 Bijia Song
Bijia Song Jun-Chao Zhu*
Jun-Chao Zhu*- Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, China
Recently, sleep has been recognized as a crucial factor for health and longevity. The daily sleep/wake cycle provides the basis of biorhythm, which controls whole-body homeostasis and homeodynamics. Sleep disturbances can contribute to several physical and psychological disorders, including cardiovascular disease, obesity, depression, and cognitive dysfunction. The clinical use of the N-methyl-D-aspartate (NMDA) receptor antagonist ketamine began in the 1970s. Over the years, physicians have used it as a short-acting anesthetic, analgesic, and antidepressant; however, in-depth research has revealed new possible applications for ketamine, such as for treating sleep disturbances and circadian rhythm disorders. The aim of this narrative review is to examine the literature on the mechanistic role of the antidepressant ketamine in affecting sleep disturbance. Additionally, we discuss the pharmacologic and pharmacokinetic mechanisms of ketamine as an antidepressant and the predictive biomarkers for ketamine’s effect on sleep and cognitive function.
Introduction
Sleep is a dynamic state characterized by behavioral, physiological, and electrophysiological changes. Behavioral changes include an increase in the sensory threshold of external stimuli, stillness, species-specific sleep posture, and reversibility. According to the electrophysiological activities of the brain and surrounding muscles, sleep is classified into two phases that alternate during the night cycle: rapid eye movement sleep (REM) and non-rapid eye movement sleep (NREM) (McCarley, 2011; Scammell et al., 2017; Eban-Rothschild et al., 2018). Some changes occur in the sleep pattern with increase in age, including reduced total sleep time and efficiency, increased sleep fragmentation, greater difficulty falling asleep, short eyesight exercise (REM) sleep and slow wave sleep (Wolkove et al., 2007; Pace-Schott and Spencer, 2011). Insufficient sleep and sleep disorders are associated with adverse health outcomes, including obesity, diabetes, chronic pain or pain sensitivity, cardiovascular disease, cancer, stress, neurocognitive dysfunctions, and psychiatric symptoms (Yaremchuk, 2018; Mehra, 2019). Moreover, basic neuroscience research has shown that prolonged chronic stress and depression-like behaviors are associated with impaired neuroplasticity. Sleep disturbances as one of the prominent symptoms of impaired neuroplasticity and are highly prevalent in mental disorders, such as major depressive disorder (MDD), bipolar disorder, post-traumatic stress disorder, and generalized anxiety disorder; additionally, sleep disturbances are associated with poor cognitive, emotional, and interpersonal function (Kahn et al., 2013; Rasch and Born, 2013). Sleep deprivation caused by psychological problems may lead to changes in the secretion of neuroinflammatory factors and neurotransmitter activity (such as adenosine) and brain hypoxia or hypoperfusion injury, and consequently potentiate neuronal apoptosis in cognition-related brain regions (Gulia et al., 2018). Ketamine, an N-methyl-D-aspartate (NMDA) antagonist has been reported to exert an antidepressant effect, which occurs as early as 40 min after administration and usually lasts for 7 days in patients with treatment-resistant major depression (TRD), therefore representing a new therapeutic option for treating depression (Moaddel et al., 2013; Kryst et al., 2020). Although studies have shown that ketamine exerts neuroprotective activity, studies on its rapid antidepressant effects on sleep disturbances and circadian dysregulation are limited. Treatments for sleep disorders may improve sleep-related diseases by aligning physical and psychological functions with circadian sleep/wake rhythm. Therefore, in this narrative review, we examine the literature on the internal relationship between sleep, cognitive function and depression (Table 1), the consequences of sleep disturbances (Table 2) and the effect of ketamine on sleep disturbances (Table 3). Additionally, pharmacology and pharmacokinetics of ketamine and predictive biomarkers of ketamine’s effect on sleep and cognitive function are also discussed (Table 3).

TABLE 1. The internal relationship between sleep, cognitive function and depression.

TABLE 2. The consequences of sleep disturbances.
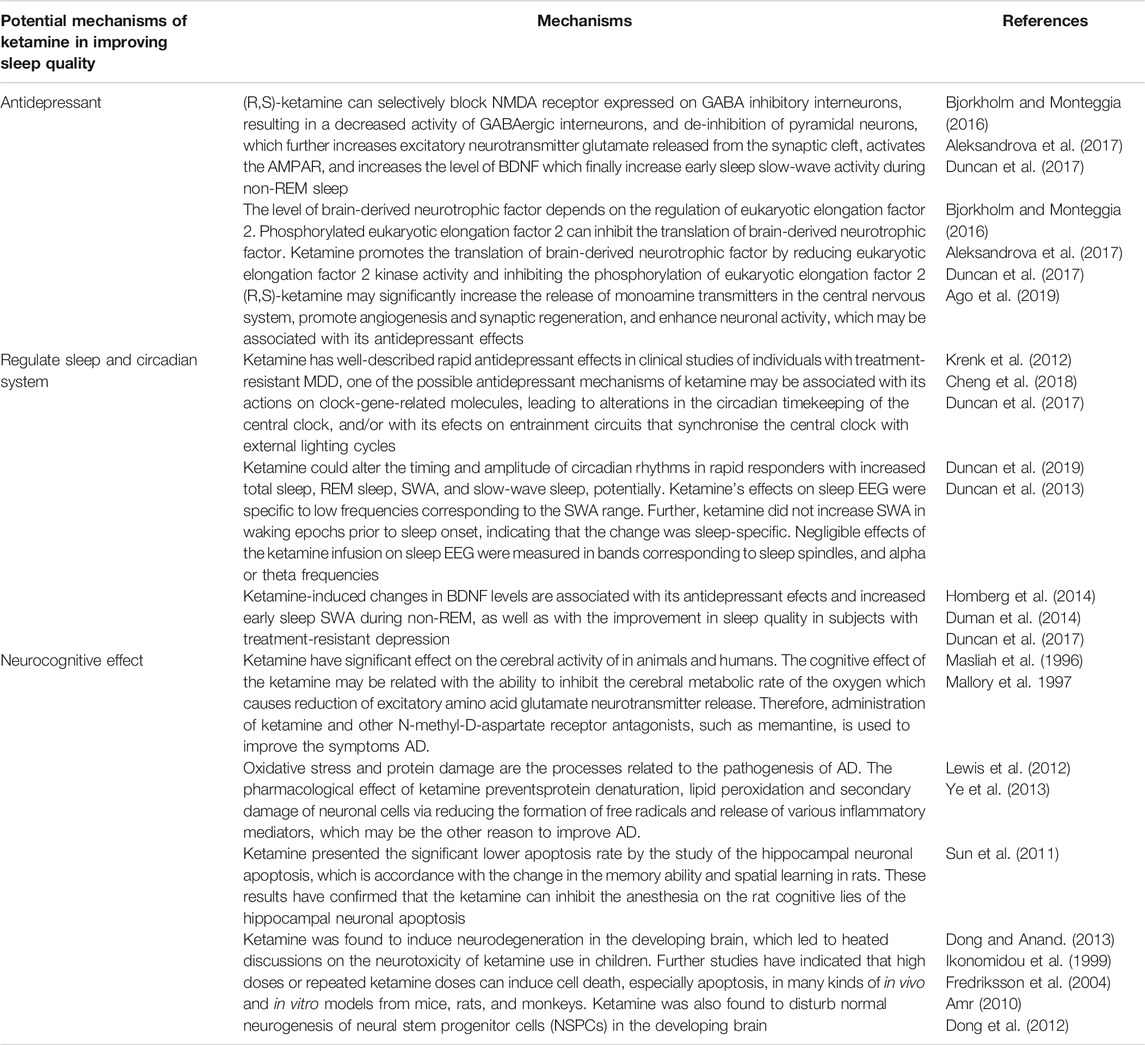
TABLE 3. Potential mechanisms of ketamine in improving sleep quality.
The Relationship Between Sleep, Cognitive Function, and Depression
Sleep Disturbances in Depression
A large and occasionally contradictory body of literature has described the association of sleep disorders with MDD. Polysomnographic studies have shown that MDD is associated with several sleep problems, including insomnia (88%), hypersomnia (27%), sleep disordered breathing, restless legs syndrome (RLS), and abnormal sleep architecture, such as prolonged sleep onset latency, frequent nocturnal awakenings, and poor sleep efficiency. Decreased REM sleep latency and prolonged REM sleep periods early in the night occur in depressed patients with sleep disturbance, leading to an overall increase in the proportion of REM sleep (Benca et al., 1992; Yates et al., 2004). The relative excess of REM sleep seems to come at the expense of stage N3 sleep, also known as slow wave sleep. Apart from less time spent in slow wave sleep in depressed patients compared with controls, there is an abnormal distribution of slow wave activity (SWA), a marker of SWS intensity, in patients with depression (Kupfer et al., 1990). Moreover, the relationship between sleep problems, and mood symptoms is bidirectional, in that poor sleep can precede an episode of major depressive disorder, and depressed mood can disrupt normal sleep pattern (Fang et al., 2019).
Sleep deficiency increases the transcription levels of IL-6 and TNF by activating nuclear factor-kappaB (NF-κB), resulting in increased levels of inflammatory cytokines (e.g., IL-6 and TNF) throughout the day (Irwin et al., 2006; Irwin et al., 2008). Moreover, studies have shown a strong relationship between inflammation and depression, as evidenced by the higher levels of inflammatory markers in depressed patients compared with non-depressed individuals, and patients with inflammatory disorder also showed a high rate of comorbid depression (Slavich and Irwin, 2014). Furthermore, the mutual effect of cholinergic and monoaminergic neurons regulates the onset of REM sleep with a rapid decrease in monoamines, such as serotonin (5-HT), norepinephrine (NE) and dopamine, and a concomitant increase in cholinergic tone. Abnormal genetic regulation of serotonergic transmission, abnormal levels of serotonin metabolites, and decrease in NE level result in fragmented REM sleep in patients with MDD (Krishnan and Nestler, 2008; Caspi et al., 2010; Wang et al., 2015). Additionally, glutamate signaling plays an important role in sleep, particularly during the thalamocortical slow oscillations of non-REM (NREM) sleep. Glutamate deficiency could lead to sleep disturbances and cell death in areas of the brain responsible for mood regulation (Peterson and Benca, 2008). Circadian rhythm is a 24-h rhythm in physiology and behavior controlled by molecular clocks in suprachiasmatic nuclei (SCN), and it plays an important role in sleep duration, continuity, and architecture. Abnormal expression of clock genes resulting from sleep disturbance and environmental factors is associated with the development of both insomnia and depression (Monteleone et al., 2011).
The Association of Cognitive Dysfunction With MDD and the Underlying Pathological Mechanism
MDD is the leading cause of disability worldwide with a high public health burden. At the molecular level, depression involves neuroplasticity failure, including neuronal atrophy and synaptic depression in the medial pre-frontal cortex (mPFC) and hippocampus (Abdullah et al., 2015; Duman et al., 2016). At the neurocognitive level, depression is defined as impaired cognitive flexibility and prefrontal inhibition, which could lead to inflexible negative biases in cognition, such as rigidly held negative beliefs (Disner et al., 2011). A comparative study involving 22 nonpsychotic MDD patients and 30 healthy participants showed significantly diminished attention span, working memory, verbal long-term memory, and verbal fluency in MDD patients compared with normal patients (Landro et al., 2001). Similarly, depressed patients scored more than 1.5 standard deviations lower in attention and working memory, 1.0 standard deviation lower in verbal long-term memory, and 0.5 standard deviation lower in verbal fluency (Beblo et al., 1999; Leuchter et al., 2004). Moreover, the relationship between MDD and global cognitive function may be modulated by the severity of depression. Rohling et al. (2002) reported greater cognitive difficulties in both automatic and effortful attention in patients hospitalized for depression. Additionally, depression may negatively impact different types of memory, including explicit, implicit, short term, long term, and working memory (Wang et al., 2006). Therefore, research into their pathological mechanisms may facilitate better insight into the neurocognitive dysfunction of depression. MDD patients with low information processing by the amygdala usually exhibit cognitive distortions (increased responsivity to negative information) (Stuhrmann et al., 2013). Increased activity of the amygdala frequently occurs in combination with increased concentration of noradrenaline and cortisol, which are responsible for memory enhancement. Additionally, the most crucial area of the cerebral cortex involved in the production of cognitive deficits is the anterior cingulate cortex (ACC), which includes ventral and dorsal anterior cingulate cortex (vACC and dACC). The ACC integrates neuronal circuits responsible for emotion processing and affects regulation. During MDD, there is a decrease in the activity of the vACC, resulting in a weaker stimulation of dopamine secretion in the limbic system, which is the major neurotransmitters in MDD; moreover, the decrease in vACC activity is due to cellular abnormalities in the cerebral and sub-cerebral structures, which disrupt monoaminergic transmission (Drevets et al., 2008). Chronic stress can facilitate the development of MDD by inducing the level of inflammatory mediators, such as IL-1β, IL-6, BDNF, and TNF-α (tumor necrosis factor), which can result in the activation of microglia (Réus et al., 2018). Moreover, microglia activation is a response for decrease in the level of CX3CL1 protein, a chemokine that regulates neuroimmunization, and participates in synaptic plasticity and cognitive function regulation. Additionally, increased expression of IL-1β is related to increased rate of nerve cell mortality, predominantly in the hippocampus, which has the highest IL-1β binding sites (Rogers et al., 2011).
Consequences of Sleep Disturbances
Effect and Mechanism of Sleep Disturbances on Neurodegenerative Disease
Neurodegenerative diseases are characterized by progressive deterioration of brain structure and function. Additionally, degeneration of selective neuron populations can lead to cognitive disorders [as observed in Alzheimer’s disease (AD) and frontotemporal dementia] or major motor incoordination [as observed in Parkinson’s disease (PD), amyotrophic lateral sclerosis, and Huntington’s disease]. There is a bidirectional link between disturbed sleep and dementia. Mazzotti et al. (2014) demonstrated that neurodegeneration was accompanied by sleep disturbances due to a reduction in the amplitude and phase changes of circadian rhythms. Two-third of patients with AD- or PD-related dementia have sleep-related problems, such as frequent daytime napping, nighttime wakefulness, and sundowning (i.e., agitation and disruptive behaviors that occur in the evening or night). Conversely, in terms of duration and quality, lack of sleep could increase neurodegenerative process and aggravate underlying clinical condition. Additionally, poor sleep and sleep disorders, such as insomnia, sleep-disordered breathing, REM sleep behavior disorder (RBD), have been linked to dementia subtypes, such as AD and PD related dementias (Guarnieri et al., 2012; Mazzotti et al., 2014). The difficulty in falling asleep, poor sleep quality, sleep loss, excessive daytime sleepiness, and sleep-disordered breathing can inhibit the inflow of apolipoprotein E (APOE) in cerebrospinal fluid (CSF) and clearance of APOE in interstitial fluid (ISF), which can reduce the removal of Aβ in CSF and increase cerebral Aβ deposition (Branger et al., 2015; Lucey et al., 2017; Carvalho et al., 2018; Lucey et al., 2018). Moreover, Aβ plaques can trigger the mislocalization of aquaporin 4 (AQP4) and decrease CSF influx, thus forming a vicious circle (Peng et al., 2016). Furthermore, apnea or OSA syndrome was related to higher levels of AD-related neuronal injury biomarkers, including P-Tau and T-Tau (Hooghiemstra et al., 2016; Diaz et al., 2017). PD dementia is caused by the aggregation of the protein α-synuclein, deposits of which are known as Lewy bodies (DLBs). Excessive daytime sleepiness, insomnia, and REM sleep behavioral disturbances are some of the most common complaints in PD patients. Moreover, sleep disturbances could increase the level of DLBs, which can aggravate PD development (Barone et al., 2009). Additionally, sleep apnea-associated intermittent brain hypo-oxygenation and inflammation may accelerate the degenerative process in already vulnerable or affected nigral dopaminergic neurons. As such, sleep apnea may exacerbate or accelerate the clinical manifestation of prodromal PD, rather than act as a primary triggering event (Bohnen et al., 2019).
Effect and Mechanism of Sleep Disturbances on Cardiovascular Disease
Long-term clinical observation shows that the frequency of adverse cardiovascular events is higher in the morning, suggesting that circadian rhythm mechanism plays a significant role in the pathogenesis of cardiovascular diseases. Irregular sleep schedules can lead to milder but chronic circadian disruption, which is widespread in all populations. Specifically, those with irregular sleep patterns may be at higher risk of cardiovascular disease due to disrupted circadian rhythm function (Huang et al., 2020). The physiological mechanisms of disturbed sleep in cardiovascular disease are described in this section. Inflammation is associated with the occurrence and development of cardiovascular diseases (Willeit et al., 2016). A systematic review involving 72 studies concluded that a short sleep time was significantly associated with increased circulating IL-6 levels, and that experimental sleep deprivation was associated with a dramatic increase in the activity of upstream pro-inflammatory molecular pathways (tumor necrosis factor-α mRNA and nuclear factor-κβ activation) (Irwin et al., 2011). Additionally, autonomic nervous system activity, which was characterized by reduced parasympathetic nerve activity and increased sympathetic nerve activity is a risk factor of cardiovascular events (Hillebrand et al., 2013). Similar to the hypothesis that short sleep duration increases the risk of cardiovascular disease through autonomic dysfunction, experimental sleep restriction has been shown to reduce parasympathetic activity and increase sympathetic activity (Tobaldini et al., 2013). Moreover, an early case-control study showed that compared with age- and sex-matched participants without insomnia, patients with insomnia had significantly lower parasympathetic nerve activity (Bonnet et al., 2010). Furthermore, chronic metabolic dysfunctions, such as insulin resistance and impaired glucose tolerance, are main risk factor of cardiovascular disease morbidity and mortality (Huang et al., 2014). A recent study of healthy adults with sleep-deprived lifestyle showed that reduced sleep is a key risk factor for obesity, diabetes, and cardiovascular diseases; moreover, extending sleep duration by approximately 1 h per night improved insulin sensitivity (Killick et al., 2015).
Effect and Mechanism of Sleep Disturbances on Cognitive Function
Several evidences have shown that sleep disorders could contribute to cognitive decline or cognitive disorders. Studies have shown that various sleep-related parameters, including long sleep latency, poor sleep efficiency or quality, excessive daytime sleepiness, sleep-disordered breathing, delayed sleep phase, and long sleep duration, were associated with cognitive impairments, such as poor attention span, working memory, processing speed, short-term memory, and reasoning, in late life (Lim and Dinges, 2010; Haba-Rubio et al., 2017). Sleep-disordered breathing (SDB), including obstructive sleep apnea (OSA) is associated with 2–6 times higher risk of mild cognitive impairment (MCI) or dementia, and early onset of MCI or dementia (Chang et al., 2013; Leng et al., 2017). Additionally, both insufficient sleep duration (less than 6 h) and excessive sleep duration (more than 9 h) are associated with cognitive impairment, and sleep fragmentation also accelerates the development of preoperative neurocognitive disorder (Ramos et al., 2016). The link between sleep disturbance and cognitive decline and dementia may be related to cortical thinning, a biomarker of several dementia subtypes. A decrease in cortical thickness in the lateral orbitofrontal cortex and inferior frontal gyrus is associated with increased sleep fragmentation, as measured by an Actigraph, and grey matter atrophy in the mPFC is associated with attenuated SOs-spindles coupling and impairment of hippocampal-dependent memory in the elderly. Moreover, short sleep duration may cause hippocampus degeneration through multiple pathways, including changes in neuronal excitability and decreased synaptic plasticity and neurogenesis, which can contribute to cognitive decline (Zimmerman and Aloia, 2006; Macey et al., 2008; Mander et al., 2013; Zarei et al., 2013; Möller et al., 2016). A study showed that sleep deprivation activates neurotoxic complement components C3a and C5a, which affects the hippocampal brain-derived neurotrophic factor (BDNF) pathway and adult neurogenesis, eventually impairing spatial memory (Kessler et al., 2019). Sleep disruption can also directly impair neuronal excitability and synaptic plasticity. Specifically, acute sleep deprivation reduces dendritic spine density in the hippocampal neurons and results in long-term memory impairment, whereas recovery sleep ameliorates spine loss and memory impairment (Havekes et al., 2016).
Potential Mechanisms of Ketamine in Improving Sleep Quality
Ketamine is a non-competitive antagonist against glutamate NMDARs and has been traditionally used as a dissociative anesthetic. Ketamine contains a mixture of two water-soluble, optical stereoisomers: S (+) and R (−)-ketamine. (Muller et al., 2016). However, whether the antidepressant mechanisms of ketamine is associated with its antagonism of NMDARs is unclear. A previous study showed that although the non-competitive, glutamatergic NMDAR antagonist (R, S)-ketamine exerts rapid and sustained antidepressant effects after a single dose in patients with depression, its use is associated with undesirable side effects. The metabolism of (R, S)-ketamine to (2S, 6S; 2R, 6R)-hydroxynorketamine (HNK) is essential for its antidepressant effects, and the (2R, 6R)-HNK enantiomer exerts behavioral, electroencephalographic, electrophysiological, and cellular antidepressant-related actions in mice. These antidepressant actions are independent of NMDAR inhibition, but involves early and sustained activation of receptors, These same issues may be also apply to sleep effects. (Zanos et al., 2016). The mechanisms through which ketamine improves sleep may be due to its antidepressant effect, its interaction with the circadian system, and its positive neurocognitive effect.
Ketamine vs. its Enantiomers in Treatment-Resistant Major Depressive Disorder
Ketamine was developed in the early 1960s, and is a popularly abused drug among young individuals and spiritualists because it produces schizophrenia-like symptoms. However, over the years, clinicians and researchers were able to discover its rapid-acting antidepressant effects (Berman et al., 2000; Domino, 2010). Researchers in the United States noticed, in the early 1990s, that patients with chronic depression experienced almost instant relief from depressive symptoms after taking (R,S)-ketamine. Subsequently, several studies replicated the robust antidepressant and anti-suicidal effects of (R,S)-ketamine in treatment-resistant patients with MDD or bipolar depression. Single administration of sub-anesthetic dose of ketamine can induce antidepressant effects in treatment-resistant patients with depression within hours of administration, lasting up to 7 days (Grunebaum et al., 2018; Wilkinson et al., 2018; Yang et al., 2019; Zhang et al., 2019). Ketamine exerts its antidepressant effects by selectively blocking the expression of NMDARs in GABA inhibitory interneurons, resulting in decreased activity of GABAergic interneurons, and de-inhibition of pyramidal neurons, which increases the release of glutamate (an excitatory neurotransmitter) from the synaptic cleft, activates AMPAR, and increases the level of BDNF. These activities result in increased early sleep slow-wave activity during non-REM sleep and improved sleep quality in subjects with treatment-resistant depression. The level of BDNF depends on the regulation of eukaryotic elongation factor 2. Phosphorylated eukaryotic elongation factor 2 can inhibit the translation of BDNF. Ketamine promotes the translation of BDNF by reducing eukaryotic elongation factor 2 kinase activity and inhibiting the phosphorylation of eukaryotic elongation factor 2 (Bjorkholm and Monteggia, 2016; Aleksandrova et al., 2017; Duncan et al., 2017). Moreover, (R, S)-ketamine may significantly increase the release of monoamine transmitters in the central nervous system, promote angiogenesis and synaptic regeneration, and enhance neuronal activity, which may be associated with its antidepressant effects (Ago et al., 2019). Regarding the two enantiomers of (R, S)-ketamine, Zhang et al. first reported that (R)-ketamine had greater potency and longer lasting antidepressant effects than (S)-ketamine in a neonatal dexamethasone-treated model of depression (Zhang et al., 2014). Moreover, (S)-ketamine can cause psychotic reactions, such as depersonalization and hallucinations, in healthy subjects, whereas the same dose of (R)-ketamine did not produce any psychotic symptoms in the same subjects (Wei Y et al., 2021). A PET study showed that (S)-ketamine markedly increased glucose utilization in the frontal cortex and thalamus, whereas (R)-ketamine significantly suppressed glucose metabolic rate in several brain areas, indicating that (S)-ketamine may be responsible for the acute side effects of ketamine (Vollenweider et al., 1997; Zanos et al., 2018). Although adverse effects, such as headache, nausea, and dissociation of (S)-ketamine were observed, intravenous infusion of (S)-ketamine (0.2 and 0.4 mg/kg for 40 min) elicited rapid-acting and sustained antidepressant actions in treatment-resistant patients with MDD (Singh et al., 2016). Another comparative study of (S)-ketamine (0.25 mg/kg by intravenous infusion) and (R, S)-ketamine (0.5 mg/kg by intravenous infusion) demonstrated that (S)-ketamine produced antidepressant effects similar to those of (R, S)-ketamine in treatment-resistant patients with depression, indicating the non-inferiority of (S)-ketamine (0.25 mg/kg) to (R, S)-ketamine (0.5 mg/kg) 24 h after a single infusion (Correia-Melo et al., 2019).
Regulatory Mechanism of Ketamine on Sleep and Circadian System
Reduced REM, slow-wave sleep and sleep fragmentation are prominent clinical biomarkers of perioperative sleep disturbances and desynchronization of the circadian rhythm (Krenk et al., 2012). For decades, depressive disorders have been related to disrupted sleep patterns and disorganised circadian rhythms. A study of 1,017 participants found that, among 162 functional connections involving sleep-related areas, 39 were also associated with depressive problem scores (Cheng et al., 2018). Ketamine has well-described rapid antidepressant effects in clinical studies of individuals with treatment-resistant MDD, one of the possible antidepressant mechanisms of ketamine may be associated with its actions on clock-gene-related molecules, leading to alterations in the circadian timekeeping of the central clock, and/or with its efects on entrainment circuits that synchronise the central clock with external lighting cycles (Duncan et al., 2017). Ketamine could alter the timing and amplitude of circadian rhythms in rapid responders with increased total sleep, REM sleep, SWA, and slow-wave sleep, potentially (Duncan et al., 2019). Ketamine’s effects on sleep EEG were specific to low frequencies corresponding to the SWA range. Further, ketamine did not increase SWA in waking epochs prior to sleep onset, indicating that the change was sleep-specific. Negligible effects of the ketamine infusion on sleep EEG were measured in bands corresponding to sleep spindles, and alpha or theta frequencies (Duncan et al., 2013). Moreover, BDNF belongs to the neurotrophin family and is produced by astrocytes, in addition to neurons, and the noradrenergic and serotonergic systems play a role in controlling BDNF synthesis (Homberg et al., 2014). Its neurotrophic functions are connected to various physiological functions in the brain particularly relevant in neuroplasticity, memory and sleep (Duman et al., 2014), suggesting its biological role over the entire life span. Duncan et al. also reported that ketamine-induced changes in BDNF levels are associated with its antidepressant efects and increased early sleep SWA during non-REM, as well as with the improvement in sleep quality in subjects with treatment-resistant depression (Duncan et al., 2017).
Ketamine’s Potential Mechanisms of Action and Potential Predictive Biomarkers Regarding Neurocognitive Function
Apart from its rapid antidepressant effects in patients with treatment-resistant depression, ketamine has been shown to exert neuroprotective effect (either intraoperatively or in the intensive care unit setting with adequate neurocognitive or neuroradiological follow-up), supporting the hypothesis that ketamine may protect cognitive functions. A single dose of ketamine (0.5 mg/Kg) at the induction of anesthesia phase might attenuate postoperative cognitive dysfunction in patients undergoing cardiac surgery and has a potential induction phase that reduces the incidence of postoperative delirium from 31 to 3% (Bell, 2017). Koffler et al. (2007) reported that deep ketamine therapy was effective in relieving chronic pain when patients reached a Ramsay score of 4–5, with ketamine levels of 250–300 µg/dl for at least 4.5 days in a medically-induced coma. Additionally, AD was characterized by glutamatergic hyperactivity receptors, and neuronal and astroglial glutamate transporter dysfunction in Alzheimer’s disease may lead to excess glutamate in the synaptic cleft and excitotoxic neuronal damage. Several investigations have confirmed that ketamine have significant effect on the cerebral activity of animals and humans. The cognitive effect of ketamine may be related to its ability to inhibit cerebral metabolic rate of oxygen, resulting in a decrease in the secretion of excitatory amino acid glutamate neurotransmitter. Therefore, administration of ketamine and other NMDA receptor antagonists, such as memantine, could be used to improve AD symptoms (Masliah et al., 1996; Mallory et al., 1997). Moreover, oxidative stress and protein damage are related to the pathogenesis of AD. Ketamine prevents protein denaturation, lipid peroxidation, and secondary damage of neuronal cells by reducing the formation of free radicals and secretion of various inflammatory mediators, which could improve AD (Lewis et al., 2012; Ye et al., 2013). The hippocampus, which is a significant structure for memory function in humans and spatial memory in rodents (including memory acquisition, consolidation, and retrieval), is mainly affected by AD. A study on the effect of sub-anesthetic dose of ketamine on memory acquisition, consolidation, and retrieval in mice showed that pre-training administration of a sub-anesthetic dose of ketamine inhibited learning and pre-probe administration of ketamine impaired performance. However, post-training administration of ketamine did not affect performance, indicating that sub-anesthetic ketamine can affect memory acquisition and retrieval, but not memory consolidation (Rosenbrock et al., 2011; Moosavi et al., 2012). Moreover, Sun et al. (2011) reported significantly lower level of hippocampal neuronal apoptosis in ketamine-treated rats, which is in accordance with the change in the memory ability and spatial learning in rats. These results confirmed that the ketamine can inhibit hippocampal neuronal apoptosis, and consequently cognitive loss in rat (Sun et al., 2011). However, the neuroprotective mechanism of ketamine in pediatric populations is still poorly understood. Dong and Anand, 2013) reviewed recent data on ketamine neurotoxicity development and proposed strategies for evaluating the safety of ketamine in pediatric patients. Ketamine was found to induce neurodegeneration in the developing brain (Ikonomidou et al., 1999), suggesting the neurotoxicity of ketamine in children. Further studies have indicated that high doses or repeated ketamine doses can induce cell death, especially apoptosis, in vitro and in mice, rat, and monkey models (Fredriksson et al., 2004; Amr, 2010). Additionally, ketamine was found to disturb normal neurogenesis of neural stem progenitor cells (NSPCs) in the developing brain (Dong et al., 2012). These findings forced scientists and physicians to reconsider the safety and toxicity of ketamine in pediatric patients. Presently, developmental neurotoxicity is being explored and clarified as a new complication in the clinical use of ketamine in pediatric patients.
Conclusion and Future Directions
Sleep has recently been recognized as a crucial factor for health and longevity. The biorhythm of daily sleep/wake cycle controls whole-body homeostasis and homeodynamics. Sleep disturbances can cause several physical and psychological disorders, including cardiovascular disease, obesity, depression, and cognitive dysfunction. Thus, improving sleep may reduce the incidence of delirium and cognitive dysfunction. Recently, there has been an increasing research interest on the antidepressant effects of ketamine in patients with treatment-resistant depression and its unique mechanism of action. Particularly, two markers of synaptic plasticity: BDNF levels and EEG sleep slow waves, have been shown to be effective in examining the ability of ketamine to increase synaptic strength. Studies on pharmacology and pharmacokinetics of ketamine and on the predictive biomarkers of ketamine’s effect on sleep and cognitive function have resulted in interesting findings worthy of further research. However, several questions regarding the correlations among sleep, slow waves, and the time course of the emotional response to ketamine are yet to be addressed. For example, sleep slow wave measurements (SWA, amplitude, and slope) seem to be related to a sharp rise in BDNF levels as well as rapid antidepressant response, with a decline 2 days into remission. Conversely, increased total sleep and improved sleep continuity on day 2 are associated with prolonged remission. Further research is needed to identify other signs of plasticity and emotional responses related to the effect of ketamine. If interventions that induce rapid antidepressant effects can increase synaptic strength, then such treatments could have similar effects on EEG sleep slow-wave measurements. Such research would be essential to reveal the key components of the rapid antidepressant response mechanism and to identify novel therapies for treating sleep disturbances.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The present study was funded by the Joint plan of key R&D plan of Liaoning Provincial Science and Technology Department (2020JH2/10300123), 345 Talent project and the Support Plan for Innovative Talents in Liaoning Higher Education Institution (Grant No. 201834).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdallah, C. G., Sanacora, G., Duman, R. S., and Krystal, J. H. (2015). Ketamine and Rapid-Acting Antidepressants: a Window into a New Neurobiology for Mood Disorder Therapeutics. Annu. Rev. Med. 66, 509–523. doi:10.1146/annurev-med-053013-062946
Ago, Y., Tanabe, W., Higuchi, M., Tsukada, S., Tanaka, T., Yamaguchi, T., et al. (2019). (R)-Ketamine Induces a Greater Increase in Prefrontal 5-HT Release Than (S)-Ketamine and Ketamine Metabolites via an AMPA Receptor-independent Mechanism. Int. J. Neuropsychopharmacol. 22 (10), 665–674. doi:10.1093/ijnp/pyz041
Aleksandrova, L. R., Phillips, A. G., and Wang, Y. T. (2017). Antidepressant Effects of Ketamine and the Roles of AMPA Glutamate Receptors and Other Mechanisms beyond NMDA Receptor Antagonism. J. Psychiatry Neurosci. 42 (4), 222–229. doi:10.1503/jpn.160175
Amr, Y. M. (2010). Multi-day Low Dose Ketamine Infusion as Adjuvant to Oral Gabapentin in Spinal Cord Injury Related Chronic Pain: a Prospective, Randomized, Double Blind Trial. Pain Physician 13, 245–249. doi:10.36076/ppj.2010/13/245
Barone, P., Antonini, A., Colosimo, C., Marconi, R., Morgante, L., Avarello, T. P., et al. (2009). The PRIAMO Study: A Multicenter Assessment of Nonmotor Symptoms and Their Impact on Quality of Life in Parkinson's Disease. Mov Disord. 24 (11), 1641–1649. doi:10.1002/mds.22643
Beblo, T., Baumann, B., Bogerts, B., Wallesch, C.-W., and Herrmann, M. (1999). Neuropsychological Correlates of Major Depression: A Short-Term Follow-Up. Cogn. Neuropsychiatry 4, 333–341. doi:10.1080/135468099395864
Bell, J. D. (2017). In Vogue: Ketamine for Neuroprotection in Acute Neurologic Injury. Anesth. Analg 124, 1237–1243. doi:10.1213/ANE.0000000000001856
Benca, R. M., Obermeyer, W. H., Thisted, R. A., and Gillin, J. C. (1992). Sleep and Psychiatric Disorders. A Meta-Analysis. Arch. Gen. Psychiatry 49 (8), 651–670. doi:10.1001/archpsyc.1992.01820080059010
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., Charney, D. S., et al. (2000). Antidepressant Effects of Ketamine in Depressed Patients. Biol. Psychiatry 47, 351–354. doi:10.1016/s0006-3223(99)00230-9
Björkholm, C., and Monteggia, L. M. (2016). BDNF - a Key Transducer of Antidepressant Effects. Neuropharmacology 102, 72–79. doi:10.1016/j.neuropharm.2015.10.034
Bohnen, N. I., and Hu, M. T. M. (2019). Sleep Disturbance as Potential Risk and Progression Factor for Parkinson's Disease. J. Parkinsons Dis. 9 (3), 603–614. doi:10.3233/JPD-191627
Bonnet, M. H., and Arand, D. L. (2010). Hyperarousal and Insomnia: State of the Science. Sleep Med. Rev. 14, 9–15. doi:10.1016/j.smrv.2009.05.002
Branger, P., Mézenge, F., André, C., de La Sayette, V., de Flores, R., Egret, S., et al. (2015). Difficulties Falling Asleep Is Associated with Higher Abeta burden in Healthy Adults. Alzheimers Dement 11, P390–P391. doi:10.1016/j.jalz.2015.06.326
Carvalho, D. Z., StLouis, E. K., Knopman, D. S., Boeve, B. F., Lowe, V. J., Roberts, R. O., et al. (2018). Association of Excessive Daytime Sleepiness with Longitudinal β-Amyloid Accumulation in Elderly Persons without Dementia. JAMA Neurol. 75, 672–680. doi:10.1001/jamaneurol.2018.0049
Caspi, A., Hariri, A. R., Holmes, A., Uher, R., and Moffitt, T. E. (2010). Genetic Sensitivity to the Environment: the Case of the Serotonin Transporter Gene and its Implications for Studying Complex Diseases and Traits. Am. J. Psychiatry 167 (5), 509–527. doi:10.1176/appi.ajp.2010.09101452
Chang, W. P., Liu, M. E., Chang, W. C., Yang, A. C., Ku, Y. C., Pai, J. T., et al. (2013). Sleep Apnea and the Risk of Dementia: a Population-Based 5-year Follow-Up Study in Taiwan. PLoS One 8 (10), e78655. doi:10.1371/journal.pone.0078655
Cheng, W., Rolls, E. T., Ruan, H., and Feng, J. (2018). Functional Connectivities in the Brain that Mediate the Association between Depressive Problems and Sleep Quality. JAMA Psychiatry 75 (10), 1052–1061. doi:10.1001/jamapsychiatry.2018.1941
Correia-Melo, F. S., Leal, G. C., Vieira, F., Jesus-Nunes, A. P., Mello, R. P., Magnavita, G., et al. (2020). Efficacy and Safety of Adjunctive Therapy Using Esketamine or Racemic Ketamine for Adult Treatment-Resistant Depression: a Randomized, Double-Blind, Non-inferiority Study. J. Affect. Disord. 264, 527–534. doi:10.1016/j.jad.2019.11.086
Díaz, M., Pulopulos, M., Baquero, M., Cuevas, A., Ferrer, I., Martn, N., et al. (2017). Sleep Disorders in Mild Cognitive Impairment. Alzheimers Dement 13.
Disner, S. G., Beevers, C. G., Haigh, E. A., and Beck, A. T. (2011). Neural Mechanisms of the Cognitive Model of Depression. Nat. Rev. Neurosci. 12, 467–477. doi:10.1038/nrn3027
Domino, E. F. (2010). Taming the Ketamine Tiger. 1965. Anesthesiology 113, 678–684. doi:10.1097/ALN.0b013e3181ed09a2
Dong, C., and Anand, K. J. (2013). Developmental Neurotoxicity of Ketamine in Pediatric Clinical Use. Toxicol. Lett. 220 (1), 53–60. doi:10.1016/j.toxlet.2013.03.030
Dong, C., Rovnaghi, C. R., and Anand, K. J. (2012). Ketamine Alters the Neurogenesis of Rat Cortical Neural Stem Progenitor Cells. Crit. Care Med. 40, 2407–2416. doi:10.1097/CCM.0b013e318253563c
Drevets, W. C., Savitz, J., and Trimble, M. (2008). The Subgenual Anterior Cingulate Cortex in Mood Disorders. CNS Spectr. 13 (8), 663–681. doi:10.1017/s1092852900013754
Duman, R. S., Aghajanian, G. K., Sanacora, G., and Krystal, J. H. (2016). Synaptic Plasticity and Depression: New Insights from Stress and Rapid-Acting Antidepressants. Nat. Med. 22, 238–249. doi:10.1038/nm.4050
Duncan, W. C., Ballard, E. D., and Zarate, C. A. (2019). “Ketamine-induced Glutamatergic Mechanisms of Sleep and Wakefulness: Insights for Developing Novel Treatments for Disturbed Sleep and Mood,” in Handbook of Experimental Pharmacology. Editors H. P. Landolt, and D. J. Dijk, 253, 337–358.
Duncan, W. C., Slonena, E., Hejazi, N. S., Brutsche, N., Yu, K. C., Park, L., et al. (2017). Motor-Activity Markers of Circadian Timekeeping Are Related to Ketamine's Rapid Antidepressant Properties. Biol. Psychiatry 82 (5), 361–369. doi:10.1016/j.biopsych.2017.03.011
Duncan, W. C., Sarasso, S., Ferrarelli, F., Selter, J., Riedner, B. A., Hejazi, N. S., et al. (2013). Concomitant BDNF and Sleep Slow Wave Changes Indicate Ketamine-Induced Plasticity in Major Depressive Disorder. Int. J. Neuropsychopharmacol. 16, 301–311. doi:10.1017/S1461145712000545
Eban-Rothschild, A., Appelbaum, L., and de Lecea, L. (2018). Neuronal Mechanisms for Sleep/wake Regulation and Modulatory Drive. Neuropsychopharmacology 43 (5), 937–952. doi:10.1038/npp.2017.294
Fang, H., Tu, S., Sheng, J., and Shao, A. (2019). Depression in Sleep Disturbance: A Review on a Bidirectional Relationship, Mechanisms and Treatment. J. Cel Mol Med 23 (4), 2324–2332. doi:10.1111/jcmm.14170
Fredriksson, A., Archer, T., Alm, H., Gordh, T., and Eriksson, P. (2004). Neurofunctional Deficits and Potentiated Apoptosis by Neonatal NMDA Antagonist Administration. Behav. Brain Res. 153, 367–376. doi:10.1016/j.bbr.2003.12.026
Grunebaum, M. F., Galfalvy, H. C., Choo, T. H., Keilp, J. G., Moitra, V. K., Parris, M. S., et al. (2018). Ketamine for Rapid Reduction of Suicidal Thoughts in Major Depression: a Midazolam-Controlled Randomized Clinical Trial. Am. J. Psychiatry 175, 327–335. doi:10.1176/appi.ajp.2017.17060647
Guarnieri, B., Adorni, F., Musicco, M., Appollonio, I., Bonanni, E., Caffarra, P., et al. (2012). Prevalence of Sleep Disturbances in Mild Cognitive Impairment and Dementing Disorders: a Multicenter Italian Clinical Cross-Sectional Study on 431 Patients. Dement Geriatr. Cogn. Disord. 33 (01), 50–58. doi:10.1159/000335363
Gulia, K. K., and Kumar, V. M. (2018). Sleep Disorders in the Elderly: a Growing challenge. Psychogeriatrics 18 (3), 155–165. doi:10.1111/psyg.12319
Haba-Rubio, J., Marti-Soler, H., Tobback, N., Andries, D., Marques-Vidal, P., Waeber, G., et al. (2017). Sleep Characteristics and Cognitive Impairment in the General Population: The HypnoLaus Study. Neurology 88 (5), 463–469. doi:10.1212/WNL.0000000000003557
Hashimoto, K. (2019). Rapid-acting Antidepressant Ketamine, its Metabolites and Other Candidates: A Historical Overview and Future Perspective. Psychiatry Clin. Neurosci. 73, 613–627. doi:10.1111/pcn.12902
Havekes, R., Park, A. J., Tudor, J. C., Luczak, V. G., Hansen, R. T., Ferri, S. L., et al. (2016). Sleep Deprivation Causes Memory Deficits by Negatively Impacting Neuronal Connectivity in Hippocampal Area CA1. Elife 5, e13424. doi:10.7554/eLife.13424
Hillebrand, S., Gast, K. B., de Mutsert, R., Swenne, C. A., Jukema, J. W., Middeldorp, S., et al. (2013). Heart Rate Variability and First Cardiovascular Event in Populations without Known Cardiovascular Disease: Meta-Analysis and Dose-Response Meta-Regression. Europace 15, 742–749. doi:10.1093/europace/eus341
Homberg, J. R., Molteni, R., Calabrese, F., and Riva, M. A. (2014). The Serotonin-BDNF Duo: Developmental Implications for the Vulnerability to Psychopathology. Neurosci. Biobehav Rev. 43, 35–47. doi:10.1016/j.neubiorev.2014.03.012
Hooghiemstra, A. M., Visser, P. J., Slot, R. E. R., Teunissen, C. E., Scheltens, P., and van der Flier, W. M. (2016). Alzheimer’s Disease Patients with Osas History Have Higher Csf Tau Levels. Alzheimers Dement 12. doi:10.1016/j.jalz.2016.06.2316
Huang, T., Mariani, S., and Redline, S. (2020). Sleep Irregularity and Risk of Cardiovascular Events: The Multi-Ethnic Study of Atherosclerosis. J. Am. Coll. Cardiol. 75 (9), 991–999. doi:10.1016/j.jacc.2019.12.054
Huang, Y., Cai, X., Chen, P., Mai, W., Tang, H., Huang, Y., et al. (2014). Associations of Prediabetes with All-Cause and Cardiovascular Mortality: A Meta-Analysis. Ann. Med. 46, 684–692. doi:10.3109/07853890.2014.955051
Ikonomidou, C., Bosch, F., Miksa, M., Bittigau, P., Vöckler, J., Dikranian, K., et al. (1999). Blockade of NMDA Receptors and Apoptotic Neurodegeneration in the Developing Brain. Science 283, 70–74. doi:10.1126/science.283.5398.70
Irwin, M. R., and Cole, S. W. (2011). Reciprocal Regulation of the Neural and Innate Immune Systems. Nat. Rev. Immunol. 11 (9), 625–632. doi:10.1038/nri3042
Irwin, M. R., Wang, M., Campomayor, C. O., Collado-Hidalgo, A., and Cole, S. (2006). Sleep Deprivation and Activation of Morning Levels of Cellular and Genomic Markers of Inflammation. Arch. Intern. Med. 166 (16), 1756–1762. doi:10.1001/archinte.166.16.1756
Irwin, M. R., Wang, M., Ribeiro, D., Cho, H. J., Olmstead, R., Breen, E. C., et al. (2008). Sleep Loss Activates Cellular Inflammatory Signaling. Biol. Psychiatry 64 (6), 538–540. doi:10.1016/j.biopsych.2008.05.004
Kahn, M., Sheppes, G., and Sadeh, A. (2013). Sleep and Emotions: Bidirectional Links and Underlying Mechanisms. Int. J. Psychophysiol 89, 218–228. doi:10.1016/j.ijpsycho.2013.05.010
Kessler, R., Knutson, K. L., Mokhlesi, B., Anderson, S. L., Shah, M., Meltzer, D. O., et al. (2019). Sleep and Activity Patterns in Older Patients Discharged from the Hospital. Sleep 42 (11), zsz153. doi:10.1093/sleep/zsz153
Killick, R., Hoyos, C. M., Melehan, K. L., Dungan, G. C., Poh, J., and Liu, P. Y. (2015). Metabolic and Hormonal Effects of 'catch-Up' Sleep in Men with Chronic, Repetitive, Lifestyle-Driven Sleep Restriction. Clin. Endocrinol. (Oxf) 83, 498–507. doi:10.1111/cen.12747
Koffler, S. P., Hampstead, B. M., Irani, F., Tinker, J., Kiefer, R. T., Rohr, P., et al. (2007). The Neurocognitive Effects of 5 Day Anesthetic Ketamine for the Treatment of Refractory Complex Regional Pain Syndrome. Arch. Clin. Neuropsychol. 22 (6), 719–729. doi:10.1016/j.acn.2007.05.005
Krenk, L., Jennum, P., and Kehlet, H. (2012). Sleep Disturbances after Fast-Track Hip and Knee Arthroplasty. Br. J. Anaesth. 109, 769–775. doi:10.1093/bja/aes252
Krishnan, V., and Nestler, E. J. (2008). The Molecular Neurobiology of Depression. Nature 455 (7215), 894–902. doi:10.1038/nature07455
Kryst, J., Kawalec, P., and Pilc, A. (2020). Efficacy and Safety of Intranasal Esketamine for the Treatment of Major Depressive Disorder. Expert Opin. Pharmacother. 21 (1), 9–20. doi:10.1080/14656566.2019.1683161
Kupfer, D. J., Frank, E., McEachranEachran, A. B., and Grochocinski, V. J. (1990). Delta Sleep Ratio. A Biological Correlate of Early Recurrence in Unipolar Affective Disorder. Arch. Gen. Psychiatry 47, 1100–1105. doi:10.1001/archpsyc.1990.01810240020004
Landro, N. I., Stiles, T. C., and Sletvold, H. (2001). Neuropsychological Function in Nonpsychotic Unipolar Major Depression. Neuropsychiatry, Neuropsychol. Behav. Neurol. 14, 233–240.
Leng, Y., McEvoy, C. T., Allen, I. E., and Yaffe, K. (2017). Association of Sleep-Disordered Breathing with Cognitive Function and Risk of Cognitive Impairment: A Systematic Review and Meta-Analysis. JAMA Neurol. 74 (10), 1237–1245. doi:10.1001/jamaneurol.2017.2180
Leuchter, A. F., Morgan, M., Cook, I. A., Dunkin, J., Abrams, M., and Witte, E. (2004). Pretreatment Neurophysiological and Clinical Characteristics of Placebo Responders in Treatment Trials for Major Depression. Psychopharmacology (Berl) 177, 15–22. doi:10.1007/s00213-004-1919-2
Lewis, L. D., Weiner, V. S., Mukamel, E. A., Donoghue, J. A., Eskandar, E. N., Madsen, J. R., et al. (2012). Rapid Fragmentation of Neuronal Networks at the Onset of Propofol-Induced Unconsciousness. Proc. Natl. Acad. Sci. U S A. 109 (49), E3377–E3386. doi:10.1073/pnas.1210907109
Li, S., Mallory, M., Alford, M., Tanaka, S., and Masliah, E. (1997). Glutamate Transporter Alterations in Alzheimer Disease Are Possibly Associated with Abnormal APP Expression. J. Neuropathol. Exp. Neurol. 56 (8), 901–911. doi:10.1097/00005072-199708000-00008
Lim, J., and Dinges, D. F. (2010). A Meta-Analysis of the Impact of Short-Term Sleep Deprivation on Cognitive Variables. Psychol. Bull. 136 (3), 375–389. doi:10.1037/a0018883
Lucey, B. P., Hicks, T. J., McLeland, J. S., Toedebusch, C. D., Boyd, J., Patterson, B. W., et al. (2017). Sleep Loss Increases Risk of Alzheimer’s Disease by Increasing CNS Ab Production. Ann. Neurol. 82, S53–S54.
Lucey, B. P., Hicks, T. J., McLeland, J. S., Toedebusch, C. D., Boyd, J., Elbert, D. L., et al. (2018). Effect of Sleep on Overnight Cerebrospinal Fluid Amyloid β Kinetics. Ann. Neurol. 83, 197–204. doi:10.1002/ana.25117
Macey, P. M., Kumar, R., Woo, M. A., Valladares, E. M., Yan-Go, F. L., and Harper, R. M. (2008). Brain Structural Changes in Obstructive Sleep Apnea. Sleep 31 (07), 967–977.
Mander, B. A., Rao, V., Lu, B., Saletin, J. M., Lindquist, J. R., Ancoli-Israel, S., et al. (2013). Prefrontal Atrophy, Disrupted NREM Slow Waves and Impaired Hippocampal-dependent Memory in Aging. Nat. Neurosci. 16 (3), 357–364. doi:10.1038/nn.3324
Masliah, E., Alford, M., DeTeresa, R., Mallory, M., and Hansen, L. (1996). Deficient Glutamate Transport Is Associated with Neurodegeneration in Alzheimer's Disease. Ann. Neurol. 40 (5), 759–766. doi:10.1002/ana.410400512
Mazzotti, D. R., Guindalini, C., Moraes, W. A., Andersen, M. L., Cendoroglo, M. S., Ramos, L. R., et al. (2014). Human Longevity Is Associated with Regular Sleep Patterns, Maintenance of Slow Wave Sleep, and Favorable Lipid Profile. Front. Aging Neurosci. 6, 134. doi:10.3389/fnagi.2014.00134
McCarley, R. W. (2011). Neurobiology of REM Sleep. Handb Clin. Neurol. 98, 151–171. doi:10.1016/B978-0-444-52006-7.00010-1
Mehra, R. (2019). Sleep Apnea and the Heart. Cleve Clin. J. Med. 86 (9 Suppl. 1), 10–18. doi:10.3949/ccjm.86.s1.03
Moaddel, R., Abdrakhmanova, G., Kozak, J., Jozwiak, K., Toll, L., Jimenez, L., et al. (2013). Sub-anesthetic Concentrations of (R,S)-ketamine Metabolites Inhibit Acetylcholine-Evoked Currents in α7 Nicotinic Acetylcholine Receptors. Eur. J. Pharmacol. 698, 228–234. doi:10.1016/j.ejphar.2012.11.023
Möller, C., Hafkemeijer, A., Pijnenburg, Y. A. L., Rombouts, S. A. R. B., van der Grond, J., Dopper, E., et al. (2016). Different Patterns of Cortical gray Matter Loss over Time in Behavioral Variant Frontotemporal Dementia and Alzheimer’s Disease. Neurobiol. Aging 38, 21–31. doi:10.1016/j.neurobiolaging.2015.10.020
Monteleone, P., Martiadis, V., and Maj, M. (2011). Circadian Rhythms and Treatment Implications in Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 35 (7), 1569–1574. doi:10.1016/j.pnpbp.2010.07.028
Moosavi, M., Yadollahi Khales, G., Rastegar, K., and Zarifkar, A. (2012). The Effect of Sub-anesthetic and Anesthetic Ketamine on Water Maze Memory Acquisition, Consolidation and Retrieval. Eur. J. Pharmacol. 677 (1-3), 107–110. doi:10.1016/j.ejphar.2011.12.021
Muller, J., Pentyala, S., Dilger, J., and Pentyala, S. (2016). Ketamine Enantiomers in the Rapid and Sustained Antidepressant Effects. Ther. Adv. Psychopharmacol. 6, 185–192. doi:10.1177/2045125316631267
Pace-Schott, E. F., and Spencer, R. M. (2011). Age-related Changes in the Cognitive Function of Sleep. Prog. Brain Res. 191, 75–89. doi:10.1016/B978-0-444-53752-2.00012-6
Peng, W., Achariyar, T. M., Li, B., Liao, Y., Mestre, H., Hitomi, E., et al. (2016). Suppression of Glymphatic Fluid Transport in a Mouse Model of Alzheimer's Disease. Neurobiol. Dis. 93, 215–225. doi:10.1016/j.nbd.2016.05.015
Peterson, M. J., and Benca, R. M. (2008). Sleep in Mood Disorders. Sleep Med. Clin. 3 (2), 231–249. doi:10.1016/j.jsmc.2008.01.009
Ramos, A. R., Tarraf, W., Daviglus, M., Davis, S., Gallo, L. C., Mossavar-Rahmani, Y., et al. (2016). Sleep Duration and Neurocognitive Function in the Hispanic Community Health Study/Study of Latinos. Sleep 39 (10), 1843–1851. doi:10.5665/sleep.6166
Rasch, B., and Born, J. (2013). About Sleep's Role in Memory. Physiol. Rev. 93, 681–766. doi:10.1152/physrev.00032.2012
Réus, G. Z., Moura, de, A. B., Silva, R. H., Resende, W. R., and Quevedo, J. (2018). Resilience Dysregulation in Major Depressive Disorder: Focus on Glutamatergic Imbalance and Microglial Activation. Curr. Neuropharmacol. 16 (3), 297–307.
Rogers, J. T., Morganti, J. M., Bachstetter, A. D., Hudson, C. E., Peters, M. M., Grimmig, B. A., et al. (2011). CX3CR1 Deficiency Leads to Impairment of Hippocampal Cognitive Function and Synaptic Plasticity. J. Neurosci. 31 (45), 16241–16250. doi:10.1523/JNEUROSCI.3667-11.2011
Rohling, M. L., Green, P., Allen, L. M., and Iverson, G. L. (2002). Depressive Symptoms and Neurocognitive Test Scores in Patients Passing Symptom Validity Tests. Arch. Clin. Neuropsychol. 17, 205–222. doi:10.1093/arclin/17.3.205
Rosenbrock, H. D-C. C., Kroker, K., and Rast, G. (2011). Effects of Specific Nicotinic Acetylcholine Receptor Agonists on Hippocampal Long-Term Potentiation. Alzheimer's Demen. J. Alzheimer's Assoc. 7 (1), S116. doi:10.1016/j.jalz.2011.05.295
Scammell, T. E., Arrigoni, E., and Lipton, J. O. (2017). Neural Circuitry of Wakefulness and Sleep. Neuron 93 (4), 747–765. doi:10.1016/j.neuron.2017.01.014
Singh, J. B., Daly, E., Xi, L., Melman, C., De Bruecker, G., Tadic, A., et al. (2016). Intravenous Esketamine in Adult Treatment-Resistant Depression: a Double-Blind, Double-Randomization, Placebo-Controlled Study. Biol. Psychiatry 80, 424–431. doi:10.1016/j.biopsych.2015.10.018
Slavich, G. M., and Irwin, M. R. (2014). From Stress to Inflammation and Major Depressive Disorder: a Social Signal Transduction Theory of Depression. Psychol. Bull. 140 (3), 774–815. doi:10.1037/a0035302
Stuhrmann, A., Dohm, K., Kugel, H., Zwanzger, P., Redlich, R., Grotegerd, D., et al. (2013). Mood-congruent Amygdala Responses to Subliminally Presented Facial Expressions in Major Depression: Associations with Anhedonia. J. Psychiatry Neurosci. 38 (4), 249–258. doi:10.1503/jpn.120060
Sun, Y. T., Hou, M., Zou, T., Liu, Y., Li, J., and Wang, Y. L. (2016). Effect of Ketamine Anesthesia on Cognitive Function and Immune Function in Young Rats. Cell. Mol. Biol. (Noisy-le-grand) 62 (4), 63–66.
Tobaldini, E., Cogliati, C., Fiorelli, E. M., Nunziata, V., Wu, M. A., Prado, M., et al. (2013). One Night On-Call: Sleep Deprivation Affects Cardiac Autonomic Control and Inflammation in Physicians. Eur. J. Intern. Med. 24, 664–670. doi:10.1016/j.ejim.2013.03.011
Wang, C. E., Halvorsen, M., Sundet, K., Steffensen, A. L., Holte, A., and Waterloo, K. (2006). Verbal Memory Performance of Mildly to Moderately Depressed Outpatient Younger Adults. J. Affect Disord. 92, 283–286. doi:10.1016/j.jad.2006.02.008
Wang, Y. Q., Li, R., Zhang, M. Q., Zhang, Z., Qu, W. M., and Huang, Z. L. (2015). The Neurobiological Mechanisms and Treatments of REM Sleep Disturbances in Depression. Curr. Neuropharmacol 13 (4), 543–553. doi:10.2174/1570159x13666150310002540
Wei, Y., Chang, L., and Hashimoto, K. (2021). Molecular Mechanisms Underlying the Antidepressant Actions of Arketamine: Beyond the NMDA Receptor. Mol. Psychiatry. doi:10.1038/s41380-021-01121-1
Wilkinson, S. T., Ballard, E. D., Bloch, M. H., Mathew, S. J., Murrough, J. W., Feder, A., et al. (2018). The Effect of a Single Dose of Intravenous Ketamine on Suicidal Ideation: a Systematic Review and Individual Participant Data Meta-Analysis. Am. J. Psychiatry 175, 150–158. doi:10.1176/appi.ajp.2017.17040472
Willeit, P., Thompson, S. G., Agewall, S., Bergström, G., Bickel, H., Catapano, A. L., et al. (2016). Inflammatory Markers and Extent and Progression of Early Atherosclerosis: Meta-Analysis of Individual-Participant-Data from 20 Prospective Studies of the PROG-IMT Collaboration. Eur. J. Prev. Cardiol. 23, 194–205. doi:10.1177/2047487314560664
Wolkove, N., Elkholy, O., Baltzan, M., and Palayew, M. (2007). Sleep and Aging: 1. Sleep Disorders Commonly Found in Older People. CMAJ 176, 1299–1304. doi:10.1503/cmaj.060792
Yang, C., Yang, J., Luo, A., and Hashimoto, K. (2019). Molecular and Cellular Mechanisms Underlying the Antidepressant Effects of Ketamine Enantiomers and its Metabolites. Transl Psychiatry 9 (1), 280. doi:10.1038/s41398-019-0624-1
Yaremchuk, K. (2018). Sleep Disorders in the Elderly. Clin. Geriatr. Med. 34 (2), 205–216. doi:10.1016/j.cger.2018.01.008
Yates, W. R., Mitchell, J., Rush, A. J., Trivedi, M. H., Wisniewski, S. R., Warden, D., et al. (2004). Clinical Features of Depressed Outpatients with and without Co-occurring General Medical Conditions in STAR*D. Gen. Hosp. Psychiatry 26 (6), 421–429. doi:10.1016/j.genhosppsych.2004.06.008
Ye, X., Lian, Q., Eckenhoff, M. F., Eckenhoff, R. G., and Pan, J. Z. (2013). Differential General Anesthetic Effects on Microglial Cytokine Expression. PLoS One 8 (1), e52887. doi:10.1371/journal.pone.0052887
Zanos, P., Moaddel, R., Morris, P. J., Georgiou, P., Fischell, J., Elmer, G. I., et al. (2016). NMDAR Inhibition-independent Antidepressant Actions of Ketamine Metabolites. Nature 533 (7604), 481–486. doi:10.1038/nature17998
Zanos, P., Moaddel, R., Morris, P. J., Riggs, L. M., Highland, J. N., Georgiou, P., et al. (2018). Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol. Rev. 70, 621–660. doi:10.1124/pr.117.015198
Zarei, M., Ibarretxe-Bilbao, N., Compta, Y., Hough, M., Junque, C., Bargallo, N., et al. (2013). Cortical Thinning Is Associated with Disease Stages and Dementia in Parkinson's Disease. J. Neurol. Neurosurg. Psychiatry 84 (08), 875–881. doi:10.1136/jnnp-2012-304126
Zhang, J. C., Li, S. X., and Hashimoto, K. (2014). R (-)-ketamine Shows Greater Potency and Longer Lasting Antidepressant Effects Than S (+)-ketamine. Pharmacol. Biochem. Behav. 116, 137–141. doi:10.1016/j.pbb.2013.11.033
Zhang, K., and Hashimoto, K. (2019). An Update on Ketamine and its Two Enantiomers as Rapid-Acting Antidepressants. Expert Rev. Neurother. 19, 83–92. doi:10.1080/14737175.2019.1554434
Keywords: sleep disturbances, ketamine, antidepressant, depression, neurocognition
Citation: Song B and Zhu J-C (2021) Mechanisms of the Rapid Effects of Ketamine on Depression and Sleep Disturbances: A Narrative Review. Front. Pharmacol. 12:782457. doi: 10.3389/fphar.2021.782457
Received: 24 September 2021; Accepted: 22 November 2021;
Published: 14 December 2021.
Edited by:
Ahmad Reza Dehpour, Tehran University of Medical Sciences, IranReviewed by:
Steven James Mennerick, Washington University in St. Louis, United StatesKenji Hashimoto, Chiba University, Japan
Copyright © 2021 Song and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Chao Zhu, emh1anVuY2hhbzFAaG90bWFpbC5jb20=