Abstract
Fructose, especially industrial fructose (sucrose and high fructose corn syrup) is commonly used in all kinds of beverages and processed foods. Liver is the primary organ for fructose metabolism, recent studies suggest that excessive fructose intake is a driving force in non-alcoholic fatty liver disease (NAFLD). Dietary fructose metabolism begins at the intestine, along with its metabolites, may influence gut barrier and microbiota community, and contribute to increased nutrient absorption and lipogenic substrates overflow to the liver. Overwhelming fructose and the gut microbiota-derived fructose metabolites (e.g., acetate, butyric acid, butyrate and propionate) trigger the de novo lipogenesis in the liver, and result in lipid accumulation and hepatic steatosis. Fructose also reprograms the metabolic phenotype of liver cells (hepatocytes, macrophages, NK cells, etc.), and induces the occurrence of inflammation in the liver. Besides, there is endogenous fructose production that expands the fructose pool. Considering the close association of fructose metabolism and NAFLD, the drug development that focuses on blocking the absorption and metabolism of fructose might be promising strategies for NAFLD. Here we provide a systematic discussion of the underlying mechanisms of dietary fructose in contributing to the development and progression of NAFLD, and suggest the possible targets to prevent the pathogenetic process.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is characterized by an excessive fat build-up in the liver without clear other causes, e.g., alcohol addiction, virus infection, and drug induction. Epidemiology investigation reveals that NAFLD has affected more than one-quarter population around the world (Loomba and Sanyal, 2013; Softic et al., 2017). Recently, NAFLD is proposed to be named as metabolic (dysfunction) associated fatty liver disease (MAFLD) due to the heterogeneous etiology and metabolic risks (Eslam et al., 2020). NAFLD has a spectrum ranging from simple fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH), related fibrosis, cirrhosis and even hepatocellular carcinoma (Williams et al., 2011). While NAFL is usually considered to be benign, patients with NASH often potentiate a high probability to further progression. Although NAFLD has been the focus of numerous studies, the pathological mechanisms are still unclear.
Fructose is a plant-derived monosaccharide, the natural form can be found in fruits, berries, and certain vegetables. The association of fructose with NAFLD can be derived from the ancient Egyptians, who fed ducks and geese-dried fruits to make foie gras. Industrial fructose, e.g., the high-fructose corn syrup, is widely used in almost all kinds of processed foods and beverages, partially driven by the huge commercial profits (Moeller et al., 2009; Vos and Lavine, 2013). During food processing, fructose undergoes a series of reactions (e.g., polymerization, condensation, etc.) upon heating to produce aldehydes, reducing ketones, and heterocyclic compounds, thus enhancing the flavor and improving the palatability of food (Maillard reaction). In the past 200 years, per capita dietary fructose intake has increased more than 100-fold (Jang et al., 2018). The average daily consumption of added sugars currently is estimated at 15% of total energy intake all over the world, and almost half is fructose (Wittekind and Walton, 2014; Newens and Walton, 2016; Powell et al., 2016). The wide application of processed foods, such as various baked food and yogurt, further exaggerate the increase of “invisible sugar” intake. Although fructose intake has significantly increased, the early warning mechanisms for the harm in humans are still immature. When excess glucose consumption raises blood glucose, insulin will be secreted to reduce the overwhelmed blood glucose, and prevents the continuous hyperglycemia. The metabolic rate of fructose is much higher than that of glucose, but no immediate feedback mechanisms to suppress its absorption or transportation. Furthermore, the transcription of glucose transporter 5 (GLUT5) increases upon fructose stimulation, which, in turn, enhances fructose transportation and absorption (Gouyon et al., 2003). Simultaneously, fructose is continuously transformed into fructose-1-phosphate (F1P) in the liver, which is an unrestricted process. High fructose-contained diets also induce more caloric intake, thus exacerbating the metabolic disorders indirectly (Lustig, 2013).
Fructose consumption is found to be positively correlated with obesity, diabetes, cardiovascular disease, NAFLD, hypertension, and cancer (Ouyang et al., 2008; Shapiro et al., 2011; White, 2013; DiNicolantonio et al., 2018). Excessive fructose-contained drinks intake is strongly related to the childhood obesity and pediatric NAFLD (Forshee and Storey, 2003). In adults ≥48 years old, daily fructose consumption increases hepatic inflammation and hepatocyte ballooning (Abdelmalek et al., 2010). By producing toxic advanced glycation end-products, fructose is also a threat to the aging process, the occurrence of diabetic complications (e.g. vascular, renal, and ocular complications), and the development of atherosclerosis (Gaby, 2005). In a prospective study involving 77,797 subjects in Sweden, overconsumption of high-sugar-containing foods is found to be a great risk of pancreatic cancer (Larsson et al., 2006). In addition, fructose is also the main cause of symptoms associated with chronic diarrhea or functional bowel disturbances (Gaby, 2005). In animals, overconsumption of fructose can assemble most metabolic features that associated with NAFLD patients, such as insulin resistance, hyperlipidemia, visceral obesity and hyperuricemia (Sánchez-Lozada et al., 2008), suggesting that fructose is a noticeable factor that drives the process of NAFLD. Therefore, we will review the recent studies that focus on the contribution of dietary fructose to NAFLD development and progression, and highlight the metabolic risks and possible drug targets.
Dietary Fructose and Intestinal Environment
Fructose and Dysbiosis
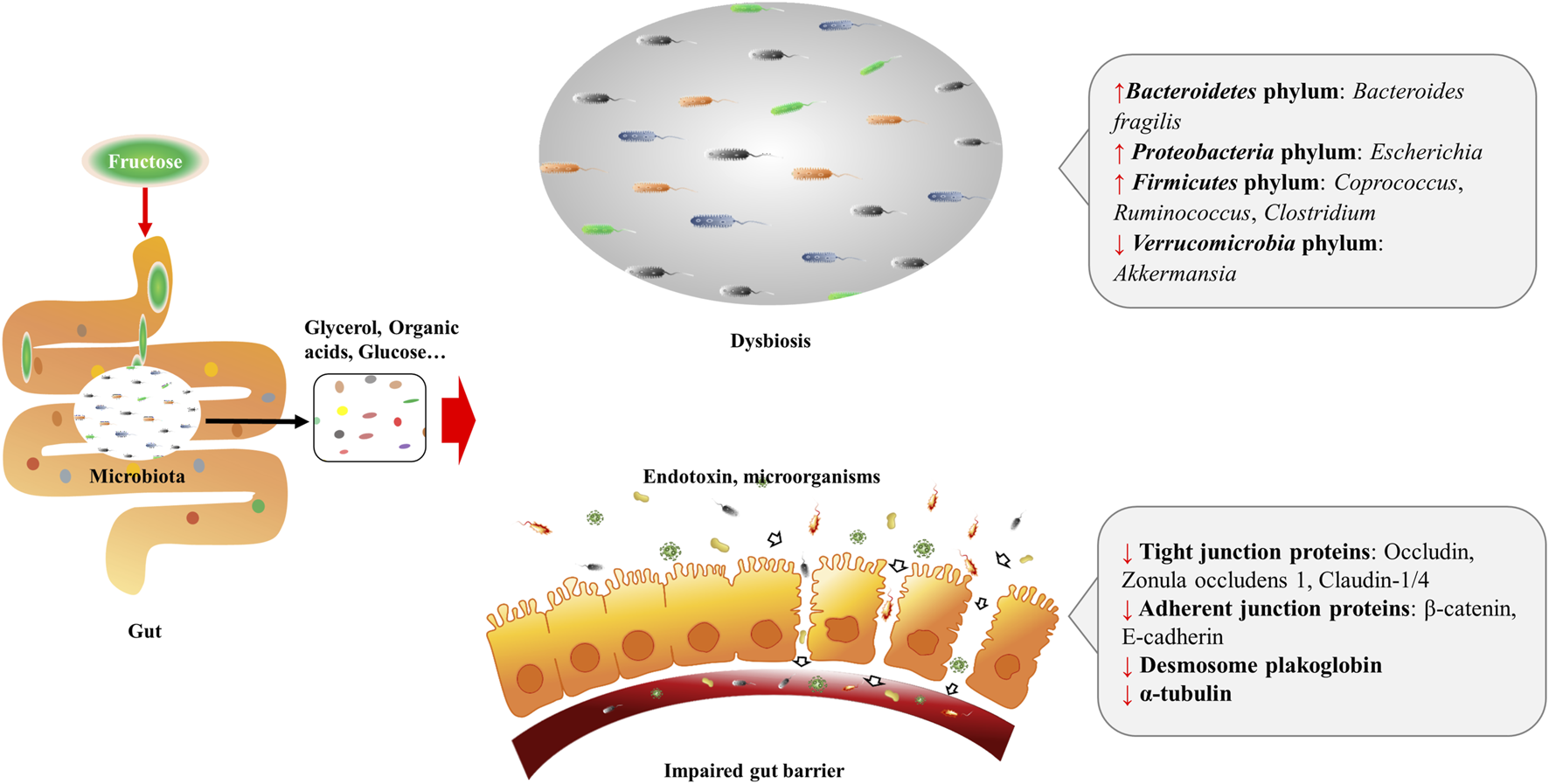
Diet is the main source of fructose, and most fructose is absorbed in the small intestine, with 25 g being the upper limit for a healthy adult. Fructose transport proteins (GLUT2 and GLUT5) that locate in the enterocytes are accounting for sensing and passively transporting fructose (Ferraris et al., 2018). Studies showed that GLUT5 transcription is specifically stimulated by fructose via cyclic adenosine monophosphate (cAMP) and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) systems (Cui et al., 2004; Cui et al., 2005), whereas GLUT2 is modulated by fructose and glucose levels, as well as systemic factors that released during their absorption (Cui et al., 2003). Absorbed fructose enters the liver via the portal vein for further metabolism. When fructose intake exceeds the absorptive capacity of enterocytes, it can transport to the colon where settles more than 100 trillion bacteria (Boulangé et al., 2016). These microbiomes are able to hydrolyze and ferment dietary polysaccharides by producing glycoside hydrolase enzymes (Xu et al., 2003; Sonnenburg et al., 2005; Patterson et al., 2016). By the action of gut microbiomes, fructose can transform into glucose, glycerol, and various organic acids, including uric acids (UA), short-chain fatty acids (SCFA) (e.g., acetate, butyric acid, butyrate and propionate), and amino acids (e.g., glutamic acid, glutamine and alanine) (Jahn et al., 2019; Qi et al., 2020; Zhao et al., 2020). These fructose metabolites either transport to the liver and serve as lipogenic substrates, or interact with the intestinal environment locally. As a monosaccharide, fructose can be the energy source for certain gut microbiomes, for instance, fructose is the single energy source of Anaerostipes caccae DSM 14662 and Roseburia intestinalis DSM 14610, the acetate-converting and butyrate-producing strains (Falony et al., 2006). High fructose diet (HFrD) is reported to affect the composition of gut microbiota characterized by decreased bacterial diversity, increased Firmicutes/Bacteriodetes ratio in Sprague-Dawley rats (Sen et al., 2017), and reduced protective commensal and bile salt hydrolase-expressing microorganisms in dextran sodium sulfate (DSS)-induced colitis mice (Montrose et al., 2021). HFrD decreases the abundance of Firmicutes phylum (Lactobacillus) and Verrucomicrobia phylum (Akkermansia), while increases the abundance of Bacteroidetes phylum (Bacteroides fragilis) and Proteobacteria phylum (Sutterella, Bilophila, and Escherichia) in rodents (Sen et al., 2017; Zubiría et al., 2017; Do et al., 2018; Cho et al., 2021). And the alteration of these gut microbiomes might contribute to the dysbiosis and possibly the impairment of gut barrier (Figure 1). Luminal fructose at the physiological level stimulates the release of glucagon-like peptide 1 (GLP-1) from L-subtype EECs in humans and animals, however, the type 2 diabetic mice are GLP-1 resistant due to gut dysbiosis (Kuhre et al., 2014; Seino et al., 2015; Grasset et al., 2017).
FIGURE 1
Excessive fructose intake leads to dysbiosis and gut barrier impairment. Fructose is absorbed in the small intestine, and transforms into glycerol, organic acids, and glucose, etc. by the action of resident microbiota. Fructose and fructose-derived metabolites may trigger dysbiosis characterized by decreased bacterial diversity and increased Firmicutes/Bacteriodetes ratio. Fructose over-intake also impairs gut barrier by decreasing protein expression of the tight junction (zonula occludens 1, occludin, claudin-1, and claudin-4) and adherent junction (β-catenin and E-cadherin) proteins, desmosome plakoglobin, and α-tubulin of enterocytes.
Fructose and Gut Barrier
When high-dose fructose saturates fructose clearance capacity in the ileum, the excessive fructose can enter the colon, and influence the gut barrier. Evidence implied that fructose acts on the intestinal barrier by regulating the transcriptional and post-translational modification of related proteins. In mammals, HFrD decreases protein expression of the tight junction (zonula occludens 1, occludin, claudin-1, and claudin-4) and adherent junction (β-catenin and E-cadherin) proteins, desmosome plakoglobin, and α-tubulin, and increases apoptotic proteins (p-JNK, Bax, cleaved caspase-3, and caspase-3 activity) of enterocytes (Cho et al., 2021). At the transcriptional level, prolonged HFrD feeding reduces genes encoding tight-junction protein-2, occluding, and different claudins in the intestine (Todoric et al., 2020) (Figure 1). In addition, HFrD feeding is reported to cause colon shortening possibly through endoplasmic reticulum (ER) stress and inflammatory reaction (Todoric et al., 2020). Recently, fructose is found to improve the survival of intestinal villus under hypoxia, resulting in the expanded nutrient absorption of the gut, thus contributing to increased body weight gain and fat accumulation in mice (Taylor et al., 2021). Nonetheless, it is still unclear whether there is an association between the extension of intestinal villi and disordered gut barrier.
Fructose also influences the intestinal barrier by regulating ethanol metabolism (Bradford et al., 1991; Uzuegbu and Onyesom, 2009; Villalobos-García et al., 2021). In alcohol-fed mice that lack alcohol dehydrogenase (ADH), fructose administration decreases the rates of ethanol metabolism by about 60% via diminishing H2O2 generation (Bradford et al., 1991). In light alcohol drinkers (<20 g/day) between 25 and 35 years old, however, fructose reduced the duration of alcohol intoxication by 30.7% through facilitating its clearance (Uzuegbu and Onyesom, 2009). At the same time, dysbiosis in fructose-exposed rodents may contribute to the endogenous ethanol production. Additionally, excess fructose intake may lead to fructose malabsorption, and unabsorbed fructose has been found to draw fluid into the intestinal lumen, resulting in abdominal pain, flatulence, diarrhea, and other digestive dysfunctions (Beyer et al., 2005; Gaby, 2005). The organic acids and gas that are produced during fructose metabolism also contribute to the aforementioned gastrointestinal symptoms (Southgate, 1995; Beyer et al., 2005).
Fructose Metabolism in the Development of NASH
Fructose Metabolism and Hepatic Lipogenesis
Fructose dose as low as 0.33 g·kgBM−1 is sufficient to stimulate de novo lipogenesis (DNL) in mice (Tran et al., 2010), while fructose <0.35 g·kgBM−1 would not appear in the portal blood as a prototype (Jang et al., 2018), implicating that gut-derived fructose metabolites, e.g., acetate, amino acid, endotoxins, SCFA, and ethanol can enter the liver and stimulate lipogenesis physiologically. The saturation of intestinal fructose catabolism would occur at about 5 g sugar intake (e.g., one-fourth of a banana) (Ferraris et al., 2018; Gonzalez and Betts, 2018). And the hepatocellular metabolism of fructose (which bypasses the rate-limiting step of glycolysis at the level of phosphofructokinase) is responsible for most of the metabolic disorders (Andres-Hernando et al., 2020). Fructose at the physiological level can be completely metabolized by the liver, overwhelming fructose consumption, however, leads to steatosis via promoting lipogenesis, dyslipidemia, visceral adiposity, and insulin resistance (Jin and Vos, 2015). In addition, fructose metabolism lacks hormonal regulation (Douard and Ferraris, 2008; Douard et al., 2013), which may explain the significant variation in peripheral fructose levels (from 0.008 to 16 mM) (Kawasaki et al., 2002; Hui et al., 2009). On the contrary, fructose malabsorption is negatively correlated to hepatic steatosis (Walker et al., 2012), suggesting that fructose is an environmental factor that favors the development of NAFLD.
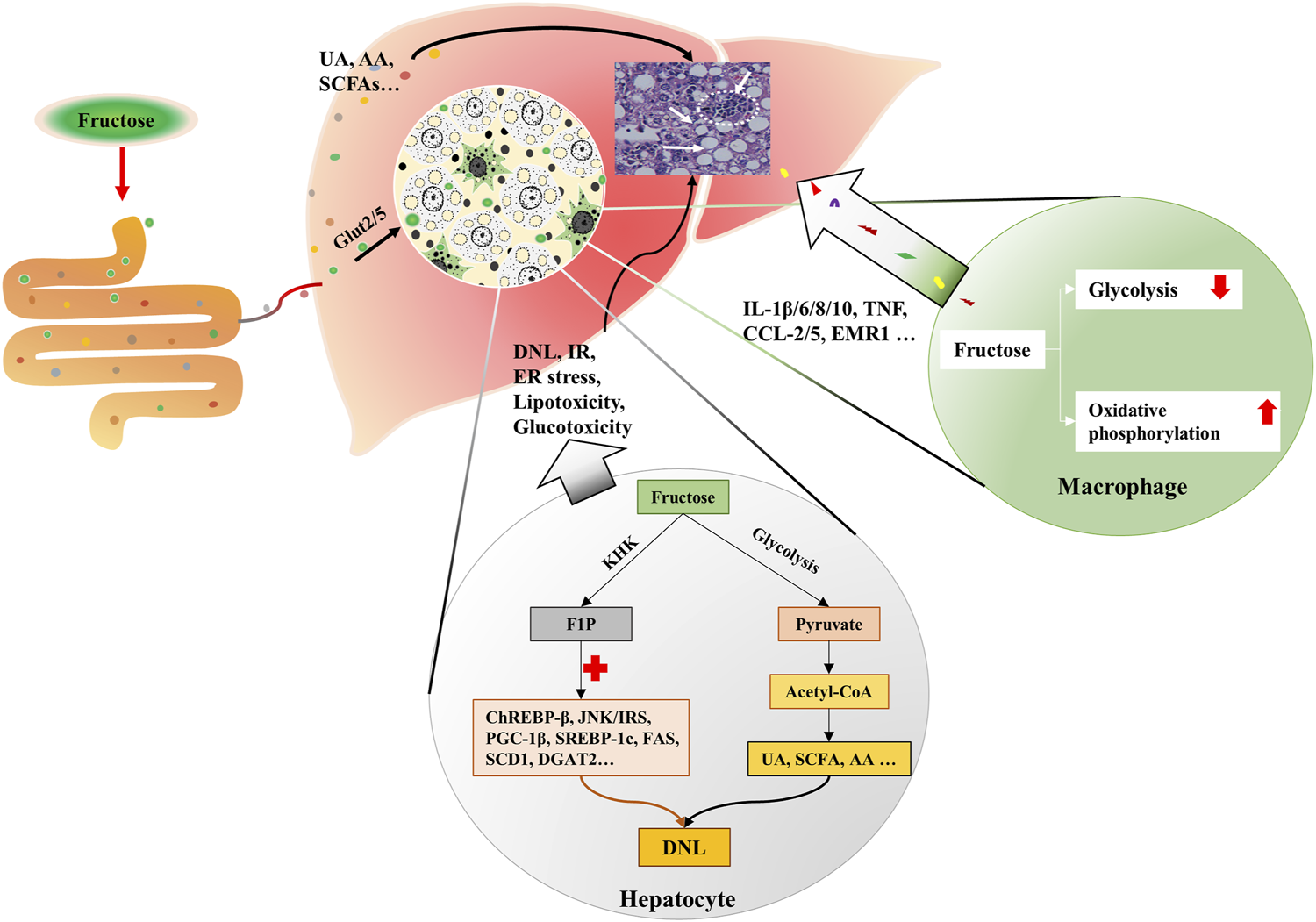
More than a quarter of the intrahepatic lipid is generated by DNL in obese and NAFLD individuals (Donnelly et al., 2005; Lim et al., 2010). Fructose enters hepatocytes in a GLUT5-mediated process, intracellular fructose transforms into Fructose-1-phosphate (F1P) by the action of fructokinase (KHK) (Mirtschink et al., 2018). Then, F1P is metabolized into pyruvate and acetyl-CoA for the tricarboxylic acid cycle (Lustig, 2013). During this process, fructose, F1P, as well as their intermediates can activate carbohydrate-responsive element-binding protein (ChREBP)-β, c-Jun N-terminal kinase/insulin receptor substrates, and peroxisome proliferator-activated receptor gamma co-activator-1β/sterol regulatory element-binding protein-1(SREBP-1)/acetyl-CoA carboxylase 1 (ACC1) and downstream DNL pathways (Lustig, 2013; Premachandran et al., 2017; Softic et al., 2017; Gonzalez and Betts, 2018; Pan et al., 2018). Concurrently, genes that encode fatty acid oxidizing enzymes are down-regulated upon fructose exposure, which further exacerbate the hepatic lipid accumulation (Premachandran et al., 2017; Pan et al., 2018). The metabolites of fructose in the intestine, such as SCFA, and amino acid also provide substrates for hepatic DNL, leading to the lipid accumulation in hepatocytes and the increase of circulating fatty acids (Jang et al., 2018; Jahn et al., 2019; Zhao et al., 2020) (Figure 2). Besides, fructolysis provides a carbon backbone for the synthesis of nucleotides, triglyceride-glycerol (glycerol-3-phosphate), and amino acids (serine, glutamate, glutamine, aspartate, and asparagine) (Liu et al., 2020).
FIGURE 2
Fructose metabolism in the liver. Fructose is mainly catabolized by hepatocytes. Fructose is absorbed by hepatocytes via Glut2/5. Intracellular fructose transforms into F1P by the action of KHK. Then, F1P is metabolized into pyruvate and acetyl-CoA for tricarboxylic acid cycle. During this process, fructose, F1P, as well as their intermediates can activate ChREBP-β, JNK/IRS, and PGC-1β/ SREBP-1/ACC1, which results in DNL. Gut-derived fructose metabolites enter the liver through portal vein, and contribute to DNL in the liver. In addition, UA, SCFA and AA produced in this process also trigger DNL. Simultaneously, fructose exposure alters the energy supply mode of macrophages, which induces oxidative stress and secretion of inflammatory cytokines. KHK, fructokinase; F1P, fructose-1-phosphate; ChREBP-β, carbohydrate-responsive element-binding protein β; JNK, c-Jun N-terminal kinase; IRS, insulin receptor substrates; PGC-1β, peroxisome proliferator-activated receptor gamma co-activator-1β; SREBP-1, sterol regulatory element-binding protein-1; ACC1, acetyl-CoA carboxylase 1; DNL, de novo lipogenesis; UA, uric acid; SCFA, short chain fatty acid; AA, amino acid.
Fructose in the liver can be transformed into glycogen (Davies et al., 1990; Warner et al., 2021), and hepatic glycogen storage also stimulates DNL (Hengist et al., 2019). In humans, fructose intake rapidly increases post-exercise liver glycogen synthesis (Décombaz et al., 2011). In rats, administration of fructose causes an immediate increase of glycogen synthase (GS) activity and initiates a fast accumulation of glycogen in the liver (Bezborodkina et al., 2014). Actually, glucose is usually used in food processing in a ratio of nearly 1:1 with fructose, high levels of glucose in the portal vein can induce the expression of hepatic aldose reductase (AR), resulting in the conversion of glucose into sorbitol, which is then metabolized to fructose by sorbitol dehydrogenase (Lanaspa et al., 2013). The production of endogenous fructose may further increase the total fructose content. Fructose contributes to hepatic lipid accumulation also by blocking fatty acid oxidation (FAO) (Lanaspa et al., 2012a; Lanaspa et al., 2012b). In high-fat high-fructose-fed mice, the activity of CTP1a (the rate-limiting enzyme of FAO) and its acylcarnitine products are decreased, and the mitochondrial fission and mitochondrial integrity are damaged (Softic et al., 2019), suggesting the blockage of FAO.
Fructose and Liver Inflammation
Fructose is believed to be an inflammatory mediator that promotes the progression of NAFLD to NASH (Jegatheesan and De Bandt, 2017). HFrD has been proven to induce insulin resistance, hepatic steatosis, ballooning degeneration and fibrosis in transgenic (Tg) MUP-uPA mice, and significantly increased the tumor necrosis factor (Tnf), interleukin (Il)-6, Il1β, Ccl2, Ccl5 and Emr1 (Todoric et al., 2020). Moreover, HFrD enhances diethyl nitrosamine-induced hepatocellular carcinoma in BL6 mice (Todoric et al., 2020).
Macrophage plays an important role in the hepatic inflammatory response. Fructose is thought to influence the metabolic phenotype of macrophages, which is a decisive factor for cellular function. Stressed macrophages present increased glycolysis, whereas oxidative metabolism primed macrophages for a less inflammatory mode (Vats et al., 2006). 2-deoxy-glucose (2-DG), an inhibitor of glycolysis, also blocks TNF-α and IL-6 production in macrophages (Wang et al., 2014). In lipopolysaccharide (LPS)-stressed macrophages, high concentrations of fructose alter the mode of energy supply, with impaired glycolysis and enhanced oxidative phosphorylation, along with glutaminolysis and oxidative stress, which result in the secretion of inflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, IL-8, IL-10, and TNF (Jones et al., 2021) (Figure 2). Considering the fact that increased glycolysis favors cell proliferation in cancer while enhanced oxidative phosphorylation supplies sufficient amounts of energy for inflammatory macrophages, the different effects of fructose on cells may be derived from cell demands in different situations. And glycolysis-produced intermediates may contribute to both cell proliferation and activation.
And fructose overconsumption is shown to induce local macrophage infiltration in the liver (Hotamisligil et al., 1993; Glushakova et al., 2008; DiNicolantonio et al., 2018). Studies also show that fructose promotes monocyte recruitment via monocyte chemoattractant protein 1 and intracellular adhesion molecule 1 (Fantuzzi and Faggioni, 2000; Shapiro et al., 2011; DiNicolantonio et al., 2018). Besides, fructose exposure in LPS-stimulated human dendritic cells provokes the secretion of pro-inflammatory cytokines from T cells (Jaiswal et al., 2019). And mice deficient in T or NK cells are prevented from developing fructose-induced NAFLD (Bhattacharjee et al., 2014). Moreover, fructose treatment influences the viability of stressed immune cells (Jones et al., 2021).
Fructose overconsumption also increases visceral adipose tissue mass and intracellular cortisol concentration (Targher et al., 2006; Kovačević et al., 2014). Adipocyte hypertrophy may trigger ER stress, disturb adipokines (leptin and adiponectin) release, and increase pro-inflammatory cytokine secretion (de Heredia et al., 2012; DiNicolantonio et al., 2018). Fructose-induced increase of intracellular cortisol level via 11beta-hydroxysteroid dehydrogenase type1 (11β-HSD1) leads to a raise of fatty acid flux out of the subcutaneous adipocytes, thus providing more substrates for visceral fat accumulation (de Heredia et al., 2012; DiNicolantonio et al., 2018). Plasminogen activator inhibitor 1 (PAI-1) is an acute-phase protein that participates in hepatic lipid transport and provokes inflammation in the liver (Bergheim et al., 2006). It is reported that fructose content is positively correlated to PAI-1 concentration, and PAI-1 knock-out mice are protected from fructose-induced steatosis and inflammation (Castrogiovanni et al., 2012).
Dietary Fructose and UA Metabolism
Dietary fructose increases both serum and intrahepatic UA levels (Lanaspa et al., 2012b). UA is primarily produced in hepatocytes, and the process of fructose metabolization to F1P consumes a large amount of intracellular ATP and phosphate, the decrease of phosphate activates adenosine monophosphate (AMP) deaminase that converts AMP to inosine monophosphate, which results in UA production (Van den Berghe, 1986). Simultaneously, fructose-derived amino acids also increase UA excretion and decrease plasma uridine (Yamamoto et al., 1999). UA is known to promote hepatic fat accumulation, and provokes mitochondrial oxidative stress by increasing superoxide generation and mitochondrial NOX4 expression, and decreasing manganese superoxide dismutase in NASH mice (Lanaspa et al., 2012b; Ishimoto et al., 2012). Meanwhile, UA can activate nuclear factor-κB and Nod-like receptor protein 3 inflammasomes (Wan et al., 2016). In patients with NAFLD/NASH, the serum UA level is positively correlated to the extent of the lobular inflammation and steatosis (Sertoglu et al., 2014). Additionally, UA may increase endogenous fructose production in a feedback way (Jensen et al., 2018). Inhibition of UA formation has been proven to be beneficial for metabolic diseases (Lanaspa et al., 2012c; Lanaspa et al., 2013; Nakatsu et al., 2015).
Fructose metabolites are found to induce renal damage (Ramezani and Raj, 2014; Tang et al., 2015). In healthy male adults, daily ingestion of 200 g fructose for 2 weeks appears to increase urinary stone formation partly via regulating urate metabolism, urinary pH, and increasing oxalate (Johnson et al., 2018). Fructose-contained beverage intake in infancy has been reported to be associated with worse outcomes in a later event of acute kidney injury and kidney damage during adolescence (García-Arroyo et al., 2020). In experimental hamsters, monosodium glutamate plus a high-fat and high-fructose (HFF) diet increases the risk of kidney injury, induces gut dysbiosis, and an increase in the amount of p-cresol sulfate (Pongking et al., 2020). At the same time, HFF can induce dyslipidemia and lipid accumulation in the kidney (Thongnak et al., 2020). Besides, fructose increases segmental artery vascular resistance by increasing serum UA and copeptin (Chapman et al., 2020), enhances angiotensin II-stimulated Na transport via activation of protein kinase C α1 in renal proximal tubules (Yang et al., 2020). Renal DNL stimulated by high-fructose supplementation may also contribute to the increase of intrahepatic triglycerides (Milutinović et al., 2020).
Possible Drug Targets in Fructose Metabolism
Glucose Transporter Family Members (GLUTs)
Glucose transporter family members (GLUTs) belong to the major facilitator superfamily, which are encoded by the SLC2 genes (Mueckler and Thorens, 2013; Nomura et al., 2015). In humans, 14 GLUTs are found to mediate the facilitative diffusion of sugar along the concentration gradient (Schmidl et al., 2020). Dietary fructose absorption is very efficient in the small intestine mediated by GLUTs transporters.
The transport of fructose across the intestinal basolateral membrane is mediated by one or more sodium-independent routes. GLUT2 is responsible for moving both fructose in and glucose out of the enterocyte across the basolateral membrane under basal conditions (Cheeseman, 1993). The malfunction or dysregulation of GLUT2 is associated with diabetes, metabolic syndrome, and cancer (Schmidl et al., 2020). GLUT2 variants increase the risks of fasting hyperglycemia, type 2 diabetes, hypercholesterolemia and cardiovascular diseases. Besides, individuals with a missense mutation in GLUT2 show a preference for sugar-containing foods (Thorens, 2015). GLUT2 inhibition is desirable for patients with fructose-induced metabolic disorders, but it may lead to insulin insensitivity (Ferraris et al., 2018), suggesting that specific inhibition of GLUT2 is necessary. GLUT5 level is increased in patients with acute myeloid leukemia and prostate cancer, and is reported to be negatively related to the prognosis of patients (Carreno et al., 2021; Jeong et al., 2021). In rodents, increased GLUT5 is associated with enhanced steatosis and pancreatic inflammation (Roncal-Jimenez et al., 2011). GLUT5 deletion could reduce fructose absorption by approximately 75% in the jejunum and decrease the concentration of serum fructose by approximately 90% in comparison to wild-type mice on excess fructose intake, suggesting that GLUT5 is required for fructose transportation, and the function cannot be compensated by GLUT2 (Barone et al., 2009; Patel et al., 2015). GLUT5 is also highly expressed in many cancers, thus promoting the proliferation of cancer cells (Su et al., 2018; Jin et al., 2019; Chen et al., 2020). Inhibiting GLUT5 is expected to be a promising strategy for fructose-associated diseases. Lately, several drugs are found to inhibit GLUT2/5. i.e., Chamomile and green tea display acute inhibition on GLUT2/5 and thus decrease fructose and glucose transportation in human Caco-2 cells and Xenopus oocytes (Villa-Rodriguez et al., 2017). Kefir, a fermented drink, is found to decrease GLUT2/5 in the liver of HFrD fed rats (Akar et al., 2021).
Fructokinase (KHK)
KHK is an essential fructose-metabolizing enzyme in the liver, which specifically catalyzes the transfer of a phosphate group from adenosine triphosphate to fructose. KHK-C, the principal isoform of KHK in the liver, is increased in NAFLD patients that consume excessive fructose-contained beverages (Ouyang et al., 2008; Ishimoto et al., 2012). Pharmacological inhibition of KHK activity with PF-06835919 is reported to prevent patients from hepatic steatosis, lipogenic, and fibrosis (Shepherd et al., 2021). KHK deficiency or inhibition (PF-06835919) protects animals from fructose-induced obesity, insulin resistance, hypertriglyceridemia, and NAFLD (Lanaspa et al., 2013; Futatsugi et al., 2020; Gutierrez et al., 2021). Knockout KHK could also decrease fructose-derived UA production and attenuate mitochondrial oxidative stress in mice (Ishimoto et al., 2012). Besides, Extracts of Angelica archangelica, Garcinia mangostana, Petroselinum crispum, and Scutellaria baicalensis are identified with inhibitory activity against ketohexokinase-C (Le et al., 2016). Interestingly, fructokinase A (KHK-A), another isoform of KHK, protects against C-mediated metabolic diseases, but its activity is relatively low due to a low fructose affinity (Ishimoto et al., 2012). Drugs such as that targeting reshaping the balance between the two types of KHK is an attractive strategy.
Triokinase (TK)
TK is a rate-limiting enzyme in fructolysis, and is essential for the induction of the lipogenic program under physiological conditions (Liu et al., 2020). TK knockdown abrogates the expression of lipogenic enzymes and results in a significant reduction in HFrD-induced hepatic steatosis, as well as liver size and plasma glucose levels (Liu et al., 2020). Meanwhile, TK is necessary for maintaining mitochondrial respiration. It is found that 5 mM fructose is sufficient to have a significant decapacitating effect on mitochondrial respiration in the TK-deficient cells, an overnight culture of mutant cells with 15 mM fructose obliterates their respiration capacity (Liu et al., 2020). Liver TK deficiency results in a more than 2-fold sensitization of hepatocytes to fructose toxicity, indicating TK might be a promising target to block fructose toxicity. Although ADP is reported to be a potent inhibitor of TK, agents that target TK inhibition are not available yet.
Aldose Reductase (AR)
AR is the key enzyme in stimulating endogenous fructose production. It can be activated under pathological conditions, including ischemia, heart failure, inflammation, and hyperuricemia (Huang et al., 2017; Nakagawa et al., 2020). Increased AR results in the conversion of blood glucose into sorbitol, which is further metabolized into endogenous fructose by sorbitol dehydrogenase (Lanaspa et al., 2013). Liver AR knockdown, or AR inhibitors can attenuate hepatic steatosis and related metabolic disorders in mice (Lanaspa et al., 2013; Nakagawa et al., 2020). Furthermore, the addition of salt to fructose in the drinking water significantly accelerates the development of NAFLD, the high-salt diet activates the AR-fructokinase pathway in the liver and hypothalamus, resulting in endogenous fructose production (Lanaspa et al., 2018). These results suggest that AR is the target of blocking endogenous fructose production. Actually, a series of extracts from natural products such as Luteolin, quercetin, apigenin, fisetin, and myricitrin are potential AR inhibitors, and found to be beneficial for NAFLD/NASH (Grewal et al., 2016). And AR inhibitors Sorbinil and Ranirestat, are under clinical trials.
Conclusions and Perspectives
The liver is the critical organ to metabolize fructose, the increasing consumption of fructose in various forms specially exaggerates liver burden and contributes to NAFLD/NASH. The rapidly metabolized fructose in the liver promotes lipogenesis, lipotoxicity, as well as the inflammatory reaction of immune cells. Meanwhile, the interaction of fructose and gut microbiota results in dysbiosis, impaired intestinal mucosa barrier, production of toxins and microbial metabolites that may further serve as substrates for liver lipogenesis, and pathogens for liver inflammation. In addition, fructose metabolism lacks hormonal regulation, making excessive fructose consumption thus a more dangerous factor to NAFLD patients.
Even so, patients with obesity, hypertension, and diabetes are encouraged to intake certain fructose contained in vegetables and specific fruits, such as blueberries, grapes, and apples (Bazzano et al., 2008; Muraki et al., 2013; Sundborn et al., 2019). The possible explanation might be that most fruits contain modest amounts of fructose (3–8 g per fruit), and the fiber, vitamin, and other constituents (flavonols, epicatechin, ascorbate, and other antioxidants) in it carries substantial metabolic benefits (Vasdev et al., 2002; Sundborn et al., 2019). Restriction of calorie intake when supplying high fructose failed to induce obesity but still triggered steatosis in rats (Roncal-Jimenez et al., 2011). Even supplement with high palm oil in high fructose diet, the rats tend to become non-obese NAFLD model (Abdelmoneim et al., 2021), suggested fructose induction may account for lean NAFLD. Lean NAFLD makes up about one-third of the NAFLD population, according to recent studies, lean NAFLD may potentiate higher metabolic risks than obese NAFLD. Thus, the association of fructose intake and lean NAFLD deserve to be systematically explored. Also, the fructose-associated NAFLD in children and adolescents needs to be highlighted. Considering the global epidemic of metabolic syndrome especially NAFLD, it is extremely important to provide alarming in controlling daily fructose intake for people, especially children and teenagers, in prevention and management of NAFLD.
Statements
Author contributions
LZ and GJ conceptualized the manuscript, SY and CL retrieved the literature, SY and CL drafted the manuscript, GJ and LZ revised the manuscript. All authors edited, revised, and approved the final version of this review.
Funding
This work is supported by the grant from Shanghai Shenkang Hospital Development Center (No. SHDC2020CR4044). The funding resources have no involvement in conceptualization, the literature collection, the writing of the review, and the decision to submit the article for publication. The fund and our institution all acknowledge open access publication fees.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
AbdelmalekM. F.SuzukiA.GuyC.Unalp-AridaA.ColvinR.JohnsonR. J.et al (2010). Increased Fructose Consumption Is Associated with Fibrosis Severity in Patients with Nonalcoholic Fatty Liver Disease. Hepatology51 (6), 1961–1971. 10.1002/hep.23535
2
AbdelmoneimD.El-AdlM.El-SayedG.El-SherbiniE. S. (2021). Protective Effect of Fenofibrate Against High-Fat-High-Fructose Diet Induced Non-obese NAFLD in Rats. Fundam. Clin. Pharmacol.35 (2), 379–388. 10.1111/fcp.12597
3
AkarF.SumluE.AlçığırM. E.BostancıA.SadiG. (2021). Potential Mechanistic Pathways Underlying Intestinal and Hepatic Effects of Kefir in High-Fructose-Fed Rats. Food Res. Int.143, 110287. 10.1016/j.foodres.2021.110287
4
Andres-HernandoA.OrlickyD. J.KuwabaraM.IshimotoT.NakagawaT.JohnsonR. J.et al (2020). Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab32 (1), 117–127.e3. 10.1016/j.cmet.2020.05.012
5
BaroneS.FussellS. L.SinghA. K.LucasF.XuJ.KimC.et al (2009). Slc2a5 (Glut5) Is Essential for the Absorption of Fructose in the Intestine and Generation of Fructose-Induced Hypertension. J. Biol. Chem.284 (8), 5056–5066. 10.1074/jbc.M808128200
6
BazzanoL. A.LiT. Y.JoshipuraK. J.HuF. B. (2008). Intake of Fruit, Vegetables, and Fruit Juices and Risk of Diabetes in Women. Diabetes Care31 (7), 1311–1317. 10.2337/dc08-0080
7
BergheimI.GuoL.DavisM. A.LambertJ. C.BeierJ. I.DuveauI.et al (2006). Metformin Prevents Alcohol-Induced Liver Injury in the Mouse: Critical Role of Plasminogen Activator Inhibitor-1. Gastroenterology130 (7), 2099–2112. 10.1053/j.gastro.2006.03.020
8
BeyerP. L.CaviarE. M.McCallumR. W. (2005). Fructose Intake at Current Levels in the United States May Cause Gastrointestinal Distress in Normal Adults. J. Am. Diet. Assoc.105 (10), 1559–1566. 10.1016/j.jada.2005.07.002
9
BezborodkinaN. N.ChestnovaA. Y.OkovityS. V.KudryavtsevB. N. (2014). Activity of Glycogen Synthase and Glycogen Phosphorylase in Normal and Cirrhotic Rat Liver During Glycogen Synthesis from Glucose or Fructose. Exp. Toxicol. Pathol.66 (2-3), 147–154. 10.1016/j.etp.2013.12.001
10
BhattacharjeeJ.KumarJ. M.ArindkarS.DasB.PramodU.JuyalR. C.et al (2014). Role of Immunodeficient Animal Models in the Development of Fructose Induced NAFLD. J. Nutr. Biochem.25 (2), 219–226. 10.1016/j.jnutbio.2013.10.010
11
BoulangéC. L.NevesA. L.ChillouxJ.NicholsonJ. K.DumasM. E. (2016). Impact of the Gut Microbiota on Inflammation, Obesity, and Metabolic Disease. Genome Med.8 (1), 42. 10.1186/s13073-016-0303-2
12
BradfordB. U.HandlerJ. A.SeedC. B.FormanD. T.ThurmanR. G. (1991). Inhibition of Ethanol Metabolism by Fructose in Alcohol Dehydrogenase-Deficient Deer Mice In Vivo. Arch. Biochem. Biophys.288 (2), 435–439. 10.1016/0003-9861(91)90217-7
13
CarreñoD. V.CorroN. B.Cerda-InfanteJ. F.EcheverríaC. E.Asencio-BarríaC. A.Torres-EstayV. A.et al (2021). Dietary Fructose Promotes Prostate Cancer Growth. Cancer Res.81 (11), 2824–2832. 10.1158/0008-5472.CAN-19-0456
14
CastrogiovanniD.AlzamendiA.OngaroL.GiovambattistaA.GaillardR. C.SpinediE. (2012). Fructose Rich Diet-Induced High Plasminogen Activator Inhibitor-1 (PAI-1) Production in the Adult Female Rat: Protective Effect of Progesterone. Nutrients4 (8), 1137–1150. 10.3390/nu4081137
15
ChapmanC. L.GrigoryanT.VargasN. T.ReedE. L.KueckP. J.PietrafesaL. D.et al (2020). High-fructose Corn Syrup-Sweetened Soft Drink Consumption Increases Vascular Resistance in the Kidneys at Rest and During Sympathetic Activation. Am. J. Physiol. Ren. Physiol318 (4), F1053–f1065. 10.1152/ajprenal.00374.2019
16
CheesemanC. I. (1993). GLUT2 Is the Transporter for Fructose Across the Rat Intestinal Basolateral Membrane. Gastroenterology105 (4), 1050–1056. 10.1016/0016-5085(93)90948-c
17
ChenW. L.JinX.WangM.LiuD.LuoQ.TianH.et al (2020). GLUT5-mediated Fructose Utilization Drives Lung Cancer Growth by Stimulating Fatty Acid Synthesis and AMPK/mTORC1 Signaling. JCI Insight5 (3), e131596. 10.1172/jci.insight.131596
18
ChoY. E.KimD. K.SeoW.GaoB.YooS. H.SongB. J. (2021). Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2e1-Mediated Oxidative and Nitrative Stress. Hepatology73 (6), 2180–2195. 10.1002/hep.30652
19
CuiX. L.JiangL.FerrarisR. P. (2003). Regulation of Rat Intestinal GLUT2 mRNA Abundance by Luminal and Systemic Factors. Biochim. Biophys. Acta1612 (2), 178–185. 10.1016/s0005-2736(03)00129-9
20
CuiX. L.SoteropoulosP.ToliasP.FerrarisR. P. (2004). Fructose-Responsive Genes in the Small Intestine of Neonatal Rats. Physiol. Genomics18 (2), 206–217. 10.1152/physiolgenomics.00056.2004
21
CuiX. L.SchlesierA. M.FisherE. L.CerqueiraC.FerrarisR. P. (2005). Fructose-induced Increases in Neonatal Rat Intestinal Fructose Transport Involve the PI3-kinase/Akt Signaling Pathway. Am. J. Physiol. Gastrointest. Liver Physiol.288 (6), G1310–G1320. 10.1152/ajpgi.00550.2004
22
DaviesD. R.DetheuxM.Van SchaftingenE. (1990). Fructose 1-Phosphate and the Regulation of Glucokinase Activity in Isolated Hepatocytes. Eur. J. Biochem.192 (2), 283–289. 10.1111/j.1432-1033.1990.tb19225.x
23
DécombazJ.JentjensR.IthM.ScheurerE.BuehlerT.JeukendrupA.et al (2011). Fructose and Galactose Enhance Postexercise Human Liver Glycogen Synthesis. Med. Sci. Sports Exerc.43 (10), 1964–1971. 10.1249/MSS.0b013e318218ca5a
24
de HerediaF. P.Gómez-MartínezS.MarcosA. (2012). Obesity, Inflammation and the Immune System. Proc. Nutr. Soc.71 (2), 332–338. 10.1017/s0029665112000092
25
DiNicolantonioJ. J.MehtaV.OnkaramurthyN.O'KeefeJ. H. (2018). Fructose-induced Inflammation and Increased Cortisol: A New Mechanism for How Sugar Induces Visceral Adiposity. Prog. Cardiovasc. Dis.61 (1), 3–9. 10.1016/j.pcad.2017.12.001
26
DoM. H.LeeE.OhM. J.KimY.ParkH. Y. (2018). High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice Without Body Weight Change. Nutrients10 (6), 761. 10.3390/nu10060761
27
DonnellyK. L.SmithC. I.SchwarzenbergS. J.JessurunJ.BoldtM. D.ParksE. J. (2005). Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Invest.115 (5), 1343–1351. 10.1172/jci23621
28
DouardV.FerrarisR. P. (2008). Regulation of the Fructose Transporter GLUT5 in Health and Disease. Am. J. Physiol. Endocrinol. Metab.295 (2), E227–E237. 10.1152/ajpendo.90245.2008
29
DouardV.SabbaghY.LeeJ.PatelC.KempF. W.BogdenJ. D.et al (2013). Excessive Fructose Intake Causes 1,25-(OH)(2)D(3)-Dependent Inhibition of Intestinal and Renal Calcium Transport in Growing Rats. Am. J. Physiol. Endocrinol. Metab.304 (12), E1303–E1313. 10.1152/ajpendo.00582.2012
30
EslamM.SanyalA. J.GeorgeJ. (2020). MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology158 (7), 1999–2014.e1. 10.1053/j.gastro.2019.11.312
31
FalonyG.VlachouA.VerbruggheK.De VuystL. (2006). Cross-feeding BBetween Bifidobacterium Longum BB536 and Acetate-Converting, Butyrate-Producing Colon Bacteria During Growth on Oligofructose. Appl. Environ. Microbiol.72 (12), 7835–7841. 10.1128/aem.01296-06
32
FantuzziG.FaggioniR. (2000). Leptin in the Regulation of Immunity, Inflammation, and Hematopoiesis. J. Leukoc. Biol.68 (4), 437–446.
33
FerrarisR. P.ChoeJ. Y.PatelC. R. (2018). Intestinal Absorption of Fructose. Annu. Rev. Nutr.38, 41–67. 10.1146/annurev-nutr-082117-051707
34
ForsheeR. A.StoreyM. L. (2003). Total Beverage Consumption and Beverage Choices Among Children and Adolescents. Int. J. Food Sci. Nutr.54 (4), 297–307. 10.1080/09637480120092143
35
FutatsugiK.SmithA. C.TuM.RaymerB.AhnK.CoffeyS. B.et al (2020). Discovery of PF-06835919: A Potent Inhibitor of Ketohexokinase (KHK) for the Treatment of Metabolic Disorders Driven by the Overconsumption of Fructose. J. Med. Chem.63 (22), 13546–13560. 10.1021/acs.jmedchem.0c00944
36
GabyA. R. (2005). Adverse Effects of Dietary Fructose. Altern. Med. Rev.10 (4), 294–306.
37
García-ArroyoF. E.Pérez-EstévezH. E.TapiaE.GonzagaG.Muñoz-JiménezI.SotoV.et al (2020). Restricted Water Intake and Hydration with Fructose-Containing Beverages During Infancy Predispose to Aggravate an Acute Renal Ischemic Insult in Adolescent Rats. Biomed. Res. Int.2020, 4281802. 10.1155/2020/4281802
38
GlushakovaO.KosugiT.RoncalC.MuW.HeinigM.CirilloP.et al (2008). Fructose Induces the Inflammatory Molecule ICAM-1 in Endothelial Cells. J. Am. Soc. Nephrol.19 (9), 1712–1720. 10.1681/asn.2007121304
39
GonzalezJ. T.BettsJ. A. (2018). Dietary Fructose Metabolism by Splanchnic Organs: Size Matters. Cel Metab27 (3), 483–485. 10.1016/j.cmet.2018.02.013
40
GouyonF.OnestoC.DaletV.PagesG.LeturqueA.Brot-LarocheE. (2003). Fructose Modulates GLUT5 mRNA Stability in Differentiated Caco-2 Cells: Role of cAMP-Signalling Pathway and PABP (Polyadenylated-Binding Protein)-Interacting Protein (Paip) 2. Biochem. J.375 (Pt 1), 167–174. 10.1042/bj20030661
41
GrassetE.PuelA.CharpentierJ.ColletX.ChristensenJ. E.TercéF.et al (2017). A Specific Gut Microbiota Dysbiosis of Type 2 Diabetic Mice Induces GLP-1 Resistance Through an Enteric NO-Dependent and Gut-Brain Axis Mechanism. Cel Metab25 (5), 1075–1090.e5. 10.1016/j.cmet.2017.04.013
42
GrewalA. S.BhardwajS.PanditaD.LatherV.SekhonB. S. (2016). Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-Diabetic Diseases. Mini Rev. Med. Chem.16 (2), 120–162. 10.2174/1389557515666150909143737
43
GutierrezJ. A.LiuW.PerezS.XingG.SonnenbergG.KouK.et al (2021). Pharmacologic Inhibition of Ketohexokinase Prevents Fructose-Induced Metabolic Dysfunction. Mol. Metab.48, 101196. 10.1016/j.molmet.2021.101196
44
HengistA.KoumanovF.GonzalezJ. T. (2019). Fructose and Metabolic Health: Governed by Hepatic Glycogen Status. J. Physiol.597 (14), 3573–3585. 10.1113/jp277767
45
HotamisligilG. S.ShargillN. S.SpiegelmanB. M. (1993). Adipose Expression of Tumor Necrosis Factor-Alpha: Direct Role in Obesity-Linked Insulin Resistance. Science259 (5091), 87–91. 10.1126/science.7678183
46
HuangZ.HongQ.ZhangX.XiaoW.WangL.CuiS.et al (2017). Aldose Reductase Mediates Endothelial Cell Dysfunction Induced by High Uric Acid Concentrations. Cell Commun. Signal.15 (1), 3. 10.1186/s12964-016-0158-6
47
HuiH.HuangD.McArthurD.NissenN.BorosL. G.HeaneyA. P. (2009). Direct Spectrophotometric Determination of Serum Fructose in Pancreatic Cancer Patients. Pancreas38 (6), 706–712. 10.1097/MPA.0b013e3181a7c6e5
48
IshimotoT.LanaspaM. A.LeM. T.GarciaG. E.DiggleC. P.MacleanP. S.et al (2012). Opposing Effects of Fructokinase C and A Isoforms on Fructose-Induced Metabolic Syndrome in Mice. Proc. Natl. Acad. Sci. U S A.109 (11), 4320–4325. 10.1073/pnas.1119908109
49
JahnD.KircherS.HermannsH. M.GeierA. (2019). Animal Models of NAFLD from a Hepatologist's Point of View. Biochim. Biophys. Acta Mol. Basis Dis.1865 (5), 943–953. 10.1016/j.bbadis.2018.06.023
50
JaiswalN.AgrawalS.AgrawalA. (2019). High Fructose-Induced Metabolic Changes Enhance Inflammation in Human Dendritic Cells. Clin. Exp. Immunol.197 (2), 237–249. 10.1111/cei.13299
51
JangC.HuiS.LuW.CowanA. J.MorscherR. J.LeeG.et al (2018). The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cel Metab27 (2), 351–361.e3. 10.1016/j.cmet.2017.12.016
52
JegatheesanP.De BandtJ. P. (2017). Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients9 (3), 230. 10.3390/nu9030230
53
JensenT.AbdelmalekM. F.SullivanS.NadeauK. J.GreenM.RoncalC.et al (2018). Fructose and Sugar: A Major Mediator of Non-Alcoholic Fatty Liver Disease. J. Hepatol.68 (5), 1063–1075. 10.1016/j.jhep.2018.01.019
54
JeongS.SavinoA. M.ChirayilR.BarinE.ChengY.ParkS. M.et al (2021). High Fructose Drives the Serine Synthesis Pathway in Acute Myeloid Leukemic Cells. Cel Metab33 (1), 145–159.e6. 10.1016/j.cmet.2020.12.005
55
JinR.VosM. B. (2015). Fructose and Liver Function-Iis This Behind Nonalcoholic Liver Disease? Curr. Opin. Clin. Nutr. Metab. Care18 (5), 490–495. 10.1097/mco.0000000000000203
56
JinC.GongX.ShangY. (2019). GLUT5 Increases Fructose Utilization in Ovarian Cancer. Onco Targets Ther.12, 5425–5436. 10.2147/ott.S205522
57
JohnsonR. J.Perez-PozoS. E.LilloJ. L.GrasesF.ScholdJ. D.KuwabaraM.et al (2018). Fructose Increases Risk for Kidney Stones: Potential Role in Metabolic Syndrome and Heat Stress. BMC Nephrol.19 (1), 315. 10.1186/s12882-018-1105-0
58
JonesN.BlagihJ.ZaniF.ReesA.HillD. G.JenkinsB. J.et al (2021). Fructose Reprogrammes Glutamine-dependent Oxidative Metabolism to Support LPS-Induced Inflammation. Nat. Commun.12 (1), 1209. 10.1038/s41467-021-21461-4
59
KawasakiT.AkanumaH.YamanouchiT. (2002). Increased Fructose Concentrations in Blood and Urine in Patients with Diabetes. Diabetes Care25 (2), 353–357. 10.2337/diacare.25.2.353
60
KovačevićS.NestorovJ.MatićG.ElakovićI. (2014). Dietary Fructose-Related Adiposity and Glucocorticoid Receptor Function in Visceral Adipose Tissue of Female Rats. Eur. J. Nutr.53 (6), 1409–1420. 10.1007/s00394-013-0644-1
61
KuhreR. E.GribbleF. M.HartmannB.ReimannF.WindeløvJ. A.RehfeldJ. F.et al (2014). Fructose Stimulates GLP-1 but Not GIP Secretion in Mice, Rats, and Humans. Am. J. Physiol. Gastrointest. Liver Physiol.306 (7), G622–G630. 10.1152/ajpgi.00372.2013
62
LanaspaM. A.CicerchiC.GarciaG.LiN.Roncal-JimenezC. A.RivardC. J.et al (2012a). Counteracting Roles of AMP Deaminase and AMP Kinase in the Development of Fatty Liver. PLoS One7 (11), e48801. 10.1371/journal.pone.0048801
63
LanaspaM. A.Sanchez-LozadaL. G.ChoiY. J.CicerchiC.KanbayM.Roncal-JimenezC. A.et al (2012b). Uric Acid Induces Hepatic Steatosis by Generation of Mitochondrial Oxidative Stress: Potential Role in Fructose-dependent and -independent Fatty Liver. J. Biol. Chem.287 (48), 40732–40744. 10.1074/jbc.M112.399899
64
LanaspaM. A.Sanchez-LozadaL. G.CicerchiC.LiN.Roncal-JimenezC. A.IshimotoT.et al (2012c). Uric Acid Stimulates Fructokinase and Accelerates Fructose Metabolism in the Development of Fatty Liver. PLoS One7 (10), e47948. 10.1371/journal.pone.0047948
65
LanaspaM. A.IshimotoT.LiN.CicerchiC.OrlickyD. J.RuzyckiP.et al (2013). Endogenous Fructose Production and Metabolism in the Liver Contributes to the Development of Metabolic Syndrome. Nat. Commun.4, 2434. 10.1038/ncomms3434
66
LanaspaM. A.KuwabaraM.Andres-HernandoA.LiN.CicerchiC.JensenT.et al (2018). High Salt Intake Causes Leptin Resistance and Obesity in Mice by Stimulating Endogenous Fructose Production and Metabolism. Proc. Natl. Acad. Sci. U S A.115 (12), 3138–3143. 10.1073/pnas.1713837115
67
LarssonS. C.BergkvistL.WolkA. (2006). Consumption of Sugar and Sugar-Sweetened Foods and the Risk of Pancreatic Cancer in a Prospective Study. Am. J. Clin. Nutr.84 (5), 1171–1176. 10.1093/ajcn/84.5.1171
68
LeM. T.LanaspaM. A.CicerchiC. M.RanaJ.ScholtenJ. D.HunterB. L.et al (2016). Bioactivity-Guided Identification of Botanical Inhibitors of Ketohexokinase. PLoS One11 (6), e0157458. 10.1371/journal.pone.0157458
69
LimJ. S.Mietus-SnyderM.ValenteA.SchwarzJ. M.LustigR. H. (2010). The Role of Fructose in the Pathogenesis of NAFLD and the Metabolic Syndrome. Nat. Rev. Gastroenterol. Hepatol.7 (5), 251–264. 10.1038/nrgastro.2010.41
70
LiuL.LiT.LiaoY.WangY.GaoY.HuH.et al (2020). Triose Kinase Controls the Lipogenic Potential of Fructose and Dietary Tolerance. Cel Metab32 (4), 605–618.e7. 10.1016/j.cmet.2020.07.018
71
LoombaR.SanyalA. J. (2013). The Global NAFLD Epidemic. Nat. Rev. Gastroenterol. Hepatol.10 (11), 686–690. 10.1038/nrgastro.2013.171
72
LustigR. H. (2013). Fructose: It's "Alcohol without the Buzz". Adv. Nutr.4 (2), 226–235. 10.3945/an.112.002998
73
MilutinovićD. V.BrkljačićJ.TeofilovićA.BursaćB.NikolićM.GligorovskaL.et al (2020). Chronic Stress Potentiates High Fructose-Induced Lipogenesis in Rat Liver and Kidney. Mol. Nutr. Food Res.64 (13), 1901141. 10.1002/mnfr.201901141
74
MirtschinkP.JangC.AranyZ.KrekW. (2018). Fructose Metabolism, Cardiometabolic Risk, and the Epidemic of Coronary Artery Disease. Eur. Heart J.39 (26), 2497–2505. 10.1093/eurheartj/ehx518
75
MoellerS. M.FryhoferS. A.OsbahrA. J.3rdRobinowitzC. B. (2009). The Effects of High Fructose Syrup. J. Am. Coll. Nutr.28 (6), 619–626. 10.1080/07315724.2009.10719794
76
MontroseD. C.NishiguchiR.BasuS.StaabH. A.ZhouX. K.WangH.et al (2021). Dietary Fructose Alters the Composition, Localization, and Metabolism of Gut Microbiota in Association with Worsening Colitis. Cell Mol Gastroenterol Hepatol11 (2), 525–550. 10.1016/j.jcmgh.2020.09.008
77
MuecklerM.ThorensB. (2013). The SLC2 (GLUT) Family of Membrane Transporters. Mol. Aspects Med.34 (2-3), 121–138. 10.1016/j.mam.2012.07.001
78
MurakiI.ImamuraF.MansonJ. E.HuF. B.WillettW. C.van DamR. M.et al (2013). Fruit Consumption and Risk of Type 2 Diabetes: Results from Three Prospective Longitudinal Cohort Studies. Bmj347, f5001. 10.1136/bmj.f5001
79
NakagawaT.LanaspaM. A.MillanI. S.FiniM.RivardC. J.Sanchez-LozadaL. G.et al (2020). Fructose Contributes to the Warburg Effect for Cancer Growth. Cancer Metab.8, 16. 10.1186/s40170-020-00222-9
80
NakatsuY.SenoY.KushiyamaA.SakodaH.FujishiroM.KatasakoA.et al (2015). The Xanthine Oxidase Inhibitor Febuxostat Suppresses Development of Nonalcoholic Steatohepatitis in a Rodent Model. Am. J. Physiol. Gastrointest. Liver Physiol.309 (1), G42–G51. 10.1152/ajpgi.00443.2014
81
NewensK. J.WaltonJ. (2016). A Review of Sugar Consumption from Nationally Representative Dietary Surveys Across the World. J. Hum. Nutr. Diet.29 (2), 225–240. 10.1111/jhn.12338
82
NomuraN.VerdonG.KangH. J.ShimamuraT.NomuraY.SonodaY.et al (2015). Structure and Mechanism of the Mammalian Fructose Transporter GLUT5. Nature526 (7573), 397–401. 10.1038/nature14909
83
OuyangX.CirilloP.SautinY.McCallS.BruchetteJ. L.DiehlA. M.et al (2008). Fructose Consumption as a Risk Factor for Non-alcoholic Fatty Liver Disease. J. Hepatol.48 (6), 993–999. 10.1016/j.jhep.2008.02.011
84
PanJ. H.KimH. S.BeaneK. E.MontalbanoA. M.LeeJ. H.KimY. J.et al (2018). IDH2 Deficiency Aggravates Fructose-Induced NAFLD by Modulating Hepatic Fatty Acid Metabolism and Activating Inflammatory Signaling in Female Mice. Nutrients10 (6), 679. 10.3390/nu10060679
85
PatelC.DouardV.YuS.GaoN.FerrarisR. P. (2015). Transport, Metabolism, and Endosomal Trafficking-dependent Regulation of Intestinal Fructose Absorption. Faseb j29 (9), 4046–4058. 10.1096/fj.15-272195
86
PattersonE.RyanP. M.CryanJ. F.DinanT. G.RossR. P.FitzgeraldG. F.et al (2016). Gut Microbiota, Obesity and Diabetes. Postgrad. Med. J.92 (1087), 286–300. 10.1136/postgradmedj-2015-133285
87
PongkingT.HaononO.DangtakotR.OnsurathumS.JusakulA.IntuyodK.et al (2020). A Combination of Monosodium Glutamate and High-Fat and High-Fructose Diets Increases the Risk of Kidney Injury, Gut Dysbiosis and Host-Microbial Co-metabolism. PLoS One15 (4), e0231237. 10.1371/journal.pone.0231237
88
PowellE. S.Smith-TaillieL. P.PopkinB. M. (2016). Added Sugars Intake across the Distribution of US Children and Adult Consumers: 1977-2012. J. Acad. Nutr. Diet.116 (10), 1543–1550.e1. 10.1016/j.jand.2016.06.003
89
PremachandranV.TarlowD.YuilleA. L.BatraD. (2017). Empirical Minimum Bayes Risk Prediction. IEEE Trans. Pattern Anal. Mach Intell.39 (1), 75–86. 10.1109/tpami.2016.2537807
90
QiY.WanM.Abd El-AtyA. M.LiH.CaoL.SheY.et al (2020). A "half" Core-Shell Magnetic Nanohybrid Composed of Zeolitic Imidazolate Framework and Graphitic Carbon Nitride for Magnetic Solid-phase Extraction of Sulfonylurea Herbicides from Water Samples Followed by LC-MS/MS Detection. Mikrochim Acta187 (5), 279. 10.1007/s00604-020-04243-5
91
RamezaniA.RajD. S. (2014). The Gut Microbiome, Kidney Disease, and Targeted Interventions. J. Am. Soc. Nephrol.25 (4), 657–670. 10.1681/asn.2013080905
92
Roncal-JimenezC. A.LanaspaM. A.RivardC. J.NakagawaT.Sanchez-LozadaL. G.JalalD.et al (2011). Sucrose Induces Fatty Liver and Pancreatic Inflammation in Male Breeder Rats Independent of Excess Energy Intake. Metabolism60 (9), 1259–1270. 10.1016/j.metabol.2011.01.008
93
Sánchez-LozadaL. G.LeM.SegalM.JohnsonR. J. (2008). How Safe Is Fructose for Persons With or Without Diabetes. Am. J. Clin. Nutr.88 (5), 1189–1190. 10.3945/ajcn.2008.26812
94
SchmidlS.Tamayo RojasS. A.IancuC. V.ChoeJ. Y.OrebM. (2020). Functional Expression of the Human Glucose Transporters GLUT2 and GLUT3 in Yeast Offers Novel Screening Systems for GLUT-Targeting Drugs. Front. Mol. Biosci.7, 598419. 10.3389/fmolb.2020.598419
95
SeinoY.OgataH.MaekawaR.IzumotoT.IidaA.HaradaN.et al (2015). Fructose Induces Glucose-dependent Insulinotropic Polypeptide, Glucagon-like Peptide-1 and Insulin Secretion: Role of Adenosine Triphosphate-Sensitive K(+) Channels. J. Diabetes Investig.6 (5), 522–526. 10.1111/jdi.12356
96
SenT.CawthonC. R.IhdeB. T.HajnalA.DiLorenzoP. M.de La SerreC. B.et al (2017). Diet-driven Microbiota Dysbiosis Is Associated with Vagal Remodeling and Obesity. Physiol. Behav.173, 305–317. 10.1016/j.physbeh.2017.02.027
97
SertogluE.ErcinC. N.CelebiG.GurelH.KayadibiH.GencH.et al (2014). The Relationship of Serum Uric Acid with Non-alcoholic Fatty Liver Disease. Clin. Biochem.47 (6), 383–388. 10.1016/j.clinbiochem.2014.01.029
98
ShapiroA.TümerN.GaoY.ChengK. Y.ScarpaceP. J. (2011). Prevention and Reversal of Diet-Induced Leptin Resistance with a Sugar-free Diet Despite High Fat Content. Br. J. Nutr.106 (3), 390–397. 10.1017/s000711451100033x
99
ShepherdE. L.SaboranoR.NorthallE.MatsudaK.OginoH.YashiroH.et al (2021). Ketohexokinase Inhibition Improves NASH by Reducing Fructose-Induced Steatosis and Fibrogenesis. JHEP Rep.3 (2), 100217. 10.1016/j.jhepr.2020.100217
100
SofticS.GuptaM. K.WangG. X.FujisakaS.O'NeillB. T.RaoT. N.et al (2017). Divergent Effects of Glucose and Fructose on Hepatic Lipogenesis and Insulin Signaling. J. Clin. Invest.127 (11), 4059–4074. 10.1172/jci94585
101
SofticS.MeyerJ. G.WangG. X.GuptaM. K.BatistaT. M.LauritzenH. P. M. M.et al (2019). Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cel Metab.30 (4), 735–753.e4. 10.1016/j.cmet.2019.09.003
102
SonnenburgJ. L.XuJ.LeipD. D.ChenC. H.WestoverB. P.WeatherfordJ.et al (2005). Glycan Foraging In Vivo by an Intestine-Adapted Bacterial Symbiont. Science307 (5717), 1955–1959. 10.1126/science.1109051
103
SouthgateD. A. (1995). Digestion and Metabolism of Sugars. Am. J. Clin. Nutr.62 (1), 203S–210S. 10.1093/ajcn/62.1.203S
104
SuC.LiH.GaoW. (2018). GLUT5 Increases Fructose Utilization and Promotes Tumor Progression in Glioma. Biochem. Biophys. Res. Commun.500 (2), 462–469. 10.1016/j.bbrc.2018.04.103
105
SundbornG.ThornleyS.MerrimanT. R.LangB.KingC.LanaspaM. A.et al (2019). Are Liquid Sugars Different from Solid Sugar in Their Ability to Cause Metabolic Syndrome?Obesity (Silver Spring)27 (6), 879–887. 10.1002/oby.22472
106
TangW. H.WangZ.KennedyD. J.WuY.BuffaJ. A.Agatisa-BoyleB.et al (2015). Gut Microbiota-Dependent Trimethylamine N-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ. Res.116 (3), 448–455. 10.1161/circresaha.116.305360
107
TargherG.BertoliniL.RodellaS.ZoppiniG.ZenariL.FalezzaG. (2006). Associations Between Liver Histology and Cortisol Secretion in Subjects with Nonalcoholic Fatty Liver Disease. Clin. Endocrinol. (Oxf)64 (3), 337–341. 10.1111/j.1365-2265.2006.02466.x
108
TaylorS. R.RamsamoojS.LiangR. J.KattiA.PozovskiyR.VasanN.et al (2021). Dietary Fructose Improves Intestinal Cell Survival and Nutrient Absorption. Nature597, 263–267. 10.1038/s41586-021-03827-2
109
ThongnakL.ChatsudthipongV.KongkaewA.LungkaphinA. (2020). Effects of Dapagliflozin and Statins Attenuate Renal Injury and Liver Steatosis in High-Fat/high-Fructose Diet-Induced Insulin Resistant Rats. Toxicol. Appl. Pharmacol.396, 114997. 10.1016/j.taap.2020.114997
110
ThorensB. (2015). GLUT2, Glucose Sensing and Glucose Homeostasis. Diabetologia58 (2), 221–232. 10.1007/s00125-014-3451-1
111
TodoricJ.Di CaroG.ReibeS.HenstridgeD. C.GreenC. R.VrbanacA.et al (2020). Fructose Stimulated De Novo Lipogenesis Is Promoted by Inflammation. Nat. Metab.2 (10), 1034–1045. 10.1038/s42255-020-0261-2
112
TranC.Jacot-DescombesD.LecoultreV.FieldingB. A.CarrelG.LêK. A.et al (2010). Sex Differences in Lipid and Glucose Kinetics After Ingestion of an Acute Oral Fructose Load. Br. J. Nutr.104 (8), 1139–1147. 10.1017/s000711451000190x
113
UzuegbuU. E.OnyesomI. (2009). Fructose-induced Increase in Ethanol Metabolism and the Risk of Syndrome X in Man. C R. Biol.332 (6), 534–538. 10.1016/j.crvi.2009.01.007
114
Van den BergheG. (1986). Fructose: Metabolism and Short-Term Effects on Carbohydrate and Purine Metabolic Pathways. Prog. Biochem. Pharmacol.21, 1–32.
115
VasdevS.GillV.ParaiS.LongerichL.GadagV. (2002). Dietary Vitamin E and C Supplementation Prevents Fructose Induced Hypertension in Rats. Mol. Cel Biochem241 (1-2), 107–114. 10.1023/a:1020835229591
116
VatsD.MukundanL.OdegaardJ. I.ZhangL.SmithK. L.MorelC. R.et al (2006). Oxidative Metabolism and PGC-1beta Attenuate Macrophage-Mediated Inflammation. Cel Metab4 (1), 13–24. 10.1016/j.cmet.2006.05.011
117
Villalobos-GarcíaD.Ayhllon-OsorioC. A.Hernández-MuñozR. (2021). The Fructose-dependent Acceleration of Ethanol Metabolism. Biochem. Pharmacol.188, 114498. 10.1016/j.bcp.2021.114498
118
Villa-RodriguezJ. A.AydinE.GauerJ. S.PynerA.WilliamsonG.KerimiA. (2017). Green and Chamomile Teas, but Not Acarbose, Attenuate Glucose and Fructose Transport via Inhibition of GLUT2 and GLUT5. Mol. Nutr. Food Res.61 (12), 1700566. 10.1002/mnfr.201700566
119
VosM. B.LavineJ. E. (2013). Dietary Fructose in Nonalcoholic Fatty Liver Disease. Hepatology57 (6), 2525–2531. 10.1002/hep.26299
120
WalkerR. W.LêK. A.DavisJ.AldereteT. L.CherryR.LebelS.et al (2012). High Rates of Fructose Malabsorption Are Associated with Reduced Liver Fat in Obese African Americans. J. Am. Coll. Nutr.31 (5), 369–374. 10.1080/07315724.2012.10720445
121
WanX.XuC.LinY.LuC.LiD.SangJ.et al (2016). Uric Acid Regulates Hepatic Steatosis and Insulin Resistance Through the NLRP3 Inflammasome-dependent Mechanism. J. Hepatol.64 (4), 925–932. 10.1016/j.jhep.2015.11.022
122
WangQ.ZhaoY.SunM.LiuS.LiB.ZhangL.et al (2014). 2-Deoxy-d-glucose Attenuates Sevoflurane-Induced Neuroinflammation Through Nuclear Factor-Kappa B Pathway In Vitro. Toxicol. Vitro28 (7), 1183–1189. 10.1016/j.tiv.2014.05.006
123
WarnerS. O.WadianA. M.SmithM.FarmerB.DaiY.SheanonN.et al (2021). Liver Glycogen-Induced Enhancements in Hypoglycemic Counterregulation Require Neuroglucopenia. Am. J. Physiol. Endocrinol. Metab.320 (5), E914–e924. 10.1152/ajpendo.00501.2020
124
WhiteJ. S. (2013). Challenging the Fructose Hypothesis: New Perspectives on Fructose Consumption and Metabolism. Adv. Nutr.4 (2), 246–256. 10.3945/an.112.003137
125
WilliamsC. D.StengelJ.AsikeM. I.TorresD. M.ShawJ.ContrerasM.et al (2011). Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology140 (1), 124–131. 10.1053/j.gastro.2010.09.038
126
WittekindA.WaltonJ. (2014). Worldwide Trends in Dietary Sugars Intake. Nutr. Res. Rev.27 (2), 330–345. 10.1017/s0954422414000237
127
XuJ.BjursellM. K.HimrodJ.DengS.CarmichaelL. K.ChiangH. C.et al (2003). A Genomic View of the Human-Bacteroides Thetaiotaomicron Symbiosis. Science299 (5615), 2074–2076. 10.1126/science.1080029
128
YamamotoT.MoriwakiY.TakahashiS.TsutsumiZ.OhataH.YamakitaJ.et al (1999). Effect of Amino Acids on the Plasma Concentration and Urinary Excretion of Uric Acid and Uridine. Metabolism48 (8), 1023–1027. 10.1016/s0026-0495(99)90200-7
129
YangN.HongN. J.GarvinJ. L. (2020). Dietary Fructose Enhances Angiotensin II-Stimulated Na+ Transport via Activation of PKC-α in Renal Proximal Tubules. Am. J. Physiol. Ren. Physiol318 (6), F1513–f1519. 10.1152/ajprenal.00543.2019
130
ZhaoS.JangC.LiuJ.UeharaK.GilbertM.IzzoL.et al (2020). Dietary Fructose Feeds Hepatic Lipogenesis via Microbiota-Derived Acetate. Nature579 (7800), 586–591. 10.1038/s41586-020-2101-7
131
ZubiríaM. G.GambaroS. E.ReyM. A.CarasiP.SerradellM. L. Á.GiovambattistaA. (2017). Deleterious Metabolic Effects of High Fructose Intake: The Preventive Effect of Lactobacillus Kefiri Administration. Nutrients9 (5), 470. 10.3390/nu9050470
Summary
Keywords
non-alcoholic fatty liver disease (NAFLD), fructose, intestinal environment, de novo lipogenesis, inflammation
Citation
Yu S, Li C, Ji G and Zhang L (2021) The Contribution of Dietary Fructose to Non-alcoholic Fatty Liver Disease. Front. Pharmacol. 12:783393. doi: 10.3389/fphar.2021.783393
Received
26 September 2021
Accepted
02 November 2021
Published
18 November 2021
Volume
12 - 2021
Edited by
Irwin Rose Alencar de Menezes, Regional University of Cariri, Brazil
Reviewed by
Almir Gonçalves Wanderley, Federal University of São Paulo, Brazil
Radosław Kowalski, University of Life Sciences of Lublin, Poland
Updates
Copyright
© 2021 Yu, Li, Ji and Zhang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang Ji, jg@shutcm.edu.cn, jiliver@vip.sina.com; Li Zhang, zhangli.hl@163.com
†These authors have contributed equally to this work
This article was submitted to Gastrointestinal and Hepatic Pharmacology, a section of the journal Frontiers in Pharmacology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.