, as indicated by the overt hyperglycaemia (blood glucose 600 mg/dl) and a lack of hyperinsulinemia, a sign of insulin resistance (plasma insulin <0.5 ng/ml) (Saito et al., 2017). Although the STAM model has been discussed in other reviews, consideration of the combination of STZ and HFD (STZ + HFD) where STZ was delivered at a later stage of the animal’s life is less frequently noted (Friedman et al., 2018a; Farrell et al., 2019; Oligschlaeger and Shiri-Sverdlov, 2020). FVB/N mice which received STZ (55 mg/kg) at 6 weeks old coupled to a HFD displayed hyperinsulinemia (Tate et al., 2019). Moreover, rats fed with a HFD before receiving STZ injection also showed hyperinsulinemia (Reed et al., 2000). The contradictory findings in the literature may be partially explained by variable susceptibility toward STZ in different mouse strains (FVB/N Vs C57BL/6) (Saito et al., 2017; Tate et al., 2019; Marshall et al., 2020). Noteworthily, the STZ model has also been criticized for its ability to damage other organs such as the kidney and the liver via DNA alkylation (Lenzen, 2008). STZ-induced liver injury is thought to be direct rather than secondary to the natural course of NASH-induced liver injury (driven by T2D and obesity) which is one of the major issues limiting the utility of the STAM model (Middleton et al., 2018; Farrell et al., 2019).
Diethylnitrosamine + HFD Model
The hepatic carcinogen DEN has been shown to induce severe hepatic injury, by inducing mutagenic DNA damage and upregulating ROS production (Williams et al., 1996). After receiving 25–30 mg/kg of DEN at 14 days old, rodents which were fed a HFD for 4–6 weeks were reported to display severe liver injury characterized by elevated inflammatory gene expression and hepatocyte ballooning (Wang et al., 2009; Park et al., 2010). An important caveat of this model is that DEN + HFD-treated animals rapidly develop hepatocellular carcinoma (HCC) due to the potential carcinogenic effects of DEN (Wang et al., 2009).
Western Diet + Carbon Tetrachloride Model
Another NASH model is induced by the hepatotoxin CCL4 which rapidly causes severe liver inflammation and fibrosis (Hellerbrand et al., 1999; Baeck et al., 2012). The use of CCL4 coupled with a WD is also reported to give rise to weight gain and severe liver histological features similar to those of NASH patients (Tsuchida et al., 2018). Although this model is capable of inducing stage 3 fibrosis after 12 weeks of HFD feeding and CCL4 treatment, CCL4 induces severe liver injury via oxidative DNA damage, which is distinctly different from the natural course of NASH (Alkreathy et al., 2014; Calzadilla Bertot and Adams, 2016).
NASH is a heterogenous disease characterized by both liver injury and systemic metabolic disruptions (Friedman et al., 2018a). Currently, although diet-induced models such as HFD and ALMN diet-induced NASH models are time consuming and only show mild liver injuries, they recapitulate the natural course of NASH development in humans (Friedman et al., 2018a). The MUP-uPA model, although not widely-used at the moment, does mimic aspects of NASH in humans as oppose to the MCD diet and some toxin-induced NASH models (Febbraio et al., 2019). A summary of all the frequently used animal models of NASH is provided in Table 2.
Current Interventions in Non-Alcoholic Steatohepatitis Management
Lifestyle Modification
Management of fatty liver diseases has been addressed by lifestyle modifications, including regular physical exercise and consuming a hypocaloric diet (Vilar-Gomez et al., 2015). Often, a reduction of ≥5–10% of the subject’s body weight is required to achieve attenuation of NASH (Vilar-Gomez et al., 2015; Younossi et al., 2018). Nevertheless, a study also noted a lack of patient compliance with the proposed exercise and nutritional recommendations after the study period (Eckard et al., 2013). More importantly, lifestyle changes alone are insufficient to stop disease progression, especially for patients who are at later stages of the disease where there are ongoing liver inflammation and fibrosis (Promrat et al., 2010). Patients with progressed fibrosis have an increased risk of developing cirrhosis and liver failure, and it is currently the second leading cause of liver transplant (Wong et al., 2015).
Pharmacological Treatments
Apart from lifestyle modifications, obeticholic acid (OCA), originally approved for the treatment of primary biliary cholangitis (PBC), is the only FDA-approved treatment for NASH (Vilar-Gomez et al., 2015; Younossi et al., 2019). OCA is a farnesoid X receptor (FXR) agonist which regulates the expression of transcription factors that reduce bile acid synthesis and hepatic steatosis (Pellicciari et al., 2002; Jiao et al., 2015). In the FLINT trial (NCT01265498), OCA has been shown to improve liver inflammation with no worsening of liver fibrosis (Neuschwander-Tetri et al., 2015). In the recent 18 months phase III clinical trial REGENERATE (NCT02548351), 23% (71/308) of the patient cohort who received 25 mg daily achieved reduction of NAS by at least one score without worsening of fibrosis compared to 12% (37/311) in the placebo group (Eslam et al., 2019). The trial results enabled OCA to be granted accelerated approval from the FDA (Younossi et al., 2019). Although, there were 19 deaths observed in PBC patients who received obeticholic acid due to incorrect dosing (Eslam et al., 2019). Within in the 19 cases of death, 8 cases were reported. The cause of death for seven patients were due to the worsening of PBC, and the other patient due to cardiovascular complications (FDA website). A safety warning has been issued by the FDA for patients and health professionals regarding the use of obeticholic acid for its potential effect of worsening liver disease in patients.
Vitamin E is an anti-oxidant which acts by reducing the ROS and inflammation-induced liver damage (Singh et al., 2005). Results from a 96 weeks multicenter, placebo-controlled trial showed improvements of liver histology such as inflammation, steatosis and ballooning in 43% (34/84) of the non-diabetic NAFLD subjects treated with 800IU vitamin E daily compared to 19% (16/83) treated with placebo (Sanyal et al., 2010). However, concerns that long-term vitamin E use may be associated with hemorrhagic stroke are also highlighted in the study (Sanyal et al., 2010). Alarmingly, in a separate study where vitamin E (400IU/d) was administered for 7–11 years showed increased risk of prostate cancer was identified in healthy men with long-term vitamin E treatment (Klein et al., 2011).
Pioglitazone primarily targets the PPARγ receptor which ameliorates insulin resistance, an independent predictor of NASH (Belfort et al., 2006). In a 96 weeks placebo-controlled trial, 30 mg of pioglitazone daily also improved the liver histology in 34% (27/80) of non-diabetic NASH patients, although an average of 4.7 kg weight gain was reported in the treatment group (Sanyal et al., 2010; Chalasani et al., 2012). Whilst a recent 18 months study showed that pioglitazone treatment combined with a hypocaloric diet (500 kcal/day deficit) improved liver histology in 51% (26/50) of patients with NASH and diabetes, its efficacy still needs to be evaluated in larger patient cohorts (Cusi et al., 2016).
Currently, both pioglitazone and vitamin E are currently prescribed in a case-by-case manner, as the risk of all-causes of liver-related mortality of these two drugs still need to be evaluated in a larger patient cohort (Sanyal et al., 2010; Chalasani et al., 2012; Younossi et al., 2018). A summary of NASH/NAFLD-related drug treatment is displayed in Table 3.
TABLE 3
| Drug name | Target | Mechanism of action | Trial phase NCT no | Trial population | Outcome (estimated completion date) | References |
|---|---|---|---|---|---|---|
| Aramchol | SCD1 inhibitor | ↓DNL synthesis | Phase III NCT04104321 | NASH | TBD | FDA website |
| June 2022 | ||||||
| ↓Steatosis | Phase II NCT01094158 | NAFLD/NASH/MS | Complete | (Safadi et al., 2014) | ||
| Reduced the percentage of liver fat at | ||||||
| Cenicriviroc | CCR2/5 dual inhibitor | ↓Inflammation | Phase III NCT03028740 | NAFLD/NASH/MS | TBD | FDA website |
| October 2021 | ||||||
| Phase II NCT02217475 | NAFLD/NASH/MS | Complete | (Friedman et al., 2018b) | |||
| Reduced fibrosis with no worsening of inflammation and steatosis | ||||||
| Elafibranor | PPARα/δ agonist | ↓Inflammation | Phase II,I NCT02704403 | NAFLD/NASH/MS | Complete (unsuccessful) | (GENFIT S.A, 2020) |
| High placebo effect, no difference between placebo arm and treatment arm | ||||||
| ↓Fibrosis | Phase IIb, NCT01694849 | NAFLD/NASH/MS | Complete | (Ratziu et al., 2016) | ||
| Well tolerated in patients. No significant change between placebo and treatment | ||||||
| ↓Insulin resistance | Phase II, NCT01275469 | Obese/pre-diabetic | Complete | (Cariou et al., 2011) | ||
| Improved insulin resistance, decreased fasting TG and blood glucose. Increased HDL | ||||||
| Emricasan | Pan-caspase inhibitor | ↓Inflammation | Phase II NCT02686762 | NAFLD/NASH/MS | Complete (unsuccessful) | (Ratziu et al., 2012; Harrison et al., 2020a) |
| No improvement in liver histology in patients with NASH, and may exhibit worsened fibrosis and ballooning | ||||||
| GR-MD-02 | Galectin-3 inhibitor | ↓Fibrosis | Phase II NCT02462967 | NASH, cirrhosis, and portal hypertension | Complete (unsuccessful) | (Chalasani et al., 2020) |
| No improvement in hepatic venous pressure and liver histology compared to placebo | ||||||
| Liraglutide | GLP-1 agonist | ↓Insulin resistance | Phase II NCT01237119 | NASH/NAFLD/MS | Complete (Preparing for phase III) | (Armstrong et al., 2016) |
| ↓Blood glucose | Reduced body weight and liver steatosis, and plasma ALT/AST | |||||
| ↓Body weight | ||||||
| Semaglutide | GLP-1 agonist | ↓Insulin resistance | Phase II NCT02453711 | Obese/Type 2 diabetic | Complete (successful, not for NASH) | (Newsome et al., 2019) |
| ↓Blood glucose | Reduced plasma ALT and significant weight loss | |||||
| ↓Body weight | ||||||
| Obeticholic acid | FXR agonist | ↓Bile acid synthesis | FDA-approved PHASEIII NCT02548351 | NASH/NAFLD/MS | Complete (successful) | (Eslam et al., 2019) |
| ↓Inflammation | 23% patients in the 25 mg group had improved fibrosis, but had increased pruritus | |||||
| ↓Fibrosis | ||||||
| Selonsertib | ASK1 inhibitor | ↓Cell apoptosis | Phase III NCT03053050NCT03053063 | NASH/NAFLD/MS | Complete (unsuccessful) | (Harrison et al., 2020b) |
| ↓Inflammation | Neither trial improved fibrosis without worsening of NASH | |||||
| ↓Fibrosis | ||||||
| Pioglitazone | PPARγ agonist | ↓Insulin resistance | NCT00063622 Phase III | Non-diabetic NAFLD | Completed (successful) | (Sanyal et al., 2010) |
| Lowered plasma ALT/AST, liver inflammation and steatosis | ||||||
| Vitamin E | Antioxidant | ↓Oxidative stress | NCT00063622 Phase III | Non-diabetic NAFLD | Completed (successful) | (Sanyal et al., 2010) |
| Lowered plasma ALT/AST, liver inflammation and steatosis |
Summary table of drugs for NASH treatment.
MS: metabolic syndrome; TG: triglyceride; HDL: high-density lipoprotein; SCD1: stearoyl CoA desaturase 1, CCR2/5: C-C chemokines receptor type 2/5, GLP-1: glucagon-like peptide-1, FXR: Farnesoid X receptor, ASK1: Apoptosis signal-regulating kinase 1, PPARγ: peroxisome proliferator-activated receptor γ.
Pharmacological Treatments Under Clinical Trial
Most phase IIb and phase III clinical trials of NASH generally have two primary clinical endpoints: 1) resolution of NASH without worsening of liver fibrosis and/or 2) improving liver fibrosis without worsening of NASH (clinicaltrials.gov). Resolution of NASH generally refers to a reduction of NAS, whereas improvement of liver fibrosis refers to reduction in fibrosis scores by liver histology (Kleiner et al., 2005). Many pharmacological treatments are undergoing clinical trials (Smeuninx et al., 2020). Drug candidates from their corresponding pharmaceutical companies and the pathways they are targeting are presented in Figure 3.
FIGURE 3
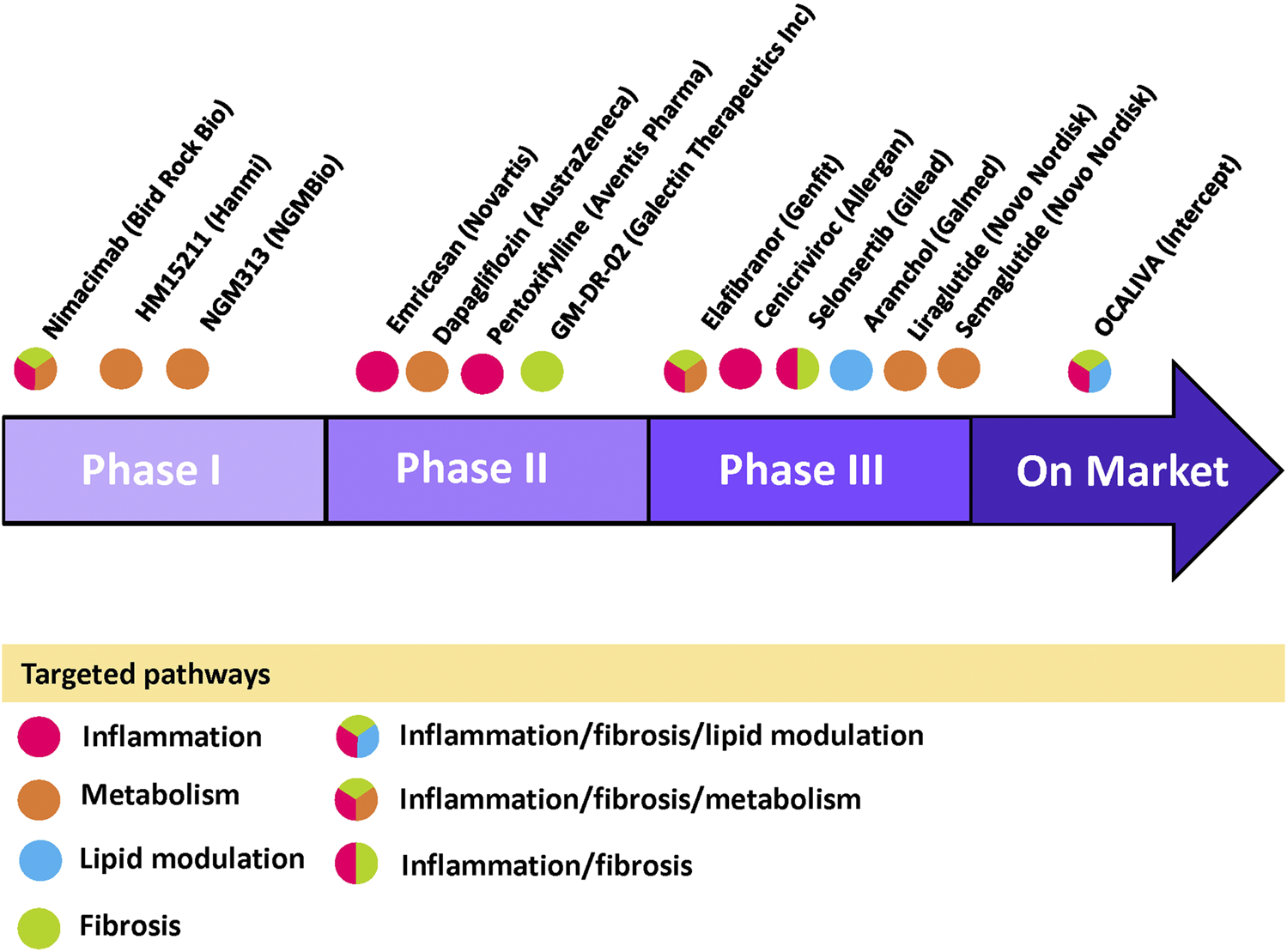
Current NASH/NAFLD pipeline drugs with targeted pathways Pipeline drugs labeled with its pharmaceutical company are placed in their corresponding trial phases. The circle color indicates its targeted pathway(s) as shown in the legend within the figure. Information are gathered from clinicaltrials.gov and pharmaceutical company websites. Figure is designed and drawn using Microsoft Powerpoint.
Glucagon-Like Peptide 1 Receptor Agonists
Synthetic long-acting glucagon-like peptide 1 (GLP-1) receptor agonists such as liraglutide and semaglutide were originally approved for treatment of type 2 diabetes (Pearson et al., 2019). Recently, both liraglutide and semaglutide have gained attention for their efficacy in attenuating insulin resistance, hyperglycemia and liver lipotoxicity in NASH patients (Armstrong et al., 2013; Armstrong et al., 2016). GLP-1, a hormone secreted by the small intestine after a meal, has been observed to restore insulin sensitivity and attenuate hyperglycemia in humans (Garber et al., 2009). Treatment of NASH with GLP-1 receptor agonists was reported to ameliorate liver steatosis in both preclinical and clinical studies (Ding et al., 2006; Armstrong et al., 2013). Novo Nordisk has completed its 48 weeks phase II clinical trial (NCT02970942) assessing the efficacy of 1.8 mg liraglutide given daily and it is preparing for its phase III clinical trial. Moreover, semaglutide, a structurally-related analogue of GLP-1 receptor agonist, significantly reduced body weight and liver enzymes in obese and T2D patients (Newsome et al., 2019). Information from a 72 weeks multicenter phase II trial for semaglutide (NCT02970942) showed that, 33 of 56 NASH patients who received 0.4 mg semaglutide had NASH resolution compared to 10 of 58 patients on placebo (Newsome et al., 2020). Semaglutide was well tolerated with the reported adverse event being gastrointestinal events (Newsome et al., 2020).
DNL Enzyme Inhibitors
De novo lipogenesis pathway enzymes are another popular target for pipeline drugs. Aramchol, is a synthetic molecule created by conjugating bile acid and arachidic acid (Safadi et al., 2014). Aramchol acts by inhibiting the SCD-1 enzyme, which is a key rate limiting enzyme that is responsible for converting FA into TG (Softic et al., 2016). Aramchol has displayed antioxidative, and anti-fibrotic effects in animal studies whilst reducing hepatic steatosis (Iruarrizaga-Lejarreta et al., 2017). A phase II clinical trial for (NCT01094158) showed that NASH patients treated with 300 mg aramchol daily had liver fat reduced by 12.6–22.1% as compared to the placebo group in which case the liver fat increased by 6.4–36.3% (Safadi et al., 2014). Aramchol is currently undergoing phase III trial (NCT04104321) with an estimated completion date of June 2022.
Anti-Inflammatory and Anti-Apoptotic Drugs
Liver inflammation, one of the hallmarks of NASH is also one of the popular targets of pipeline drugs. Several agents targeting inflammation, such as emricasan, a pan caspase inhibitor were observed to be unsuccessful in meeting the primary clinical trial endpoints (Harrison et al., 2020a). Similarly, apoptosis signal-regulating kinase 1 (ASK1) inhibitor selonsertib which acts to prevent hepatocyte apoptosis, displayed promising results in reversing fibrosis and lowering liver inflammation in various preclinical models (Alexander et al., 2019; Challa et al., 2019). However, selonsertib did not reach its primary clinical endpoint, i.e., reversing fibrosis, in either of its phase III trials (STELLAR3: NCT03053050, STELLAR4: NCT03053063). It is worth noting that animal models have limited life-span compared to humans. It is difficulty to accurately determine whether the treatment in animal models is reducing fibrosis or merely delaying its progression.
Cenicriviroc, a CCR2/CCR5 dual-inhibitor is currently undergoing phase III trial with an estimated completion date around October 2021 (NCT03028740). CCR2 is one of the major mechanisms for the recruitment of extrahepatic inflammatory cells (Karlmark et al., 2009; Miura et al., 2012). Inhibition of CCR2 has been shown to exhibit anti-inflammatory effects in the liver in animal studies (Baeck et al., 2012; Krenkel et al., 2018). More importantly, 20% (23/145) of patients receiving 150 mg of cenicriviroc daily had reduced fibrosis as opposed to 10% (14/144) of subjects receiving placebo in its phase II clinical trial (Friedman et al., 2018b; Lefere et al., 2020). Overall, the level of inflammation was reduced in patients receiving cenicriviroc compared to controls (Friedman et al., 2018b; Lefere et al., 2020).
PPAR Agonist
Elafibranor, a peroxisome proliferator-activated receptor α/δ (PPARα/δ) dual agonist, was one of the drugs that demonstrated efficacy in improving NASH histology in its phase II trial with 274 patients (Ratziu et al., 2016). Preclinical models used for validation of elafibranor include db/db mice, CCL4-induced liver fibrosis model and hApoE2-KI mice coupled to WD (Staels et al., 2013). PPARα activation improves NASH by increasing FFA ß-oxidation (Stienstra et al., 2007) and lowering inflammation via negative cross-talk with NF-κB (Delerive et al., 1999). PPARδ is responsible for improving hepatic and systemic insulin sensitivity (Lee et al., 2006). Elafibranor attenuated fibrosis in the CCL4-induced liver fibrosis model (Staels et al., 2013; Tsuchida et al., 2018). Moreover, elevated TG, VLDL and inflammatory gene expression exhibited by the WD-fed hApoE2-KI model were also normalized by elafibranor (Shiri-Sverdlov et al., 2006; Staels et al., 2013). However, neither of the CCL4 and the WD + hApoE2-KI models exhibit obesity or hyperglycemia (Shiri-Sverdlov et al., 2006; Tsuchida et al., 2018). Elafibranor’s efficacy in improving glucose homeostasis and insulin sensitivity was separately demonstrated in obese db/db mice (Hanf et al., 2014). Nevertheless, elafibranor did not achieve its primary clinical endpoint in its recently completed 72 weeks phase III trial (RESOLVE-IT: NCT02704403). Results and interim analysis of the RESOLVE-IT trial showed no significant difference between the placebo arm and the treatment arm (120 mg/daily) (GENFIT S.A, 2020). Although, the full dataset will not be released until the second half of 2020 at an international hepatology congress (GENFIT S.A, 2020). While many reasons may have contributed to the failure for candidate drugs to successfully move from pre-clinical studies to the clinic, the use of animal models that are only partially mimicking the NASH phenotype (as highlighted in the models’ section) may be an important factor. Nevertheless, the full dataset from the phase III clinical study of elafibranor will not be released until the second half of 2020 at an international hepatology congress (GENFIT S.A, 2020). Further analysis of the existing clinical data is required to determine the therapeutic effect of long-term elafibranor treatment in a large trial population.
Plant-Based Natural Products
In recent years, there has been growing interests in using plant-based natural products or extracts for the treatment of NASH (Jadeja et al., 2014). Many of these products are widely-used as traditional Chinese medicine and are now being investigated for their potential beneficial effect for NASH in preclinical models (Jadeja et al., 2014; Sun et al., 2017). Plants including Acanthopanax senticosus (Siberian Ginseng) (Park et al., 2006) and glycyrrhizic acid (Sun et al., 2017) showed reduced hepatic de novo lipogenesis and improved insulin sensitivity in mouse models of NASH. Likewise, a series of natural-product-derived analogues are also being tested for therapeutic potential in mice with diet-induced NASH and have been shown to lower hepatic lipogenesis as well as ER stress and oxidative stress (Rao et al., 2015; Rao et al., 2019; Rao et al., 2020). Moreover, the use of herbal medicine for the induction of autophagy as a treatment for NASH/NAFLD has been thoroughly reviewed by Zhang et al. (2018). Nevertheless, large-scale clinical trials involving participants from multiple ethnic background are required to confirm the therapeutic potential of plant-based natural products for counteracting NASH.
Perspectives
In order to further bridge the gap between preclinical and clinical studies, animal studies should exploit publicly-available gene profiling data derived from biopsies of healthy controls and NASH patients to verify the animal models (Morrison et al., 2018). The “multiple-omits” approach, incorporating proteomics and lipidomics into the preclinical studies may give an integrated understanding of the animal model and can better assess its translatability as a preclinical model for human NASH (Hasin et al., 2017). Currently, proteomics and lipidomic analyses have often been used for the identification of potential non-invasive biomarkers of NASH in humans, which can also potentially shed light on patient stratification (Puri et al., 2007; Puri et al., 2009; Niu et al., 2019). In addition, validation of non-invasive diagnostic tools such as MRE for its ability to quantify liver fibrosis in a larger patient population would also largely benefit the assessment of clinical trial outcome and longitudinal follow-up studies (Allen et al., 2020).
Furthermore, clinical relevance of a disease model can be further validated by proven efficacy of pharmacological interventions that have shown to be beneficial in clinical trials. Currently, pipeline drugs such as aramchol, GLP-1 receptor agonists, and cenicriviroc as well as OCA have all shown efficacy in improving NASH liver histology in NASH patients (Safadi et al., 2014; Armstrong et al., 2016; Friedman et al., 2018b; Eslam et al., 2019). The development of preclinical models can also leverage on clinical trial results where ideally, the use of these drugs in the animal model should display similar effects to those that are observed in the human studies. The converse is equally important, whereby drugs failing to show efficacy in human trials should ideally also fail in animal models that have both sensitivity and specificity.
Epidemiological studies indicate that the incidence of NAFLD and NASH is estimated to rise to 101 million and 27 million cases respectively by 2030 (Estes et al., 2018). Patients with progressed NASH have an increased risk of developing cirrhosis and liver failure, which is currently the second leading cause of liver transplantation (Wong et al., 2015). The enormous discrepancy between clinical trial results and preclinical data remain a prominent issue in the field of NASH research. The complexity of NASH disease pathology warrants the development of a clinically relevant NASH model for studying the mechanism of pathogenesis and drug evaluation.
Funding
This work was supported in part by the CASS Foundation (CXQ), Victoria Medical Acceleration Grant (RHR), and the Victorian Government's Operational Infrastructure Support Program. RHR was supported by the National Health and Medical Research Council (NHMRC) of Australia (ID1059960, ID1158013), and CXQ is Australia National Heart Foundation Future Fellow. Victorian Medical Research Acceleration Fund (RR, CXQ, AGS).
Statements
Author contributions
CP and CXQ were responsible for the design and draft of the manuscript, RHR, OLW and AGS provided critical review and revision of the manuscript. All authors provide approval for publication of the content.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AguirreV.UchidaT.YenushL.DavisR.WhiteM. F. (2000). The c-Jun NH2-terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser307. J. Biol. Chem.275, 9047–9054. 10.1074/jbc.275.12.9047
2
AjmeraV.PeritoE. R.BassN. M.TerraultN. A.YatesK. P.GillR.et al (2017). Novel plasma biomarkers associated with liver disease severity in adults with nonalcoholic fatty liver disease. Hepatology65, 65–77. 10.1002/hep.28776
3
AlexanderM.LoomisA. K.LeiJ. van. der.Duarte-SallesT.Prieto-AlhambraD.AnsellD.et al (2019). Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: real-world study of 18 million patients in four European cohorts. BMC Med.17, 95. 10.1186/s12916-019-1321-x
4
AlkreathyM. M.KhanA. A.KhanR. R.SahreenS. (2014). CCl4 induced genotoxicity and DNA oxidative damages in rats: hepatoprotective effect of Sonchus arvensis. BMC Compl. Alternative Med.14, 2–8. 10.1186/1472-6882-14-452
5
AllenA. M.ShahV. H.TherneauT. M.VenkateshS. K.MounajjedT.LarsonJ. J.et al (2020). Multiparametric magnetic resonance elastography improves the detection of nash regression following bariatric surgery. Hepatol. Commun.4, 185–192. 10.1002/hep4.1446
6
AnsteeQ. M.,GoldinR. D. (2006). Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol.87, 1–16. 10.1111/j.0959-9673.2006.00465.x
7
AragnoM.TomasinelliC. E.VercellinattoI.CatalanoM. G.CollinoM.FantozziR.et al (2009). SREBP-1c in nonalcoholic fatty liver disease induced by Western-type high-fat diet plus fructose in rats. Free Radic. Biol. Med.47, 1067–1074. 10.1016/j.freeradbiomed.2009.07.016
8
ArmstrongM. J.BartonD.GauntP.HullD.GuoK.StockenD.et al (2013). Liraglutide efficacy and action in non-alcoholic steatohepatitis (LEAN): study protocol for a phase II multicentre, double-blinded, randomised, controlled trial. BMJ Open3, e003995. 10.1136/bmjopen-2013-003995
9
ArmstrongM. J.HullD.GuoK.BartonD.HazlehurstJ. M.GathercoleL. L.et al (2016). Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol.64, 399–408. 10.1016/j.jhep.2015.08.038
10
AsgharpourA.CazanaveS. C.PacanaT.SeneshawM.VincentR.BaniniB. A.et al (2016). A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol.65, 579–588. 10.1016/j.jhep.2016.05.005
11
BachemM. G.MeyerD.MelchiorR.SellK. M.GressnerA. M. (1992). Activation of rat liver perisinusoidal lipocytes by transforming growth factors derived from myofibroblastlike cells. A potential mechanism of self perpetuation in liver fibrogenesis. J. Clin. Invest.89, 19–27. 10.1172/jci115561
12
BaeckC.WehrA.KarlmarkK. R.HeymannF.VucurM.GasslerN.et al (2012). Pharmacological inhibition of the chemokines CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut61, 416–426. 10.1136/gutjnl-2011-300304
13
BaffyG. (2009). Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J. Hepatol.51, 212–223. 10.1016/j.jhep.2009.03.008
14
BleischV. R.MayerJ.DickieM. M. (1952). Familial diabetes mellitus in mice, associated with insulin resistance, obesity, and hyperplasia of the islands of langerhans. Am. J. Pathol.28, 369–385
15
BelfortR.HarrisonS. A.BrownK.DarlandC.FinchJ.HardiesJ.et al (2006). A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med.355, 2297–2307. 10.1056/nejmoa060326
16
BolandM. L.OróD.TølbølK. S.ThraneS. T.NielsenJ. C.CohenT. S.et al (2019). Towards a standard diet-induced and biopsy-confirmed mouse model of non-alcoholic steatohepatitis: impact of dietary fat source. WJG25, 4904–4920. 10.3748/wjg.v25.i33.4904
17
BruckbauerA.BanerjeeJ.FuL.LiF.CaoQ.CuiX.et al (2016). A combination of leucine, metformin, and sildenafil treats nonalcoholic fatty liver disease and steatohepatitis in mice. Int. J. Hepatol.2016, 9185987. 10.1155/2016/9185987
18
BruntE. M.KleinerD. E.WilsonL. A.BeltP.Neuschwander-TetriB. A., and Nash Clinical Research Network (CRN) (2011). Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology53, 810–820. 10.1002/hep.24127
19
BuzzettiE.PinzaniM.TsochatzisE. A. (2016). The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism65, 1038–1048. 10.1016/j.metabol.2015.12.012
20
Caballeria BertotL.AdamsL. (2016). The natural course of non-alcoholic fatty liver disease. IJMS17, 774. 10.3390/ijms17050774
21
CaballeroF.FernándezA.MatíasN.MartínezL.FuchoR.ElenaM.et al (2010). Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem.285, 18528–18536. 10.1074/jbc.m109.099333
22
CanbayA.TaimrP.TorokN.HiguchiH.FriedmanS.GoresG. J. (2003). Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Invest.83, 655–663. 10.1097/01.lab.0000069036.63405.5c
23
CariouB.ZaïrY.StaelsB.BruckertE. (2011). Effects of the new dual PPARα/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care34, 2008–2014. 10.2337/dc11-0093
24
CarlssonB.LindellK.GabrielssonB.KarlssonC.BjarnasonR.WestphalO.et al (1997). Obese (ob) gene defects are rare in human obesity. Obes. Res.5, 30–35. 10.1002/j.1550-8528.1997.tb00280.x
25
ChalasaniN.AbdelmalekM. F.Garcia-TsaoG.VuppalanchiR.AlkhouriN.RinellaM.et al (2020). Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology158, 1334–1345.e5. 10.1053/j.gastro.2019.11.296
26
ChalasaniN.YounossiZ.LavineJ. E.DiehlA. M.BruntE. M.CusiK.et al (2012). The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology55, 2005–2023. 10.1002/hep.25762
27
ChallaT. D.WueestS.LucchiniF. C.DedualM.ModicaS.BorsigovaM.et al (2019). Liver ASK1 protects from non‐alcoholic fatty liver disease and fibrosis. EMBO Mol. Med.11, 1–17. 10.15252/emmm.201810124
28
CharltonM.KrishnanA.VikerK.SandersonS.CazanaveS.McConicoA.et al (2011). Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol.301, G825–G834. 10.1152/ajpgi.00145.2011
29
ChenF.EsmailiS.RogersG. B.BugianesiE.PettaS.MarchesiniG.et al (2020). Lean NAFLD: a distinct entity shaped by differential metabolic adaptation. Hepatology71, 1213–1227. 10.1002/hep.30908
30
ChenH.CharlatO.TartagliaL. A.WoolfE. A.WengX.EllisS. J.et al (1996). Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell84, 491–495. 10.1016/s0092-8674(00)81294-5
31
Chinchilla-LópezP.Ramírez-pérezO.Cruz-ramónV.Canizales-quinterosS.Domínguez-lópezA.Ponciano-rodríguezG.et al (2019). More evidence for the genetic susceptibility of mexican population to nonalcoholic fatty liver disease through PNPLA3. Ann. Hepatol.17, 250–255. 10.5604/01.3001.0010.8644
32
ClapperJ. R.HendricksM. D.GuG.WittmerC.DolmanC. S.HerichJ.et al (2013). Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol.305, G483–G495. 10.1152/ajpgi.00079.2013
33
CsakT.GanzM.PespisaJ.KodysK.DolganiucA.SzaboG. (2011). Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology54, 133–144. 10.1002/hep.24341
34
CusiK.OrsakB.BrilF.LomonacoR.HechtJ.Ortiz-LopezC.et al (2016). Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus. Ann. Intern. Med.165, 305. 10.7326/m15-1774
35
DasK.,ChowdhuryA. (2013). Lean NASH: distinctiveness and clinical implication. Hepatol. Int.7, 806–813. 10.1007/s12072-013-9477-5
36
DavisJ. N.LêK.-A.WalkerR. W.VikmanS.Spruijt-MetzD.WeigensbergM. J.et al (2010). Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am. J. Clin. Nutr.92, 1522–1527. 10.3945/ajcn.2010.30185
37
DayC. P.,JamesO. F. W. (1998). Steatohepatitis: a tale of two “hits”?Gastroenterology114, 842–845. 10.1016/s0016-5085(98)70599-2
38
DeleriveP.De BosscherK.BesnardS.Vanden BergheW.PetersJ. M.GonzalezF. J.et al (1999). Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J. Biol. Chem.274, 32048–32054. 10.1074/jbc.274.45.32048
39
DemiragM. D.OnenH. I.KaraoguzM. Y.DoganI.KarakanT.EkmekciA.et al (2007). Apolipoprotein E gene polymorphism in nonalcoholic fatty liver disease. Dig. Dis. Sci.52, 3399–3403. 10.1007/s10620-007-9740-5
40
DiehlA. M.,DayC. (2017). Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med.377, 2063–2072. 10.1056/nejmra1503519
41
DingX.SaxenaN. K.LinS.GuptaN.AnaniaF. A.AnaniaF. A. (2006). Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology43, 173–181. 10.1002/hep.21006
42
DongiovanniP. (2013). PNPLA3 I148M polymorphism and progressive liver disease. WJG19, 6969. 10.3748/wjg.v19.i41.6969
43
DonnellyK. L.SmithC. I.SchwarzenbergS. J.JessurunJ.BoldtM. D.ParksE. J. (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest.115, 1343–1351. 10.1172/jci23621
44
DooleyS.DelvouxB.LahmeB.Mangasser-StephanK.GressnerA. M. (2000). Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology31, 1094–1106. 10.1053/he.2000.6126
45
DoychevaI.WattK. D.AlkhouriN. (2017). Nonalcoholic fatty liver disease in adolescents and young adults: the next Frontier in the epidemic. Hepatology65, 2100–2109. 10.1002/hep.29068
46
DysonJ. K.AnsteeQ. M.McPhersonS. (2014). Non-alcoholic fatty liver disease: a practical approach to diagnosis and staging. Frontline Gastroenterol.5, 211–218. 10.1136/flgastro-2013-100403
47
EckardC.ColeR.LockwoodJ.TorresD. M.WilliamsC. D.ShawJ. C.et al (2013). Prospective histopathologic evaluation of lifestyle modification in nonalcoholic fatty liver disease: a randomized trial. Therap. Adv. Gastroenterol.6, 249–259. 10.1177/1756283x13484078
48
EkstedtM.HagströmH.NasrP.FredriksonM.StålP.KechagiasS.et al (2015). Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology61, 1547–1554. 10.1002/hep.27368
49
EslamM.AlvaniR.ShihaG. (2019). Obeticholic acid: towards first approval for NASH. Lancet394, 2131–2133. 10.1016/s0140-6736(19)32963-0
50
EslamM.ValentiL.RomeoS. (2018). Genetics and epigenetics of NAFLD and NASH : clinical impact. J. Hepatol.68, 268–279. 10.1016/j.jhep.2017.09.003
51
EstesC.RazaviH.LoombaR.YounossiZ.SanyalA. J. (2018). Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology67, 123–133. 10.1002/hep.29466
52
FaehD.MinehiraK.SchwarzJ.-M.PeriasamyR.ParkS.TappyL.et al (2005). Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes54, 1907–1913. 10.2337/diabetes.54.7.1907
53
FarrellG.SchattenbergJ. M.LeclercqI.YehM. M.GoldinR.TeohN.et al (2019). Mouse models of nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology69, 2241–2257. 10.1002/hep.30333
54
FebbraioM. A.ReibeS.ShalapourS.OoiG. J.WattM. J.KarinM. (2019). Preclinical models for studying NASH-driven HCC: how useful are they?Cell Metabol.29, 18–26. 10.1016/j.cmet.2018.10.012
55
FeldsteinA. E.CharatcharoenwitthayaP.TreeprasertsukS.BensonJ. T.EndersF. B.AnguloP. (2009). The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut58, 1538–1544. 10.1136/gut.2008.171280
56
FengR.-N.DuS.-S.WangC.LiY.-C.LiuL.-Y.GuoF.-C.et al (2014). Lean-non-alcoholic fatty liver disease increases risk for metabolic disorders in a normal weight Chinese population. WJG20, 17932–17940. 10.3748/wjg.v20.i47.17932
57
FujitaK.NozakiY.YonedaM.WadaK.TakahashiH.KirikoshiH.et al (2010). Nitric oxide plays a crucial role in the development/progression of nonalcoholic steatohepatitis in the Choline-Deficient, l-Amino Acid-defined diet-fed rat model. Alcohol Clin. Exp. Res.34, 18–24. 10.1111/j.1530-0277.2008.00756.x
58
FriedmanJ. M.,HalaasJ. L. (1998). Leptin and the regulation of body weight in mammals. Nature395, 763–770. 10.1038/27376
59
FriedmanJ. M.LeibelR. L.SiegelD. S.WalshJ.BaharyN. (1991). Molecular mapping of the mouse ob mutation. Genomics11, 1054–1062. 10.1016/0888-7543(91)90032-a
60
FriedmanS. L.Neuschwander-TetriB. A.RinellaM.SanyalA. J. (2018a). Mechanisms of NAFLD development and therapeutic strategies. Nat. Med.24, 908–922. 10.1038/s41591-018-0104-9
61
FriedmanS. L.RatziuV.HarrisonS. A.AbdelmalekM. F.AithalG. P.CaballeriaJ.et al (2018b). A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology67, 1754–1767. 10.1002/hep.29477
62
FrithJ.DayC. P.HendersonE.BurtA. D.NewtonJ. L. (2009). Non-alcoholic fatty liver disease in older people. Gerontology55, 607–613. 10.1159/000235677
63
FujiiM.ShibazakiY.WakamatsuK.HondaY.KawauchiY.SuzukiK.et al (2013). A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol.46, 141–152. 10.1007/s00795-013-0016-1
64
GaddV. L.SkoienR.PowellE. E.FaganK. J.WinterfordC.HorsfallL.et al (2014). The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology59, 1393–1405. 10.1002/hep.26937
65
GarberA.HenryR.RatnerR.Garcia-HernandezP. A.Rodriguez-PattziH.Olvera-AlvarezI.et al (2009). Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet373, 473–481. 10.1016/s0140-6736(08)61246-5
66
GehrkeN.,SchattenbergJ. M. (2020). Metabolic inflammation-a role for hepatic inflammatory pathways as drivers of comorbidities in nonalcoholic fatty liver disease?Gastroenterology158, 1929–1947. 10.1053/j.gastro.2020.02.020
67
GENFIT S.A. (2020). GENFIT: announces results from interim analysis of RESOLVE-IT phase 3 trial of elafibranor ina adults with NASH and fibrosis. Paris, France: GENFIT.
68
GoyalN. P.,SchwimmerJ. B. (2016). The progression and natural history of pediatric nonalcoholic fatty liver disease. Clin. Liver Dis.20, 325–338. 10.1016/j.cld.2015.10.003
69
GrecoD.KotronenA.WesterbackaJ.PuigO.ArkkilaP.KiviluotoT.et al (2008). Gene expression in human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol.294, G1281–G1287. 10.1152/ajpgi.00074.2008
70
HaczeyniF.PoekesL.WangH.MridhaA. R.BarnV.Geoffrey HaighW.et al (2017). Obeticholic acid improves adipose morphometry and inflammation and reduces steatosis in dietary but not metabolic obesity in mice. Obesity25, 155–165. 10.1002/oby.21701
71
HanfR.MillattL. J.CariouB.NoelB.RigouG.DelatailleP.et al (2014). The dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 exerts anti-diabetic effects in db/db mice without peroxisome proliferator-activated receptor gamma-associated adverse cardiac effects. Diabetes Vasc. Dis. Res.11, 440–447. 10.1177/1479164114548027
72
HardingS. E.ZhangY.RonD. (1999). Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature397, 271–305. 10.1038/16729
73
HarrisonS. A.GoodmanZ.JabbarA.VemulapalliR.YounesZ. H.FreilichB.et al (2020a). A randomized, placebo-controlled trial of emricasan in patients with NASH and F1–F3 fibrosis. J. Hepatol.72, 816–827. 10.1016/j.jhep.2019.11.024
74
HarrisonS. A.WongV. W.-S.OkanoueT.BzowejN.VuppalanchiR.YounesZ.et al (2020b). Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J. Hepatol.73, 26–39. 10.1016/j.jhep.2020.02.027
75
HasinB. R.SeldinM.LusisA. (2017). Multi-omits approaches to disease. Genome Biol.18, 83. 10.1186/s13059-017-1215-1
76
HaukelandJ. W.DamåsJ. K.KonopskiZ.LøbergE. M.HaalandT.GoverudI.et al (2006). Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol.44, 1167–1174. 10.1016/j.jhep.2006.02.011
77
HellerbrandC.StefanovicB.GiordanoF.BurchardtE. R.BrennerD. A. (1999). The role of TGFβ1 in initiating hepatic stellate cell activation in vivo. J. Hepatol.30, 77–87. 10.1016/s0168-8278(99)80010-5
78
HildebrandtM. A.HoffmannC.Sherrill–MixS. A.KeilbaughS. A.HamadyM.ChenY. Y.et al (2009). High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology137, 1716–1724. 10.1053/j.gastro.2009.08.042
79
HotamisligilG. S.ArnerP.CaroJ. F.AtkinsonR. L.SpiegelmanB. M. (1995). Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Invest.95, 2409–2415. 10.1172/jci117936
80
HotamisligilG.ShargillN.SpiegelmanB. (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science259, 87–91. 10.1126/science.7678183
81
HuangY.,MahleyR. W. (2014). Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis.72, 3–12. 10.1016/j.nbd.2014.08.025
82
HuberY.BoyleM.HallsworthK.TiniakosD.StraubB. K.LabenzC.et al (2019). Health-related quality of life in nonalcoholic fatty liver disease associates with hepatic inflammation. Clin. Gastroenterol. Hepatol.17, 2085–2092. 10.1016/j.cgh.2018.12.016
83
InagakiY.MamuraM.KanamaruY.GreenwelP.NemotoT.TakeharaK.et al (2001). Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J. Cell. Physiol.187, 117–123. 10.1002/1097-4652(2001)9999:9999<00::aid-jcp1059>3.0.co;2-s
84
Iruarrizaga-LejarretaM.Varela-ReyM.Fernández-RamosD.Martínez-ArranzI.DelgadoT. C.SimonJ.et al (2017). Role of aramchol in steatohepatitis and fibrosis in mice. Hepatology Communications1, 911–927. 10.1002/hep4.1107
85
JadejaR.DevkarR. V.NammiS. (2014). Herbal medicines for the treatment of nonalcoholic steatohepatitis: current scenario and future prospects. Evid. base Compl. Alternative Med.2014, 1–18. 10.1155/2014/648308
86
JiangM.LiC.LiuQ.WangA.LeiM. (2019). Inhibiting ceramide synthesis attenuates hepatic steatosis and fibrosis in rats with non-alcoholic fatty liver disease. Front. Endocrinol.10, 1–8. 10.3389/fendo.2019.00665
87
JiaoY.LuY.LiX.-y. (2015). Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin.36, 44–50. 10.1038/aps.2014.116
88
KapsL.LabenzC.GalleP. R.Weinmann-MenkeJ.KostevK.SchattenbergJ. M. (2020). Non-alcoholic fatty liver disease increases the risk of incident chronic kidney disease. United European Gastroenterology Journal8, 942–948. 10.1177/2050640620944098
89
KarlmarkK. R.WeiskirchenR.ZimmermannH. W.GasslerN.GinhouxF.WeberC.et al (2009). Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosisHepatic recruitment of the inflammatory Gr1+monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology50, 261–274. 10.1002/hep.22950
90
KewM. C. (2000). Serum aminotransferase concentration as evidence of hepatocellular damage. Lancet355, 591–592. 10.1016/s0140-6736(99)00219-6
91
KimR. S.HasegawaD.GoossensN.TsuchidaT.AthwalV.SunX.et al (2016). The XBP1 arm of the unfolded protein response induces fibrogenic activity in hepatic stellate cells through autophagy. Sci. Rep.6, 1–9. 10.1038/srep39342
92
KimS. Y.JeongJ.-M.KimS. J.SeoW.KimM.-H.ChoiW.et al (2017). Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun.8, 2247. 10.1038/s41467-017-02325-2
93
KimW. R.FlammS. L.Di BisceglieA. M.BodenheimerH. C. (2008). Serum activity of alanine aminotransferase (alt) as an indicator of health and disease. Hepatology47, 1363–1370. 10.1002/hep.22109
94
KishidaN.MatsudaS.ItanoO.ShinodaM.KitagoM.YagiH.et al (2016). Development of a novel mouse model of hepatocellular carcinoma with nonalcoholic steatohepatitis using a high-fat, choline-deficient diet and intraperitoneal injection of diethylnitrosamine. BMC Gastroenterol.16, 61. 10.1186/s12876-016-0477-5
95
KleinE. A.ThompsonI. M.TangenC. M.CrowleyJ. J.LuciaM. S.GoodmanP. J.et al (2011). Vitamin E and the risk of prostate cancer: the selenium and vitamin E cancer prevention trial (SELECT). J. Am. Med. Assoc.306, 1549–1556. 10.1001/jama.2011.1437
96
KleinerD. E.BruntE. M.Van NattaM.BehlingC.ContosM. J.CummingsO. W.et al (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology41, 1313–1321. 10.1002/hep.20701
97
KohliR.KirbyM.XanthakosS. A.SofticS.FeldsteinA. E.SaxenaV.et al (2010). High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology52, 934–944. 10.1002/hep.23797
98
KojimaH.SakuraiS.UemuraM.FukuiH.MorimotoH.TamagawaY. (2007). Mitochondrial abnormality and oxidative stress in nonalcoholic steatohepatitis. Alcohol Clin. Exp. Res.31, S61–S66. 10.1111/j.1530-0277.2006.00288.x
99
KoliakiC.SzendroediJ.KaulK.JelenikT.NowotnyP.JankowiakF.et al (2015). Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in Steatohepatitis. Cell Metabol.21, 739–746. 10.1016/j.cmet.2015.04.004
100
KoppeS. W. P.EliasM.MoseleyR. H.GreenR. M. (2009). Trans fat feeding results in higher serum alanine aminotransferase and increased insulin resistance compared with a standard murine high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol.297, G378–G384. 10.1152/ajpgi.90543.2008
101
KrenkelO.PuengelT.GovaereO.AbdallahA. T.MossanenJ. C.KohlheppM.et al (2018). Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology67, 1270–1283. 10.1002/hep.29544
102
KumashiroN.YoshimuraT.CantleyJ. L.MajumdarS. K.Guebre‐EgziabherF.KursaweR.et al (2013). Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology57, 1763–1772. 10.1002/hep.26170
103
KuoT.-F.TatsukawaH.MatsuuraT.NagatsumaK.HiroseS.KojimaS. (2012). Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell. Physiol.227, 1130–1137. 10.1002/jcp.22833
104
LabenzC.HuberY.MichelM.NagelM.GalleP. R.KostevK.et al (2020). Impact of NAFLD on the incidence of cardiovascular diseases in a primary care population in Germany. Dig. Dis. Sci.65, 2112–2119. 10.1007/s10620-019-05986-9
105
LakeA. D.NovakP.HardwickR. N.Flores-KeownB.ZhaoF.KlimeckiW. T.et al (2014). The Adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol. Sci.137, 26–35. 10.1093/toxsci/kft230
106
LambertJ. E.Ramos–RomanM. A.BrowningJ. D.ParksE. J. (2014). Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology146, 726–735. 10.1053/j.gastro.2013.11.049
107
LebeaupinC.ValléeD.HazariY.HetzC.ChevetE.Bailly-MaitreB. (2018). Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol.69, 927–947. 10.1016/j.jhep.2018.06.008
108
LeclercqI. A.FarrellG. C.FieldJ.BellD. R.GonzalezF. J.RobertsonG. R. (2000). CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Invest.105, 1067–1075. 10.1172/jci8814
109
LeclercqI. A.FarrellG. C.SchriemerR.RobertsonG. R. (2002). Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol.37, 206–213. 10.1016/s0168-8278(02)00102-2
110
LeclercqI. A.FarrellG. C.SempouxC.PeñaA. d.HorsmansY. (2004). Curcumin inhibits NF-κB activation and reduces the severity of experimental steatohepatitis in mice. J. Hepatol.41, 926–934. 10.1016/j.jhep.2004.08.010
111
LefereS.DevisscherL.TackeF. (2020). Targeting CCR2/5 in the treatment of nonalcoholic steatohepatitis (NASH) and fibrosis: opportunities and challenges. Expet Opin. Invest. Drugs29, 89–92. 10.1080/13543784.2020.1718106
112
LeeC.-H.OlsonP.HevenerA.MehlI.ChongL.-W.OlefskyJ. M.et al (2006). PPARδ regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. Unit. States Am.103, 3444–3449. 10.1073/pnas.0511253103
113
LenzenS. (2008). The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia51, 216–226. 10.1007/s00125-007-0886-7
114
LoguercioC.De GirolamoV.de SioI.TuccilloC.AscioneA.BaldiF.et al (2001). Non-alcoholic fatty liver disease in an area of southern Italy: main clinical, histological, and pathophysiological aspects. J. Hepatol.35, 568–574. 10.1016/s0168-8278(01)00192-1
115
LomonacoR.Ortiz-LopezC.OrsakB.WebbA.HardiesJ.DarlandC.et al (2012). Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology55, 1389–1397. 10.1002/hep.25539
116
LudwigJ.ViggianoT. R.McGillD. B.OhB. J. (1980). Nonalcoholic steatohepatitis: mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc.55, 434–438
117
LuukkonenP. K.SädevirtaS.ZhouY.KayserB.AliA.AhonenL.et al (2018). Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugars. Diabetes Care41, 1732–1739. 10.2337/dc18-0071
118
MarshallJ. D.CollinG. B.NaggertJ. K.NaggertK. (2011). Alström syndrome: genetics and clinical overview. Curr. Genom.12, 225–235. 10.2174/138920211795677912
119
MachadoM. V.MichelottiG. A.XieG.AlmeidaT. P. D.BoursierJ.BohnicB.et al (2015). Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One10, 1–16. 10.1371/journal.pone.0127991
120
MagkosF.SuX.BradleyD.FabbriniE.ConteC.EagonJ. C.et al (2012). Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology142, 1444–1446. 10.1053/j.gastro.2012.03.003
121
MahdessianH.TaxiarchisA.PopovS.SilveiraA.Franco-CerecedaA.HamstenA.et al (2014). TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. Unit. States Am.111, 8913–8918. 10.1073/pnas.1323785111
122
MaherJ. J.,McGuireR. F. (1990). Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J. Clin. Invest.86, 1641–1648. 10.1172/jci114886
123
MakK. M.,MeiR. (2017). Basement membrane type IV collagen and laminin: an overview of their biology and value as fibrosis biomarkers of liver disease. Anat. Rec.300, 1371–1390. 10.1002/ar.23567
124
MakkonenJ.PietiläinenK. H.RissanenA.KaprioJ.Yki-JärvinenH. (2009). Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: a study in monozygotic and dizygotic twins. J. Hepatol.50, 1035–1042. 10.1016/j.jhep.2008.12.025
125
MalhiH.BronkS. F.WerneburgN. W.GoresG. J. (2006). Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem.281, 12093–12101. 10.1074/jbc.m510660200
126
MarshallS. A.QinC. X.JelinicM.O’SullivanK.DeoM.WalshJ.et al (2020). The novel small-molecule annexin-a1 mimetic, compound 17b, elicits vasoprotective actions in streptozotocin-induced diabetic mice. IJMS21, 1384. 10.3390/ijms21041384
127
MasaroneM.RosatoV.DallioM.GravinaA. G.AglittiA.LoguercioC.et al (2018). Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid. Med. Cell. Longev.2018, 9547613. 10.1155/2018/9547613
128
MelloT.ZanieriF.CeniE.GalliA. (2016). Oxidative stress in the healthy and wounded hepatocyte: a cellular organelles perspective. Oxid. Med. Cell. Longev., 2016, 8327410. 10.1155/2016/8327410
129
MiddletonS. A.RajpalN.CutlerL.ManderP.RiojaI.PrinjhaR. K.et al (2018). BET inhibition improves NASH and liver fibrosis. Sci. Rep.8, 17257. 10.1038/s41598-018-35653-4
130
MiuraK.YangL.RooijenN.van, OhnishiH.SekiE. (2012). Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol.302, 1310–1321. 10.1152/ajpgi.00365.2011
131
MontandonS. A.SommE.Loizides-MangoldU.VitoC. de.DibnerC.JornayvazF. R. (2019). Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci. Rep.9, 1–14. 10.1038/s41598-019-53346-4
132
MorignyP.HoussierM.MouiselE.LanginD. (2016). Adipocyte lipolysis and insulin resistance. Biochimie125, 259–266. 10.1016/j.biochi.2015.10.024
133
MorrisonM. C.KleemannR.KoppenA. van.HanemaaijerR.VerschurenL. (2018). Key inflammatory processes in human NASH are reflected in Ldlr-/-.Leiden mice: a translational gene profiling study. Front. Physiol.9, 1–13. 10.3389/fphys.2018.00132
134
MotaM.BaniniB. A.CazanaveS. C.SanyalA. J. (2016). Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism65, 1049–1061. 10.1016/j.metabol.2016.02.014
135
MridhaA. R.WreeA.RobertsonA. A. B.YehM. M.JohnsonC. D.Van RooyenD. M.et al (2017). NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol.66, 1037–1046. 10.1016/j.jhep.2017.01.022
136
MulderP.MorrisonM. C.WielingaP. Y.van DuyvenvoordeW.KooistraT.KleemannR. (2016). Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non-alcoholic steatohepatitis in obesity. Int. J. Obes.40, 675–684. 10.1038/ijo.2015.226
137
NakagawaH.UmemuraA.TaniguchiK.Font-BurgadaJ.DharD.OgataH.et al (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Canc. Cell26, 331–343. 10.1016/j.ccr.2014.07.001
138
Neuschwander-TetriB. A. (2010). Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology52, 774–788. 10.1002/hep.23719
139
Neuschwander-TetriB. A.LoombaR.SanyalA. J.LavineJ. E.Van NattaM. L.AbdelmalekM. F.et al (2015). Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet385, 956–965. 10.1016/s0140-6736(14)61933-4
140
NewsomeP.FrancqueS.HarrisonS.RatziuV.Van GaalL.CalannaS.et al (2019). Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharmacol. Ther.50, 193–203. 10.1111/apt.15316
141
NewsomeP. N.BuchholtzK.CusiK.LinderM.OkanoueT.RatziuV.et al (2020). A Placebo‐Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 10.1056/NEJMoa2028395
142
NingJ.HongT.WardA.PiJ.LiuZ.LiuH.-Y.et al (2011). Constitutive role for IRE1α-XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology152, 2247–2255. 10.1210/en.2010-1036
143
NiuL.GeyerP. E.Wewer AlbrechtsenN. J.GluudL. L.SantosA.DollS.et al (2019). Plasma proteome profiling discovers novel proteins associated with non‐alcoholic fatty liver disease. Mol. Syst. Biol.15, 1–16. 10.15252/msb.20188793
144
OgawaY.ImajoK.HondaY.KessokuT.TomenoW.KatoS.et al (2018). Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci. Rep.8, 1–14. 10.1038/s41598-018-29735-6
145
OligschlaegerY.,Shiri-SverdlovR. (2020). NAFLD preclinical models: more than a handful, less of a concern?. Biomedicines8, 28. 10.3390/biomedicines8020028
146
OrimeK.ShirakawaJ.TogashiY.TajimaK.InoueH.NagashimaY.et al (2016). Lipid-lowering agents inhibit hepatic steatosis in a non-alcoholic steatohepatitis-derived hepatocellular carcinoma mouse model. Eur. J. Pharmacol.772, 22–32. 10.1016/j.ejphar.2015.12.043
147
OzH. S.ChenT. S.NeumanM. (2008). Methionine deficiency and hepatic injury in a dietary steatohepatitis model. Dig. Dis. Sci.53, 767–776. 10.1007/s10620-007-9900-7
148
ParkE. J.LeeJ. H.YuG.-Y.HeG.AliS. R.HolzerR. G.et al (2010). Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell140, 197–208. 10.1016/j.cell.2009.12.052
149
ParkS. H.LeeS. G.KangS. K.ChungS. H. (2006). Acanthopanax senticosus reverses fatty liver disease and hyperglycemia in ob/ob mice. Arch Pharm. Res.29, 768–776. 10.1007/bf02974078
150
PeñadelaA.LeclercqI.FieldJ.GeorgeJ.JonesB.FarrellG. (2005). NF-κB activation, rather than tnf, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology129, 1663–1674. 10.1053/j.gastro.2005.09.004
151
PearsonS.KietsirirojeN.AjjanR. A. (2019). Oral semaglutide in the management of type 2 diabetes: a report on the evidence to date. Dmso12, 2515–2529. 10.2147/dmso.s229802
152
PellicciariR.FiorucciS.CamaioniE.ClericiC.CostantinoG.MaloneyP. R.et al (2002). 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem.45, 3569–3572. 10.1021/jm025529g
153
PeraldiP.HotamisligilG. S.BuurmanW. A.WhiteM. F.SpiegelmanB. M. (1996). Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J. Biol. Chem.271, 13018–13022. 10.1074/jbc.271.22.13018
154
PettaS.MieleL.BugianesiE.CammàC.RossoC.BocciaS.et al (2014). Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS One9, e87523. 10.1371/journal.pone.0087523
155
PromratK.KleinerD. E.NiemeierH. M.JackvonyE.KearnsM.WandsJ. R.et al (2010). Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology51, 121–129. 10.1002/hep.23276
156
PuriP.BaillieR. A.WiestM. M.MirshahiF.ChoudhuryJ.CheungO.et al (2007). A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology46, 1081–1090. 10.1002/hep.21763
157
PuriP.MirshahiF.CheungO.NatarajanR.MaherJ. W.KellumJ. M.et al (2008). Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology134, 568–576. 10.1053/j.gastro.2007.10.039
158
PuriP.WiestM. M.CheungO.MirshahiF.SargeantC.MinH.-K.et al (2009). The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology50, 1827–1838. 10.1002/hep.23229
159
RaoY.LiuH.GaoL.YuH.OuT.-M.TanJ.-H.et al (2015). Synthesis and biological evaluation of novel bouchardatine derivatives as potential adipogenesis/lipogenesis inhibitors for antiobesity treatment. J. Med. Chem.58, 9395–9413. 10.1021/acs.jmedchem.5b01566
160
RaoY.LuY. T.LiC.SongQ. Q.XuY. H.XuZ.et al (2019). Bouchardatine analogue alleviates non-alcoholic hepatic fatty liver disease/non-alcoholic steatohepatitis in high-fat fed mice by inhibiting ATP synthase activity. Br. J. Pharmacol.176, 2877–2893. 10.1111/bph.14713
161
RaoY.XuZ.HuY.-T.LiC.XuY.-H.SongQ.-Q.et al (2020). Discovery of a promising agent IQZ23 for the treatment of obesity and related metabolic disorders. Eur. J. Med. Chem.192, 112172. 10.1016/j.ejmech.2020.112172
162
RatziuV.HarrisonS. A.FrancqueS.BedossaP.LehertP.SerfatyL.et al (2016). Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology150, 1147–1159.e5. 10.1053/j.gastro.2016.01.038
163
RatziuV.SheikhM. Y.SanyalA. J.LimJ. K.ConjeevaramH.ChalasaniN.et al (2012). A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology55, 419–428. 10.1002/hep.24747
164
ReedM. J.MeszarosK.EntesL. J.ClaypoolM. D.PinkettJ. G.GadboisT. M.et al (2000). A new rat model of type 2 diabetes: the fat-fed, streptozotocin-treated rat. Metabolism49, 1390–1394. 10.1053/meta.2000.17721
165
RensenS. S.SlaatsY.NijhuisJ.JansA.BieghsV.DriessenA.et al (2009). Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol.175, 1473–1482. 10.2353/ajpath.2009.080999
166
RethorstC. D.BernsteinI.TrivediM. H. (2014). Inflammation, obesity, and metabolic syndrome in depression: analysis of the 2009-2010 national health and nutrition examination survey (NHANES). J. Clin. Psychiatr.75, e1428–e1432. 10.4088/jcp.14m09009
167
RinellaM. E.EliasM. S.SmolakR. R.FuT.BorensztajnJ.GreenR. M. (2008). Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res.49, 1068–1076. 10.1194/jlr.m800042-jlr200
168
RinellaM. E.,GreenR. M. (2004). The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol.40, 47–51. 10.1016/j.jhep.2003.09.020
169
RollaS.AlcheraE.ImarisioC.BardinaV.ValenteG.CappelloP.et al (2016). The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin. Sci.130, 193–203. 10.1042/cs20150405
170
RomeoS.KozlitinaJ.XingC.PertsemlidisA.CoxD.PennacchioL. A.et al (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet.40, 1461–1465. 10.1038/ng.257
171
RooyenD. M.LarterC. Z.HaighW. G.YehM. M.IoannouG.KuverR.et al (2011). Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology141, 1393–1395. 10.1053/j.gastro.2011.06.040
172
RossoC.KazankovK.YounesR.EsmailiS.MariettiM.SaccoM.et al (2019). Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J. Hepatol.71, 1012–1021. 10.1016/j.jhep.2019.06.031
173
RudermanN.ChisholmD.Pi-SunyerX.SchneiderS. (1998). The metabolically obese, normal-weight individual revisited. Diabetes47, 699–713. 10.2337/diabetes.47.5.699
174
SabioG.Cavanagh-KyrosJ.KoH. J.JungD. Y.GrayS.JunJ. Y.et al (2009). Prevention of steatosis by hepatic JNK1. Cell Metabol.10, 491–498. 10.1016/j.cmet.2009.09.007
175
SafadiR.KonikoffF. M.MahamidM.Zelber-SagiS.HalpernM.GilatT.et al (2014). The fatty acid–bile acid conjugate aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol.12, 2085–2091. 10.1016/j.cgh.2014.04.038
176
SahaiA.MalladiP.Melin-AldanaH.GreenR. M.WhitingtonP. F.GreenR. M.et al (2004). Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol.287, G264–G273. 10.1152/ajpgi.00199.2004
177
SaitoT.MuramatsuM.IshiiY.SaigoY.KonumaT.ToriniwaY.et al (2017). Pathophysiological analysis of the progression of hepatic lesions in STAM mice. Physiol. Res.66, 791–799. 10.33549/physiolres.933592
178
SaklayenM. G. (2018). The global epidemic of the metabolic syndrome. Curr. Hypertens. Rep.20, 12. 10.1007/s11906-018-0812-z
179
SamuelV. T.LiuZ.-X.QuX.ElderB. D.BilzS.BefroyD.et al (2004). Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem.279, 32345–32353. 10.1074/jbc.m313478200
180
SanyalA. J.Campbell–SargentC.MirshahiF.RizzoW. B.ContosM. J.SterlingR. K.et al (2001). Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology120, 1183–1192. 10.1053/gast.2001.23256
181
SanyalA. J.ChalasaniN.KowdleyK. V.McCulloughA.DiehlA. M.BassN. M.et al (2010). Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med.362, 1675–1685. 10.1056/nejmoa0907929
182
SazciA.AkpinarG.AygunC.ErgulE.SenturkO.HulaguS. (2008). Association of apolipoprotein E polymorphisms in patients with non-alcoholic steatohepatitis. Dig. Dis. Sci.53, 3218–3224. 10.1007/s10620-008-0271-5
183
ScaglioniF.CicciaS.MarinoM.BedogniG.BellentaniS. (2011). ASH and NASH. Dig. Dis.29, 202–210. 10.1159/000323886
184
SchattenbergJ. M.SinghR.WangY.LefkowitchJ. H.RigoliR. M.SchererP. E.et al (2006). JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology43, 163–172. 10.1002/hep.20999
185
SchierwagenR.MaybüchenL.ZimmerS.HittatiyaK.BäckC.KleinS.et al (2015). Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep.5, 1–14. 10.1038/srep12931
186
SchützeS.WiegmannK.MachleidtT.KrönkeM. (1995). TNF-induced activation of NF-κB. Immunobiology193, 193–203. 10.1016/s0171-2985(11)80543-7
187
SchwimmerJ. B.CeledonM. A.LavineJ. E.SalemR.CampbellN.SchorkN. J.et al (2009). Heritability of nonalcoholic fatty liver disease. Gastroenterology136, 1585–1592. 10.1053/j.gastro.2009.01.050
188
SekiE.,BrennerD. A. (2008). Toll-like receptors and adaptor molecules in liver disease: update. Hepatology48, 322–335. 10.1002/hep.22306
189
SeversonT. J.BesurS.BonkovskyH. L. (2016). Genetic factors that affect nonalcoholic fatty liver disease: a systematic clinical review. WJG22, 6742–6756. 10.3748/wjg.v22.i29.6742
190
ShalapourS.LinX.-J.BastianI. N.BrainJ.BurtA. D.AksenovA. A.et al (2017). Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature551, 340–345. 10.1038/nature24302
191
Shiri-SverdlovR.WoutersK.GorpP. J. v.GijbelsM. J.NoelB.BuffatL.et al (2006). Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol.44, 732–741. 10.1016/j.jhep.2005.10.033
192
SiddiquiM. S.HarrisonS. A.AbdelmalekM. F.AnsteeQ. M.BedossaP.CasteraL.et al (2018). Case definitions for inclusion and analysis of endpoints in clinical trials for nonalcoholic steatohepatitis through the lens of regulatory science. Hepatology67, 2001–2012. 10.1002/hep.29607
193
SinghU.DevarajS.JialalI. (2005). Vitamin E, oxidative stress, and inflammation. Annu. Rev. Nutr.25, 151–174. 10.1146/annurev.nutr.24.012003.132446
194
SmagrisE.BasuRayS.LiJ.HuangY.LaiK. m. V.GromadaJ.et al (2015). Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology61, 108–118. 10.1002/hep.27242
195
SmeuninxB.BoslemE.FebbraioM. A. (2020). Current and future treatments in the fight against non-alcoholic fatty liver disease. Cancers12, 1714. 10.3390/cancers12071714
196
SofticS.CohenD. E.KahnC. R. (2016). Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci.61, 1282–1293. 10.1007/s10620-016-4054-0
197
SongG.LiuJ.ZhaoZ.YuY.TianH.YaoS.et al (2011). Simvastatin reduces atherogenesis and promotes the expression of hepatic genes associated with reverse cholesterol transport in apoE-knockout mice fed high-fat diet. Lipids Health Dis.10, 8. 10.1186/1476-511x-10-8
198
SoonR. K.YanJ. S.GrenertJ. P.MaherJ. J. (2010). Stress signaling in the methionine-choline-deficient model of murine fatty liver disease. Gastroenterology139, 1730–1739. 10.1053/j.gastro.2010.07.046
199
StaelsB.RubenstrunkA.NoelB.RigouG.DelatailleP.MillattL. J.et al (2013). Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology58, 1941–1952. 10.1002/hep.26461
200
StienstraR.MandardS.PatsourisD.MaassC.KerstenS.MüllerM. (2007). Peroxisome proliferator-activated receptor α protects against obesity-induced hepatic inflammation. Endocrinology148, 2753–2763. 10.1210/en.2007-0014
201
SunX.DuanX.WangC.LiuZ.SunP.HuoX.et al (2017). Protective effects of glycyrrhizic acid against non-alcoholic fatty liver disease in mice. Eur. J. Pharmacol.806, 75–82. 10.1016/j.ejphar.2017.04.021
202
SutterA. G.PalanisamyA. P.LenchJ. H.JessmoreA. P.ChavinK. D. (2015). Development of steatohepatitis in ob/ob mice is dependent on Toll-like receptor 4. Ann. Hepatol.14, 735–743. 10.1016/s1665-2681(19)30769-0
203
TajiriK.ShimizuY.TsuneyamaK.SugiyamaT. (2009). Role of liver-infiltrating CD3+ CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol.21, 673–680. 10.1097/meg.0b013e32831bc3d6
204
TargherG.DayC. P.BonoraE. (2010). Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med.363, 1341–1350. 10.1056/nejmra0912063
205
TateM.PrakosoD.WillisA. M.PengC.DeoM.QinC. X.et al (2019). Characterising an alternative murine model of diabetic cardiomyopathy. Front. Physiol.10, 1–15. 10.3389/fphys.2019.01395
206
ThomasE. L.HamiltonG.PatelN.O’DwyerR.DoréC. J.GoldinR. D.et al (2005). Hepatic triglyceride content and its relation to body adiposity: a magnetic resonance imaging and proton magnetic resonance spectroscopy study. Gut54, 122–127. 10.1136/gut.2003.036566
207
TilgH.,MoschenA. R. (2006). Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol.6, 772–783. 10.1038/nri1937
208
Tosello-TrampontA.-C.LandesS. G.NguyenV.NovobrantsevaT. I.HahnY. S. (2012). Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem.287, 40161–40172. 10.1074/jbc.m112.417014
209
Trak-SmayraV.ParadisV.MassartJ.NasserS.JebaraV.FromentyB. (2011). Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int. J. Exp. Pathol.92, 413–421. 10.1111/j.1365-2613.2011.00793.x
210
TsuchidaT.,FriedmanS. L. (2017). Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol.14, 397–411. 10.1038/nrgastro.2017.38
211
TsuchidaT.LeeY. A.FujiwaraN.YbanezM.AllenB.MartinsS.et al (2018). A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol.69, 385–395. 10.1016/j.jhep.2018.03.011
212
TurpinS. M.NichollsH. T.WillmesD. M.MourierA.BrodesserS.WunderlichC. M.et al (2014). Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metabol.20, 678–686. 10.1016/j.cmet.2014.08.002
213
Vilar-GomezE.Martinez-PerezY.Calzadilla-BertotL.Torres-GonzalezA.Gra-OramasB.Gonzalez-FabianL.et al (2015). Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology149, 367–378.e5. 10.1053/j.gastro.2015.04.005
214
WangB.ChandrasekeraP.PippinJ. (2014). Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. CDR10, 131–145. 10.2174/1573399810666140508121012
215
WangD.WeiY.PagliassottiM. J. (2006). Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology147, 943–951. 10.1210/en.2005-0570
216
WangX.WangY.AntonyV.SunH.LiangG. (2020). Metabolism-associated molecular patterns (MAMPs). Trends Endocrinol. Metab.31 (10) 712–724. 10.1016/j.tem.2020.07.001
217
WangY.AusmanL. M.GreenbergA. S.RussellR. M.WangX.-D. (2009). Nonalcoholic steatohepatitis induced by a high-fat diet promotes diethylnitrosamine-initiated early hepatocarcinogenesis in rats. Int. J. Canc.124, 540–546. 10.1002/ijc.23995
218
WeglarzT. C.DegenJ. L.SandgrenE. P. (2000). Hepatocyte transplantation into diseased mouse liver: kinetics of parenchymal repopulation and identification of the proliferative capacity of tetraploid and octaploid hepatocytes. Am. J. Pathol.157, 1963–1974. 10.1016/s0002-9440(10)64835-3
219
WeisbergS. P.McCannD.DesaiM.RosenbaumM.LeibelR. L.FerranteA. W. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest.112, 1796–1808. 10.1172/jci200319246
220
WelshJ. A.KarpenS.VosM. B. (2013). Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J. Pediatr.162, 496–500. 10.1016/j.jpeds.2012.08.043
221
WhiteM. F. (1997). The insulin signalling system and the IRS proteins. Diabetologia40, S2–S17. 10.1007/s001250051387
222
WilliamsC. D.StengelJ.AsikeM. I.TorresD. M.ShawJ.ContrerasM.et al (2011). Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology140, 124–131. 10.1053/j.gastro.2010.09.038
223
WilliamsG. M.IatropoulosM. J.WangC. X.AliN.RivensonA.PetersonL. A.et al (1996). Diethylnitrosamine exposure-responses for DNA damage, centrilobular cytotoxicity, cell proliferation and carcinogenesis in rat liver exhibit some non-linearities. Carcinogenesis17, 2253–2258. 10.1093/carcin/17.10.2253
224
WongR. J.AguilarM.CheungR.PerumpailR. B.HarrisonS. A.YounossiZ. M.et al (2015). Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology148, 547–555. 10.1053/j.gastro.2014.11.039
225
WorthamM.HeL.GyamfiM.CoppleB. L.WanY.-J. Y. (2008). The transition from fatty liver to NASH associates with SAMe depletion in db/db mice fed a methionine choline-deficient diet. Dig. Dis. Sci.53, 2761–2774. 10.1007/s10620-007-0193-7
226
WreeA.EguchiA.McgeoughM. D.PenaC. A.JohnsonC. D.CanbayA.et al (2014). NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology59, 898–910. 10.1002/hep.26592
227
XiaoG.ZhangT.YuS.LeeS.Calabuig-NavarroV.YamauchiJ.et al (2013). ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J. Biol. Chem.288, 25350–25361. 10.1074/jbc.m113.470526
228
XuH.BarnesG. T.YangQ.TanG.YangD.ChouC. J.et al (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest.112, 1821–1830. 10.1172/jci200319451
229
YesilovaZ.YamanH.OktenliC.OzcanA.UygunA.CakirE.et al (2005). Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol.100, 850–855. 10.1111/j.1572-0241.2005.41500.x
230
YounossiZ. M.RatziuV.LoombaR.RinellaM.AnsteeQ. M.GoodmanZ. (2019). Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet394, 2184–2196. 10.1016/S0140-6736(19)33041-7
231
YounossiZ. M.BlissettD.BlissettR.HenryL.StepanovaM.YounossiY.et al (2016a). The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology64, 1577–1586. 10.1002/hep.28785
232
YounossiZ. M.KoenigA. B.AbdelatifD.FazelY.HenryL.WymerM. (2016b). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology64, 73–84. 10.1002/hep.28431
233
YounossiZ. M.LoombaR.RinellaM. E.BugianesiE.MarchesiniG.Neuschwander-TetriB. A.et al (2018). Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology68, 361–371. 10.1002/hep.29724
234
YounossiZ. M.StepanovaM.RafiqN.MakhloufH.YounoszaiZ.AgrawalR.et al (2011). Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology53, 1874–1882. 10.1002/hep.24268
235
ZangS.WangL.MaX.ZhuG.ZhuangZ.XunY.et al (2015). Neutrophils play a crucial role in the early stage of nonalcoholic steatohepatitis via neutrophil elastase in mice. Cell Biochem. Biophys.73, 479–487. 10.1007/s12013-015-0682-9
236
ZeiselS. H.,da CostaK.-A. (2009). Choline: an essential nutrient for public health. Nutr. Rev.67, 615–623. 10.1111/j.1753-4887.2009.00246.x
237
ZhanS.-S.JiangJ. X.WuJ.HalstedC.FriedmanS. L.ZernM. A.et al (2006). Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology43, 435–443. 10.1002/hep.21093
238
ZhangL.YaoZ.JiG. (2018). Herbal extracts and natural products in alleviating non-alcoholic fatty liver disease via activating autophagy. Front. Pharmacol.9, 1–7. 10.3389/fphar.2018.01459
239
ZhangY.ProencaR.MaffeiM.BaroneM.LeopoldL.FriedmanJ. M. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature372, 425–432. 10.1038/372425a0
240
ZhengS.HoosL.CookJ.TetzloffG.DavisH.van HeekM.et al (2008). Ezetimibe improves high fat and cholesterol diet-induced non-alcoholic fatty liver disease in mice. Eur. J. Pharmacol.584, 118–124. 10.1016/j.ejphar.2008.01.045
241
ZinsznerH.KurodaM.WangX.BatchvarovaN.LightfootR. T.RemottiH.et al (1998). CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev.12, 982–995. 10.1101/gad.12.7.982
Summary
Keywords
animal models, pharmacological treatments, metabolic syndrome, obesity, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, steatosis
Citation
Peng C, Stewart AG, Woodman OL, Ritchie RH and Qin CX (2020) Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front. Pharmacol. 11:603926. doi: 10.3389/fphar.2020.603926
Received
08 September 2020
Accepted
19 October 2020
Published
03 December 2020
Volume
11 - 2020
Edited by
Sabata Pierno, University of Bari Aldo Moro, Italy
Reviewed by
Fabio Vivarelli, University of Bologna, Italy
Jörn M. Schattenberg, Johannes Gutenberg University Mainz, Germany
Updates
Copyright
© 2020 Peng, Stewart, Woodman, Ritchie and Qin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Xue Qin, helena.qin@monash.edu
This article was submitted to Translational Pharmacology, a section of the journal Frontiers in Pharmacology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.