 Oana-Maria Thoma
Oana-Maria Thoma Markus F. Neurath
Markus F. Neurath Maximilian J. Waldner
Maximilian J. Waldner- 1Department of Medicine 1, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
- 2German Center for Immunotherapy (DZI), University Hospital Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
- 3Erlangen Graduate School in Advanced Optical Technologies (SAOT), Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
Cyclin-dependent kinases (CDKs) are key players in cell cycle regulation. So far, more than ten CDKs have been described. Their direct interaction with cyclins allow progression through G1 phase, transitions to S and G2 phase and finally through mitosis (M). While CDK activation is important in cell renewal, its aberrant expression can lead to the development of malignant tumor cells. Dysregulations in CDK pathways are often encountered in various types of cancer, including all gastrointestinal (GI) tract tumors. This prompted the development of CDK inhibitors as novel therapies for cancer. Currently, CDK inhibitors such as CDK4/6 inhibitors are used in pre-clinical studies for cancer treatment. In this review, we will focus on the therapeutic role of various CDK inhibitors in colorectal cancer, with a special focus on the CDK4/6 inhibitors.
Cyclin-Dependent Kinases and Their Role in Cell Cycle Progression
Cell cycle is defined as the process through which the cell replicates all its genomic material and divides into two identical cells (Alberts et al., 2002). It consists of four phases: gap 1 (G1), where the cell grows in size and transcribes the RNA and protein necessary during cell division; synthesis or S phase, where all chromosomes are being replicated; gap 2 (G2), where cell growth and protein synthesis continue; and mitosis or M phase, where the cell restructures its membrane and organizes the newly synthesized chromosomes and then divides into two daughter cells. Before entering cell cycle, highly proliferative cells such as stem cells and lymphocytes are in a reversible cell cycle arrest, known as quiescence or gap 0 (G0). However, other cells such as neurons or adipocytes are irreversibly arrested in G0 phase, a phenomenon often described as cellular senescence. Senescence is also predominant in highly damaged cells, acting as a protective mechanism during the DNA damage response (DDR) (Terzi et al., 2016).
Each cell cycle phase, as well as transitions from one phase to the other, are tightly regulated by interactions between cyclins and cyclin-dependent kinases (CDKs) (Johnson and Walker, 1999). In general, cyclins directly bind CDKs and induce the formation of cyclin—CDK complexes. This promotes CDK activity and therefore ensures activation of specific transcriptional programs that allow cell cycle progression. More than ten CDKs are known to be involved in various events during cell cycle. From these, CDK1, 2, 3, 4, and 6 directly mediate cell cycle progression.
Transition from quiescence or G0 phase in G1 phase is modulated by growth factor signals or mitogenic stimulation. These result in the upregulation of Cyclin D, which binds to and activates CDK4 and CDK6 to promote cell commitment to enter G1 phase (Jinno et al., 1999; Lea et al., 2003). High CDK4/6 expression and activation ensures cell progression through G1 phase (Mende et al., 2015; Topacio et al., 2019).
On the molecular level, CDK4 and 6 phosphorylate Retinoblastoma (Rb) and promote the accumulation of E2F, a direct regulator of genes necessary during DNA synthesis. Furthermore, CDK4 and CDK6 activation initiates cell growth through activation of mammalian target of rapamycin complex 1 (mTORC1) (Romero-Pozuelo et al., 2020). Besides, CDK4 and 6 are involved in the control of DNA replication mechanisms (Braden et al., 2008). Along with CDK4/6, CDK2 and CDK3 are also activated during G1 phase. Rb phosphorylation, and therefore the accumulation of E2F during G1 phase, directly mediate the upregulation of Cyclin E in late G1 phase, which binds and activates CDK2. Formation of CDK2/Cyclin E complex maintains Rb phosphorylated in order to promote G1/S phase transition (Massague 2004; Horiuchi et al., 2012). However, CDK3 upregulation during late G1 phase seems to be independent of Cyclin D, E or A binding (Braun et al., 1998). Interestingly, the upregulation of CDK2 has been also shown to be important during the G1/S checkpoint in response to DNA damage. For example, knocking-down CDK2 in the HCT116 tumor cell line significantly reduced p53 phosphorylation in response to hydroxyurea (HU) and suppressed G1/S cell cycle arrest (Bacevic et al., 2017). Some recent studies also described a role of CDK2 directly after mitosis, as an intermediate level will remain in the cells that continue proliferating, while those that lack CDK2 can enter quiescence or so called gap 0 (G0) (Spencer et al., 2013; Gookin et al., 2017). On the other hand, high levels of Cyclin C/CDK3 have been reported to directly mediate quiescence (Ren and Rollins 2004).
The beginning of S phase is marked by increasing levels of Cyclin A, which binds CDK2. The complex formed by Cyclin A/CDK2 drives the cells through S phase and promotes DNA replication. During late S/G2 phase, increased levels of Cyclin A induce CDK1 activation, which drives entry into mitosis (Gavet and Pines, 2010; Kalous et al., 2020). Later, the formation of CDK1/Cyclin B complex triggers progression through M phase. Along with its important role in successful cell mitosis (Vassilev et al., 2006), CDK1 can also influence the remodeling of cell adhesion complexes during G1, S and G2 cell cycle phases (Jones et al., 2018) and promotes protein synthesis during proliferation (Haneke et al., 2020). Interestingly, CDK1 is reported to be the only necessary cyclin-dependent kinase during cell cycle, being able to bind to all cyclins and drive all events during cell division (Santamaria et al., 2007).
Several other CDKs are known to be involved in cell cycle progression as well. CDK7, for example, is an important cell cycle regulator. Its binding to Cyclin H and mating-type 1 protein (Mat1) induces the formation of CDK-activating kinase (CAK) complex. CAK activity is crucial to promote CDK2 and CDK1 binding to cyclins, therefore allowing cell division (Fisher and Morgan, 1994; Larochelle et al., 2007; Olson et al., 2019). CDK5 upregulation is mostly observed in, but not limited to, neurons, and is often correlated to cell apoptosis. Nevertheless, it can also regulate the cell cycle by phosphorylating Rb and interacting with E2F during G1 phase (Zhang et al., 2010; Chang et al., 2012; Futatsugi et al., 2012). CDK8 is a partner of Cyclin C and its expression has been shown to be important in stabilizing Cyclin C activity during cell cycle (Tassan et al., 1995; Barette et al., 2001). Interestingly, CDK8 and Cyclin C, as well as CDK19/Cyclin C complex, are strongly required during p53-dependent p21 transcriptional activation, for cell cycle arrest in response to DNA damage (Donner et al., 2007; Audetat et al., 2017). Last, cyclin-dependent kinases such as CDK9 and CDK13 are not directly controlling cell cycle phase transitions, but are rather involved in transcription mechanisms, by associating with Cyclin T or Cyclin K (Garriga et al., 2003; Yu et al., 2010; Greifenberg et al., 2016).
To summarize, entry into cell cycle depends on mitogenic or growth factor signals. CDK4/6/Cyclin D complex formation promotes Rb phosphorylation and accumulation of free E2F, which ensures progression through G1 phase. CDK5 activity also increases E2F levels during G1. High levels of E2F during late G1 induce CDK2/Cyclin E complex that in return further phosphorylates Rb and promotes G1/S transition. At the beginning of S phase, Cyclin E levels decrease and CDK2 forms a complex with the increasing Cyclin A, which not only ensures progression through S phase, but also transition into G2 phase. CDK2/Cyclin A complex is especially regulated by the CDK7/Cyclin H/Mat1 complex, also described as CAK. CAK also regulates CDK1/Cyclin A complex formation during late G2 and Cyclin B binding to CDK1 during mitosis. Any disturbances to the cell cycle machinery will result in cell cycle arrest. CDK2 and CDK3 are especially important in mediating either quiescence or senescence. Indirectly, CDK8, 9, 13, and 19 also mediate cell cycle, being involved in the transcription machinery, while CDK5 can directly modulate apoptosis as well. A schematic representation of the important role of CDKs in cell cycle is shown in Figure 1. While normal cells are able to activate the necessary mechanisms for cell cycle arrest when the DNA is damaged, these pathways are usually suppressed or non-existent in tumor cells, enabling them to continue progression through cell cycle. The following sections will address the CDK’s role in the tumor cell division and how therapies targeting CDKs can modulate CRC development.
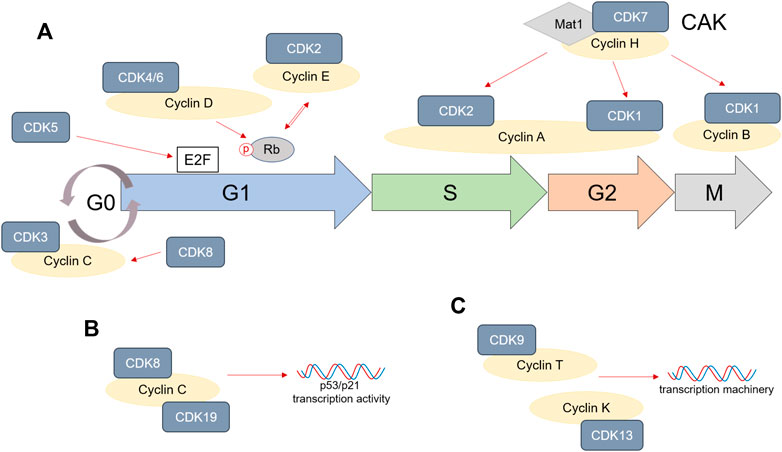
FIGURE 1. Cyclins and cyclin-dependent kinase (CDK) role in cell cycle. (A) CDK1, 2, 3, 4/6, and 7 are directly involved in progression through cell cycle phases by associating themselves with various Cyclins. CDK5 can have a direct impact on E2F accumulation, especially in cancer, while CDK8 activation stabilizes Cyclin C. (B,C) CDK8, 9, 13, and 19 are not directly involved in cell cycle progression, but are involved in either p53/p21 transcription (CDK8/19) or the DNA transcription machinery (CDK9/13).
Cyclin-Dependent Kinase Expression in Human Colorectal Cancer
Changes in the regulatory mechanisms that control cell division are often related to accumulation of mutations and/or epigenetic dysregulations of cancer related genes and can contribute to the molecular mechanisms of colorectal cancer (CRC). CRC tissue often shows changes in genes related to cell cycle arrest (p16 and p21), apoptosis (p53) or proliferation (PCNA) (Yue et al., 2003; Kruschewski et al., 2011). Multiple other mutations have also been described to be involved in CRC development. As a result, CDKs expression can be changed in tumor cells.
When looking at the signature of differentially expressed genes (DEGs) in patients with CRC compared to normal colon tissue, an upregulation in CDK1 gene expression is often observed (Zhao et al., 2019; Ding et al., 2020; Li et al., 2020). Interestingly, the expression of CDK1 in the nucleus and cytoplasm has been used as a marker to describe patterns in the overall survival of patients with CRC (Sung et al., 2014). Staining of over 164 cancer samples from primary CRC revealed that CDK1 is expressed in both cell nucleus and cytoplasm to a certain degree. The evaluation of nuclear/cytoplasm (N/C) ratio on these samples showed that high N/C expression is often found in patients with overall worse survival and a N/C > 1.5 can be considered a risk factor. Furthermore, high CDK1 expression is predominant in patients with resistance to 5-fluorouracil (5-Fu), a common CRC treatment, and it seems to reduce the effect of chemotherapy (Zhu et al., 2020). An upregulation of CDK1 in CRC has been also observed in response to other drugs such as: betaxol, penbutolol and propofol amongst others (Mastrogamvraki and Zaravinos, 2020).
CDK2, 4 and 6 levels in CRC are closely related to the Rb protein hyperphosphorylation, which seems to promote cancer progression. CDK4/6 is usually amplified in colon tumors compared to healthy epithelium (Mastrogamvraki and Zaravinos, 2020; Jardim et al., 2021). Abundant levels of CDK4 are especially observed in CRC patients with enhanced dysplasia and are correlated to increased tumor cell proliferation (Zhang et al., 1997; Bartkova et al., 2001). Some CDK2 expression is normally found in healthy epithelium. However, its upregulation can be predominantly observed human CRC tissue samples (Yamamoto et al., 1995). Interestingly, CDK2 overexpression in primary CRC tumors is also linked to lymph nodes metastasis, but not liver metastasis (Li et al., 2001; McCurdy et al., 2017). Nevertheless, a certain CDK2 activity has been reported to improve recurrence-free survival (RFS) of patients after surgery (Yamamoto et al., 1995). A similar pattern to CDK2 expression in CRC is observed in CDK3 levels as well. Its overexpression has been linked to metastasis and tumor cell invasion, where it seems to be promoting epithelial to mesenchymal transitions (Lu et al., 2016).
CDK5 expression is also reported to be much higher in CRC cells compared to normal epithelium and it correlates to increased tumor growth and poor prognosis (Zhuang et al., 2016; de Porras et al., 2019). Most important, CDK5 is directly involved in the degradation of the cell cycle inhibitor p21 and can enhance CDK2 activity, which might further promote tumor cell growth (Huang et al., 2016). Decreased survival rates are also observed in CRC patients with high CDK9 and CDK13 levels (Kim et al., 2012; Wang et al., 2019). Interestingly, high CDK9 expression in CRC tissue was negatively correlated with cytotoxic CD8+ T cell infiltration. Furthermore, these infiltrated cells showed increased cell exhaustion in CDK9-high tumors, which might further affect patient outcome (Wang et al., 2019). Last, CDK8 overexpression in CRC is also considered as a marker for poor patient prognosis, being directly linked to β-catenin activation amongst others and therefore promoting cancer growth (Firestein et al., 2008; Firestein et al., 2010; Seo et al., 2010). Overall, cyclin-dependent kinase activation is often observed in colorectal cancer and seems to promote tumor progression and an overall worse survival of patients, as summarized in Table 1.
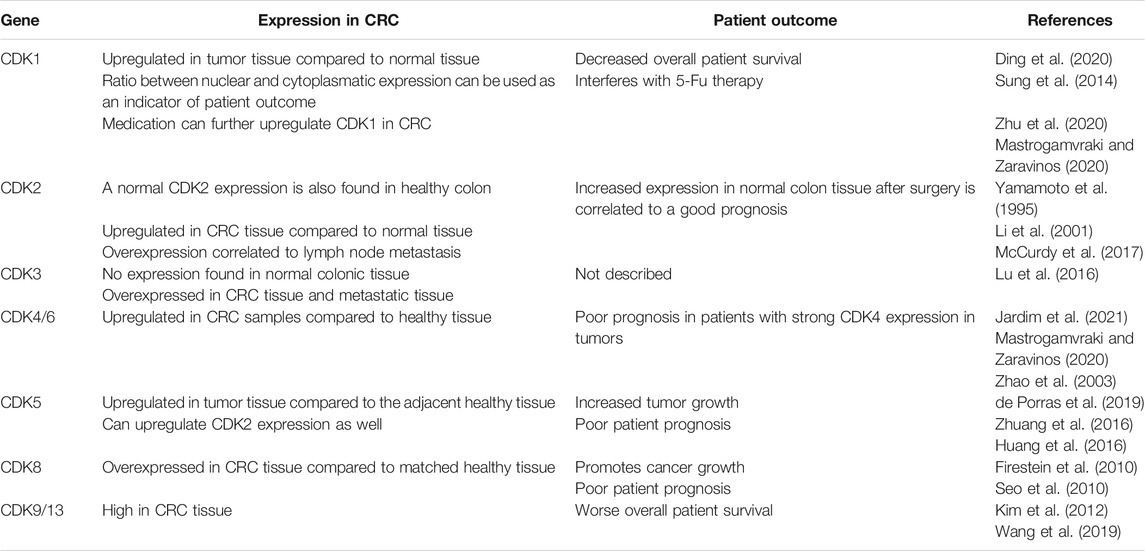
TABLE 1. Effects of increased CDK expression in patients with colorectal cancer.
The Functional Role of CDKs in CRC
Basic research using murine knockout models or in vitro gene silencing in tumor colon cancer cell lines also provided some understanding for the relevance on CDKs in CRC development. Since CDKs are vital components of the cell cycle, creating knockout mouse models is usually unsuccessful. This is because most CDKs (e.g. CDK1, 4, 6, 9, and 13) are critical during embryonic development, as is summarized in (Campbell et al., 2020). Similarly, conditional knockout models often show severe impairments.
Nevertheless, some fundamental research data in regards to the role of CDKs in colorectal cancer are available. For example, it is know that CDK4 activation in CDK4R24C/R24CApc+/min mice leads to significant increased in tumor vascularity in comparison to CDK4+/+Apc+/min mice or APC+/min mice (Abedin et al., 2010), while knocking out CDK4 in APC+/min mice reduces adenoma development (Karim et al., 2013). CDK5 silencing via transfection can directly reduce the proliferation of human HCT116 and SW480 tumor cell lines (Zhuang et al., 2016). Similarly, knocking down CDK9 in HCT116 and HT29 tumor cell lines induced their apoptosis by Caspase 7 cleavage (Rahaman et al., 2019). Furthermore, it reduced Cyclin D1 protein expression, suggesting cell cycle arrest induction in these cells.
Stable silencing of CDK8 and CDK19 in Colo205 human colon cancer cells reduced β-catenin/TCF-dependent transcription (Dale et al., 2015). A direct link between CDK8 and β-catenin regulation in tumor cell proliferation and death has also been described, where inactivation of CDK8 by siRNA transfection in HCT116 cells significantly reduced the RNA and protein levels of β-catenin (He et al., 2011). Generally, silencing CDK genes in colon cancer cells reduces their proliferation and induces cell death, which makes them an attractive target for the development of new inhibitory therapies.
CDK Inhibitors as a Potential CRC Treatment
CDK inhibitors are also often used in basic research to understand molecular mechanisms of CDK activation in cell cycle regulation or tumor cell proliferation. This section describes the current understanding on the potential use of various CDK inhibitors to mediate colorectal cancer development.
CDK7-Specific Inhibitors
Samuraciclib and SY-1365 are inhibitors of CDK7 activity. Interestingly, the colon cell line HCT116 is particulary sensitive to Samuraciclib, which induces their apoptosis and cell cycle arrest (Patel et al., 2018). Its mechanism of action is mostly based on inhibition of phosphorylation of CDK7 substrates like CDK1 and 2. One important advantage of Samuraciclib is its availability as an oral drug that can accumulate at the tumor site upon multiple doses, as shown by the in vivo HCT116 murine tumor xenograf model. CDK7 inhibition was also successful when using SY-1365, in more than 26 types of cancer types, including colon cell lines (Hu et al., 2019).
CDK1/2-Specific Inhibitors
SU9516 and CVT-313 are known to directly inhibit CDK2 activity. The use of SU9516 for in vitro treatment of HT29, RKO and SW480 human colon carcinoma cell lines revealed that it can successfully induce their apoptosis and cell cycle arrest (Lane et al., 2001; Yu et al., 2002). CDK2 inhibition also significantly decreases free E2F, but increases E2F/Rb complexes, therefore arresting the tumor cells. This effect was dependent on the duration of the treatment, since more E3F/Rb complexes were observed after 48 h than after 24 h in HT29 cell line. Inhibition of CDK2 in patient-derived human cell lines using CVT-313 has minimal effect on cell death (Somarelli et al., 2020). Nevertheless, combined therapy using CDK2 and 9 inhibitors significantly increased the numbers of cells arrested in G2/M.
RO-3306 is a CDK1-specific inhibitor can be used to induce apoptosis in a specific type of BRAF-mutated colorectal cancer cells (Zhang et al., 2018). Interestingly, this inhibitor induced Caspase 8-regulated cell death when combined with the MEK inhibitor, cobimetinib, while most CDK inhibitors promote apoptosis via Caspase 3 cleavage.
CDK5, 8/19, and 9-Specific Inhibitors
CP668863 or 20-223 is a CDK5 inhibitor whose cytotoxic potential has been evaluated in CRC settings as well (Robb et al., 2018). Interestingly, 20-223 is 65-fold more potent for cell growth inhibition than the pan CDK inhibitor AT7519. Its cytotoxicity has been evaluated on SW620, DLD1 and HT29 tumor cell lines. 20-223 also significantly inhibited tumor growth in xenograf models and reduced the migration of colon cancer cells, which shows its potential for CRC therapy.
The development of MSC2530818 was fine tuned to specifically inhibit CDK8/19 (Czodrowski et al., 2016). This compound can be orally administered and it is well tolerated by mice. Treatment with MSC2530818 of mice subjected to an in vivo xenograft model using SW620 human colon cell line showed its potential to reduce tumor growth. CDK8/19 inhibition by MSC2530818 it is known to directly reduce STAT1 phosphorylation, further proving its efficacy.
CDKI-73 is a potent CDK9 inhibitor, which shows increased cytotoxicity against the HT29 and HCT116 human carcinoma cell lines (Rahaman et al., 2019). In vitro treatment of these cell lines revealed that CDKI-73 reduces the expression of survival genes. Its effect has also been tested in in vivo HT116 xenograf models. CDKI-73 significantly reduced tumor growth without being over toxic to the mice.
Purvanalol and Roscovitine
Purvanalol and Roscovitine (Celiciclib or CYC202) are common CDK inhibitors effective against CDK2, 4, and 5 activity. Purvanalol is known to induce apoptosis and autophagy of HCT116 colon tumor cells by activating endoplasmatic reticulum (ER) stress (Coker-Gurkan et al., 2015). Its effect is nevertheless limited to wildtype HCT116, while Bax-deficient HCT116 cells are resistant against this treatment. This effect can be overcome by combining of Purvanalol with 3-MA, an inhibitor of autophagy, which promotes Purvanalol-induced apoptosis in Bax−/− HCT116 as well (Coker-Gurkan et al., 2014). Roscovitine has a similar effect on apoptosis induction in HCT116 tumor cells, but on a weaker scale than Purvanalol (Gurkan et al., 2013; Coker-Gurkan et al., 2015). Analysis of Roscovitine-induced apoptosis using Raman spectroscopy revealed changes in amide I and III bands, common of protein and DNA alterations (Akyuz et al., 2011). HCT116 cell death in presence of Roscovitine has been shown to be enhanced during polyamine depletion or phosphatase nuclear targeting subunit (PNUTS) knockdown (De Leon et al., 2010; Arisan et al., 2012). More important, the effect of Roscovitine is especially higher in combination to current chemotherapeutic drugs such as 5-Fu or doxorubicine, as shown by the experiments done with SW48, SW116 and SW837 colon cancer cell lines (Abaza et al., 2008).
Wogonin
Wogonin is a flavone isolated from Scutellaria baicalensis known to inhibit CDK2, 4, 8, and 9. Nevertheless, its effect is not specific to only CDKs, but it also downregulates activation of PI3K/Akt and Stat3 signaling pathways (Wang et al., 2014; Tan et al., 2019). Along with its role in inducing apoptosis and autophagy of colorectal tumor cells, Wogonin can also induce cell cycle arrest in both G1 and G2/M cell cycle phases (He et al., 2013; Tan et al., 2019). Interestingly, Wogonin treatment of wildtype mice subjected to AOM/DSS tumor model reduces tumor growth by facilitating nuclear translocation of tumor suppresor p53 (Feng et al., 2018).
Flavopiridol
Flavopiridol or Alvocidib is effective in inhibiting most CDKs: CDK1, 2, CDK4/6 and 9, by inducing cell cycle arrest and apoptosis of human colon tumor cell lines (Sausville et al., 2000; Kim et al., 2003; Okada et al., 2017). Treatment of CRC cell lines with flavopiridol enhances cell death when used in combination with chemotherapeutic agent gemcitabine or γ-radiation (Jung et al., 2001; Jung et al., 2003). Furthermore, a combination of docetaxal, flavopiridol and 5-Fu is described to more effective in inhibiting tumor growth and inducing increased apoptosis in HCT116 tumor cells, than any of the drugs alone (Guo et al., 2006). Phase I and phase II studies in patients with untreated advanced colorectal cancer showed little efficacy and was terminated early (Aklilu et al., 2003). Overall, it appears that Flavopiridol works best when coupled with other chemotherapeutic drugs.
Other Pan CDK Inhibitors
Along with Purvanalol, Roscovitine, Wogonin and Flavopiridol, various other molecules have been described to inhibit multiple CDKs. For example, AT7519 is able to inhibit CDK1, 2, 4/6, and 9 and therefore induce colon cancer cell death. Its potency has been observed in xenograf mouse models using HCT116 and HT29, where tumor regression was observed upon multiple doses (Squires et al., 2009). Nevertheless, other CDK inhibitors such as 20-223 seem to be more effective than AT7519 (Robb et al., 2018). Pan CDK inhibitor AG-012986 has been shown to significantly reduce the colony formation of HCT116 colon carcinoma in a concentration-dependent manner, by inducing arrest into G1 phase (Zhang et al., 2008). Indirubin derivates are also known to reduce proliferation of DLD1 and HT29 tumor cell lines (Kim et al., 2009). Last, SNS-032 or BMS-387032, a specific inhibitor against CDK2, 7, and 9, was used to significantly reduce the intestinal tumor burden of Ink4/Arf-null Min mice (Boquoi et al., 2009). All in all, these data provide important insight on the effectiveness of CDK inhibitors in colorectal cancer therapy.
CDK4/6 Inhibitors Use in CRC
When thinking about preventing cell cycle progression and proliferation of tumor cells, CDK4/6 inhibitors are very efficient. The most commonly used are Ribociclib, Palbociclib, Abemaciclib and Trilaciclib. CDK4/6 inhibitors are especially effective at treating breast cancer amongst others, many of them being nowadays tested in phase I and II clinical trials (Wu et al., 2020). Nevertheless, they are also being tested as therapy for colorectal cancer. A schematic representation of the mechanism of action of CDK4/6 inhibitors is shown in Figure 2.
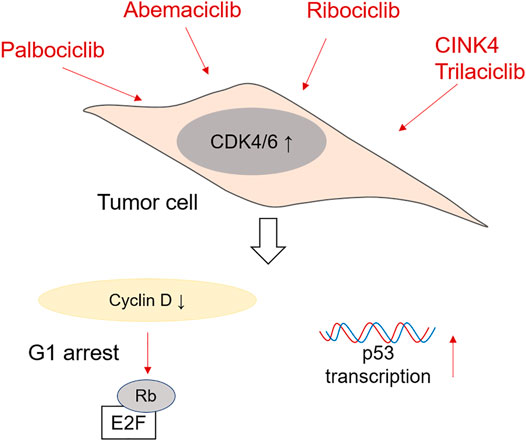
FIGURE 2. Mechanism of action of CDK4/6 inhibitors in CRC cells. Palbociclib, Ademaciclib, Ribociclib, CINK4 and Trilaciclib are able to prevent the formation of Cyclin D/CDK4/6 complexes, which reduces Retinoblastoma phosphorylation and induces G1 cell cycle arrest. Palbociclib has been shown to be effective in promoting p53 transcription after irradiation as well.
CINK4 and Trilaciclib
Small molecule CINK4 is a triaminopyrimidine derivative specially designed to inhibit the activity of CDK4 in tumor cells. In vitro treatment of HCT116 colon tumor cell line with CINK4 prevented their cell growth by reducing Cyclin D/CDK4 complexes and Rb phosphorylation (Soni et al., 2001). Furthermore, intraperitoneal injection of CINK4 every 12 h was successful in reducing tumor growth in an in vivo mouse xenograf model using HCT116 tumor cells. Trilaciclib (CoselaTM) is known to directly induce reversible G1 cell cycle arrest and inhibit the formation of complexes between CDK4/6 and Cyclin D. As of 2021, Trilaciclib is used in a multinational trial (ClinicalTrials.gov Identifier: NCT04607668 in United States) in treating microsatelite stable metastatic CRC, in patients treated with FOLFOXIRI and Bevacizumab (Dhillon 2021). This clinical study has been recently approved and is at the moment recruiting participants in USA, Europe (Hungary, Italy, Poland, Slovakia, Spain, Ukraine, United Kingdom) and China.
Abemaciclib
Patients with advanced and metastatic breast cancer can be treated with the CDK4/6 inhibitor Abemaciclib (also known as LY2835219, Verzenio, Verzenios, Ramiven). This inhibitor is also involved in various clinical trials for treating other advanced solid tumors such as melanoma or lung cancer (Shapiro et al., 2013; Fujiwara et al., 2016). The potential of Abemaciclib to treat colorectal cancer has been tested in mice with human tumor xenographs using Colo205 and A375 (Tate et al., 2014). The mice were treated orally in a concentration-dependent manner. The authors suggest that a constant level of 200 ng/ml Abemaciclib in plasma are necessary to arrest the tumor cells in G1 phase, as shown by Rb phosphorylation data. This shows that treatment using multiple doses might promote tumor cell cycle arrest in humans as well. Indeed Abemaciclib therapy in CRC patient cohort during a clinical trial induced stable disease even in a patient with KRAS and p53 mutated tumor cells (Patnaik et al., 2016). At the moment, Abemaciclib, in combination with LY3214996 (ERK1/2 inhibitor) and Cetuximab (EGFR inhibitor), is undergoing evaluation in Phase I and Phase II clinical trials in patients with metastatic CRC (ClinicalTrials.gov Identifier: NCT04616183). Recruiting phase is set to be completed in December 2021.
Palbociclib
The efficacy of Palbociclib (PD-0332991) in inhibiting CDK4/6 activity has been assessed in human colon carcinoma cell lines as well (Li et al., 2014). Palbociclib successfully arrested various tumor cells (HT29, Colo205 and DLD1 amongst others) in G1 cell cycle phase, by reducing the phosphorylation of Rb. Interestingly, its therapeutic effect does depend on Rb presence (Heijink et al., 2011). Nevertheless, in vivo administration of Palbociclib in ApcMin mice successfully reduced tumor cell proliferation without affecting normal epithelial cells. It is very important to remark that Palbociclib mechanism of action directly targets the transcriptional activity of p53 after exposure to radiation and therefore, its efficacy might be limited to p53-expressing CRC tumors (Fernandez-Aroca et al., 2019). Palbociclib is also involved in a phase II clinical trial (ClinicalTrials.gov Identifier: NCT03981614), where it is used in combination with chemotherapeutic drug TAS-102 for KRAS/NRAS metastatic or unresectable CRC. First phase of the study has been recently completed (June 2021), but no data are momentarily available.
Ribociclib
Treatment of HT29 and SW480 colon tumor cell lines with Ribociclib (or LEE011) significantly decreases their viability and induces G1 cell cycle arrest in concentration dependent manner (Lin et al., 2020). Similarly to the other CDK4/6 inhibitors, Ribociclib also reduces the phosphorylation of Retinoblastoma protein. Furthermore, used in combination with 5-FU, it increases significantly p53 phosphorylation. Ribociclib treatment was also used in a study case on a young female diagnosed with desmoid tumors (DT) (Santti et al., 2019). She underwent colectomy and various other surgeries to remove the tumors, as well as irradiation therapy. Unfortunately, the treatment with cytotoxic drugs usually used to treat these cancers did not reduced the tumors. The addition of Ribociclib, together with goserelin and letrozole therapy, stabilized temporarily the tumors and gave symptomatic relief. A Phase I clinical trial for treating selected malignancies, including CRC, using Ribociclib in combination with TNO155 (SPH2 inhibitor) is currently running (ClinicalTrials.gov Identifier: NCT04000529). Patients are still being recruited in this clinical trial.
Future Perspectives in CDK4/6 Inhibitor Therapy in CRC
There is no doubt that targeting cell cycle machinery, and especially cyclin-dependent kinase activity of tumor cells, offers new opportunities to treat patients with advanced colorectal cancer. Nevertheless, cancer itself is a multifactorial disease and therefore the treatment with just one drug is not always successful.
CDK4/6 inhibitor therapy in particular shows promising results in the relief and stabilization of the patients, but its effect is amplified when used in combination with other treatments. More recent studies have focused on evaluating therapeutic potential of CDK4/6 inhibitors when coupled with other drugs in treating CRC. For example, when treating tumors in patient-derived Rb+ colorectal xenograph models, the authors found that a combination of MEK inhibitor Trametinib with Palbociclib significantly reduces tumor volume in comparison to monotherapy. Furthermore, KRAS-mutated cells were especially sensitive to this treatment (Lee et al., 2016; Ziemke et al., 2016). Similiar results were obtained when using a Raf inhibitor (LY3009120) in combination with Abemaciclib, where Ras- and Braf-mutated CRC was especially sensitive to this treatment (Chen et al., 2018). Last, the combination of checkpoint inhibitors like anti-PD1 therapy (SHR-1210) with CDK4/6 inhibitor (SHR6390) is currently evaluated in Phase I and II clinical trial for advanced colorectal cancer (ClinicalTrials.gov Identifier: NCT03601598), but no data have been published yet.
Further studies are necessary for understanding the potential of targeting CDK4/6, together with other genes involved in cell cycle machinery. For example, tumor cells depend on high telomerase activity, which enables them to preserve the telomeres during extensive proliferation. Inducing telomere dysfunctions in tumor cells, using the telomere-specific inhibitor 6-thio-dG, potentiates antitumor responses in mice bearing MC38 tumors (Mender et al., 2020). Therefore, combining CDK4/6 inhibitors for cell cycle arrest and 6-thio-dG might provide a more efficient tumor targeted therapy.
One significant challenge raised by the use of CDK4/6 inhibitors is its effect on normal cells, and especially on the highly proliferating cells, such as activated immune cells found in the tumor microenvironment. Targeting CDKs might disrupt the function of upstream genes involved in the cell cycle, such as sirtuins, in normal cells. Modifications in sirtuin 1 (SIRT1) function are especially important. Even though SIRT1 is also upregulated in the CRC tissue compared to the normal one and it has been linked to tumor size and invasion (Chen et al., 2014; Yu et al., 2016), its function in haematopoiesis is nevertheless crucial (Rimmele et al., 2012). Dysfunctions in SIRT1 in normal cells due to CDK4/6 inhibitor use might therefore potentiate cellular senescence and premature aging in various cellular compartments (Sasaki et al., 2006).
Overall, CDK inhibitors are efficient in preventing colon tumor cells from proliferating by inducing cell cycle arrests, and, in some cases, even apoptosis, making them useful for developing new potential therapeutic strategies for CRC. Nevertheless, a comprehensive analysis on how CDK inhibitors might affect normal cells, as well as the antitumor response of immune cells to CRC, would enhance our understanding on this novel therapy.
Author Contributions
O-MT and MW prepared the concept, wrote and reviewed the manuscript. MN provided valuable input during the review process of this manuscript.
Funding
The authors gratefully acknowledge funding by German Research Foundation (DFG) within the Forschergruppe 2438 (FOR 2438), as well as by Deutsche Krebshilfe (DKH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abaza, M. S., Bahman, A. M., and Al-Attiyah, R. J. (2008). Roscovitine Synergizes with Conventional Chemo-Therapeutic Drugs to Induce Efficient Apoptosis of Human Colorectal Cancer Cells. World J. Gastroenterol. 14, 5162–5175. doi:10.3748/wjg.14.5162
Abedin, Z. R., Ma, Z., Reddy, E. P., and Litvin, J. (2010). Increased Angiogenesis in Cdk4(R24C/R24C):Apc(+/Min) Intestinal Tumors. Cell Cycle 9, 2456–2463. doi:10.4161/cc.9.12.12055
Aklilu, M., Kindler, H. L., Donehower, R. C., Mani, S., and Vokes, E. E. (2003). Phase II Study of Flavopiridol in Patients with Advanced Colorectal Cancer. Ann. Oncol. 14, 1270–1273. doi:10.1093/annonc/mdg343
Akyuz, S., Ozel, A. E., Balci, K., Akyuz, T., Coker, A., Arisan, E. D., et al. (2011). Raman Micro-spectroscopic Analysis of Cultured HCT116 colon Cancer Cells in the Presence of Roscovitine. Spectrochim Acta A. Mol. Biomol. Spectrosc. 78, 1540–1547. doi:10.1016/j.saa.2011.01.046
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, E., and Walter, P. (2002). Molecular Biology of the Cell. 4th Edition. New York: Garland Science Publishing.
Arisan, E. D., Coker, A., and Palavan-Unsal, N. (2012). Depletion Enhances the Roscovitine-Induced Apoptosis through the Activation of Mitochondria in HCT116 colon Carcinoma Cells. Amino Acids 42, 655–665.
Audetat, K. A., Galbraith, M. D., Odell, A. T., Lee, T., Pandey, A., Espinosa, J. M., et al. (2017). A Kinase-independent Role for Cyclin-dependent Kinase 19 in P53 Response. Mol. Cell Biol 37, 37. doi:10.1128/MCB.00626-16
Bacevic, K., Lossaint, G., Achour, T. N., Georget, V., Fisher, D., and Dulic, V. (2017). Strengthens the Intra-S Checkpoint and Counteracts Cell Cycle Exit Induced by DNA Damage. Sci. Rep. 7, 13429.
Barette, C., Jariel-Encontre, I., Piechaczyk, M., and Piette, J. (2001). Human Cyclin C Protein Is Stabilized by its Associated Kinase Cdk8, Independently of its Catalytic Activity. Oncogene 20, 551–562. doi:10.1038/sj.onc.1204129
Bartkova, J., Thullberg, M., Slezak, P., Jaramillo, E., Rubio, C., Thomassen, L. H., et al. (2001). Aberrant Expression of G1-phase Cell Cycle Regulators in Flat and Exophytic Adenomas of the Human colon. Gastroenterology 120, 1680–1688. doi:10.1053/gast.2001.24880
Boquoi, A., Chen, T., and Enders, G. H. (2009). Chemoprevention of Mouse Intestinal Tumorigenesis by the Cyclin-dependent Kinase Inhibitor SNS-032. Cancer Prev. Res. (Phila) 2, 800–806. doi:10.1158/1940-6207.CAPR-09-0053
Braden, W. A., McClendon, A. K., and Knudsen, E. S. (2008). Cyclin-dependent Kinase 4/6 Activity Is a Critical Determinant of Pre-replication Complex Assembly. Oncogene 27, 7083–7093. doi:10.1038/onc.2008.319
Braun, K., Hölzl, G., Soucek, T., Geisen, C., Möröy, T., and Hengstschläger, M. (1998). Investigation of the Cell Cycle Regulation of Cdk3-Associated Kinase Activity and the Role of Cdk3 in Proliferation and Transformation. Oncogene 17, 2259–2269. doi:10.1038/sj.onc.1202145
Campbell, G. J., Hands, E. L., and Van de Pette, M. (2020). The Role of CDKs and CDKIs in Murine Development. Int. J. Mol. Sci. 21, 21. doi:10.3390/ijms21155343
Chang, K. H., Vincent, F., and Shah, K. (2012). Deregulated Cdk5 Triggers Aberrant Activation of Cell Cycle Kinases and Phosphatases Inducing Neuronal Death. J. Cell Sci 125, 5124–5137. doi:10.1242/jcs.108183
Chen, S. H., Gong, X., Zhang, Y., Van Horn, R. D., Yin, T., Huber, L., et al. (2018). RAF Inhibitor LY3009120 Sensitizes RAS or BRAF Mutant Cancer to CDK4/6 Inhibition by Abemaciclib via superior Inhibition of Phospho-RB and Suppression of Cyclin D1. Oncogene 37, 821–832. doi:10.1038/onc.2017.384
Chen, X., Sun, K., Jiao, S., Cai, N., Zhao, X., Zou, H., et al. (2014). High Levels of SIRT1 Expression Enhance Tumorigenesis and Associate with a Poor Prognosis of Colorectal Carcinoma Patients. Sci. Rep. 4, 7481. doi:10.1038/srep07481
Coker-Gürkan, A., Arisan, E. D., Obakan, P., Akalın, K., Özbey, U., and Palavan-Unsal, N. (2015). Purvalanol Induces Endoplasmic Reticulum Stress-Mediated Apoptosis and Autophagy in a Time-dependent Manner in HCT116 colon Cancer Cells. Oncol. Rep. 33, 2761–2770. doi:10.3892/or.2015.3918
Coker-Gurkan, A., Arisan, E. D., Obakan, P., Guvenir, E., and Unsal, N. P. (2014). Inhibition of Autophagy by 3-MA Potentiates Purvalanol-Induced Apoptosis in Bax Deficient HCT 116 colon Cancer Cells. Exp. Cell Res 328, 87–98. doi:10.1016/j.yexcr.2014.07.022
Czodrowski, P., Mallinger, A., Wienke, D., Esdar, C., Pöschke, O., Busch, M., et al. (2016). Structure-Based Optimization of Potent, Selective, and Orally Bioavailable CDK8 Inhibitors Discovered by High-Throughput Screening. J. Med. Chem. 59, 9337–9349. doi:10.1021/acs.jmedchem.6b00597
Dale, T., Clarke, P. A., Esdar, C., Waalboer, D., Adeniji-Popoola, O., Ortiz-Ruiz, M. J., et al. (2015). A Selective Chemical Probe for Exploring the Role of CDK8 and CDK19 in Human Disease. Nat. Chem. Biol. 11, 973–980. doi:10.1038/nchembio.1952
De Leon, G., Cavino, M., D’Angelo, M., and Krucher, N. A. (2010). PNUTS Knockdown Potentiates the Apoptotic Effect of Roscovitine in Breast and colon Cancer Cells. Int. J. Oncol. 36, 1269–1275. doi:10.3892/ijo_00000611
Ding, X., Duan, H., and Luo, H. (2020). Identification of Core Gene Expression Signature and Key Pathways in Colorectal Cancer. Front. Genet. 11, 45. doi:10.3389/fgene.2020.00045
Donner, A. J., Szostek, S., Hoover, J. M., and Espinosa, J. M. (2007). CDK8 Is a Stimulus-specific Positive Coregulator of P53 Target Genes. Mol. Cell 27, 121–133. doi:10.1016/j.molcel.2007.05.026
Feng, Q., Wang, H., Pang, J., Ji, L., Han, J., Wang, Y., et al. (2018). Prevention of Wogonin on Colorectal Cancer Tumorigenesis by Regulating P53 Nuclear Translocation. Front. Pharmacol. 9, 1356. doi:10.3389/fphar.2018.01356
Fernández-Aroca, D. M., Roche, O., Sabater, S., Pascual-Serra, R., Ortega-Muelas, M., Sánchez Pérez, I., et al. (2019). P53 Pathway Is a Major Determinant in the Radiosensitizing Effect of Palbociclib: Implication in Cancer Therapy. Cancer Lett. 451, 23–33. doi:10.1016/j.canlet.2019.02.049
Firestein, R., Bass, A. J., Kim, S. Y., Dunn, I. F., Silver, S. J., Guney, I., et al. (2008). CDK8 Is a Colorectal Cancer Oncogene that Regulates Beta-Catenin Activity. Nature 455, 547–551. doi:10.1038/nature07179
Firestein, R., Shima, K., Nosho, K., Irahara, N., Baba, Y., Bojarski, E., et al. (2010). CDK8 Expression in 470 Colorectal Cancers in Relation to Beta-Catenin Activation, Other Molecular Alterations and Patient Survival. Int. J. Cancer 126, 2863–2873. doi:10.1002/ijc.24908
Fisher, R. P., and Morgan, D. O. (1994). A Novel Cyclin Associates with MO15/CDK7 to Form the CDK-Activating Kinase. Cell 78, 713–724. doi:10.1016/0092-8674(94)90535-5
Fujiwara, Y., Tamura, K., Kondo, S., Tanabe, Y., Iwasa, S., Shimomura, A., et al. (2016). Phase 1 Study of Abemaciclib, an Inhibitor of CDK 4 and 6, as a Single Agent for Japanese Patients with Advanced Cancer. Cancer Chemother. Pharmacol. 78, 281–288. doi:10.1007/s00280-016-3085-8
Futatsugi, A., Utreras, E., Rudrabhatla, P., Jaffe, H., Pant, H. C., and Kulkarni, A. B. (2012). Cyclin-dependent Kinase 5 Regulates E2F Transcription Factor through Phosphorylation of Rb Protein in Neurons. Cell Cycle 11, 1603–1610. doi:10.4161/cc.20009
Garriga, J., Bhattacharya, S., Calbó, J., Marshall, R. M., Truongcao, M., Haines, D. S., et al. (2003). CDK9 Is Constitutively Expressed throughout the Cell Cycle, and its Steady-State Expression Is Independent of SKP2. Mol. Cell Biol 23, 5165–5173. doi:10.1128/mcb.23.15.5165-5173.2003
Gavet, O., and Pines, J. (2010). Progressive Activation of CyclinB1-Cdk1 Coordinates Entry to Mitosis. Dev. Cell 18, 533–543. doi:10.1016/j.devcel.2010.02.013
Gookin, S., Min, M., Phadke, H., Chung, M., Moser, J., Miller, I., et al. (2017). A Map of Protein Dynamics during Cell-Cycle Progression and Cell-Cycle Exit. Plos Biol. 15, e2003268. doi:10.1371/journal.pbio.2003268
Greifenberg, A. K., Hönig, D., Pilarova, K., Düster, R., Bartholomeeusen, K., Bösken, C. A., et al. (2016). Structural and Functional Analysis of the Cdk13/Cyclin K Complex. Cell Rep 14, 320–331. doi:10.1016/j.celrep.2015.12.025
Guo, J., Zhou, A. W., Fu, Y. C., Verma, U. N., Tripathy, D., Frenkel, E. P., et al. (2006). Efficacy of Sequential Treatment of HCT116 colon Cancer Monolayers and Xenografts with Docetaxel, Flavopiridol, and 5-fluorouracil. Acta Pharmacol. Sin 27, 1375–1381. doi:10.1111/j.1745-7254.2006.00421.x
Gürkan, A. C., Arisan, E. D., Obakan, P., and Palavan-Ünsal, N. (2013). Inhibition of Polyamine Oxidase Prevented Cyclin-dependent Kinase Inhibitor-Induced Apoptosis in HCT 116 colon Carcinoma Cells. Apoptosis 18, 1536–1547. doi:10.1007/s10495-013-0885-8
Haneke, K., Schott, J., Lindner, D., Hollensen, A. K., Damgaard, C. K., Mongis, C., et al. (2020). CDK1 Couples Proliferation with Protein Synthesis. J. Cell Biol 219, 219. doi:10.1083/jcb.201906147
He, L., Lu, N., Dai, Q., Zhao, Y., Zhao, L., Wang, H., et al. (2013). Wogonin Induced G1 Cell Cycle Arrest by Regulating Wnt/β-Catenin Signaling Pathway and Inactivating CDK8 in Human Colorectal Cancer Carcinoma Cells. Toxicology 312, 36–47. doi:10.1016/j.tox.2013.07.013
He, S. B., Yuan, Y., Wang, L., Yu, M. J., Zhu, Y. B., and Zhu, X. G. (2011). Effects of Cyclin-dependent Kinase 8 Specific siRNA on the Proliferation and Apoptosis of colon Cancer Cells. J. Exp. Clin. Cancer Res. 30, 109. doi:10.1186/1756-9966-30-109
Heijink, D. M., Fehrmann, R. S., de Vries, E. G., Koornstra, J. J., Oosterhuis, D., van der Zee, A. G., et al. (2011). A Bioinformatical and Functional Approach to Identify Novel Strategies for Chemoprevention of Colorectal Cancer. Oncogene 30, 2026–2036. doi:10.1038/onc.2010.578
Horiuchi, D., Huskey, N. E., Kusdra, L., Wohlbold, L., Merrick, K. A., Zhang, C., et al. (2012). Chemical-genetic Analysis of Cyclin Dependent Kinase 2 Function Reveals an Important Role in Cellular Transformation by Multiple Oncogenic Pathways. Proc. Natl. Acad. Sci. U S A. 109, E1019–E1027. doi:10.1073/pnas.1111317109
Hu, S., Marineau, J. J., Rajagopal, N., Hamman, K. B., Choi, Y. J., Schmidt, D. R., et al. (2019). Discovery and Characterization of SY-1365, a Selective, Covalent Inhibitor of CDK7. Cancer Res. 79, 3479–3491. doi:10.1158/0008-5472.CAN-19-0119
Huang, P. H., Chen, M. C., Peng, Y. T., Kao, W. H., Chang, C. H., Wang, Y. C., et al. (2016). Cdk5 Directly Targets Nuclear p21CIP1 and Promotes Cancer Cell Growth. Cancer Res. 76, 6888–6900. doi:10.1158/0008-5472.CAN-15-3253
Jardim, D. L., Millis, S. Z., Ross, J. S., Woo, M. S., Ali, S. M., and Kurzrock, R. (2021). Cyclin Pathway Genomic Alterations across 190,247 Solid Tumors: Leveraging Large-Scale Data to Inform Therapeutic Directions. Oncologist 26, e78–e89. doi:10.1634/theoncologist.2020-0509
Jinno, S., Hung, S. C., and Okayama, H. (1999). Cell Cycle Start from Quiescence Controlled by Tyrosine Phosphorylation of Cdk4. Oncogene 18, 565–571. doi:10.1038/sj.onc.1202347
Johnson, D. G., and Walker, C. L. (1999). Cyclins and Cell Cycle Checkpoints. Annu. Rev. Pharmacol. Toxicol. 39, 295–312. doi:10.1146/annurev.pharmtox.39.1.295
Jones, M. C., Askari, J. A., Humphries, J. D., and Humphries, M. J. (2018). Cell Adhesion Is Regulated by CDK1 during the Cell Cycle. J. Cell Biol 217, 3203–3218. doi:10.1083/jcb.201802088
Jung, C., Motwani, M., Kortmansky, J., Sirotnak, F. M., She, Y., Gonen, M., et al. (2003). The Cyclin-dependent Kinase Inhibitor Flavopiridol Potentiates Gamma-Irradiation-Induced Apoptosis in colon and Gastric Cancer Cells. Clin. Cancer Res. 9, 6052–6061.
Jung, C. P., Motwani, M. V., and Schwartz, G. K. (2001). Flavopiridol Increases Sensitization to Gemcitabine in Human Gastrointestinal Cancer Cell Lines and Correlates with Down-Regulation of Ribonucleotide Reductase M2 Subunit. Clin. Cancer Res. 7, 2527–2536.
Kalous, J., Jansová, D., and Šušor, A. (2020). Role of Cyclin-dependent Kinase 1 in Translational Regulation in the M-phase. Cells 9, 9. doi:10.3390/cells9071568
Karim, B. O., Rhee, K. J., Liu, G., Zheng, D., and Huso, D. L. (2013). Chemoprevention Utility of Silibinin and Cdk4 Pathway Inhibition in Apc(-/+) Mice. BMC Cancer 13, 157. doi:10.1186/1471-2407-13-157
Kim, D. M., Koo, S. Y., Jeon, K., Kim, M. H., Lee, J., Hong, C. Y., et al. (2003). Rapid Induction of Apoptosis by Combination of Flavopiridol and Tumor Necrosis Factor (TNF)-alpha or TNF-Related Apoptosis-Inducing Ligand in Human Cancer Cell Lines. Cancer Res. 63, 621–626.
Kim, H. E., Kim, D. G., Lee, K. J., Son, J. G., Song, M. Y., Park, Y. M., et al. (2012). Frequent Amplification of CENPF, GMNN and CDK13 Genes in Hepatocellular Carcinomas. PLoS One 7, e43223. doi:10.1371/journal.pone.0043223
Kim, S. H., Choi, S. J., Kim, Y. C., and Kuh, H. J. (2009). Anti-tumor Activity of noble Indirubin Derivatives in Human Solid Tumor Models In Vitro. Arch. Pharm. Res. 32, 915–922. doi:10.1007/s12272-009-1614-2
Kruschewski, M., Mueller, K., Lipka, S., Budczies, J., Noske, A., Buhr, H. J., et al. (2011). The Prognostic Impact of P53 Expression on Sporadic Colorectal Cancer Is Dependent on P21 Status. Cancers (Basel) 3, 1274–1284. doi:10.3390/cancers3011274
Lane, M. E., Yu, B., Rice, A., Lipson, K. E., Liang, C., Sun, L., et al. (2001). A Novel Cdk2-Selective Inhibitor, SU9516, Induces Apoptosis in colon Carcinoma Cells. Cancer Res. 61, 6170–6177.
Larochelle, S., Merrick, K. A., Terret, M. E., Wohlbold, L., Barboza, N. M., Zhang, C., et al. (2007). Requirements for Cdk7 in the Assembly of Cdk1/cyclin B and Activation of Cdk2 Revealed by Chemical Genetics in Human Cells. Mol. Cell 25, 839–850. doi:10.1016/j.molcel.2007.02.003
Lea, N. C., Orr, S. J., Stoeber, K., Williams, G. H., Lam, E. W., Ibrahim, M. A., et al. (2003). Commitment point during G0-->G1 that Controls Entry into the Cell Cycle. Mol. Cell Biol 23, 2351–2361. doi:10.1128/mcb.23.7.2351-2361.2003
Lee, M. S., Helms, T. L., Feng, N., Gay, J., Chang, Q. E., Tian, F., et al. (2016). Efficacy of the Combination of MEK and CDK4/6 Inhibitors In Vitro and In Vivo in KRAS Mutant Colorectal Cancer Models. Oncotarget 7, 39595–39608. doi:10.18632/oncotarget.9153
Li, C., Qi, L., Bellail, A. C., Hao, C., and Liu, T. (2014). PD-0332991 Induces G1 Arrest of Colorectal Carcinoma Cells through Inhibition of the Cyclin-dependent Kinase-6 and Retinoblastoma Protein axis. Oncol. Lett. 7, 1673–1678. doi:10.3892/ol.2014.1957
Li, J., Wang, Y., Wang, X., and Yang, Q. (2020). CDK1 and CDC20 Overexpression in Patients with Colorectal Cancer Are Associated with Poor Prognosis: Evidence from Integrated Bioinformatics Analysis. World J. Surg. Oncol. 18, 50. doi:10.1186/s12957-020-01817-8
Li, J. Q., Miki, H., Ohmori, M., Wu, F., and Funamoto, Y. (2001). Expression of Cyclin E and Cyclin-dependent Kinase 2 Correlates with Metastasis and Prognosis in Colorectal Carcinoma. Hum. Pathol. 32, 945–953. doi:10.1053/hupa.2001.27116
Lin, P. M., Lee, H. M., Huang, C. I., Tai, T. S., Chen, J. H., Chen, C. I., et al. (2020). Synergistic Antiproliferative Effect of Ribociclib (LEE011) and 5-Fluorouracil on Human Colorectal Cancer. Anticancer Res. 40, 6265–6271. doi:10.21873/anticanres.14647
Lu, J., Zhang, Z. L., Huang, D., Tang, N., Li, Y., Peng, Z., et al. (2016). Cdk3-promoted Epithelial-Mesenchymal Transition through Activating AP-1 Is Involved in Colorectal Cancer Metastasis. Oncotarget 7, 7012–7028. doi:10.18632/oncotarget.6875
Mastrogamvraki, N., and Zaravinos, A. (2020). Signatures of Co-deregulated Genes and Their Transcriptional Regulators in Colorectal Cancer. NPJ Syst. Biol. Appl. 6, 23. doi:10.1038/s41540-020-00144-8
McCurdy, S. R., Pacal, M., Ahmad, M., and Bremner, R. (2017). A CDK2 Activity Signature Predicts Outcome in CDK2-Low Cancers. Oncogene 36, 2491–2502. doi:10.1038/onc.2016.409
Mende, N., Kuchen, E. E., Lesche, M., Grinenko, T., Kokkaliaris, K. D., Hanenberg, H., et al. (2015). CCND1-CDK4-mediated Cell Cycle Progression Provides a Competitive Advantage for Human Hematopoietic Stem Cells In Vivo. J. Exp. Med. 212, 1171–1183. doi:10.1084/jem.20150308
Mender, I., Zhang, A., Ren, Z., Han, C., Deng, Y., Siteni, S., et al. (2020). Telomere Stress Potentiates STING-dependent Anti-tumor Immunity. Cancer Cell 38, 400–e6. doi:10.1016/j.ccell.2020.05.020
Okada, Y., Kato, S., Sakamoto, Y., Oishi, T., and Ishioka, C. (2017). Synthetic Lethal Interaction of CDK Inhibition and Autophagy Inhibition in Human Solid Cancer Cell Lines. Oncol. Rep. 38, 31–42. doi:10.3892/or.2017.5684
Olson, C. M., Liang, Y., Leggett, A., Park, W. D., Li, L., Mills, C. E., et al. (2019). Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem Biol 26, 792–e10. doi:10.1016/j.chembiol.2019.02.012
Patel, H., Periyasamy, M., Sava, G. P., Bondke, A., Slafer, B. W., Kroll, S. H. B., et al. (2018). ICEC0942, an Orally Bioavailable Selective Inhibitor of CDK7 for Cancer Treatment. Mol. Cancer Ther. 17, 1156–1166. doi:10.1158/1535-7163.MCT-16-0847
Patnaik, A., Rosen, L. S., Tolaney, S. M., Tolcher, A. W., Goldman, J. W., Gandhi, L., et al. (2016). Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 6, 740–753. doi:10.1158/2159-8290.CD-16-0095
Rahaman, M. H., Lam, F., Zhong, L., Teo, T., Adams, J., Yu, M., et al. (2019). Targeting CDK9 for Treatment of Colorectal Cancer. Mol. Oncol. 13, 2178–2193. doi:10.1002/1878-0261.12559
Ren, S., and Rollins, B. J. (2004). Cyclin C/cdk3 Promotes Rb-dependent G0 Exit. Cell 117, 239–251. doi:10.1016/s0092-8674(04)00300-9
Rimmele, P., Bigarella, C. L., Escamard, V. D., and Izac, B. (2012). Deacetylase Is Essential for Hematopoietic Stem Cell Activity via Regulation of Foxo3. Blood 120, 2315. doi:10.1182/blood.v120.21.2315.2315
Robb, C. M., Kour, S., Contreras, J. I., Agarwal, E., Barger, C. J., Rana, S., et al. (2018). Characterization of CDK(5) Inhibitor, 20-223 (Aka CP668863) for Colorectal Cancer Therapy. Oncotarget 9, 5216–5232. doi:10.18632/oncotarget.23749
Romero-Pozuelo, J., Figlia, G., Kaya, O., Martin-Villalba, A., and Teleman, A. A. (2020). Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep 31, 107504. doi:10.1016/j.celrep.2020.03.068
Ruiz de Porras, V., Bystrup, S., Cabrero-de Las Heras, S., Musulén, E., Palomero, L., Alonso, M. H., et al. (2019). Tumor Expression of Cyclin-dependent Kinase 5 (Cdk5) Is a Prognostic Biomarker and Predicts Outcome of Oxaliplatin-Treated Metastatic Colorectal Cancer Patients. Cancers (Basel) 11, 11. doi:10.3390/cancers11101540
Santamaría, D., Barrière, C., Cerqueira, A., Hunt, S., Tardy, C., Newton, K., et al. (2007). Cdk1 Is Sufficient to Drive the Mammalian Cell Cycle. Nature 448, 811–815. doi:10.1038/nature06046
Santti, K., Beule, A., Rönty, M., Ihalainen, H., Tarkkanen, M., and Blomqvist, C. (2019). The CDK 4/6 Inhibitor Ribociclib Has Activity in the Treatment of Inoperable Desmoid Tumor. A Case Report. Acta Oncol. 58, 897–900. doi:10.1080/0284186X.2019.1588992
Sasaki, T., Maier, B., Bartke, A., and Scrable, H. (2006). Progressive Loss of SIRT1 with Cell Cycle Withdrawal. Aging Cell 5, 413–422. doi:10.1111/j.1474-9726.2006.00235.x
Sausville, E. A., Johnson, J., Alley, M., Zaharevitz, D., and Senderowicz, A. M. (2000). ‘Inhibition CDKs as a Ther. modality’, Colorectal Cancer New Aspects. Mol. Biol. Immunol. Their Clin. Appl. 910, 207–222.
Seo, J. O., Han, S. I., and Lim, S. C. (2010). Role of CDK8 and Beta-Catenin in Colorectal Adenocarcinoma. Oncol. Rep. 24, 285–291.
Shapiro, G., Rosen, L. S., Tolcher, A. W., Goldman, J. W., Gandhi, L., Papadopoulos, K. P., et al. (2013). First-In-Human Phase I Study of the CDK4/6 Inhibitor, LY2835219, for Patients with Advanced cancer. J. Clin. Oncol. 31, 25. doi:10.1200/jco.2013.31.15_suppl.2500
Somarelli, J. A., Roghani, R. S., Moghaddam, A. S., Thomas, B. C., Rupprecht, G., Ware, K. E., et al. (2020). A Precision Medicine Drug Discovery Pipeline Identifies Combined CDK2 and 9 Inhibition as a Novel Therapeutic Strategy in Colorectal Cancer. Mol. Cancer Ther. 19, 2516–2527. doi:10.1158/1535-7163.MCT-20-0454
Soni, R., O’Reilly, T., Furet, P., Muller, L., Stephan, C., Zumstein-Mecker, S., et al. (2001). Selective In Vivo and In Vitro Effects of a Small Molecule Inhibitor of Cyclin-dependent Kinase 4. J. Natl. Cancer Inst. 93, 436–446. doi:10.1093/jnci/93.6.436
Spencer, S. L., Cappell, S. D., Tsai, F. C., Overton, K. W., Wang, C. L., and Meyer, T. (2013). The Proliferation-Quiescence Decision Is Controlled by a Bifurcation in CDK2 Activity at Mitotic Exit. Cell 155, 369–383. doi:10.1016/j.cell.2013.08.062
Squires, M. S., Feltell, R. E., Wallis, N. G., Lewis, E. J., Smith, D. M., Cross, D. M., et al. (2009). Biological Characterization of AT7519, a Small-Molecule Inhibitor of Cyclin-dependent Kinases, in Human Tumor Cell Lines. Mol. Cancer Ther. 8, 324–332. doi:10.1158/1535-7163.MCT-08-0890
Sung, W. W., Lin, Y. M., Wu, P. R., Yen, H. H., Lai, H. W., Su, T. C., et al. (2014). High Nuclear/cytoplasmic Ratio of Cdk1 Expression Predicts Poor Prognosis in Colorectal Cancer Patients. BMC Cancer 14, 951. doi:10.1186/1471-2407-14-951
Tan, H., Li, X., Yang, W. H., and Kang, Y. (2019). A Flavone, Wogonin from Scutellaria Baicalensis Inhibits the Proliferation of Human Colorectal Cancer Cells by Inducing of Autophagy, Apoptosis and G2/M Cell Cycle Arrest via Modulating the PI3K/AKT and STAT3 Signalling Pathways. J. BUON 24, 1143–1149.
Tassan, J. P., Jaquenoud, M., Léopold, P., Schultz, S. J., and Nigg, E. A. (1995). Identification of Human Cyclin-dependent Kinase 8, a Putative Protein Kinase Partner for Cyclin C. Proc. Natl. Acad. Sci. U S A. 92, 8871–8875. doi:10.1073/pnas.92.19.8871
Tate, S. C., Cai, S., Ajamie, R. T., Burke, T., Beckmann, R. P., Chan, E. M., et al. (2014). Semi-mechanistic Pharmacokinetic/pharmacodynamic Modeling of the Antitumor Activity of LY2835219, a New Cyclin-dependent Kinase 4/6 Inhibitor, in Mice Bearing Human Tumor Xenografts. Clin. Cancer Res. 20, 3763–3774. doi:10.1158/1078-0432.CCR-13-2846
Terzi, M. Y., Izmirli, M., and Gogebakan, B. (2016). The Cell Fate: Senescence or Quiescence. Mol. Biol. Rep. 43, 1213–1220. doi:10.1007/s11033-016-4065-0
Topacio, B. R., Zatulovskiy, E., Cristea, S., Xie, S., Tambo, C. S., Rubin, S. M., et al. (2019). Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol. Cell 74, 758–e4. doi:10.1016/j.molcel.2019.03.020
Vassilev, L. T., Tovar, C., Chen, S., Knezevic, D., Zhao, X., Sun, H., et al. (2006). Selective Small-Molecule Inhibitor Reveals Critical Mitotic Functions of Human CDK1. Proc. Natl. Acad. Sci. U S A. 103, 10660–10665. doi:10.1073/pnas.0600447103
Wang, H., Zhao, L., Zhu, L. T., Wang, Y., Pan, D., Yao, J., et al. (2014). Wogonin Reverses Hypoxia Resistance of Human colon Cancer HCT116 Cells via Downregulation of HIF-1α and Glycolysis, by Inhibiting PI3K/Akt Signaling Pathway. Mol. Carcinog 53, E107–E118. doi:10.1002/mc.22052
Wang, J., Liu, J., Tian, F., Zhan, Y., and Kong, D. (2019). Cyclin-dependent Kinase 9 Expression and its Association with CD8+ T Cell Infiltration in Microsatellite-Stable Colorectal Cancer. Oncol. Lett. 18, 6046–6056. doi:10.3892/ol.2019.10970
Wu, Y., Zhang, Y., Pi, H., and Sheng, Y. (2020). Current Therapeutic Progress of CDK4/6 Inhibitors in Breast Cancer. Cancer Manag. Res. 12, 3477–3487. doi:10.2147/CMAR.S250632
Yamamoto, H., Monden, T., Ikeda, K., Izawa, H., Fukuda, K., Fukunaga, M., et al. (1995). Coexpression of Cdk2/cdc2 and Retinoblastoma Gene Products in Colorectal Cancer. Br. J. Cancer 71, 1231–1236. doi:10.1038/bjc.1995.238
Yu, B., Lane, M. E., and Wadler, S. (2002). SU9516, a Cyclin-dependent Kinase 2 Inhibitor, Promotes Accumulation of High Molecular Weight E2F Complexes in Human colon Carcinoma Cells. Biochem. Pharmacol. 64, 1091–1100. doi:10.1016/s0006-2952(02)01264-9
Yu, D. F., Jiang, S. J., Pan, Z. P., Cheng, W. D., Zhang, W. J., Yao, X. K., et al. (2016). Expression and Clinical Significance of Sirt1 in Colorectal Cancer. Oncol. Lett. 11, 1167–1172. doi:10.3892/ol.2015.3982
Yu, D. S., Zhao, R., Hsu, E. L., Cayer, J., Ye, F., Guo, Y., et al. (2010). Cyclin-dependent Kinase 9-cyclin K Functions in the Replication Stress Response. EMBO Rep. 11, 876–882. doi:10.1038/embor.2010.153
Yue, S. Q., Yang, Y. L., Dou, K. F., and Li, K. Z. (2003). Expression of PCNA and CD44mRNA in Colorectal Cancer with Venous Invasion and its Relationship to Liver Metastasis. World J. Gastroenterol. 9, 2863–2865. doi:10.3748/wjg.v9.i12.2863
Zhang, C., Lundgren, K., Yan, Z., Arango, M. E., Price, S., Huber, A., et al. (2008). Pharmacologic Properties of AG-012986, a Pan-cyclin-dependent Kinase Inhibitor with Antitumor Efficacy. Mol. Cancer Ther. 7, 818–828. doi:10.1158/1535-7163.MCT-07-0440
Zhang, J., Li, H., Yabut, O., Fitzpatrick, H., D’Arcangelo, G., and Herrup, K. (2010). Cdk5 Suppresses the Neuronal Cell Cycle by Disrupting the E2F1-DP1 Complex. J. Neurosci. 30, 5219–5228. doi:10.1523/JNEUROSCI.5628-09.2010
Zhang, P., Kawakami, H., Liu, W., Zeng, X., Strebhardt, K., Tao, K., et al. (2018). Targeting CDK1 and MEK/ERK Overcomes Apoptotic Resistance in BRAF-Mutant Human Colorectal Cancer. Mol. Cancer Res. 16, 378–389. doi:10.1158/1541-7786.MCR-17-0404
Zhang, T., Nanney, L. B., Luongo, C., Lamps, L., Heppner, K. J., DuBois, R. N., et al. (1997). Concurrent Overexpression of Cyclin D1 and Cyclin-dependent Kinase 4 (Cdk4) in Intestinal Adenomas from Multiple Intestinal Neoplasia (Min) Mice and Human Familial Adenomatous Polyposis Patients. Cancer Res. 57, 169–175.
Zhao, P., Hu, Y. C., and Talbot, I. C. (2003). Expressing Patterns of P16 and CDK4 Correlated to Prognosis in Colorectal Carcinoma. World J. Gastroenterol. 9, 2202–2206. doi:10.3748/wjg.v9.i10.2202
Zhao, Z. W., Fan, X. X., Yang, L. L., Song, J. J., Fang, S. J., Tu, J. F., et al. (2019). The Identification of a Common Different Gene Expression Signature in Patients with Colorectal Cancer. Math. Biosci. Eng. 16, 2942–2958. doi:10.3934/mbe.2019145
Zhu, Y., Li, K., Zhang, J., Wang, L., Sheng, L., and Yan, L. (2020). Inhibition of CDK1 Reverses the Resistance of 5-Fu in Colorectal Cancer. Cancer Manag. Res. 12, 11271–11283. doi:10.2147/CMAR.S255895
Zhuang, K., Zhang, J., Xiong, M., Wang, X., Luo, X., Han, L., et al. (2016). CDK5 Functions as a Tumor Promoter in Human Colorectal Cancer via Modulating the ERK5-AP-1 axis. Cell Death Dis 7, e2415. doi:10.1038/cddis.2016.333
Keywords: cyclin-dependent kinases (CDKs), CDK inhibitors, CDK4/6 cell cycle inhibitors, colorectal cancer, CRC therapy, cell cycle
Citation: Thoma O-M, Neurath MF and Waldner MJ (2021) Cyclin-Dependent Kinase Inhibitors and Their Therapeutic Potential in Colorectal Cancer Treatment. Front. Pharmacol. 12:757120. doi: 10.3389/fphar.2021.757120
Received: 11 August 2021; Accepted: 26 November 2021;
Published: 21 December 2021.
Edited by:
Ester Pagano, University of Naples Federico II, ItalyReviewed by:
Elisa Herráez Aguilar, University of Salamanca, SpainIan James Martins, University of Western Australia, Australia
Copyright © 2021 Thoma, Neurath and Waldner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oana-Maria Thoma, b2FuYS1tYXJpYS50aG9tYUB1ay1lcmxhbmdlbi5kZQ==