 Giselle A. Borges e Soares1†
Giselle A. Borges e Soares1† Tanima Bhattacharya2,3*†
Tanima Bhattacharya2,3*† Shivalingrao Mamledesai4†
Shivalingrao Mamledesai4† Zhaoquan Ai2
Zhaoquan Ai2 Alexandru Madalin Hasan5
Alexandru Madalin Hasan5 Simona Cavalu5*
Simona Cavalu5*- 1Department of Medicinal and Biological Chemistry, University of Toledo, Toledo, OH, United States
- 2Hubei Collaborative Innovation Centre for Advanced Organic Chemical Materials, Hubei University, Wuhan, China
- 3Innovation, Incubation and Industry (i-cube) Laboratory, Techno India NJR Institute of Technology, Udaipur, India
- 4Department of Pharmaceutical Chemistry, PES Rajaram and Tarabai Bandekar College of Pharmacy, Goa, India
- 5Faculty of Medicine and Pharmacy, University of Oradea, Oradea, Romania
12 analogs bearing a structural similarity to Linomide, a bonafide anticancer agent were synthesized wherein cyclization of substituted dianilides rendered 4-hydroxyquinolin-2(1H)-ones that were subjected to a Mannich reaction to yield 4-hydroxy-3-(substituted-1-ylmethyl) quinolin-2(1H)-one analogs. Characterization was performed using IR, 1H nuclear magnetic resonance and 13C NMR spectral analysis. Subsequently, in vitro anticancer studies revealed that Compound 4b showed maximum cytotoxicity with IC50 values of 1.539 μM/ml and 1.732 μM/ml against A549 and K562 cell lines respectively. This, however, is lower in comparison with standard Paclitaxel (IC50 values of 0.3 μM/ml for both cell lines). Surprisingly, docking studies at the active site of EGFRK revealed Compound 4b possessed a MolDock Score of -110.2253 that is highly comparable to the standard 4-anilinoquinazoline (MolDock Score of -112.04). Our computational and biological data thus provides an insight on the cytotoxicity of these derivatives and warrants future research that can possibly lead to the development of potent anticancer therapeutics.
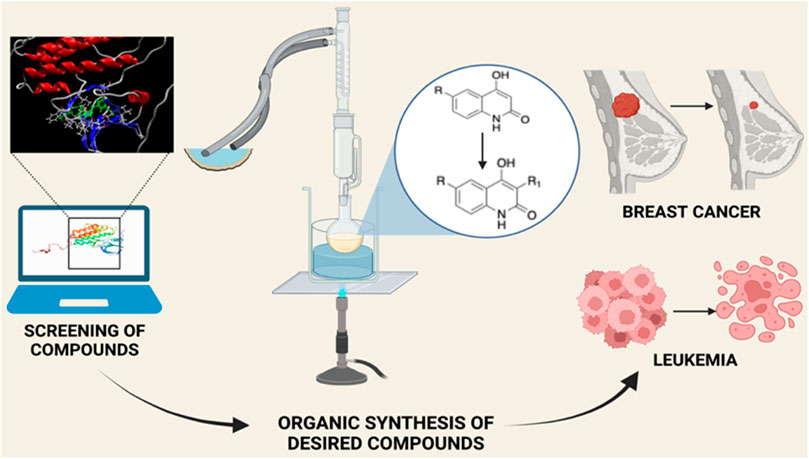
GRAPHICAL ABSTRACT
Introduction
Cancer, a major cause of death world-wide refers to the uncontrollable growth of cells that can metastasize. The localization of cancer at the primary site, is coupled with a good prognosis, however, metastasis, the term used to describe the escape of these cells from the primary site to other distant organs is difficult to treat (Zubair and Ahmad, 2017; Smaranda, 2020) According to the WHO, approx. 10 million lives were lost in 2020 due to cancer, from which lung cancer accounted for 2.21 million cases (WHO, 2021). Due to a better understanding of genetics, cancer pathology and the immune system, several therapies for cancer diagnostics and treatment have been discovered. However, in most instances, treatment failure is observed either due to pharmacological and toxicity issues or drug resistance (Maeda and Khatami, 2018; Islam, et al., 2022).
Linomide, a quinoline-3-carboxamide (Figure 1), an immunomodulatory drug has been shown to possess antibacterial, antiangiogenic, and anticancer activity (Gaude et al., 2017). As the mechanism of action of Linomide is elusive till today, world-wide, researchers have attempted to develop analogs using Linomide as a lead molecule to understand its pharmacological action and biological target. Yang et al. developed Linomide analogs that showed better and a more selective anti-angiogenic activity on endothelial cells as compared to Linomide. These results led the authors to conclude that the selectivity is due to an alternative unknown pathway not involving VEGF/KDR and that requires further investigation (Yang et al., 2005). Ma et al. developed novel histone deacetylase inhibitors for hepatocellular carcinoma using Linomide as a lead molecule. Compounds showed cytotoxicity in Hep G2, and HuH-7 cell lines, the best compound showing an IC50 of 1.53 and 3.06 μM respectively (Ma et al., 2021). Another study involved the replacement of the anilido moiety of Linomide at C-3 position with a sulfamoyl acyl, N-methyl, and N-phenyl functionality by Priolkar et al. (2020). These compounds showed superior inhibitory activity through well conserved H-bonding with amino acids at the EGFRK active site observed through docking studies. Moreover, this study indicated that a phenyl ring system at C-3 rendered superior IC50 when tested on A549 and K562 cell lines.
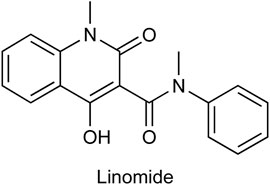
FIGURE 1. Chemical structure of Linomide, a known anticancer agent and the lead molecule of our current paper.
In keeping with the above-mentioned study and to further understand the mechanism of action of Linomide, we decided to use Linomide as a “lead-molecule” and to synthesize 12 derivatives bearing a structural resemblance to it through modification at the C-3 position of the quinoline-2-one nucleus using various heterocyclic ring systems. We then performed molecular docking studies which allowed us to conclusively determine the binding site of these analogs thus allowing the prediction and determination of the binding site of Linomide. Following synthesis, we characterized these compounds using Fourier transform infrared spectrometry (FT-IR), 1H nuclear magnetic resonance (NMR) and 13C NMR spectral analysis in terms of docking results. Finally, to evaluate the anticancer ability of these compounds, we performed the MTT assay.
Hence, by using computational and biological data, the aim of our work was to establish the mechanism by which Linomide behaves as an anticancer drug and warrants future in vitro and in vivo studies on Linomide derivatives that can be further developed into potent anticancer agents.
Rationale of Molecular Design
The discovery of the 4-anilinoquinazoline class of EGFRK-targeting anti-cancer drugs was a major step forward in drug discovery as these agents brought about a potent and selective inhibition of tyrosine kinase through competitive binding at the enzyme’s ATP site. The SAR of 4-anilinoquinazoline revealed the requirement of a small lipophilic electron withdrawing group at position three of the aniline ring along with electron donating groups at position six and seven of the quinazoline ring system. Moreover, SAR studies indicated that the -N atom on position one of the quinazoline nucleus is significant for H-bonding formation with Met-769 (Rewcastle et al., 1998). Investigation of the pharmacophoric features shared by EGFRK inhibitors indicated that two features are significant for activity:
1) The presence of a heterocyclic aromatic ring system that takes part in hydrogen bond formation with amino acids in the active site of EGFRK (Ma et al., 2021).
2) A hydrophobic side chain that interacts with the hydrophobic pocket of EGFRK (Rewcastle et al., 1998; Priolkar et al., 2020).
In keeping with this observation, with respect to the synthesis of all Linomide derivatives described in this paper, we decided to replace the -CH3 group attached to the -N atom at position one of Linomide with a -H atom to determine its role in -H bonding with amino acids at the EGFRK groove. Moreover, we also decided to modify position C-3 of Linomide and introduce bulky heterocyclic ring systems such as morpholine, piperidine and pyrrolidine to determine if these groups could improve interactions at the active site of EGFRK. Since the presence of an electron-withdrawing group was also found to be necessary for activity, we decided to synthesize analogs bearing groups such as -Cl, -Br, -F at the C-6 position to determine the significance of the C-6 position as well as the role of these functionalities in bringing about an inhibition of EGFRK.
Results and Discussion
Chemistry
The strategy implemented for the synthesis of 4-hydroxy-3-(substituted-1-ylmethyl) quinolin-2(1H)-one derivatives is described in Scheme 1. The starting material constituting of dianilides N1, N3- diphenyl/bis(4-chlorophenyl)/bis (4- fluorophenyl)/di-(p-tolyl) malonamide (1a/1b/1c/1d) were synthesized as described in the literature (Mokrov et al., 2019). Table 1 illustrates the structures of the intermediates and synthesized compounds along with their IUPAC names.
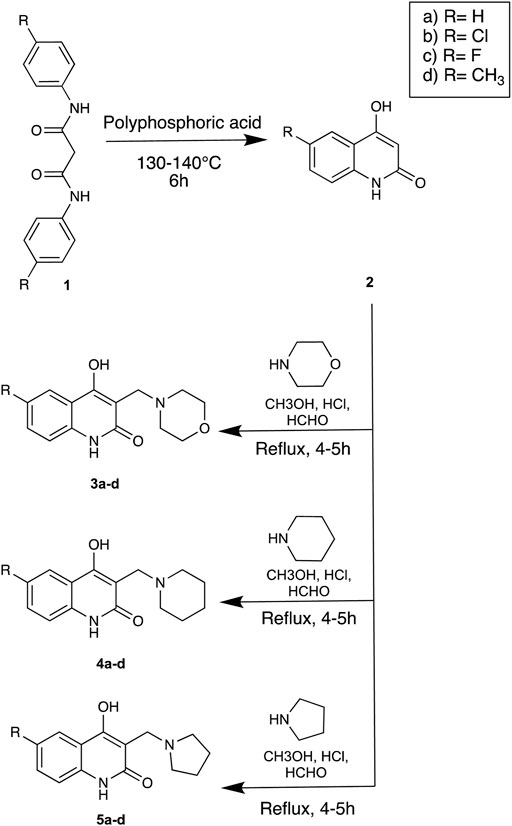
SCHEME 1. Strategy for the synthesis of Linomide derivatives described in this paper.
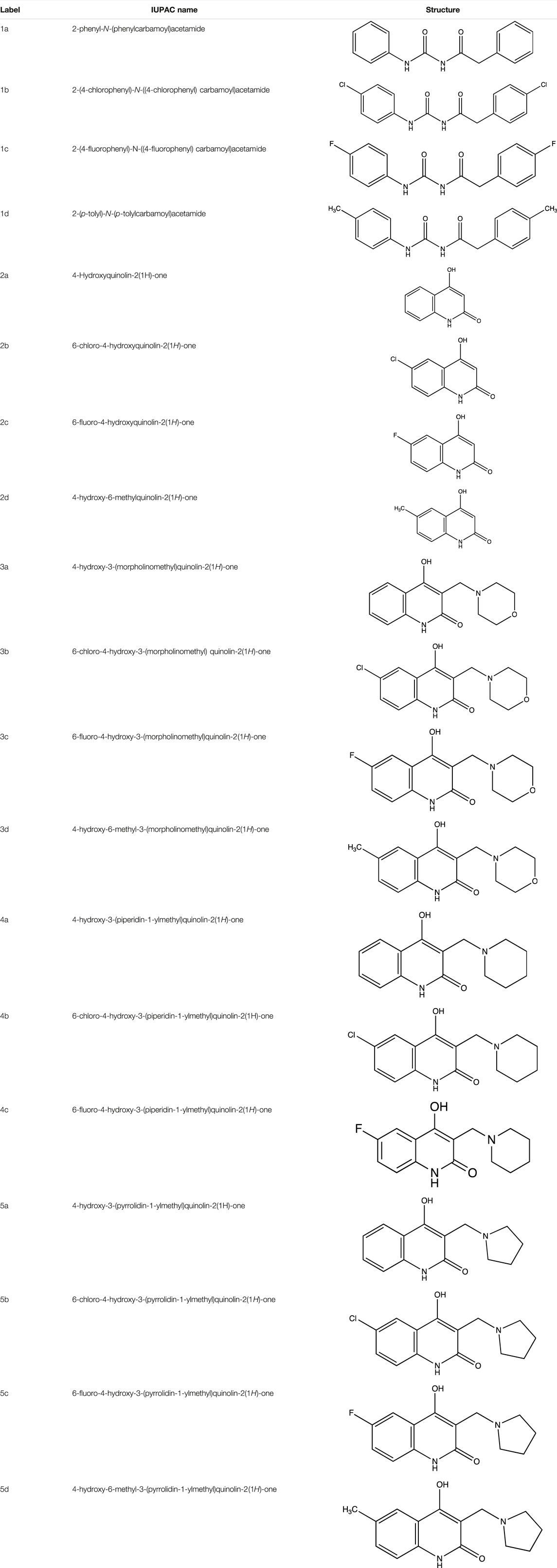
TABLE 1. Summary of the reagents, intermediates, synthesized compounds, their labels, IUPAC names and functional groups.
In Silico Studies
Molecular Docking Results
The results obtained upon performing docking studies using the MoleGro virtual docking software is summarized in Table 2. Docking scores of all 12 synthesized derivatives with the EGFRK enzyme was obtained and were compared to the score obtained by the reference ligand 4-anilinoquinazoline (4-AQ) (a known inhibitor of EGFRK) upon binding to EGFRK as illustrated in Figure 2A and Figure 2B. MolDock scores of the synthesized compounds ranged from -138.886 to -84.6748 while the MolDock score of 4-AQ was -112.04.
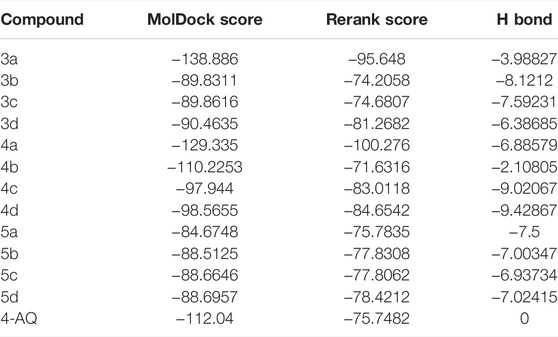
TABLE 2. MolDock scores of synthesized compounds (3a-d, 4a-d, 5a-d).
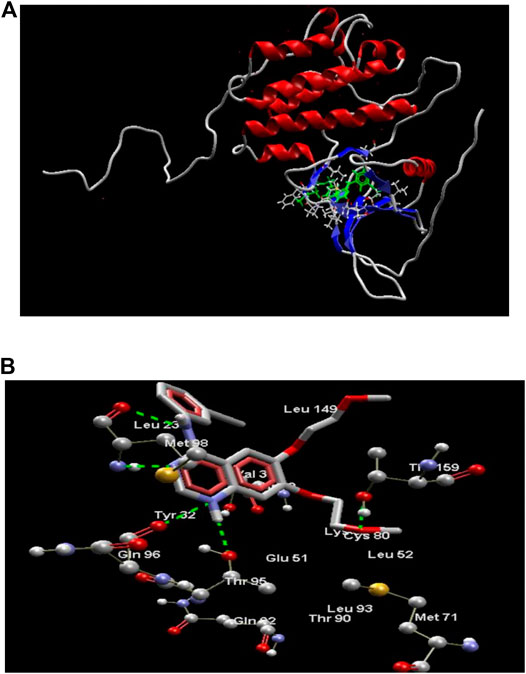
FIGURE 2. (A) Structure of EGFRK domain complexed with 4-AQ (PDB ID: 1m17). (B) 4-AQ docked in its best conformation (pose) into the active site of EGFRK.
As illustrated in Figure 2B, the binding of 4-AQ to EGFK involves four interactions:
1) H-bond formation between -NH at position one of quinazoline and -C=O of Gln96 and -OH of Thr95.
2) H-bond interaction between nitrogen atom at position three of quinazoline and -NH of Met98.
3) H-bond interaction between -NH at position four of quinazoline and -C=O of Met98.
4) Interaction between oxygen from the terminal methoxy group of the side chain with -OH of Thr159.
Amongst all compounds docked, compound 4b had a dock score of -110.2253, a score that was comparable with that obtained with 4-AQ. Upon further analysis, we observed an-NH group interaction of the quinolin-2-one nucleus with both Thr159 and -C=O of Asp160 as illustrated in Figure 3, indicating a good fit. A good fit and a strong non-covalent interaction of the molecule at the EGFRK site indicates its potential to be highly therapeutic and effective as a pharmacological agent.
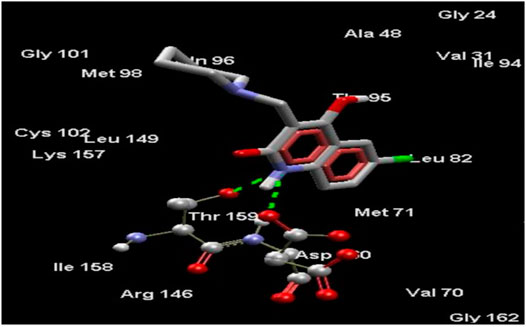
FIGURE 3. Compound 4b docked in its best conformation (pose) into the binding site of EGFRK.
Based on the molecular docking scores, we performed the MTT assay of representative derivatives that had highest, lowest, and comparable docking scores to that of 4-AQ to determine the correlation between docking results obtained and in vitro studies as well as to determine their ability to behave as potential anticancer agents.
Biological Evaluation
MTT Assay
Cell line of A549 (Millipore Sigma, 86012804) cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermofischer, Cat. No: 11965-092) supplemented with 10% heat inactivated fetal bovine serum (HI-FBS) (Gibco, Invitrogen, Cat. No: 10270106) and cell line of K562 (Millipore Sigma, 89121407) cultured in RPMI (HiMedia Laboratories, Cat. No: AL180A) were supplemented with 1% antibiotic-antimycotic 100X solution (Thermofischer Scientific, Cat. No: 15240062) and then seeded at a density of approximately 5 × 103 cells/well in a 96 well flat bottom microplate and maintained overnight at 37°C ± 5% CO2 and 95% humidity. Synthesized compounds dissolved in 1X PBS were then used for treatment in concentrations of 400, 300, 200, 100, 50 and 25 μg/ml and cells were further incubated for 48 h. The cells were washed twice with 1X PBS followed by the addition of 20 μL of the MTT staining solution and incubation at 37 °C. After 4 h, 100 μL of di-methyl sulfoxide (DMSO) was added to dissolve the formazan crystals and the absorbance was recorded at 570 nm using microplate reader. The formula used to determine surviving cells and inhibitory cells was as follows:
IC50 of compounds 3a (MolDock score -138.886), 4a (MolDock score -129.335), 5a (MolDock score -84.6748) and 4b (MolDock score -110.2253) were measured using the MTT Assay which are illustrated in Table 3 for the A549 cell line and Table 4 for the K562 cell line. These compounds were compared with standard Paclitaxel that showed an IC50 of 0.3 μM in both A549 and K562 cell lines.

TABLE 3. IC50 values of synthesized compounds against A549 cell line.

TABLE 4. IC50 values of synthesized compounds against K562 cell line.
SAR of Synthesized Linomide Analogs
Our in-silico data as well as the data we obtained through the biological assay performed on A549 and K562 cell lines sheds light on the significance of certain positions of the quinoline-2-one nucleus of Linomide as well as the presence of certain functionalities. Here, in this paper, we report the significance of N-1, C-3, and C-6 of the quinoline-2-one nucleus.
Upon performing docking studies on all the synthesized compounds, it was revealed that the -N atom at position one plays a very important role in H-bonding interactions. As all our analogs possessed a hydrogen atom bonded to N-1 of the quinoline-2-one nucleus, all the compounds synthesized showed two hydrogen-bond interactions with Thr159 and Asp160 present at the active site of EGFRK. This is illustrated in Figure 3 with compound 4b, the most potent of the series that showed a MolDock score of -110.2253 which is comparable to that of the standard 4-AQ with a MolDock score of -112.04.
Presence of bulky heterocyclic ring systems at C-3 seems to improve the efficacy and anticancer activity. We observed that analogs possessing the 6-membered morpholine ring system or the piperidine ring system showed better docking scores that were comparable with the standard while analogs bearing a substitution with a 5-membered ring system such as pyrrolidine at C-3 showed diminished MolDock scores. These scores coupled with our results obtained when the analogs were evaluated using the MTT assay reveal that a 6-membered ring system offers better anticancer activity than a 5-membered system.
To determine the significance of the C-6 position of the quinoline-2-one nucleus, we synthesized analogs bearing electron withdrawing groups and electron donating groups. We observed that a substitution of -H or -Cl at C-6 rendered analogs that showed superior MolDock scores and possessed better anticancer activity. However, substitution with -F or -CH3 showed inferior in silico and MTT assay results.
Methodology
Chemicals and Apparatus
Reagents and solvents used for experimentation were of laboratory grade (SD Fine- ChemLimited, Mumbai; Molychem, Mumbai, India). Reaction monitoring was per-formed using thin-layer chromatography (TLC) with pre-coated Silica gel-G plates. Purification was performed through recrystallization. Thiele’s melting point apparatus was used to determine the melting points of all synthesized compounds. FT-IR spectra of all derivatives were recorded with Shimadzu IR AFFINITY-1 spectrophotometer using the KBr pellet technique. 1H NMR and 13C spectra data was obtained on Bruker Advance II 400 NMR Spectrometer using CDCl3 or DMSO-d6 as a solvent. Chemical shifts are expressed as δ (ppm) values.
Experimental
General Procedure for the Synthesis of 2a-d
The dianilide [1a-d] (1 mol) was placed in a round bottom flask and treated with 10 times (by weight) of polyphosphoric acid. The resultant viscous material was heated at 130–140°C for 6 h, followed by pouring on to crushed ice which was made alkaline with 4N NaOH leading to formation of precipitate which was filtered off. The filtrate was washed with diethyl ether (3 × 5 ml). The aqueous phase was collected and acidified with 6N HCl to pH one in an ice bath, resulting in the precipitation of the substituted four- hydroxyquinolin-2(1H)-one product (2a-d). The product was filtered, washed with water, and dried in a desiccator over P2O5 for 24 h. This product was used without further purification.
General Procedure for the Synthesis of 3a-d, 4a-d, 5a-d
The solution of secondary amine (1 mmol) and formaldehyde (10 mmol) was stirred for 5 min, and a solution of compound [2a-d] (0.01 mmol) in methanol was added and refluxed for 4–5 h. The solution was then kept overnight to precipitate the product, filtered using suction, and recrystallized using ethanol. Upon treatment of 2a-d with secondary amines and formaldehyde (Mannich reaction), Mannich bases 3a-d, 4a-d, 5a-d were formed.
4-Hydroxyquinolin-2(1H)-One (2a)
C9H7NO2. Yield: 31.57%. m. p.: >300°C. FT-IR (KBr, cm−1): 3093.82 (-NH), 2953.02 (aromatic, -CH), 2906.73 (aromatic, -CH), 2860.43(aromatic, -CH) and 2821.86 (aromatic, -CH), 1662.64 and 1633.71 (-C=O). 1H NMR (DMSO-d6, δ ppm): 11.1817 (s, 1H, -OH), 8.1777 (s, 1H, -NH), 7.0975-7.8063 (m, 4H, -Ar), 5.7947 (s, 1H, -CH).
6-Chloro-4-Hydroxyquinolin-2(1H)-One (2b)
C9H6NO2Cl. Yield: 25.61%. m. p.: >300°C. FT-IR (KBr, cm−1): 3250.50 (-NH), 3122.75, 3100.50 (-Aromatic–CH), 2933.73 (-CH), 2854.65 (-CH), 1649.14 (-C=O), 1610.56 (-C=O). 1H NMR (DMSO-d6, δ ppm): 11.8215 (s, 1H, -OH), 11.3188 (s, 1H, -NH), 7.2655-7.7340 (m, 3H, -Ar), 5.8117 (s, 1H, -CH). 13C (DMSO-d6, δ ppm): 163.32 (1C, -C=O), 161.44 (1C, -C-OH), 137.87 (1C, -C=C- of aromatic ring) 130.65 (1C, -C-Cl aromatic ring), 125.06 (1C, -C=CH aromatic ring), 121.72 (1C, -C=CH aromatic ring), 117.08 (1C, -C=C- aromatic ring), 116.36 (1C, -C=CH aromatic ring), 99.11 (1C, -C=C).
6-Fluoro-4-Hydroxyquinolin-2(1H)-One (2c)
C9H6NO2F. Yield: 25.42%. m. p.: >300°C. IR (KBr, cm−1): 3088.03 (Aromatic–CH), 2947.23(-CH), 2899.01 (-CH), 1658.78(-C=O), 1604.77 (-C=O). 1H NMR (DMSO-d6, δ ppm): 11.7305 (s, 1H, -OH), 11.2719 (s, 1H, -NH), 8.2135 (s, 1H, -Ar), 7.2744-7.4805 (m, 2H, -Ar), 5.8015 (s, 1H, -CH). 13C (DMSO-d6, δ ppm): 163.26 (1C, -C=O), 161.62, 161.60 (1C, -C-OH), 157.88 (1C, -C=C- of aromatic ring), 135.83 (1C, -C-F of aromatic ring), 117.58 (1C, -C=CH of aromatic ring), 114.05 (1C, -C=CH of aromatic ring), 113.00 (1C, -C=C- of aromatic ring), 111.06 (1C, -C=CH of aromatic ring), 99.06 (1C, -C=C).
6-Methyl-4-Hydroxyquinolin-2(1H)-One (2d)
C10H9NO2. Yield: 30.09%. m. p: >300°C. IR (KBr, cm−1): 3138.18 (Aromatic–CH), 3024.38 (Aromatic–CH) 2976.16 (-CH), 2920.23 (-CH), 2862.36 (-CH), 1670.35(-C=O), 1608.63 (-C=O).
4-Hydroxy-3-(Morpholinomethyl) Quinolin-2(1H)-One (3a)
C14H16N2O3. Yield: 83.10%. m. p.: >300°C. IR (KBr, cm−1): 3078.39 (Aromatic–CH), 2943.37 (-Aliphatic–CH), 2872.01 (-Aliphatic–CH), 1656.85 (-C=O), 1606.70 (-C=O). 1H NMR (DMSO-d6, δ ppm): 12.7076 (s, 1H, -OH), 12.0977 (s, 1H, -NH), 7.2234-7.9370 (m, 4H, -Ar), 3.8284 (s, 2H, -CH2), 2.5167- 2.5344 (m, 8H, -morpholine). 13C (DMSO-d6, δ ppm): 166.52 (1C, -C=O), 160.53 (1C, -C-OH), 136.82 (1C, -C=C-), 130.88 (1C, -CH = CH of aromatic ring), 122.86, 122.84 (1C, -CH = CH of aromatic ring), 115.98, 115.96 (2C, -C-CH, -C=C- of aromatic ring), 109.15 (1C, -C=C- of aromatic ring), 70.04, 70.02 (2C, -CH2, morpholine), 62.98, 62.96 (2C, -CH2, morpholine), 55.97 (1C, -CH2).
6-Chloro-4-Hydroxy-3-(Morpholinomethyl) Quinolin-2(1H)-One (3b)
C14H15N2O3Cl. Yield: 86.47%. m. p.: >300°C. IR (KBr, cm−1): 3093.82 (Aromatic–CH) 2997.38(Aliphatic–CH), 2956.87(Aliphatic–CH), 2808.36(Aliphatic–CH), 2644.41 (Aliphatic–CH), 1654.92(-C=O)., 1606.70 (-C=O).1H NMR (DMSO-d6, δ ppm): 12.6207 (s, 1H, -OH), 12.2501 (s, 1H, -NH), 7.3891-7.8623 (m, 3H, -Ar), 3.2204 (s, 2H, -CH2), 2.4984-2.5166 (t, 8H, -morpholine).
6-Fluoro-4-Hydroxy-3-(Morpholinomethyl) Quinolin-2(1H)-One (3c)
C14H15N2O3F. Yield: 36.058%. m. p.: >300°C. IR (KBr, cm−1): 2954.95(-CH stretch), 2924.09(-CH stretch), 2852.72 (-CH stretch), 1658.78(-C=O), 1612.49 (-C=O).
4-Hydroxy-6-Methyl-3-(Morpholinomethyl) Quinolin-2(1H)-One (3d)
C15H18N2O3. Yield: 49.18%. m. p.: >300°C. IR (KBr, cm−1): 2954.95(-CH stretch), 2924.09(-CH stretch), 2854.65 (-CH stretch), 1656.85 (-C=O).
4-Hydroxy-3-(Piperidin-1-Ylmethyl) Quinolin-2(1H)-One (4a)
C15H18N2O2. Yield: 52.0%. m. p.: >300°C. IR (KBr, cm−1): 3076.46 (Aromatic–CH), 2941.44(Aliphatic–CH), 2906.73(Aliphatic–CH), 2872.01 (Aliphatic–CH), 1651.07(-C=O),1606.70 (-C=O). 1H NMR (DMSO-d6, δ ppm): 12.6865 (s, 1H, -OH), 12.0186 (s, 1H, -NH), 7.2037- 7.9460 (m, 4H, -Ar), 3.8852 (s, 2H, -CH2), 2.5381-2.5552 (m, 8H, CH2-piperidine), 2.2280-2.3151 (m, 2H, CH2 -piperidine). 13C (DMSO-d6, δ ppm): 163.26 (1C, -C=O), 159.70 (1C, -C-OH), 135.63 (1C, -C=CH of aromatic ring), 133.03 (1C, -C-CH of aromatic ring), 121.64, 121.61 (2C, -CH = CH of aromatic ring), 114.52, 114.53 (2C, -CH = CH of aromatic ring) 109.93 (1C, C=C), 63.04, 63.01 (2C, -CH2, piperidine), 55.0 (1C, -CH2), 27.02, 27.00 (2C, -CH2 piperidine), 21.61 (1C, -CH2 piperidine).
6-Chloro-4-Hydroxy-3-(Piperidin-1-Ylmethyl) Quinolin-2(1H)-One (4b)
C15H17N2O2Cl. Yield: 69.19%. m. p.: >300°C. IR (KBr, cm−1): 2954.95(-CH stretch), 2924.09(-CH stretch), 2854.65 (-CH stretch) 1658.78(-C=O), 1606.70 (-C=O). 1H NMR (DMSO-d6, δ ppm): 12.7102 (s, 1H, -OH), 12.0608 (s, 1H, -NH), 8.0805 (s, 1H, -Ar), 7.2822-7.3555 (m, 2H, -Ar), 4.5228 (s, 2H, -CH2), 2.8535-2.8880 (m, 4H, CH2-piperidine), 2.5424-2.7893 (m, 5H, CH2-piperidine), 2.4080-2.5386 (m, 1H, CH2-piperidine). 13C (DMSO-d6, δ ppm): 165.58 (1C, -C=O), 160.04 (1C, -C-OH), 140.17 (1C, -C=CH of aromatic ring), 130.11, 130.19 (2C, -C-CH of aromatic ring), 125.02, 125.15 (2C, -CH = CH of aromatic ring), 115.06 (1C, -CH = CH of aromatic ring) 99.17 (1C, C=C), 74.55, 74.57 (2C, -CH2, piperidine), 64.03 (1C, -CH2), 26.02, 26.09 (2C, -CH2 piperidine), 21.55 (1C, -CH2 piperidine).
6-Fluoro-4-Hydroxy-3-(Piperidin-1-Ylmethyl) Quinolin-2(1H)-One (4c)
C15H17N2O2F. Yield: 56.438%. m. p.: >300°C. IR (KBr, cm−1): 2956.87 (-CH stretch), 2924.09 (-CH stretch), 2854.65 (-CH stretch), 2821.86 (-CH stretch), 2644.41 (-CH stretch), 1658.78(-C=O), 1616.35 (-C=O).
4-Hydroxy-6-Methyl-3-(Piperidin-1-Ylmethyl) Quinolin-2(1H)-One (4d)
C16H20N2O2. Yield: 35.07%. m. p.: >300°C. IR (KBr, cm−1): 2954.95 (-CH stretch), 2922.16 (-CH stretch), 2854.65 (-CH stretch) 1656.85 (-C=O).
4-Hydroxy-3-(Pyrrolidin-1-Ylmethyl) Quinolin-2(1H)-One (5a)
C14H16N2O2. Yield: 70.0%. m. p.: >300°C. IR (KBr, cm−1): 3076.46 (Aromatic–CH stretch) 2941.44(-CH stretch), 2872.01(-CH stretch), 2752.42(-CH stretch), 2636.69 (-CH stretch), 1651.07(-C=O stretch), 1606.70 (-C=O stretch).
6-Chloro-4-Hydroxy-3-(Pyrrolidin-1-Ylmethyl) Quinolin-2(1H)-One (5b)
C14H15N2O2Cl. Yield: 67.91%. m. p.: >300°C. IR (KBr, cm−1): 2954.95(-CH stretch), 2924.09(-CH stretch), 2852.72 (-CH stretch), 1658.78(-C=O stretch), 1606.70 (-C=O stretch).
6-Fluoro-4-Hydroxy-3-(Pyrrolidin-1-Ylmethyl) Quinolin-2(1H)-One (5c)
C14H15N2O2F. Yield: 42.198%. m. p.: >300°C. IR (KBr, cm−1): 2954.95 (-CH stretch), 2924.09(-CH stretch), 2852.72 (-CH stretch), 1658.78 (-C=O), 1620.21 (-C=O).
4-Hydroxy-6-Methyl-3-(Pyrrolidin-1-Ylmethyl) Quinolin-2(1H)-One (5d)
C15H18N2O2. Yield: 30.189%. m. p.: >300°C. IR (KBr, cm−1): 2954.95(-CH stretch), 2924.09 (-CH stretch), 2854.65 (-CH stretch), 1658.78 (-C=O stretch), 1606.70 (-C=O stretch).
Conclusion
Twelve derivatives of Linomide with a modification at C-3 of the quinoline-2-one nucleus were designed, synthesized, and characterized by FT-IR, 1H-NMR, and 13C-NMR. Anticancer activity of the final compounds was evaluated using the MTT Assay. Docking studies using the EGFRK binding site revealed compound 4b containing a -Cl group at position six of the quinoline-2-one nucleus along with a bulky piperidine group at position three of the quinoline-2-one nucleus, was the most active within the series, with a MolDock score of -110.2253 that was comparable to that of the standard inhibitor 4-AQ with a MolDock score of -112.04. Moreover, it also showed the most cytotoxicity with IC50 values of 1.539 μM/ml and 1.732 μM/ml against A549 and K562 cell lines, while all the synthesized compounds were found to be less active than the reference standard paclitaxel (IC50 = 0.3 μM/ml). Collectively, as our in silico and in vitro results agree with each other, we conclude that Linomide and the C-3 modified derivatives reported in this paper interact with the EGFRK binding site in a similar manner as that of 4-AQ. Future in vivo studies and investigations will therefore allow for the deeper understanding of the essential pharmacophoric features that are required for inhibiting EGFRK and will thus lead to the development of promising pharmacologically active anticancer therapeutics.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author Contributions
Conceptualization: GABeS, TB, SM, ZA, AMH, and SC. Methodology: GABeS and ZA. Investigation: GABeS, TB, SM, AMH, and ZA. Writing and reviewing: GABeS, TB, and SM. Supervision: SM, ZA, and SC.
Funding
The APC fees for this manuscript were supported by University of Oradea, Romania.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would sincerely like to thank Dr. Kishore Bhat, Professor and Head, Department of Microbiology, Maratha Mandal’s NGH Institute of Dental Sciences & Research Centre, Belgaum, Karnataka, India for providing the facilities and technologies needed to perform the biological assays. We would also like to appreciate Dr. Mahesh Palkar, Department of Pharmaceutical Chemistry, KLEU’s College of Pharmacy, Hubli, Karnataka, India for assisting with the docking studies of the synthesized derivatives and the authorities of Sophisticated Analytical Instrumentation Facility, Panjab University, Chandigarh, India for assisting in the characterization of our derivatives. The graphical abstract of this paper was created using Biorender.com while chemical structures and schemes were drawn using ChemDraw.
References
Gaude, T. T., Soares, G. A. B. e., Priolkar, R. N. S., Biradar, B., and Mamledesai, S. (2017). Synthesis of 4-Hydroxy-1-(phenyl/methyl)-3-[3-(substituted Amino)-2nitropropanoyl]quinolin-2(1h)-Ones as Antimicrobial and Antitubercular Agents. Indian J. Heterocycl. Chem. 27 (Jul-Sep 2017), 223–228.
Islam, M. R., Islam, F., Nafady, M. H., Akter, M., Mitra, S., Das, R., et al. (2022). Natural Small Molecules in Breast Cancer Treatment: Understandings from a Therapeutic Viewpoint. Molecules 27, 2165. doi:10.3390/molecules27072165
Ma, C., Taghour, M. S., Belal, A., Mehany, A. B. M., Mostafa, N., Nabeeh, A., et al. (2021). Design and Synthesis of New Quinoxaline Derivatives as Potential Histone Deacetylase Inhibitors Targeting Hepatocellular Carcinoma: In Silico, In Vitro, and SAR Studies. Front. Chem. 9, 725135. doi:10.3389/fchem.2021.725135
Maeda, H., and Khatami, M. (2018). Analyses of Repeated Failures in Cancer Therapy for Solid Tumors: Poor Tumor-Selective Drug Delivery, Low Therapeutic Efficacy and Unsustainable Costs. Clin. Transl. Med. 7 (1), 11. doi:10.1186/s40169-018-0185-6
Mokrov, G. V., Litvinova, S. A., Voronina, T. A., Nerobkova, L. N., Kutepova, I. S., Kovalev, I. G., et al. (2019). Design, Synthesis, and Anticonvulsant Evaluation of 4-GABA-3-Nitrocoumarines, 1-thiocoumarines, Quinolone-2-Ones, and Their Derivatives. Med. Chem. Res. 28, 1901–1911. doi:10.1007/s00044-019-02422-5
Priolkar, R. N. S., Shingade, S., Palkar, M., and Desai, S. M. (2020). Design, Synthesis, and Characterization of Novel Linomide Analogues and Their Evaluation for Anticancer Activity. Curr. Drug Discov. Technol. 17 (2), 203–212. doi:10.2174/1570163815666181008151037
Rewcastle, G. W., Murray, D. K., Elliott, W. L., Fry, D. W., Howard, C. T., Nelson, J. M., et al. (1998). Tyrosine Kinase Inhibitors. 14. Structure-Activity Relationships for Methylamino-Substituted Derivatives of 4-[(3-Bromophenyl)amino]-6-(methylamino)-Pyrido[3,4-D]pyrimidine (PD 158780), a Potent and Specific Inhibitor of the Tyrosine Kinase Activity of Receptors for the EGF Family of Growth Factors. J. Med. Chem. 41 (5), 742–751. doi:10.1021/jm970641d
WHO (2021). Cancer [Online]. World Health Organization. Available at: https://www.who.int/news-room/fact-sheets/detail/cancer (Accessed December 24, 2021).
Yang, Y., Meadows, S., Li, P.-K., Davidorf, F. H., and Abdel-Rahman, M. H. (2005). Anti–Angiogenic Effect of Linomide Analogue. Investigative Ophthalmol. Vis. Sci. 46 (13), 4166.
Keywords: Mannich reaction, molecular docking, quinolin-2-ones, Linomide, anticancer
Citation: Borges e Soares GA, Bhattacharya T, Mamledesai S, Ai Z, Hasan AM and Cavalu S (2022) Identification of Linomide Derivatives as Potential Anticancer Therapeutics using Molecular Docking Studies. Front. Pharmacol. 13:892914. doi: 10.3389/fphar.2022.892914
Received: 09 March 2022; Accepted: 20 May 2022;
Published: 16 June 2022.
Edited by:
Christian Celia, University of Studies G. d'Annunzio Chieti and Pescara, ItalyReviewed by:
Mohamed A. Abdelgawad, Al Jouf University, Saudi ArabiaMuhammad Wahab Amjad, University of Pittsburgh, United States
Copyright © 2022 Borges e Soares, Bhattacharya, Mamledesai, Ai, Hasan and Cavalu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanima Bhattacharya, YnRhbmltYTE5ODdAZ21haWwuY29t; Simona Cavalu, c2ltb25hLmNhdmFsdUBnbWFpbC5jb20=
†These authors have contributed equally to this work